
International Graduate School of Neurosciences (IGSN)€¦ · International Graduate School of...
119
i International Graduate School of Neurosciences (IGSN) Ruhr Universität Bochum Transcriptional and translational regulation of zebrafish Connexin genes, zfCx55.5 & zfCx52.6. Doctoral Dissertation Mahboob-ul-hussain Department of Neuroanatomy and Molecular Brain Research Thesis advisor: Dr. Rolf Dermietzel Bochum, Germany (31.03.05)
Transcript of International Graduate School of Neurosciences (IGSN)€¦ · International Graduate School of...

i
International Graduate School of Neurosciences (IGSN) Ruhr Universität Bochum
Transcriptional and translational regulation of zebrafish Connexin genes, zfCx55.5 & zfCx52.6.
Doctoral Dissertation
Mahboob-ul-hussain
Department of Neuroanatomy and Molecular Brain Research
Thesis advisor: Dr. Rolf Dermietzel
Bochum, Germany (31.03.05)

ii
Table of CONTENTS
Acknowledgement……………........................................................................viii
Abbreviations.......................................................................................................x
Abstract…………………………………………………………..……..………1 INTRODUCTION………………………………………………………..…….2 Gap-junction Proteins: The Connexins……….…………..…….......…………2.1 Structure of Gap-junctions…………………………………………......……2.1.1 Topology of connexins……………………………………………...…….....2.1.2 Eukaryotic Transcription…………………………………………………...….2.2
Transcription factors………………………………………………………....2.2.1
Transcription of connexins…………………………………………………….2.3 Transcription of Cx32…………………………………………………...…..2.3.1 Transcription of Cx43……………………………………………….…...….2.3.2 Role of methylation in the transcription of connexion genes……............…..2.3.3 Eukaryotic Translation…………………………………………………...……2.4 Cap-dependent V/S Cap-independent Translation………………………..…2.4.1 IRES Elements…………………………………………………………….....2.4.2 How widespread are IRES elements...............................................................2.4.3 Molecular events underlying IRES function………………………………...2.4.4 IRES element in Connexin genes…………………………..…………...…...2.4.5 Connexin functions without junctions………………………………………....2.5 Zebrafish as an animal model……………………………………………...…..2.6

iii
Retina as a system to study connexin expression……………………….…...2.6.1 Connexin expression in Horizontal cells of retina……………………….….2.6.2 Aims and Objectives of the thesis work……………………………………….2.7 Materials and Methods……………………………………………………...…3 Plasmid construction………………………………………………………......3.1 For promoter study of zfCx52.6………. ……………….…………………...3.1.1 For promoter study of zfCx55.5……………………………………………..3.1.2 To generate transgenic zebrafish……………………………………..……...3.1.3 For Translational study………………………………………………………3.1.4 Cell Culture…………………………………………………………………....3.2 Transient transfections…………………………………………………………3.3 Reporter assay………………………...…………………………………….....3.4 Extraction of cytosolic and nuclear protein……………………..……………..3.5 Immunoblot analysis…………………………………………………...……...3.6 Northern blot analysis…………………………………………………….……3.7 RNA analysis………………………………………………………………..…3.8 EGFP-fluorescence analysis………………………………………………..….3.9 Immunocytochemistry……………………………………………………..…3.10 Protein expression and purification……………………………………….….3.11 In-Vitro transcription…………………………………………………………3.12 RNA-EMSA………………………………………………………………….3.13

iv
UV-crosslinking……………………………………………………………...3.14 DNA-EMSA………………………………………………………………….3.15 RESULTS…………………………………………………………………….…4 Identification of putative promoter elements in zfCx55.5………………..........4.1 Confirmation of the zfCx55.5 promoter specificity
in transgenic fish……………………………………………………….……...4.2
Specific protein complex binds to promoter element I and promoter
element II of zfCx55.5 and the promoter element of the zfCx52.6………...…4.3
Preliminary evidence for the binding of CCAAT binding protein (CBP)
and OCT-1 to the promoter element of zfCx52.6……………………………..4.4
In-vitro evidence of splicing of small exon I to main exon II of zfCx55.5
and the possible existence of an IRES element upstream ……………….. .4.5
Full length zfCx55.5 and a portion of its carboxy-terminal
domain are co-translated……………………………………………………….4.6
The carboxy-terminal protein (p11-CT) is translated from the zfCx55.5
transcript via internal translation…………………………….………………...4.7 An IRES element in the coding region of zfCx55.5 is responsible for the
expression of the p11-CT protein……………………………...………………4.8
Increased expression of the second cistron in the Di-cistronic assay is
due to the IRES activity and not to a cryptic promoter……………..…………4.9

v
The p11-CT protein is not expressed from a monocistronic mRNA……...….4.10
The p11-CT product can translocate to the nucleus………………………….4.11
In vivo evidence for the nuclear staining of
zfCx55.5 in the Horizontal cells of fish retina……………………………….4.12
Zebrafish connexin 55.5, zfCx55.5, internal IRES elements activity is
determined by two polypyrimidine tracts……………………………...……..4.13
Polypyrimidine tract binding protein (PTB) plays an essential role in the
IRES activity through its influence on the PPT1 and PPT……………..…….4.14
Specific ribonucleic-protein complex (RNP) assembles on the ~360 nt
zfCx55.5 IRES element……………………………………………………..4.15
Purified GST-PTB fusion protein is able to bind the IRES element……...….4.16
Secondary structure prediction……………………………………….………4.17
DISCUSSION……………………………………………………………..….…5
Promoter elements of zfCx55.5 and zfCx52.6…………………...……………5.1
Putative DNA binding proteins of the promoter
elements of zfCx52.6 and zfCx55.5………………………………..……….....5.2
Extension of the N-terminus of zfCx55.5………………………...……………5.3

vi
Internal translation of the CT of zfCx55.5……………………………..….......5.4
Functional motifs and trans-acting factor(s) of the zfCx55.5
internal IRES element……………………………………………………….…5.5
BIBLIOGRAPHY……………………………....………………………………6
VECTOR MAPS………………………………………….……………………7
Curriculum Vitae

vii
To my....., Father, Mother, Grandmother.

viii
Acknowledgement My foremost thank goes to my thesis adviser Dr. Rolf Dermietzel. Without him,
this dissertation would not have been possible. I thank him for his patience and
encouragement that carried me on through all times, and for his insights and
suggestions that helped to shape my research skills. He was always ready to
discuss my work, but let me free to pursue my own goals in my own way. It has
been a great pleasure to have him as a supervisor.
I am grateful to Dr. Georg Zoidl, who introduced and helped me to start my
graduate student life in the lab. It was always nice to discuss work with him and
to get inputs for shaping the experiment to its best form. I appreciate the friendly
atmosphere he created in the lab.
I would like to thank the “Architect” of my carrier, Dr.Khursheed I Andrabhi.
He was always as source of inspiration for me. It was he who introduced me to
world of science. His encouraging words used to lift my sagging spirits always.
He is the one that I can always count on to discuss the tiniest details of a
problem I am also thankful to entire teaching staff of my MSc course for their
hard work.
I am thankful to Marian Kremer who made my life comfortable in lab. I learned
a lot from her experience in molecular biology. Her visionary thoughts and
energetic working style have influenced me greatly as a graduate student. I
would love to imbibe the determination she used to show during the work.
Thanks also go to Christina Zoidl for her help and the way she used to keep the
lab things intact and infusing in us the sense of cleanness.
I am grateful to Dr.Patresh Parwez Elizabeth and her husband Dr. Parwez for
their smiles. Hans Werner Habes unconditional help is equally commendable.

ix
I am grateful to Frau Becker for her help with the official work and the efforts
she used to put to convey anything to me in English.
I am grateful to Helga Schulz for helping with Photoshop.
My friends where always a source of comfort to me. Sameer is one of them with
whom I spend most important part of my life. His friendship will remain a
pleasant memory for the rest of my life.
In this part of lonely world, it was kaoushik and Ismail who provided the
needful friendship. I will remember the evening hour chats with koushik where
we use to discuss the vicissitudes of daily life. I will really miss their company.
My friends of M.Sc are the best gifts I can ever think off. Tanveer, Bashir,
Younis, Mushataq, Amjad, Jamal, Farooq, Rouf, Samina, Refiqa, the memories
of who always use to make me feel good. I would like to thank my M.Sc juniors
especially Abhar and Asia and Dr. Talib for their friendship. Thanks also go to
my JNU friends Vikas, Sarub, Azhar, Veenu, Amjad, Prerna, Sarita, vandana,
Jai.
Special thanks goes to my friends back home, Javid, Haneef, Zahoor, Ajaz,
Tariq, Ghulam, Ayub, Yousuf, Farooq.
My relatives always make me feel special to them. Thanks go to all my relatives
for their caring attitude towards me. I am grateful to Mr. Gh. Mohi-ud-Din
Malik for always being there with me when I needed it most, and for supporting
me through all these years.
Last but not least I would like to thank to my parents and grandmother for their
unwavering dedication, patience and support.
Mahboob-ul-hussain

x
Abbreviations
ATP, adenosine triphosphate
bp, basepair
Cx, connexion
c-AMP, cyclic adenosine mono phosphate
CTP, Cytidine Triphosphate
Ci, curie(s)
cpm, counts per minute
CBF, CCAAT binding factor
CT, carboxy-terminal
CMV, cytomegalovirus
EGFP, enhanced green fluorescent protein
EMSA, electromobility shift assay
FL-CT, full length carboxy-terminal
F-WT, frameshifted wild type
FLuc, Firefly Luciferase
GTP, guanosine 5 -triphosphate
IRES, internal ribosome binding site
IR, IRES element
kD, kilodalton
Oct-1, octamer binding protein
PPT, polypyrimidine tract
PAGE, polyacryalamide gel electrophoresis
PTB, polypyrimidine tract binding protein
rRNA, ribosomal RNA
Rluc, Rennila Luciferase
SDS, sodium dodecyl sulphate
SEM, standard error of the mean
SV40, Simian Virus 40

xi
Tris, tris (hydroxymethyl) aminomethane
UV, ultraviolet
UTP, uridine 5 -triphosphate
UTR, untranslated region
WT, wild type
Zf., zebrafish
µCi, microcurie(s)

1
1. Abstract Zebrafish connexins 55.5 (zfCx55.5) and connexin 52.6 (zfCx52.6) show highly
restricted expression pattern in the nervous system. Both connexins are confined to
subsets of neurons in the fish retina. In order to get the initial answers to the question
of their cell specific expression in horizontal cells, we elucidated the molecular
mechanism at the transcriptional level. For the same purpose, basal promoter regions
of these connexins were identified using the reporter gene luciferase assay in N2A
cells. Luciferase activity showed the presence of two putative promoter elements in
zfCx55.5 and a promoter element in zfCx52.6. The efficacy of these promoter
elements was confirmed by generating a transgenic fish (in collaboration with Dr.
Marteen Kamermann) having EGFP gene expression under the control of these
putative promoter elements. Exclusive EGFP expression from the horizontal cell
layer of the transgenic fish retina confirmed the role of these promoter elements in
imparting the site restricted expression to these connexins. Electromobility shift assay
using the N2A nuclear extract showed that a number of specific proteins bind to the
promoter region of zfCx55.5 and zfCx52.6. Initial results indicate that CCAAT
binding factor (CBF) and Oct-1 binding protein are part of the complex which binds
to the promoter element of zfCx52.6. Moreover, in pursuit of the molecular
mechanism which may shed light on the “functions without junctions” property of
connexins, we here provide first evidence that the carboxy-terminal domain of
zfCx55.5 can be internally translated from the main zfCx55.5 mRNA. An IRES
element in the coding region of zfCx55.5 mRNA was found to be responsible for the
separate expression of a carboxy-terminal domain (here named p11-CT).
Interestingly, our in-vitro and in-vivo data indicate that this internally translated
product can translocate to cell nucleus. We were successful in identifying two cis-
acting elements called polypyrimidine tracts (PPT1 and PPT2) and a trans-acting
factor called polypyrimidine tract binding protein (PTB) as important constituents of
the IRES mediated internal translation of p11-CT.

2
2. Introduction
Multicellular organisms with complex tissue systems have evolved over a period of
time from simple unicellular organisms. As opposed to unicellular organisms, which
carry out most of their biological processes within a single cell, individual cells
within a multicellular organization need to communicate with each other for the
successful exchange of nutrients and signals, necessary for the maintenance of the
organization. Organisms have evolved multiple strategies to achieve this goal, which
include long-range interactions mediated by neural or endocrine mechanisms or
short-range interactions that include direct physical or cell-cell contact. This is
accomplished in a variety of ways, mostly by the formation of a series of pores, or
communicating channels which can facilitate cell-cell communication. In animal cell
system, gap junctions between cells form one such communication system. The
fundamental function of two or more cells coupled by gap junctions is clearly to
“communicate”. While humans communicate with other humans via words, body
language, and touch, cells communicate with each other in a multicellular organism
via chemical signals. The major physiological role of gap junctions is to synchronize
metabolic or electronic signals between cells in a tissue. Cells have only four basic
functions, namely (a) to proliferate; (b) to differentiate; (c) to apoptose or die by
programmed cell death; and (d) to adaptively respond if it is already terminally
differentiated. In multicelluar organism, a delicate coordination or orchestration of
these four cellular functions must occur. Growth, differentiation, apoptosis, wound
healing, and homeostatic control of differentiated functions must occur in a single
space and this is done by coupling the cells within a tissue/organ mainly through
gap junctions.
2.1 Gap-junction proteins: The connexins
Gap junctions are specialized areas of the cell membranes that connect neighbouring
cells. They are organized collections of protein channels that allow ions and small
molecules to traverse between the connected cells. These allow for the
communicating cells to equilibrate critical regulatory ions and small molecules (e.g.,
Ca++, c-AMP, glutathione), as well as macro-molecular substrates (amino-acids,

3
sugars, nucleotides). These protein channels that make up the gap junctions consist
of two hemi-channels or connexons. One connexon resides in the membrane of one
cell and it aligns and joins the connexon of the neighbouring cell, forming continuous
aqueous pathways by which these ions and small molecules can pass from one cell to
the other, as shown in the Fig 2.1.
Fig. 2.1: Gap junction channel and connexin structure. A) Gap junction channels assemble in plaques containing few to several hundred single channels. Each cell contributes one hemichannel called connexon that consists of six connexin proteins. The gap junction channels span a small gap (3.5nm) between the cell membranes and connect the cytoplasm of neighbouring cells (drawing by H. Schulze).
Each hemi-channel or connexon consists of six proteins (hexamer) called connexins
(Cx). Gap junctions have been typically described as relatively non-selective,
permeable to a wide range of molecules smaller than ~1200 dalton (Simpson et
al.,1977). However, experiments carefully examining the movement of ions and dyes
between cells expressing different connexins have revealed that there are connexin-
dependent differences in the permeation of intercellular channels (Veenstra et al.,
1996; Elfgang et al., 1995; Cao et al., 1998).
Apart from few terminally differentiated cells, such as skeletal muscle, erythrocytes,
and circulating lymphocytes, most cells in normal tissues generally communicate via
gap junctions. These junctions exist in almost all animals, both vertebrates and
invertebrates, and higher plant cells utilize a similar mechanism for cell-cell
communication via plasmodesmata structures.

4
2.1.1 The structure of gap junctions
Techniques such as freeze-etch electron microscopy, labelled site–specific antibodies,
selective protease cleavage, and X-ray diffraction studies have been successfully used
to determine the structure of gap junctions (Yeager et al., 1998; Makowski et al., 1977;
Unwin et al., 1984). Gap junctions exhibit a hierarchy of assembly. The principal
structural component, the membrane protein connexin, is organized into the basic
unit of structure, the connexon, which is a hexameric structure with a toroid
appearance in negative-stained preparations. An individual connexon from one cell
docks or associates with a corresponding connexon on a neighbouring cell to form a
gap junction channel, and multiple channels, in turn, cluster or aggregate in the
plane of the membrane to form gap junction plaques. The properties of the gap
junction channels are defined by the connexins. Structural and biophysical studies
are being used to define the mechanism by which connexins function.
Connexins are the principal protein component of gap junctions. There is much
evidence to support the fact that the connexins alone (assembled in a lipid bilayer)
are responsible for the generation of gap junctional channels. This evidence includes
the following: connexin sequences are consistent with an integral membrane protein
that has a transmembrane domain containing polar amino acids that would
contribute to the formation of a channel lining; reconstitution of purified connexins
into artificial membranes yields functional channels (Buehler et al., 1995) expression
of connexin cDNAs in heterologous systems (including yeast) yields not only
functional gap junction channels, but also gap junctions that are ultra structurally
identical to those occurring naturally in vivo; electron microscopic
immunocytochemical studies localize connexins to gap junction plaques; and the
distribution of connexins observed in vivo can be related to gap junctional
communication pathways.
2.1.2 Topology of Connexins
Each of the connexins appears to fit the general topological model for gap junction
protein (Figure 2.2).

5
Figure 2.2 Topological model of a connexin protein. The cylinders represent transmembrane domains (M1-M4). The loops between the first and the second, as well as the third and fourth transmembrane domains are predicted to be extracellular (E1 and E2), each with three conserved cysteine residues (adapted from Kumar and Gilula, 1996).
In this model, the polypeptide traverses the lipid bilayer four times, with both the N-
and C-termini facing the cytoplasm (Milks et al., 1988; Yeager et al., 1992). Analysis
of the different connexins indicates that one of the transmembrane domains, M3, has
an amphipathic character, suggesting that it contributes to the lining of the channel.
The two extracellular loops (E1 and E2) are thought to be involved in initiating the
interaction between connexons in adjacent cells. A set of three cysteine residues
exists in each of the extracellular loops with a characteristic arrangement that is a
signature of connexins. These may help to maintain the rigid tertiary structure that
enables two opposing connexons to dock with each other. The regions between the
transmembrane domains M2 and M3, as well as the C-termini of the connexins, are
highly variable among the different connexins and are, therefore, thought to be
important for the regulation of the channel.
It has been suggested that the folding pattern for the connexins corresponds to an
antiparallel arrangement of four transmembrane domains that associate to form a
left-handed bundle which is consistent with the known structural and permeability
properties of gap junctions. X-ray (Tibbitts et al., 1990) and circular dichroism studies
(Cascio et al., 1995) are consistent with the high helical content of the transmembrane
domains of the connexin predicted by this model. Much progress has yet to be made
in obtaining some structural information on the gap junction connexins at the atomic
level.
The oligomeric arrangement of connexins has been indicated in structural studies on
gap junctions where 6-fold symmetry has been used as a constraint in the image

6
analysis. Independent evidence has been provided by chemical cross-linking studies
on purified rat liver gap junction connexons to indicate that each connexon consists
of six subunits to form hexameric connexons.
2.2 Eukaryotic transcription
Gene expression can be regulated at a number of levels, starting from the chromatin
remodelling, transcription, RNA splicing, RNA degradation, translation and post
translational modification. Transcription of genes serves a primary control of
regulating gene expression.
Eukaryotic transcription is more complex than prokaryotic transcription and, until
recently, it has seemed that every eukaryotic gene was unique requiring its own
transcription machinery. However, it is now possible to simplify the story somewhat.
1) The promoters for different genes are different; 2) each promoter contains a
combination of sites to which specific protein factors bind. 3) All of these factors help
RNA polymerase to bind in the correct place and to initiate transcription. However,
the repertoire of transcription factors and transcription factor binding sites is not
unlimited. Eukaryotic RNA polymerases cannot find or bind to a promoter by
themselves. Each requires the binding of assembly factors and a positional factor to
locate the promoter and to orient the polymerase correctly. The positional factor is
the same in all cases. All genes that are transcribed and expressed via mRNA are
transcribed by RNA polymerase II. Until recently, it was common to think of
eukaryotic transcription (and particularly mRNA synthesis) as taking place in
discrete steps: transcription, capping, tailing, splicing and export from the nucleus
for translation. The contemporary view of eukaryotic gene expression entails
simultaneous transcription and processing. Recent discoveries have revealed that
many of the protein factors required for these individual steps do, in fact, interact
with one another. This makes sense for it allows the cell to coordinate and regulate
the complete process more efficiently.
Promoters used by RNA polymerase II have different structures depending upon the
particular combination of transcription factors that are required to build a functional
transcriptional complex at each promoter. Nevertheless, these different structures

7
can be viewed as a combination of a relatively limited number of specific sequence
elements.
Some of the common elements that have been described in class II eukaryotic
promoters are the following:
• The TATA Box located approximately 25 bp upstream of the start-point of
transcription is found in many promoters. The consensus sequence of this
element is TATAAAA. The TATA box appears to be more important for
selecting the start point of transcription (i.e. positioning the enzyme) than for
defining the promoter.
• The Initiator is a sequence that is found in many promoters and defines the
start point of transcription.
• The GC box is a common element in eukaryotic class II promoters. Its
consensus sequence is GGGCGG. It may be present in one or more copies
which can be located between 40 and 100 bp upstream of the start point of
transcription. The transcription factor Sp1 binds to the GC box.
• The CAAT box - consensus sequence CCAAT - is also often found between 40
and 100 bp upstream of the start point of transcription. The transcription
factor CTF or NF1 binds to the CAAT box.
In addition to the above elements, Enhancers may be required for full expression.
These elements are not part of the promoter per se. They can be located upstream
or downstream of the promoter and may be quite far away from it. The
mechanism by which they work is not known. They may provide an entry point
for RNA polymerase or they may bind other proteins that assist RNA polymerase
to bind to the promoter region
The transcriptional complex
When it was first purified and characterized, it was found that RNA polymerase
II can transcribe mRNA in vitro as long as a suitable template -- such as a nicked
dsDNA or ssDNA -- is provided. The fact that the enzyme could not initiate
transcription correctly on a dsDNA template indicated that RNA polymerase II
could not function alone in the cell nucleus and a search was begun for additional
transcription factors. At least six general (or basal) transcription factors (TFIIA,
TFIIB, TFIID, TFIIE, TFIIF, TFIIH) have been characterized. In the presence of

8
these transcription factors, the enzyme is able to initiate transcription at
promoters correctly. However, even in the presence of transcription factors, the
enzyme complex is unable to recognize and respond to regulatory signals.
In addition to the general transcription factors, the transcriptional complex will
also be affected by the presence of a promoter-proximal regulatory sequences and
the presence of transcription factors that bind to those sequences. Such factors
may be present in some cells/tissues but not in others. For example, the octamer
motif binds two different transcription factors: Oct-1 and Oct-2. Oct-1 is
ubiquitous but Oct-2 is expressed only in lymphoid cells where it activates
immunoglobulin k light chain gene transcription. A simple schematic view of
above facts is provided in the Figure 2.3 below.
Figure 2.3. Proteins at Typical Eukaryotic Promoter Activators (red, green)) bound to enhancer elements stimulate transcription via activation domains by protein–protein interactions (arrow) with components of TFIID and the pol II holoenzyme (purple). Of the basic factors defined by in vitro transcription, TFIIA is considered here as part of the TFIID group, whereas TFIIB, TFIIE, TFIIF, TFIIH, and core pol II are considered part of the pol II holoenzyme (adapted from Struhl, K; cell 84, 179-182)
2.2.1 Transcription factors
As of the latest release of TRANSFAC, a transcription factor database, in 2001, it
contained 2785 entries. Many of these are homologous proteins from different
species; nevertheless this number is indicative of the vast number of transcription
factors now known that regulate the expression of eukaryotic genes. Transcription
factors are the ultimate targets of cell-signalling pathways. Whenever cells need to
response to an extracellular signal such as a hormone, the response is mediated by a

9
change in gene expression that comes about, most often as the result of a change in
the phosphorylation state of a transcription factor.
2.3 Transcription of connexin genes
Changes in the level of connexin expression play an important role in controlling
gap-junctional cellular communication (Bennett et al., 1991). However, the
mechanisms that modulate the expression of the different connexin genes are not
well known. The genomic organization of the majority of the connexin genes is very
similar, with two exons, the short one forming the part of 5´ UTR and the second
with rest of the untranslated region and the encoding sequence (Miller et al., 1988;
Hennemann et al., 1992; Yu et al., 1994). In spite of this similarity at the level of
genomic organization, the expression of the different connexin genes is regulated at
different levels and the expression of connexin vary in different tissues or same tissue
at different spatio-temporal points.
Transcriptional control of many connexin genes has been well studied and the initial
evidence depicts that transcription of connexins is controlled by multiple promoters
and is far more complex then previously thought.
2.3.1 Transcription of Cx32
The transcriptional control of connexin 32 serves the best example of transcriptional
complexity in the connexin family. Cx32 is the major connexin expressed by
heptocytes and is also expressed in neural, renal, testicular, and other tissues (Paul et
al., 1986). Hepatic cell lines also demonstrate cell-specific connexin expression. The
well differentiated rat hepatoma cell line, MH1C1, expresses Cx32 but not Cx43 (Ren
et al., 1994). Promoters of most conexin genes is located upstream of exon 1. In Cx32
gene three promoters have been identified. One is located upstream of the first exon,
lacks a TATA box, contains CCAAT box elements and positively acting Sp1 elements,
and is active in adult liver (Miller et al., 1988; Bai et al., 1993; Bai et al., 1995). Two
additional promoters are located within the intron, contain TATA boxes, and are
active in neural and embryonic tissue but are inactive in adult liver (Neuhaus et al.,
1995; Neuhaus et al., 1996). Liver cell-specific expression of Cx32 is regulated by
positively and negatively acting transcription factors. These include Sp1, HNF-1,

10
proteins of the B2 complex, and perhaps others. In addition, Cx32 transcriptional
control in non-hepatic cells is thought to be regulated through the cell-specific use of
alternative promoters. These regulatory mechanisms may play a role in the reduced
expression of Cx32 that has been observed frequently in human and rodent
hepatocellular carcinomas (Krutovskikh et al., 1997).
2.3.2 Transcription of Cx43
Similarly connexin 43 transcription, an abundant expressing connexin, is well
documented and new evidences are emerging which may explain the diversity in the
expression of this connexins. Cx43 promoter activity has been mapped with in the 5´
upstream region of the first exon (De Leon et al., 1994; Fernandez-Cobo et al., 1998).
The proximal promoters for the mouse, human and rat Cx43 genes have been
mapped in several Cx43-expressing cell types to an evolutionary conserved region of
approximately 150 nucleotides up- and downstream of the TIS (Echetebu et al., 1999;
Chen et al., 1995; Teunissen et al., 2003). Within this region, four evolutionary
conserved Sp-binding sites (Bruzzone et al., 1996; Saez et al., 2003; Gros et al., 1996;
Van Kempen et al., 1996), and one AP1-binding element are located; the rat Cx43
promoter contains an additional AP1-binding element which is absent in the mouse
and human genes. In myometrial smooth muscle cells, both a positive and a negative
regulatory DNA element have been identified in the mouse Cx43 promoter, which
was capable of binding to as yet, unidentified nuclear proteins. For the human Cx43
proximal promoter, it was demonstrated by promoter/reporter assays and Sp1/AP1
over-expression studies that both Sp1 and AP1 are necessary as transcriptional
activators for optimal promoter activity. The rat Cx43 proximal promoter has been
extensively studied in rat primary neonatal cardio-myocytes, thoracic aorta smooth
muscle and normal kidney cells which all are known to express Cx43. Each of the Sp-
and AP1-binding sites was shown to contribute to promoter activity and to bind the
transcription factors Sp1/Sp3 or AP1, respectively. In trans-activation assays, Sp1
and Sp3 were both able to activate the rat Cx43 promoter. Within the rat Cx43
promoter, a negative regulatory element, as detected in the mouse, was not
identified; however, the mouse "activator" might very well correspond with one of
the Sp1/Sp3-binding elements in the rat promoter. Altogether these results indicate

11
that rat Cx43 proximal promoter activity is determined by the transcription factors
Sp1, Sp3 and AP1. Interestingly, rat proximal promoter activity could hardly be
detected in mouse neuroblastoma cells, which correlated with the lack of
endogenous Cx43 RNA and protein expression in these cells, suggesting some cell
type-specificity. These results may be explained by the absence of Sp1, Sp3 and/or
AP1 expression or the presence of a neural-specific repressor in the neuroblastoma
cells. Because of the similarities in proximal promoter regulation by ubiquitously
expressed transcription factors (Sp1, Sp3, AP1) in different Cx43-expressing cell types
(including cardiomyocytes), it is likely that cell type-specific expression of Cx43
depends on additional activators or repressors. Several studies have provided
evidence that Nkx2.5 may serve such an additional role for Cx43 expression in the
heart. As for Cx40, reduced Cx43 protein and RNA levels were noticed in mice over-
expressing a putative dominant-negative mutant of Nkx2.5 in the heart, suggesting
an activating role for this homeoprotein (Kasahara et al., 2001). However, mice over
expressing wild type Nkx2.5 in the heart also displayed reduced Cx43 expression,
suggesting that Nkx2.5 may act as a transcriptional repressor of Cx43 as well
(Kasahara et al., 2003; Akazawa et al., 2003). It has also been shown that adenoviral-
mediated over-expression of Nkx2.5 in rat neonatal ventricular myocytes results in a
dramatic decrease of endogenous Cx43 protein and RNA levels and a two-fold drop
in rat Cx43 proximal promoter activity (Teunissen et al., 2003). The drop in promoter
activity could not completely account for the observed reduction in protein/RNA,
suggesting the involvement of more distal regulatory regions as well. Thus, Nkx2.5
appears to be able to act as an activator as well as a repressor of Cx43 expression, but
the precise molecular mechanism has not been elucidated yet. Transcriptional
cofactors, such as members of the T-box gene family, may determine whether Nkx2.5
acts as an activator or as a repressor. Indeed, Tbx2 has been identified as a negative
regulator of Cx43 expression at the transcriptional level (Borke et al., 2003; Chen et
al., 2001), but the effect of other T-box family members on Cx43 expression has not
been reported.
Besides knowledge on Cx43 gene structure and proximal promoter regulation insight
has also been gained on signalling events affecting Cx43 transcription in cardiac and
non-cardiac cells. In human primary myometrial cells, Cx43 transcription and

12
proximal promoter activity were increased upon activation of protein kinase C with
the phorbol ester TPA suggesting the involvement of the protein kinase C pathway
in the up-regulation of myometrial Cx43 at the onset of labor (Geinomen et al., 1996).
TPA was further shown to up-regulate and activates c-jun and c-fos, the molecular
constituents of AP1, which exert their inducing effect on Cx43 proximal promoter
activity through AP1-binding site 2. Responsiveness of Cx43 transcription and/or
proximal promoter activity has also been shown to prostaglandin E2, parathyroid
hormone and 8Br-cAMP in osteoblastic cells, to the thyroid hormones T3 and T4 in
liver cells, to the Ras-signaling pathway in fibroblasts and to the Wnt1-signaling
pathway in neural crest-derived cells (Civitelli et al., 1998; Mitchel et al., 2001; Stock
et al., 2000; Carystinos et al., 2003; Van der Heyden et al., 1998). The responsive
element for parathyroid hormone has been mapped to the (−31,+1) region, relative to
the TIS, and the responsiveness in bone cells of both endogenous Cx43 and its
proximal promoter was confirmed in transgenic mice carrying a 1.8-kb Cx43
proximal promoter/reporter construct. The thyroid hormone responsive element has
been characterized as the (−480,−464) region, and binding of a heterodimer of the
retinoid X receptor thyroid hormone receptor to this element was demonstrated
(Stock et al., 2000). In mouse fibroblasts, the (+149, +158) region has been identified
as the binding site for the heat shock protein HSP90 and c-myc, which mediate the
transcriptional up-regulation of Cx43 by the Ras-Raf-MAPK pathway (Carystinos et
al., 2003). In cardiac myocytes, activation of the Wnt-signaling pathway and
dibutyryl-cAMP were shown to induce Cx43 protein and RNA expression (Darrow
et al., 1996; Ai et al., 2000). These responses appear to be transcriptionally regulated,
since Cx43 proximal promoter activity increases correspondingly with treatment.
Although the Cx43 proximal promoter contains evolutionary conserved cyclic AMP
and TCF/LEF (the transcriptional effectors of Wnt-signaling) binding elements, the
precise molecular mechanism for induction has not been elucidated. In contrast,
activation of c-jun N-terminal kinase (JNK) resulted in the down-regulation of Cx43
RNA and protein, both in transgenic mouse hearts and cultured cardiomyocytes
(Petrich et al., 2002). As mentioned above, AP1 is not only an activator of the Cx43
proximal promoter in several different cell types, but also well known as a
downstream target of JNK. Further studies are evidently necessary to resolve this

13
discrepancy. Altogether these studies illustrate that the Cx43 gene regulatory region
is the target of diverse signalling events in different cell types, but that the precise
molecular mechanisms and signal transduction molecules involved still have to be
elucidated.
2.3.3 Role of methylation in the transcription of connexin genes
Further level of transcriptional control was found to dependent upon the
methylation status of promoter regions of connexin genes. For example, Cx26 has
been implicated as a tumour suppressor gene (Lee et al., 1992; Locke et al., 1998) and
its expression was shown to be possible in normal human mammary epithelial but
not in breast cancer cells. Significantly, it was established that lack of expression was
not due to any physical loss of the gene, but due to hypermethylation of promoter
region of Cx26 (Tan et al., 2002). Similarly, the activities of transiently transfected rat
Cx32 and Cx43 promoters are reported different in Cx32-expressing and Cx43-
expressing liver cells (Piechocki et al., 1999). The Cx32 promoter was found to be
four-fold more active in Cx32-expressing MH1C1 cells than in Cx43-expressing WB-
F344 cells. It has been also shown that cytosine residues in the Cx32 promoter and
intron are methylated in WB-F344 cells, but not in MH1C1 cells and that the opposite
was seen for the Cx43 promoter. It has been shown that trans-activating factors and
DNA methylation contribute to differential connexin expression of this connexin
gene. Methylation of promoter associated CpG islands is a mechanism for the
transcriptional silencing of number of tumour suppressor genes (Jones et al., 1999;
Baylin et al., 1998).
2.4 Eukaryotic translation
Second step of gene expression control is at the level of translation. Translation is the
process by which the information contained in the nucleotide sequence of mRNA
instructs the synthesis of a particular polypeptide. This process, outlined in Fig. 2.4,
has been divided into three phases: initiations, elongation, and termination, and it is
regulated by soluble proteins called (appropriately) initiation factors, elongation
factors, and termination factors (Hershey et al., 1989; No authors listed). Initiation is
the rate limiting step of translation and consists of the reactions wherein the first

14
aminoacyl-transfer RNA and the mRNA are bound to the ribosome. The classical
way of translational initiation, called cap-dependent is initiated by the recognition of
the 7-methyl guanosine cap by the host of initiation factors which helps in the
recruitment of 40S ribosomal subunit to the 5´ UTR of the m-RNA (Shatkin, 1976;
Shatkin, 1985). In accordance with the scanning model, the 40S ribosomal subunit
then travels down the message until it reaches an AUG codon in the proper context
called Kozak sequence, with the “optimum" sequence of ACCAUGG (Kozak, 1986).
Figure 2.4 Schematic representation of the events of eukaryotic translation. The initiation steps bring
together the 40S and 60S ribosomal subunits, mRNA, and the initiator tRNA, which is complexed to the
amino acid methionine (Met). During elongation, amino acids are brought to the polysome, and peptide
bonds are formed between the amino acids. The sequence of amino acids in the growing protein is directed
by the sequence of nucleic acid codons in the mRNA. After the last peptide bond of the protein has been
made, one of the codons UAG, UGA, or UAA signals the termination of translation. The ribosomal
subunits and message can be reutilized (adapted from a book, Developmental Biology (Seventh Edition, by
Scott F. Gilbert)
2.4.1 Cap-dependent V/S cap-independent translation
Understanding of the full potential of the genome coding capacity demands a deep
knowledge of the different pathways that control gene expression. Translation
initiation in eukaryotic mRNAs is a highly regulated process that accounts for the
last step of gene expression control. For a majority of the eukaryotic mRNAs, the
ribosome associates with the mRNA by virtue of the cap-structure, a 7-methyl-
guanylic acid residue at the 5´ terminus. Then this cap-binding complex scannes the

15
5´´ UTR till it finds the start codon. This classical mechanism, as simplified below in
Fig. 2.5 is termed as cap-dependent translation.
Cap-dependent translation
Figure 2.5 The 40 S ribosomal subunit, together with certain eukaryotic initiation factors (eIF‘s)
binds to the 5‘ terminal m7GpppN and scans the untranslated region (UTR) until the AUG (protein
synthesis initiation) codon is reached. The joining of the 60 S subunit, results in the formation of
the 80 S initiation complex, which includes the initiator tRNA.
While most mRNAs initiate translation by the above discussed mechanism, a
growing number of mRNAs appear to follow different rules, wherein certain cis-
elements present in the mRNA was found enough to recruit the translational
machinery without the need of a cap structure, as depicted below in Fig. 2.6, hence
named as cap-independent translation. These cis-acting are termed as internal
ribosome entry site (IRES).
Cap-
independent Translation
Figure 2.6 Certain mRNAs reveal internal ribosomal entry sites (IRES), usually in the 5‘- UTR. These IRES
containing mRNAs are not subject to some of the complex regulatory mechanisms involved in cap-
dependent translation. IRES mediated translation initiation is typically found in mRNAs translated under
conditions of cellular stress. (F stands for additional protein factor(s), binding to IRES, involved in internal
initiation.)

16
2.4.2 Definition of IRES elements
IRES elements as the name indicates are the Internal Ribosome Entry Sequence which
bypass the cap-dependent translation and thus recruit the translational machinery
directly (without the need of cap structure) to the mRNA sequence. This alternative
way to initiate translation allows the use of internal start codons, sometimes located
several hundred of residues away from the 5'end of the mRNA, bypassing strong
RNA structures. Therefore, they represent a strategy to increase genetic diversity
without increasing genome length. The IRES sequences found in viral and cellular
mRNAs do not show overall sequence similarity, albeit they perform a similar
function. IRES elements in viral mRNAs constitute an efficient method to distinguish
its own mRNA from that of the host, and thus facilitate its survival when cellular
protein synthesis is impaired. Viral IRES exploit different strategies to recruit the
translational machinery, including direct ribosome binding, eIF3 or eIF4G-mediated
mechanism. Cellular IRES mediated-translation represents a regulatory mechanism
that helps the cell to cope with transient stress. They may be grouped according to
common tropism, stimulation by similar situations and expression of specific targets
in differentiated cells. Protein mediated-ribosome binding is likely to enhance the
efficiency of cellular IRES sequences under specific environments.
2.4.3 How widespread are IRES elements?
Studies on viral gene translation were essential for the initial discovery of internal
entry of ribosome. Unlike their cellular counterparts, picornaviral mRNAs are
naturally uncapped at their 5' end. Their 5' UTRs also have complex features
predicted to impair ribosome recruitment and linear scanning: (i) a long leader
sequence; (ii) stable secondary structures; and (iii) potential upstream initiation
codons. Nevertheless, these 5' UTRs confer efficient 40S joining. The poliovirus and
encephalomyocarditis virus (EMCV) 5' UTRs were the first to be described to 'break
the rule' of translation initiation (Jackson, 1988; Pelletier et al., 1988). Bicistronic
RNAs with two non-overlapping open reading frames (ORFs) were shown to be
good models to test cap-independent translation initiation. This was first shown for
poliovirus, where inserting a segment of the 5' UTR of a poliovirus genome between
the two ORFs allows translation of the downstream cistron, independent of the cap-

17
mediated translation of the first cistron. This strategy can be considered the 'gold
standard' for characterizing IRESs (Sachs, 2000) if one considers the presence of
cryptic RNA processing signals or promoter sequences in the intercistronic space as
having been ruled out (Kozak, 2001). Using this assay as the basis for defining IRESs,
these elements have been found in all picornavirus genera. Their presence in viruses
as diverse as flaviviruses, retroviruses and even DNA viruses such as the Kaposi's
sarcoma-associated herpesvirus reveals the widespread nature of these RNA
elements. As is the case for many viral mRNAs, a number of cellular mRNAs possess
structural features in their 5' UTRs that make them unlikely to be translated by a 5'
cap-dependent ribosome-scanning mechanism. Moreover, a few cellular mRNAs are
translated preferentially when cap-dependent initiation of translation is impaired.
These discoveries argued for an alternative mechanism such as the internal entry of
ribosomes. Indeed, the first cellular IRES was identified in the 220-nucleotide-long 5'
UTR of the immunoglobulin heavy chain-binding protein (BiP) mRNA, whose
translation is maintained in poliovirus-infected cells at a time when cap-dependent
translation is severely inhibited (Macejak et al., 1991). Since then, and particularly
over the last three years, IRES activities have been detected in a restricted but
increasing number of cellular mRNAs from yeast, Drosophila, birds and mammals,
showing that the internal ribosome entry process is far more extensive than
previously thought.
2.4.4 Molecular events underlying IRES function
Natural IRESs have developed complex interaction networks. Various attempts to
define cis-elements required for IRES activity revealed that the three-dimensional
RNA fold, rather than its primary sequence, is the major determinant of IRES
function. To operate as IRES, RNA should form a structural scaffold in which
precisely positioned RNA tertiary structures contact the 40S ribosomal subunit
through a number of specific intermolecular interactions. In a reconstituted
translation initiation system, purified 40S ribosomal subunits are able to form a
binary complex with the hepatitis C virus (HCV) IRES, even in the absence of the
canonical translation initiation factors (Pestova et al, 1998). One site that had
previously been defined as a contact point between the 40S subunit and the HCV

18
IRES is the ribosomal protein S9. However, mutations that reduce S9 binding do not
affect binary complex formation suggesting that multiple contact point’s act together
to stabilize the complex. On the other hand, a more recent study showed the
ribosomal protein S5, but not S9, to interact with the HCV IRES (Fukushi et al., 2001).
In other IRESs, such as those of cricket paralysis-like viruses, translation is initiated
at non-AUG codons without the help of any proteins and even without initiator Met-
tRNA (Sasaki et al., 2000; Wilson et al., 2000) suggesting a strong dependence on
RNA structure (Spahn et al., 2001). Indeed, phylogenetic and mutational analyses
have identified a pseudoknot structure to be essential for IRES function (Kanamori et
al., 2001). One might expect that when mRNA is not correctly folded to establish
contacts with ribosomal proteins or RNA, some non-ribosomal cofactors are required
either to create additional interactions with the 40S subunit or to act as RNA
chaperones controlling the functional configuration of the IRES. Studies on
picornaviral IRESs have revealed unexpected mRNA-binding properties for various
canonical translation initiation factors including eIF3 and eIF4G. Non-canonical
translation initiation factors with known functions in other processes were shown to
interact with various IRESs. The functional roles of these additional IRES trans-acting
factors (ITAFs) are generally assessed by in vitro translation assays of IRES-
containing reporter constructs supplemented with recombinant proteins. Such assays
have led to the assignment of heterogeneous nuclear ribo-nucleoprotein (hnRNP)
I/PTB (Kaminski et al., 1998), a polypyrimidine-tract-binding protein known for its
role as a splicing regulator, hnRNP E2/PCBP2 (Hunt et al., 1999), La (Meerovitch et
al., 1993; Holcik et al., 2000)-an autoantigen with diverse RNA metabolism activities,
unr (Hunt et al., 1999) upstream of N-ras, ITAF45/Mpp1 (Pilipenko et al., 2000) a
protein whose expression is up-regulated in response to mitogen stimulation and is
not detectable in differentiated cells, and DAP5/NAT1/p97 (Henis-Korenblit et al.,
2000) an eIF4G homolog and nucleolin (Izumi et al., 2001) as ITAFs. These ITAFs are
not active on all IRESs and they act either alone or in combination to mediate IRES-
dependent translation (Hunt et al., 1999; Pilipenko et al., 2000; Mitchell et al., 2001).
However, in vivo assays are required to confirm their ITAF function. Indeed,
disruption of the DAP5 gene in mouse embryonic stem cells does not affect the IRES
activities of various bicistronic transfected genes (Yamanaka et al., 2000).

19
Interestingly, the predominant nuclear localization of several ITAFs led to the
hypothesis that either their binding to IRES-containing mRNAs is a nuclear process
or they relocalize to the cytoplasm to bind their target mRNAs. The observation that
several cellular, but not viral, IRES-containing mRNAs are translated only when
expressed within the nucleus suggest that there is a requirement for a 'nuclear
history' in the functionality of certain cellular IRESs (Stoneley et al., 2000). However,
the ability of some ITAFs, e.g. hnRNPs (PTB), to shuttle between the nucleus and the
cytoplasm could also reflect their putative role in translation initiation. Discoveries of
new ITAFs and the definition of the complexes involved in IRES-dependent
translation will help in the precise understanding of the initiation process. Whereas
the biochemical purification of a complex on such a long and incompletely defined
RNA is not an easy task, the recent discovery of IRESs in Saccharomyces cerevisiae
(Zhou et al., 2001) makes a genetic approach possible and this will certainly speed up
the discovery process.
2.4.5. IRES elements in connexin genes
Recently, translational initiation in connexin genes was regarded mainly as cap-
dependent. However, recent reports on Cx43 and Cx32 have shown that these
connexins possess functional IRES elements in their 5´ UTR. The unusual long 5´
UTR of Cx 43 suggest being involved in translational regulation in different tissues
(Schiavi et al., 1999). Moreover, Cx32 5´ UTR was shown to posses a functional IRES
element. More interestingly, point mutation in this element results in less
translational efficiency of this connexin in neurons and this has been linked to the
Charcot-Marie-Tooth disease (Hudder et al., 2000). As discussed previously, presence
of multiple promoters in connexin would result in the different 5´ UTRs. The
difference of 5´ UTRs will have effect on the translational efficiency of the connexins
and presence of IRES elements would provide the additional control mechanism for
the differential expression of connexins. Interestingly, recent finding of the presence
of IRES elements in the coding region of certain genes has opened gates for the
separate expression of carboxy-terminal domains of proteins (Cornelis et al., 2000).
These observations are of great importance for certain properties of connexins for

20
which the gap-junction communication seems to be dispensable, they are discussed
below.
2.5 Connexin functions without junctions
Are connexins involved in functions not directly associated with their channel
forming ability? Several lines of evidence suggest they are. Since most transformed
cells do not establish gap junctions, it was suggested many years ago that junctional
communication might influence proliferation. Subsequently, many studies have
correlated the suppression of growth in transformed cells with restoration of
communication, typically by connexin transfection (Qin et al., 2002). Paradoxically, it
appears that in some cases connexin expression alone, without establishment of
intercellular channels, might be enough to achieve this goal. In one example,
retroviral delivery of Cx43 or Cx26 to MDA-MB-231 cells does not restore
intercellular communication or even cause the establishment of gap junctional
plaques but does dramatically suppress tumour growth when cells are implanted in
nude mice. Although the mechanism is not clear, exogenous connexin expression
down-regulates at least one growth factor receptor and up-regulates an anti-
angiogenic agent (Qin et al., 2003). In another example, expression of the C-terminal
region of Cx43, a non-channel-forming domain, is sufficient to suppress HeLa cell
growth (Dang et al., 2003). The C-terminal domain becomes partially localized in the
nucleus, although it is not clear if this localization is necessary for growth inhibition.
Together, these data suggest that growth suppression by connexins might involve a
mechanism that is independent of either intercellular or hemichannel activity.
Another channel-independent function of connexins could be related to resistance to
injury. Recently, it was shown that expression of Cx43 protects cultured glial cells
from certain apoptotic stimuli as effectively as expression of bcl-2 (Lin et al., 2003).
Surprisingly, the protective effect is not eliminated by sparse plating of cells to limit
the formation of gap junctions or by connexin channel ‘blockers’. Furthermore,
exogenous expression of mutant connexins incapable of forming intercellular
channels also confers resistance to injury. The study correlated increased resistance
with a connexin-mediated cytoskeletal re-organization and faster normalization of
cytotoxic elevations of calcium which enabled connexin expressing cells to survive an

21
otherwise lethal injury. These studies conclude that the connexin expression has a
very significant impact on cellular injury resistance by a process independent of gap-
junction coupling.
2.6 Zebrafish as an animal model to study connexin expression
The zebrafish, Danio rerio, has emerged as a novel vertebrate model system that is
amenable to mutagenesis and transgenesis. High fecundity, rapid oviparous
development, and a translucent embryo make zebrafish a prolific experimental
model (Streisinger et al., 1981). Furthermore, the zebrafish eye possesses distinct
advantages for studying the development, function, and inherited diseases of the
retina in relation to the expression of genes. Eye ontogenesis proceeds rapidly,
completing the laminae of the adult retina by 3 days post fertilization (Branchek et
al., 1984). The zebrafish eye is relatively large and accessible, and the position and
morphology of the rod and cone classes are readily distinguishable (Raymond et al.,
1993). Finally, the integrity of visual system structure and function can be evaluated
by morphological, behavioural, and electrophysiological methods (Brockerhoff et al.,
1997; Malicki et al., 1996; Neuhauss et al., 1999).
2.6.1 Retina as a system to study connexin expression
The retina is a highly ordered laminar structure, comprising three compact layers of
neurons separated by two synaptic layers, which has proven a valuable model to
study gap junctions and cell specific expression patterns of connexins in neuronal
tissues (Sohl et al., 2000; White et al., 2000). Gap junction-mediated dye transfer is
found between nearly all cell types that form the neuronal retinal matrix (Becker et
al., 1998) and a diversity of coupling patterns that is so far unmatched in any other
part of the brain (Vaney et al., 1991; Vaney et al., 1993). More recently, direct
demonstration of electrical and metabolic communication between different classes
of retinal neurons has been obtained (Vaney et al., 1998; Guldenagel et al., 2001;
Veruki et al., 2002; Veruki et al., 2002; Deans et al., 2002). The selective nature of
neuronal coupling and its differential regulation by neuromodulators (Piccolino et
al., 1982; Lasater et al., 1987; De Vries et al., 1989; Miyachi et al.,; Hampson et al.,
1992; Qian et al., 1992; Lu et al., 1999), as shown recently for the amacrine AII cells

22
(Hampson et al., 1994; Mills et al., 1995), supports the idea that multiple types of
connexins may exist within the neuronal populations of this tissue.
2.6.2 Connexin expression in horizontal cells of retina
Extensive analysis of retinal gap junctions has concentrated on horizontal cells, a
population of retinal neurons that is endowed with extensive gap junction coupling.
Dual cell recording experiments and dye-transfer studies in parallel with freeze-
fracture investigation have made horizontal cells by far the best studied class of
coupled neurons in the CNS (Dowling et al., 1966; Piccolino et al., 1982; Lasater et al.,
1987; De Vries et al., 1989; Vaney et al., 1993; Weiler et al., 1996; Janssen-Bienhold at
al., 2001).
Visual processing in the retina information is partly accomplished by laterally
orientated horizontal cells. These second-order neurons are postsynaptic to
photoreceptors and modulate the transfer of information between photoreceptors
and bipolar cells in the outer plexiform layer (OPL) by exhibiting lateral feedback
inhibition on to the presynaptic cones. Horizontal cells of all vertebrate retina form
extensively coupled networks and therefore electrical coupling and its
neuromodulation have been most intensively studied in this context. Recent reports
suggest functional hemichannels at horizontal cell dendrites involving Cx26 in carp
and turtle retina (Kamermans et al., 2001; Pottek et al., 2003). Very recently, Cx52.6
has been shown to be expressed in zebrafish horizontal cells and to form Ca2+-gated
hemichannels after ectopic expression in Xenopus oocytes (Zoidl et al., 2004).
Sequence analysis of zfCx55.5 and zfCx52.6 has revealed only limited homology of
these connexins to other connexins from fish and higher vertebrates. ZfCx52.6 shows
around ~57% amino-acid homology with zfCx55.5, where as the zfCx55.5 has been
shown to share about 50% homology with the mouse Cx57. The striking feature of
these connexins is their long carboxy-terminal domain with least amino-acid
homology. Furthermore, there is a striking abundance of serine in the C-terminal
domain of both of these connexins along with numerous putative phosphorylation
sites.

23
2.7 Aims and objectives of this work
As it goes by the title of this thesis work, transcriptional and translational control of
zfCx55.5 and zfCx52.6, we try to unravel the molecular mechanism at the
transcription and translation level of two zebrafish connexins, zfCx55.5 and Cx52.6.
The motivation behind the transcriptional study of these connexins was there
peculiar expression pattern. Both of these connexins have been found to show highly
restricted expression in the horizontal cells of zebrafish retina (Zoidl et al., 2004;
Dermietzel et al., 2000). Tissue specific expression of connexin is a rare observation
with most of connexins showing broad expression pattern. To get the initial answers
about the mechanism of their restricted expression, we investigated the promoter
elements and other regulatory elements of these two connexin.
Our second aim was based on the fact that connexins perform various functions for
which gap-junction communication seems to be dispensable. Mostly these functions,
as discussed previously, have been attributed to carboxy-terminal domain of the
connexins. Molecular mechanism behind these observations has remained enigmatic.
Keeping these concepts in view, we propose a mechanism which can endow the
connexins with the ability to perform functions without the need of gap-junctional
communication.

24
3. Materials and Methods 3.1 Plasmid construction.
3.1.1. For promoter study of zfCx52.6.
A zebrafish genomic clone of zfCx52.6 at XbaI site in pBluescripit II KS+ ( Stratagene,
Amsterdam, Netherlands) was used as a template for the amplication of various
upstream DNA fragments. A ~1905 bp upstream region of zfCx52.6 (relative to
translational start site) was PCR amplified using the T3 sense primer, 5´ AAT TAA
CCC TCA CTA AAG GC 3´ (Corresponding to the T3 promoter sequence present in
multiple cloning site of pBluescripit vector) and antisense primer, 5´ GTG GAA TTC
ACG GAA AAA CTG 3´, starting form -135nt relative to start codon. A PCR product
of ~1.9Kb was separated on a 1.2% agarose gel and gel purified using the Qiaex-II
Gel extraction Kit, (Qiagen, Hilden, Germany). After digestion with SacI (MBI
Fermantas GMBH, St. Leon-Rot, Germany), this fragment was ligated at the SacI/
SmaI site of promoterless pGL3-Basic vector (Promega, Madison, WI, USA) to get -
1905 /-135 pGL3-Basic construct. A DNA fragment of ~1127 bp from -1905 to -778
was PCR amplified using the above T3 sense primer and an anti-sense primer, 5´
TAA GCA CAA TTT TGA AAT TTT GAA GGC 3´. A PCR product of ~1.1Kb was
separated on 1.2% agarose gel and gel purified using the Qiaex-II Gel extraction Kit,
(Qiagen). After digestion with SacI (MBI Fermantas), this fragment was ligated at the
SacI/ SmaI site of promoterless pGL3-Basic vector (Promega) to get the -1905 /-778
pGL3-Basic construct. a DNA fragment from -1905 to -1095 was PCR amplified using
above sense T3 primer and an antisense primer, 5´ GAC TGA TGG CTA AAT GTT
GC 3´. A PCR product of ~810bps was separated on 1.2% agarose gel and gel purified
using the Qiaex-II Gel extraction Kit, (Qiagen). After digestion with SacI (MBI
Fermantas) and Hind III (MBI Fermantas), this fragment was ligated at the SacI/
Hind III site of a promoterless pGL3-Basic vector (Promega) to get the -1905 /-1095
pGL3-Basic construct. Similarly a DNA fragment from -1162 to -135 was PCR
amplified using sense primer, 5´ TAA ATG TGT TTT ACA GGA G 3´ and anti- sense
primer 5´ GTG GAA TTC ACG GAA AAA CTG 3´. A PCR product of ~847bps was
separated on 1.2% agarose gel and gel purified using the Qiaex-II Gel extraction Kit,

25
(Qiagen) and ligated at the Sma I site of promoterless pGL3-Basic vector (Promega)
to get -1162 /-315 pGL3-Basic construct.
3.1.2. For promoter study of zfCx55.5.
Zebrafish genomic clones of zfCx55.5 at Sac I site in pBluescripit II KS+ ( Stratagene)
were used as a template for the amplication of various upstream Cx55.5 DNA
fragments. A DNA fragment from +134 to -881 (relative to the translational start site)
was PCR amplified using the sense primer 5´ AGT GTG TAG ATG CAG GAT GGG
C 3´ and anti sense primer 5´TTC CAC ACA TCC TCC GCT GC 3´. A PCR product of
~1014bps was separated on 1.2% agarose gel and gel purified using the Qiaex-II Gel
extraction Kit, (Qiagen) and ligated at the EcoRV site of pBluescripit II KS+
(Stratagene). After confirmation of orientation, this DNA fragment was digested
using SacI/ XhoI restriction enzymes (MBI Fermatas). After separation on 1.2%
agarose gel, gel purified using the Qiaex-II Gel extraction Kit, (Qiagen), it was ligated
at SacI / XhoI restriction sites of the promoterless pGL3-Basic vector (Promega) to
get a -2004 /+134 pGL3-Basic construct. One more upstream DNA fragment from -
2507 to -664 was PCR amplified using sense primer 5´ TAT ACG ACA CCA TCA
ACC CG 3´ and anti-sense primer 5´ CTG AAA TAC AAT TAC AGC AAG C 3´. A
PCR product of ~1843bps was separated on a 1.2% agarose gel and gel purified using
the Qiaex-II Gel extraction Kit, (Qiagen) and ligated at the EcoRV site of pBluescripit
II KS+ (Stratagene). After confirmation of orientation, this DNA fragment was
digested using SacI/ XhoI restriction enzymes (MBI Fermantas), separated on 1.2%
agarose gel, gel purified using the Qiaex-II Gel extraction Kit, (Qiagen) and ligated at
SacI / XhoI restriction sites of the promoterless pGL3-Basic vector (Promega) to get a
-2507/-644 pGL3-Basic construct. A DNA fragment from -1261 to -195 was PCR
amplified using sense primer 5´ CTT CAT GTT GAT AGT GGA GC 3´ and anti-sense
primer 5´ CAG TAA CCT CAC ACA AAT ATG C 3´. A PCR product of ~1067bps
was separated on 1.2% agarose gel and gel purified using the Qiaex-II Gel extraction
Kit, (Qiagen) and ligated at the EcoRV site of pBluescripit II KS+ (Stratagene). After
confirmation of orientation, this DNA fragment was digested using KpnI/ SmaI
restriction enzymes (MBI Fermatas), separated on 1.2% agarose gel, gel purified
using the Qiaex-II Gel extraction Kit, (Qiagen) and ligated at KpnI/ SmaI restriction

26
sites of the promoterless pGL3-Basic vector (Promega) to get -1261/-195 pGL3-Basic
construct. A further ~1911 bp upstream DNA fragment of Cx55.5 from -3915 to -2004
was obtained by restriction digesting the genomic clone in pBluescripit II KS+ with
the Hind III restriction enzyme (MBI Fermantas). A ~1911 bp fragment was
separated on 1.2% agarose gel, gel purified using the Qiaex-II Gel extraction Kit,
(Qiagen) and ligated at Hind III restriction site of the promoterless pGL3-Basic vector
(Promega) to get the -3915/-2004 pGL3-Basic construct.
The -3915 /-2004 pGL3-Basic construct was used to further narrow down the ~1911
bp promoter element. A ~449 bp from the 5´ end of the 1911 bp fragment were cut
using the AflII unique restriction site at position (1753nt) in the fragment and Sma I
site of the vector. After digestion with Sma I and AflII (MBI Fermantas), the AflII
restriction site was Klenow filled using Klenow fragment of DNA polymerase I (MBI
Fermantas). The resulting construct was separated on 1.2%agarose gel, purified using
the Qiaex-II Gel extraction Kit, (Qiagen), and re-ligated to get the -3166/-2004 pGL3-
Basic construct. Moreover, ~450bp 5´ DNA fragment of the -3915 /-2004 construct
was obtained by digesting the -3915/-2004 construct with SmaI and AflII and
subsequently the AflII site was Klenow filled. After gel purification using the Qiaex-
II gel extraction kit (Qiagen), it was ligated at the SmaI site of pGL3-Basic vector to
get the -3915 /-3166 pGL3-Basic construct.
3.1.3 Plasmid construction of zfCx52.6 and zfCx55.5 to generate transgenic
zebrafish.
For the construction of transgenic zebrafish of the putative promoter elements of
zfCx52.6 and zfCx55.5, zebrafish genomic clones of the zfCx52.6 in pBlueScript II
SK(+) (Stratagene) was used as a template for the amplication of the 5´ upstream
DNA fragment of zfCx52.6 (from position -135 to -1905 relative to the translational
start site). The PCR was performed using a sense primer with a SacI restriction site at
5´ end (5` GGC, GAG, CTC, AAT, CAA, TTT, CCG, TTT, GC 3`) and the antisense
primer with an EcoRI restriction site at the 5´ end (5´GTG, GAA, TTC, ACG, GAA,
AAA, CTG 3`). A PCR product of ~1.7kb was separated on 1.2% agarose gel and gel
purified using the Qiaex-II Gel extraction Kit, (Qiagen). After digestion with the SacI
and EcoRI restriction endonucleases (MBI Fermantas), this fragment was ligated into

27
the SacI/EcoRI restriction sites of the promoterless pEGFP-1 vector (BD Biosciences
Clontech, CA, USA).
Similarly, the 5´ upstream DNA region of zfCx55.5 (from -14 to – 4538, relative to
translational start site), was obtained from the zebrafish genomic clone of zfCx55.5 in
pBlueScript II SK (+) (Stratagene) by restriction digesting the 5´ upstream DNA
region of the zfCx55.5 with Bgl II (- 4538) and AvaI (-14) restriction enzymes (MBI
Fermantas). Restriction digested AvaI site was Klenow filled (Fermentas) so as to
make it blunt. A DNA fragment of ~4.5kb was separated on 1 % agarose gel and gel
purified using the Qiaex-II Gel extraction Kit, (Qiagen). This fragment was ligated
into the Bgl II/Sma I restriction sites of the promoterless pEGFP-1 vector (BD
Biosciences).
Full length coding main exon II of zfCx55.5 was obtained by PCR amplifying from a
genomic clone in pBluescripit (Stratagene), using sense primer with EcoR-I site (5’
CCG GAA TTC GTT CAT GTT TCT TTC TTC TTA 3`) and antisense primer (5’-ATC
GGA TCC AAT TTG TAA GTG TGT GGG AGC -3’) with BamHI site in place of the
stop-codon. A PCR product of ~1.5Kb was separated on 1.2% agarose gel and gel
purified using the Qiaex-II Gel extraction Kit, (Qiagen). After digestion with EcoRI
and BamHI (MBI Fermantas), this fragment was ligated in-frame into the
EcoRI/BamHI site of pEGFP-N3 (BD Biosciences Clontech, CA, USA) to get the
zfCx55.5 exon II EGFP construct. To include the small exon 1 and the intervening
intron (present upstream of main AUG start codon) with the main coding exon II of
zfCx55.5, PCR was performed using the sense primer (ahead of exon I)
GAGGGGGTCACAAAAGTTTAGG (hypothetical) and the same anti-sense primer
as above. A PCR product of ~1.5Kb was separated on 1.2% agarose gel and gel
purified using the Qiaex-II Gel extraction Kit, (Qiagen). After digestion with BamHI
(MBI Fermantas), this fragment was ligated in-frame into the BglII
(Klenowed)/BamHI site of pEGFP-N3 (BD Biosciences Clontech) to get exon I/exon
II EGFP construct. The ~333bp, present in the intronic region of exon I /exon II EGFP
construct, were removed by using unique XbaI (4576) and Ava I (4909) restriction
sites. After digestion with Xba I and Ava I restriction enzymes (MBI Fermantas) and
subsequently Klenow filled using Klenow fragment of DNA polymerase I, the
resulting construct was separated on 1.2%agarose gel, purified using the Qiaex-II Gel

28
extraction Kit, (Qiagen), and re-ligated to get the construct deleted exon I/exon II
EGFP construct.
3.1.4. For translational study.
Full length zfCx55.5 was obtained by PCR amplifying the coding region from a
genomic clone in pBluescripit (Stratagene, Amsterdam, Netherlands), using sense
primer with EcoR-I site (5’ CCG GAA TTC GTT CAT GTT TCT TTC TTC TTA 3`)
and antisense primer (5’-ATC GGA TCC AAT TTG TAA GTG TGT GGG AGC -3’)
with BamHI site in place of the stop-codon. A PCR product of ~1.5Kb was separated
on 1.2% agarose gel and gel purified using the Qiaex-II Gel extraction Kit, (Qiagen,
Hilden, Germany). After digestion with EcoRI and BamHI (MBI Fermantas GMBH,
St. Leon-Rot, Germany), this fragment was ligated in-frame into the EcoRI/BamHI
site of pEGFP-N3 (BD Biosciences Clontech, CA, USA). The full length carboxyl
terminal domain (634bp to 1497bp) was PCR amplified using sense primer (5’-TCT
TCA TGG TGT TCA TGC AAT GC- 3’) and the same antisense primer as that of the
full length zfCx55.5. A PCR product of ~863bp was obtained and gel purified using
the Qiaex-II Gel extraction Kit, (Qiagen). After digestion with BamHI (Fermantas),
this fragment was ligated in-frame at the SmaI/BamHI site of pEGFP-N3 (BD
Biosciences). N-terminal truncated carboxyl-terminal domain (946bp to 1497bp) was
PCR amplified using sense primer (5-’GCC TGT TCA GGG TGA TTT ACC AG- 3’)
and the same anti-sense primer as above. PCR product of ~551bp was gel purified
digested with BamHI (Fermantas) and ligated in-frame at the SmaI/BamHI site of
pEGFP-N3 (BD Biosciences). The full length zfCx55.5 pEGFP-N3 plasmid was used
to perform site directed mutagenesis of the in-frame internal start AUG (1202bp) in
the carboxyl terminal domain to GCG codon using the Transformer site directed
mutagenic kit (Clontech East Meadow Circle, Palo Alto, CA, USA). The sequence of
the mutagenic primer was (5´-CTC ATC CAG CGC GGT AAA GAA ACC -3). A
frameshift mutation was introduced at position 1179 of the zfCx55.5 protein coding
region by addition of single nucleotide (T) between positions 1179 and 1180 using the
following mutagenic primer (5´-CAC ACC AGA GAA TTC ATC TCA TGC CTC-3´),
with the nucleotide added underlined. Eventually the addition of “T” resulted in the

29
creation of the EcoRI restriction site (used for screening the mutants) and hence this
modification created a frame shift at position 1179.
Di-cistronic vector (pRL-Di-cis) comprising Renilla luciferase as first cistron and
Firefly luciferase as second cistron was a kind gift from Dr. Rudolf Werner
(Department of Biochemistry and Molecular Biology, University of Miami, School of
Medicine). The expression of the Renilla cistron was driven by a CMV promoter with
stable hairpin structures at the start and end of the Renilla gene to inhibit cap-
dependent translation and read-through from the first cistron. The zfCx55.5 coding
region from 631bp to 1201bp (CT-region) was PCR amplified using sense primer (5`-
CCG GAA TTC TTC ATG GTG TTC ATG CAA-3`) having an EcoRI site and
antisense primer (5` CCG CTC GAG GCT GGA TAA GGC ATG 3`) having an XhoI
site. A PCR product of ~510bp was separated on 1.2% agarose gel and gel purified
using Qiaex-II Gel extraction Kit (Qiagen). After digestion with EcoRI and XhoI
(Fermantas) this fragment was ligated into the EcoRI/XhoI inter-cistronic region of
the pRF Di-cis vector to get the pRF-IR1 construct. 211bp from the 3’ end of the pRF-
IR1 vector were removed by digesting it with ScaI/XhoI. Both digested restriction
sites were Klenow filled (Fermantas) and the resulting construct was separated on
1.2%agarose gel purified using the Qiaex-II Gel extraction Kit, (Qiagen), and re-
ligated to get the construct pRF-IR2.
The pRF Di-cistronic vector was modified by inserting an EGFP gene in place of the
luciferase gene (pR-GFP Di-cis vector). For this purpose, the EGFP fragment was
isolated from the pEGFP-N3 vector (BD Biosciences) using the XhoI/Not-I restriction
enzymes (Fermantas). The 790bp XhoI/Not-I fragment was separated on 1.2%
agarose gel, and gel purified using Qiaex-II Gel extraction Kit (Qiagen). This
fragment was ligated into the XhoI/Not-I digested pRF Di-cis vector, to get a
modified Di-cis vector (pR-GFP). To engineer the promoterless Di-cistronic
constructs, the CMV promoter, including the chimeric intron and hairpin structure,
was removed by digesting the respective Di-cis constructs by BglII/NheI. The
resulting Di-cis constructs were gel purified, Klenow filled (Fermantas) to blunt both
digested sites and religated. All constructs were confirmed by sequencing.
Di-cistronic vector pRF-IR having zebrafish connexin IRES element sub-cloned at
EcoRI /XhoI restriction site, as described previously, was used for various mutagenic

30
experiments. Deletion of 9 bps (TCCTCCTTT) of the polypyrimidine tract 1 (PPT-1)
was performed using the Transformer site directed mutagenic kit (Clontech East
Meadow Circle, Palo Alto, CA, USA). The sequence of mutagenic primer used was 5`
CCT GAT GCC TAG ATT AAC CCA TCC 3`. The resultant mutagenic IRES
containing di- cis vector was designated as pRF-IR (del.PPT1). 14 bp second
polypyrimidine tract (PPT-2) was deleted using the following site directed mutagenic
strategy. A unique Sma I restriction site was created at the immediate 5´ flanking of
the PPT-2 using the following mutagenic primer 5´ CCT TTG ATT AAC CCG GGA
TCC TCT GCT TTC 3. Similarly a unique Pvu II restriction was created at the
immediate 3´ flanking of PPT-2 using the following mutagenic primer 5` CTG CTT
TCT TGC AGC TGT TCA GGG TG 3`. After confirmation of the creation of Sma I and
Pvu II site, the pRF-IR di-cis construct was first restriction digested with the Sma I
restriction enzyme and subsequently with Pvu II restriction enzyme (MBI Fermantas
GMBH, St. Leon-Rot, Germany). The double digested pRF-IR vector was separated
on 1.2% agarose gel, gel purified using the Qiaex-II Gel extraction Kit, (Qiagen,
Hilden, Germany) and religated to get a construct without polypyrimidine tract 2,
pRF-IR (del.PPT-2). For the simultaneous deletion of both Polypyrimidine tracts i.e
PPT-1 and PPT-2 (and intervening sequence of 11 bps), a unique EcoRV site was
created at the immediate 5´ flanking of the PPT-1 using the following mutagenic
primer 5` TGG AGC TGC CGA TAT CTT CTT TTG 3` and the already existing Pvu II
site at the 3´ immediate flanking of PPT-2. The resultant construct was double
digested with the EcoRV and Pvu II restriction enzymes (Fermantas). The double
digested pRF-IR vector was separated on 1.2% agarose gel; gel purified using the
Qiaex-II Gel extraction Kit, (Qiagen) and religated to get the construct without both
PPT-1 and PPT-2 (including the intervening sequence), pRF-IR (del.PPT 1-2). To sub-
clone the wild type IRES element (IR) and its various deletion mutants in the di-cis
vector, pR-EGFP (having EGFP instead of firefly luciferase as second cistron), the
respective pRF-IR, pRF-IR (del.PPT-1), pRF-IR (del.PPT-2) and pRF-IR (del.PPT 1-2)
constructs were restriction digested using the EcoRI and Xho I restriction sites. The
respective wild type IRES fragment and its various deletion mutant DNA fragments
(~360bp) were separated on 1.2% agarose gel and gel purified using the Qiaex-II Gel
extraction Kit, (Qiagen). After gel purification, the Xho I restriction site was blunt

31
ended using the Klenow fragment of DNA polymeraseI (Fermantas) and sub-cloned
at EcoRI / Sma I restriction site in the intercistroinc region of pR-EGFP vector.
GST-human polypyrimidine tract binding protein fusion vector, pGEX2TK (huPTB)
was a kind gift from Dr M. Garcia-Blanco, Durham, N.C, USA. This vector was used
to get the entire PTB coding DNA sequence (1594 bp) using the EcoRI restriction site.
This DNA fragment was separated on 1.2% agarose gel and gel purified using the
Qiaex-II Gel extraction Kit, (Qiagen) and subsequently ligated at the EcoRI site of
pEGFP-C1 vector (BD Biosciences Clontech, CA, USA) to get pEGFP-C1-PTB
construct. This construct was further manipulated to remove the EGFP gene at the N-
terminus of the PTB gene. For that purpose, it was restriction digested using the Nhe
I site (5´ flanking of EGFP gene) and the Bgl II site (3´ flanking of EGFP gene). A ~750
bp EGFP DNA fragment was removed from the rest of construct by gel separation
using 1.2% agarose gel and rest of the construct was gel purified using the Qiaex-II
Gel extraction Kit, (Qiagen). After gel purification, the Nhe I and Bgl II sites were
blunt ended using the Klenow fragment of the DNA polymeraseI (Fermantas) and
subsequently religated to get pC1-PTB construct (without EGFP).
To in-vitro transcribe the wild type IRES sequence and its various deletion mutants
from the T7 promoter, the wild type di-cis IRES construct , pRF-IR and the deletion
constructs were manupilated in such a way so as to make them monocistronic by
removing the Rennila luciferase gene (RLuc, 5´ flanking of IRES element). For this
purpose, the di-cis constructs were restriction digested with the Nhe I (5´ flanking of
RLuc gene) and the EcoRI (3´ flanking of RLuc gene) restriction enzymes
(Fermantas). A ~750 bp RLuc DNA fragment was removed from the rest of construct
by gel separation using 1.2% agarose gel and rest of the construct was gel purified
using the Qiaex-II Gel extraction Kit, (Qiagen). After gel purification, the Nhe I and
EcoRI sites were blunt ended using the Klenow fragment of the DNA polymerase I
(Fermantas) and subsequently religated to get pT7-IR, pT7-IR (del.PPT-1), pT7-IR
(del.PPT-2) and pT7-IR (del. PPT 1-2) mono-cistronic constructs.

32
3.2 Cell culture.
HeLa, NIH3T3 and Neuro2A cells purchased from the ATCC collection (Manassas,
VA, USA) were grown in 10-cm tissue culture dishes (Becton Dickinson, Heidelberg,
Germany) in high glucose (4,500 mg/l) Dulbecco's Modified Eagles medium
(DMEM) supplemented with 10% serum, penicillin and 100 µg/ml streptomycin
(Gibco Life Technologies) in a humidified atmosphere of 5% CO2/95% air at 37°C.
Cells were routinely passaged twice a week.
3.3 Transient transfections.
Transient transfections where performed using plasmid DNA of various constructs
and the Effectene® transfection protocol (Qiagen, Hilden, Germany). Day before
transfection, cells where seeded according to the scheme given in table 1 below.
On the day of transfection the amount of plasmid constructs and the different
reagents used were according to the scheme given in table 2 below. Briefly, required
amount of plasmid DNA was diluted in buffer EC. To the diluted DNA buffer mix,
enhancer was added and the mixture was vortexed for 1 sec. The mixture was
incubated at room temperature for the 5 minutes. Required amount of Effectene
Transfection reagent was added to the DNA-Enhancer mixture. Mixture was
vortexed for 10 sec. and then

33
in
gr
gr
ad
3.4
Pr
Re
ve
w
tra
Ef
lu
D
as
w
m
de
Lu
10
*In case of transfections performed in 96 well, Effectene Reagent was diluted with buffer EC to a total volume of 20µl before addition to the diluted DNA-Enhancer mixture.
cubated for 10 minutes at room temperature. To the adherent monolayer of cells,
owth media was aspired and cells were washed once with cold PBS. Finally
owth media was added to the transfection mixture complex and immediately
ded drop-wise to the cell monolayer.
Reporter assay.
omoter activity was determined using Firefly luciferase as a reporter gene, while
nilla luciferase vector, pRL-TK (Promega) in the ratio of 1: 20 to experimental
ctor was used as co-reporter in each transfection. 2×104 N2A and HeLa cells
ere plated in 96 well flat bottom plates (Becton Dickinson). After twelve hours,
nsient transfections were performed using 100 ng plasmid DNA and the
fectene® transfection protocol (Qiagen). 48 hours after transfection, Firefly
ciferase activity was measured in an Orion II Micro plate Luminometer (Berthold
etection Systems, Pforzhein, Germany), using the Dual-Luciferase Reporter
say system (Promega). Briefly, media was aspirated and cell monolayer were once
ashed with 1x PBS. Cells were lysed in 20µl of 1x Passive Lysis Buffer (PLB) for 30
inutes at room temperature on a rocking platform. Firefly luciferase activity was
termined using 100µl of LAR II substrate (promega) in the Micro plate
minometer (Berthold Detection Systems). To measure Renilla luciferase activity,
0µl of Stop & Glow (to stop the Firefly luciferase activity and start the Renilla

34
lucifesae activity) assay reagent was added to the above mixture. The Renilla
luciferase activity measurement was used to normalize for differences in transfection
efficiencies between individual transfections. Each experiment was performed 5
times with all constructs tested in triplicates. Data are expressed as mean ± SEM.
For determination of IRES activity, 2×104 N2A and NIH3T3 cells were plated in 96
well flat bottom plates (Becton Dickinson). After twelve hours, transient
transfections were performed using 100 ng plasmid DNA and the Effectene®
transfection protocol (Qiagen). 48 hours after transfection, luciferase activity was
measured as above. IRES activity was expressed as the ratio of Firefly
luciferase/Renilla luciferase (FLuc/RLuc) with the activity of the control vector (pRF-
Di-cis.) set to “1”. Each experiment was performed 5 times with all constructs tested
in triplicates. Data are expressed as mean ± SEM.
3.5 Extraction of cytosolic and nuclear proteins from N2A, NIH3T3, and HeLa cells.
Cytosolic and nuclear proteins were extracted from transiently transfected N2A,
NIH3T3 cells using Active Motif Nuclear Extraction kit (Active Motif Nuclear
Extraction kit, Rixensart, Belgium). Media was aspirated from the cell monolayer and
washed with ice-cold PBS. Cells were gently scraped from the wells and transferred
to 1.5ml eppendorf tube. Cells were collected by centrifugation for 5 minutes at 500
rpm in pre-cooled centrifuge. Various reagents and buffers for the extraction of
cytosolic and nuclear fraction used were according to the scheme shown in table 3
below.

35
Cytoplasmic fraction was obtained by resuspended the cells in 1x hypotonic buffer
by pipetting the cells several times followed by incubated for 15 minutes on ice. 25µl
of detergent was added each to 500µl of hypotonic buffer and vortexed for 10
seconds at highest setting. Suspension was centrifuged for 30 seconds at 14,000 x g in
a microcentrifuge pre-cooled at 4º C and supernatant was transfered (cytoplasmic
fraction) into a pre-chilled microcentrifuge tube. For nuclear fraction, nuclear pellet
was resuspended in complete lysis buffer by pipetting up and down and vortexed
for 10 seconds at highest setting. Suspension was incubated for 30 minutes on ice on
a rocking plate set at 150 rpm. Before centrifugation, it was vortexed for 30 more
seconds at highest setting and then centrifuged for 10 minutes at 14,000 x g in a
microcentrifuge pre-cooled at 4ºC. The supernatant (nuclear fraction) was transferred
into a pre-chilled microcentrifuge tube. Protein estimation was performed with
Bradfords method using BSA as standard.
3.6 Immunoblot Analysis.
For immunoblots, 2x105 N2A, NIH3T3 cells were grown in 12 well plates (Becton
Dickinson). After twelve hours, transient transfections were performed using 300 ng
plasmid DNA and the Effectene® transfection protocol (Qiagen). 48 hours after
transfection, cytosolic and nuclear extracts were prepared according to the
manufactures protocol (Active Motif Nuclear Extraction kit). 20 µg of each protein
fraction was separated on 10% SDS PAGE and then transfered to Hybond N by semi-
dry electroblotting. Membranes were washed three times with PBS for 10 minutes
each and blocked for 60 min with 0.5% low background blocking solution (Roche
Molecular Biochemicals, Mannheim, Germany). Primary antibodies were diluted
1:2000 (anti-GFP; Roche), 1:2000 (anti-PTB; Zymed) 1:7500 (anti-Beta actin; Sigma
Immunochemicals, St. Louis, USA), 1:1000 (anti-Cx43; Zymed) in 0.2% low
background solution and incubated overnight at 4°C on a rocking plate. Thereafter,
membranes were washed three times with PBS-T (PBS with 0.05% Tween 20),
followed by an incubation for 1 hr at RT with a 1:7000 dilution of peroxidase labelled
anti-mouse secondary antibody or 1:2000 dilution of peroxidase labelled anti-rabbit
(ECL-Kit, Amersham-Pharmacia, Buckinghamshire, England). After subsequent

36
washes with PBS-T, antibody binding was visualized by enhanced chemiluminiscent
detection (ECL, Amersham-Pharmacia).
For PTB detection, anti-PTB antibody was included along with the anti-EGFP
antibody. For the loading control, anti-β-actin antibody was used to re-probe the
same membrane. For that purpose, blots were stripped in stripping buffer (100mM 2-
Mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl pH 6.7) and incubated for 30 minutes at
50 oC with occasional agitation. Membranes were washed for 2 x 10 minutes in PBS-T
at room temperature using large volumes of washing buffer. Membranes were then
blocked and processed the same way as above.
3.7 Northern blot analysis.
For Northen blot analysis, 2x106 N2A cells were seeded in 6-well plate (Becton
Dickinson). After 12 hours transient transfection was performed using 600 ng of
various Di-cistronic constructs with the Effectene® transfection protocol (Qiagen).
After 48 hours, the total RNA was extracted using the RNeasy mini kit (Qiagen)
according to manufactures instructions. 10 µg of total RNA was denatured in
formaldehyde and separated on a 1.2% agarose gel in the presence of formaldehyde
and morpholinepropanesulfonic acid (MOPS) buffer. RNAs was transferred onto a
nylon membrane (Amersham Life Science) by the capillary blot procedure. The filter
was cross linked using a UV Stratalinker apparatus (Stratagene, La Jolla, CA USA).
The blots were hybridized with a α 32p-labeled Firefly luciferase DNA probe (856 bp)
using ULTRAhyb hybridization solution (Ambion, Inc., Austin TX, USA). The DNA
luciferase probe was obtained from pGL3-control vector by cleaving it with Nco-I
and Dra-II and labelled using Prime-It RmT Random Primer Labelling Kit
(Stratagene) and 32p dCTP (10µCi/µl; 3000 Ci/nmole) (Amersham).
3.8 RNA analysis.
RT-PCR was performed as a test for mRNA splicing in N2A cells following DNA
transfection of control di-cis vector (pRF-di-cis), IR-1 IRES containing di-cis construct
(pRF-IR-1 di-cis) and IR-2 IRES containing di-cis construct (pRF-IR-2 di-cis). Total
RNA was isolated from transiently transfected N2A cells using the RNeasy mini kit
(Qiagen) according to manufactures instructions. First strand cDNA synthesis was

37
carried out with 1 g total RNA pre-treated with DNase I (Invitrogen) in a 50- l
reaction mixture containing 50 mM Tris-HCl, pH 8.3 (at room temperature, RT), 40
mM KCl, 6 mM MgCl2, 10 mM DTT, 0.5 mM dNTPs, 20 ng random hexamer primers,
and 200U RNA H-Reverse Transcriptase (Superscript II, Invitrogen). After incubation
for 90 min at 42°C, the reaction was terminated by incubation at 70°C for 15 min.
PCR was performed from the C-DNA using the following primers, a´) 5´ GCA GAA
GTT GGT CGT GAG GC 3´ (Sense ) upstream of chimeric intron present in the di-cis
vector, b´) 5´ AGG CTA GCC AAC ATG ACT TCG 3´ (sense) corresponding to the 5`
UTR of Renilla luciferase gene and c´) 5´ GGC GTC TTC CAT GGT GGC CTC 3´
(anti-sense) corresponding to the Firefly luciferase coding region. Cycling conditions
were: one initial cycle for 2 min at 94°C, then 30 cycles for 30 sec at 94°C, 30 sec at
55°C, 2 minute at 72°C, followed by 10 min at 72°C and held at 4°C. Volumes of 15 l
of each amplification reaction were separated on 1% agarose gels and recorded on a
gel documentation system (Imagemaster, Amersham-Pharmacia, Piscataway, NJ).
3.9 EGFP-fluorescence analysis.
For EGFP- fluorescence analysis 2x105 NIH3T3 and N2A cells were grown on 12-mm
poly-L-lysine-coated glass coverslips in 24 well plates. After twelve hours, transient
transfections were performed using 200ng plasmid DNA and the Effectene®
transfection protocol as recommended by the manufacturer (Qiagen). 48 hours later,
sub-confluent cell monolayers were washed once with PBS (Dulbecco's, pH 7.4) and
fixed with 4% paraformaldehyde in PBS for 20 min at 4°C. Cells were washed with
ice-cold PBS and then incubated for 5 min at room temperature with Hoechst 33248
solution (Sigma-Aldrich, St. Louis, MO, USA) diluted 1:10,000 in PBS. Cells were
washed again with cold PBS and mounted with Prolong® Antifade Mounting
Medium (Molecular Probes, Leiden, NL). Fluorescence was documented using a
confocal laser scanning microscope (Zeiss LSM 510, Zeiss, Jena, Germany) equipped
with a krypton/argon laser and a 63x oil objective (1.4 numerical aperture).
3.10 Immunocytochemistry
For the immunofluorescence analysis of FLAG tagged carboxy-terminal domain
(P11-CT) of zfCx55.5, 2x105 NIH3T3 and N2A cells were grown on 12-mm poly-L-

38
lysine-coated glass coverslips in 24 well plates. Transient transfections with FLAG
tagged P11-CT were performed as described above. 48 hour post transfection, cell
monolayers were washed once with PBS (Dulbecco's, pH 7.4) and then fixed with 4%
paraformaldehyde in PBS for 20 min at 4°C. Cells were gently washed twice with
PBS-T for 5 min. Nonspecific binding sites was blocked using 10% normal Fetal calf
serum (NGS) and 0.1% Triton X-100 in PBS for 60 min at RT. Cells were then
incubated with a dilution of anti- FLAG (1:400, Sigma) in 5% NGS for 1 hr at RT.
After several washes with PBS-T cells were incubated with the Alexa 568nm-
conjugated goat-anti-mouse IgG antiserum (Molecular Probes, Eugene, OR) for 1 hr
at RT. Cells were then washed extensively in PBS-T, briefly rinsed in PBS, and
mounted in Prolong® Antifade Mounting Medium (Molecular Probes, Leiden, NL).
Fluorescence was documented using confocal imaging microscopy (Zeiss LSM 510
inverted confocal microscope, argon/krypton and HeNe laser). Cells were imaged
using Plan Apochromat 63× oil (1.4 numerical apertures) objectives (Zeiss).
3.11 Protein expression and purification
Constructs pGEX2TK(huPTB) and the parental control plasmid pGEX6P2 were
transformed into the BL21 host strain (Stratagene) and fusion protein expression was
induced for 16hrs at 30 oC with 1mM IPTG. Bacteria were collected at 5,000g and cell
lysates prepared using the French Press 2-FA-031 (Thermo Spectronic, Rochester,
NY, USA). Precleared lysates were subjected to affinity chromatography using the
Äkta-LC System, GST-Trap FF columns and standard conditions as recommended by
the manufacturer (Amersham Biosciences). Peak fractions were desalted using
HITrap desalting columns (Amersham Biosciences) and concentrated using Amicon
Ultra-4 columns (Millipore) and SDS-PAGE was used to asses the purity of the
protein.
3.12 In-Vitro Transcription
For in-vitro transcription, pT7-IR, pT7-IR (del. PPT-1), pT7-IR (del. PPT2) and pT7-IR
(del.PPT 1-2) were linerazied 3´ to the IRES element using Xho I restriction site. For
internally labelling RNA, in-vitro transcription was performed using MAXIscript T7
Kit (Ambion, Inc., Austin TX, USA) in accordance to the manufactures instructions.

39
Briefly, reaction was set in 20µl volume using 1µg of template DNA, 2µl of 10 x
Transcription buffer, 10 mM each of ATP, GTP, UTP and 1 mM of CTP. 5µl of α32P
CTP (10µCi/µl; 3000 Ci/nmole) (Amersham) was included in the reaction mixture.
The mixture was incubated at 37 oC for 1 hour and 1µl of DNase I (2U/µl) was added
to the reaction mixture and incubated at 37 oC for 15 minutes. Labelled RNA probes
were purified using the G-50 sephedex columns (Amersham). Unlabelled competitor
RNA was synthesized using MEGAscript™ T7 Kit (Ambion).
3.13 RNA-EMSA
Internally labelled wild type RNA IRES and its various deletion mutants were used
for elctromobility shift assay (EMSA). Approximately 20,000 cpm of the radio probe
were mixed with 30µg of cytosolic protein prepared from N2A cells or 0.3µg of
purified GST-PTB fusion protein, in a buffer mix containing 2µl of 5x binding buffer
(100mM Hepes (7.4), 3mM MgCl2, 100mM KCl, 1.3mM ATP, 1mM DTT and 6% v/v
glycerol), 40U of RNase inhibitor (Fermantas), 1.5 µl of t-RNA (10mg/ml) or 0.5µl of
t-RNA in case of the purified GST-PTB fusion protein, in a reaction volume of 10µl at
room temperature for 30 minutes. For competition, five minutes after the addition of
ribonucleic probe, unlabelled RNA was added to the reaction mixture. The
ribonucleic-protein complexes were electrophorised at 200V for about 3 hours in a
4% non-denaturing poly acrylamide gel using 1x TBE (0.045 M Tris-borate, 0.001 M
EDTA). After electrophoresis the gel was transferred to Wattmann paper, dried
under vacuum for 30 minutes in gel dryer (Bio Rad) and then visualized by
autoradiography.
3.14 UV-cross linking
RNA-protein complex for UV-cross linking were prepared as described above. For
cold competition, unlabeled RNA was added 5 minutes after the addition of the
radio probe. After 30 minutes of incubation at room temperature the samples were
transferred to ELISA plates and irradiated with UV light in a UV stratalinker
(Stratagen) for 30 minutes. RNase cocktail, 2µl of RNase A (10mg/ml) and 1µl of
RNase T1 (100 units) (Fermantas), was added and the samples were incubated at 37 oC for 30 minutes. RNA-protein complexes were then resolved on 10% by sodium-

40
dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE) for 3 hours at 200V.
Subsequently the gel was dried under vacuum and visualized by autoradiography.
3.15 DNA-EMSA
For the DNA mobility shift assay, specific primers corresponding to putative
promoter regions of zfCx52.6 and zfCx55.5 were used. Genomic sequences
corresponding to specific parts in the 5′-flanking region of the zfCx52.6 were PCR
amplified using the primer pairs as shown in table 4 below.
For competition with CCAAT and Oct-1 oligos, the following DNA oligos were used:
Sense CCAAT oligo 5´ ACACACCAATCAGCT 3´ and anti-sense CCAAT oligo 5´
AGCTGATTGGTGTGT 3´. Sense Oct-1 oligo 5´ TACATTGAAATGTATACA 3´ and
anti-sense Oct-1 oligo 5´ TGTATACATTTCAATGTA 3´. The respective oligo were
mixed in equal proposition in Tris-EDTA buffer and boiled for 5 minutes. The oligo
were let to anneal at 37 oC for two hours and then at room temperature for two more
hours.
The PCR products were separated on 1.5% agarose gel and gel purified using the
Qiaex-II Gel extraction Kit, (Qiagen). The purified PCR products were digested either
with NheI or BglII restriction enzymes (Fermantas) and then ethanol precipitated.
For end labelling, 100ng of digested PCR products were Klenow filled with Klenow
fragment of DNA polymerase I (Fermantas) using 32P dCTP (10µCi/µl;
3000Ci/mmole) (Amersham). The labelled DNA fragments were purified by a
Sephadex G-50 column chromatography (Amersham). Nuclear extracts from N2A
and HeLa cells were prepared using Active Motif nuclear extraction Kit as described
previously. DNA-protein binding reactions were carried out in 10µl of 2x binding
buffer (containing 4% glycerol, 2.5mM MgCl2, 0.5 mMEDTA, 0.5 mM dithiothreitol
(DTT), 75mM KCl, 25mM Hepes (pH 7.6), 1µl of poly dI.dC. poly dI.dC (1µg/µl) and
20µg of nuclear proteins. Where appropriate, an unlabeled homologous ds-DNA was
added to the reaction mixture at 100-fold molar excess. The reaction mixtures were
incubated for 5 min at 4 oC on ice, after which 15,000 to 20,000 cpm (0.1 to 0.4 ng) of
end labelled probe was added to each tube and tubes were further incubated at 4 oC
on ice for further 25 minutes.

41
The resultant protein-DNA complexes were resolved by electrophoresis through 4%
native polyacrylamide gels in 44.5 mM Tris base (pH 8.0), 44.5 mM boric acid1 mM
EDTA. Gels were pre-run at 50 mA for 30 min, after which the samples were run at
the same current for 1.5 to 2 h, dried on Whatman 3MM paper, and visualized by
autoradiography.

42
4. Results 4.1. Identification of putative promoter elements in zf Cx55.5 and zfCx52.6
In order to elucidate the molecular mechanism responsible for the site restricted
expression of the zfCx55.5 and zfCx52.6, we investigated the regulation at the
primary level, i.e basal transcription of these genes. First step towards this finding is
to characterize their promoter elements. For this purpose, upstream region of
zfCx55.5 (~3.5kb) was screened for potential promoter elements. Different
upstream DNA fragments of zfCx55.5 were sub-cloned in pGL3-Basic vector
and transiently transfected in HeLa and N2A cells. 48 hour post transfection,
Firefly luciferase activity were measured. Luciferase activity showed the presence
of two putative DNA constructs which showed enhanced luciferase activity
relative to the pGL3-Basic vector, as shown in Fig 4.1
Fig 4.1 Promoter activity of zfCx55.5 upstream DNA fragments in HeLa (white bars)
and N2A (black bars) cell lines. Promoter activity of different DNA fragments is depicted as the luciferase activity. Luciferase activity of
control vector (pGL3-Basic) was set to “1”and the luciferase activity of different DNA fragments is
represented as fold of control vector pGL3-Basic. Each number of the DNA fragments represents the
nucleotide position relative to the translation start site. The Renilla luciferase activity measurement
were used to normalize for differences in transfection efficiencies between individual transfections.

43
Each experiment was performed 5 times with all constructs tested in triplicates. Data are expressed as
mean ± SEM. DNA fragment, -3915/-2004 ( ) showed 13 fold higher activity as compared to control
pGL3-Basic vector while DNA fragment -881/+134 ( ) showed 8-fold higher activity then control vector.
One of the DNA construct, - 88 / +134 ( relative to translation start site )
showed approximately 8-fold higher activity than the pGL3-Basic vector, while
the DNA construct, (- 3915 /- 2004) showed approximately 13 fold higher
activity relative to pGL3-Basic vector. Promoter element - 881/ +134 (designated
here as promoter II) was located flanking the translational start site of coding
exon II, while the stronger promoter element (designated here as promoter I) was
located upstream of the short exon I.
Promoter element I (- 3915/-2004) was further characterized to find the minimal
DNA sequence required for its activity. Different deletion constructs were prepared
as described in Materials and Methods. Deletion construct (-3166/-2004 pGL3-basic)
having approximately ~750bp deleted at the 5´ end of promoter I construct (- 3915
/- 2004) showed an increases the promoter activity from 13-fold to 22-fold
relative to pGL3-Basic (Fig 4.2). This hints towards the possibility of a repressor
element located in the 5´ end of -3915 /-2004 promoter element.
Fig 4.2. Characterization of promoter element I of zfCx55.5. Promoter element I was further delineated into 3- fragments for the minimal promoter activity. Promoter
activity of different DNA fragments is depicted as luciferase activity. Luciferase activity of control vector
(pGL3-Basic) was set to “1”and the luciferase activity of different DNA fragments was represented as fold
of control vector pGL3-Basic. Each number of the DNA fragments represents the nucleotide position
relative to the translation start site. The Renilla luciferase activity measurement were used to

44
normalize for differences in transfection efficiencies between individual transfections. Each experiment
was performed 5 times with all constructs tested in triplicates. Data are expressed as mean ± SEM. By
removing the ~750bp (-3915/-3166) from promoter element I (-3915/-2004), the activity increased from 13
fold to 22 fold (-3166/-2004).
4.2. Confirmation of the zfCx55.5 promoter specificity in transgenic fish.
To investigate the efficacy of the promoter element of zfCx55.5 in the zebrafish,
transgenic fish was generated (in collaboration with Marteen Kamermann group)
using the 5´ upstream DNA fragment of zfCx55.5 (-4919nt to -1nt, relative to
translation start site) sub-cloned in the promoterless pGAL4 vector. Fertilized eggs
were double injected with pGAL4 zfCX55.5 and pUAS EGFP. The expression was
monitored as an indication of promoter activity. As shown in Fig 4.3, 96 hour post
fertilization EGFP expression was exclusively detectable in the eyes of transgenic fish
(A). Confocal laser scanning microscopy of sections of the retina revealed that EGFP
expression was confined to a band of cells at the border between the INL and
OPL/ONL (B). The specificity of EGFP protein was confirmed by
immunocytochemistry using EGFP specific anti-body (C). The localization and
morphology is indicative for the horizontal cells. This result is consistent with our
previous expression analysis in vivo.
Fig.4.3. Specificity of zfCx55.5 promoter element in zebrafish retina. Transgenic fish was
generated using the EGFP reporter gene, driven by the putative promoter element of zfCx55.5. A) Dorsal
lateral view of 96 hour post fertilization transgenic zebrafish eye (bar=500µm) B) EGFP fluorescennce
from the horizontal cell layer of transgenic zebrafish. C) Immmunohistochemistry of transgenic fish
retina using anti-GFP as primary anti-body showing the exclusive labeling of horizontal cell layer. D)
Merge of B & C (bar=10µm) (in collaboration with Marteen Kameramm)

45
Similarly, a 1.9kb upstream region of zf.Cx52.6 was screened for the existence of a
putative promoter. Various upstream DNA fragments were sub-cloned in the pGL3-
Basic vector. Transient transfections in HeLa and N2A cells were performed and 48
hour post-transfection Firefly luciferase activities were measured. Subsequent
luciferase readings showed that there are different requirements, in terms of
length of DNA fragments for the promoter activity in HeLa and N2A cells, as
shown in Fig 4.4.
Fig 4.4. Promoter activity of zfCx52.6 upstream DNA fragments in HeLa (White bars)
and N2A (Black bars) cell lines. Promoter activity of different DNA fragments was depicted as luciferase activity. Luciferase activity of
control vector (pGL3-Basic) was set to “1”and the luciferase activity of different DNA fragments was
represented as fold of control vector pGL3-Basic. Each number of the DNA fragments represents the
nucleotide position relative to the translation start site. The Renilla luciferase activity measurement
were used to normalize for differences in transfection efficiencies between individual transfections.
Each experiment was performed 5 times with all constructs tested in triplicates. Data are expressed as
mean ± SEM. In HeLa cells, DNA construct - 1905 /-135 showed 14-fold higher activity
relative to the pGL3-basic vector. The DNA constructs, -1905/-778 and -1162/-
315 showed lower activity than the entire fragment of -1905/-135. In N2A
cells, DNA construct, 1905/-135 was completely silent, while only 5´ flanking

46
region of this fragment -1905/-778 is required for the full promoter activity.
These results indicate the possibility of a putative repressor element in the DNA
fragment -778 to-135 which is specific in N2A cells only and not in HeLa cells.
Above data led us to investigate the possible transcription factors which are
responsible for the basal transcription of these genes.
4.3. Specific protein complex binds to promoter element I and promoter element II
of zfCx55.5 and the promoter element of the zfCx52.6
To get an insight into the protein factors which recognize the promoter elements of
zfCx55.5, DNA mobility shift assays were performed. For this purpose, end labelled
DNA probes of promoter element I and promoter element II were incubated with
nuclear extract prepared from N2A cells, as described in Material and Methods. As
shown in Fig 4.5, DNA fragment -3179/-3029 showed two retarded DNA- protein
bands.
Fig 4.5. DNA mobility shift assay of the promoter element I and II of zfCx55.5. Nuclear extract from the Neuroblastoma N2A cell line was prepared as described in Material and
Methods. A 20µg amount of nuclear proteins was incubated with 15,000 to 20,000 cpm of the 32 P-end
labeled DNA probes. Protein-DNA complexes were resolved by electrophoresis through 4% native
polyacrylamide gel and visualized by autoradiography. Where appropriate, an unlabeled homologous ds-
DNA was added to the reaction mixture at 100-fold molar excess as competitor. Promoter element I: DNA
fragment -3179/-3029 showed two specific DNA-protein-complexes (lane 2), similarly DNA fragment -
2910/-2788 showed two specific DNA-protein complexes (lane 5), whereas DNA fragment -2794/-2658
showed one specific complex (lane 8). Promoter element II: DNA fragment -199/-49 showed one specific
DNA-protein-complexes (lane 11) (NE= nuclear extract; “arrow” indicates specific banding and
“arrowheads non-specific banding)

47
Similarly two retarded DNA-protein bands were detected with the DNA fragment -
2910/-2788. DNA fragment -2794/-2658 showed a single DNA-protein retarded
band. Formations of all these complexes were effectively inhibited by the inclusion
of 100-fold molar excess of homologous unlabeled competitor DNA.
In case of promoter element II of zfCx55.5, DNA fragment -199/-49 showed two
retarded DNA-protein complexes which can be competed out by adding the
unlabeled homologous DNA.
The promoter element of zfCx52.6 was also studied to identify the protein complexes
which bind to this element using nuclear extract from N2A cells. As shown in Fig 3.6,
a DNA fragment -1433/-1302 (lane 2) showed single retarded band of DNA-protein
complex and similarly the DNA fragment -1215/-1096 (lane 5) showed similar
retarded DNA-protein complex. Formations of these complexes were effectively
inhibited by the inclusion of 100-fold molar excess of homologous unlabeled
competitor DNA.
Fig 4.6. DNA mobility shift assay of the promoter element of zfCx52.6. Nuclear extract from the Neuroblastoma N2A cell line was prepared as described in Material and
Methods. A 20µg amount of nuclear proteins was incubated with 15,000 to 20,000 cpm of the 32 P-end
labeled DNA probes. Protein-DNA complexes were resolved by electrophoresis through 4% native
polyacrylamide gel and visualized by autoradiography. Where appropriate, an unlabeled homologous ds-
DNA was added to the reaction mixture at 100-fold molar excess as competitor. DNA fragment -1433/-
1302 showed one specific DNA-protein-complexes (lane 2), similarly DNA fragment -1215/-1096 showed
one specific DNA-protein complexes (lane 5). (“arrow” denotes specific binding, “arrow head” denotes
non-specific binding)

48
4.4. Preliminary evidence for the binding of CCAAT binding protein (CBP) and
OCT-1 to the promoter element of zfCx52.6
Sequence analysis of zfCx52.6 DNA fragments showed the presence of consensus
CCAAT binding site in the DNA fragment -1433/-1302 and an OCT-1 binding site in
the DNA fragment -1215/-1096. To check the possibility of binding of these proteins
to these DNA fragments, small oligos corresponding to CCAAT binding site and an
OCT-1 binding site were synthesized. As shown in Fig 4.7, DNA-protein complex
formed by the DNA fragment -1433/-1302 was competed out using 50- fold molar
excess of CCAAT oligo. Similarly the DNA-protein complex formed by the DNA
fragment -1215/-1096 was effectively competed out using 50- fold molar excess of
OCT-1 oligo. Further experiments, like the super-shift assay using the specific
antibody against these factors and mutational analysis of binding sites will further
unravel the role of these factors in the gene regulation.
Fig 4.7. DNA mobility shift assay of the potential CCAAT and Oct-1 binding sites of
promoter element of zfCx52.6. Sequence analysis of DNA fragment -1433/-1302 of promoter element
of zfCx52.6 showed a potential CBP site and the DNA fragment -1215/-1096 showed potential Oct-1
protein binding site. To check the possibility of the binding of these transcription factors, EMSA was
performed with the N2A nuclear extract and the specific DNA-protein complexes were competed out
using CCAAT oligos and Oct-1 oligos, as described in Material and Methods. DNA fragment -1433/-1302
binds a protein complex (lane 2) which was effectively competed out using 50 fold molar excess of CCAAT
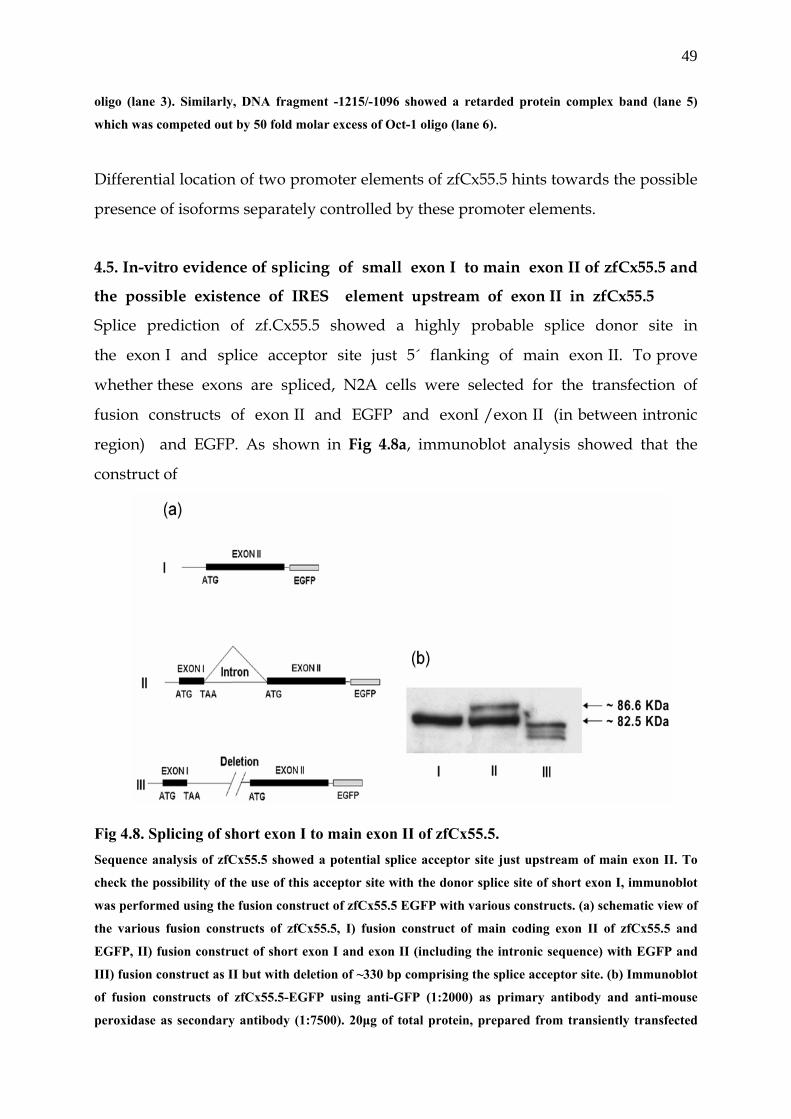
49
oligo (lane 3). Similarly, DNA fragment -1215/-1096 showed a retarded protein complex band (lane 5)
which was competed out by 50 fold molar excess of Oct-1 oligo (lane 6).
Differential location of two promoter elements of zfCx55.5 hints towards the possible
presence of isoforms separately controlled by these promoter elements.
4.5. In-vitro evidence of splicing of small exon I to main exon II of zfCx55.5 and
the possible existence of IRES element upstream of exon II in zfCx55.5
Splice prediction of zf.Cx55.5 showed a highly probable splice donor site in
the exon I and splice acceptor site just 5´ flanking of main exon II. To prove
whether these exons are spliced, N2A cells were selected for the transfection of
fusion constructs of exon II and EGFP and exonI /exon II (in between intronic
region) and EGFP. As shown in Fig 4.8a, immunoblot analysis showed that the
construct of
Fig 4.8. Splicing of short exon I to main exon II of zfCx55.5. Sequence analysis of zfCx55.5 showed a potential splice acceptor site just upstream of main exon II. To
check the possibility of the use of this acceptor site with the donor splice site of short exon I, immunoblot
was performed using the fusion construct of zfCx55.5 EGFP with various constructs. (a) schematic view of
the various fusion constructs of zfCx55.5, I) fusion construct of main coding exon II of zfCx55.5 and
EGFP, II) fusion construct of short exon I and exon II (including the intronic sequence) with EGFP and
III) fusion construct as II but with deletion of ~330 bp comprising the splice acceptor site. (b) Immunoblot
of fusion constructs of zfCx55.5-EGFP using anti-GFP (1:2000) as primary antibody and anti-mouse
peroxidase as secondary antibody (1:7500). 20µg of total protein, prepared from transiently transfected

50
N2A cells, was resolved on 10% SDS polyacrylamide gel and immunodetected using anti-GFP antibody.
Lane I) fusion construct of exon II with EGFP showing the expected fusion protein band of ~82.5 kDa
(55.5 kDa + 27kDa), lane II) fusion construct of exon I and exon II (including the intronic sequence)
showing the fusion protein band of~82.5 kda and an additional fusion protein band of ~86.6 kDa. Lane
III) deletion construct, having ~330 bp deleted from the intronic sequence. Note that after deletion of the
~330bp fragment some higher mobility protein bands became visible.
exon II and EGFP resulted in an expected protein band of ~ 82.5 KDa ( 55.5
exon II + 27 EGFP ), while the construct of exon I /exon II EGFP showed two
protein bands, one of which migrated at the same level as that of exon II band
( 82.5 KDa ), and a higher band of approximately ~ 86.8 KDa. This band can be
explained only when exon I is spliced to exon II. Moreover, the co- expression
of a exon II in the exon I / exon II EGFP construct can be mechanistically due
to the presence of a promoter element or IRES element in the 5´ flanking end
of main exon II. The presence of the splice site and possible promoter/ IRES
elements in the 5´ flanking sequence of exon II was confirmed by deleting ~
330 bp (intronic region) upstream of the ATG of exon II. Immunoblot detection
showed that by deleting the ~330bp from the exon I/ exon II EGFP construct,
the expression of both isoforms was abolished Fig 4.8b. Few immuno-reactive
bands of higher mobility found in the deletion construct may be due to leaky
scanning from downstream ATGs present in the coding region of main exon II.
In summary, the basal transcription of zfCx55.5 seems to be controlled by two
promoter elements with the possible two isoforms generated by splicing. The basal
promoter of zfCx52.6 seems to be regulated by CCAAT binding protein and Oct-1
transcription factor.
Translation study of zfcx55.5
Most of the connexins have their coding information present in single exon, but still
they posses’ number of short exons which form part of 5´ un-translated region (UTR)
of these genes. Presence of variable 5´ UTR is a feature of those genes which are
under strict translational control. Moreover, immunoblot of zfCx55.5 detects not only
the main protein but also some higher mobility bands which hint towards the
possible translational regulation of this connexin.

51
4.6. Full length zfCx55.5 and a portion of its carboxy-terminal domain are co-
translated.
To investigate the possible molecular mechanism which is responsible for the
generation of higher mobility bands of zfCx55.5, we engineered a fusion construct of
the coding region of zfCx55.5 with EGFP using the pEGFP-N3 vector. After transient
transfection into N2A cells, a whole cell extract was prepared 48 hours post
transfection. 20µg of total protein was separated on a 10%SDS gel. The zfCx55.5EGFP
fusion protein with a calculated molecular weight of 82.5kDa (55.5kDa for zfCx55.5 +
27kDa EGFP) was detected with a monoclonal anti-GFP antibody. Repeatedly higher
mobility bands became apparent aside of the expected protein including a fusion
protein band of ~38 kDa (11kDa + 27kDa EGFP) (Fig 4.9C, lane I). A DNA sequence
analysis of the coding region of zfCx55.5 showed the presence of several in frame
AUG codons of which one was present at the beginning of the carboxy-terminal
domain (CT; bp634) and another one at nucleotide position 1202 with a near perfect
Kozak sequence (Fig 4.9A). We proposed that the fusion protein of ~38kda is
translated from the in-frame AUG codon at nucleotide position 1202 in the CT
domain of zfCx55.5 on the basis of the near perfect start codon (ccagcATGG). To
further confirm that this protein is indeed derived from the CT portion of zfCx55.5,
we made two additional fusion proteins, one of which corresponds to the full length
CT (bp634 to bp1497), having its own in-frame AUG codon and a second construct
that starts from bp946 to bp1497 (including the 5` sequence of the AUG codon at
position 1202) (Fig 4.9B). Transient transfections into N2A cells and subsequent
Western blot detection with the anti-GFP antibody showed a band at ~60kDa (33kDa
CT-domain + 27kDa EGFP) corresponding to the full length CT and an additional
prominent band at ~38kDa (Fig.4.9C, lane II). The DNA construct with the zfCx55.5
sequence from nt946 to nt1497 showed the expected fusion protein band of ~38kDa
(Fig.4.9C, lane III). In all constructs a double protein band corresponding to ~38kDa
was detected. The expression of the ~38kDa fusion protein (in the following termed
p11-CT referring to the calculated molecular weight of the zfCx55.5 CT domain) fits
the criteria of internal translation from an in-frame AUG codon at nt1202 in the
coding region of zfCx55.5.

52
Fig 4.9. Simultaneous expression of zfCx55.5 and its carboxy-terminal domain: (A) Nucleotide sequence of the 5` end of the carboxy-terminal domain, with in-frame AUG codons at
nucleotide positions 634 and 1202 shown in bold. (B) Schematic representation of the EGFP-fusion protein
constructs of full length zfCx55.5 (I) (nt1 to nt1497), carboxy-terminal domain of zfCx55.5 FL-CT (II)
(nt634 to nt1497) and 3` half of carboxy-terminal domain p11-CT (III) (nt946 to nt1497). (C) Western blot
analysis of transiently transfected N2A cells with EGFP fusion constructs: (lane I) full length zfCx55.5,
(lane II) full length carboxy-terminal domain (FL-CT) (lane III) 3´ half of the carboxy-terminal domain
(p11-CT). Immunodetection was performed using anti-GFP as the primary antibody and a peroxidase
labelled anti-mouse IgG antibody for ECL detection. Note: Full length zfCx55.5 construct (lane I), besides
82.6kDa and 38kda fusion protein bands, a few N-terminally truncated zfCx55.5 protein bands are also
visible.
4.7. The carboxy-terminal protein (p11-CT) is translated from the zfCx55.5
transcript via internal translation.
To elucidate the molecular mechanism responsible for the expression of the p11-CT

53
protein, we introduced a frame shift mutation at bp 1179 in the zfCx55.5 coding
region. This construct was transiently transfected in N2A cells. 48 hours post
transfection, cell extracts were prepared and separated on 10% SDS PAGE. Western
blot detection using the anti-GFP antibody showed that by creating the frame shift,
translation of full length zfCx55.5 was completely abolished, when compared with
the non-mutated full length zfCx55.5 (Fig. 2B, lane I) while the p11-CT protein can be
still detected (Fig. 4.10B, lane II).
The disappearance of the full length protein and the persistent expression of p11-CT
is a clear indication that a cleavage mechanism cannot be responsible for the
generation of this carboxy-terminal protein. In fact, above results indicate that the
p11-CT protein is translated from the in-frame AUG codon at nucleotide position
1202. To further prove this concept, we modified the in frame AUG (nt1202) codon to
GCG and expressed the mutation in N2A cells. Immunoblot detection using anti-GFP
antibody indicated the presence of full length zfCx55.5, while the expression of the
p11-CT protein was completely abolished (Fig 4.10B, lane III)
Fig 4.10. A part of the carboxy-terminal domain of zfCx55.5 is translated from an
internal translation site within the coding region of zfCx55.5: (A) Schematic view of wild type full length WT (I), frameshift mutated (position 1179) full length, F-WT
(II), and in-frame AUG replaced by GCG (position 1202) of full length zfCx55.5 construct (III). (B)
Western blot of transiently transfected N2A cells with: (lane I) Wild type (WT), (lane II) frameshift

54
mutated F-WT, and (lane III) AUG replaced by GCG of full length zfCx55.5. Immunodetection was
performed using anti-GFP as primary antibody and peroxidase labeled anti-mouse antibody for ECL
detection.
4.8. An IRES element in the coding region of zfCx55.5 is responsible for the
expression of the p11-CT protein.
To uncover the possible mechanism of internal translation of p11-CT, we sub-cloned
a fragment of the coding region from nucleotide position 631 to1201 which
constitutes a fragment of 510bp ahead of the in-frame AUG start codon at nt1202 into
the di-cistronic vector pRF Dicis. The construct, pRF-IR1 (carrying the coding region
from nt631 to nt1201 subcloned in the inter-cistronic region of the dicistronic vector)
along with the control pRF Di-cis vector (Fig. 4.11A, I, II) was transiently transfected
into N2A cells. Renilla and Firefly luciferase activity were measured 48 hours post
transfection. Luciferase activity readings depicted that the fragment was able to
enhance the expression of the downstream located Firefly luciferase cistron by ~15
fold as compared to the control vector pRF Di-cis (Fig 4.11 B). IRES activity was
calculated as the ratio of the activity of the Firefly luciferase to the activity of the
Renilla luciferase. The pRF-IR1 construct was transiently transfected into HeLa and
NIH3T3 cell lines to prove whether this putative IRES element is active in other cell
lines. 48 hours after transfection, luciferase activity was similar in HeLa and N2A
cells, while in NIH3T3 cells the activity was increased ~ 25 fold compared to the
control pRF-Di-cis vector (Fig 4.11 B).

55
Fig 4.11. Identification of an IRES element in the coding region of zfCx55.5 using Di-cis
vectors. (A) Schematic map of the (I) pRF –Di-cis vector having Renilla luciferase (RLuc) as the first
cistron and Firefly luciferase (FLuc) as the downstream cistron, with two stable stem-loops or hairpins
(HP), one at the 5´ end and another at 3` end of Renilla luciferase gene. (II) pRF-IR1 Di-cis construct
having coding region (from nt631 to nt1202) of the CT in the intercistronic region, and (III) pRF-IR2
construct having the coding region (from nt631 to nt990) of the CT in the intercistronic region. (B). IRES
activity of the above three constructs in N2A, HeLa and NIH3T3 cells. IRES activity is represented as
ratio of Firefly to Renilla luciferase activity (FLuc / RLuc) with the activity of the control vector, pRF-Di-
cis set is at 1. Each construct was tested five times and each experiment was done in triplicate. Data are
expressed as mean ± SEM
To further delineate the putative IRES element, the pRF-IR1 construct was truncated
by removing a ~200bp fragment from the 3` end of the pRF-IR1 construct. This new
construct pRFIR2 (Fig. 4.11A, III) with a shortened zfCx55.5 CT domain (nt631 to
nt990) along with the construct carrying the entire fragment (pRF-IR1) and the
control pRF-Di-cis vector were transiently transfected into N2A, HeLa and NIH3T3
cells. Subsequent luciferase activity determination indicated a substantial overall
increase of IRES activity. The increase over control levels was, ~34 fold in HeLa, ~35
fold in N2A and ~77 fold in NIH3T3 cells (Fig 4.11B). This observation indicates that
the DNA sequence immediately upstream of the in frame (nt1202) AUG codon
exhibits a regulative function on the IRES activity.
3.9. Increased expression of the second cistron in the Di-cistronic assay is due to
the IRES activity and not to a cryptic promoter.
To rule out the possibility that the increased expression of the second cistron in the
Dicistronic assay is due to cryptic promoter activity, promoterless Di-cistronic
constructs [pRF-Dicis (-P), pRF-IR1 (-P) and pRF-IR2 (-P)] were prepared from pRF,
pRF-IR1 and pRF-IR2 constructs by removing the CMV promoter (Fig 4.12A).
Each promoterless Di-cis construct was transiently transfected into N2A, HeLa and
NIH3T3 cells. The ratio of Firefly to Renilla luciferase activity showed a marginal 4 to
8 fold increase as compared to the control vector indicating that the IRES element
and not a cryptic promoter activity is responsible for the increased expression (Fig
4.12B, C)

56
An additional confirmation of the presence of a putative IRES element or cryptic
promoter element was done by Western blot analysis. For this purpose pRF-Di-cis,
pRF-IR1 and pRF-IR2 vectors were modified by removing the Firefly luciferase
cistron and replacing it with the EGFP gene in the position of the second cistron (Fig
4.13A). All constructs were transiently transfected into N2A cells and cell lysates
were prepared 48 hours after transfection. Immunodection with anti-GFP antibodies
showed a ~10fold enhanced expression of EGFP in the presence of the IRES element
(pR-EGFPIR- 2; lane III) as compared to control vector pR-EGFP (lane I). The
promoterless control vector, pR-EGFP (-P) (lane II) showed a faint expression of
EGFP while the promoterless IRES vector, pR-EGFP-IR2 (-P) (lane IV) showed no
expression at all (Fig 4.13B).

57
Fig 4.12. IRES activity versus cryptic promoter activity of the coding region of zfCx55.5. (A) Schematic view of the Di-cis constructs with the respective promoterless Di-cis constructs. (B) and (C)
IRES activity and cryptic promoter activity of the above constructs in N2A and HeLa cells (B), and
NIH3T3 (C) cells. IRES activity is represented as the ratio of Firefly to Renilla luciferase (FLuc / RLuc)
with the activity of the control vector, pRF-Di-cis, set at “1”. Each construct was tested three times and
each experiment was done in triplicate. Data are expressed as mean ± SEM
Fig 4.13. Confirmation of the IRES element by Western blot analysis (A) Schematic
representation of the Di-cis vectors having EGFP as a second cistron, with pR-EGFP as control vector (I),
pR-EGFP(-P) as promoterless control vector (II), pR-EGFP-IR2 having IRES element IR2 in the inter-
cistronic region (III) and its promoterless construct(IV). (B) Western blot of the Di-cis constructs
transiently transfected in N2A cells: lane I) control vector (pR-EGFP), lane II) promoterless control
vector pR-EGFP(-P), lane III) having the IRES element IR2 (nt631 to nt990) in the inter-cistronic region
of the Di-cis vector (pR-EGFP-IR2) and lane IV) promoterless IR2 Di-cis vector (pR-EGFP-IR2 (-P). 10µg
of total protein was loaded on 10 % SDS gel and immunoblot detection was done by using anti-GFP
(1:2,000) as primary antibody and peroxidase labeled anti- mouse IgG (1:7,500) as secondary antibody.
(C) Western blot of β-actin as loading control.

58
4.10. The p11-CT protein is not expressed from a monocistronic mRNA
Next, we have excluded the possibility that the expression of the second cistron was
derived from a monocistronic mRNA generated either by a specific ribonuclease
cleavage of the IRES element or by cryptic splicing mechanism. A Northern blot
analysis of the Di-cistronic vectors with and without the IRES element was
performed. As a positive control, pGL3-control vector with the SV40 promoter was
used to detect the Firefly mono-cistronic mRNA. As shown no monocistronic
message comparable to the positive control was identified excluding the possibility
of mRNA cleavage as the primary cause for the formation of the p11-CT protein
product (Fig 4.14A).
To detect possible splice variants which evade Northern detection, total RNA from
the transiently transfected N2A cells was subjected to RT-PCR as described in the
Material and Methods. PCR products were separated on 1% agarose gel and
visualized by ethidium bromide (EtBr) staining (Fig 3.13D). PCR using the C-DNA
from control di-cis vector with the primer pairs, b´/c´, resulted in a single PCR
product (~1100 bps) (Fig 4.14D, b´/c´ lane I). Similarly, PCR from the C-DNA of IR-1
and IR-2 di-cis constructs using the primer pairs, b´/c´, resulted in a single PCR
product (~1600 bps) corresponding to the IR-1 construct (Fig 4.14D, lane II) and a
single PCR product (~1400 bps) corresponding to the IR-2 construct (Fig 4.14D, lane
III). Primer pairs, a´/c´ (corresponding to the 5´upstream of intron), did not result in
any amplification of PCR product from the three constructs (Fig 4.14D, a´/c` C-DNA,
lane I, II, III), while using the same primer pairs (a´/c`) for the PCR amplication
from the plasmid DNA of the three constructs resulted in single PCR product (Fig
4.14D, a´/c´ DNA, lane I, II, III). The most feasible explanation for the lack of any
amplicon from the C-DNA of the above three constructs using the primer pair a´/c`
is that the transcriptional start site of the full di-cistronic mRNA starts after chimeric
intron, thus excluding the intron from the mRNA. A PCR product of around 450bps
from IR-2 construct (b´/c´ C-DNA, lane III) seems to be unlikely a splice out product
as same band was also observed in the DNA sample (a´/c´ DNA, lane III).

59
Fig. 4.14. Northern blot and RT-PCR analysis of Di-cis constructs: (A) Northern blot
analysis of various Di-cistronic mRNA expressions in N2A cells transiently transfected with the pGL3
control vector (lane I), control Di- cis vector (pRF-Di-cis; lane II), IR-1 (nt631 to nt1201) Di-cis construct
(pRF- Di-cis-IR1; lane III), and IR-2 (nt631 to nt990) Di-cis construct (pRF-Di-cis-IR2; lane IV). 5µg total
RNA was loaded per lane. (Lane I) pGL3-control vector as a positive control for the detection of the
monocistroinc message. The Northern blot was hybridized with a Firefly luciferase probe. Note: each
construct shows some cross-reactivity of the Luciferase hybridization probe with both 28S and 18S
ribosomal RNA(*) (rRNA)
(B) Ethidum bromide stained agarose gel of (A) demonstrating a comparative loading of total RNA, with
the 18S and 28S rRNA bands marked.
(C) Schematic view of various di-cis constructs used for the RT-PCR analysis. , represents the
chimeric intron, while broken arrows represents the primers used for RT-PCR analysis.
(D) N2A cells were transiently transfected with control di-cis vector, pRF di-cis (I), pRF-IR1 di-cis
construct (II) and pRF-IR-2 di-cis vector (III). 24 post transfection, total RNA was isolated and subjected
to RT-PCR, and PCR products were separated on 1% agarose / EtBr gel and visualized by UV
illumination with 100bp DNA ladder on the extreme left of the gel. NTC, denotes no template control, (**),
represents non-specific amplicon and the template DNA.

60
4.11. The p11-CT product can translocate to the nucleus.
Finally, we have analyzed the intracellular localization of the p11-CT protein. It was
fused in-frame with EGFP using the pEGFP-N3 vector. N2A and NIH3T3 cells,
grown on polylysine coated cover slips, were transiently transfected with the above
construct. 48 hours post transfection, cells were analyzed by confocal laser scanning
microscopy. The subcellular distribution of the EGFP-fluorescence showed the
presence of the fusion protein product in the cell nucleus and cytoplasm (Fig 4.15A).
In order to confirm the presence of the p11-CT product in the nucleus Western-blot
analysis was performed on nuclear extracts prepared from transiently transfected
N2A cells. A band of ~38 kDa was detected correlating molecular identity with
subcellular distribution (Fig 4.15B).
Fig 4.15. Localization of the carboxy-terminal domain in the nucleus of N2A and
NIH3T3 cells. (A) Confocal laser scanning imaging of the p11-CT-EGFP fusion protein in the nucleus
of NIH3T3 cells and N2A cells. A single optical section of 0.7µm was recorded. (B) Western blot analysis
of a nuclear extract of p11-CT-EGFP (nt946 to nt1497). The nuclear extract was prepared from
transiently transfected N2A cells. 10µg of total nuclear protein was separated on a 10 % SDS gel and
blotted onto a nitrocellulose membrane. Immunoblot detection was done using anti-GFP (1:2,000) as
primary antibody and peroxidase labeled anti- mouse IgG (1:7,500) as secondary antibody.

61
In order to rule out the effect of EGFP tag on the nuclear localization of p11-CT, we
replaced the EGFP gene by a 6-amino acid FLAG tag at the carboxy-terminal end of
zfCx55.5-CT. N2A and NIH3T3 cells, grown on polylysine coated cover slips, were
transiently transfected with the above p11-CT-FLAG construct. 48 hours post
transfection, cells were analyzed by immunofluorescence using primary anti-FLAG
antibody. Immunofluorescence detection showed that p11-CT-FLAG fusion protein
can be detected in the cytosol and cell nucleus of NIH3T3 and N2A cells (Fig. 4.16A).
In order to confirm the localization of the p11-CT product in the cell cytosol and
nucleus , Western-blot analysis was performed on the cytosolic and nuclear extracts
prepared from transiently transfected N2A cells and NIH3T3 cells As shown in
Fig.4.16B, the p11-CT-FLAG fusion protein can be detected by the anti-FLAG
antibody in both cell nucleus and cytosol, confirming the above result.
Fig. 4.16. Additional confirmation for the sub-cellular distribution of p11-CT. A) A FLAG
tagged p11-CT construct was transiently transfected in NIH3T3 and N2A cells. 48 hour post tranfection
expression of p11-CT-FLAG was detected using anti-FLAG antibody as primary antibody (1:400) and
Alexa 568nm-conjugated goat-anti-mouse IgG antiserum as secondary antibody (1:2000). The distribution
of immunofluorescent signals in transiently transfected NIH3T3 cells (upper panel) and N2A cells (lower
panel) demonstrated immunreactivity in the cytosol and cell nucleus, red arrows indicate nuclear
distribution (bar = 10µm). B) Western blot of transiently transfected NIH3T3 and N2A cells with p11-CT-
FlAG construct supported a localization of p11-CT-FLAG in the cytosol and cell nucleus. 10µg either of
cytosolic and nuclear fraction were loaded on 10 % SDS gel and immunoblot detection was done by using
anti-FLAG (1:2,000) as primary antibody and peroxidase labeled anti- mouse IgG (1:7,500) as secondary
antibody (C and N represents Cytosolic and Nuclear fraction respectively).

62
4.12. In vivo evidence for the nuclear staining of zfCx55.5 in the Horizontal cells of
fish retina.
In order to provide the in-vivo evidence for the existence of p11-CT fragment of
zfCx55.5 and its nuclear translocation, a specific polyclonal antibody against the
carboxy-terminal domain of zfCx55.5 was generated (Zoidl et.,al; unpublished data).
Western blot of the total protein extract prepared from the fish retina showed only
the faint protein band corresponding to the zfCx55.5 (data not shown). This result is
of no surprise keeping in view the fact that Horizontal cells in fish retina resemble a
small fraction of the total cell number. To overcome this problem,
immunohistochemistry was performed on the adult fish retina using the above
mentioned zfCx55.5 antibody. In immunofluorescence analysis using confocal
microscopy zfCx55.5 immunoreactivity was exclusively detectable in a single cell
layer at the border between inner nuclear layer and outer plexiform layer/outer
nuclear layer (Fig.4.17 a-c). Distribution and morphology of labelled cells was
indicative for Horizontal cells. This observation is consistent with our previously
reported mRNA localization (Zoidl et al., 2004). The staining was prominent in the
perinuclear region and cellular processes. Most importantly, we were able to detect
nuclear immunoreacitivity in a fraction of HCs. In order to confirm this result
immunoelectron microscopy was performed and the localization of zfCx55.5 studied
at the ultrastructural level. This analysis, which was performed in collaboration with
the Group of Dr. M. Kamermans in Amsterdam, supported the data obtained by
LSM (Fig. 4.17 d-e) (Schields et al., in preparation). Interestingly, zfCx55.5
immunoreactivity appeared localized in a small number of clusters within the cell
nucleus. No immunoreactivity was seen with preimmunserum or with the specific
antibody fraction in cells that were not HCs.

63
FiG. 4.17. Nuclear localization signal of zfCx55.5 from horizontal cells of fish retina: a-c) Confocal laser scanning microscopy of fisg retina using zfCx55.5 specific antibody. a) Immunofluorescence from the horizontal cell layer of fish retina showing typical membrane staning and in some cells some nuclear singnal. b) Shows the corresponding popidium iodide stained nuclei and Cx55.5 and c) combines plane 7 pseudocoloured to highlight signals with the Z-stack through the Cx55.5 positive nuclei (“open arrows” indicate nuclear signal, “filled arrows” indicate negative nuclei). d-e) electron micrograph of three different horizontal cells of fish retina showing the isolated signal from the nuclei. bars= (a-c) 20µm, (d, e) 300nm, (f) 100nm.
4.13. Zebrafish connexin 55.5, zfCx55.5, internal IRES elements activity is
determined by two polypyrimidine tracts.
In the above we have shown that a carboxy-terminal domain (P11-CT) of zebrafish
connexin 55.5 can be internally translated from an IRES element present in the coding
region of main zfCx55.5. Sequence analysis of this IRES element showed the presence
of two stretches of polypyrimidine tracts named as polypyrimidine tract 1
(TCCTCCTTT) (PPT1) and polypyrimidine tract 2 (TCCTCTGCTTTCTT) (PPT2) (Fig
4.18a). Polypyrimidine tracts, as the name suggests are cis-acting elements of C and T
nucleotides present in the RNA molecule which are found to play regulatory role
during RNA splicing or IRES mediated functions. To investigate the role of these

64
tracts in the internal translation of P11-CT of zfcx55.5, mutational analysis was
carried out.
Deletion of PPT1 and PPT2, separately or in combination, of the wild type IRES
element (IR) containing di-cis vector, pRF-IR was performed as described in
Materials and Methods. Control di-cis vector, pRF, along with wild type IRES di-cis
vector, pRF-IR and the various deletion mutant construct of IRES element, pRF-IR
(del. PPT1), pRF-IR (del. PPT2) and pRF-IR (del. PPT1-2) (Fig 4.18b), were transiently
transfected into the N2A cells. Rennila and Firefly luciferase activity were measured
48 hours post transfection. Luciferase activity readings depicted that the wild type
IRES element was able to enhance the expression of the downstream located Firefly
luciferase cistron by ~20 fold as compared to the control vector pRF (Fig 4.18c). IRES
di-cis construct having PPT1 deleted, pRF-IR (del. PPT1), showed ~8 fold luciferase
activity as compared to control vector, while deletion construct of PPT2, pRF-IR (del.
PPT2), alone and in combination with PPT1, pRF-IR (del. PPT1-2), resulted in
complete lost of luciferase activity , comparable to that of control vector. IRES
activity in each case was calculated as the ratio of Firefly luciferase to Rennila
lucifease activity (FLuc / RLuc).

65
Fig 4.18. Sequence elements which determine the IRES activity : a) partial DNA sequence of
zfCx55.5 IRES element with polypyrimidine tract 1 (PPT1) (TCCTCCTTT) and polypyrimidine tract 2
(PPT2) (TCCTCTGCTTTCTT) underlined. b) Schematic representation of various di-cis constructs: I)
pRF, control vector having Rennila luciferase as first cistron and Firefly luciferase as downstream cistron
with first cistron under the control of CMV promoter. II) pRF-IR wild type IRES containing di-cis
construct, III) pRF-IR (del. PPT1) PPT1 deleted di-cis construct, IV) pRF-IR (del. PPT2) PPT2 deleted di-
cis construct and V) pRF-IR (del. PPT1-2) deleted di-cis construct. c) IRES activity of above constructs in
transiently transfected N2A cells. IRES activity is represented as the ratio of Firefly to Rennila luciferase
activities (FLuc / RLuc) with the activity of control vector set to “1”. Each construct was tested 4-times
and each experiment was done in triplicates. Data is represented as mean of ± SEM.
Western blot confirms the absolute requirement of polypyrimidine tract 2 (PPT2)
for the IRES activity.
To further confirm the role of PPT1 and PPT2 sequences on the IRES activity, wild
type IRES element (IR) and its various deletion mutants were sub-cloned in the pR-
EGFP di cis vctor having EGFP gene as downstream cistron, as described in material
and methods.

66
Fig 4.19.Western blot of wild type IRES element and its deletion mutations: a)
Schematic representation of di-cis constructs used for the Western blots I) pR-EGFP
control vector having Rennila luciferase as first cistron and EGFP as downstream cistron, II) pR-EGFP-
IR wild type IRES containing di-cis construct, III) pR-EGFP-IR (del. PPT1) PPT1 deleted IRES
construct, IV) pR-EGFP-IR (del. PPT2) PPT2 deleted IRES construct and V) pR-EGFP-IR (del. PPT1-2)
PPT1-2 deleted IRES construct. b) Western blot of above constructs transiently transfected into N2A cells.
30µg of cytosolic total proteins were resolved on 10% SDS gel. Immunodetection was done by using anti-
GFP (1:2000) as primary antibody and peroxidase labeled anti-mouse IgG (1:7,500) as secondary
antibody. c) Western blot β-actin as loading control Wild type IRES construct, pR-EGFP-IR, and the various deletion constructs, pR-
EGFP-IR (del. PPT1), pR-EGFP-IR (del. PPT2) and pR-EGFP-IR (del. PPT1-2), along
with the control vector pR-EGFP (Fig 4.19a), were transiently transfected into the
N2A cells. 48 hour post transfection cytosolic cell extract was prepared and 30µg of
the total protein was separated on 10 % SDS gel. Immunodection with the anti-GFP
antibody showed ~10 fold enhanced expression of EGFP in wild type IRES element
construct (Fig 4.19b, lane II) as compared to the control vector (lane I). Deletion
construct with PPT1 deletion showed appreciable decrease in the expression of EGFP
(lane III) as compared to wild type, while the deletion of PPT2 alone (lane IV) or
deletion of both PPT1-2 (lane V) constructs resulted in the complete lost of
expression of EGFP.
4.14. Polypyrimidine tract binding protein (PTB) plays essential role in the IRES
activity through its influence on the PPT1 and PPT2
Essential requirement of the polypyrimidine tract 2 (PPT2) for the IRES activity lead
us to study the potential role of the polypyrimidine tract binding protein (PTB) in the
IRES activity. Polypyrimidine tract binding protein (PTB) is known to bind the
polypyrimidine tracts and this interaction has been found to be important for the
IRES activity. For this purpose, we over-expressed the human PTB (hnPTB) in the
N2A cells by co-transfecting the C1-PTB vector with the wild type IRES constructs,
pRF-IR, along with the various deletions constructs. 48 hour after transient
transfection into the N2A cells, rennila and firefly luciferase activity was measured.
Luciferase reading showed that by co-transfecting the pC1-PTB vector, the luciferase
activity of wild type IRES element containing di-cis construct, pRF-IR, increases from

67
~20 to ~60 fold as compared to control vector pRF. Similarlay the PPT1 deletion
construct, pRF-IR (del. PPT1) showed an increase in the luciferase activity from ~8
fold to ~45 fold, while the PPT2 deletion construct, pRF-IR (del. PPT2) and the
combination of PPT1-2 deletion construct, pRF-IR (de. PPT1-2), did not show any
effect by co-transfection of the PTB (Fig 4.20a). To further confirm that there exists a
direct correlation between the expression of PTB and the activity of the IRES element,
we performed Western blots using EGFP.
Fig 4.20. Over-expression of PTB results in enhancement of IRES activity: a) IRES activity
of various di-cis constructs (follow fig1b) transiently transfected into N2A cells either in the presence of
endogenous expression of PTB or over expressed PTB by co-transfection of pC1-PTB construct. IRES
activity is represented as the ratio of Firefly to Rennila luciferase (FLuc / RLuc), with the activity of
control vector pRF set to “1”. Each construct was tested 4-times and each experiment was done in
triplicates. Data are expressed as mean ± SEM. b) Western blot of various di-cis constructs (follow fig 2a)
transiently transfected in the N2A cells in the presence of either endogenous expression of PTB (-) or over-
expressed PTB (+). 30µg of total cytosolic proteins were resolved on 10% SDS gel and immunodetection
was done by using anti-GFP (1:2000) as primary antibody and peroxidase labeled anti-mouse IgG

68
(1:7,500) as secondary antibody. c) Western blot of endogenous expression of PTB and over-expressed
PTB from transiently transfected N2A cells. Immuniodetection was done by using anti-PTB (1:1000) as
primary antibody and peroxidase labeled anti-mouse IgG (1:7,500) as secondary antibody. Note: both
endogenous and over-expressed PTB is detected as doublet. d) Western blot β-actin as loading control. containing di-cis constructs. Wild type IRES construct and its various deletions
constructs were transiently transfected into the N2A cells along with the co-
transfection of the pC1-PTB vector where ever indicated (Fig 4.20b). 48 hour post
transfection, cytosolic cell extract was prepared and 30µg of the total protein was
separated on 10 % SDS gel. Immunodection with the anti-GFP antibody and anti-PTB
antibody showed that by overexpression of PTB along with the wild type IRES
element construct resulted in the ~3fold increase in the expression of EGFP as
compared to endogenous expression of PTB (Fig 4.20b, II). Similarly PPT1 deletion
construct showed an increase in the expression of the EGFP by the over expression of
PTB (III), albeit less than the wild type. PPT2 deletion construct showed a slight
increase in the expression of EGFP by over- expressing the PTB (IV), while the PPT1-
2 deletion construct showed very little effect of over-expressing PTB (V). The
expression of EGFP for the control vector did not show any effect by over expression
of PTB (I).
4.15. Specific ribonucleic-protein complex (RNP) assembles on the ~360 nt
zfCx55.5 IRES element.
We used RNA-electromobility shift assay (RNA-EMSA) to determine whether
cellular proteins recognize this IRES element. For this purpose, internally labeled
RNA probe was incubated with the S10 cytosolic N2A protein extract, as described in
Material and Methods. As shown in Fig 4.21 (lane II), cytoplasmic S10 extract
retarded the migration of RNA probe, leading to the formation of a single dominant
RNA-protein complex as compared to protein less control RNA probe (lane I).
Formation of this complex was effectively inhibited by the inclusion of 50-fold molar
excess of homologous unlabeled competitor RNA (lane III). Similar RNA-protein
complexes were also observed with PPT1 (lane IV), PPT2 (lane V) and PPT1-2 (lane
VI) deleted RNA probes.
To provide insight into the nature of proteins (in terms of Mol. Wt.) that are part of
the RNA-protein complex which assemble on the IRES element, we performed the

69
UV cross-linking experiment of the wild type IRES probe and its deletion mutants
with the S10 cytosolic N2A protein extract. Following separation on SDS- PAG, many
distinct RNA-Protein bands were detected (Fig 4.21b lane II).
Fig 4.21. Specific formation of RNA-protein complex on the IRES element: a) RNA-EMSA
of wild type IRES. Internally labeled 32 P RNA-probes were incubated with S10 N2A extract. RNA-protein
complex was resolved on 4% non-denaturing polyacryalamide gel and visualized by autoradiography.
Lane I) only RNA probe, lane II) wild type IRES RNA probe plus S10 N2A extract, lane III) same as II,
but in presence of 50 fold molar excess of unlabelled homologous competitor RNA. b) RNA-EMSA of wild
type IRES and its various deletion mutants. Internally labeled 32 P RNA-probes were incubated with S10
N2A extract. RNA-protein complex was resolved on 4% non-denaturing polyacryalamide gel and
visualized by autoradiography. Lane I) wild type IRES probe without protein as control, lane II) wild type

70
IRES element in presence of S10 N2A extract, lane III) PPT1, lane IV) PPT-2 and lane V) PPT1-2 deleted
IRES RNA probe plus S10 N2A extract. c) UV cross-linking of RNA probe with S10 N2A extract: RNA-
protein complex were formed as in (a) and then the samples were subjected to UV cross-linking,
subsequently RNase treated and resolved on 10% SDS gel. Lane I) only RNA probe, lane II) wild type
IRES element in presence of N2A S10 extract, lane III) same as (II) but in presence of 50 fold molar excess
of unlabelled homologous RNA. Lane IV) PPT1, lane V) PPT2 and lane VI) PPT1-2 deleted IRES RNA
probe. Arrows on the left represent specific RNA-protein complexes and the numbers on right represent
molecular weight marker in kilodalton, kDa.
The cross-linked complexes have apparent molecular masses of about 100, 80, 60, 55,
42 kilo Dalton. The formations of the cross-linked complexes were prevented by the
inclusion of the 50 fold molar excess of homologous unlabelled competitor RNA
(lane III). Almost similar RNA-protein complexes were also detected with the PPT1
(lane IV), PPT2 (lane V) and PPT1-2 (lane VI) deletion constructs.
4.16. Purified GST-PTB fusion protein is able to bind the IRES element.
From the above UV cross-linking experiment, an apparent RNA-Protein band
around ~57kDa (Molecular weight of PTB is ~57 kDa) led us to explore the
possibility of binding of purified PTB to the IRES element. To this end, we used GST-
PTB purified protein for the RNA-EMSA. Wild type IRES RNA probe and its various
deletion mutants were incubated with GST alone or GST-PTB fusion proteins. As
shown is Fig 4.22a, GST-PTB is able to retard the migration of RNA probe (lane II),
while GST alone doesn’t show any retardation of RNA probe (lane I). The formations
of the GST-PTB retardation band was prevented by the inclusion of 20 fold molar
excess of homologous unlabelled competitor RNA (lane III). Furthermore, UV cross-
linking of GST-PTB fusion protein to wild type IRES element resulted in the
formation of RNA-protien complex of ~86 kDa (Fig 4.22b, lane II). This complex was
effectively competed out by adding 20 fold molar excess of unlabelled homologous
RNA (lane III). No such RNA-protein complex was formed by UV cross-linking GST
alone to the IRES element (lane I). All the deletion mutants also showed same
molecular weight RNA-protein complex (lanes IV, V and VI). Above data indicate
that PPT1 and PPT2 are important for the IRES structure and PTB seems to act as an
RNA chaperon to stabilize the structure around IRES element.

71
Fig 4.22. Recombinant GST-PTB fusion is able to bind the IRES element:a) RNA-EMSA of wild type 32 P
labeled IRES RNA with, lane I) ~50µg of purified GST alone, lane II) 0.3µg of purified GST/PTB fusion
protein and lane III) cold competition of RNA-protein complex formed in (II) by 20 fold molar excess of
unlabelled RNA. b) UV cross-linking of GST- PTB to IRES element. Lane I) GST plus wild type IRES
element, lane II) GST-PTB fusion protein plus wild type IRES element, lane III) cold competition of (II)
using 50 fold molar excess of unlabeled RNA. Lane IV) PPT1, lane V) PPT2 and lane VI) PPT1-2 deleted
IRES mutants. A RNA-protein complex of about 84 kDa was detected in all cases, which corresponds to
the PTB (57 kDa) plus GST (27 kDa). Number on right side represent molecular weight marker in
kilodalton, kDa.
4.17. Secondary structure prediction.
From the above results it seems that the secondary structure of RNA is important for
the IRES activity. To check the predicted structure of the IRES element, the Zucker
algorithm, using the default parameters was used to predict the secondary structure
of the wild type IRES element and its various deletion mutants by determining free
energies (dG). As shown in Figure 4.23(a), the predicted secondary structure of the

72
Fig 4.23 Predicted structure of the zfCx55.5 IRES element and its deletion mutants: The most thermodynamically stable structure predicted by the Zucker algorithm using default
parameters. a) Wild type IRES (WT-IR) showing extended stem-loop structure with dG= -86.41. a`) shows
the magnified part of (a) where PPT1 forms a small hairpin loop and PPT2 forms extended internal stem
loops. b) PPT1 deleted IRES (IR (del.PPT1) structure with dG=-88.53, b´) with the hairpin loop formed by
PPT1 in (a) missing and c) PPT2 deleted IRES structure (IR (del.PPT2) with dG= 82.85. Note the
complete remodeling of structure upon deletion of PPT2.
wild type IRES element exhibiting the most negative dG value
dG=86.41) showed a Y-shaped structure with several hairpin loops, buldges, internal
loops and junctions with PPT1 forming a small internal hairpin loop and PPT2
mostly involved in stem loop structure. The IRES element with PPT1 deleted showed
similar secondary structure as that of the wild type IRES element (dG = -88.53) except
that the internal hairpin loop structure does no more exists (b). Deletion of PPT2
resulted in complete remodelling of the secondary structure with energetically less
stable structure (dG = -82.85) than that of wild type IRES element (c). A reasonable
explanation of the above prediction is that the internal stem structure formed by the
PPT2 is essential to maintain the energetically most favourable structure of the IRES
element.

73
5. Discussion
5.1 Promoter elements of zfCx55.5 and zfCx52.6.
Zebrafish connexin zfCx55.5 and zfCx52.6 show highly restricted expression pattern.
Both of these connexins have been found to be exclusively expressed in horizontal
cells of the fish retina. Transcriptional control serves for the primary control for gene
expression and this may play a critical role in the site restricted expression of these
connexins. To check this possibility, we characterized the promoter elements of both
connexins. A 3.5 kb upstream region of the zfCx55.5 was screened for the potential
promoter elements. To this end, different DNA fragments encompassing the entire
upstream region of zfCx55.5 were sub-cloned in the pGL3-Basic vector with Firefly
luciferase as reporter gene. Luciferase data showed that two differently located
upstream DNA fragments of zfCx55.5 enhance the expression of the reporter gene,
one of which was proximal to the translational start site (-881/+134, promoter
element II) and the other distal to it (-3915/-2004, promoter element II). Promoter
element I enhances the expression of reporter gene by ~13 fold, while the promoter
element II did it by ~8-fold. These results point toward the possible presence of two
promoter elements which may control the expression of zfCx55.5. Moreover
promoter element II was 5´ flanking to main coding exon II, while the promoter
element I was 5´ flanking to upstream small exon I. Multiple promoters is the
emerging discovery in many genes and in particular the connexin genes. Since the
main coding information for the zfCx55.5 is present in a single exon, the presence of
multiple promoters will give rise to different 5´ UTRs with different translational
efficiencies. A similar promoter organization has been found in other connexins: for
example two different Cx32 transcripts have been detected, each controlled
separately by two promoters. The larger transcript is expressed in liver and the
promoter activity has been localized in the 5´ flanking region of the first exon. The
shorter transcript lacks the first exon and the promoter activity is composed within
the two exons. (Bai et al., 1995; Bai et al., 1993). Moreover, the expression pattern of
Cx43 is found to be highly variable in different tissues. Recently it was shown that
nine different mRNA species are expressed in mouse tissues, each having distinct 5´
UTRs (Pfeifer et al., 2004). This can be explained by the differential promoter usage

74
and alternate splicing. Thus presence of multiple promoters in connexins seems to be
an important regulatory mechanism for the control of their differential expression
during development and in different tissues. To further characterize the promoter
element I, we made different deletion constructs of this element. Interestingly,
Luciferase data showed that by deleting ~750bp from the 5´ end of the DNA
fragment -3915/-2004, the luciferase activity increased from 13 fold to ~20 fold. This
data gives hint about the possible presence of repressor element in the promoter
element I of zfCx55.5. The presence of such repressor elements has been already
reported in the 5´ flanking promoter region of Cx43 gene. Evidently, such kind of
regulatory elements are important for the fine tuning of the expression of the
zfCx55.5 gene.
Similarly, 1.9kb upstream region of zf.Cx52.6 was screened for the existence of
a putative promoter. Luciferase data from transient transfectrd HeLa and N2A
cells showed that there are different requirements in terms of length of DNA
fragments for the promoter activity in HeLa and N2A cells. These results
indicate that the DNA fragment -778 to-135 contains a putative repressor
element which is active in N2A cells only and not in HeLa cells. Evidently,
there seems to be a complex interplay between various cis-acting elements in
zfCx 52.6.
5.2. Putative DNA binding proteins of the promoter elements of zfCx52.6 and
zfCx55.5.
The expression of eukaryotic genes is governed to a large extent by sequence-specific
interactions between a promoter and specific DNA-binding proteins which facilitate
the activity of polymerase II (Tjian et al., 1994). Most promoters of class II genes have
canonical DNA elements, such as the TATA and CAT motifs, which bind
transcription factors essential for mRNA initiation (Buratowski et al., 1994).
However, other class II promoters lack TATA elements and initiate transcription by
using alternative mechanisms to associate with the TFIID complex (Pugh et al., 1990).
The promoter of zfCx52.6 seems to fall to former class. ZfCx52.6 promoter element
sequence was investigated for the potential binding of transcription factors. Sequence
analysis showed the presence of the binding sites of various known transcription

75
factors. A TATA signal was found at -1283nt position (TATAAA), CBF consensus
sequence at position -1411 (CCAAT) and OCT-1 binding site at position -720nt
(TACATTGAAATGTA). Using DNA-EMSA a specific DNA-protein complex was
detected with the DNA fragment (-1433/-1302) which posses the CCAAT binding
sequence motif. To this end, we investigated the possibility of CBF binding to this
complex or at least be a part of it. For this purpose, the specific DNA-protein
complex formed by the DNA fragment (-1433/-1302) was successfully competed out
using 50-fold molar excess of CCAAT oligo. CCAAT box is a widespread regulatory
sequence found in promoters and enhancers of several genes. Among the proteins
reported to bind to this sequence, only NF-Y (also termed as CBF) has an absolute
requirement for these 5 nucleotides. NF-Y is an ubiquitous heteromeric protein
formed by 3 sub-units, NF-YA, NF-YB, NF-YC, all necessary for DNA binding (Kim
et al., 1990; Sinha et al., 1995).
Moreover, DNA fragment -1215/-1096 also binds a specific protein complex.
Sequence analysis shows binding sites for the transcription factors, OCT-1 and
GATA-1. To check the possibility of whether these two proteins are part of this
complex, we performed electromobility shift assays of this DNA fragment and did
the cold competition using either an OCT-1 oligo or a GATA-1 oligo. From our data it
became clear that OCT-1 was able to compete out the binding.
Protein Oct-1, which is probably contained in all proliferating eukaryotic cells, is one
of the multifunctional molecules. Oct-1 belongs to transcription factors of the POU
family (Ryan et al., 1997; Phillips et al., 2000; Herr at al., 19888; Sturm et al., 1988;
Hinkley et al., 1992). Since it is virtually ubiquitous and has binding sites in the
promoters of the histone H2B gene and the genes for snRNA U2, U6, and 7SK , Oct-1
has been assumed to act as a constitutive transcription factor and regulates
expression of housekeeping genes. Yet its functions in the cell proved to be far more
complex and diverse. It is now known that: first; Oct-1 controls not only the
housekeeping but also numerous tissue-specific genes. The latter include the genes
for interleukins (IL) 2 (Ullman et al., 1991), the granulocyte-macrophagal colony-
stimulating factor (Kaushansky et al., 1994). Tissue specificity of Oct-1 is of interest
keeping in view the site restricted expression of zfcx52.6. Interestingly, Oct-1
activates transcription in some cases and suppresses it in some others. Thus our

76
preliminary results indicate that CCAAT binding protein and OCT-1 binding protein
forms part of DNA-protein complex which binds the promoter element of zfCx52.6.
It is of interest to note that there are many examples where both OCT-1 and CCAAT
binding proteins are shown to be critical for the basal promoter activity and the
regulation of promoters (Guimond et al., 2002; Jin et al., 2001; Bellorini et al., 1997;
Fan et al., 2002; Wright et al., 1994).
Sequence analysis of zfCx55.5 shows that it lacks the putative TATA signal and is
highly AT rich. To explore the transcription factors which bind to the promoter
element of zfCx55.5, EMSA was performed with various DNA fragments of
promoter element I and II. DNA fragment -3179/-3029 and -2910/-2788 showed that
two specific protein complexes bind to these elements and a single protein complex
can be detected with the DNA fragment -2794/-2658. Sequence analysis of these
DNA fragments using transcription factor binding prediction programme depicts the
potential binding of various transcription factors with high precedence binding of
CdxA- a homeodomain transcription factor. Many putative consensus binding sites
(A, A/T, T, A/T, A, T, A/G) of this factor were found in the DNA fragments. CdxA
belongs to a family of homeobox gene which regulates developmental decisions
during embryogenesis (McGinnis et al., 1992). A common feature of these genes is a
183 bp long sequence conserved in evolution, the homeobox (McGinnis et al., 1984;
Scott et al., 1984). Regulation of connexins during development is well documented.
During development a role for intercellular communication through gap-junctions is
implicated in cell migration, neuronal differentiation and circuit formation, for
review (see Dermietzel and Meir, in press) (Naus et al., 1998). The developmental
regulation of connexins in numerous regions of CNS has been observed (Nadarajah
et al., 1997; Condorelli et al., 2000; Leung et al., 2002) including the retinal ganglion
cells (Becker et al., 2002). Keeping in view the tissue specific expression of zfCx55.5, it
will be of interest to find whether these homeodomain transcription factors regulate
the expression of this gene and whether the expression of this connexin changes
during retinal development.

77
5.3. Extension of the N-terminus of zfCx55.5.
Genomic organization of connexins is defined by single coding exons which contain
all information for the translation of the protein and some smaller exons upstream of
main exon which forms part of the 5´ UTRs of these genes. New reports are emerging
that indicates that the upstream region of these genes contains many more small
exons then previously thought. ZfCx55.5 coding information lies in the single main
exon II of ~1450 bps. However sequence analysis showed the presence of a small
open reading frame (~100bp) just upstream of main exon II. Splice prediction
indicate a highly efficient donor splice site in the small exon I and an acceptor site
flanking of main exon II. To check whether this short exon forms part of the zfCx55.5
protein, immunoblot was performed. Immunoblot dectection showed an immuno-
reactive band of ~82.5 kDa corresponding to main zfCx55.5 and an additional lower
mobility band of ~86.6 kDa. The detection of this protein band can only be explained
when the small exon I is being spliced to main exon II. This was further confirmed by
deleting a DNA fragment just 5´ flanking of the main exon which includes the splice
acceptor site. Deletion resulted in complete absence of this additional protein band.
Functional significance of this extension of N-terminus remains to be established. It is
of interest to note that the N-terminus of gap junctions has been shown to have
properties of acting as a trans-junctional voltage sensor (155Purnick et al., 2000).
Moreover it has been reported that charge substitution in the N-terminus reverses
the gating polarity of Cx32 (Purnick et al., 2000). ZfCx55.5 has been shown to have
unique electrophysiological properties by forming rectifying junctions in a
heterotypic setting. Thus by extension of the N-terminal region, the
electrophysiological properties of this isoform may be different than that of the main
connexin. However, whether these splice sites, which result in the new isoform of
zfCx55.5, are used in, the zebrafish is under investigation.
5.4. Internal translation of the CT of zfCx55.5
Translation serves as an important spatiotemporal control mechanism for the
expression of genes. The effect of translational regulation on gene expression is fast
as compared to transcriptional regulation. Thus translational regulation serves as a
prime target for certain physiological mechanisms which need fast response in the

78
expression of genes at a particular time window. Keeping in view the differential
lengths of 5´ UTRs of connexin genes, it becomes obvious that connexin too are
targets of translational regulation. ZfCx55.5 belongs to the class of connexins
exhibiting an extended carboxy-tail (~288 aa) similar to the mammalian Cx57 and
Cx59 (Manthey et al., 1999). Our observation from immunoblots of the fusion protein
of zfCx55.5-EGFP detects the expected protein band of 82.6 Kda. In addition, some
higher mobility bands are apparent. We argued against a proteolytic degradation of
the proteins during extraction as we included strict protease inhibition in our
extraction protocol. Moreover, identical bands were obtained when performing
Western blots with transiently transfected HeLa cells (data not shown). An
alternative explanation for the existence of truncated fragments would be regulatory
mechanisms either at the transcriptional or translational level.
By performing sequence analyses of the coding region of zfCx55.5 we found several
in frame AUG codons. Two in-frame AUG codons exist in the long carboxy-terminal
tail of zfCx55.5, one of which was found immediate to the start of the carboxy-
terminal tail (nt634) and a second one in the middle of the CT-tail at nucleotide
position 1202 which would give rise to a protein of 11kDa in case of internal
translation. Sequence analyses indicated that this fragment is preceded by a near
perfect Kozak sequence. The translation of the p11-CT domain was confirmed by
Western blot from the full length CT-tail and from a construct starting from an in-
frame AUG at nucleotide 1202. Moreover, the p11-CT protein was always detected as
a double band. Keeping in view the number of possible phosphorylation sites
present in this protein, we proposed that one band could represent a
hyperphosphorylated form of the p11-CT protein product.
From the above observation it became evident that the p11-CT portion of zfCx55.5 is
internally translated from the zfCx55.5 transcript. To rule out the possibility of a
specific proteolytic cleavage, we created a frame shift ahead of the in-frame AUG
start codon. By Western blot analysis, we were able to detect p11-CT only but no full
length zfCx55.5 protein. The most reasonable explanation for this phenomenon is the
existence of an internal initiation site in the coding region which can recruit the
translational machinery directly to the coding region of zfCx55.5 and drive the
expression of the p11-CT domain. To further confirm that the in-frame AUG codon at

79
position 1202 is used as a start codon for the translation of the p11-CT protein, we
altered this codon. Western blot detection showed that by mutating the AUG codon
to GCG, the expression of the p11-CT protein was completely abolished while the
expression of full length zfCx55.5 remained unaltered.
On the basis of the above observations we hypothesized that the possible candidate
for an internal translation is an IRES element in the coding region of zfCx55.5. To
identify the presence of the IRES element we used the classical Di-cistronic approach,
in which the 5`coding sequence of the CT-domain was sub-cloned in the inter-
cistronic region. After transient transfection into N2A cells and subsequent luciferase
activity determination, the expression of the downstream Firefly luciferase cistron
was increased to ~15 fold as compared to the control vector. Similar IRES activity
was also measured in HeLa cells, while the IRES activity in NIH3T3 was found to be
increased by ~ 25 fold, much higher than in N2A and HeLa cells. A deletion of
~200bp immediately 5` of the in-frame AUG start codon at position 1202 resulted in
an overall increase in the IRES activity from 15 fold to 34 fold in N2A and HeLa cells,
and from 25 fold to 77 fold in NIH3T3 cells. A possible explanation for the
differences of IRES activities in the cell lines used may be due to different levels of
endogenous trans-acting factors (Pickering et al., 2003; Jopling et al., 2001; Stoneley et
al., 2000), which are regarded to play a critical role in IRES mediated internal
translation.
To date the only IRES elements reported in connexin genes are IRES elements in the
5`UTR of Cx43 and Cx32 (Schiavi et al., 1999; Hudder et al., 2000). Many more have
been reported in the 5`UTR of other eukaryotic genes (Yang et al., 1997; Nanbru et al.,
1997; Huez et al., 1998; Oumard et al., 2000; Shiroki et al., 2002; Hellen et al., 2001).
The existence of IRES elements in the coding region of eukaryotic genes is still a rare
observation with only a few reports in the literature (Cornelis et al., 2000; Pyronnet et
al., 2000; Maier et al., 2002), where in some examples similarly to the p11-CT the
carboxy-terminal domain has been described to be internally translated (Lauring et
al., 2000).
Recently, the use of Di-cistronic vectors to study IRES activity has been criticized on
the basis that putative IRES elements may contain cryptic promoter elements which
can give rise to mono-cistronic messages which evade detection by Northern blot

80
analysis (Kozak, 2003). To approach this problem, we created a promoterless Di-
cistronic constructs by removing the CMV promoter ahead of the first cistron.
Transient transfection in N2A, HeLa and NIH3T3 cells and subsequent luciferase
activity reading showed only 4 to 8 fold increases in the expression of the
downstream cistron as compared to control levels of the promoterless Di-cistronic
vector. This is in sharp contrast to the IRES activity which we measured with normal
Di-cis vectors. The small increase in the activity of the downstream cistron in the
promoterless Di-cis vector can be explained by the presence of some leaky message
transcribed from unknown regulatory sequences in the vector backbone. This
explanation was further confirmed by modifying the Di-cis constructs by replacing
the Firefly luciferase with EGFP. Western blot analysis showed a ~10 fold enhanced
expression of the EGFP protein in the IRES containing construct as compared to the
control vector. No expression was evident from the promoterless IRES containing
construct, while the promoterless Di-cis control vector yielded some very low
expression close to the detection limit. The presence of the low EGFP expression in
the promoterless pR-GFP (-P) led credence to our explanation of some leaky
transcription from some unknown regulatory sequences in the vector backbone
which may be responsible for some IRES activity. The use of a promoterless Di-cis
vector has been recently regarded as an authentic test to differentiate between the
IRES element and cryptic promoter activities (Han et al., 2002).
The biological significance of a separate expression of the carboxy-terminal domain
via IRES mediated internal translation is still to be established. A fusion protein of
p11-CT with EGFP was found to be localized in the nucleus when transfected in N2A
and NIH3T3 cells. The nuclear localization of a carboxy-terminal domain of a
connexin has been already reported in case of Cx43. A nuclear translocation of
carboxy-terminal domains would be of paramount importance as far as gap junction
biology is concerned. The existence of a molecular mechanism initiating an internal
expression of CT fragments could provide first insight into connexin properties not
readily explainable by the channel properties of gap junctions. A separate expression
of biologically active CT-domains and the nuclear translocalization can endow
connexins with the capability of modulating the gene expression directly in response
to various physiological and pathophysiological conditions. Here, the basic

81
properties of the p11-CT domain with a calculated pI of >10 may resemble properties
comparable to the basic DNA binding domains of some transcription factor gene
families (Marchler-bauer et al., 2003). The manipulation of connexin genes (gene
deletion or over-expression) has been shown to affect the transcription of many genes
in various tissues (Huang et al., 2002; Nicholson et al., 2001; Vozzi et al., 1995).
Recently, it has been shown that gap junctions modulate transcription of an
osteoblast promoter by altering the recruitment of the transcription factors Sp1 and
Sp3 to connexin-response elements (Stains et al., 2003). Although this phenomenon
has been attributed to the passage of some unknown molecular cues from gap
junctions, a separate expression of carboxy-terminal domains could explain a direct
impact of connexins in such functions. Further studies have to address whether the
described IRES mediated internal translation of a CT domain is a common
phenomenon among the connexin protein family or restricted to a subgroup of
connexins and if so which form of signal transduction is achieved by its nuclear
translocation.
5.5. Functional motifs and trans-acting factor(s) of the zfCx55.5 internal IRES
element.
Internal ribosome entry sites (IRES) are complex RNA structure with extensive
secondary structures. Several conserved motifs have been described to be essential
for IRES activities (Martinez-Salas, 1999) among which polypyrimidine tracts are
well documented. In continuation of our previous study, we analyzed the putative
IRES element of zfCx55.5 for the sequence elements or motifs and the trans-acting
factor(s) which are important for the functioning of this element. On the assumption
that conserved motifs often correspond to essential parts of the molecule, mutational
analysis has been carried out on many IRES elements to define the precise sequences
required for activity. Sequence analyses showed the presence of two stretches of
polypyrimidine tracts, PPT1 and PPT2. To investigate the role of these
polypyrimidine tracts on the activity of the IRES element, we deleted PPT1 and PPT2
either singly or in combination. Subsequent luciferase and Western blot analyses
showed that the deletion of PPT1 exerts an appreciable effect on the IRES activity,
while deletion of PPT2 results in complete abolishing of IRES activity, equivalent to

82
that of control vector. This result indicates that 14bp stretch of polypyrimidine tract 2
serves an important element in determining the IRES activity. Polypyrimidine tracts
are well know elements in the regulation of mRNA metabolism and the importance
of such kind of oligopyrimidine sequences have already been reported upstream of
AUG initiation codon in picornaviruses (Jackson et al., 1990; Oh et al., 1993) and in
the hepatitis C virus (Pellerin et al., 2002).
In addition to their requirements for eukaryotic initiation factors, the efficiency of
most of the IRES elements is augmented by the noncanonical initiation factors know
as ITAFs (Internal initiation trans-acting factors). Since above results indicate the
involvement of polypyrimidine tracts in defining the IRES activity, we further
pursued the possible role of the polypyrimidine tract binding (PTB) protein. To
address this possibility, we studied the IRES activity of the wild type IRES element
and its deletion mutants in the presence of endogenous levels of PTB or over
expressed levels of PTB. Over-expression of PTB resulted in an increase of IRES
activity of the wild type IRES element from 20 fold to 60 fold as compared to the
control vector. Deletion mutant PPT1 showed also enhancement of the IRES activity
from 8 fold to 45 fold. More strikingly, there was no effect of over-expression of PTB
on the IRES activity when PPT2 alone or in combination with the PPT1 (PPT1-2) was
deleted. Similar results from Western blot confirmed the importance of PPT2 and
PTB. These results indicate that the polypyrimidine tract 2 (PPT2) is crucial for the
IRES activity and PTB has a definite a role to play in the IRES activity of zfCx55.5.
PTB protein is very well known in regulating the IRES activity of both viral IRES
elements (Anwar et al., 2000; Zang et al., 2001) and cellular IRES elements (Mitchell
et al., 2001). This data is of particular interest keeping in view the fact that PTB is
primarily a nuclear protein, where it plays role in the regulation of splicing of
eukaryotic mRNAs, but to perform the IRES related functions it needs to shuttle from
cell nucleus to cytoplasm. Recent evidence of the involvement of IRES element in the
internal translation of biologically active domains of proteins makes it essential that
this processes needs to be regulated. Shuttling event of PTB from cell nucleus to
cytoplasm can be one of the steps subjected to regulation by various internal or
external stimuli. Interestingly, recently it was shown that protein kinase A
phosphorylation modulates transport of the polypyrimidine tract-binding protein

83
and phosphorylation of particular serine residue of PTB results in the increase of the
cytoplasmic transport of PTB from the nucleus (Xie et al., 2003). These results couple
the cAMP-dependent protein kinase pathway with the shuttling of PTB. It will be
interesting to examine whether natural inducers of protein kinase A in the zebrafish
retina, such as the neurotransmitters dopamine and serotonin (induced by various
physiological stimuli like light-dark cycle) or various stress induced activation of
protein kinase A, can stimulate PTB phosphorylation and whether this regulation of
PTB results in the increased translation of carboxy-terminal domain (p11-CT) of
zfCx55.5 and how these signals effect downstream cellular functions.
Translation from IRES elements requires, in addition of canonical initiation factors,
some non-canonical initiation factors as reviewed in (Martinez-Salas et al., 2001). To
get an insight into the protein complex which binds to this IRES element, we
performed RNA-EMSA with the wild type IRES element and its deletion mutants.
RNA-EMSA detected a major RNA-protein complex whose specificity was confirmed
by unlabelled homologous competitor RNA. Deletion mutants also showed similar
RNA-protein complex. In order to resolve this protein complex into individual
protein factors, we performed UV cross-linking experiment with the N2A cytosolic
proteins. UV cross-linking revealed the presence of a number of protein factors
whose molecular masses fall into the range of ~100 kDa, ~55 to ~57 kDa, ~35 to ~40
kDa. Some of these molecular masses fall within the range of proteins which are
already known to bind some of the IRES elements (Kim et al., 2001). It is of
interesting to note that the sequence of the IRES element showed a number of GCAC
sequence motifs which are regarded potential binding site for La protein (Pudi et al.,
2004) whose molecular weight (~52 kDa) falls within the range we obtained with UV
cross-linking experiment. However it remains to be established whether this protein
is part of this complex or not.
Luciferase data and Western blot confirmation of the functional involvement of the
PTB and the detection of the RNA-protein complex of ~57 kDa in the UV cross-
linking experiment made it intriguing to investigate the potential of binding of PTB
to the IRES element. RNA-EMSA and UV cross-linking with the purified GST-PTB
fusion protein showed that GST-PTB was able to bind specifically to the IRES
element. To our surprise PPT1 and PPT2 deletion mutants were also able to bind the

84
GST-PTB protein. From the above observation, it became clear that PPT1 and PPT2
are not directly involved in the binding of PTB, but PTB seems to exert its effect on
the overall IRES activity indirectly through PPT1 and PPT2. PPT1 seems to play an
auxiliary role in the functioning of the IRES element, while requirement of PPT2 for
the IRES activity seems to be indispensable. A plausible explanation for the critical
role of PPT2 either is that it binds to crucial trans acting factor(s) which are further
important for the recruitment of the ribosomal translational machinery or it directly
acts as an ribosome entry window site with complementarity between
polypyrimdine tract and the 3´ end of 18S rRNA which results in the direct
recruitment of 40S ribosomal subunit (Yang et al., 2003). A complementarity between
the polypyrimidine tract of IRES and 3´ OH end of the 18S rRNA has been already
observed. A base pairing between these two sequences seems to contribute to select
the initiation codon to use (Scheper et al., 1994). The role of PTB seems to stabilize
this interaction by maintaining the active confirmation of the IRES element by
binding to the RNA scaffold (Kaminiski et al., 1998). Thus the role of PTB seems to
act as an “RNA chaperone”to stabilize the structure of the IRNA, as depicted in the
following cartoon.
Model for RNA–RNA and RNA–protein interactions
within the IRES
adapted from Belsham & Sonenberg (2000), Trends in Microbiology 8, 330-335
eIF4GeIF4A
RNA chaperones(La, PTB, PCB2)
AUG
40S SU, eIF3, TC, ?
PTB: poly- pyrimidine-tract binding proteinPCB2: poly(rC) binding protein 2
Interestingly, the secondary structure prediction using mFold algorithm of the wild
type IRES element and its deletion mutants revealed that wild type IRES of zfCx55.5

85
has an extended stem-loop structure with semi-conserved Y-like structure, described
for other IRES elements (Le et al., 1997; Le et al., 1998). Deletion mutant PPT1 showed
similar structure as that of wild type with only the absence of small stem loop. The
importance of such loops in IRES is a striking phenomenon that suggests that the
recognition of these particular structures at given position in the IRES by cellular
proteins might be more important than precise consensus primary sequences to elicit
the biological effect. Deletion mutant PPT2 showed complete remodelling of
structure which was predicted to be energetically less stable as compared to wild
type IRES. The importance of secondary structure to IRES function is underscored by
the studies of genetic drift in highly infectious viruses. It has been shown that
sequence substitutions within the IRES element are accompanied by the
compensatory mutations that act to maintain the RNA secondary structure.
Furthermore, mutational analysis has identified structural domains and short
sequence motifs located in apical loops and internal buldges that are vital to IRES
function (Le et al., 1998). However, direct experimental evidences are required to
unravel the role of such elements in defining the overall structure of IRES element
and subsequent IRES activity (Spahn et al., 2004).

86
6. Bibliography
• Ai Z, Fischer A, Spray DC, Brown AM, Fishman GI. (2000). Wnt-1 regulation
of connexin43 in cardiac myocytes. J Clin Invest. 105, 161.
• Akazawa H, Komuro I. Too much Csx/Nkx2-5 is as bad as too little? (2003). J
Mol Cell Cardiol. 35, 227-229.
• Anwar A, Ali N, Tanveer R, Siddiqui A. Demonstration of functional
requirement of polypyrimidine tract-binding protein by SELEX RNA during
hepatitis C virus internal ribosome entry site-mediated translation initiation.
(2000). J Biol Chem. 275, 34231-34235.
• Bai S, Spray DC, Burk RD. Identification of proximal and distal regulatory
elements of the rat connexin32 gene. (1993). Biochim Biophys Acta. 1216, 197-
204.
• Bai S, Schoenfeld A, Pietrangelo A, Burk RD. Basal promoter of the rat
connexin 32 gene: identification and characterization of an essential element
and its DNA-binding protein. (1995). Mol Cell Biol. 15, 1439-1445.
• Bai S, Spray DC, Burk RD. Identification of proximal and distal regulatory
elements of the rat connexin32 gene. (1993). Biochim Biophys Acta. 1216, 197-
204.
• Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA
methylation: a fundamental aspect of neoplasia. (1998). Adv Cancer Res. 72,
141-196.
• Becker D, Bonness V, Mobbs P. Cell coupling in the retina: patterns and
purpose. (1998). Cell Biol Int. 22, 781-792.
• Becker DL, Bonness V, Catsicas M, Mobbs P. Changing patterns of ganglion
cell coupling and connexin expression during chick retinal development.
(2002). J Neurobiol. 52, 280-293.
• Bellorini M, Lee DK, Dantonel JC, Zemzoumi K, Roeder RG, Tora L,
Mantovani R. CCAAT binding NF-Y-TBP interactions: NF-YB and NF-YC
require short domains adjacent to their histone fold motifs for association with
TBP basic residues. (1997). Nucleic Acids Res. 25, 2174-2181.

87
• Bennett MV, Barrio LC, Bargiello TA, Spray DC, Hertzberg E, Saez JC. Gap
junctions: new tools, new answers, new questions. (1991). Neuron 6, 305-320.
• Borke JL, Yu JC, Isales CM, Wagle N, Do NN, Chen JR, Bollag RJ. Tension-
induced reduction in connexin 43 expression in cranial sutures is linked to
transcriptional regulation by TBX2. (2003). Ann Plast Surg. 51, 499-504.
• Branchek T, Bremiller R. The development of photoreceptors in the zebrafish,
Brachydanio rerio. I. Structure. (1984). J Comp Neurol. 224, 107-115.
• Brockerhoff SE, Hurley JB, Niemi GA, Dowling JE. A new form of inherited
red-blindness identified in zebrafish. (1997). J Neurosci. 17, 4236-4242.
• Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular
basis of direct intercellular signaling. (1996). Eur J Biochem. 238, 1-27.
• Buehler LK, Stauffer KA, Gilula NB, Kumar NM. Single channel behavior of
recombinant beta 2 gap junction connexons reconstituted into planar lipid
bilayers. (1995). Biophys J. 68, 1767-1775.
• Buratowski S. The basics of basal transcription by RNA polymerase II. (1994).
Cell 77, 1-3.
• Cao F, Eckert R, Elfgang C, Nitsche JM, Snyder SA, H-ulser DF, Willecke K,
Nicholson BJ. A quantitative analysis of connexin-specific permeability
differences of gap junctions expressed in HeLa transfectants and Xenopus
oocytes. (1998). J Cell Sci. 111, 31-43.
• Cascio M, Kumar NM, Safarik R, Gilula NB. Physical characterization of gap
junction membrane connexons (hemi-channels) isolated from rat liver. (1995). J
Biol Chem. 270, 18643-18648.
• Carystinos GD, Kandouz M, Alaoui-Jamali MA, Batist G. Unexpected
induction of the human connexin 43 promoter by the ras signaling pathway is
mediated by a novel putative promoter sequence. (2003). Mol Pharmacol. 63,
821-831.
• Chen ZQ, Lefebvre D, Bai XH, Reaume A, Rossant J, Lye SJ. Identification of
two regulatory elements within the promoter region of the mouse connexin 43
gene. (1995). J Biol Chem. 270, 3863-3868.

88
• Chen J, Zhong Q, Wang J, Cameron RS, Borke JL, Isales CM, Bollag RJ.
Microarray analysis of Tbx2-directed gene expression: a possible role in
osteogenesis. (2001). Mol Cell Endocrinol. 177, 43-54.
• Civitelli R, Ziambaras K, Warlow PM, Lecanda F, Nelson T, Harley J, Atal N,
Beyer EC, Steinberg TH. Regulation of connexin43 expression and function by
prostaglandin E2 (PGE2) and parathyroid hormone (PTH) in osteoblastic cells.
(1998). J Cell Biochem. 68, 8-21.
• Condorelli DF, Belluardo N, Trovato-Salinaro A, Mudo G. Expression of Cx36
in mammalian neurons. (2000). Brain Res Brain Res Rev. 32, 72-85.
• Cornelis S, Bruynooghe Y, Denecker G, Van Huffel S, Tinton S, Beyaert R.
Identification and characterization of a novel cell cycle-regulated internal
ribosome entry site. (2000). Mol. Cell 5, 597-605.
• Dang X, Doble BW, Kardami E. The carboxy-tail of connexin-43 localizes to the
nucleus and inhibits cell growth. (2003). Mol Cell Biochem. 242, 35-38.
• Darrow BJ, Fast VG, Kleber AG, Beyer EC, Saffitz JE. Functional and structural
assessment of intercellular communication. Increased conduction velocity and
enhanced connexin expression in dibutyryl cAMP-treated cultured cardiac
myocytes. (1996). Circ Res. 79, 174-183.
• Deans MR, Volgyi B, Goodenough DA, Bloomfield SA, Paul DL. Connexin36
is essential for transmission of rod-mediated visual signals in the mammalian
retina. (2002). Neuron. 36, 703-712.
• De Leon JR, Buttrick PM, Fishman GI. Functional analysis of the connexin43
gene promoter in vivo and in vitro. J Mol Cell Cardiol. 1994 Mar;26(3):379-89.
• Dermietzel R, Kremer M, Paputsoglu G, Stang A, Skerrett IM, Gomes D,
Srinivas M, Janssen-Bienhold U, Weiler R, Nicholson BJ, Bruzzone R, Spray
DC. Molecular and functional diversity of neural connexins in the retina.
(2000). J Neurosci. 20, 8331-8343.
• DeVries SH, Schwartz EA. Modulation of an electrical synapse between
solitary pairs of catfish horizontal cells by dopamine and second messengers.
(1989). J Physiol. 414, 351-375.
• Dowling JE, Brown JE, Major D. Synapses of horizontal cells in rabbit and cat
retinas. (1966). Science 153, 1639-1641.

89
• Echetebu CO, Ali M, Izban MG, MacKay L, Garfield RE. Localization of
regulatory protein binding sites in the proximal region of human myometrial
connexin 43 gene. (1999). Mol Hum Reprod. 5, 757-766.
• Elfgang C, Eckert R, Lichtenberg-Frate H, Butterweck A, Traub O, Klein RA,
Hulser DF, Willecke K. Specific permeability and selective formation of gap
junction channels in connexin-transfected HeLa cells. (1995). J Cell Biol. 129,
805-817.
• Fernandez-Cobo M, Gingalewski C, De Maio A. Expression of the connexin 43
gene is increased in the kidneys and the lungs of rats injected with bacterial
lipopolysaccharide. (1998). Shock 10, 97-102.
• Fukushi S, Okada M, Stahl J, Kageyama T, Hoshino FB, Katayama K.
Ribosomal protein S5 interacts with the internal ribosomal entry site of
hepatitis C virus. (2001). J Biol Chem. 276, 20824-20826.
• Geimonen E, Jiang W, Ali M, Fishman GI, Garfield RE, Andersen J. Activation
of protein kinase C in human uterine smooth muscle induces connexin-43
gene transcription through an AP-1 site in the promoter sequence. (1996). J
Biol Chem. 271, 23667-23674.
• Gros DB, Jongsma HJ. Connexins in mammalian heart function. (1996).
Bioessays 18, 719-730.
• Guldenagel M, Ammermuller J, Feigenspan A, Teubner B, Degen J, Sohl G,
Willecke K, Weiler R. Visual transmission deficits in mice with targeted
disruption of the gap junction gene connexin36. (2001). J Neurosci. 21, 6036-
6044.
• Guimond J, Devost D, Brodeur H, Mader S, Bhat PV. Characterization of the
rat RALDH1 promoter. A functional CCAAT and octamer motif are critical for
basal promoter activity. (2002). Biochim Biophys Acta. 1579, 81-91.
• Hampson EC, Vaney DI, Weiler R. Dopaminergic modulation of gap junction
permeability between amacrine cells in mammalian retina. (1992). J Neurosci.
12, 4911-4922.
• Hampson EC, Weiler R, Vaney DI. pH-gated dopaminergic modulation of
horizontal cell gap junctions in mammalian retina. (1994). Proc R Soc Lond B
Biol Sci. 255, 67-72.

90
• Han B, Zhang JT. Regulation of gene expression by internal ribosome entry
sites or cryptic promoters: the eIF4G story. (2002). Mol Cell Biol. 22, 7372-7384.
• Hellen CU, Sarnow P. Internal ribosome entry sites in eukaryotic mRNA
molecules. (2001). Genes Dev. 15, 1593-1612.
• Henis-Korenblit S, Strumpf NL, Goldstaub D, Kimchi A. A novel form of
DAP5 protein accumulates in apoptotic cells as a result of caspase cleavage
and internal ribosome entry site-mediated translation. (2000). Mol Cell Biol.
20, 496-506.
• Hennemann H, Kozjek G, Dahl E, Nicholson B, Willecke K. Molecular cloning
of mouse connexins26 and -32: similar genomic organization but distinct
promoter sequences of two gap junction genes. (1992). Eur J Cell Biol. 58, 81-
89.
• Herr W, Sturm RA, Clerc RG, Corcoran LM, Baltimore D, Sharp PA, Ingraham
HA, Rosenfeld MG, Finney M, Ruvkun G, et al. The POU domain: a large
conserved region in the mammalian pit-1, oct-1, oct-2, and Caenorhabditis
elegans unc-86 gene products. (1988). Genes Dev. 2, 1513-1516.
• Hershey JW. Protein phosphorylation controls translation rates. (1989). J Biol
Chem. 264, 20823-20826.
• Hinkley C, Perry M. Histone H2B gene transcription during Xenopus early
development requires functional cooperation between proteins bound to the
CCAAT and octamer motifs. (1992). Mol Cell Biol. 12, 4400-4411.
• Holcik M, Korneluk RG. Functional characterization of the X-linked inhibitor
of apoptosis (XIAP) internal ribosome entry site element: role of La
autoantigen in XIAP translation. (2000). Mol Cell Biol. 20, 4648-4657.
• Huang R, Lin Y, Wang CC, Gano J, Lin B, Shi Q, Boynton A, Burke J, Huang
RP. Connexin 43 suppresses human glioblastoma cell growth by down-
regulation of monocyte chemotactic protein 1, as discovered using protein
array technology. (2002). Cancer Res. 62, 2806-2812.
• Hudder A, Werner R. Analysis of a Charcot-Marie-Tooth disease mutation
reveals an essential internal ribosome entry site element in the connexin-32
gene. (2000). J Biol Chem. 275, 34586-34591.

91
• Huez I, Creancier L, Audigier S, Gensac MC, Prats AC, Prats H. Two
independent internal ribosome entry sites are involved in translation initiation
of vascular endothelial growth factor mRNA. (1998). Mol Cell Biol. 18, 6178-
6190.
• Hunt SL, Hsuan JJ, Totty N, Jackson RJ. unr, a cellular cytoplasmic RNA-
binding protein with five cold-shock domains, is required for internal
initiation of translation of human rhinovirus RNA. (1999). Genes Dev. 13, 437-
448.
• Izumi RE, Valdez B, Banerjee R, Srivastava M, Dasgupta A. Nucleolin
stimulates viral internal ribosome entry site-mediated translation. (2001).
Virus Res. 76, 17-29.
• Jackson RJ. RNA translation. Picornaviruses break the rules. (1988). Nature
334, 292-293.
• Jackson RJ, Howell MT, Kaminski A. The novel mechanism of initiation of
picornavirus RNA translation. (1990). Trends Biochem Sci. 15, 477-483.
• Janssen-Bienhold U, Schultz K, Gellhaus A, Schmidt P, Ammermuller J, Weiler
R. Identification and localization of connexin26 within the photoreceptor-
horizontal cell synaptic complex. (2001). Vis Neurosci. 18, 169-178.
• Jin S, Fan F, Fan W, Zhao H, Tong T, Blanck P, Alomo I, Rajasekaran B, Zhan
Q. Transcription factors Oct-1 and NF-YA regulate the p53-independent
induction of the GADD45 following DNA damage. (2001). Oncogene 20, 2683-
2690.
• Jones PA, Laird PW. Cancer epigenetics comes of age. (1999). Nat Genet. 21,
163-167.
• Jopling CL, Willis AE. N-myc translation is initiated via an internal ribosome
entry segment that displays enhanced activity in neuronal cells. (2001).
Oncogene 20, 2664-2670.
• Kamermans M, Fahrenfort I, Schultz K, Janssen-Bienhold U, Sjoerdsma T,
Weiler R. Hemichannel-mediated inhibition in the outer retina. (2001). Science
292, 1178-1180.

92
• Kaminski A, Jackson RJ. The polypyrimidine tract binding protein (PTB)
requirement for internal initiation of translation of cardiovirus RNAs is
conditional rather than absolute. (1998). RNA 4, 626-638.
• Kanamori Y, Nakashima N. A tertiary structure model of the internal
ribosome entry site (IRES) for methionine-independent initiation of
translation. (2001) RNA 7, 266-274.
• Kasahara H, Ueyama T, Wakimoto H, Liu MK, Maguire CT, Converso KL,
Kang PM, Manning WJ, Lawitts J, Paul DL, Berul CI, Izumo S. Nkx2.5
homeoprotein regulates expression of gap junction protein connexin 43 and
sarcomere organization in postnatal cardiomyocytes. (2003). J Mol Cell
Cardiol. 35, 243-256.
• Kasahara H, Wakimoto H, Liu M, Maguire CT, Converso KL, Shioi T, Huang
WY, Manning WJ, Paul D, Lawitts J, Berul CI, Izumo S. Progressive
atrioventricular conduction defects and heart failure in mice expressing a
mutant Csx/Nkx2.5 homeoprotein. (2001). J Clin Invest. 108, 189-201.
• Kaushansky K, Shoemaker SG, O'Rork CA, McCarty JM. Coordinate
regulation of multiple human lymphokine genes by Oct-1 and potentially
novel 45 and 43 kDa polypeptides. (1994). Immunol. 152, 1812-1820.
• Kim CG, Sheffery M. Physical characterization of the purified CCAAT
transcription factor, alpha-CP1. (1990). J Biol Chem. 265, 13362-13369.
• Kim YK, Back SH, Rho J, Lee SH, Jang SK. La autoantigen enhances translation
of BiP mRNA. (2001). Nucleic Acids Res. 29, 5009-5016.
• Kozak M. Point mutations defines a sequence flanking the AUG initiator
codon that modulates translation by eukaryotic ribosomes. (1986). Cell 44, 283-
292.
• Kozak M. New ways of initiating translation in eukaryotes? (2001). Mol Cell
Biol. 21, 1899-1907.
• Kozak M. Alternative ways to think about mRNA sequences and proteins that
appear to promote internal initiation of translation. (2003). Gene 318, 1-23.
• Krutovskikh V, Yamasaki H. The role of gap junctional intercellular
communication (GJIC) disorders in experimental and human carcinogenesis.
(1997). Histol Histopathol. 12, 761-768.

93
• Lasater EM. Retinal horizontal cell gap junctional conductance is modulated
by dopamine through a cyclic AMP-dependent protein kinase. (1987). Proc
Natl Acad Sci U S A 84, 7319-7323.
• Lauring AS, Overbaugh J. Evidence that IRES within the Notch2 coding region
can direct expression of a nuclear form of the protein. (2000). Mol Cell 6, 939-
945.
• Lee SW, Tomasetto C, Paul D, Keyomarsi K, Sager R. Transcriptional
downregulation of gap-junction proteins blocks junctional communication in
human mammary tumor cell lines. (1992). J Cell Biol. 118, 1213-1221.
• Le SY, Maizel JV Jr. Evolution of a common structural core in the internal
ribosome entry sites of picornavirus. (1998). Virus Genes. 16, 25-38.
• Le SY, Maizel JV Jr. A common RNA structural motif involved in the internal
initiation of translation of cellular mRNAs. (1997). Nucleic Acids Res. 25, 362-
369.
• Leung DS, Unsicker K, Reuss B. Expression and developmental regulation of
gap junction connexins cx26, cx32, cx43 and cx45 in the rat midbrain-floor.
(2002). Int J Dev Neurosci. 20, 63-75.
• Lin JH, Yang J, Liu S, Takano T, Wang X, Gao Q, Willecke K, Nedergaard M.
Connexin mediates gap junction-independent resistance to cellular injury.
(2003). J Neurosci. 23, 430-441.
• Locke D. Gap junctions in normal and neoplastic mammary gland. (1998). J
Pathol. 186, 343-349.
• Lu C, Zhang DQ, McMahon DG. Electrical coupling of retinal horizontal cells
mediated by distinct voltage-independent junctions. (1999). Vis Neurosci. 16,
811-818.
• Macejak DG, Sarnow P. Internal initiation of translation mediated by the 5'
leader of a cellular mRNA. (1991). Nature 353, 90-94.
• Maier D, Nagel AC, Preiss A. Two isoforms of the Notch antagonist Hairless
are produced by differential translation initiation. (2002). Proc Natl Acad Sci U
S A 99, 15480-15485.

94
• Makowski L, Caspar DL, Phillips WC, Goodenough DA. Gap junction
structures. II. Analysis of the x-ray diffraction data. (1977). J Cell Biol. 74, 629-
645.
• Malicki J, Neuhauss SC, Schier AF, Solnica-Krezel L, Stemple DL, Stainier DY,
Abdelilah S, Zwartkruis F, Rangini Z, Driever W. Mutations affecting
development of the zebrafish retina. (1996). Development 123, 263-273.
• Manthey D, Bukauskas F, Lee CG, Kozak CA, Willecke K. Molecular cloning
and functional expression of the mouse gap junction gene connexin-57 in
human HeLa cells. (1999). J Biol Chem. 274, 14716-14723.
• Marchler-Bauer A, Anderson JB, DeWeese-Scott C, Fedorova ND, Geer LY, He
S, Hurwitz DI, Jackson JD, Jacobs AR, Lanczycki CJ, Liebert CA, Liu C, Madej
T, Marchler GH, Mazumder R, Nikolskaya AN, Panchenko AR, Rao BS,
Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Vasudevan S, Wang Y,
Yamashita RA, Yin JJ, Bryant SH. CDD: a curated Entrez database of
conserved domain alignments. (2003) Nucleic Acids Res. 31, 383-387.
• Martinez-Salas E. Internal ribosome entry site biology and its use in
expression vectors. (1999). Curr Opin Biotechnol. 10, 458-464.
• Martinez-Salas E, Ramos R, Lafuente E, Lopez de Quinto S. Functional
interactions in internal translation initiation directed by viral and cellular IRES
elements. (2001). J Gen Virol. 82, 973-984.
• McGinnis W, Krumlauf R. Homeobox genes and axial patterning. (1992). Cell
68, 283-302.
• McGinnis W, Levine MS, Hafen E, Kuroiwa A, Gehring WJ. A conserved DNA
sequence in homoeotic genes of the Drosophila Antennapedia and bithorax
complexes. (1984). Nature 308, 428-433.
• Meerovitch K, Svitkin YV, Lee HS, Lejbkowicz F, Kenan DJ, Chan EK, Agol VI,
Keene JD, Sonenberg N. La autoantigen enhances and corrects aberrant
translation of poliovirus RNA in reticulocyte lysate. (1993). J Virol. 67, 3798-
3807.
• Miller T, Dahl G, Werner R. Structure of a gap junction gene: rat connexin-32.
(1988). Biosci Rep. 8, 455-464.

95
• Milks LC, Kumar NM, Houghten R, Unwin N, Gilula NB. Topology of the 32-
kd liver gap junction protein determined by site-directed antibody
localizations. (1988). EMBO J. 7, 2967-2975.
• Mills SL, Massey SC. Differential properties of two gap junctional pathways
made by AII amacrine cells. (1995). Nature 377, 734-737.
• Mitchell JA, Ou C, Chen Z, Nishimura T, Lye SJ. Parathyroid hormone-
induced up-regulation of connexin-43 messenger ribonucleic acid (mRNA) is
mediated by sequences within both the promoter and the 3'untranslated
region of the mRNA. (2001). Endocrinology 142, 907-915.
• Mitchell SA, Brown EC, Coldwell MJ, Jackson RJ, Willis AE. Protein factor
requirements of the Apaf-1 internal ribosome entry segment: roles of
polypyrimidine tract binding protein and upstream of N-ras. (2001). Mol Cell
Biol. 21, 3364-3374.
• Miyachi E, Murakami M. Decoupling of horizontal cells in carp and turtle
retinae by intracellular injection of cyclic AMP.
• Nadarajah B, Jones AM, Evans WH, Parnavelas JG. Differential expression of
connexins during neocortical development and neuronal circuit formation.
(1997). J Neurosci. 17, 3096-3111.
• Nanbru C, Lafon I, Audigier S, Gensac MC, Vagner S, Huez G, Prats AC.
Alternative translation of the proto-oncogene c-myc by an internal ribosome
entry site. (1997). J Biol Chem. 272, 32061-32066.
• Naus CC, Bani-Yaghoub M. Gap junctional communication in the developing
central nervous system. (1998). Cell Biol Int. 22, 751-763.
• Neuhaus IM, Bone L, Wang S, Ionasescu V, Werner R. The human connexin32
gene is transcribed from two tissue-specific promoters. (1996). Biosci Rep. 16,
239-248.
• Neuhaus IM, Dahl G, Werner R. Use of alternate promoters for tissue-specific
expression of the gene coding for connexin32. (1995). Gene 158, 257-262.
• Neuhauss SC, Biehlmaier O, Seeliger MW, Das T, Kohler K, Harris WA, Baier
H. Genetic disorders of vision revealed by a behavioral screen of 400 essential
loci in zebrafish. (1999). J Neurosci. 19, 8603-8615.

96
• Nicholson SM, Gomes D, de Nechaud B, Bruzzone R. Altered gene expression
in Schwann cells of connexin32 knockout animals. (2001). J Neurosci Res. 66,
23-36.
• Oh SK, Sarnow P. Gene regulation: translational initiation by internal
ribosome binding. (1993). Curr Opin Genet Dev. 3, 295-300.
• Oumard A, Hennecke M, Hauser H, Nourbakhsh M. Translation of NRF
mRNA is mediated by highly efficient internal ribosome entry. (2000). Mol
Cell Biol. 20, 2755-2759.
• Paul DL. Molecular cloning of cDNA for rat liver gap junction protein. (1986).
J Cell Biol. 103, 123-134.
• Pellerin C, Lefebvre S, Little MJ, McKercher G, Lamarre D, Kukolj G. Internal
initiation sites of de novo RNA synthesis within the hepatitis C virus
polypyrimidine tract. (2002). Biochem Biophys Res Commun. 295, 682-688.
• Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA
directed by a sequence derived from poliovirus RNA. (1988). Nature 334, 320-
325.
• Pestova TV, Shatsky IN, Fletcher SP, Jackson RJ, Hellen CU. A prokaryotic-
like mode of cytoplasmic eukaryotic ribosome binding to the initiation codon
during internal translation initiation of hepatitis C and classical swine fever
virus RNAs. (1998). Genes Dev. 12, 67-83.
• Petrich BG, Gong X, Lerner DL, Wang X, Brown JH, Saffitz JE, Wang Y. c-Jun
N-terminal kinase activation mediates downregulation of connexin43 in
cardiomyocytes. (2002). Circ Res. 91, 640-647.
• Pfeifer I, Anderson C, Werner R, Oltra E. Redefining the structure of the
mouse connexin43 gene: selective promoter usage and alternative splicing
mechanisms yield transcripts with different translational efficiencies. (2004).
Nucleic Acids Res. 32, 4550-4562.
• Phillips K, Luisi B. The virtuoso of versatility: POU proteins that flex to fit.
(2000). J Mol Biol. 302, 1023-1039.
• Piccolino M, Neyton J, Witkovsky P, Gerschenfeld HM. gamma-Aminobutyric
acid antagonists decrease junctional communication between L-horizontal
cells of the retina. (1982). Proc Natl Acad Sci U S A 79, 3671-3675.

97
• Piechocki MP, Burk RD, Ruch RJ. Regulation of connexin32 and connexin43
gene expression by DNA methylation in rat liver cells. (1999). Carcinogenesis
20, 401-406.
• Pickering BM, Mitchell SA, Evans JR, Willis AE. Polypyrimidine tract binding
protein and poly r(C) binding protein 1 interact with the BAG-1 IRES and
stimulate its activity in vitro and in vivo. (2003). Nucleic Acids Res. 31, 639-
646.
• Pilipenko EV, Pestova TV, Kolupaeva VG, Khitrina EV, Poperechnaya AN,
Agol VI, Hellen CU. A cell cycle-dependent protein serves as a template-
specific translation initiation factor. (2000). Genes Dev. 14, 2028-2045.
• Pottek M, Hoppenstedt W, Janssen-Bienhold U, Schultz K, Perlman I, Weiler
R. Contribution of connexin26 to electrical feedback inhibition in the turtle
retina. (2003). J Comp Neurol. 466, 468-477.
• Pudi R, Srinivasan P, Das S. La protein binding at the GCAC site near the
initiator AUG facilitates the ribosomal assembly on the hepatitis C virus RNA
to influence internal ribosome entry site-mediated translation. (2004). J Biol
Chem. 279, 29879-2988.
• Pugh BF, Tjian R. Mechanism of transcriptional activation by Sp1: evidence for
coactivators. (1990). Cell 61, 1187-1197.
• Purnick PE, Benjamin DC, Verselis VK, Bargiello TA, Dowd TL. Structure of
the amino terminus of a gap junction protein. (2000). Arch Biochem Biophys.
381, 181-190.
• Purnick PE, Oh S, Abrams CK, Verselis VK, Bargiello TA. Reversal of the
gating polarity of gap junctions by negative charge substitutions in the N-
terminus of connexin 32. (2000). Biophys J. 79, 2403-2415.
• Pyronnet S, Pradayrol L, Sonenberg N. A cell cycle-dependent internal
ribosome entry site. (2000). Mol Cell 5, 607-616.
• Qian H, Ripps H. Receptive field properties of rod-driven horizontal cells in
the skate retina. (1992). J Gen Physiol. 100, 457-478.
• Qin H, Shao Q, Curtis H, Galipeau J, Belliveau DJ, Wang T, Alaoui-Jamali MA,
Laird DW. Retroviral delivery of connexin genes to human breast tumor cells
inhibits in vivo tumor growth by a mechanism that is independent of

98
significant gap junctional intercellular communication. (2002). J Biol Chem.
277, 29132-29138.
• Qin H, Shao Q, Igdoura SA, Alaoui-Jamali MA, Laird DW. Lysosomal and
proteasomal degradation play distinct roles in the life cycle of Cx43 in gap
junctional intercellular communication-deficient and -competent breast tumor
cells. (2003). J Biol Chem. 278, 30005-30014.
• Raymond PA, Barthel LK, Rounsifer ME, Sullivan SA, Knight JK. Expression
of rod and cone visual pigments in goldfish and zebrafish: a rhodopsin-like
gene is expressed in cones. (1993). Neuron 10, 1161-1174.
• Ren P, de Feijter AW, Paul DL, Ruch RJ. Enhancement of liver cell gap
junction protein expression by glucocorticoids. (1994). Carcinogenesis 15,
1807-1813.
• Ryan AK, Rosenfeld MG. POU domain family values: flexibility, partnerships,
and developmental codes. (1997). Genes Dev. 11, 1207-1225.
• Sachs AB. Cell cycle-dependent translation initiation: IRES elements prevail.
(2000). Cell 101, 243-245.
• Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma
membrane channels formed by connexins: their regulation and functions.
(2003). Physiol Rev. 83, 1359-1400.
• Sasaki J, Nakashima N. Methionine-independent initiation of translation in the
capsid protein of an insect RNA virus. (2000). Proc Natl Acad Sci U S A 97,
1512-1515.
• Scheper GC, Voorma HO, Thomas AA. Base pairing with 18S ribosomal RNA
in internal initiation of translation. (1994). FEBS Lett. 352, 271-275.
• Schiavi A, Hudder A, Werner R. Connexin43 mRNA contains a functional
internal ribosome entry site. (1999). FEBS Lett. 464, 118-122.
• Scott MP, Weiner AJ. Structural relationships among genes that control
development: sequence homology between the Antennapedia, Ultrabithorax,
and fushi tarazu loci of Drosophila. (1984). Proc Natl Acad Sci U S A 81, 4115-
4119.
• Shatkin AJ. Capping of eucaryotic mRNAs. (1976). Cell 9, 645-653.

99
• Shatkin AJ. m-RNA cap binding proteins: essential factors for initiating
translation. (1985). Cell 40, 223-224.
• Shiroki K, Ohsawa C, Sugi N, Wakiyama M, Miura K, Watanabe M, Suzuki Y,
Sugano S. Internal ribosome entry site-mediated translation of Smad5 in vivo:
requirement for a nuclear event. (2002). Nucleic Acids Res. 30, 2851-2861.
• Simpson I, Rose B, Loewenstein WR. Size limit of molecules permeating the
junctional membrane channels. (1977). Science 195, 294-296.
• Sinha S, Maity SN, Lu J, de Crombrugghe B. Recombinant rat CBF-C, the third
subunit of CBF/NFY, allows formation of a protein-DNA complex with CBF-
A and CBF-B and with yeast HAP2 and HAP3. (1995). Proc Natl Acad Sci U S
A 92, 1624-1628.
• Sohl G, Guldenagel M, Traub O, Willecke K. Connexin expression in the
retina. (2000). Brain Res Brain Res Rev. 32, 138-145.
• Spahn CM, Jan E, Mulder A, Grassucci RA, Sarnow P, Frank J. (2004) Cryo-EM
visualization of a viral internal ribosome entry site bound to human
ribosomes: the IRES functions as an RNA-based translation factor. (2004). Cell
118, 465-475.
• Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA, Frank J.
Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s
ribosomal subunit. (2001). Science 291, 1959-1962.
• Stains JP, Lecanda F, Screen J, Towler DA, Civitelli R. Gap junctional
communication modulates gene transcription by altering the recruitment of
Sp1 and Sp3 to connexin-response elements in osteoblast promoters. (2003). J
Biol Chem. 278, 24377-24387.
• Stoneley M, Subkhankulova T, Le Quesne JP, Coldwell MJ, Jopling CL,
Belsham GJ, Willis AE. Analysis of the c-myc IRES; a potential role for cell-
type specific trans-acting factors and the nuclear compartment. (2000). Nucleic
Acids Res. 28, 687-694.
• Stock A, Sies H. Thyroid hormone receptors bind to an element in the
connexin43 promoter. (2000). Biol Chem. 381, 973-979.

100
• Streisinger G, Walker C, Dower N, Knauber D, Singer F. Production of clones
of homozygous diploid zebra fish (Brachydanio rerio). (1981). Nature 291, 293-
296.
• Sturm RA, Das G, Herr W. The ubiquitous octamer-binding protein Oct-1
contains a POU domain with a homeo box subdomain. (1988). Genes Dev. 2,
1582-1599.
• Tan LW, Bianco T, Dobrovic A. Variable promoter region CpG island
methylation of the putative tumor suppressor gene Connexin 26 in breast
cancer. (2002). Carcinogenesis 23, 231-236.
• Teunissen BE, Jansen AT, van Amersfoorth SC, O'Brien TX, Jongsma HJ,
Bierhuizen MF Analysis of the rat connexin 43 proximal promoter in neonatal
cardiomyocytes. (2003). Gene 322, 123-136.
• Tibbitts TT, Caspar DL, Phillips WC, Goodenough DA. Diffraction diagnosis
of protein folding in gap junction connexons. Biophys J. 1990 May;57(5):1025-
36.
• Tjian R, Maniatis T. Transcriptional activation: a complex puzzle with few
easy pieces. (1994). Cell 77, 5-8.
• Ullman KS, Flanagan WM, Edwards CA, Crabtree GR. Activation of early
gene expression in T lymphocytes by Oct-1 and an inducible protein, OAP40.
(1991). Science 254, 558-562.
• Unwin PN, Ennis PD. Two configurations of a channel-forming membrane
protein. (1984). Nature 307, 609-613.
• Van der Heyden MA, Rook MB, Hermans MM, Rijksen G, Boonstra J, Defize
LH, Destree OH. Identification of connexin43 as a functional target for Wnt
signalling. (1998). J Cell Sci. 111, 1741-1749.
• Vaney DI. Many diverse types of retinal neurons show tracer coupling when
injected with biocytin or Neurobiotin. (1991). Neurosci Lett. 125, 187-190.
• Vaney DI. The coupling pattern of axon-bearing horizontal cells in the
mammalian retina. (1993). Proc R Soc Lond B Biol Sci. 252, 93-101.
• Vaney DI, Nelson JC, Pow DV. Neurotransmitter coupling through gap
junctions in the retina. (1998). J Neurosci. 18, 10594-10602.

101
• Van Kempen MJ, Vermeulen JL, Moorman AF, Gros D, Paul DL, Lamers WH.
Developmental changes of connexin40 and connexin43 mRNA distribution
patterns in the rat heart. (1996). Cardiovasc Res. 32, 886-900.
• Veenstra RD. Size and selectivity of gap junction channels formed from
different connexins. (1996). J Bioenerg Biomembr. 28, 327-337.
• Veruki ML, Hartveit E. Electrical synapses mediate signal transmission in the
rod pathway of the mammalian retina. (2002). J Neurosci. 22, 10558-10566.
• Veruki ML, Hartveit E. AII (Rod) amacrine cells form a network of electrically
coupled interneurons in the mammalian retina. (2002). Neuron 33, 935-946.
• Vozzi C, Ullrich S, Charollais A, Philippe J, Orci L, Meda P. Adequate
connexin-mediated coupling is required for proper insulin production. (1995).
J Cell Biol. 131, 1561-1572.
• Weiler R, Schultz K, Janssen-Bienhold U. Ca(2+)-dependency of spinule
plasticity at dendrites of retinal horizontal cells and its possible implication for
the functional role of spinules. (1996). Vision Res. 36, 3891-3900.
• White TW, Bruzzone R. Intercellular communication in the eye: clarifying the
need for connexin diversity. (2000). Brain Res Brain Res Rev. 32, 130-137.
• Wilson JE, Pestova TV, Hellen CU, Sarnow P. Initiation of protein synthesis
from the A site of the ribosome. (2000). Cell 102, 511-520.
• Wright KL, Vilen BJ, Itoh-Lindstrom Y, Moore TL, Li G, Criscitiello M,
Cogswell P, Clarke JB, Ting JP. CCAAT box binding protein NF-Y facilitates in
vivo recruitment of upstream DNA binding transcription factors. (1994).
EMBO J. 13, 4042-4053.
• Xie J, Lee JA, Kress TL, Mowry KL, Black DL. Protein kinase A
phosphorylation modulates transport of the polypyrimidine tract-binding
protein. (2003). Proc Natl Acad Sci U S A 100, 8776-8781.
• Yamanaka S, Zhang XY, Maeda M, Miura K, Wang S, Farese RV Jr, Iwao H,
Innerarity TL. Essential role of NAT1/p97/DAP5 in embryonic differentiation
and the retinoic acid pathway. (2000). EMBO J. 19, 5533-5541.
• Yang D, Cheung P, Sun Y, Yuan J, Zhang H, Carthy CM, Anderson DR,
Bohunek L, Wilson JE, McManus BM. A shine-dalgarno-like sequence

102
mediates in vitro ribosomal internal entry and subsequent scanning for
translation initiation of coxsackievirus B3 RNA. (2003). Virology 305, 31-43.
• Yang Q, Sarnow P. Location of the internal ribosome entry site in the 5' non-
coding region of the immunoglobulin heavy-chain binding protein (BiP)
mRNA: evidence for specific RNA-protein interactions. (1997). Nucleic Acids
Res. 25, 2800-2807.
• Yeager M. Structure of cardiac gap junction intercellular channels. (1998). J
Struct Biol. 121, 231-245.
• Yeager M, Gilula NB. Membrane topology and quaternary structure of cardiac
gap junction ion channels. (1992). J Mol Biol. 223, 929-948.
• Yu W, Dahl G, Werner R. The connexin43 gene is responsive to oestrogen.
(1994). Proc R Soc Lond B Biol Sci. 255, 125-132.
• Zang WQ, Li B, Huang PY, Lai MM, Yen TS. Role of polypyrimidine tract
binding protein in the function of the hepatitis B virus posttranscriptional
regulatory element. (2001). J Virol. 75, 10779-10786.
• Zhou W, Edelman GM, Mauro VP. Transcript leader regions of two
Saccharomyces cerevisiae mRNAs contain internal ribosome entry sites that
function in living cells. (2001). Proc Natl Acad Sci U S A 98, 1531-1536.
• Zoidl G, Bruzzone R, Weickert S, Kremer M, Zoidl C, Mitropoulou G, Srinivas
M, Spray DC, Dermietzel R. Molecular cloning and functional expression of
zfCx52.6: a novel connexin with hemichannel-forming properties expressed in
horizontal cells of the zebrafish retina. (2004). J Biol Chem. 279, 2913-2921.

103
7. Vector maps. I) Vector map of pGL3-Basic. pGL3-Basic Vector Sequence Reference Points: SV40 Promoter…………………(none) SV40 Enhancer…………………(none) Multiple cloning region……….......1–58 Luciferase gene ( luc+)…………88–1740 GLprimer2 binding site……….. 89–111 SV40 late poly(A) signal……….1772–1993 RVprimer4 binding site………..2080–2061 ColE1-derived plasmid replication origin…………………2318 β-lactamase gene (Ampr)…….3080–3940 f1 origin………………………….4072–4527 Synthetic poly(A) signal………...4658–4811 RVprimer3 binding………………4760–4779.
II) pBluescripit vector KS II f1 (–) origin……………………………………21–327 β-galactosidase α-fragment…………………460–816 multiple cloning site………………………….653–760 lac promoter…………………………………...817–938 pUC origin…………………………………….1158–1825 ampicillin resistance (bla) ORF……………..1976–2833

104
III) pEGFP-1 MCS...............................................................12–89 Enhanced green fluorescent protein (EGFP) gene………………………..97-816 SV40 early mRNA polyadenylation signal……………970–975 & 999–1004 mRNA 3' ends...............................................1008 & 1020 Kanamycin/neomycin resistance gene……2047-2841
Multiple cloning site III) pEGFP-N3 Human cytomegalovirus (CMV) immediate early promoter…………………1-589 MCS………………………………………….591-665 Enhanced green fluorescent protein gene………………………………675-1394 SV40 early mRNA polyadenylation signal………1548-1553 & 1577-1582 Kanamycin/neomycin resistance gene..........................................2625-3419
Multiple cloning site

105
IV) pRL-CMV CMV enhancer and immediate early promoter……………………….. 7–803 Chimeric intron……………………….860–996 T7 promoter (–17 to +2)……………. 1040–1058 T7 promoter transcription start site…………………………………1057 R lucreporter gene…………………….1068–2003 SV40 late polyadenylation Signal…………………………………..2045–2246 β-lactamase (Ampr) coding region………………………….2393–3253 V) pGEX-2TK Glutathione S-transferase gene MCS.....................................930-945 Beta-lactamase gene......1356-2214

106
Curriculum Vitae
Mahboob ul Hussain
[email protected] Dept. of Neuroanatomy and Molecular Brain Research, Ruhr University Bochum, Bochum, Germany. Universitätstrasse 150, D44801 Telephone 49-(0) 234-32-24408 Fax 49-(0) 234-32-25004
Education/Training August 2002-Present: Graduate student, International Graduate School of
Neurosciences, Ruhr University Bochum, Bochum, Germany. Doctoral thesis: “Transcriptional and translational regulation of zebrafish connexin genes, zfCx55.5 and zfCx52.6”.
Thesis advisor: Dr. Rolf Dermietzel. August 1999-July 2002: Junior Research Fellow, Centre for Biotechnology, Jawaharlal Nehru University (JNU), New Delhi, India. 1997-1999: Master student, Department of Biochemistry, University of Kashmir, India. (Degree, Masters in Biochemistry) Fellowships: Research fellowship from the “Council of Scientific and Industrial Research”, Govt. of India (CSIR/NET).
Publications:
M-U-Hussain, M. Kremer, G.Zoidl, R.Dermietzel. 2003. Transcriptional and translational regulation of zebrafish connexin 55.5 (zf.Cx.55.5) and connexin 52.6 (zf.Cx52.6). Cell Commun Adhes. 2003 Jul-Dec;10(4-6):227-31.

107
V. Handa, M-u-Hussain, N. Pati, U. Pati. 2002. Multiple liver-specific factors bind to a 64-bp element and activate apo(a) gene. Biochem Biophys Res Commun. 2002 Mar 22;292(1):243-9. M-U-Hussain, G.Zoidl, R.Dermietzel. Evidence for the internal translation of carboxy-terminal domain of zfCx55.5: IRES element in the coding region makes the translation possible. (Under submission) M-U-Hussain, G.Zoidl, M. DeStafino, R.Dermietzel. Characterization of IRES element of zfCx55.5: Functional implication of polypyrimidine tract binding protein (PTB). (Under submission) CR Shields, J Klooster, Y Claassen, G Zoidl, M-U-Hussain, R Dermietzel, M Kamermans. Two Connexins Expressed in Zebrafish Retinal Horizontal Cells (in preparation).
Conferences and Presentations 23rd – 28th August, 2003. International Gap junction conference, St John’s College, University of Cambridge, Cambridge, UK. Platform presentation “Transcriptional and Translational regulation of zfCx55.5 and zfCx52.6. 9th – 10th Oct, 2003. Retinal connexin meeting, Research and Retinal Signal processing. The Netherlands Ophthalmic Research Institute, Amsterdam, The Netherlands. 17th – 22th July, 2004. As a co-author of the poster “Two connexins expressed in zebrafish retinal Horizontal cells”, FASEB Summer Research Conference Retinal Neurobiology and visual Processing, Miami, USA. 4th – 6th Nov, 2004. Presented poster “IRES mediated internal translation of zfCx55.5”, SFB509, Ruhr University Bochum, Germany.

108
References Rolf Dermietzel, M.D., Ph.D. Department of Neuroanatomy and Molecular Brain Research, Ruhr University Bochum, Universitätstrasse 150; MA 6/159 D-44801 Bochum, Germany Telephone: 0049-234-322-5003 Fax: 0049-234-321-4655 E-mail: [email protected] Georg Zoidl, Ph.D. Department of Neuroanatomy and Molecular Brain Research, Ruhr University Bochum, Universitätstrasse 150; MA 6/159 D-44801 Bochum, Germany Telephone: 0049-234-322-5003 Fax: 0049-234-321-4655 E-mail: [email protected] Khurshid I Andrabhi (Ph.D) Department of Biotechnology, University of Kashmir, Srinagar, Kashmir, India 190006 E-mail: [email protected]


















