
Interaction of eosinophils with endothelial cells is modulated by prostaglandin EP4 receptors
11
Interaction of eosinophils with endothelial cells is modulated by prostaglandin EP4 receptors Viktoria Konya 1 , Sonia Philipose 1 , Zolta ´n Ba ´lint 2 , Andrea Olschewski 2 , Gunther Marsche 1 , Eva M. Sturm 1 , Rudolf Schicho 1 , Bernhard A. Peskar 1 , Rufina Schuligoi 1 and Akos Heinemann 1 1 Institute of Experimental and Clinical Pharmacology, Medical University of Graz, Graz, Austria 2 Experimental Anesthesiology, Department of Anesthesia and Intensive Care Medicine, Medical University of Graz, Graz, Austria Eosinophil extravasation across the endothelium is a key feature of allergic inflammation. Here, we investigated the role of PGE 2 and its receptor, E-type prostanoid receptor (EP)-4, in the regulation of eosinophil interaction with human pulmonary microvascular endothelial cells. PGE 2 and the EP4 receptor agonist ONO AE1-329 significantly reduced eotaxin- induced eosinophil adhesion to fibronectin, and formation of filamentous actin and gelsolin-rich adhesive structures. These inhibitory effects were reversed by a selective EP4 receptor antagonist, ONO AE3-208. PGE 2 and the EP4 agonist prevented the activation and cell-surface clustering of b2 integrins, and L-selectin shedding of eosinophils. Under physiological flow conditions, eosinophils that were treated with the EP4 agonist showed reduced adhesion to endothelial monolayers upon stimulation with eotaxin, as well as after TNF-a-induced activation of the endothelial cells. Selective activation of EP1, EP2, and EP3 receptors did not alter eosinophil adhesion to endothelial cells, whereas the EP4 antagonist prevented PGE 2 from decreasing eosinophil adhesion. Finally, eosinophil transmigration across thrombin- and TNF-a-activated endothelial cells was effectively reduced by the EP4 agonist. These data suggest that PGE 2 –EP4 signaling might be protective against allergic responses by inhibiting the interaction of eosinophils with the endothelium and might hence be a useful therapeutic option for controlling inappropriate eosinophil infiltration. Keywords: Adhesion . Endothelium . Eosinophils . Migration . Prostaglandins Supporting Information available online Introduction Eosinophil granulocytes are important effector cells in allergic inflammation and are recruited to the tissue by chemotactic signals [1–5]. The infiltrating eosinophils release several toxic and proinflammatory mediators which induce epithelial injury, airway hyper-responsiveness, and airway remodeling [6–8]. Therefore, eosinophils are regarded as potential therapeutic targets in allergic diseases and asthma [9]. Prostaglandins (PGs) play diverse roles in inflammation, depending on the cellular context, the type of PG being released, and the differential expression of PG receptors [10, 11]. While PGD 2, which is the predominant PG formed in mast cells, exerts proinflammatory effects by regulating the recruitment of eosino- Correspondence: Dr. Akos Heinemann e-mail: [email protected] & 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu Eur. J. Immunol. 2011. 41: 2379–2389 DOI 10.1002/eji.201141460 Leukocyte signaling 2379
-
Upload
viktoria-konya -
Category
Documents
-
view
212 -
download
0
Transcript of Interaction of eosinophils with endothelial cells is modulated by prostaglandin EP4 receptors

Interaction of eosinophils with endothelial cellsis modulated by prostaglandin EP4 receptors
Viktoria Konya1, Sonia Philipose1, Zoltan Balint2, Andrea Olschewski2,
Gunther Marsche1, Eva M. Sturm1, Rudolf Schicho1,
Bernhard A. Peskar1, Rufina Schuligoi1 and Akos Heinemann1
1 Institute of Experimental and Clinical Pharmacology, Medical University of Graz, Graz, Austria2 Experimental Anesthesiology, Department of Anesthesia and Intensive Care Medicine, Medical
University of Graz, Graz, Austria
Eosinophil extravasation across the endothelium is a key feature of allergic inflammation.
Here, we investigated the role of PGE2 and its receptor, E-type prostanoid receptor (EP)-4, in
the regulation of eosinophil interaction with human pulmonary microvascular endothelial
cells. PGE2 and the EP4 receptor agonist ONO AE1-329 significantly reduced eotaxin-
induced eosinophil adhesion to fibronectin, and formation of filamentous actin and
gelsolin-rich adhesive structures. These inhibitory effects were reversed by a selective EP4
receptor antagonist, ONO AE3-208. PGE2 and the EP4 agonist prevented the activation and
cell-surface clustering of b2 integrins, and L-selectin shedding of eosinophils. Under
physiological flow conditions, eosinophils that were treated with the EP4 agonist showed
reduced adhesion to endothelial monolayers upon stimulation with eotaxin, as well as
after TNF-a-induced activation of the endothelial cells. Selective activation of EP1, EP2, and
EP3 receptors did not alter eosinophil adhesion to endothelial cells, whereas the EP4
antagonist prevented PGE2 from decreasing eosinophil adhesion. Finally, eosinophil
transmigration across thrombin- and TNF-a-activated endothelial cells was effectively
reduced by the EP4 agonist. These data suggest that PGE2–EP4 signaling might be
protective against allergic responses by inhibiting the interaction of eosinophils with the
endothelium and might hence be a useful therapeutic option for controlling inappropriate
eosinophil infiltration.
Keywords: Adhesion . Endothelium . Eosinophils . Migration . Prostaglandins
Supporting Information available online
Introduction
Eosinophil granulocytes are important effector cells in allergic
inflammation and are recruited to the tissue by chemotactic
signals [1–5]. The infiltrating eosinophils release several toxic
and proinflammatory mediators which induce epithelial injury,
airway hyper-responsiveness, and airway remodeling [6–8].
Therefore, eosinophils are regarded as potential therapeutic
targets in allergic diseases and asthma [9].
Prostaglandins (PGs) play diverse roles in inflammation,
depending on the cellular context, the type of PG being released,
and the differential expression of PG receptors [10, 11]. While
PGD2, which is the predominant PG formed in mast cells, exerts
proinflammatory effects by regulating the recruitment of eosino-Correspondence: Dr. Akos Heinemanne-mail: [email protected]
& 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
Eur. J. Immunol. 2011. 41: 2379–2389 DOI 10.1002/eji.201141460 Leukocyte signaling 2379

phils, basophils, and Th2 lymphocytes to the sites of allergic
inflammation [12], PGE2 seems to attenuate inflammatory
responses and reduce tissue injury in airways [13]. PGE2 was found
to exert bronchoprotective effects in patients with asthma [14]. In
rats, the ovalbumin-induced early and late phase airway responses
were inhibited by intratracheally administered PGE2 [15].
The activity of PGE2 is mediated by four subtypes of E-type
prostanoid receptors (EP), EP1, EP2, EP3, and EP4 [16]. These G
protein-coupled receptors have distinct biological imprints
depending on their affinity with synthetic agonists and the acti-
vated intracellular signaling pathways. Activation of the
Gq-coupled EP1 receptor leads to intracellular Ca21 increase and
protein kinase C activation. The EP2 and EP4 receptors are
G protein-coupled and elevate intracellular cyclic adenosine
monophosphate (cAMP) which activates protein kinase A. The
EP3 receptor is coupled to the Gi protein and its activation
increases the intracellular levels of Ca21. The EP3 receptor itself
has several splice variants exhibiting some constitutive activity
[16, 17]. At the cellular level and in murine allergic inflamma-
tion, PGE2 was found to inhibit eosinophil trafficking via EP2
receptor activation [18]. In mouse BM, PGE2 decreased the
number of eosinophil peroxidase-positive cells and reduced the
expression of a4 integrin in eosinophils [19].
The role of PGE2 in endothelial function has not been inves-
tigated in detail. Particularly, it is unknown how PGE2 modulates
the interaction of eosinophils with the endothelium, which is a
pivotal step in eosinophil extravasation to the sites of allergic
inflammation. In this study, we report that PGE2 potently
attenuates the adhesion of human peripheral blood eosinophils to
pulmonary microvascular endothelial cells and their trans-
endothelial migration by negatively regulating eosinophil polar-
ization and activation of adhesion molecules.
Results
PGE2 reduces eosinophil adhesion to fibronectin viaEP4 receptor activation
The effect of PGE2 and the selective EP4 agonist ONO AE1-329 on
eosinophil adhesion was investigated using fibronectin-coated
plates (Fig. 1A). Eosinophils were preincubated with vehicle,
PGE2, or EP4 agonist (30 nM each) for 10 min at 371C. Eotaxin
induced concentration-dependent (0.3–3 nM) adhesion of eosi-
nophils to fibronectin which was significantly reduced by PGE2
and the EP4 agonist (Fig. 1A). The specific involvement of the
EP4 receptor was demonstrated by preincubating the cells with
an EP4-selective antagonist (ONO AE3-208; 300 nM) which
completely prevented the inhibitory effect of PGE2 on eosinophil
adhesion. Consistently, eosinophils showed membrane-associated
EP4 receptor expression as detected by laser-scanning confocal
microscopy (Fig. 1B and Supporting Information Fig. 1).
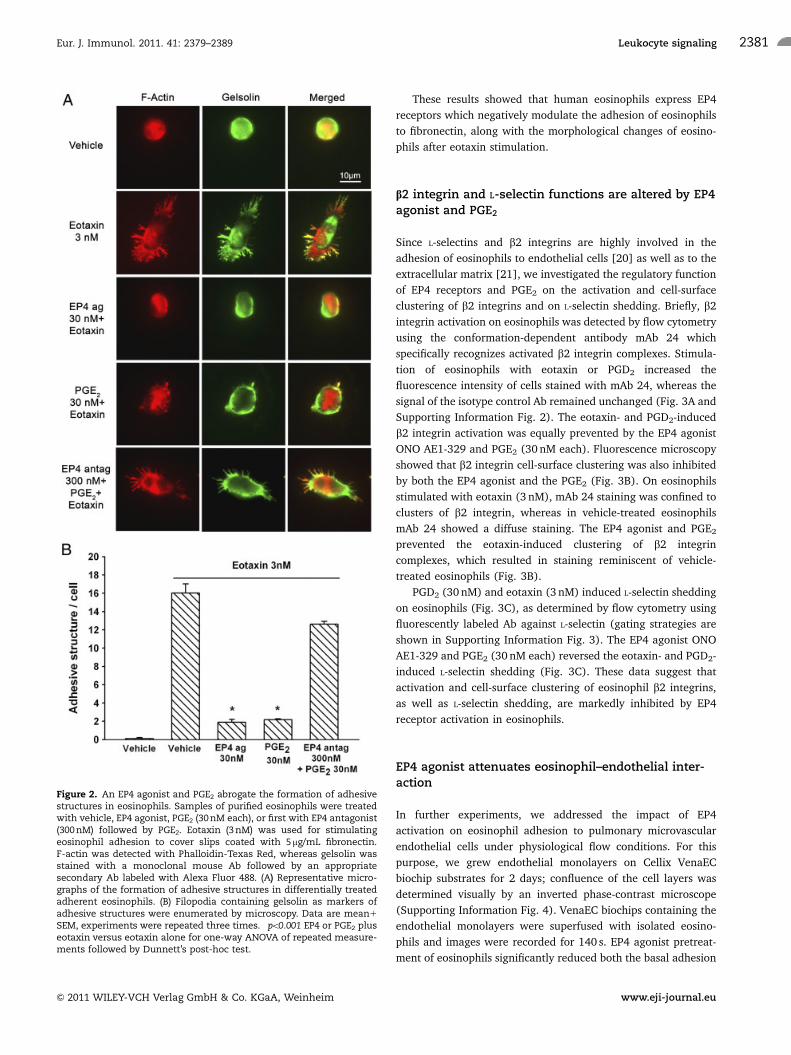
Formation of adhesive structures or protrusions is a key
element in eosinophil adhesion and migration. The effect of the
EP4 agonist and PGE2 on adhesive structure formation was
analyzed by preincubating eosinophils with vehicle, EP4 agonist,
or PGE2, 30 nM each (Fig. 2A and B). Adhesive structure
formation was induced by stimulation of eosinophils with eotaxin
(3 nM). Fluorescence microscopy revealed dramatic differences
in the cell shape and filamentous actin (F-actin) staining between
eosinophils stimulated in the presence of vehicle or EP4 agonist
and PGE2 respectively. The eotaxin-induced cellular polarization
and the development of lamellipodia/filopodia that contained
highly polymerized F-actin fibers were inhibited by the EP4
agonist and PGE2 (Fig. 2A). Gelsolin, an actin-severing protein,
was evenly distributed in vehicle-stimulated eosinophils but was
relocated to the cell periphery after eotaxin stimulation. In
eotaxin-stimulated eosinophils, gelsolin strongly colocalized with
F-actin as shown in the merged images (Fig. 2A). The colocali-
zation of gelsolin with F-actin was prevented by both the EP4
agonist and the PGE2. Importantly, the specific EP4 antagonist
largely reversed the inhibitory effect of PGE2 on adhesive struc-
ture formation and restored the colocalization of gelsolin with
F-actin (Fig. 2A). On average, eotaxin-stimulated eosinophils
exhibited 16 protrusions per cell; both the EP4 agonist and the
PGE2 reduced the number of adhesive structures to two to three
per cell (Fig. 2B) and the EP4 antagonist reversed the inhibitory
effect of PGE2 to 12 protrusions per cell (Fig. 2B).
Figure 1. The EP4 receptor is expressed on human eosinophils andinhibits the eotaxin-induced eosinophil adhesion to fibronectin.(A) Human-purified eosinophils were preincubated with the specificEP4 antagonist (antag) ONO AE3-208 or its vehicle, and then treatedwith vehicle, 30 nM of the EP4-selective agonist (ag) ONO AE1-329 orPGE2. In the presence of serial dilutions (0.3–3 nM) of eotaxin,eosinophils were allowed for 30 min to adhere to 96-well plates coatedwith 5mg/mL fibronectin. Results are mean1SEM percent of adherentcells relative to total eosinophils added. Experiments were performedfour times. �po0.05 versus vehicle for one-way ANOVA of repeatedmeasurements followed by Dunnett’s post-hoc test. (B) EP4 receptorexpression was determined by immunofluorescence microscopy. Thetop panel shows staining with the isotype control Ab, the bottom paneldisplays eosinophils stained with the EP4 Ab. The insert in the bottompanel is a laser-scanning micrograph showing cell-surface expressionof EP4 receptors on one single eosinophil (Z-stacks of the image areshown in Supporting Information). The secondary Ab was labeled withAlexa Fluor 647, and the nuclei are stained with DAPI. The micrographsare representative of eosinophil staining of three different donors.
Eur. J. Immunol. 2011. 41: 2379–2389Viktoria Konya et al.2380
& 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
These results showed that human eosinophils express EP4
receptors which negatively modulate the adhesion of eosinophils
to fibronectin, along with the morphological changes of eosino-
phils after eotaxin stimulation.
b2 integrin and L-selectin functions are altered by EP4agonist and PGE2
Since L-selectins and b2 integrins are highly involved in the
adhesion of eosinophils to endothelial cells [20] as well as to the
extracellular matrix [21], we investigated the regulatory function
of EP4 receptors and PGE2 on the activation and cell-surface
clustering of b2 integrins and on L-selectin shedding. Briefly, b2
integrin activation on eosinophils was detected by flow cytometry
using the conformation-dependent antibody mAb 24 which
specifically recognizes activated b2 integrin complexes. Stimula-
tion of eosinophils with eotaxin or PGD2 increased the
fluorescence intensity of cells stained with mAb 24, whereas the
signal of the isotype control Ab remained unchanged (Fig. 3A and
Supporting Information Fig. 2). The eotaxin- and PGD2-induced
b2 integrin activation was equally prevented by the EP4 agonist
ONO AE1-329 and PGE2 (30 nM each). Fluorescence microscopy
showed that b2 integrin cell-surface clustering was also inhibited
by both the EP4 agonist and the PGE2 (Fig. 3B). On eosinophils
stimulated with eotaxin (3 nM), mAb 24 staining was confined to
clusters of b2 integrin, whereas in vehicle-treated eosinophils
mAb 24 showed a diffuse staining. The EP4 agonist and PGE2
prevented the eotaxin-induced clustering of b2 integrin
complexes, which resulted in staining reminiscent of vehicle-
treated eosinophils (Fig. 3B).
PGD2 (30 nM) and eotaxin (3 nM) induced L-selectin shedding
on eosinophils (Fig. 3C), as determined by flow cytometry using
fluorescently labeled Ab against L-selectin (gating strategies are
shown in Supporting Information Fig. 3). The EP4 agonist ONO
AE1-329 and PGE2 (30 nM each) reversed the eotaxin- and PGD2-
induced L-selectin shedding (Fig. 3C). These data suggest that
activation and cell-surface clustering of eosinophil b2 integrins,
as well as L-selectin shedding, are markedly inhibited by EP4
receptor activation in eosinophils.
EP4 agonist attenuates eosinophil–endothelial inter-action
In further experiments, we addressed the impact of EP4
activation on eosinophil adhesion to pulmonary microvascular
endothelial cells under physiological flow conditions. For this
purpose, we grew endothelial monolayers on Cellix VenaEC
biochip substrates for 2 days; confluence of the cell layers was
determined visually by an inverted phase-contrast microscope
(Supporting Information Fig. 4). VenaEC biochips containing the
endothelial monolayers were superfused with isolated eosino-
phils and images were recorded for 140 s. EP4 agonist pretreat-
ment of eosinophils significantly reduced both the basal adhesion
Figure 2. An EP4 agonist and PGE2 abrogate the formation of adhesivestructures in eosinophils. Samples of purified eosinophils were treatedwith vehicle, EP4 agonist, PGE2 (30 nM each), or first with EP4 antagonist(300 nM) followed by PGE2. Eotaxin (3 nM) was used for stimulatingeosinophil adhesion to cover slips coated with 5mg/mL fibronectin.F-actin was detected with Phalloidin-Texas Red, whereas gelsolin wasstained with a monoclonal mouse Ab followed by an appropriatesecondary Ab labeled with Alexa Fluor 488. (A) Representative micro-graphs of the formation of adhesive structures in differentially treatedadherent eosinophils. (B) Filopodia containing gelsolin as markers ofadhesive structures were enumerated by microscopy. Data are mean1
SEM, experiments were repeated three times. �po0.001 EP4 or PGE2 pluseotaxin versus eotaxin alone for one-way ANOVA of repeated measure-ments followed by Dunnett’s post-hoc test.
Eur. J. Immunol. 2011. 41: 2379–2389 Leukocyte signaling 2381
& 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

(Fig. 4A, top) and the eotaxin-induced (3 nM) adhesion to the
endothelial layer (Fig. 4A, middle). Preactivation of the
endothelial layers with TNF-a (10 pM) for 4 h greatly enhanced
the number of adherent eosinophils. Similarly, eosinophil
adhesion to TNF-a-activated endothelial cells was reduced by
more than 50% in the presence of the EP4 agonist (Fig. 4A,
bottom). Corresponding quantitative data were obtained from
computerized image analysis of adherent eosinophils (Fig. 4B).
Representative video footage can be viewed on the journal
homepage. On the contrary, the EP1/EP3 agonist (17-phenyl
trinor PGE2, 30 nM), EP2 agonist (butaprost, 30 nM), or EP3
agonist (sulprostone, 30 nM) were found to have no effect on
eosinophil adhesion to TNF-a-stimulated endothelial monolayers
(Fig. 4C). Treatment with PGE2 mimicked the action of the EP4
agonist, and the inhibitory effect of PGE2 was prevented by the
selective EP4 receptor antagonist (Fig. 4C). Collectively, these
data demonstrated that the activation of EP4 receptors negatively
regulates the adhesion of eosinophils to endothelial cells.
Next, we tested the possibility that EP4 receptor activation
was capable of reversing adhesion, resulting in detachment of
eosinophils from endothelial cells. In Fig. 4D, eosinophils were
allowed to adhere to TNF-a-stimulated endothelial layers for
140 s and were then superfused with vehicle or EP4 agonist
(30 nM) for further 10 min. Images were acquired before and at
5–10 min after the start of the vehicle/EP4 agonist treatment.
While the number of adherent eosinophils remained stable when
superfused with vehicle, the EP4 agonist led to a time-dependent
detachment of adherent eosinophils from the endothelial mono-
layer (Fig. 4D).
Finally, we investigated the effect of the EP4 agonist on
eosinophil transmigration across monolayers of pulmonary
microvascular endothelial cells (Fig. 5). Endothelial cells were
grown in transwell inserts until confluence, as confirmed by
measuring transendothelial electrical resistance. Transendothe-
lial migration was calculated as the percent of total eosinophils
added to each well. Eosinophil migration was more than six-fold
enhanced when eotaxin (3 nM) was present in the bottom well.
After pretreatment of the endothelial layer with thrombin (0.5 U/
mL) for 15 min, eosinophil migration was further increased to
almost 15-fold of baseline (Fig. 5A). Pretreatment of eosinophils
with 30 nM of the EP4 agonist reduced the eotaxin-triggered
transendothelial migration of eosinophils from approximately 35
to 12% (Fig. 5A). Similar observations were made when endo-
thelial monolayers were activated with TNF-a for 4 h which
resulted in a 20-fold increase of eosinophil migration (Fig. 5B).
The EP4 agonist reduced eosinophil transmigration across
TNF-a-stimulated endothelial layers from 57 to 29% of input
eosinophils (Fig. 5B). These data suggested that EP4 agonists are
able to control transendothelial migration of eosinophils.
Discussion
In the present study, we show that PGE2 and EP4 receptors
potently regulate eosinophil interaction with pulmonary endothe-
lial cells, as PGE2 and the EP4 agonist ONO AE1-329 significantly
reduced adhesion of eosinophils to endothelial cells under
physiological flow conditions and blocked eosinophil trans-
endothelial migration. Visualization of eosinophil adhesion to
fibronectin revealed that PGE2 and the EP4 agonist deranged
the polarization and formation of cellular adhesive structures in
eosinophils. Furthermore, activation and cell-surface clustering of
b2 integrins and L-selectin shedding were inhibited by PGE2 and
the EP4 agonist of that ilk. Eosinophil EP4 receptor expression on
the cell membrane was confirmed by laser-scanning confocal
Figure 3. Eosinophil b2 integrin activation and cell-surface clusteringand L-selectin shedding are inhibited by an EP4 agonist and PGE2.(A) Purified human eosinophils were treated with the EP4 agonist (ag)ONO AE1-329 or PGE2 (30 nM each) and stimulated with eotaxin (3 nM)or PGD2 (30 nM). The activation of b2 integrins was thereafterdetermined by indirect immunofluorescence staining using mAb 24and flow cytometry. Data are mean1SEM fluorescence intensity,experiments were performed four times. �po0.001 EP4 agonist orPGE2 versus vehicle for one-way ANOVA of repeated measurementsfollowed by Dunnett’s post-hoc test. (B) Clustering of activatedb2 integrins was detected by staining with mAb 24 and immuno-fluorescence microscopy. Micrographs are representative of threeexperiments. (C) Polymorphonuclear leukocyte preparations wereincubated with vehicle, EP4 agonist (ONO AE1-329) or PGE2 (30 nMeach). L-selectin shedding was induced by eotaxin (3 nM) or PGD2
(30 nM) and was determined using flow cytometry. Eosinophils weredistinguished from neutrophils as CD16neg cells. Data are presented asmean1SEM fluorescence intensity, experiments were repeated fourtimes. �po0.001 EP4 agonist or PGE2 versus vehicle for one-way ANOVAof repeated measurements followed by Dunnett’s post-hoc test.
Eur. J. Immunol. 2011. 41: 2379–2389Viktoria Konya et al.2382
& 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

microscopy. Therefore, EP4-mediated attenuation of eosinophil
–endothelial interaction might afford a novel therapeutic
approach to preventing eosinophil recruitment at the sites of
allergic inflammation.
In detail, we found that selective activation of the EP4
receptor significantly inhibited the chemokine-induced adhesion
of eosinophils to fibronectin. One of the initial steps in the
extravasation process is the capture and adhesion of eosinophils
to the endothelium, via the molecular interaction of eosinophil
selectins and integrins with endothelial adhesion molecules.
Integrins also mediate adhesion to extracellular matrix compo-
nents, such as fibronectin, vitronectin, laminin, or collagen
[21, 22]. In the latter case, integrins recognize the RGD (Arg-Gly-
Asp) amino acid sequence of the matrix macromolecules which is
the recognition and binding site for the a5b1 and aMb2 integrins
[23]. Addition of PGE2 elicited the same inhibitory effect on
eosinophil adhesion to fibronectin as the EP4 agonist. Morpho-
logical analysis of eosinophils adhering to fibronectin showed
that the actin cytoskeleton was reorganized upon eotaxin
stimulation, an event that was coupled to cellular polarization
and formation of lamellipodia/filopodia and F-actin/gelsolin-rich
adhesive structures. Treatment with PGE2 and the EP4 selective
agonist prevented these morphological changes. The highly
dynamic F-actin-rich protrusions contain large amounts of
integrins, and actin-binding and actin-severing proteins such as
gelsolin. The major function of gelsolin is to rapidly depolymerize
Figure 4. EP4 receptor activation suppresses eosinophil adhesion to endothelial cells under flow conditions. Eosinophils treated with vehicle orthe EP4 agonist ONO AE1-329 (30 nM) were flowed over human pulmonary microvascular endothelial cells grown on VenaEC biochips (Cellix).Eosinophil adhesion was induced by stimulating eosinophils with eotaxin (3 nM) or by activating the endothelial monolayer with TNF-a (10 pM).(A) Representative images taken 2 min after the start of the superfusion with eosinophils. (B) Images were quantified by computerized imageanalysis. Data are shown as mean1SEM of four experiments with different donors. �po0.001, EP4 agonist versus vehicle for one-way ANOVA ofrepeated measurements followed by Dunnett’s post-hoc test. (C) Eosinophils were treated with EP1/EP3 agonist (EP1, 17-phenyl trinor PGE2, 30 nM),EP2 agonist (butaprost, 30 nM), EP3 agonist (sulprostone, 30 nM), EP4 agonist (ONO AE1-329, 30 nM), or PGE2 (30 nM). Alternatively, eosinophils werefirst pretreated with EP4 antagonist (ONO AE3-208, 300 nM) followed by PGE2 (30 nM). Cells were then flowed over TNF-a-stimulated endothelial cellsand their adherence was recorded 2 min later. Results are shown as mean1SEM of four experiments with different donors. �po0.001, EP4 agonist andPGE2 versus vehicle, or EP4 antagonist plus PGE2 versus PGE2 alone for one-way ANOVA of repeated measurements followed by Dunnett’s post-hoctest. (D) Vehicle-treated eosinophils were flowed over TNF-a-stimulated endothelial cells and the attached eosinophils were then superfused withvehicle or EP4 agonist (ONO AE1-329, 30 nM) for an additional 5 or 10 min. Data are presented as mean1SEM of four experiments with differentdonors. �po0.05, EP4 agonist after 5 or 10 min versus before for one-way ANOVA of repeated measurements followed by Dunnett’s post-hoc test.
Eur. J. Immunol. 2011. 41: 2379–2389 Leukocyte signaling 2383
& 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

F-actin, and thus gelsolin sustains the dynamics of protrusions
and withdrawal which is essential for directed locomotion
[24, 25].
Adhesion sites contain clustered integrins that transmit
mechanical forces in the migration process and are involved in
signaling cascades essential for cell survival and morphogenesis
[26]. Eotaxin- and PGD2-induced b2 integrin activation and
clustering was markedly reduced by the EP4 agonist and PGE2.
Therefore, it is very likely that the PGE2/EP4-induced blockade of
b2 integrin activation and cell-surface clustering might take a
profound effect on eosinophil recruitment and function at the
sites of inflammation. A potential limitation of our clustering
experiments may be that we used the activation-sensitive mAb 24
for detecting b2 integrin complexes. Since a recent report showed
that integrin activation and clustering can occur independently
[26], it is possible that we missed detecting the lateral clustering
of nonactivated b2 integrins. L-selectin mediates the capture and
rolling of eosinophils on the endothelium. Chemoattractant-
induced activation of eosinophils can lead to shedding of
L-selectin due to metalloproteinase activity. The EP4 agonist and
PGE2 prevented the eotaxin- and PGD2-stimulated L-selectin
shedding, which seems consistent with the ability of EP4 recep-
tors to reverse eosinophils activation. Since these changes in
eosinophil function might also translate to compromised eosi-
nophil–endothelial interaction, we examined the impact of EP4
receptor activation on eosinophil adhesion to human pulmonary
microvascular endothelial cells under flow conditions. In fact,
eosinophils that had been pretreated with EP4 agonist showed
reduced adhesion to resting as well as TNF-a-activated endo-
thelial cells. On the contrary, other PGE2 receptors, i.e. EP1, EP2,
and EP3, do not seem to play a role since agonists of these
receptors did not mimic the inhibitory effects of the EP4 agonist
or PGE2. Interestingly, the activation of EP4 receptors was suffi-
cient to deactivate eosinophils that had previously become
adherent, leading to detachment from endothelial cells.
TNF-a, a predominant proinflammatory cytokine, has been
shown to be a chemoattractant for both neutrophils and eosino-
phils [27]. Furthermore, TNF-a has been suggested to play an
important role in severe refractory asthma, a rare type of asthma
which affects 5–10% of all asthmatic patients. Being largely
unresponsive to inhaled corticosteroids, this subgroup of
asthmatics urgently requires novel therapies [28–30]. Anti-TNF-atherapy has been proved to improve lung function, airway hyper-
responsiveness, and exacerbation rates in some but not all
refractory asthma patients [31–34]. However, a potential risk of
anti-TNF-a therapy is the development of malignancies and
increased susceptibility for infections which can limit the clinical
use of TNF-a blockers [35]. Importantly, the present study
suggests a possible application of EP4-selective agonists in
treating severe refractory asthma, as an EP4 agonist markedly
reduced the adhesion of eosinophils to TNF-a-treated pulmonary
microvascular endothelial cells.
In additional interaction studies, we investigated eosinophil
transmigration across endothelial monolayers activated with
TNF-a or thrombin. Eosinophil chemotaxis was triggered by
eotaxin. In further support of the inhibitory role of the EP4
receptor in eosinophil trafficking, we observed that the EP4
agonist markedly reduced the transendothelial migration of
eosinophils through thrombin-activated as well as TNF-a-stimu-
lated endothelial monolayers. The involvement of thrombin in
bronchial asthma is supported by several reports; increased levels
of thrombin were found in asthmatic airways [36–38]. Moreover,
thrombin was shown to exert chemotactic activity on eosinophils
via activation of protease-activated receptor-1 [39]. In our assay,
TNF-a and thrombin were present in the experimental setup
throughout the 4 h migration period and could hence exert their
effects both on endothelial cells and on eosinophils.
Importantly, the reduced responsiveness of eosinophils after
EP4 receptor activation was not due to downregulation of the
respective C–C chemoattractant receptors (CCR3 and chemo-
attractant receptor homologous molecule expressed on Th2
lymphocytes) or reduced viability or a proapoptotic effect of the
EP4 agonist (Supporting Information Figs. 5 and 6). Moreover,
PGE2 showed the same potency in all assays tested at inhibiting
eosinophil responses as the EP4 agonist, and the effects of PGE2
were completely prevented by EP4 antagonist. This included
eosinophil adhesion to fibronectin, adhesion to endothelial
monolayers, polarization of eosinophils, and formation of adhe-
sive structures, which suggests that PGE2 might exert inhibitory
effects on eosinophil–endothelial interaction also physiologically
through EP4 receptors.
Collectively, the results of the current study are in agreement
with our previous findings that PGE2 decreases the migration of
eosinophils through uncoated filters toward PGD2 and eotaxin.
This effect was at least partially reversed by the EP4 antagonist
[18], although EP2 receptors were also found to be involved in
this setup. In further experiments, we could substantiate the
Figure 5. EP4 agonist inhibits eosinophil transendothelial migrationacross thrombin- and TNF-a-activated endothelial monolayers.Endothelial cells were grown in Transwell inserts and were activatedby (A) 0.5 U/mL thrombin or (B) 10 pM TNF-a. Eosinophils werepretreated with vehicle or the EP4 agonist ONO AE1-329 (30 nM), andtransmigration was stimulated with eotaxin (3 nM) or vehicle for 4 h.Results are shown as the mean1SEM percentage of transmigratedeosinophils relative to total eosinophils added. Experiments wereperformed four times, �po0.05 EP4 agonist versus vehicle for one-wayANOVA of repeated measurements followed by Dunnett’s post-hoctest.
Eur. J. Immunol. 2011. 41: 2379–2389Viktoria Konya et al.2384
& 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

inhibitory role of EP4 receptors in eosinophils in assays of CD11b
upregulation, production of reactive oxygen species and Ca21
mobilization [40]. These findings have now fundamentally been
extended by the current study as we found that the regulatory
role of EP4 receptors translates to preventing the interaction of
eosinophils with the endothelium, which is probably the most
crucial step in eosinophil recruitment to the sites of inflamma-
tion. Interestingly, we recently identified the EP4 receptor as a
potent anti-aggregatory regulator. Inhibition of platelet aggre-
gation by a selective EP4 agonist was characterized by attenuated
Ca21 flux, inhibition of glycoprotein IIb/IIIa, and downregulation
of P-selectin. Moreover, adhesion of platelets to fibrinogen under
flow conditions and in vitro thrombus formation were similarly
prevented by the EP4 agonist [41]. Other anti-inflammatory
actions of EP4 receptors reported previously include inhibition of
cytokine production in monocytes and macrophages [42–44],
and mucus secretion in airway epithelial cells [45]. From our
current data, we cannot exclude the possibility that parts of the
inhibitory effects of PGE2 and the EP4 agonist also involve EP4
receptors expressed on the endothelial cells. Therefore, the role
of EP4 receptors in endothelial function awaits further investi-
gation.
In summary, increased levels and protective effects of PGE2 in
pulmonary inflammatory diseases have been described previously
[14, 46]. The accumulation of eosinophils in asthmatic airways
greatly contributes to the outcome of the disease by releasing
cytotoxic mediators, leading to airway remodeling and angiogen-
esis in the chronically inflamed pulmonary tissue [7]. The present
study provides an explanation for the protective role of PGE2 in
allergic inflammation, by (i) identifying the EP4 subtype as key
binding site for PGE2 and (ii) revealing the cellular mechanisms by
which the EP4 receptor is involved in controlling eosinophil
interaction with the endothelium. Furthermore, our findings
suggest that EP4-selective agonists might be useful therapeutic
agents for treating otherwise uncontrolled bronchial asthma.
Materials and methods
Chemicals
Laboratory reagents were purchased from Sigma-Aldrich
(Vienna, Austria), unless specified. Human eotaxin/CCL11 and
TNF-a were from Peprotech (London, UK). PGE2, PGD2, and the
selective agonists for EP1/EP3, 17-phenyl trinor PGE2, EP2,
butaprost, and EP3, sulprostone were purchased from Cayman
(Ann Arbor, MI, USA). The monoclonal mouse anti-EP4 receptor
Ab was purchased from SantaCruz (Heidelberg, Germany). ONO
AE1-329 and ONO AE3-208 were kind gifts from ONO
Pharmaceutical (Osaka, Japan). The EP4 agonist ONO AE1-329
(2-[[2-[2-(2-methylnaphthalen-1-yl) propanoylamino] phenyl]-
methyl]benzoic acid) has been shown to selectively bind to EP4
receptors (Ki 5 35 nM) relative to the EP1, EP2, and EP3
receptors (Ki 5 3000, 2000, and 43000 nM respectively) [47].
The Ki values of the EP4 antagonist ONO AE3-208
(2-[[2-[2-(2-methylnaphthalen-1-yl)propanoylamino]phenyl]-
methyl]benzoic acid) are 1.3, 30, 790, 2400 nM for EP4, EP3, FP,
and TP, respectively, and more than 10 000 nM for other
prostanoid receptors [48]. Anti-rabbit and anti-mouse IgG
secondary Abs conjugated with Alexa Fluor 488 and 647 were
supplied by Invitrogen. mAb 24 (activation-sensitive b2 integrin
Ab) was from Hycult Biotechnology (Uden, The Netherlands).
Phalloidin-Texas Red was purchased from Molecular Probes
(Eugene, OR, USA). Vectashield/DAPI mounting medium was
from Vector Laboratories (Burlingame, CA, USA). Ultra V Blocking
solution was purchased from Lab Vision (Fremont, CA, USA) and
Ab diluent from Dako (Carpinteria, CA, USA). Anti-CD62L (FITC),
anti-CD1 (PE), CellFix, and FACS-Flow were obtained from BD
(Vienna, Austria). Assay buffer was prepared from Dulbecco-
modified PBS (with 0.9 mmol/L Ca21 and 0.5 mmol/L Mg21;
Invitrogen, Vienna, Austria), 0.1% BSA, 10 mmol/L HEPES, and
10 mmol/L glucose, pH 7.4 [49]. Fixative solution was made by
adding 9 mL of distilled water and 30 mL of FACS-Flow to 1 mL of
CellFix [50]. Drugs were dissolved in distilled water, ethanol, or
dimethyl sulfoxide and further diluted in assay buffer to have a
final concentration of the solvents ofo0.1%.
Preparation of human peripheral blood eosinophils
Blood was drawn from healthy nonatopic volunteers, according
to a protocol approved by the Ethics Committee of the Medical
University of Graz. Polymorphonuclear leukocytes (PMNLs,
containing eosinophils and neutrophils) were isolated by dextran
sedimentation of erythrocytes then by centrifugation on Histo-
paque gradients as described previously [51, 52]. Eosinophils
were purified from polymorphonuclear leukocyte using negative
magnetic selection with an Ab cocktail (CD2, CD14, CD16, CD19,
CD56, and glycophorin A) and colloidal magnetic particles from
StemCell Technologies (Vancouver, Canada) [53]. All separation
steps were performed at room temperature. The eosinophil
preparations showed more than 95% purity and viability.
Culture of endothelial cells
Human lung microvascular endothelial cells (HMVEC-L, Lonza,
Verviers, Belgium) were cultivated in EGM-2 MV Bullet kit
medium (Lonza) with 5% FCS. Endothelial cell attachment and
growth was promoted by precoating with 1% gelatin for 1 h at
371C. Fresh medium was added every 2 days and cells were
passaged after reaching 90% confluence (5–6 days); the cells
were used between 5 and 10 passages [54, 55].
Eosinophil adhesion to fibronectin
Adhesion of purified eosinophils to fibronectin was determined by
incubating the eosinophils (6� 104/well) for 30 min at 371C in flat-
Eur. J. Immunol. 2011. 41: 2379–2389 Leukocyte signaling 2385
& 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

bottom 96-well plates that had been precoated with 5mg/mL
fibronectin for 2 h [55]. Eosinophils were treated with vehicle, EP4
agonist (ONO AE1-329, 30 nM), or PGE2 (30 nM) or first
preincubated with EP4-selective antagonist (ONO AE3-208,
100 nM) for 15 min at 371C and then treated with PGE2 (30 nM).
Adhesion of eosinophils was induced by increasing concentrations
of eotaxin (0.3–1.3 nM). After 30 min incubation, first the non-
adherent cells were collected, and thereafter the adherent eosino-
phils were harvested with 100mL trypsin/EDTA. Both nonadherent
and adherent cells were enumerated by means of flow cytometry.
Eosinophil adhesion was calculated as the percentage of adherent
eosinophils with respect to the total cells added.
L-selectin shedding
Polymorphonuclear leukocyte preparations were incubated with
vehicle or EP4 agonist (ONO AE1-329, 30 and 300 nM) for 5 min
at 371C. L-selectin shedding was induced by eotaxin (3 nM) or
PGD2 (30 nM) for 10 min at 371C. Cells were then stained with
anti-CD62L (FITC) and anti-CD16 (PE) Abs for 30 min at 41C.
CD62L expression on CD16neg eosinophils was quantified by flow
cytometry [56].
Immunofluorescence staining of adherent eosinophils
For double staining of F-actin and gelsolin in eosinophils, chamber
slides were precoated with 5mg/mL fibronectin for 2 h, and
purified eosinophils were incubated for 12 min at 371C. Eosino-
phils were pretreated in Eppendorf tubes with vehicle, ONO AE1-
329 (30 nM), or PGE2 (30 nM) or first with EP4 antagonist
(300 nM) or its vehicle for 15 min and subsequently with PGE2
(30 nM). Eosinophil adhesion was stimulated with eotaxin (3 nM).
After the removal of nonadherent cells, adherent eosinophils were
fixed with 3.7% formaldehyde for 10 min, permeabilized with
0.1% Triton X-100 for 10 min, and nonspecific binding sites were
blocked with 1% BSA for 20 min. Eosinophils were first incubated
with anti-gelsolin primary Ab (1:100) for 1 h, then with a mixture
of phalloidin-Texas Red (5 U/mL) and the second Ab labeled with
AF-488 (1:500) for 30 min in the dark. All steps were performed at
room temperature [57]. The slides were then mounted with
Vectashield mounting medium.
Activation and clustering of b2 integrins
Activation of b2 integrins in eosinophils was determined by
means of indirect immunofluorescence flow cytometry with
mAb 24, which recognizes only the activated conformation of
b2 integrins (CD18) [58]. Purified eosinophils were treated with
vehicle or 30 nM ONO AE1-329 for 10 min, activated with vehicle,
eotaxin (3 nM), or PGD2 (30 nM) for 12 min at 371C in the
presence of mAb 24 (1:50) or isotype control Ab followed by
staining with Alexa Fluor 488-labeled secondary Ab (1:500). The
stained samples were either directly measured by flow cytometry
[59] or were cytospun onto microscope slides (600 rpm 3 min,
Cytospin3, Shandon) and observed with a Olympus IX70 fluor-
escence microscope and UPlanFI – 60� /1.20 lens; images were
acquired using a Hamamatsu ORCA-ER digital camera [60].
Immunofluorescence staining of EP4 receptor oneosinophils
EP4 expression of purified human eosinophils was detected by
immunofluorescence and laser-scanning microscopy. The mono-
clonal mouse Ab used targets the C-terminal intracellular tail of the
EP4 receptor, and thus the cells were fixed and permeabilized, and
nonspecific binding sites were blocked with Ultra V Blocking
solution for 30 min at room temperature. Samples were then
incubated with the specific mouse EP4 primary Ab or an irrelevant
mouse IgG control Ab (4mg/mL each) for 1 h at room temperature.
Finally, 4mg/mL goat anti-mouse IgG secondary Ab conjugated
with Alexa Fluor 647 was added, cells were fixed, cytospun
(600 rpm 3 min, Cytospin3, Shandon) to microscope slides and
analyzed on a laser-scanning confocal microscope, LSM 510 Meta
(Zeiss), with the following Ex/Em settings: 405/BP420-480
(DAPI); 633/679-754 (Alexa Fluo 647). The images were taken
with a Zeiss 100� oil immersion objective and 0.6mm Z-slice
thickness over the whole cell. Typically, 12–15 slices were taken
from one cell with 1024� 1024 resolution; these images were
reconstituted to a 3-D image with the software of the instrument.
Eosinophil adhesion to endothelial cells under flowconditions
Endothelial cells (4� 105/substrate) were grown on VenaEC
biochips (Cellix, Dublin, Ireland). After reaching confluence
(usually within 2 days), endothelial layers were incubated with
vehicle or TNF-a (10 pM) for 4 h. Endothelial monolayers were
superfused with suspensions of 3� 106/mL eosinophils at
0.5 dyne/cm2 for 2 min and 20 s at 371C in a OKOLAB H201-
T1-heated cage. Eosinophils were treated with vehicle or ONO
AE1-329 (30 nM) for 10 min at 371C. In some cases, eosinophil
adhesion was stimulated with eotaxin treatment (3 nM for 5 min
at 371C). Eosinophils were treated with 17-phenyl trinor PGE2,
butaprost, sulprostone, ONO AE1-329, or PGE2 (each 30 nM) for
10 min at 371C. Alternatively, eosinophils were first incubated
with the EP4-selective antagonist (ONO AE3-208, 300 nM for
15 min) followed by PGE2. In other cases, the EP4 agonist
(30 nM) or its vehicle was superfused over eosinophils already
attached to TNF-a-stimulated endothelial cells as post-treatment.
Cell adhesion was monitored by phase contrast on a Zeiss
Axiovert 40 CFL microscope and Zeiss A-Plan 10� /0.25 Ph1
lens, using a Hamamatsu ORCA-03G digital camera and Cellix
VenaFlux software. Computerized image analysis was performed
by DucoCell analysis software (Cellix, Dublin), where adherent
eosinophils were quantified on each single image [41, 61].
Eur. J. Immunol. 2011. 41: 2379–2389Viktoria Konya et al.2386
& 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Eosinophil transendothelial migration
Endothelial cells (3.5� 104/insert) were grown to confluence on
5mm filters in 6.5 mm Transwell inserts (Corning, NY, USA).
Confluence of the monolayers (usually 3 days after seeding) was
confirmed by measuring transendothelial electrical resistance
with an Endohm device (WPI, Sarasota, Fla) [54, 55]. The
monolayers grown in the inserts were activated by 0.5 U/mL
thrombin for 15 min or by 10 pM TNF-a for 4 h at 371C and placed
into 24-well plates containing eotaxin (3 nM) or vehicle in the
bottom wells. Eosinophils were preincubated with vehicle or the
EP4 agonist ONO AE1-329 for 10 min at 371C prior to being
added to the top wells (2� 105/well). The plates were incubated
at 371C in a humidified incubator for 4 h, and the nonmigrated
and transmigrated eosinophils were collected separately and
were enumerated by flow cytometry. Transendothelial migration
was calculated as the percentage of transmigrated eosinophils
with respect to total cells added to the filters [55, 62].
Statistical analyses
Data are shown as means1SEM for n observations. Statistical
analysis was performed with SigmaPlot 11.0 software using one-
way ANOVA or two-way ANOVA for repeated measurements
followed by Dunnett’s post-hoc test. p-Values of o0.05 were
considered as statistically significant.
Acknowledgements: V. Konya and S. Philipose were funded by
the Ph.D. Program Molecular Medicine of the Medical University
of Graz. This work was supported by the Start Funding Program
of the Medical University of Graz (ASO109000101 to V. Konya),
Jubilaumsfonds of the Austrian National Bank (12552 to
A. Heinemann and 13487 to R. Schuligoi), the Austrian Science
Fund FWF (Grants P19424 and P22521 to A. Heinemann, P21004
to G. Marsche, and P22771 to R. Schicho). The expert technical
help of Wolfgang Platzer, Martina Ofner and Gerald Parzmair is
highly appreciated.
Conflict of interest: The authors declare financial or commercial
conflict of interest.
References
1 Jose, P. J., Griffiths-Johnson, D. A., Collins, P. D., Walsh, D. T., Moqbel, R.,
Totty, N. F., Truong, O. et al., Eotaxin: a potent eosinophil chemoattrac-
tant cytokine detected in a guinea pig model of allergic airways
inflammation. J. Exp. Med. 1994. 179: 881–887.
2 Ebisawa, M., Yamada, T., Bickel, C., Klunk, D. and Schleimer, R. P.,
Eosinophil transendothelial migration induced by cytokines. III. Effect of
the chemokine RANTES. J. Immunol. 1994. 153: 2153–2160.
3 Garcia-Zepeda, E. A., Combadiere, C., Rothenberg, M. E., Sarafi, M. N.,
Lavigne, F., Hamid, Q., Murphy, P. M. et al., Human monocyte
chemoattractant protein (MCP)-4 is a novel CC chemokine with activities
on monocytes, eosinophils, and basophils induced in allergic and
nonallergic inflammation that signals through the CC chemokine
receptors (CCR)-2 and -3. J. Immunol. 1996. 157: 5613–5626.
4 Uden, A. M., Palmblad, J., Lindgren, J. A. and Malmsten, C., Effects of
novel lipoxygenase products on migration of eosinophils and neutrophils
in vitro. Int. Arch. Allergy Appl. Immunol. 1983. 72: 91–93.
5 Monneret, G., Gravel, S., Diamond, M., Rokach, J. and Powell, W. S.,
Prostaglandin D2 is a potent chemoattractant for human eosinophils that
acts via a novel DP receptor. Blood 2001. 98: 1942–1948.
6 Jacobsen, E. A., Taranova, A. G., Lee, N. A. and Lee, J. J., Eosinophils:
singularly destructive effector cells or purveyors of immunoregulation?
J. Allergy Clin. Immunol. 2007. 119: 1313–1320.
7 Aceves, S. S. and Broide, D. H., Airway fibrosis and angiogenesis due to
eosinophil trafficking in chronic asthma. Curr. Mol. Med. 2008. 8: 350–358.
8 Navarro, S., Aleu, J., Jimenez, M., Boix, E., Cuchillo, C. M. and Nogues,
M. V., The cytotoxicity of eosinophil cationic protein/ribonuclease 3 on
eukaryotic cell lines takes place through its aggregation on the cell
membrane. Cell. Mol. Life Sci. 2008. 65: 324–337.
9 Foster, P. S., Rosenberg, H. F., Asquith, K. L. and Kumar, R. K., Targeting
eosinophils in asthma. Curr. Mol. Med. 2008. 8: 585–590.
10 Schuligoi, R., Grill, M., Heinemann, A., Peskar, B. A. and Amann, R.,
Sequential induction of prostaglandin E and D synthases in inflamma-
tion. Biochem. Biophys. Res. Commun. 2005. 335: 684–689.
11 Park, G. Y. and Christman, J. W., Involvement of cyclooxygenase-2 and
prostaglandins in the molecular pathogenesis of inflammatory lung
diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006. 290: L797–L805.
12 Schuligoi, R., Sturm, E., Luschnig, P., Konya, V., Philipose, S., Sedej, M.,
Waldhoer, M. et al., CRTH2 and D-type prostanoid receptor antagonists as
novel therapeutic agents for inflammatory diseases. Pharmacology 2010.
85: 372–382.
13 Vancheri, C., Mastruzzo, C., Sortino, M. A. and Crimi, N., The lung as a
privileged site for the beneficial actions of PGE2. Trends Immunol. 2004. 25:
40–46.
14 Aggarwal, S., Moodley, Y. P., Thompson, P. J. and Misso, N. L.,
Prostaglandin E2 and cysteinyl leukotriene concentrations in sputum:
association with asthma severity and eosinophilic inflammation. Clin.
Exp. Allergy 2010. 40: 85–93.
15 Martin, J. G., Suzuki, M., Maghni, K., Pantano, R., Ramos-Barbon, D.,
Ihaku, D., Nantel, F. et al., The immunomodulatory actions of prosta-
glandin E2 on allergic airway responses in the rat. J. Immunol. 2002. 169:
3963–3969.
16 Alfranca, A., Iniguez, M. A., Fresno, M. and Redondo, J. M., Prostanoid
signal transduction and gene expression in the endothelium: role in
cardiovascular diseases. Cardiovasc. Res. 2006. 70: 446–456.
17 Norel, X., Prostanoid receptors in the human vascular wall. Sci. World J.
2007. 7: 1359–1374.
18 Sturm, E. M., Schratl, P., Schuligoi, R., Konya, V., Sturm, G. J.,
Lippe, I. T., Peskar, B. A. et al., Prostaglandin E2 inhibits eosinophil
trafficking through E-prostanoid 2 receptors. J. Immunol. 2008. 181:
7273–7283.
19 Gaspar-Elsas, M. I., Queto, T., Vasconcelos, Z., Jones, C. P., Lannes-Vieira,
J. and Xavier-Elsas, P., Evidence for a regulatory role of alpha
4-integrins in the maturation of eosinophils generated from the bone
marrow in the presence of dexamethasone. Clin. Exp. Allergy 2009. 39:
1187–1198.
Eur. J. Immunol. 2011. 41: 2379–2389 Leukocyte signaling 2387
& 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

20 Nourshargh, S., Hordijk, P. L. and Sixt, M., Breaching multiple barriers:
leukocyte motility through venular walls and the interstitium. Nat. Rev.
Mol. Cell. Biol. 2010. 11: 366–378.
21 Aota, S. and Yamada, K. M., Fibronectin and cell adhesion: specificity of
integrin-ligand interaction. Adv. Enzymol. Relat. Areas Mol. Biol. 1995. 70:
1–21.
22 Mohri, H., Fibronectin and integrins interactions. J. Investig. Med. 1996. 44:
429–441.
23 Lishko, V. K., Yakubenko, V. P. and Ugarova, T. P., The interplay between
integrins alphaMbeta2 and alpha5beta1 during cell migration to fibro-
nectin. Exp. Cell. Res. 2003. 283: 116–126.
24 Kumar, N. and Khurana, S., Identification of a functional switch
for actin severing by cytoskeletal proteins. J. Biol. Chem. 2004. 279:
24915–24918.
25 Barthel, S. R., Johansson, M. W., McNamee, D. M. and Mosher, D. F., Roles
of integrin activation in eosinophil function and the eosinophilic
inflammation of asthma. J. Leukoc. Biol. 2008. 83: 1–12.
26 Saltel, F., Mortier, E., Hytonen, V. P., Jacquier, M. C., Zimmermann, P.,
Vogel, V., Liu, W. et al., New PI(4,5)P2- and membrane proximal integrin-
binding motifs in the talin head control beta3-integrin clustering. J. Cell.
Biol. 2009. 187: 715–731.
27 Lukacs, N. W., Strieter, R. M., Chensue, S. W., Widmer, M. and Kunkel,
S. L., TNF-alpha mediates recruitment of neutrophils and eosinophils
during airway inflammation. J. Immunol. 1995. 154: 5411–5417.
28 Proceedings of the ATS workshop on refractory asthma: current
understanding, recommendations, and unanswered questions.
American Thoracic Society. Am. J. Respir. Crit. Care. Med. 2000. 162:
2341–2351.
29 Moore, W. C. and Peters, S. P., Severe asthma: an overview. J. Allergy Clin.
Immunol. 2006. 117: 487–494; quiz 495.
30 Chanez, P., Wenzel, S. E., Anderson, G. P., Anto, J. M., Bel, E. H., Boulet,
L. P., Brightling, C. E. et al., Severe asthma in adults: what are the
important questions? J. Allergy Clin. Immunol. 2007. 119: 1337–1348.
31 Howarth, P. H., Babu, K. S., Arshad, H. S., Lau, L., Buckley, M., McConnell,
W., Beckett, P. et al., Tumour necrosis factor (TNFalpha) as a novel
therapeutic target in symptomatic corticosteroid dependent asthma.
Thorax 2005. 60: 1012–1018.
32 Berry, M. A., Hargadon, B., Shelley, M., Parker, D., Shaw, D. E., Green,
R. H., Bradding, P. et al., Evidence of a role of tumor necrosis factor alpha
in refractory asthma. N. Engl. J. Med. 2006. 354: 697–708.
33 Erin, E. M., Leaker, B. R., Nicholson, G. C., Tan, A. J., Green, L. M.,
Neighbour, H., Zacharasiewicz, A. S. et al., The effects of a monoclonal
antibody directed against tumor necrosis factor-alpha in asthma. Am. J.
Respir. Crit. Care. Med. 2006. 174: 753–762.
34 Morjaria, J. B., Chauhan, A. J., Babu, K. S., Polosa, R., Davies, D. E. and
Holgate, S. T., The role of a soluble TNFalpha receptor fusion protein
(etanercept) in corticosteroid refractory asthma: a double blind, rando-
mised, placebo controlled trial. Thorax 2008. 63: 584–591.
35 Rennard, S. I., Fogarty, C., Kelsen, S., Long, W., Ramsdell, J., Allison, J.,
Mahler, D. et al., The safety and efficacy of infliximab in moderate to
severe chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care.
Med. 2007. 175: 926–934.
36 Terada, M., Kelly, E. A. and Jarjour, N. N., Increased thrombin activity
after allergen challenge: a potential link to airway remodeling? Am. J.
Respir. Crit. Care. Med. 2004. 169: 373–377.
37 Kanazawa, H. and Yoshikawa, T., Up-regulation of thrombin activity
induced by vascular endothelial growth factor in asthmatic airways. Chest
2007. 132: 1169–1174.
38 Schouten, M., Van de Pol, M. A., Levi, M., Van der Poll, T. and Van der Zee,
J. S., Early activation of coagulation after allergen challenge in patients
with allergic asthma. J. Thromb. Haemost. 2009. 7: 1592–1594.
39 Feistritzer, C., Mosheimer, B. A., Kaneider, N. C., Riewald, M., Patsch, J. R.
and Wiedermann, C. J., Thrombin affects eosinophil migration via
protease-activated receptor-1. Int. Arch. Allergy Immunol. 2004. 135: 12–16.
40 Luschnig-Schratl, P., Sturm, E. M., Konya, V., Philipose, S., Marsche, G.,
Frohlich, E., Samberger, C. et al., EP4 receptor stimulation down-regulates
human eosinophil function. Cell. Mol. Life. Sci. 2011. in press: doi:10.1007/
s00018-011-0642-5.
41 Philipose, S., Konya, V., Sreckovic, I., Marsche, G., Lippe, I. T., Peskar,
B. A., Heinemann, A. et al., The prostaglandin E2 receptor EP4 is
expressed by human platelets and potently inhibits platelet aggregation
and thrombus formation. Arterioscler. Thromb. Vasc. Biol. 2010. 30:
2416–2423.
42 Ulcar, R., Peskar, B. A., Schuligoi, R., Heinemann, A., Kessler, H. H.,
Santner, B. I. and Amann, R., Cyclooxygenase inhibition in human
monocytes increases endotoxin-induced TNF alpha without affecting
cyclooxygenase-2 expression. Eur. J. Pharmacol. 2004. 501: 9–17.
43 Takahashi, H. K., Iwagaki, H., Mori, S., Yoshino, T., Tanaka, N. and
Nishibori, M., Prostaglandins E1 and E2 inhibit lipopolysaccharide-
induced interleukin-18 production in monocytes. Eur. J. Pharmacol. 2005.
517: 252–256.
44 Minami, M., Shimizu, K., Okamoto, Y., Folco, E., Ilasaca, M. L., Feinberg,
M. W., Aikawa, M. et al., Prostaglandin E receptor type 4-associated
protein interacts directly with NF-kappaB1 and attenuates macrophage
activation. J. Biol. Chem. 2008. 283: 9692–9703.
45 Hattori, R., Shimizu, S., Majima, Y. and Shimizu, T., EP4 agonist inhibits
lipopolysaccharide-induced mucus secretion in airway epithelial cells.
Ann. Otol. Rhinol. Laryngol. 2008. 117: 51–58.
46 Gauvreau, G. M., Watson, R. M. and O’Byrne, P. M., Protective effects of
inhaled PGE2 on allergen-induced airway responses and airway inflam-
mation. Am. J. Respir. Crit. Care. Med. 1999. 159: 31–36.
47 Billot, X., Chateauneuf, A., Chauret, N., Denis, D., Greig, G., Mathieu,
M. C., Metters, K. M. et al., Discovery of a potent and selective agonist of
the prostaglandin EP4 receptor. Bioorg. Med. Chem. Lett. 2003. 13:
1129–1132.
48 Kabashima, K., Saji, T., Murata, T., Nagamachi, M., Matsuoka, T., Segi, E.,
Tsuboi, K. et al., The prostaglandin receptor EP4 suppresses colitis,
mucosal damage and CD4 cell activation in the gut. J. Clin. Invest. 2002.
109: 883–893.
49 Sturm, G. J., Schuligoi, R., Sturm, E. M., Royer, J. F., Lang-Loidolt, D.,
Stammberger, H., Amann, R. et al., 5-Oxo-6,8,11,14-eicosatetraenoic acid
is a potent chemoattractant for human basophils. J. Allergy Clin. Immunol.
2005. 116: 1014–1019.
50 Schuligoi, R., Schmidt, R., Geisslinger, G., Kollroser, M., Peskar, B. A. and
Heinemann, A., PGD2 metabolism in plasma: kinetics and relationship
with bioactivity on DP1 and CRTH2 receptors. Biochem. Pharmacol. 2007.
74: 107–117.
51 Heinemann, A., Ofner, M., Amann, R. and Peskar, B. A., A novel assay to
measure the calcium flux in human basophils: effects of chemokines and
nerve growth factor. Pharmacology 2003. 67: 49–54.
52 Hartnell, A., Heinemann, A., Conroy, D. M., Wait, R., Sturm, G. J.,
Caversaccio, M., Jose, P. J. et al., Identification of selective basophil
chemoattractants in human nasal polyps as insulin-like growth factor-1
and insulin-like growth factor-2. J. Immunol. 2004. 173: 6448–6457.
53 Schratl, P., Sturm, E. M., Royer, J. F., Sturm, G. J., Lippe, I. T., Peskar, B. A.
and Heinemann, A., Hierarchy of eosinophil chemoattractants: role of
p38 mitogen-activated protein kinase. Eur. J. Immunol. 2006. 36: 2401–2409.
Eur. J. Immunol. 2011. 41: 2379–2389Viktoria Konya et al.2388
& 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

54 Sedgwick, J. B., Menon, I., Gern, J. E. and Busse, W. W., Effects of
inflammatory cytokines on the permeability of human lung microvas-
cular endothelial cell monolayers and differential eosinophil transmigra-
tion. J. Allergy Clin. Immunol. 2002. 110: 752–756.
55 Konya, V., Sturm, E. M., Schratl, P., Beubler, E., Marsche, G., Schuligoi, R.,
Lippe, I. T. et al., Endothelium-derived prostaglandin I(2) controls the
migration of eosinophils. J. Allergy Clin. Immunol. 2010. 125: 1105–1113.
56 Stubbs, V. E., Schratl, P., Hartnell, A., Williams, T. J., Peskar, B. A.,
Heinemann, A. and Sabroe, I., Indomethacin causes prostaglandin D(2)-
like and eotaxin-like selective responses in eosinophils and basophils.
J. Biol. Chem. 2002. 277: 26012–26020.
57 Johansson, M. W., Lye, M. H., Barthel, S. R., Duffy, A. K., Annis, D. S. and
Mosher, D. F., Eosinophils adhere to vascular cell adhesion molecule-1 via
podosomes. Am. J. Respir. Cell. Mol. Biol. 2004. 31: 413–422.
58 Kamata, T., Tieu, K. K., Tarui, T., Puzon-McLaughlin, W., Hogg, N. and
Takada, Y., The role of the CPNKEKEC sequence in the beta(2) subunit I
domain in regulation of integrin alpha(L)beta(2) (LFA-1). J. Immunol. 2002.
168: 2296–2301.
59 Heinemann, A., Sturm, G. J., Ofner, M., Sturm, E. M., Weller, C., Peskar,
B. A. and Hartnell, A., Stem cell factor stimulates the chemotaxis,
integrin upregulation, and survival of human basophils. J. Allergy Clin.
Immunol. 2005. 116: 820–826.
60 Grill, M., Heinemann, A., Hoefler, G., Peskar, B. A. and Schuligoi, R., Effect
of endotoxin treatment on the expression and localization of spinal
cyclooxygenase, prostaglandin synthases, and PGD2 receptors. J. Neuro-
chem. 2008. 104: 1345–1357.
61 Choi, E. Y., Chavakis, E., Czabanka, M. A., Langer, H. F., Fraemohs, L.,
Economopoulou, M., Kundu, R. K. et al., Del-1, an endogenous leukocyte-
endothelial adhesion inhibitor, limits inflammatory cell recruitment.
Science 2008. 322: 1101–1104.
62 Yamamoto, H., Nagata, M. and Sakamoto, Y., CC chemokines and
transmigration of eosinophils in the presence of vascular cell adhesion
molecule 1. Ann. Allergy Asthma Immunol. 2005. 94: 292–300.
Abbreviations: EP: E-type prostanoid receptor � F-actin: filamentous
actin � PG: prostaglandin
Full correspondence: Dr. Akos Heinemann, Institute of Experimental
and Clinical Pharmacology, Medical University of Graz,
Universitaetsplatz 4. A-8010 Graz, Austria
Fax: 143-316-380-9645
e-mail: [email protected]
Received: 28/1/2011
Revised: 18/4/2011
Accepted: 18/5/2011
Accepted article online: 17/6/2011
& 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
Eur. J. Immunol. 2011. 41: 2379–2389 Leukocyte signaling 2389











![OBE022, an Oral and Selective Prostaglandin F Receptor Antagonist · specific prostaglandin synthases], and metabolism via pros-taglandin dehydrogenase enzymes. Prostaglandin E 2](https://static.fdocuments.us/doc/165x107/612431e6b1d2d8488c3d852e/obe022-an-oral-and-selective-prostaglandin-f-receptor-antagonist-specific-prostaglandin.jpg)




![RoleofPGE inAsthmaandNonasthmatic EosinophilicBronchitis2) by COXs, and metabolism of prostaglandin H 2 to prostaglandin E 2 via prostaglandin E synthase [12]. There are three enzymes](https://static.fdocuments.us/doc/165x107/60d522031e41432a8f254505/roleofpge-inasthmaandnonasthmatic-eosinophilicbronchitis-2-by-coxs-and-metabolism.jpg)

