
Introduction to Electronic Structure Calculations Electron Correlation
12
Introduction to Electronic Structure Calculations Electron Correlation
-
Upload
kai-rosario -
Category
Documents
-
view
47 -
download
0
description
Introduction to Electronic Structure Calculations Electron Correlation. Molecular properties. Transition States Reaction coords. Ab initio electronic structure theory Hartree-Fock (HF) Electron Correlation (MP2, CI, CC, etc.). Spectroscopic observables. Geometry prediction. - PowerPoint PPT Presentation
Transcript of Introduction to Electronic Structure Calculations Electron Correlation

Introduction to Electronic Structure Calculations
Electron Correlation
Introduction to Electronic Structure Calculations
Electron Correlation

Ab initio electronic structure theoryHartree-Fock (HF)
Electron Correlation (MP2, CI, CC, etc.)
Molecular properties
Geometry prediction
Benchmarks for parameterization
Transition StatesReaction coords.
Spectroscopicobservables
ProddingExperimentalists
Goal: Insight into chemical phenomena.

The ProblemThe Problem
Vij V (rij )qiq j40rij
qiq jrij
rij
qi
qj
eeee
N
i
N
ij ji
N
i
M
A Ai
AN
ii
ee
eeneee
eenennen
EH
rr
q
Rr
qZm
H
constVVTH
VVVTTH
ˆ
2ˆ
.ˆˆˆˆ
ˆˆˆˆˆˆ
1
2
1 11
22
Moleküle bestehen aus Kernen undElektronen jeweils mit elektrischer Ladung qi.Der totale Hamiltonoperator (Energie) ist die Summe aller kinetischer und potentieller Energien. Da die Kerne viel schwerer als die Elektronen sind kann i.a. die Born-Oppenheimer Näherung angewendet werden. Daraus ergibt sich der elektronische Hamiltonoperator oder „das elektronische Problem“.

BisherBisher3. Semester:• Hartree-Fock Näherung; Lösung des elektronischen Problems ohne Korrelation.• Basissätze
4. Semester:• Behandlung von Metallen mittels effective core Potentials.
Formal:
Lösung durch eine Slater-Determinante
Dabei sind die cAcA ionische Terme (beide Elektronen beim Kern „A“ resp. „B“) und cAcB kovalente Terme. In dieser Behandlung sind ionische und kovalente Terme gleich wichtig – im Gegensatz zur Erwartung ionische Terme sollten geringer sein.
HEEH
zyxzyxrEH NNN
ˆˆ
,,,..,,,;ˆ111
ABBABBAABABA
2211
21212121
122122
11
0
11110
111111
110

NeuNeuAusser der Slater-Determinante gibt es noch viele angeregte Zustände. Werden diese „beigemischt“ so werden die ionischen und kovalenten Terme besser balanciert.
Zum Beispiel
Dieser Ansatz wird als Konfigurations-Wechselwirkung (Configuration Interaction) bezeichnet. Dem Grundzustand werden etwa 1% höher angeregte Zustände beigemischt.
Je nach Ansatz werden verschiedene Grade der Anregung betrachtet:
CI Full CI selten verwendet; sehr viele Anregungen; konvergiert langsamMP2 Moller Plesset Störungstheorie; Einfach- und zweifachanregungenMP4 MP4(SDQ) enthält alle Einfach- zweifach- und vierfachanregungen; MP4(SDTQ) auch dreifachCCSD Coupled cluster; ähnlich wie MP – konvergiert aber besser.
Rechenaufwand:SCF n4
MP2 n5
MP4 n6
CCSD(T) n7
cov202200cov2
cov0
2
~~
2211
cccc ionion
ion
ABBABBAABABA
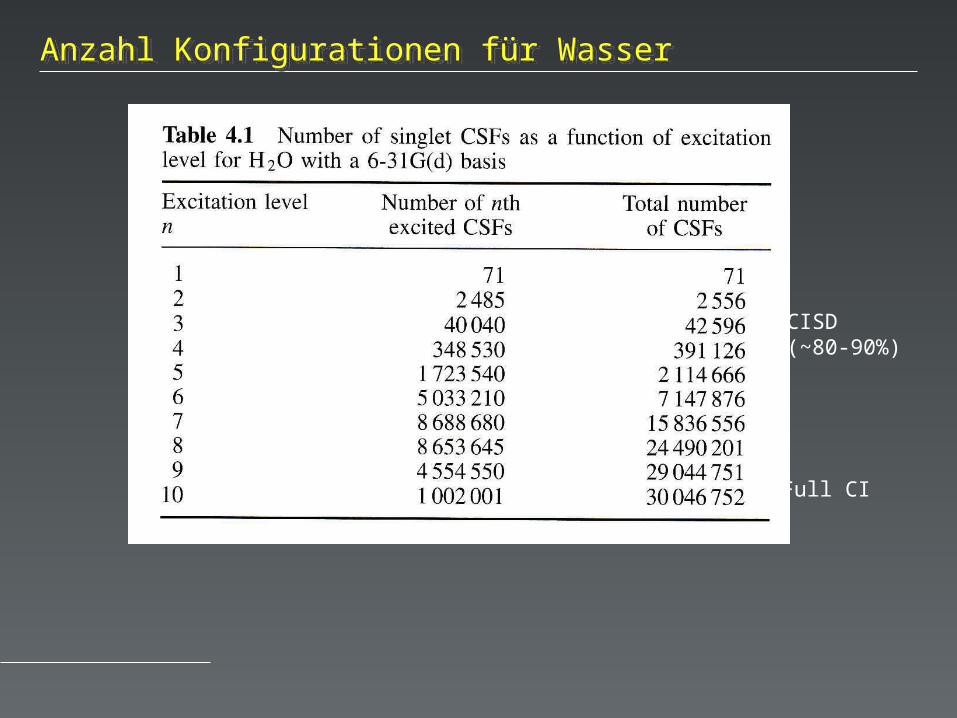
Full CI
(19 basis functions)
CISD(~80-90%)
Anzahl Konfigurationen für WasserAnzahl Konfigurationen für Wasser

Andere BetrachtungsweiseAndere BetrachtungsweiseHartree-Fock (HF) – oder SCF – Methode geht von unabhängigen Elektronen im gemittelten Feld aller andern Elektronen aus. Diese Lösungen sind jedoch nicht Eigenfunktionen des wahren Hamiltonoperators.
Die Differenz zwischen der exakten und genäherten Energie wird als Korrelationsenergie bezeichnet:
Dies ist eine Definition und nicht eine „Vorschrift“ wie die Korrelationsenergie zu berechnen ist.
Die HF Methode besteht darin die „beste WF“ mittels eines Variationsprinzips zu finden.
Da sie jedoch nur eine Elektronenkonfiguration enthält, vernachlässigt sie die Elektronenkorrelation.
Diese wird dann mit Hilfe von post-HF Rechnungen bestimmt.
Die Korrelationsenergie macht i.a. weniger als 1% der Gesamtenergie eines Systems aus. Sie ist jedoch für die meisten wichtigen chemischen Effekte wie zwischenmolekulare WW, Polarisierbarkeit oder Uebergangszustände (chem. Reaktionen) verantwortlich.
HFexactcorr EEE

Nachteil von ab initio Rechnungen ist ihre Skalierung mit der Anzahl Elektronen (s.o.)
Deshalb sind solche Methoden nur auf Systeme mit weniger als etwa 1000 Elektronen anwendbar.
Für Peptide, Protein, etc. gelangen deshalb Kraftfelder zur Anwendung. Mit ihnen lässt sich auch die nukleare Dynamik charakterisieren. Kraftfelder sind “Federmodelle” chemischer Verbindungen (siehe Beilage).
Im Praktikumsversuch wenden Sie Kraftfelder auf das Strecken von deka-Alanin an. Dabei geht es darum, ein force-extension Mikroskop zu simulieren. Damit lassen sich Rückschlüsse auf die Sekundärstruktur einer Aminosäuresequenz ziehen.
Details zu Kraftfeldrechnungen werden in der Vertiefungsvorlesung (VTV) “Computational Chemistry” vermittelt.
Kraftfelder: Alternativer Ansatz für chemische SystemeKraftfelder: Alternativer Ansatz für chemische Systeme

Big breakthrough making FC simulations practical:reactive force fields based on QMDescribes: chemistry,charge transfer, etc. For metals, oxides, organics.
Accurate calculations for bulk phases and moleculesChemical Reactions (P-450 oxidation)
time
distance
hours
millisec
nanosec
picosec
femtosec
Å nm micron mm m
MESO
Continuum(FEM)
QM
MD
ELECTRONS MOLECULES GRAINS GRIDS
Deformation and FailureProtein Structure and Function
Micromechanical modelingProtein clusters
simulations real devices full cell (systems biology)
To connect 1st Principles (QM) to Macro work use an overlapping hierarchy of methods (paradigms) (fine scale to coarse) so that parameters of coarse level
are determined by fine scale calculations. Thus all simulations are first-principles based
Motivation: Physics, Chemistry, Biology, MaterialsMotivation: Physics, Chemistry, Biology, Materials
CompChem 2011
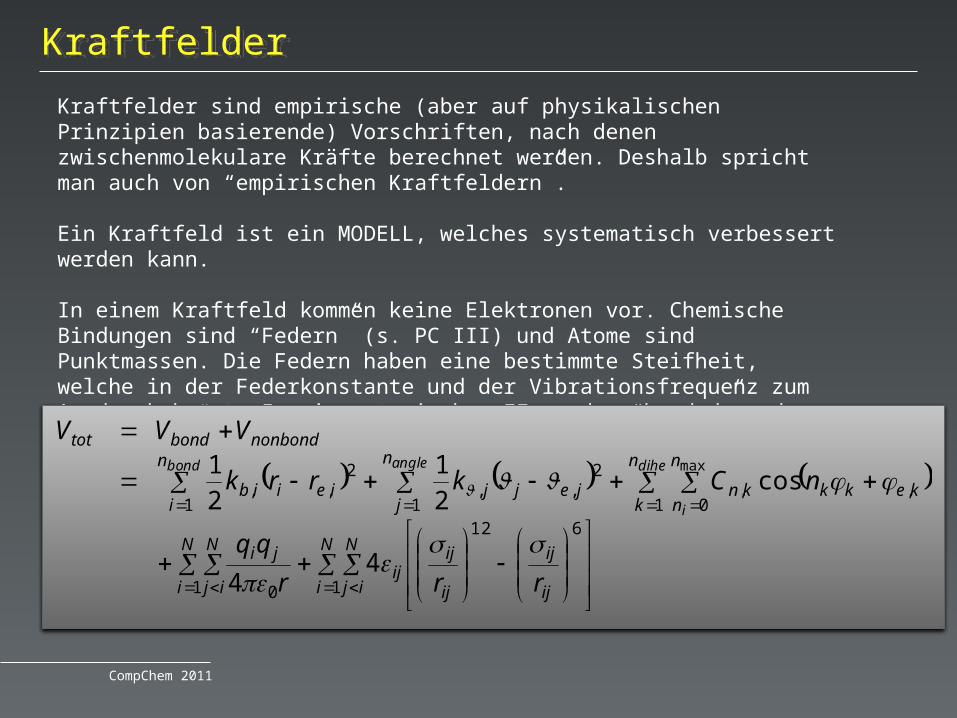
KraftfelderKraftfelder
Kraftfelder sind empirische (aber auf physikalischen Prinzipien basierende) Vorschriften, nach denen zwischenmolekulare Kräfte berechnet werden. Deshalb spricht man auch von “empirischen Kraftfeldern”.
Ein Kraftfeld ist ein MODELL, welches systematisch verbessert werden kann.
In einem Kraftfeld kommen keine Elektronen vor. Chemische Bindungen sind “Federn” (s. PC III) und Atome sind Punktmassen. Die Federn haben eine bestimmte Steifheit, welche in der Federkonstante und der Vibrationsfrequenz zum Ausdruck kommt. In einem typischen FF werden “bonded” und “nonbonded” Wechselwirkungen unterschieden.
N
i
N
ij
N
i
N
ij ij
ij
ij
ijij
ji
n
k
n
nkekkkn
n
jjejj
n
iieiib
nonbondbondtot
rrr
nCkrrk
VVVdihe
i
anglebond
1 1
612
0
1 0,,
1
2,,
1
2,,
44
cos21
21 max
CompChem 2011

Molecular DynamicsMolecular Dynamics
Die Dynamik der Atome wird durch die Newton’schen Bewegungsgleichungen beschrieben. Dabei ist V(x) das Kraftfeld.
Für N Teilchen “i” bedeutet dies:
CompChem 2011
dx
xdVtxmF
i
ii dx
xdVtxm

Molecular Dynamics: The Verlet MethodMolecular Dynamics: The Verlet MethodDer Verlet Algorithmus ist einer der bewährtesten Ansätze, um die Newton’schen Bewegungsgleichungen zu lösen:
Das Resultat einer solchen Molekulardynamik Simulation ist eine Trajektorie x(t), welche die Position jedes Teilchens zu jedem Zeitpunkt beschreibt. Die Statistische Mechanik (PC IV) beschäftigt sich damit, aus einer Trajektorie physikalisch und chemisch relevante Information (z. B. freie Energien, Diffusionskoeffizienten, etc.) zu berechnen.
CompChem 2011
22
2
2
2
22
2
2
12
1
ttattxtxttxttxtxttx
ttxtxttxttx
ttxttxtxttx
ttxttxtxttx


















