
Intravenous induction agents
91
Dr. Deepali Jamgade Dr.Pradeepa Chevala Guided by: DR . S MANIMALA RAO
-
Upload
deepali-jamgade -
Category
Health & Medicine
-
view
98 -
download
0
Transcript of Intravenous induction agents

Dr. Deepali Jamgade
Dr.Pradeepa Chevala
Guided by: DR . S MANIMALA RAO

1872
•PIERRE-CYPRIEN ORE’ (French Surgeon) used chloral hydrate
1864
•ADOLF VON BAYER – On Saint Barbara’s Day discovered barbituricacid. Coined the term Barbitute as acombination of Barbara and urea. (no sedative properties)
•1n 1903 – Emil fischer discovered hexobarbital. Helmut Weesestudied it
1934
•John Lundy of clinic of mayo studied sod .thiopental – balanced anesthesia
• In 1941 – pearl harbour – many deaths with use of STP due to cardiovascular depressant effects.

1956 - Replacements like
• HYDROXYDIONE (steroid; thrombophlebitis)
• ALTHESIN (mixture of alphaxolone and alphadolone; has rapid onset and recovery; rejected because of hypersensitivty reactions)
• PROPANIDID (solubilized in Cremephor EL; rejected because of hypersensitivity reactions)
In 1973 – Etomidate – minimal hemodynamic depression . Used use in pts with severe CVS disease
1962 – 1978
• Ketamine – unique as it does not cause CVS depression but post op hallnications are present
• Benzodiazepines were studied for anxiolysis without same degree of sedation as thiopentone ;1963 – diazepam 1978 - midazolam
1977
• Propofol was discovered
• Alkylphenol compound with antiemetic properties and depressionof laryngeal reflexes so easy placement of supraglottic airways



What are i.v. induction agent ?
Agent cause a rapid reversible loss of consciousness.
TIME :- “one arm-brain (A→B)circulation time” this time
also depend on cardiac output and ejection fraction .
Normal A → B circulation time is 15-20 sec.
They are used:
To induce anaesthesia prior to other drugs being given to
maintain anaesthesia.
To maintain anaesthesia for longer procedures by
intravenous infusion. To provide sedation. Use for day care
/ short / opd procedure

Drug conc. In CNS decreased and patient becomes awake (due to redistribution of drug)
Repeat intermittent bolus Continuous sedation required
Unbound high lipid soluble and unionized drug crosses BBB quickly
Drug conc. Increases in VRG organs
Slowly sec. uptake by diffussionto other tissue
Drug injected in vein reaches the blood
Some % bind to protein Rest % of drug free/unbound



Several factors :-
Route of administration
Age ( ↓ with age )
Lean body mass ( fat free ) (muscular > fatty
)
↓ in low cardiac output state( body
compensate to accordingly to maintain
cerebral perfusion )
↓ in Hypoprotenemia ( nutritional,
nephropathy , PIH )

Rapid onset and offset
Analgesia at subanesthetic dose
Minimal cardio respiratory depression
No emetic effect
No excitatory and emergence phenomenon
No interaction with N-M blocking agent
No pain on injection
No venous sequel ( venous thrombosis)
No toxic effect on other organs
No release of histamine ( bronchospasm )
Water soluble formulation and long self life
No hypersensitivity
No stimulation of porphyria
No adrenocortical suppression

GABAA RECEPTOR
HIGHEST NUMBER IN OLFACTORY BULB, CEREBRAL CORTEX, CEREBELLUM,
HIPPOCAMPUS, SUBSTANTIA NIGRA, INF. COLLICULUS
LOWER DENSITY IN STRIATUM , LOWER BRAIN STEM AND SPINS
ALPHA 1 – CAUSES
Sedation ,
anterograde amnesia
, and anticonvulsant
properties.
ALPHA 2 – causes
anxiolysis ms.
Relaxant

Major inhibitory Neuro-transmitter in the CNS
= GABA
Active GABA A receptor => Cl - influx =>
Hyperpolarisation
Propofol & barbiturates slow GABA A receptor
dissociation
Benzodiazepines increase GABA A to receptor
coupling
Ketamine acts at NMDA receptor
These effects lead to sedative & hypnotic
effects

Increasing dose => sedation => hypnosis
All iv anesthetics affect other organ systems
Potential for respiratory depression
Potential for CVS depression
Potential for altered CBF/ICP
Hypovolemia => severe hemodynamic effects
seen due to decreased blood pool
Use lower doses!

Rapid onset and rapid offset , no excitatory effect .
Yellow amorphous powder , in atmosphere of nitrogen
Ultra short acting barbiturate.
@C5
-aryl or alkyl group (hypnotic)
-phenyl gr. (anti convulsant)
@ C2
-O2 (oxybarb.)
-Sulfur (thiobarbiturate)
@C1-
Replacement of oxygen at C2 with sulphur.
Diluted to 2.5 % solution ,can be stored for 48 hr ↓refrigerator , Concentration >5 % cause pain
Highly Alkaline pH 10.5, contain 6 % NaHCO3
↓ in alkalinity cause ppt of solution , so avoid to dilute in acidic solution , RL .
Co-adm of vec, atra , midaz , alfentanilform ppt in I.V. line and occlude the vein

Sedation & Hypnosis by interaction with
inhibitory neurotransmitters GABA on GABAA
receptor.
GABA facilitatory & GABA mimetic action.
GABAA receptor has 5 glycoprotein sub unit.
Increases GABA mediated transmembrane conductance
of Cl– ion, Causes hyperpolarization & inhibition of
post synaptic neuron.

At low concentrations, barbiturates enhance the effects of GABA,
decreasing the rate of dissociation of GABA from its receptor–
sedative-hypnotic effects of the barbiturates.
At higher concentrations, the barbiturates directly activate the
chloride channels without the binding of GABA, acting as the
agonist itself.
The GABA-mimetic effect at slightly higher concentrations may
be responsible for what is termed barbiturate anesthesia.

Onset of action of i.v. injection - 10-20 sec.
peak 30-40 sec. duration for awakening 5-15 min.
Prompt awakening after single i.v. inj. is due to rapid redistribution to lean body tissue (muscle)
Volume of distribution is 2.5 Lit. per Kg.
Ultimate elimination due to hepatic metabolism.
Effect site equilibration time is rapid.
Brain – 30 Sec. Muscle – 15 Min. Fat > 30 Min.
Context sensitive half life is prolonged.

Excretion-< 1% excreted unchanged in the urine
Volume of distribution – 2.5L/Kg
Rapid distribution half life – 8.5 Min
Slow distribution half life – 62.7 Min
Elimination Half Life 11.6 Hours
Clearance(3.4ml/kg/min)
Prolonged in obese patient,elderly,pregnancy.
Short in paediatric patient.

1) Redistribution
- Lipid solubility (most important factor)
High Lipid Solubility makes it to cross blood brain
barrier & lean body tissue rapidly.
- Protein Binding
Highly bound to albumin & other plasma protein 72 – 86% Binding.
Only unbound fraction crosses Blood-Brain-Barrier.
-Ionization
Only non-ionized part crosses BBB.
Thiopentone has pKA 7.6 so 61% of it is non-ionized at physiologic PH
-Conc. Gradient between CSF and Plasma

2)Metabolism
By liver microsomal enzymes mainly, Slightly in CNS & kidney. 10
– 24 % Metabolised each hour.
OXIDATION of Aryl, Alkyl or Phenyl moeity @ C5
N-DEALKYLATION
DESULFURATION
DESTRUCTION of Barbituric ring
Conjugation with glucoronide to hydroxythiopentol & carboxylic
acid derivatives to form water soluble metabolites.
Excreted in urine

Act on GABA a receptor lead to Cl influx –hyperpolarisation of cell membrane -↑threshold of excitability of post synaptic neuron .
This is highly lipid soluble drug cross BBB –fast onset of axon
AT plasma pH around 50 % unionized drug ; in acidosis condition unionized % ↑; dose requirement ↓
Dose dependent↓ CBF, ↓ ICP, ↓ CMRO 2
CPP= (MAP-ICP ) but { ↓ICP > ↓MAP } ; CPP preserve

Sedation and loss of consciousness
retrograde amnesia and depression of vasomotor centre.
Induction and maintenance of anesthesia
Rate of adm α onset
Termination of effect take 5-10 min to awake ( after bolus )
Awakening depend on :-
Volume of distribution
Plasma concentration
Redistribution and Clearance
Alteration in metabolism
CNS sensitivity ↑ with age

Pupil and eye :- initially pupil contract but then dilate .
Pupillary response is lost with surgical anaesthesia .
loss of eyelash reflex is commonly used as endpoint for adequate induction dose .
Following traumatic brain injury, infusion of thiopental to produce a “barbiturate coma” lowers intracranial pressure and may improve neurological outcome.
Anticonvulsant property
Thiopental have no analgesic action and may be antianalgesic in low dose .
Burst suppression of EEG can be induced with high doses when used in treatment of status epilepticus or intractable rise in ICP following head injury .

Dose related resp depression ,peak resp depression after (1-1.5 min) after adm of bolus dose .
More susceptible patient ch lung disease , Airway obst
Apnea :- transient apnea for 25 sec only in 20 % cases.
Double apnea :- 1 st during adm of drug > transient >after 4-5 breath 2 nd apnea last for longer period .
during this period ventilation must be assessed –controlled ventilation .
↓minute ventilation , ↓ sensitivity to raised CO2
Airway reflexes preserved not suitable for LMA insertion ,may cause coughing and laryngospasm
C/I in St. asthmaticus

When choosing an induction agent, the
primary goals are as follows:
(1) to preserve maternal blood pressure,
cardiac output, and uterine blood flow;
(2) to minimize fetal and neonatal
depression;
(3) to ensure maternal hypnosis and
amnesia.

Umbilical blood and maternal blood concentration equal by the time of delivery
At this dose fetal brain suppression does not occur
Fetal CNS depression occur at >= 8mg/kg
Haemodynamic effect unlikely at this dose in normal pregnant women
Within 30 sec drug can be found in umbilical cord
Umbilical venous blood concentration peaks in 1 min
Thiopental <4 mg/kg; prompt,reliable induction
Reach in maternal blood, induce patient
Readily cross the placenta

First effect dose dependent peripheral vasodilatation
- ve inotropic effect - ↓ Ca to myocardial fiber
↓BP
↓ CO (↓venous return , vasodilatation, -veinotropic effect , ↓CNS symp outflow )
Tachycardia ( 10-36 %) Via baroreceptormediated symp reflex in response to ↓ CO & BP
CAD patient on induction ↑HR - ↑myocardial demand of O2
ECG changes :-prolonged QT , flattened T wave ,vent arrhythmia
eg - acidotic patient ,long term dialysis , Cirrhosis

Indications Induction of anaesthesia
Control convulsions
Decreased ICP
Neuroprotection
Contraindications ABSOLUTE COPD Severe asthama porphyria Previous hypersensitivity Allergy to sulphur
PRECAUTIONS : Stenotic valvular disease Severe hepatic disease Renal impairment

Anticonvulsant
for rapid control of status epilepticus
dose 0.5 – 2 mg/kg. repeated as needed
Treatment of increased intracranial pressure
CMRO2 by 55%, CBF
dose 1 – 4 mg/kg i.v.
CPP is maintained

ADVANTAGE :-
Rapid induction
Don’t sensitize myocardium to adrenaline
No nausea and vomiting
Other uses Anticonvulsant In psychiatric patient Narcoanalysis
DISADVANTAGE :-Pharyngeal and laryngeal reflex persist →apnea –controlled ventilation
Resp depression Hypotension
Poor analgesic and muscle relaxant
Gangrene and necrosis
Shivering and delirium

Stop injection immediately ,
leave the canula insitu , and dilute with
immediate inj of saline
Give intra-arterial inj of LA + vasodilator
Lidocaine 50 mg ( 5 ml of 1 %) + phenoxybenzamine( α blocker)0.5 mg bolus
or 50-200 µg/min infusion.
Consider systemic papaverine 40-80 mg
Consider sympathetic block ( brachial plexus
block or stellate ganglion block )
Start i.v heparin infusion
Give intra arterial hydrocort Postpone
surgery Consult vascular surgeon

•diazepam was synthesised by Sternbach in 1959
DIAZEPAM
•by Bell in 1961
OXAZEPAM,
•by Fryer and Walser in 1976
MIDAZOLAM
•existence of BZR was first discussed in Milan in 1971
•isolation and receptor-ligand interaction were demonstrated in 1977
•this has resulted in the generation of a number of new ligands and a specific antagonist
BENZODIAZEPINE RECEPTOR (BZR)

DIAZEPAM MIDAZOLAM
COMPOSITION milliliter of diazepam
solution (5 mg)
contains propylene
glycol 0.4 mL,
alcohol 0.1 mL,
benzyl alcohol
0.015 mL, and
sodium
benzoate/benzoic
acid in water for
injection
(pH 6.2 to 6.9)
solution (1 or
5 mg/mL) contains
0.8% sodium chloride
and 0.01% disodium
edetate,
with 1% benzyl
alcohol as a
preservative.
The pH is adjusted to
3 with hydrochloric
acid and sodium
hydroxide

DIAZEPAM MIDAZOLAM
MOLECULAR
WEIGHT
284.7 362
PKa 3.3 6.2
WATER SOLUBLE NO YES
LIPID SOLUBLE YES
HIGHLY LIPOPHILIC
YES
HIGHLY LIPOPHILIC
(DUE TO IMIDAZOLE
RING)

DIAZEPAM MIDAZOLAM
EQUIVALENT DOSE
(mg)
0.3 – 0.5 0.15 - 0.3
VOLUME OF
DISTRIBUTION
(L/KG)
1 - 1.5 0.3 – 0.5
PROTEIN BINDING 96 -98 % 96 -98 %
CLEARANCE
(ml/kg/min)
0.2 - 0.5 6 - 8

DIAZEPAM MIDAZOLAM
MECHANISM Oxidation of methylene
group of diazepine ring;
Finally glucuronidation of
metabolite
Oxidation at imidazole
ring;
And further glucuronidation
METABOLITES 1.Desmethyldiazepam
2.Oxazepam
3.temazepam
1. 1-hyddroxymidazolam
2. 4-hydroxymidazolam
ELIMINATION Kidney
(e.t1/2 21-37 hours)
Increased in cimetidine
use, old age , cirrhosis of
liver
Kidney
(e.t1/2 1-4 hours)
Increased in cimetidine
,erythromycin ,CC Blockers
, old age , cirrhosis of liver
(mainly by hepatic microsomal oxidation and glucoronide conjugation)


CNS EFFECTS RESPIRATORY EFFECTS
Dose dependent↓ CBF, ↓CMRO 2 (ratio maintained by midazolam)
Increase in seizure initiation threshold
20% - Anxiolysis 30-50% - sedation 60% - unconsciousness
SLEEP CYCLE-
alpha activity is decreased
increase in low voltage, fast activity, especially beta
the amplitude of somato-sensory EP's is reduced
"pre-anaesthetic" doses-↓alveolar ventilation
the peak onset of ventilatory depression following midazolam (0.15 -0.3 mg/kg) is at ~ 3 min and lasts for ~ 15 mins
in patients with obstructive pulmonary disease - respiratory depression, CO2 retention and narcosis
decreases the MAC of inhalational anaesthetics

in "pre-anaesthetic" doses they decrease the BP and increase HR
decrease peripheral resistance - flunitrazepam - midazolam
decrease LV work and cardiac output - diazepam – lorazepam
baroreceptor reflexes generally remain intact, though, there is
some depression
the hypotensive effect is minimal and usually less than that seen
with thiopentone
the effect is possibly slightly greater with midazolam and is dose
related
in patients with elevated cardiac filling pressures, both
midazolam and diazepam produce a "nitroglycerine like" effect,
reducing preload and increasing cardiac output
diazepam increases coronary blood flow in man, possibly by
increasing interstitial concentrations of adenosine

INTRAVENOUS SEDATION
ORAL SEDATION
INDUCTION OF ANAESTHESIA
DIAZEPAM MIDAZOLAM
INDUCTION 0.3 – 0.5 mg/kg
(Given in 5 to 15
sec; induction in
39 sec)
0.05 – 0.15 mg/kg
(Given in 5 to 15
sec; induction in
28 sec)
MAINTAINENCE 0.1 mg/kg 0.05 mg/kg
SEDATION 2 mg 0.5 - 1 mg

ADVANTAGES DISADVANTAGES
Better amnesia ( 1-2
hours)
Smoother
haemodynamic course
(in healthy patients)
Less opioid
requirement
Less dosage required
if used with opioids
Accumulate in blood
Prolonged arousal time compared to other I.vinduction agents
In hemodynamicallycompromised patients cvsdepression.
Dosage affected by age, sex , gender , obesity , enzyme induction , hepatic and renal diseases
Longer context sensitive half life.


Ketamine is a phencyclidine derivative
Rapid onset 30-60 sec ;
high lipid soluble ( 5× thiopental )
Hypnosis ,amnesia Dissociative anaesthesia , intense analgesic ( SOMATIC > VISCERAL ), ,rapid clearance
Cardio stimulation property
Minimal effect on resp system
Sympathomimetic effect
IOA choice for ASA – IV and hemodynamic compromised state the possibility of emergence delirium limits the clinical usefulness of ketamine.
Ketamine has advantages over Propofol and etomidate in being water soluble

NMDA Receptors antagonist :-
Opioid Receptors:-
Muscarinic Receptors:-
The fact that ketamine produces anticholinergicsymptoms (emergence delirium, Bronchodilation, sympathomimetics action) suggests that an antagonist effect of ketamine at muscarinicreceptors is more likely than an agonist effect.
Sodium Channels:- Consistent with its mild local anesthetic-like properties, ketamine interacts with voltage-gated sodium channels sharing a binding site with local anesthetics

Induction of general anesthesia 0.5-2 mg/kg IV;
4-6 mg/kg IM
Maintenance of general
anesthesia
0.5-1 mg/kg IV with N2O 50% in
O2
15-45 µg/kg/min IV with N2O 50-
70% in O2
30-90 µg/kg/min IV without N2O
Sedation and analgesia 0.2-0.8 mg/kg IV over 2-
3 min 2-4 mg/kg IM
Preemptive/preventive analgesia 0.15-0.25 mg/kg IV
Intra thecal ketamine 0.5-0.75 mg/kg

Emergence phenomenon (psychadelic effect) visual , auditory, propioceptive and confusional illusion ,delirium and cortical blindness
Bodily detachment / dissociative anesthesia
Preventive measures
LOC in 30-60 sec i.v and 2-4 min after i.m (end point Nystagmus in horizontal gaze)
Consciousness regain in (10-20)min
Full orientation in (60-90)min
Ketamine water soluble; consider dose dependending factor
1-2mg/kg i.v and 4-6 mg/kg i.m Apnea rare but can be there

↑CMRO2, ↑ ICP(d/t ↑symp tone ),↑IOT,↑ CBF (↑CBF> ↑CMRO2)
Dissociative anaesthesia ( cataleptic state )
Corneal , cough , swallow reflex +nt
Amnesia not prominent as compare with BZD ↑muscle tone , purposeless movement , Ө wave on EEG , petit mal type seizure activity in hippocampus
Primary site of axon in CNS thalamoneocorticalprojection system .
Depress cortical and thalamus function Stimulate limbic and hippocampal function

Associated with vivid dreaming , sense of floating of body, illusion , ext sensory experience , excitement , confusion , euphoria , fear .
Occur with ketamine due to depression of auditory and visual relay nuclei .
The loss of skin and musculoskeletal sensations results in a decreased ability to perceive gravity, thereby producing a sensation of bodily detachment or floating in space.
These feature last for 1 hr.
Factor affecting emergence reaction
Age ( adult > child )
Gender( female > male )
Dose (↑)
Concurrent drug ( BZD priming 5 min before ketamine )
Preop counseling

Sympathomimetic action ↑BP, ↑HR , ↑ CO
↑ SBP is 20 to 40 mm Hg, with a slightly increase in DBP, increases progressively during the first 3 to 5 minutes after an intravenous injection of ketamine and then decreases to predrug levels over the next 10 to 20 minutes.
↑ myocardial O2 demand – provided by adequate CO &↓ coronary vascular resistance .
These effect are more apparent in 1 st bolus dose than 2 nd dose .
Ketamine ↑ pul artery pressure – caution use in left side stenoticvalvular lesion .
Tachycardia and hypertension by ketamine can be prevented by premedication with BZD or continuous inhalational agent
Cautiously use in IHD
Useful in pt of cong heart Ds even in whom propensity for R-L shunt exist

Min effect on central resp drive
Transient (1-3 ) min ↓ in minute ventilation
Large dose produce apnea
Bronchial muscle relaxant { when given in patient of bronchospasm – pul compliance increased }
Bronchodilation make this a potentially useful drug for the rapid intravenous induction of anesthesia in patients with asthma.
Ketamine as effective as halothane
Resp problem in children are due to ↑ secretion ( salivation )-cause upper airway obstruction – laryngospasm
Increase pulmonayl artery pressure
Preserve cough and upper airway reflex so not useful with LMA

When choosing an induction agent, the
primary goals are as follows:
(1) to preserve maternal blood pressure,
cardiac output, and uterine blood flow;
(2) to minimize fetal and neonatal
depression; and
(3) to ensure maternal hypnosis and amnesia.

Fetal outcome same in both induction with ketamine or thio
Provide both analgesia and hypnosis,maternal awareness less as compared to thiopentone alone
Ketamine rapidly cross the placenta and at max conc in fetal blood in 1.5 to 2 mins
Large dose (>1 mg/kg) increased uterine tone
Ketamine excellent choice for i.v induction in LSCS at 1mg/kg
Rapid onset, sympathomimetic Best in hypovolemia and asthama patient

ADVANTAGE
increase HR,BP,CO
In asthmatic
For short procedure
Combination with BZD can use in cardiac catheterization and angiography .
In OPD surgical procedure
Good analgesic property
DISADVANTAGE
limb movement and Nystagmus
Emergence phenomenon in 50 %
Hypertensive
Increased ICP , IOT
Uterine stimulation
Schizophrenia , psychosis
Poor muscle relaxation

INDICATION
CVS except IHD and Resp. disorder
Hemodynamic compromised ( pericarditis , cardiac tamponade , CM , shock )
Traumatic and septic shock
As component in TIVA with midaz and propofol provide better hemodynamic stability
In cancer patient , neuropathy
Phantom or ischaemic limb pain
Fibromyalgia , visceral pain
Migraine
CONTRAINDICATION
↑ ICP , SOL brain
Large size Infarct
Ophthalmic injury
IHD
Vascular aneurysm
Schizophrenia

Most frequently use I.V. anaesthetic drug today
Milky white ;
pH 7.0 - 8.5 ;
isotonic to plasma
Fospropofol prodrug
Stable at room temp ;
Not light sensitive
Dilution :- water insoluble ;
Compatible in DNS Dilution cause cracking of emulsion , spontaneous degradation
Concern regarding microbial growth in emulsion – disodium edetate (0.005%) added to retard the bact. Growth
Sedation in and outside of OT
Concern regarding induction and emergence myoclonic ,jerk
Painful injection in small vessel take care of it

2,6 –di-isopropylphenol
Rapid onset 15-45 sec and offset , rapid offset even after prolonged infusion ( small context sensitive half time )
Metabolize in liver with Glucuronide and sulfateconjugation .
Extra hepatic metabolism + lung ; inactive metabolite Mainly excreted by kidney
Propofol causes the most marked fall in blood pressure of all the induction drugs. This is mainly due to systemic vasodilatation. There may be an accompanying slight increase in heart rate. The fall in blood pressure is dose dependent and is most marked in the elderly and in shocked patients. This can be minimized by slow injection – avoiding inadvertent overdose.

By the removal of Cremophor consists of 1%
(wt/ vol ) Propofol 10% soybean oil 2.25%
glycerol 1.2% purified egg phosphatide To
prevent microbial growth in the emulsion,
disodium edetate (0.005%) was added as a
retardant of bacterial growth.
If a dilute solution of propofol is required, it
is compatible with 5% dextrose in water.
Fospropofol (Aquavan)soon going to be
approved by FDA, a phosphorylated prodrug
of Propofol,

Clinical use Dose
Induction of general
anaesthesia
•1-2.5mg/kg,dose reduced with increasing
age,induction dose in 2 yr(2.9mg/kg),in 6-
12 yr(2.2 mg/kg)
Maintainance of
general anaesthesia
100-200mcg/kg/min without N2O & opiates
50-150mcg/kg/min with n2O & opiate
Sedation(with little
analgesic & amnesic)
25-75 mcg/kg/min I.V.,concious sedation
Antiemetic 10-20mg I.V.can repeat every 5-10 min
orstart 10mcg/kg/min infusion

Dose and therapeutic conc dependent action
Hypnotic action by enhancing GABA induced chloride current
Onset with 2.5 mg/kg 15-30 sec with peak effect in 90-100 sec.
Duration of hypnosis 5-10 min depending on redistribution and Vd
Subhypnotic dose – sedation and amnesia infusion @2mg/kg/hr
Propofol have shown direct depressant effect on neuron of spinal cord
sense of well being ( ↑dopamine conc in nucleus accumbence- phenomenon seen in drug abuser and pleasure seeking behavior.
Antiemetic action may be explained by ↓in serotonin level .

↓ ICP , acutely ↓ IOP -(propofol >Thio )effective in preventing raised IOP with scolin and intubation response
Neuroprotective role ↓ controversies ;due to antioxidant axn by inhibiting lipid peroxidation
Just or 1 hr after to ischemic insult produce reduction in size of infarct at sedation dose @ 25-75 µg/kg/min as compared to awake control with intralipid.
Burst suppression @blood level > 8µg/ml –better neurological outcome and less brain injury
EEG effect – ( α → ϒ → Ө ) wave Seizure like activity reported mainly on induction and emergence . Dose dependent anticonvulsant activity +nt

On induction dose and rate of adm dependent↓ BP (25-40 %) in comparable dose (propofol >Thiopental)
↓ SBP and DBP , ↓ MAP
↓ CO, ↓ SV , ↓ SVR ( 15-25 %)
HR ↓(-10 +_10 % ) to baseline; Propofol either may reset or may inhibit the baroreflex, reducing the tachycardia response to hypotension
MAP ↓ ( -10-40 %)
Propofol at high concentrations (10 µg/mL) abolishes the inotropic effect of α but not β adrenoreceptorstimulation, and enhances the lusitropic (relaxation) effect of β stimulation
CNS induced ↓ sympathetic drive on heart - cardio depression

In patient with valvular lesion ↓( PA and PCWP ) – due to ↓ pre and afterload .
cardio depression ( bolus > infusion )
Continuous Infusion cause significant ↓ in myocardial blood flow and oxygen demand
For better hemodynamic stability use one or more additive induction agent ( fentanyl , Midazolam )with propofol .
Bradycardia-related Death:-Profound bradycardiaand asystole after the administration of Propofoldespite prophylactic Anticholinergics. thus suggesting that Propofol induce a suppression of sympathetic nervous system activity. The treatment of Propofol-induced bradycardia may require the administration of a β-agonist, such as isoproterenol.

Profound resp depressant
Apnea occur depend on dose , speed of injection, concomitant premedication
Occur in 25- 30 % cases ,may last for >30 sec ,may ↑by adding opiate in premedication
Apnea risk max in this agent than other
Apnea precedes with marked ↓ TV and tachypnoea.
A maintenance infusion (100 µg/kg/min) results in a 40% ↓TV and a 20% ↑ RR ,
Propofol (50 to 120 µg/kg/min) also depresses the Ventilatory response to hypoxia, presumably by a direct action on carotid body chemoreceptor
Reduces airway and pharyngeal reflexes- use with LMA
Bronchoconstriction ( thiopental > propofol),Bronchodilation prevent intraop wheeze.

Proconvulsant Activity:-The majority of reported Propofol-induced seizures during the induction of anesthesia or emergence from anesthesia reflect spontaneous excitatory movements of sub cortical origin.
Abuse Potential :-Intense dreaming activity, amorous behaviour, and hallucinations have been reported during recovery from the effects of Propofol.
Bacterial Growth:-growth of Escherichia coli and Pseudomonas aeruginosa.
Pain on Injection:-As little as 0.2 mg/kg of lidocaine(mixed with the propofol) is effective in reducing but not eliminating this discomfort.
Mini – Bier Block –APPLY tourniquet give 1mg /kg of lidocaine 15-20 sec before propofol adm then remove tourniquet

Complication in prolonged infusion patient
Propofol infusion syndrome(PIS) in (pediatric> adult) patients receiving prolonged high-dose infusions of Propofol (>75 µg/kg per minute) for longer than 24 hours.
Associated with :-
metabolic acidosis,
lipidaemia,
cardiac arrhythmias
Unexpected tachycardia
Increase mortality

Significant bradycardia with scoline
Induction with propofol@1-2mg/kg
Marked decrease in B.P Decrease uteroplacental circulation

Advantages
Rapid induction
Anti emetic effect
TIVA
Agent of choice for
day care surgery
Disadvantages
Induction apnoea
Hypotension
Dose dependant bradycardia
Dose dependant respdepression
Pain during injection
It is also euphorigenicbut does not have residual psychotic effects like Ketamine

Etomidate is a carboxylated imidazole & prepared as a fat emulsion,
its effects on GABA A receptors
Etomidate A→B circulation time 1 min,
The clearance of etomidate is about five times that for thiopental;
Likewise, the context-sensitive half-time of etomidate is less likely to be increased by continuous infusion, as compared with thiopental.
Etomidate (0.2 to 0.4 mg/kg IV) IOC especially in the presence of an unstable cardiovascular system.
Involuntary myoclonic movements are common during the induction due to alterations in the balance of inhibitory and excitatory influences on the thalamocortical tract.
Awakening after a single intravenous dose of etomidate is more rapid than after barbiturates.
The principal limiting factor in the clinical use of etomidate for the induction of anesthesia is the ability of this drug to transiently depress adrenocortical function
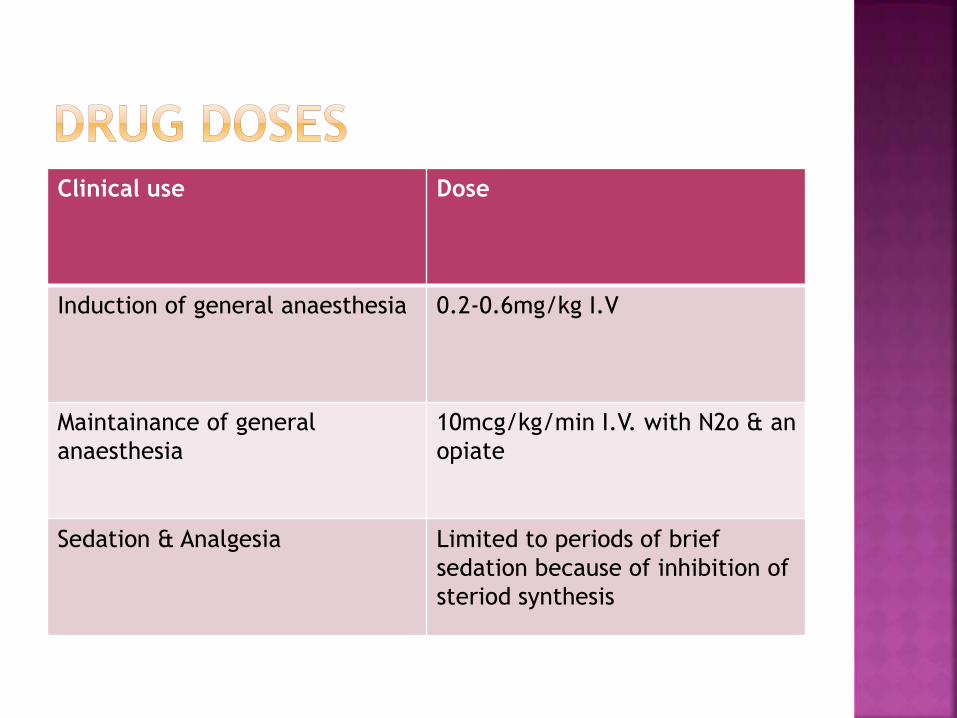
Clinical use Dose
Induction of general anaesthesia 0.2-0.6mg/kg I.V
Maintainance of general
anaesthesia
10mcg/kg/min I.V. with N2o & an
opiate
Sedation & Analgesia Limited to periods of brief
sedation because of inhibition of
steriod synthesis

↓ CBF , ↓ CMR02 , ↓ ICP
Myoclonus (spontaneous movements) occurs
in 50% to 80% of patients receiving etomidate
in the absence of premedication. etomidate-
induced Myoclonus appears to be
disinhibition of subcortical structures that
normally suppress extra pyramidal motor
activity.

Cardiovascular stability (minimal changes in
heart rate, stroke volume, cardiac output) is
characteristic of induction of anesthesia
using 0.3 mg/kg IV of etomidate So it may
differ from most other intravenous
anesthetics in that depressive effects on
myocardial contractility are minimal at the
concentrations needed for the production of
anesthesia.

The depressant effects of etomidate on
ventilation seem to be less than those of
barbiturates, although apnea may
occasionally accompany a rapid intravenous
injection of the drug.

Limitation of etomidate Etomidate causes
adrenocortical suppression by producing a
dose-dependent inhibition of the conversion
of cholesterol to cortisol

properties PROPOFOL THIOPENTONE KETAMINE
chemistry ALKYLPHENOL THIOBARBITURATE ARYLCYCLOHEXYL
AMINE
consistency Emulsion milky
white
Sodium salts(6%
sodium
carbonate)yellow
amorphous powder
Clear aquaous
solution
solubility Lipid soluble Lipid soluble Lipid soluble
ph 7 10.5 of 2.5% 3.5-5.5
pka 11 7.6 7.5
Unionised % 99.97% 61% 55.7%
onset One arm –brain
time15-30 sec
10-15 30-60
peak 90-100 sec 90-100 sec 90-100sec
awakening 5-10 min 5-10 min 10-20 min
Rapid fall in
plasma conc.
After bolus
Redistribution &
elimination
redistribution redistribution

properties PROPOFOL THIOPENTONE KETAMINE
Protein binding 98% 85% 60%
Metabolism &
excretion
LIVER
Glucuronite
sulphate
KIDNEY
LIVER
Oxidation
N-dealkylation
Desulfuration
Destruction of
barbituric acid
ring
KIDNEY & BILE
LIVER
Norketamine
Hydroxynorketam
ine
KIDNEY
metabolite Inactive pentabarbital Norketamine 20-
30%
Extrahepatic
metabolism
lung absent absent
Clearance
ml/kg/min
20-30 3-4 12-14

properties PROPOFOL THIOPENTONE KETAMINE
Content
sensitive half
time(for
infusion lasting
upto 8 hrs
<40 min <150min <40 min
MOA GABA GABA NMDA(thalamoc
ortical &
limbic)
Induction 1-2.5 mg/kg 3-5 adult
5-6 children
6-7 infant
0.5-2 mg/kg
maintenance 50-
150ug/kg/min
15-
45ug/kg/min
sedation 25-
75ug/kg/min
0.2-0.8mg/kg
analgesia 25-
75ug/kg/min
Conscious
sedation
0.2-0.8mg/kg

properties PROPOFOL THIOPENTONE KETAMINE
Neuroprotective Reduce infarct
size when adm
immediately or
1 hr after
ischemic insult
Decrese 02
demand
Preserve CPP
ROBINHOOD
phenomenon
Free radical
scavenging
Improve
perfusion in
incomplete
cerebral
ischemia
Dissociative
anasthesia
- -- +
Emergence
reaction
+ -- +
Upper airway
reflexes
-- -- +
Salivation &
lacrimation
-- -- ++
antiemetic + -- -
Sk musle tone -- -- increase
relaxant -- -- +
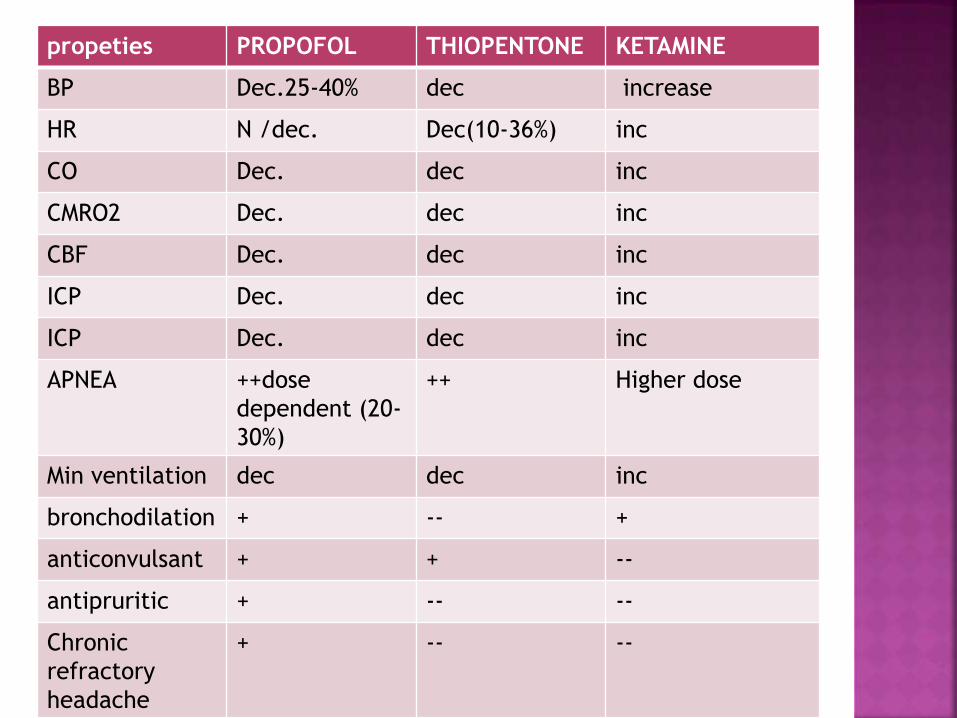
propeties PROPOFOL THIOPENTONE KETAMINE
BP Dec.25-40% dec increase
HR N /dec. Dec(10-36%) inc
CO Dec. dec inc
CMRO2 Dec. dec inc
CBF Dec. dec inc
ICP Dec. dec inc
ICP Dec. dec inc
APNEA ++dose
dependent (20-
30%)
++ Higher dose
Min ventilation dec dec inc
bronchodilation + -- +
anticonvulsant + + --
antipruritic + -- --
Chronic
refractory
headache
+ -- --

properties PROPOFOL THIOPENTONE KETAMINE
Pain on
injection
++ + --
Hypotension ++ + --
apnea ++ + +/-
bronchospasm -- + --
Allergic
reaction
+ + --
thrombophlebit
is
+ ++ --

A patient with intestinal obstruction requires
an emergency laparotomy. Which induction
drug would you use?

Any patient with intestinal obstruction must
be assumed to have a potentially full
stomach.
Traditionally a rapid sequence induction
would be performed with preoxygenation ,
cricoid pressure, thiopental and
suxamethonium.
Thiopental is chosen due to its rapid, well
defined onset at a predetermined dose. This
is also the method of choice of induction for
caesarean section.

A patient requires a burns dressing change.
Which induction drug would you use?

Ketamine is an ideal drug to be used for minor procedures.
For burns dressing changes, a sub-anaesthetic dose can be used. It will provide sedation and analgesia, preserving the protective airway reflexes.
Ketamine is often combined with benzodiazepine premedication to reduce the dose requirement and emergence reactions, and sometimes an anti-sialagogue (e.g. glycopyrrolate, glycopyrronium bromide) to reduce airway secretions

A patient with a history of heart failure
requires a general anaesthetic. Which
induction drug would you choose?

drug of choice would be etomidate due to its limited effect on the cardiovascular system. However some anaesthetists avoid etomidate all together due to its effect on steroid synthesis.
Ketamine could also be considered due to its relative cardiovascular stability.
Propofol and thiopental are also options, but potentially cause more cardiac depression. The important issue is that which ever induction drug is used, the lowest possible dose is given, it is given slowly and it is titrated to effect.
Intra-arterial blood pressure monitoring should be considered if available.

A patient with porphyria comes for an
inguinal hernia repair and is requesting a
general anaesthetic. Which induction drug
would you use?

A patient with porphyria comes for an inguinal hernia repair and is requesting a general anaesthetic. Which induction drug would you use?
The porphyrias are a group of disease characterised by overproduction and excretion of porphyrins(intermediate compounds produced during haemoprotein synthesis).
Acute attacks may be precipitated by drugs, stress, infection, alcohol, pregnancy and starvation.
Propofol would be the ideal induction drug to use in this case – being safe to use in patients with porphyria.
Thiopental and etomidate should be avoided as they can precipitate a porphyric crisis

An adult patient requires sedation on the
intensive care unit. Which of the induction
drugs would be appropriate to run as an
infusion?

A propofol infusion would be appropriate.
Midazolam could also be given in addition, or
as an alternative to propofol.
Thiopental should be avoided due to
accumulation, and etomidate should be
avoided due its effect on adrenal steroid
hormone synthesis.

THANK YOU














![Case Report Therapeutic Drug Monitoring of Meropenem in ...downloads.hindawi.com/journals/criid/2016/6207487.pdf · intravenous therapy with broad-spectrum antimicrobial agents [].](https://static.fdocuments.us/doc/165x107/5f1d3c61e345b33c50666db7/case-report-therapeutic-drug-monitoring-of-meropenem-in-intravenous-therapy.jpg)



