
Increased4R … Human-Tau Mouse Model ... Hana N. Dawson,4 C. Frank Bennett, 2Frank Rigo, ... tau...
18
Report Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model Highlights d Antisense oligonucleotide-mediated MAPT splicing is successful in vivo d Increasing 4R-tau induces tau phosphorylation and exacerbates seizures d Reducing 4R-tau is effective in human tau-expressing mouse models Authors Kathleen M. Schoch, Sarah L. DeVos, Rebecca L. Miller, ..., C. Frank Bennett, Frank Rigo, Timothy M. Miller Correspondence [email protected] In Brief Schoch et al. employ antisense oligonucleotide technology to manipulate tau isoforms, demonstrating that increased four-repeat tau drives toxic changes in a human tau mouse model. Reducing four-repeat tau was also achieved, suggesting application in human tauopathies. Schoch et al., 2016, Neuron 90, 1–7 June 1, 2016 ª 2016 Elsevier Inc. http://dx.doi.org/10.1016/j.neuron.2016.04.042
-
Upload
trinhthuan -
Category
Documents
-
view
216 -
download
0
Transcript of Increased4R … Human-Tau Mouse Model ... Hana N. Dawson,4 C. Frank Bennett, 2Frank Rigo, ... tau...

Report
Increased 4R-Tau Induces
Pathological Changes in aHuman-Tau Mouse ModelHighlights
d Antisense oligonucleotide-mediated MAPT splicing is
successful in vivo
d Increasing 4R-tau induces tau phosphorylation and
exacerbates seizures
d Reducing 4R-tau is effective in human tau-expressing mouse
models
Schoch et al., 2016, Neuron 90, 1–7June 1, 2016 ª 2016 Elsevier Inc.http://dx.doi.org/10.1016/j.neuron.2016.04.042
Authors
Kathleen M. Schoch, Sarah L. DeVos,
Rebecca L. Miller, ..., C. Frank Bennett,
Frank Rigo, Timothy M. Miller
In Brief
Schoch et al. employ antisense
oligonucleotide technology to manipulate
tau isoforms, demonstrating that
increased four-repeat tau drives toxic
changes in a human tau mouse model.
Reducing four-repeat tau was also
achieved, suggesting application in
human tauopathies.

Please cite this article in press as: Schoch et al., Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model, Neuron (2016),http://dx.doi.org/10.1016/j.neuron.2016.04.042
Neuron
Report
Increased 4R-Tau Induces PathologicalChanges in a Human-Tau Mouse ModelKathleen M. Schoch,1,5 Sarah L. DeVos,1,5 Rebecca L. Miller,1 Seung J. Chun,2 Michaela Norrbom,2 David F. Wozniak,3
Hana N. Dawson,4 C. Frank Bennett,2 Frank Rigo,2 and Timothy M. Miller1,*1Department of Neurology, Hope Center for Neurological Disorders, Washington University in St. Louis, St. Louis, MO 631102Ionis Pharmaceuticals, Carlsbad, CA 920103Taylor Family Institute for Innovative Psychiatric Research, Department of Psychiatry, Washington University in St. Louis, St. Louis,
MO 631104Department of Neurology, Duke University Medical Center, Durham, NC 277105Co-first author*Correspondence: [email protected]
http://dx.doi.org/10.1016/j.neuron.2016.04.042
SUMMARY
Pathological evidence for selective four-repeat (4R)tau deposition in certain dementias and exon 10-positioned MAPT mutations together suggest a4R-specific role in causing disease. However, directassessments of 4R toxicity have not yet been accom-plished in vivo. Increasing 4R-tau expression withoutchange to total tau in human tau-expressing miceinduced more severe seizures and nesting behaviorabnormality, increased tau phosphorylation, andproduced a shift toward oligomeric tau. Exon 10skipping could also be accomplished in vivo,providing support for a 4R-tau targeted approachto target 4R-tau toxicity and, in cases of primaryMAPT mutation, eliminate the disease-causing mu-tation.
INTRODUCTION
Cognitive disorders including Alzheimer’s disease (AD) and pri-
mary tauopathies are typified by the abnormal intracellular depo-
sition of the microtubule-associated protein tau. Tau can be
expressed with three-repeat (3R) or four-repeat (4R) domains,
which arise from alternative splicing of MAPT exon 10 mRNA.
Normal adult humans express approximately equal 3R-tau and
4R-tau; however, select sporadic tauopathies exhibit an imbal-
ance of 4R-tau isoform deposition within neurofibrillary tangles
(NFTs) (Arai et al., 2001; de Silva et al., 2003). In patients with
frontotemporal dementia with parkinsonism linked to chromo-
some 17 (FTDP-17), mutations identified within MAPT exon/
intron 10 result in an abnormal increase in 4R-tau expression
without disruption of its microtubule binding characteristics
(D’Souza et al., 1999; Hong et al., 1998).
While some in vitro studies demonstrate that 4R-tau or an
altered isoform ratio induces tau aggregation (Adams et al.,
2010; von Bergen et al., 2001), testing 3R-tau versus 4R-tau
toxicity in animal models has been challenging because mice
do not recapitulate human 4R:3R ratios, and different transgenes
are not directly comparable. We developed tau isoform switch-
ing antisense oligonucleotides (ASOs) (DeVos and Miller,
2013a) for use in hTau mice, which lack endogenous mouse
tau and express mostly 3R human tau (Andorfer et al., 2003)
without early onset behavioral deficits or tau pathology (Polydoro
et al., 2009). By comparingmice of the same genetic background
but greater ASO-driven 4R-tau, we were able to test whether
increased 4R-tau led to increased pathology and changes in
behavior. In addition, we demonstrate the feasibility of a 4R-
reducing therapeutic approach using ASOs.
RESULTS
Characterization of 3R to 4R ASO Treatment inhTau MiceIn order to test whether 4R-tau is a prime mediator of tauopathy,
we developed ASOs that target humanMAPTmRNA to increase
the 4R:3R tau ratio. hTau mice were treated with continuous
intraventricular infusion of saline, scrambled ASO, or 3R to 4R
MAPT mRNA splicing ASO for 28 days via osmotic pump. One
month following cessation of infusion, mRNA analysis revealed
a nearly 2-fold increase in 4R human MAPT mRNA with splicing
ASO treatment (n = 19) compared to saline-treated or scrambled
ASO-treated mice (n = 9/group; p < 0.005 versus saline and
scrambled controls, Figure 1A). Importantly, there was no alter-
ation of total human tau (Figures 1B and 1C), indicative of an iso-
form switch rather than a 4R-tau increase alone. ASO-mediated
MAPT alteration also occurred in the absence of off-target bind-
ing (see Figure S1 available online). 4R, 3R, and total tau protein
expression closely corresponded to MAPT mRNA levels,
revealing an increase in 4R-tau and decrease in 3R-tau following
3R to 4R splicing treatment (Figure 1D) with similar total tau
levels (Figures 1E and 1F).
3R to4RMAPTmRNASplicing in hTauMiceModifies TauPhosphorylation, Aggregation, and Solubility3R to 4R splicing ASO-treated hTau mice were evaluated for tau
phosphorylation using the AT8 antibody, which detects S202/
T205 phosphorylation events and is frequently associated with
tau pathology (Braak and Braak, 1995). Following scrambled
ASO treatment (n = 5), faint neuronal AT8 immunoreactivity
Neuron 90, 1–7, June 1, 2016 ª 2016 Elsevier Inc. 1

Figure 1. Total Tau and Tau Isoform mRNA and Protein Expression in hTau Mice following 3R to 4R MAPT mRNA Splicing ASO Treatment
(A and B) (A) Relative 4R human MAPT mRNA was significantly increased following 3R to 4R splicing ASO administration, while (B) total MAPT mRNA levels
remained similar to controls (n = 19 splicing ASO, n = 9/saline and scrambled controls). MAPT mRNA data were normalized to GAPDH and the mean + SEM
calculated relative to saline controls; **p < 0.005 compared to saline and scrambled control values.
(C) 3R and 4RMAPTmRNA products visualized following RT-PCR confirm a shift in isoform composition following 3R to 4R splicing ASO treatment compared to
mice treated with saline or scrambled ASO.
(D and E) (D) Immunoblots for 3R-tau and 4R-tau show greater 3R-tau protein compared to 4R-tau in brain homogenates obtained from saline- and scrambled-
treatedmice, ipsilateral to the catheter placement. With 3R to 4R splicing ASO administration, the ratio of tau isoforms is altered, increasing the expression of 4R-
tau and decreasing expression of 3R, (E) without changing total tau protein (HT7).
(F) Total human tau protein expression quantified by human tau-specific ELISA closely paralleled totalMAPTmRNA results, showing no change in total human tau
protein after treatment. Data are expressed as mean + SEM relative to saline controls.
Please cite this article in press as: Schoch et al., Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model, Neuron (2016),http://dx.doi.org/10.1016/j.neuron.2016.04.042
was identified within the entorhinal cortex (Figure 2A) and amyg-
dala (Figure 2D) and throughout the cortex (Figure 2G). However,
with 3R to 4R splicing ASO treatment (n = 7), the area of positive
immunoreactivity increased (Figures 2B, 2E, and 2H), showing
significantly higher percentages of staining within select areas
(p < 0.05; Figures 2C, 2F, and 2I). Exacerbated AT8 reactivity
within known regions of tau spread throughout the brain may
be predictive of increased pathological tau burden with greater
4R-tau expression.
Tau aggregation into larger, more insoluble species is also a
characteristic of fibrillar structures and increasing toxicity (Spill-
antini and Goedert, 2013). Therefore, cortical brain tissue from
12-month-old hTau mice treated with scrambled ASO (n = 3) or
3R to 4R splicing ASO (n = 4) was analyzed for high-molecular-
weight (HMW) tau forms. Greater tau reactivity was evident
within HMW regions with 3R to 4R splicing ASO treatment
compared to controls (Figure 2J). Not only was this result indic-
ative of aggregated tau proteins with increased 4R-tau, but also
regions of HMW tau were determined to be more heavily phos-
phorylated (p < 0.05; Figure 2K; Figure S2). To further confirm
4R-tau-driven pathology, soluble and insoluble tau species
were identified by fractionation analysis of cortical brain tissue
from treated hTau mice. While the majority of tau within hTau
mice was found within the RAB-soluble fraction, increased 4R-
2 Neuron 90, 1–7, June 1, 2016
tau was associated with a moderate increase RIPA-soluble
tau, indicating a decline in solubility (Figure 2L). Insoluble tau
detected within the formic acid fraction did not change. Hu-
man-tau-specific ELISA analysis mirrored the increase in RIPA-
soluble tau (Figure 2M). Both SDD-AGE and fractionation results
are consistent with phosphorylated tau histology, collectively
suggesting human tau is becoming more phosphorylated,
aggregated, and less soluble with increased 4R-tau expression.
Seizure Severity Is Increased with 3R to 4R SplicingChange in hTau MiceEmerging evidence strongly suggests a role for tau inmodulating
synaptic activity. Tau reduction, both genetically (Ittner et al.,
2010; Roberson et al., 2007) and by ASO (DeVos et al., 2013),
reduced seizure severity following pentylenetetrazole (PTZ) in-
jection. At 4 months, hTau mice were treated with saline,
scrambled ASO, or 3R to 4R splicing ASO infusion (n = 18–19/
treatment) and, 1 month later, injected with PTZ to evoke a
generalized seizure. Saline and scrambled ASO-treated mice
were not statistically significant (mean seizure stage 3.64 ±
2.29 versus 4.00 ± 2.70 SD, respectively) and, therefore, com-
bined for analyses. Compared to control treatments, 3R to 4R
splicing ASO-treated hTau mice experienced significantly more
severe seizures (p < 0.05; Figure 3A) and reached more severe
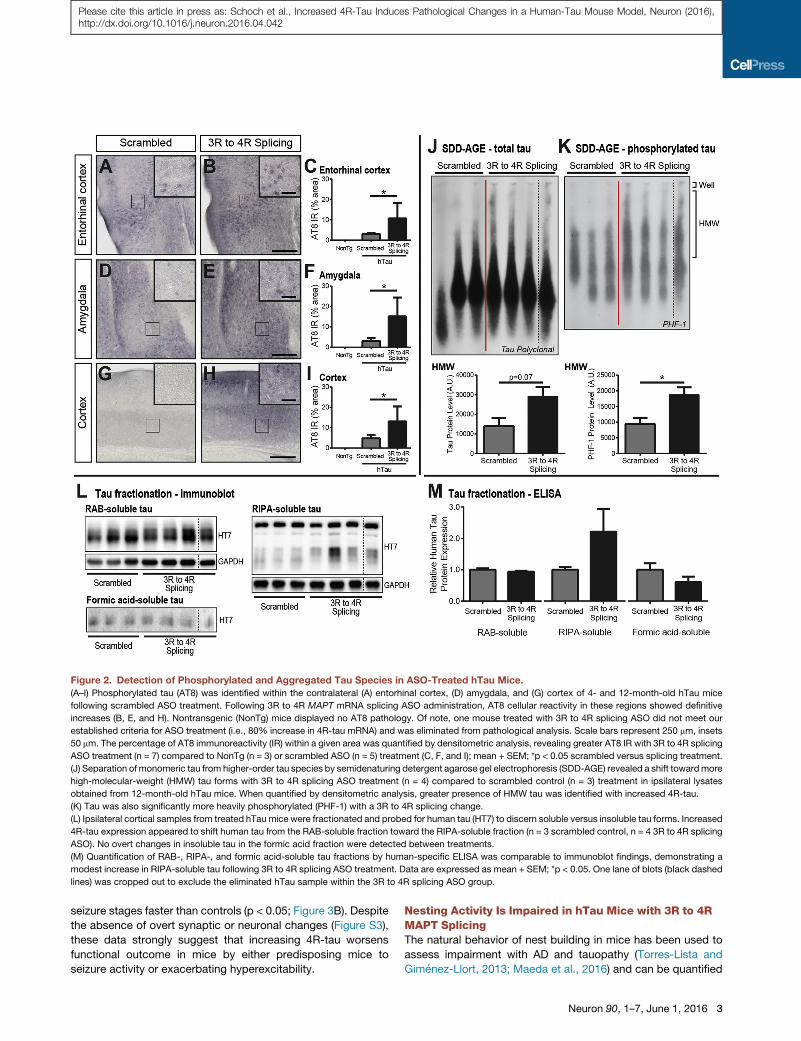
Figure 2. Detection of Phosphorylated and Aggregated Tau Species in ASO-Treated hTau Mice.
(A–I) Phosphorylated tau (AT8) was identified within the contralateral (A) entorhinal cortex, (D) amygdala, and (G) cortex of 4- and 12-month-old hTau mice
following scrambled ASO treatment. Following 3R to 4R MAPT mRNA splicing ASO administration, AT8 cellular reactivity in these regions showed definitive
increases (B, E, and H). Nontransgenic (NonTg) mice displayed no AT8 pathology. Of note, one mouse treated with 3R to 4R splicing ASO did not meet our
established criteria for ASO treatment (i.e., 80% increase in 4R-tau mRNA) and was eliminated from pathological analysis. Scale bars represent 250 mm, insets
50 mm. The percentage of AT8 immunoreactivity (IR) within a given area was quantified by densitometric analysis, revealing greater AT8 IR with 3R to 4R splicing
ASO treatment (n = 7) compared to NonTg (n = 3) or scrambled ASO (n = 5) treatment (C, F, and I); mean + SEM; *p < 0.05 scrambled versus splicing treatment.
(J) Separation of monomeric tau from higher-order tau species by semidenaturing detergent agarose gel electrophoresis (SDD-AGE) revealed a shift towardmore
high-molecular-weight (HMW) tau forms with 3R to 4R splicing ASO treatment (n = 4) compared to scrambled control (n = 3) treatment in ipsilateral lysates
obtained from 12-month-old hTau mice. When quantified by densitometric analysis, greater presence of HMW tau was identified with increased 4R-tau.
(K) Tau was also significantly more heavily phosphorylated (PHF-1) with a 3R to 4R splicing change.
(L) Ipsilateral cortical samples from treated hTau mice were fractionated and probed for human tau (HT7) to discern soluble versus insoluble tau forms. Increased
4R-tau expression appeared to shift human tau from the RAB-soluble fraction toward the RIPA-soluble fraction (n = 3 scrambled control, n = 4 3R to 4R splicing
ASO). No overt changes in insoluble tau in the formic acid fraction were detected between treatments.
(M) Quantification of RAB-, RIPA-, and formic acid-soluble tau fractions by human-specific ELISA was comparable to immunoblot findings, demonstrating a
modest increase in RIPA-soluble tau following 3R to 4R splicing ASO treatment. Data are expressed as mean + SEM; *p < 0.05. One lane of blots (black dashed
lines) was cropped out to exclude the eliminated hTau sample within the 3R to 4R splicing ASO group.
Please cite this article in press as: Schoch et al., Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model, Neuron (2016),http://dx.doi.org/10.1016/j.neuron.2016.04.042
seizure stages faster than controls (p < 0.05; Figure 3B). Despite
the absence of overt synaptic or neuronal changes (Figure S3),
these data strongly suggest that increasing 4R-tau worsens
functional outcome in mice by either predisposing mice to
seizure activity or exacerbating hyperexcitability.
Nesting Activity Is Impaired in hTau Mice with 3R to 4RMAPT SplicingThe natural behavior of nest building in mice has been used to
assess impairment with AD and tauopathy (Torres-Lista and
Gimenez-Llort, 2013; Maeda et al., 2016) and can be quantified
Neuron 90, 1–7, June 1, 2016 3

Figure 3. Functional Deficits Measured by
Seizure and Nesting Activity in 3R to
4R MAPT mRNA Splicing ASO-Treated
hTau Mice
(A and B) (A) Following intraperitoneal PTZ injec-
tion, hTau mice treated with 3R to 4RMAPTmRNA
splicing ASO exhibited a significant increase in
seizure severity and (B) a reduction in seizure la-
tency compared to mice treated with saline or
scrambled ASO (combined as controls) (n = 18–19/
treatment). Final seizure score data are plotted as
mean stage + SEM and seizure latencies are mean
time ± SEM across each seizure stage; *p < 0.05
versus controls.
(C–E) (C) Representative images of nests con-
structed by hTau mice. Nesting activity was as-
sessed using a modified scoring criteria to rate the
quality of nest construction and amount of torn
nestlet material. hTau mice treated with 3R to 4R
splicing ASO (n = 5) exhibited (D) significantly lower
nestlet scores and (E) significantly greater untorn
nestlet weights indicative of poor nesting activity
compared to control (saline and scrambled ASO)
treated hTau mice (n = 5). Nestlet scores and
weights are shown as mean + SEM; *p < 0.05
versus controls.
Please cite this article in press as: Schoch et al., Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model, Neuron (2016),http://dx.doi.org/10.1016/j.neuron.2016.04.042
based on published criteria (Figure 3A). Compared to male hTau
mice treated with either saline or scrambled ASO (n = 4), hTau
mice given 3R to 4R splicing ASO (n = 5) exhibited significantly
lower nestlet scores (p < 0.05; Figure 3B) and greater weights
of untorn nestlet (p < 0.05; Figure 3C), implicating increased
4R-tau in impaired nesting ability.
MAPT mRNA Splicing Alteration from 4R to 3R IsEffective in Human Tau-Expressing MiceOur data and others’ suggest 4R-tau to be more toxic, yet, to
date, no tau splicing strategy has been implemented in vivo
despite the overwhelming number of tau mutations within exon
10. In order to validate the use of an exon-skipping strategy in hu-
mans, we developed ASOs designed to exclude exon 10, thus
decreasing 4R-tau without change to total tau levels, to test in
hTau mice. Compared to saline treatment (n = 3), hTau mice
that received 4R to 3R splicing ASO (n = 3) exhibited a robust
decrease in exon 10 inclusionwithin the cortex (p < 0.005), hippo-
campus (p<0.05), andspinal cord (p<0.005; Figure4A). Total tau
levels remainedunchanged (Figure 4B). To further address the ef-
ficacy and feasibility of a 4R to 3R splicing strategy, we applied
this exon 10-skipping strategy to mice that recapitulate FTDP-
17 tauopathy with mutant (TauN279K) 4R-tau overexpression
(Dawson et al., 2007). Intraventricular administration of 4R to
3R splicing ASO resulted in a substantial decrease in 4R human
MAPT mRNA concomitant with an increase in 3R MAPT mRNA
4 Neuron 90, 1–7, June 1, 2016
compared tocontrols (p<0.05;Figure4C).
Again, total tau levels were not altered
following ASO treatment (Figure 4D) with
clear selectively toward human MAPT
mRNA (Figure S4). Thus, antisense-medi-
ated 4R to 3RMAPT splicing is effective in
full-length human tau-expressing mice, lending support for the
feasibility of a 4R isoform target in human dementias.
DISCUSSION
In hTaumice treated with ASO to increase 4R-tau, we identified a
significant increase in phosphorylated tau and more-aggre-
gated, less-soluble tau species. Seizures both were more severe
and occurred at earlier time points with increased 4R-tau in hTau
mice. While it is possible an imbalanced ratio of 4R-tau to 3R-tau
or other off-target toxicity may have led to increased pathology,
it remains likely that 4R-tau is more pathogenic. This is consis-
tent with previous reports citing mitochondrial axonal transport
defects (Stoothoff et al., 2009), microtubule instability (Bunker
et al., 2004), and increased polymerization (Combs et al., 2011)
of 4R-tau compared to 3R-tau. These altered characteristics
may be key to the selective deposition of 4R-tau in progressive
supranuclear palsy (PSP), corticobasal degeneration, and
FTDP-17; however, other factors may influence the 3R-tau
deposition seen in Pick’s disease and the mixed deposition of
both 3R-tau and 4R-tau in AD.
We also show in vivo application of the converse ASO
approach by lowering 4R-tau in two mouse models of human
tau expression. To date, nearly half of the 53 known MAPT mu-
tations are located within exon/intron 10 (Ghetti et al., 2015) and
account for 70% of the total number of published families

Figure 4. Validation of 4R to 3R Human MAPT mRNA Splicing ASOs In VivoIn both hTau and Tau N279K mice, a reverse MAPT splicing strategy was investigated that would induce exon 10 skipping and bias toward 3R-tau.
(A) In hTau mice, which exhibit tau isoform expression similar to humans, 4R to 3R MAPT splicing ASO induced a significant and widespread reduction of 4R
mRNA levels in the cortex, hippocampus, and spinal cord compared to saline (n = 3/treatment).
(B) No change in total tau mRNAwas detected with either treatment. To further assess the potency of exon 10 exclusion, 4R to 3R splicing ASOwas administered
to Tau N279K mice and the ipsilateral temporoparietal cortex analyzed for MAPT isoform mRNA.
(C and D) (C) Radiolabeled 4R and 3R PCR products demonstrate a clear shift toward 3R MAPT expression in mice treated with 4R to 3R splicing ASO (n = 3
saline, n = 5 ASO). When quantified, mRNA levels revealed that treatment with 4R to 3R splicing ASO afforded a significant decrease in 4RMAPTmRNA level and
increase in 3R mRNA, in contrast to saline treatment, while (D) total human MAPT mRNA levels remained unchanged. 4R mRNA levels were calculated as a
percent of exon 10 inclusion normalized to total tau, and total taumRNA levels to GAPDH. Data are expressed as the percent of exon 10 inclusion relative to saline
treatment (mean + SEM); analysis by unpaired t test (saline versus ASO) within each region, *p < 0.05, **p < 0.005.
Please cite this article in press as: Schoch et al., Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model, Neuron (2016),http://dx.doi.org/10.1016/j.neuron.2016.04.042
affected by MAPT mutations (Cruts et al., 2012). The 4R to 3R
splicing ASO strategy described here would not only change
4R-tau to 3R-tau but would also remove the exon 10-containing
amino acid missense mutation in the mature protein, without
changing the total levels of tau. For PSP and other 4R-tauopa-
thies without a primary tau mutation, exon 10 skipping may
also be an advantageous therapy, since 4R-tau selectively de-
posits and is predicted to more readily seed 4R-tau (Dinkel
et al., 2011; Sanders et al., 2014). Both exon 10-containing
alleles would be targeted by our current ASO approach, although
the concept of mutant allele-specific targeting has been suc-
cessful in other models (Hu et al., 2009; Miller et al., 2003).
Overall, these data provide compelling evidence for the pres-
ence of tau isoform-mediated neurodegeneration and introduce
exon 10-targeted ASOs that may be an effective therapy for
tauopathies. A similar ASO-mediated exon skipping strategy
has been successfully employed in Duchenne’s muscular dys-
trophy (DMD). Exon skipping of a nonsense mutation restored
dystrophin expression and improved motor function in mdx
dystrophic mouse (Lu et al., 2003), leading to a dystrophin-tar-
geting ASO (drisapersen) clinical trial in human DMD patients
(GlaxoSmithKline, 2014). The bench-to-bedside success of
exon skipping in DMD in addition to the excellent safety profile
of centrally delivered ASO for amyotrophic lateral sclerosis
(Miller et al., 2013) and spinal muscular atrophy (Ionis Pharma-
ceuticals, 2016) emphasizes the promise of other ASO-mediated
splicing strategies for the treatment of degenerative diseases.
Alternative tau-targeted approaches, including tau clearance
and total tau reduction, show promise for therapeutic applica-
tions but do not address the individual effect of tau isoforms in
mediating tauopathies. And, in contrast to a total tau reduction
approach, a MAPT splicing strategy would not affect total tau
levels, thereby avoiding potential subtle or unforeseen con-
founds associated with a loss of tau (Lei et al., 2012; Ma et al.,
2014; Dawson et al., 2010). In order to advance MAPT splicing
ASOs toward human clinical trials, additional information will
be necessary, including evaluation in preclinical tauopathy
models, greater understanding of the 3R and 4RmRNA and pro-
tein composition in human tauopathies, and insight on what
causes selective 4R deposition. Our experiments provide initial
support for an ASO therapeutic strategy in primary taumutations
and potentially other 4R tauopathies.
Neuron 90, 1–7, June 1, 2016 5

Please cite this article in press as: Schoch et al., Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model, Neuron (2016),http://dx.doi.org/10.1016/j.neuron.2016.04.042
EXPERIMENTAL PROCEDURES
Experimental Animals
Male and female hTau transgenic (Andorfer et al., 2003; RRID: IMSR_JAX:
005491) or tau N279K transgenic mice (Dawson et al., 2007) and their non-
transgenic littermates were aged to 3–4 months prior to experimental treat-
ment with the exception of biochemical and phosphorylated tau analyses in
hTau mice (4 or 12 months of age). Ages/genders were matched across treat-
ment groups. All husbandry and surgical procedures were approved by the
Washington University Animal Studies Committee in accordance with federal
standards. For more details on the animals used, refer to Supplemental
Information.
Antisense Oligonucleotides
Splicing ASOs were designed with a phosphorothioate backbone, uniformly
modified with 20-O-methoxyethyl nucleotides to enhance binding to the
MAPT target and prevent RNaseH-mediated degradation of MAPT mRNA
(DeVos and Miller, 2013a). A scrambled ASO control, designed with the
same modifications as the MAPT splicing ASO but without target specificity,
was included in experimental treatments to account for any potential toxicity
or off-target effects of the ASO backbone. All ASOs were synthesized by Ionis
Pharmaceuticals (Carlsbad, CA) and generously provided for use. ASO
sequence information can be found in Supplemental Information.
Intraventricular Delivery of ASOs
ASOs were continuously administered to the right lateral ventricle via osmotic
pump (ALZET, Cupertino, CA) as previously described (DeVos and Miller,
2013b). For a detailed surgical procedure and osmotic pump preparation,
see Supplemental Information.
Euthanasia and Tissue Dissection
Mice were euthanized by transcardial perfusion with cold heparinized phos-
phate buffered saline (PBS) and decapitated. The right brain, ipsilateral to
the catheter placement, was microdissected into temporoparietal and occip-
ital regions (�15–30 mg), snap frozen in liquid nitrogen, and stored at �80�Cuntil processing. The left brain was drop fixed into 4% paraformaldehyde for
24 hr followed by dehydration in 30% sucrose. Tissue was then frozen in
�35�C to �25�C methylbutanes and stored at �80�C until sectioning for
histology.
mRNA Isolation and Analysis
mRNA was isolated from temporoparietal right hemi-brain tissue. Total and
MAPT isoform mRNA from hTau mice was readily detectable by standard
quantitative real-time PCR methods. A modified RT-PCR protocol was used
to amplify and detect human 3R and 4R MAPT isoforms in Tau N279K mice.
Detailed methodology, including primer and probe sequences, is included
under Supplemental Information.
Tissue Homogenization and Tau Protein Analyses by ELISA and
Immunoblot
Right hemibrain tissue designated for protein analysis was homogenized and
prepared for analysis by ELISA and immunoblot as described in Supplemental
Information.
Detection of Tau by SDD-AGE
Semi-denaturing detergent agarose gel electrophoresis (SDD-AGE) was car-
ried out as previously described (Sanders et al., 2014, Halfmann and Lindquist,
2008) with minor modifications (see Supplemental Information).
Immunohistochemistry of Phosphorylated Tau
Left hemibrain tissue from treated hTau mice was used for immunohistochem-
ical detection of phosphorylated tau. Procedures, antibody information, and
analyses are detailed in Supplemental Information.
Seizure Induction and Monitoring
Seizure activity was induced by intraperitoneal injection of the GABA receptor
antagonist, pentylenetetrazole (PTZ, Sigma), at a dose of 55 mg/kg and
6 Neuron 90, 1–7, June 1, 2016
analyzed as previously published (DeVos et al., 2013) and further described
in Supplemental Information.
Nesting Activity
Following the nocturnal period, untorn nesting material was weighed, and
nests were photographed for scoring on a 0–7 ordinal scale modified from
published criteria (Deacon, 2006, Gheyara et al., 2014) to assess both the per-
centage of chewed nestlet and shape of the nest. Additional detail has been
provided in Supplemental Information.
Statistics
Data were graphed asmean ± SEM and analyzed using GraphPad Prism 6 sta-
tistical software (GraphPad Software, La Jolla, CA). Individual statistical tests
are described within Supplemental Information.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and Supplemental Experi-
mental Procedures and can be found with this article at http://dx.doi.org/10.
1016/j.neuron.2016.04.042.
AUTHOR CONTRIBUTIONS
Conceptualization, K.M.S., S.L.D., C.F.B., F.R., and T.M.M.; Methodology,
K.M.S., S.L.D., D.F.W., H.N.D., C.F.B., F.R., and T.M.M.; Investigation,
K.M.S., S.L.D., R.L.M., S.J.C., and M.N.; Formal Analysis, K.M.S., S.L.D.,
D.F.W., F.R., and T.M.M.; Writing – Original Draft, K.M.S., S.L.D. and
T.M.M.; Writing – Review & Editing, K.M.S., S.L.D., R.L.M., D.F.W., H.N.D.,
C.F.B., F.R., and T.M.M.; Funding acquisition, K.M.S. and T.M.M.; Supervi-
sion, C.F.B, F.R., and T.M.M.
ACKNOWLEDGMENTS
We wish to recognize and thank Dr. Bradley Hyman for the resources neces-
sary to complete the biochemical tau experiments. We also thank the behav-
ioral expertise provided by the Animal Behavior Core at Washington University
in St. Louis, Dr. Peter Davies for his generous gift of the PHF-1 tau antibody,
and Elena Fisher for her assistance in editing and preparing this manuscript.
This work was supported by the Tau Consortium (T.M.M.), The National Insti-
tutes of Health (P50 AG05681 to T.M.M., J.C. Morris, PI; National Institute of
Neurological Disorders and Stroke R01NS078398 to T.M.M. and F32
NS089225 to K.M.S.; and National Institute on Aging R21AG044719-01 to
T.M.M.), Cure PSP (T.M.M.), and a Paul B. Beeson Career Development Award
(NINDS K08NS074194 to T.M.M.). Microscopy was supported by the Hope
Center Alafi Neuroimaging Laboratory and P30 Neuroscience Blueprint Inter-
disciplinary Center Core award to Washington University (P30 NS057105).
Additional support was provided by the Gene Johnson Weston Brain Institute
Advisor Fellowship to K.M.S. Antisense oligonucleotides used for experi-
mental studies were generously provided by Ionis Pharmaceuticals. Washing-
ton University in St. Louis has filed patents in conjunction with Ionis
Pharmaceuticals regarding use of Tau ASOs in neurodegenerative syndrome.
S.J.C., M.N., C.F.B., and F.R. are paid employees of Ionis Pharmaceuticals.
Received: August 6, 2015
Revised: November 19, 2015
Accepted: April 26, 2016
Published: May 19, 2016
REFERENCES
Adams, S.J., DeTure, M.A., McBride, M., Dickson, D.W., and Petrucelli, L.
(2010). Three repeat isoforms of tau inhibit assembly of four repeat tau fila-
ments. PLoS ONE 5, e10810.
Andorfer, C., Kress, Y., Espinoza, M., de Silva, R., Tucker, K.L., Barde, Y.A.,
Duff, K., and Davies, P. (2003). Hyperphosphorylation and aggregation of tau
in mice expressing normal human tau isoforms. J. Neurochem. 86, 582–590.

Please cite this article in press as: Schoch et al., Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model, Neuron (2016),http://dx.doi.org/10.1016/j.neuron.2016.04.042
Arai, T., Ikeda, K., Akiyama, H., Shikamoto, Y., Tsuchiya, K., Yagishita, S.,
Beach, T., Rogers, J., Schwab, C., and McGeer, P.L. (2001). Distinct isoforms
of tau aggregated in neurons and glial cells in brains of patients with Pick’s dis-
ease, corticobasal degeneration and progressive supranuclear palsy. Acta
Neuropathol. 101, 167–173.
Braak, H., and Braak, E. (1995). Staging of Alzheimer’s disease-related neuro-
fibrillary changes. Neurobiol. Aging 16, 271–278, discussion 278–284.
Bunker, J.M., Wilson, L., Jordan, M.A., and Feinstein, S.C. (2004). Modulation
of microtubule dynamics by tau in living cells: implications for development
and neurodegeneration. Mol. Biol. Cell 15, 2720–2728.
Combs, B., Voss, K., and Gamblin, T.C. (2011). Pseudohyperphosphorylation
has differential effects on polymerization and function of tau isoforms.
Biochemistry 50, 9446–9456.
Cruts, M., Theuns, J., and Van Broeckhoven, C. (2012). Locus-specific muta-
tion databases for neurodegenerative brain diseases. Hum. Mutat. 33, 1340–
1344.
D’Souza, I., Poorkaj, P., Hong, M., Nochlin, D., Lee, V.M., Bird, T.D., and
Schellenberg, G.D. (1999). Missense and silent tau genemutations cause fron-
totemporal dementia with parkinsonism-chromosome 17 type, by affecting
multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci.
USA 96, 5598–5603.
Dawson, H.N., Cantillana, V., Chen, L., and Vitek, M.P. (2007). The tau N279K
exon 10 splicing mutation recapitulates frontotemporal dementia and parkin-
sonism linked to chromosome 17 tauopathy in a mouse model. J. Neurosci.
27, 9155–9168.
Dawson, H.N., Cantillana, V., Jansen, M., Wang, H., Vitek, M.P., Wilcock,
D.M., Lynch, J.R., and Laskowitz, D.T. (2010). Loss of tau elicits axonal degen-
eration in a mouse model of Alzheimer’s disease. Neuroscience 169, 516–531.
deSilva,R., Lashley, T.,Gibb,G.,Hanger,D.,Hope,A.,Reid,A.,Bandopadhyay,
R., Utton,M.,Strand,C., Jowett, T., et al. (2003). Pathological inclusionbodies in
tauopathies contain distinct complements of tau with three or four microtubule-
binding repeat domains as demonstrated by new specific monoclonal anti-
bodies. Neuropathol. Appl. Neurobiol. 29, 288–302.
Deacon, R.M. (2006). Assessing nest building in mice. Nat. Protoc. 1, 1117–
1119.
DeVos, S.L., andMiller, T.M. (2013a). Antisense oligonucleotides: treating neu-
rodegeneration at the level of RNA. Neurotherapeutics 10, 486–497.
DeVos, S.L., and Miller, T.M. (2013b). Direct intraventricular delivery of drugs
to the rodent central nervous system. J. Vis. Exp. http://dx.doi.org/10.3791/
50326.
DeVos, S.L., Goncharoff, D.K., Chen, G., Kebodeaux, C.S., Yamada, K.,
Stewart, F.R., Schuler, D.R., Maloney, S.E., Wozniak, D.F., Rigo, F., et al.
(2013). Antisense reduction of tau in adult mice protects against seizures.
J. Neurosci. 33, 12887–12897.
Dinkel, P.D., Siddiqua, A., Huynh, H., Shah, M., and Margittai, M. (2011).
Variations in filament conformation dictate seeding barrier between three-
and four-repeat tau. Biochemistry 50, 4330–4336.
Ghetti, B., Oblak, A.L., Boeve, B.F., Johnson, K.A., Dickerson, B.C., and
Goedert, M. (2015). Invited review: Frontotemporal dementia caused bymicro-
tubule-associated protein tau gene (MAPT) mutations: a chameleon for neuro-
pathology and neuroimaging. Neuropathol. Appl. Neurobiol. 41, 24–46.
Gheyara, A.L., Ponnusamy, R., Djukic, B., Craft, R.J., Ho, K., Guo, W.,
Finucane, M.M., Sanchez, P.E., and Mucke, L. (2014). Tau reduction prevents
disease in a mouse model of Dravet syndrome. Ann. Neurol. 76, 443–456.
GlaxoSmithKline (2014). Phase II Doubleblind Exploratory Study of
GSK2402968 in Ambulant Subjects With Duchenne Muscular Dystrophy
[Online]. Published online August 21, 2014. https://www.clinicaltrials.gov/
ct2/show/NCT01153932.
Halfmann, R., and Lindquist, S. (2008). Screening for amyloid aggregation by
semi-denaturing detergent-agarose gel electrophoresis. J. Vis. Exp. http://
dx.doi.org/10.3791/838.
Hong, M., Zhukareva, V., Vogelsberg-Ragaglia, V., Wszolek, Z., Reed, L.,
Miller, B.I., Geschwind, D.H., Bird, T.D., McKeel, D., Goate, A., et al. (1998).
Mutation-specific functional impairments in distinct tau isoforms of hereditary
FTDP-17. Science 282, 1914–1917.
Hu, J., Matsui, M., Gagnon, K.T., Schwartz, J.C., Gabillet, S., Arar, K., Wu, J.,
Bezprozvanny, I., and Corey, D.R. (2009). Allele-specific silencing of mutant
huntingtin and ataxin-3 genes by targeting expanded CAG repeats in
mRNAs. Nat. Biotechnol. 27, 478–484.
Ionis Pharmaceuticals (2016). A Study to Assess the Efficacy and Safety of
ISIS-SMN Rx in Infants With Spinal Muscular Atrophy [Online]. Published on-
line May 6, 2016. Available: https://clinicaltrials.gov/ct2/show/NCT02193074.
Ittner, L.M., Ke, Y.D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., Wolfing,
H., Chieng, B.C., Christie, M.J., Napier, I.A., et al. (2010). Dendritic function of
tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell
142, 387–397.
Lei, P., Ayton, S., Finkelstein, D.I., Spoerri, L., Ciccotosto, G.D., Wright, D.K.,
Wong, B.X., Adlard, P.A., Cherny, R.A., Lam, L.Q., et al. (2012). Tau deficiency
induces parkinsonism with dementia by impairing APP-mediated iron export.
Nat. Med. 18, 291–295.
Lu, Q.L., Mann, C.J., Lou, F., Bou-Gharios, G., Morris, G.E., Xue, S.A.,
Fletcher, S., Partridge, T.A., and Wilton, S.D. (2003). Functional amounts of
dystrophin produced by skipping the mutated exon in the mdx dystrophic
mouse. Nat. Med. 9, 1009–1014.
Ma, Q.L., Zuo, X., Yang, F., Ubeda, O.J., Gant, D.J., Alaverdyan, M., Kiosea,
N.C., Nazari, S., Chen, P.P., Nothias, F., et al. (2014). Loss of MAP function
leads to hippocampal synapse loss and deficits in the Morris Water Maze
with aging. J. Neurosci. 34, 7124–7136.
Maeda, S., Djukic, B., Taneja, P., Yu, G.Q., Lo, I., Davis, A., Craft, R., Guo, W.,
Wang, X., Kim, D., et al. (2016). Expression of A152T human tau causes age-
dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 17,
530–551.
Miller, V.M., Xia, H., Marrs, G.L., Gouvion, C.M., Lee, G., Davidson, B.L., and
Paulson, H.L. (2003). Allele-specific silencing of dominant disease genes.
Proc. Natl. Acad. Sci. USA 100, 7195–7200.
Miller, T.M., Pestronk, A., David, W., Rothstein, J., Simpson, E., Appel, S.H.,
Andres, P.L., Mahoney, K., Allred, P., Alexander, K., et al. (2013). An antisense
oligonucleotide against SOD1 delivered intrathecally for patients with SOD1
familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man
study. Lancet Neurol. 12, 435–442.
Polydoro, M., Acker, C.M., Duff, K., Castillo, P.E., and Davies, P. (2009). Age-
dependent impairment of cognitive and synaptic function in the htau mouse
model of tau pathology. J. Neurosci. 29, 10741–10749.
Roberson, E.D., Scearce-Levie, K., Palop, J.J., Yan, F., Cheng, I.H., Wu, T.,
Gerstein, H., Yu, G.Q., and Mucke, L. (2007). Reducing endogenous tau ame-
liorates amyloid beta-induced deficits in an Alzheimer’s diseasemousemodel.
Science 316, 750–754.
Sanders, D.W., Kaufman, S.K., DeVos, S.L., Sharma, A.M., Mirbaha, H., Li, A.,
Barker, S.J., Foley, A.C., Thorpe, J.R., Serpell, L.C., et al. (2014). Distinct tau
prion strains propagate in cells and mice and define different tauopathies.
Neuron 82, 1271–1288.
Spillantini, M.G., and Goedert, M. (2013). Tau pathology and neurodegenera-
tion. Lancet Neurol. 12, 609–622.
Stoothoff, W., Jones, P.B., Spires-Jones, T.L., Joyner, D., Chhabra, E.,
Bercury, K., Fan, Z., Xie, H., Bacskai, B., Edd, J., et al. (2009). Differential effect
of three-repeat and four-repeat tau on mitochondrial axonal transport.
J. Neurochem. 111, 417–427.
Torres-Lista, V., andGimenez-Llort, L. (2013). Impairment of nesting behaviour
in 3xTg-AD mice. Behav. Brain Res. 247, 153–157.
von Bergen, M., Barghorn, S., Li, L., Marx, A., Biernat, J., Mandelkow, E.M.,
andMandelkow, E. (2001). Mutations of tau protein in frontotemporal dementia
promote aggregation of paired helical filaments by enhancing local beta-struc-
ture. J. Biol. Chem. 276, 48165–48174.
Neuron 90, 1–7, June 1, 2016 7

Neuron, Volume 90
Supplemental Information
Increased 4R-Tau Induces Pathological
Changes in a Human-Tau Mouse Model
Kathleen M. Schoch, Sarah L. DeVos, Rebecca L. Miller, Seung J. Chun, MichaelaNorrbom, David F. Wozniak, Hana N. Dawson, C. Frank Bennett, FrankRigo, and Timothy M. Miller

1
Supplemental Figures and Legends
Figure S1: Exclusion of off-target mRNA alteration by 3R to 4R MAPT splicing ASO within the mouse genome. (Related to
Figure 1)
NCBI BLAST analysis of the scrambled and 3R to 4R human MAPT targeting ASO sequences was performed against the mouse
genome to identify potential off-target mRNAs. While no mouse genome sequences produced a significant alignment with the ASO
query (i.e. E value < 0.05), the best aligned mouse mRNAs by maximum score were analyzed in our treated hTau samples via
quantitative real-time PCR. These genes included: A) Pcsk5 (proprotein convertase subtilisin/kexin type 5), B) St3gal3 (ST3 beta-
galactoside alpha-2,3-sialyltransferase 3), and C) Tgfa (transforming growth factor, alpha). 3R to 4R MAPT splicing ASO treatment
did not significantly alter the mRNA profile of Pcsk5, St3gal3, or Tgfa, suggesting that the increased pathology and functional deficits
identified in treated hTau mice is not likely attributed to off-target effects. A fourth gene, Serpinb9c (serine (or cysteine) peptidase
inhibitor, clade B, member 9c), was also analyzed but not appreciably detected in brain lysates. Based on mRNA analysis through
NCBI BLAST and UCSC Genome Browser, we can also confirm that the ASO alignments are not predicted to occur at alternative
splice sites, excluding the possibility that our ASOs are affecting off-target isoform expression. Data are expressed as mean + SEM relative to scrambled controls and analyzed by unpaired t-test.

2
Figure S2: Contribution of phosphorylation to high molecular weight tau species in SDD-AGE analysis. (Related to Figure 2)
A) To identify the contribution of phosphorylation status to the higher molecular weight shift seen with 3R to 4R splicing ASO
treatment, an hTau 3R to 4R splicing ASO sample was either untreated (-λ) or incubated with lambda phosphatase (+λ) to
dephosphorylate tau and run on an SDS-PAGE gel for analysis of total tau (HT7) and phospho-tau (p396 p-Tau). Densitometric
analysis of phospho-tau normalized to total tau confirmed successful dephosphorylation with lambda phosphatase treatment. B) The
untreated and lambda-treated hTau 3R to 4R splicing ASO sample was then analyzed via SDD-AGE. Semi-quantitation of the high
molecular weight (HMW) banding area revealed a small reduction in HMW tau with lambda phosphatase treatment, suggesting
phosphorylation contributes to some but not all of the shift in tau species. Thus, increased HMW seen with increased 4R-tau in hTau
mice (Figure 3) is likely due to both phosphorylated and oligomeric tau. Data are expressed as a mean value; A.U. arbitrary units.

3
Figure S3: Immunohistochemical analysis of synapses and neurons of ASO-treated hTau mice. (Related to Figure 3)
Contralateral tissue sections obtained from hTau mice treated with scrambled ASO (n=5) and 3R to 4R MAPT splicing ASO (n=7)
were evaluated for gross synaptic structure using the antibodies, post-synaptic density-95 (PSD-95) and synaptophysin, and for
neuronal density by the antibody, NeuN. Markers of A-C) PSD-95, D-F) synaptophysin, or G-I) NeuN were not statistically different between treatments as measured in the cortex and CA1 and CA3 regions of the hippocampus. Fluorescent immunoreactivity was
quantified as a mean fluorescent intensity + SEM and illustrated relative to scrambled ASO treatment. An unpaired t-test was used to
compare treatments.

4
Figure S4: Specificity of 4R to 3R human MAPT splicing ASO in Tau N279K mice. (Related to Figure 4)
A) RT-PCR analysis of mouse MAPT mRNA in 4R to 3R MAPT splicing ASO-treated samples revealed no change in exon 10
splicing of mouse MAPT mRNA compared to saline treatment. Adult mouse and fetal mouse brain samples were included as size
controls for 4R and 3R MAPT, respectively. Total mouse MAPT (i.e. 4R) mRNA bands were measured by densitometric analysis and expressed as arbitrary units (A.U.). B) Similarly, total mouse MAPT mRNA levels measured by qPCR analysis were not significantly
altered by treatment, confirming splicing specificity for human MAPT. Total MAPT mRNA data are expressed as a percent relative to
saline treatment (mean + SEM).

5
Supplemental Experimental Procedures
Experimental animals
Human tau transgenic mice As described in Andorfer et al., 2003, generation of the human tau (hTau) transgenic mouse line involved insertion of a
human tau cDNA-containing PAC vector into female donor mice. The resulting mice (known as 8c mice) were bred to mouse tau
knockout (mTau-/-) mice and backcrossed to a C57BL/6J background to obtain mice exclusively expressing human tau. RT-PCR
analysis of mouse brains using human-specific primers confirmed the presence of MAPT isoforms 0N, 1N, and 2N as well as isoforms
with and without exon 10 (4R and 3R, respectively) (Andorfer et al., 2003). hTau transgenic mice were purchased (Jackson
Laboratories, Bar Harbor, ME) and maintained in-house through breeding between hTau+/- mTau-/- mice and mTau-/- mice. Mice
were housed in a controlled, single-barrier facility with 12:12 light:dark photoperiod and chow diet and water ad libitum. Human tau
genotyping was performed by PCR amplification of tail DNA using the primers 5’-ACT TTG AAC CAG GAT GGC TGA GCC C-3’
and 5’-CTG TGC ATG GCT GTC CAC TAA CCT T-3’.
Human N279K mutant tau transgenic mice
Human N279K mutant tau mice as described in Dawson et al., 2007 were created by microinjection of a human MAPT minigene construct, containing both intron and exon sequences and N279K FTDP-17 mutation within exon 10, into mice of mixed
C57BL/6//SJL background to produce transgenic mice expressing human tauN279K under the MAPT gene promoter. Human-specific
RT-PCR primers amplifying exons 2 and 3 and exon 10 were used to demonstrate the exclusive presence of 2N4R MAPT mRNA (441
bp) in N279K transgenic mice, albeit at levels well below mouse tau (Dawson et al., 2007). Thus, expression of the N279K-mediated
mRNA splicing aberration under the endogenous human tau promoter in this model closely parallels the genetics of human FTDP-17.
Tau N279K mice were obtained from Duke University and maintained at Washington University as heterozygotes by breeding Tau
N279K transgenic mice with C57BL/6 mice (Jackson Laboratories). Mice positive for the N279K human tau transcript were identified
by PCR amplification of tail DNA using the same primer sequences listed for hTau genotyping.
Antisense oligonucleotides (ASOs) ASO sequences used for experiments are as follows. For 3R to 4R MAPT splicing in hTau mice: 5’-GGC GCA TGG GAC
GTG TGA-3’; and scrambled control: 5’-TCA TTT GCT TCA TAC AGG-3’. For 4R to 3R MAPT splicing in Tau N279K mice: 5’-
CAG ATC CTG AGA GCC CAA-3’; and scrambled control: 5’-CCT TCC CTG AAG GTT CCT CC-3’. Although similar in structure
and modification to the above splicing oligonucleotides (Rigo et al., 2014), the 4R to 3R MAPT splicing ASO used in hTau transgenic
mice targeted a different region of the human tau sequence (5’-GGA CGT GTG AAG GTA CTC-3’). Due to the ASO specificity for
human MAPT, mouse tau remains unaltered. An in silico analysis was performed with the 3R to 4R and 4R to 3R MAPT ASOs to
ensure their specificity toward human tau mRNA. The program Bowtie (Langmead et al., 2009) was used to determine the predicted
off-targets for the MAPT ASOs in the mouse transcriptome (pre- and m-RNA). This analysis confirmed that the MAPT ASOs do not
bind any RNA other than MAPT with full. Gene downregulation events are not anticipated given that ASOs that modulate splicing do
not induce RNaseH- or Ago2-mediated mRNA degradation.
Intraventricular delivery of ASOs
Mice were anesthetized by 2-3% inhalant isoflurane and stabilized within a stereotaxic head frame (Kopf, Tujunga, CA).
While receiving constant isoflurane flow, a scalp incision was made and a subcutaneous pocket created along the left side of the body.
The osmotic pump, with plastic tubing and catheter attachment, was then placed within the pocket. The metal catheter was positioned
-1.1mm M/L, -0.5mm A/P, -2.5mm D/V from bregma (Franklin and Paxinos, 2013) for insertion into the intraventricular space and
directly driven into the skull. A small amount of super glue (Loctite, Westlake, OH) placed on the plastic cannula base ensured
immobilization of the catheter within the lateral ventricle. The incision was closed and mice were placed on a 37°C warming recovery
pad until ambulatory. Following the surgical procedure, all mice were individually housed.
Osmotic pumps were assembled according to manufacturer’s instructions (ALZET) and allowed to equilibrate in a 37°C
water bath overnight prior to surgical implantation. Drug delivery occurred over the course of 28 days at a rate of 6 μl of saline or
ASO per day. Intraventricular infusion of ASOs previously has been shown to result in widespread distribution of the compound throughout the cortex, hippocampus, thalamus, brainstem, and cerebellum (DeVos et al., 2013, Kordasiewicz et al., 2012) with both
neuronal and non-neuronal uptake (Kordasiewicz et al., 2012); therefore, we anticipated adequate delivery of ASOs throughout the
CNS of experimental mice. The dose of ASO for splicing studies was empirically determined to achieve effective splicing without
overt adverse effects: 15 μg/day for 3R to 4R MAPT splicing ASO and 25 μg/day for 4R to 3R MAPT splicing ASO. Scrambled
ASOs were given at the identical rate and dose for respective experiments. Behavioral assessments and euthanasia occurred two
months following the cessation of ASO infusion.

6
mRNA isolation and analysis following MAPT splicing
mRNA was extracted from tissues using RNeasy® Mini Kit (Qiagen, Venlo, Netherlands) and its concentration determined
by Nanodrop spectrophotometer (Thermo Scientific, Waltham, MA). MAPT mRNA levels from the hTau mouse model were readily
detectable by standard quantitative real-time PCR methods using EXPRESS One-Step Superscript qRT-PCR Universal Kit
(Invitrogen, Carlsbad, CA) for reverse transcription and amplification and ABI PRISM 7500 Fast Real-Time PCR System for
comparative analysis by the ΔΔCt method (Applied Biosystems, Waltham, MA). Given that human N279K MAPT mRNA levels were much lower in comparison to endogenous mouse MAPT mRNA (Dawson et al., 2007), a modified RT-PCR protocol was used.
Isolated RNA from Tau N279K mice was reverse transcribed (SuperScript II Reverse Transcriptase; Invitrogen). The cDNA was then
run in a PCR reaction using a PCR enhancer system kit (Invitrogen) and Platinum Taq DNA Polymerase (Invitrogen) with
radiolabeled 32-P-dCTP (PerkinElmer, Waltham, MA). PCR products were digested using HincII to discriminate mouse versus human
tau products. The radiolabeled PCR samples were run on a 6% native polyacrylamide gel under denaturing conditions, allowing for
size separation of human 4R (496bp digested; 745bp undigested) and 3R (403bp digested; 652bp undigested) MAPT cDNA, and
detected on a STORM optical scanner (GE Healthcare Bio-Sciences, Pittsburgh, PA). Individual 3R and 4R bands were quantified by
ImageJ (National Institutes of Health, Bethesda, MD) densitometric analysis. The signal intensity of each cDNA band was normalized
according to its G+C content. The extent of exon 10 inclusion was calculated as a percentage of the total amount of spliced mRNA.
Primer/probe sequences used for detection of total human MAPT mRNA following 3R to 4R MAPT splicing treatment were:
forward 5’-AGA AGC AGG CAT TGG AGA C- 3’, reverse 5’-TCT TCG TTT TAC CAT CAG CC-3’, probe 5’-/56-FAM/ACG
GGA CTG GAA GCG ATG ACA AAA /3IABkFQ/-3’; for human 4R MAPT mRNA: forward 5’-CAT GCC AGA CCT GAA GAA TG-3’, reverse 5’-GAC TGG ACG TTG CTA AGA TC-3’, probe 5’-/56-FAM/ CCA CTG AGA ACC TGA AGC ACC AGC
/3IABkFQ/-3’ (Integrated DNA Technologies, Coralville, IA). As an internal control, GAPDH primer/probe sequences were: forward
5’-TGC CCC CAT GT TGT GAT G-3’, reverse 5’-TGT GGT CAT GAG CCC TTC C-3’, probe 5’-/56-FAM/ AAT GCA TCC TGC
ACC ACC AAC TGC TT /3IABkFQ/-3’ (Integrated DNA Technologies). For hTau mice treated with a 4R to 3R MAPT splicing
ASO, total human MAPT mRNA within brain and spinal cord tissue was detected by the following primer/probe sequences: forward
5’-AAG ATT GGG TCC CTG GAC AAT-3’, reverse 5’-AGC TTG TGG GTT TCA ATC TTT TTA TT-3’, probe 5’-TTA ATT ATC
TGC ACC TTC CCG CCT CC-3’; for human 4R MAPT mRNA: forward 5’-CAC TGA GAA CCT GAA GCA CC-3’, reverse 5’-
GGA CGT TGC TAA GAT CCA GCT-3’, probe 5’-TTA ATT ATC TGC ACC TTC CCG CCT CC-3’; and for GAPDH internal
control: forward 5’-GGC AAA TTC AAC GGC ACA GT-3’, reverse 5’-GGG TCT CGC TCC TGG AAG AT-3’, and probe 5’-AAG
GCC GAG AAT GGG AAG CTT GTC ATC-3’.
Primer sequences used for detection of 3R and 4R MAPT mRNA isoforms in Tau N279K mice were: forward 5’-AAC GAA GAT CGC CAC ACC-3’ and reverse 5’-CGA CTT GTA CAC GAT CTC C-3’, complimentary to only human tau. To confirm the
human specificity of 4R to 3R MAPT splicing ASO, mouse MAPT mRNA obtained from treated Tau N279K mice was amplified by
OneStep RT-PCR kit (Qiagen) using primers: forward 5’-GAA CCA CCA AAA TCC GGA GA -3’, reverse 5’-CTC TTA CTA GCT
GAT GGT GAC-3’ and probe 5’-/56-FAM/CCA AGA AGG TGG CAG TGG TCC/3IABkFQ/- 3’; and for RT-PCR: forward 5’-
GAA GAT CGC CAC ACC TCG-3’ and reverse 5’-GGT GAC TTA TAC ACA ATT TCT G-3’. RT-PCR products were visualized
on an agarose gel, using ImageJ to semi-quantitatively measure the 3R and 4R band densities.
Tissue homogenization and tau protein analyses by immunoblot and ELISA
Brain tissue designated for protein analysis (temporoparietal region ipsilateral to the catheter) was homogenized in RAB
buffer (750mM sodium chloride, 100mM MES, 20mM sodium fluoride, 1mM EDTA, 1mM sodium orthovanadate, 0.5mM
magnesium sulfate heptahydrate) supplemented with protease inhibitor cocktail (Sigma, St. Louis, MO) and phosphatase inhibitors (PhosSTOP; Roche, San Francisco, CA) and centrifuged at 21,000 x g for 15 minutes at 4°C. For tau fractionation experiments, brain
tissue was homogenized and ultracentrifuged in RAB buffer, RIPA buffer (150mM NaCl, 50mM Tris, 0.5% deoxycholic acid, 1%
Triton X-100, 0.5% sodium dodecyl sulfate, 25mM EDTA, pH 8.0), and 70% formic acid as previously described in detail (Yamada et
al., 2011). RAB- and RIPA-soluble supernatants were analyzed by BCA protein assay and formic acid fractions by Bradford assay
(Thermo Scientific) for protein concentration.
Immunoblots for total tau and tau isoforms were used to visualize tau protein expression and tau isoform changes following
ASO treatment in mice. Protein homogenates were run on 12% SDS-PAGE gels, transferred to PVDF membrane, and blocked for 1
hour in 5% dry milk/1X PBS/0.05%Tween-20. Following overnight incubation in primary antibody, membranes were incubated in
HRP-conjugated secondary antibody and detected using ECL 2 substrate (Thermo Scientific) on G:Box Chemi XT4 Imaging System
(Syngene, Frederick, MD). Primary antibodies were diluted in 5% dry milk/1X PBS/0.05% Tween-20 and included total human tau
(mouse monoclonal HT7, 1:1000, Thermo Fisher Scientific Cat# MN1000 RRID: AB_223454), 3R-tau (RD3 1:1000; Millipore Cat# 05-803 RRID: AB_310013), and 4R-tau (RD4 1:500; Millipore Cat# 05-804 RRID: AB_11211556) with anti-mouse IgG HRP
secondary (1:5000; GE Healthcare Cat# NA931-1ml RRID: AB_772210). Membranes were washed and re-probed for GAPDH (rabbit
monoclonal 1:5000; Cell Signaling Technology Cat# 2118L RRID: AB_1031003) and anti-rabbit IgG HRP secondary antibody
(1:5000; GE Healthcare Cat# NA934-1ml RRID: AB_772206) for a loading control.
Quantification of total human tau was performed by enzyme-linked immunosorbent assay (ELISA) with tau-5 (20 μg/mL;
Millipore Cat# 577801-100UG RRID: AB_212534) coating antibody and human-specific HT7 (0.3 μg/mL, biotin-conjugated; Thermo
Fisher Scientific Cat# MN1000B RRID: AB_223453) capture antibody. Following an hour incubation in blocking reagent (4% bovine
serum albumin (BSA) diluted in 1X PBS), recombinant human tau standard (2N4R; rPeptide, Bogart, GA) and protein samples were

7
diluted in ELISA buffer (300mM Tris, 0.25% BSA, 1X protease inhibitor cocktail) and applied to half-well plates for overnight
incubation. Human tau was detected using HT7 capture antibody followed by streptavidin poly-HRP40 conjugate (1:4000; Fitzgerald,
Acton, MA) and 3,3’,5,5’-tetramethylbenzidine liquid substrate, Super Slow (Sigma) reagent and read by microplate
spectrophotometer (Biotek Epoch, Winooski, VT).
Detection of tau by SDD-AGE
In preparation of SDD-AGE analysis, occipital brain lysate was thawed on ice and a 1.5% agarose gel was prepared by
dissolving agarose in buffer G (20mM Tris-Base, 200mM glycine) with the addition of sodium dodecyl sulfate (SDS) to a final
concentration of 0.02%. Brain protein lysate (100 μg) was incubated with 0.02% SDS sample buffer at room temperature for 7
minutes prior to loading. Gel electrophoresis was done in Laemmli buffer (Buffer G with 0.1% SDS) at 35V for 14 hours. Protein was
then transferred via capillary action using 20 pieces of thick Whatmann (GB 005) and 8 pieces medium Whatmann (GB 003) to
Immobilon PVDF membrane (Millipore) at 4°C for 36 hours. Membranes were blocked in 3% BSA 1X TBS-Tween 20 (TBS-T) for 1
hour and then probed for total tau using rabbit polyclonal anti-tau ab64193 (1:4000; Abcam Cat# ab64193 RRID: AB_1143333)
overnight at 4°C. Membranes were washed three times with 1X TBS-T, probed with goat anti-rabbit HRP (1:2000; Bio-Rad
Laboratories, AbD Serotec Cat# 170-6515 RRID:AB_11125142) for 1.5 hours at room temperature, and washed three times with 1X
TBS-T. Membranes were developed using ECL (Thermo Scientific Pierce) for 3 minutes and exposed to film (GE Healthcare Bio-
Sciences). To re-probe for phosphorylated tau, the blot was incubated with PHF-1 (1:5000; a gift from P. Davies, The Feinstein Institute for Medical Research; New York; USA Cat# PHF1 RRID: AB_2313687) diluted in 3% BSA in 1X TBS-T with 0.01%
sodium azide overnight at 4°C. Once sufficiently washed in 1X TBS-T, the blot was incubated with goat anti-mouse HRP (1:2000;
Bio-Rad Laboratories, AbD Serotec Cat# 170-6516 RRID: AB_11125547) and developed in ECL as previously described above.
Lambda phosphatase treatment
Protein lysate (120 μg) was incubated at 30°C for 1.5 hours with 1200 units of lambda phosphatase (New England BioLabs)
to dephosphorylate tau. Following incubation, samples were immediately placed on ice and prepared for SDS-PAGE analysis. Lysates
(20 μg) were boiled at 95°C for 7 minutes, cooled to room temperature, loaded onto a 4-12% Bis-Tris polyacrylamide gel (NuPAGE,
Life Technologies) and run at 120V. Proteins were transferred to PVDF membrane and detection of total tau (HT7, 1:2500) and
phospho-tau (p396 p-Tau, 1:2500; Millipore Cat# AB9658 RRID:AB_11213701) occurred overnight at 4°C. Goat anti-mouse IRDye680CW (1:2000; LI-COR Biosciences Cat# 926-32220 RRID: AB_621840) and goat anti-rabbit IRDye800CW (1:2000; LI-
COR Biosciences Cat# 926-32211 RRID: AB_621843) secondary antibodies were used to detect total tau and phospho-tau,
respectively. Membranes were visualized on an Odyssey LI-COR imaging system (LI-COR Biosciences, Lincoln, NE). SDD-AGE
analysis was performed as described in “Experimental Procedures.”
Histology
Immunohistochemistry of phosphorylated tau
Fixed, frozen brain tissue contralateral to the catheter placement was sectioned on an HM 430 freezing microtome (Thermo
Scientific) at a thickness of 50μm. Serial coronal sections taken approximately 600 μm apart were placed in cryoprotectant solution
(30% ethylene glycol, 15% 0.2M phosphate buffer, and 15% w/v sucrose) and stored at -20°C until use. For identification of phosphorylated tau in hTau brain tissue, free-floating tissue were rinsed in PBS. Sections were then incubated in 0.3% v/v hydrogen
peroxide to quench endogenous peroxidases, washed, and blocked in 3% nonfat dry milk diluted in PBS containing 0.25% Triton-X.
Sections were incubated at 4°C overnight in mouse monoclonal primary antibody (AT8 biotin-conjugated, 1:500; Thermo Fisher
Scientific Cat# MN1020B RRID: AB_223648) to detect phosphorylated tau. The biotinylated signal was amplified by incubation in an
avidin-biotin solution (Vectastain Elite ABC kit; Vector Laboratories) and detected using diaminobenzidine.
Tissue sections were mounted onto slides and dried overnight. Slides were dehydrated in an ethanol gradient and
coverslipped using Cytoseal™ XYL mounting media (Thermo Scientific). Stained tissues were imaged on Olympus NanoZoomer 2.0-
HT microscope system (Hamamatsu, Hamamatsu City, Japan) and captured using NDP.scan 2.5 and NDP.view software
(Hamamatsu). Image analysis was performed in a blinded fashion using ImageJ in which regions of positive immunoreactivity were
identified above an assigned pixel size and threshold value that were kept constant across all treatment groups for each brain area.
Immunofluorescence imaging and analysis of neuronal and synaptic markers
Free-floating tissue sections were rinsed in 1X Tris-buffered saline (TBS) prior to blocking in 5% normal horse serum (NHS)
diluted in TBS containing 0.1% Triton X-100. Sections were incubated in primary antibody (NeuN 1:1000, Millipore Cat# MAB377B
RRID: AB_177621; synaptophysin 1:1000, Cell Signaling Technology Cat# 5461S RRID: AB_10698743; PSD-95 1:1000, Millipore
Cat# AB9708 RRID: AB_2092543) at 4°C overnight and, following an additional blocking incubation, were incubated with
fluorescently tagged secondary antibody (anti-rabbit DyLight 488 1:250, Thermo Fisher Scientific Cat# SA5-10038 RRID:
AB_2556618 or anti-rabbit AlexaFluor 594 1:250, Jackson ImmunoResearch Labs Cat# 711-585-152 RRID: AB_2340621). Sections
were mounted and sealed with Fluoromount media (Southern Biotech, Birmingham, AL). The Olympus NanoZoomer 2.0-HT

8
microscope system was used for fluorescent image analysis with quantification in ImageJ. For CA1 and CA3 regions, a rectangle was
drawn over the region of interest (ROI) and duplicated across multiple images for equivalent ROIs. For cortical measurements, a
region was manually drawn around the entire cortex. The mean fluorescence intensity (MFI) and integrated density measurements
were recorded from ImageJ and corrected for background fluorescence using the formula: Corrected MFICA1 = (IntDenROI -(AreaROI *
MFIBackground))/ AreaROI.
Seizure monitoring
For monitoring of PTZ seizure activity, hTau mice were placed in a clean cage and recorded for 15 min following PTZ
injection. Videos were later viewed to record the time at which a seizure stage was reached. Stages were characterized by immobility
(stage 1); spasm, twitch, or tremor (stage 2); tail extension (stage 3); forelimb clonus (stage 4); generalized clonus (stage 5);
jumping/running seizures (stage 6); tonic extension (stage 7); and death (stage 8) (DeVos et al., 2013). Video recording and viewing
was done under blinded conditions such that genotype and treatment were unknown to the observer.
Nesting activity
Mice were individually housed and previous nesting material was removed. An intact 3.0 g nestlet (Ancare, Bellmore, NY)
was placed within the cages. Approximately 14 h later, following the nocturnal period, the resulting nest was photographed for qualitative scoring and any untorn nestlet was weighed. All nest images and weights were analyzed by an individual blinded to
genotype and treatment.
Statistics
Quantifiable mRNA and protein expression data for both 3R to 4R and 4R to 3R splicing experiments were evaluated by one-
way ANOVA with Dunn’s multiple comparison post hoc analysis. A Kruskal-Wallis one-way ANOVA with Dunn’s post hoc was
used to analyze categorical nestlet scores and seizure severity scores induced by PTZ; however, an unpaired t-test was used to evaluate
nesting activity by untorn nestlet weight and a repeated measures two-way ANOVA (treatment x time) with Bonferroni post hoc was
performed on seizure latency data. Histological quantifications of phosphorylated tau used a one-way ANOVA with Dunn’s post hoc
for multiple group comparisons. In select experimental analyses, saline- and scrambled-treated controls were not statistically different and were subsequently pooled for comparison with splicing treatments. A value of p<0.05 was deemed significant for all analyses.

9
Supplemental References
FRANKLIN, K. B. J. & PAXINOS, G. 2013. Paxinos and Franklin's The mouse brain in stereotaxic coordinates, Amsterdam,
Academic Press, an imprint of Elsevier.
KORDASIEWICZ, H. B., STANEK, L. M., WANCEWICZ, E. V., MAZUR, C., MCALONIS, M. M., PYTEL, K. A., ARTATES, J.
W., WEISS, A., CHENG, S. H., SHIHABUDDIN, L. S., HUNG, G., BENNETT, C. F. & CLEVELAND, D. W. 2012. Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron, 74, 1031-44.
LANGMEAD, B., TRAPNELL, C., POP, M. & SALZBERG, S. L. 2009. Ultrafast and memory-efficient alignment of short DNA
sequences to the human genome. Genome Biol, 10, R25.
YAMADA, K., CIRRITO, J. R., STEWART, F. R., JIANG, H., FINN, M. B., HOLMES, B. B., BINDER, L. I., MANDELKOW, E.
M., DIAMOND, M. I., LEE, V. M. & HOLTZMAN, D. M. 2011. In vivo microdialysis reveals age-dependent decrease of
brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci, 31, 13110-7.


















