
In vitrosynthesis of the iron–molybdenum cofactor of ... filepurified Nif proteins, providing...
6
In vitro synthesis of the iron–molybdenum cofactor of nitrogenase from iron, sulfur, molybdenum, and homocitrate using purified proteins Leonardo Curatti, Jose A. Hernandez, Robert Y. Igarashi, Basem Soboh, Dehua Zhao, and Luis M. Rubio* Department of Plant and Microbial Biology, University of California, Berkeley, CA 94720 Edited by Sydney Kustu, University of California, Berkeley, CA, and approved September 25, 2007 (received for review April 2, 2007) Biological nitrogen fixation, the conversion of atmospheric N 2 to NH3 , is an essential process in the global biogeochemical cycle of nitrogen that supports life on Earth. Most of the biological nitro- gen fixation is catalyzed by the molybdenum nitrogenase, which contains at its active site one of the most complex metal cofactors known to date, the iron–molybdenum cofactor (FeMo-co). FeMo-co is composed of 7Fe, 9S, Mo, R-homocitrate, and one unidentified light atom. Here we demonstrate the complete in vitro synthesis of FeMo-co from Fe 2 ,S 2 , MoO 4 2 , and R-homocitrate using only purified Nif proteins. This synthesis provides direct biochemical support to the current model of FeMo-co biosynthesis. A minimal in vitro system, containing NifB, NifEN, and NifH proteins, together with Fe 2 ,S 2 , MoO 4 2 , R-homocitrate, S-adenosyl methionine, and Mg-ATP, is sufficient for the synthesis of FeMo-co and the activation of apo-dinitrogenase under anaerobic-reducing condi- tions. This in vitro system also provides a biochemical approach to further study the function of accessory proteins involved in nitro- genase maturation (as shown here for NifX and NafY). The signif- icance of these findings in the understanding of the complete FeMo-co biosynthetic pathway and in the study of other complex Fe-S cluster biosyntheses is discussed. FeMo-co nitrogen fixation nif Fe-S proteins B iological nitrogen fixation, the conversion of atmospheric N 2 to NH 3 , is an essential process of the global biogeochemical cycle of nitrogen that supports life on Earth (1). Biological nitrogen fixation provides an alternative solution to the heavy use of chemically synthesized nitrogen fertilizers and, thus, is important to achieve sustainable agricultural practices (2). The major part of biological nitrogen fixation is catalyzed by the molybdenum nitrogenase enzyme according to the following reaction: N 2 8e 16 MgATP 8H 3 2 NH 3 H 2 16 MgADP 16 Pi (3). Remarkably, this reaction produces H 2 as a by-product and has regained attention over the last few years as a promising source of clean energy (4). The molybdenum nitrogenase has two component proteins: dinitrogenase (NifDK) and dinitrogenase reductase (NifH). Dinitrogenase carries at its active site one of the most complex biological metalloclusters known to date, the iron–molybdenum cofactor (FeMo-co), composed of seven Fe, nine S, one Mo, one R-homocitrate, and one light unidentified atom (5, 6). The systematic phenotypic analyses of Azotobacter vinelandii and Klebsiella pneumoniae strains with mutations in nitrogen fixation (nif ) genes and the subsequent biochemical analyses have led to the identification of at least 11 genes (nifUS-BQ- ENX-V-H-Y and nafY) encoding proteins involved in FeMo-co biosynthesis and its insertion into a FeMo-co-deficient apo- NifDK protein (7–9). Fig. 1 depicts an integrative model of the current understanding of the pathway for FeMo-co biosynthesis. It is important to emphasize that this model is mostly based on the ability to isolate nif mutant strains. Several nif genes were shown to be essential for FeMo-co synthesis in vivo (e.g., nifB, nifU, nifS, nifH, nifN, nifE, and nifV), whereas others seemed to be required only under certain metabolic conditions (e.g., nifQ, nifX, nifY, and nafY). The identification and isolation of in vivo synthesized FeMo-co precursors, such as NifB cofactor (NifB-co) and the VK cluster † (10–12), entailed very important progress and allowed the in vitro reconstitution of the late steps of the FeMo-co biosynthetic pathway in reactions that contained only purified Nif proteins, providing direct biochemical evidence to support those steps in the current model. Much less is known about the early steps of the FeMo-co biosynthetic pathway, especially at the interface with the path- way for the biosynthesis of more simple [2Fe-2S] and [4Fe-4S] clusters, which involves the activities of the cysteine desulfurase NifS and the scaffolding protein NifU (13, 14). Pioneering studies on nitrogenase maturation by Dean and colleagues (reviewed in ref. 13) uncovered the existence of a complex cellular machinery involved in the assembly of ubiquitous [2Fe- 2S] and [4Fe-4S] clusters that, at present, is recognized to be very similar for a diversity of Fe-S proteins in all organisms. It is generally accepted that the biosynthesis of the complex FeMo-co uses common [2Fe-2S] and/or [4Fe-4S] cluster as precursors, but support from direct biochemical evidence is currently lacking. Genetic evidence has revealed the critical function of the nifB gene product for the in vivo accumulation of NifB-co and the VK cluster. The NifB protein has been recently purified and used to activate apo-NifDK when added to cell-free extracts of a NifB- deficient A. vinelandii mutant strain. This NifB-dependent in vitro activation of apo-NifDK required ferrous iron, sulfide, S-adenosyl methionine (SAM), molybdate, sodium dithionite (DTH), Mg-ATP, and R-homocitrate (15). It is still unknown whether other gene products (in combination with NifB) are required for the synthesis of NifB-co. All currently known proteins that are critical for FeMo-co biosynthesis are available in purified form. This fact has given us the unprecedented opportunity to attempt to reconstitute in vitro the complete FeMo-co biosynthesis pathway. The present study establishes a minimal and composition-defined reaction mixture showing that FeMo-co can be synthesized in vitro from Fe, S, Mo, Author contributions: L.C. and L.M.R. designed research; L.C., J.A.H., R.Y.I., B.S., and D.Z. performed research; L.C., J.A.H., and L.M.R. analyzed data; and L.C. and L.M.R. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. Abbreviations: FeMo-co, iron–molybdenum cofactor; NifB-co, NifB cofactor; DTH, sodium dithionite; SAM, S-adenosyl methionine. *To whom correspondence should be addressed. E-mail: [email protected]. † NifB-co, the metabolic product of NifB activity, is an isolatable [Fe-S] cluster of unknown structure that serves as precursor to FeMo-co. NifB-co has also been proposed to be a precursor of the iron–vanadium and iron-only cofactors of the alternative nitrogenases (29). The VK cluster is a precursor to FeMo-co that accumulates on NifEN in a nifH mutant strain and is thought to represent an intermediate after NifB-co in FeMo-co synthesis. This article contains supporting information online at www.pnas.org/cgi/content/full/ 0703050104/DC1. © 2007 by The National Academy of Sciences of the USA 17626 –17631 PNAS November 6, 2007 vol. 104 no. 45 www.pnas.orgcgidoi10.1073pnas.0703050104
Transcript of In vitrosynthesis of the iron–molybdenum cofactor of ... filepurified Nif proteins, providing...

In vitro synthesis of the iron–molybdenum cofactorof nitrogenase from iron, sulfur, molybdenum,and homocitrate using purified proteinsLeonardo Curatti, Jose A. Hernandez, Robert Y. Igarashi, Basem Soboh, Dehua Zhao, and Luis M. Rubio*
Department of Plant and Microbial Biology, University of California, Berkeley, CA 94720
Edited by Sydney Kustu, University of California, Berkeley, CA, and approved September 25, 2007 (received for review April 2, 2007)
Biological nitrogen fixation, the conversion of atmospheric N2 toNH3, is an essential process in the global biogeochemical cycle ofnitrogen that supports life on Earth. Most of the biological nitro-gen fixation is catalyzed by the molybdenum nitrogenase, whichcontains at its active site one of the most complex metal cofactorsknown to date, the iron–molybdenum cofactor (FeMo-co). FeMo-cois composed of 7Fe, 9S, Mo, R-homocitrate, and one unidentifiedlight atom. Here we demonstrate the complete in vitro synthesis ofFeMo-co from Fe2�, S2�, MoO4
2�, and R-homocitrate using onlypurified Nif proteins. This synthesis provides direct biochemicalsupport to the current model of FeMo-co biosynthesis. A minimalin vitro system, containing NifB, NifEN, and NifH proteins, togetherwith Fe2�, S2�, MoO4
2�, R-homocitrate, S-adenosyl methionine,and Mg-ATP, is sufficient for the synthesis of FeMo-co and theactivation of apo-dinitrogenase under anaerobic-reducing condi-tions. This in vitro system also provides a biochemical approach tofurther study the function of accessory proteins involved in nitro-genase maturation (as shown here for NifX and NafY). The signif-icance of these findings in the understanding of the completeFeMo-co biosynthetic pathway and in the study of other complexFe-S cluster biosyntheses is discussed.
FeMo-co � nitrogen fixation � nif � Fe-S proteins
B iological nitrogen fixation, the conversion of atmospheric N2to NH3, is an essential process of the global biogeochemical
cycle of nitrogen that supports life on Earth (1). Biologicalnitrogen fixation provides an alternative solution to the heavyuse of chemically synthesized nitrogen fertilizers and, thus, isimportant to achieve sustainable agricultural practices (2). Themajor part of biological nitrogen fixation is catalyzed by themolybdenum nitrogenase enzyme according to the followingreaction: N2 � 8e� � 16 MgATP � 8 H�3 2 NH3 � H2 � 16MgADP � 16 Pi (3). Remarkably, this reaction produces H2 asa by-product and has regained attention over the last few yearsas a promising source of clean energy (4). The molybdenumnitrogenase has two component proteins: dinitrogenase (NifDK)and dinitrogenase reductase (NifH). Dinitrogenase carries at itsactive site one of the most complex biological metalloclustersknown to date, the iron–molybdenum cofactor (FeMo-co),composed of seven Fe, nine S, one Mo, one R-homocitrate, andone light unidentified atom (5, 6).
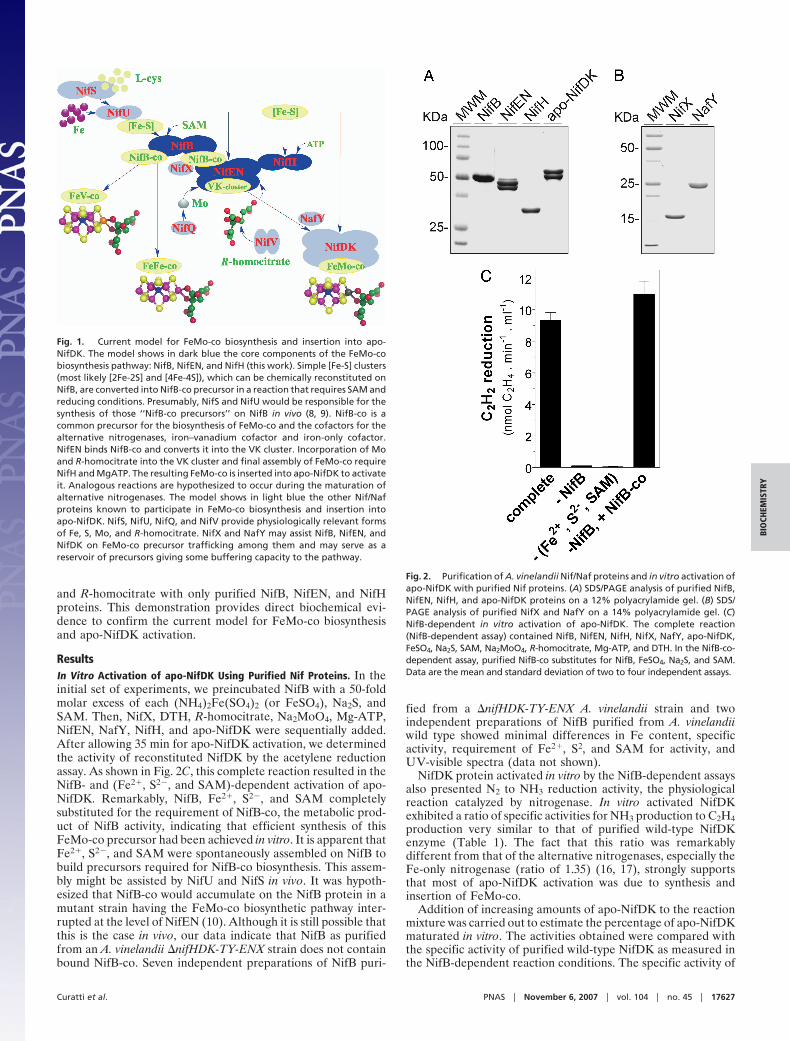
The systematic phenotypic analyses of Azotobacter vinelandiiand Klebsiella pneumoniae strains with mutations in nitrogenfixation (nif ) genes and the subsequent biochemical analyseshave led to the identification of at least 11 genes (nifUS-BQ-ENX-V-H-Y and nafY) encoding proteins involved in FeMo-cobiosynthesis and its insertion into a FeMo-co-deficient apo-NifDK protein (7–9). Fig. 1 depicts an integrative model of thecurrent understanding of the pathway for FeMo-co biosynthesis.It is important to emphasize that this model is mostly based onthe ability to isolate nif mutant strains. Several nif genes wereshown to be essential for FeMo-co synthesis in vivo (e.g., nifB,nifU, nifS, nifH, nifN, nifE, and nifV), whereas others seemed to
be required only under certain metabolic conditions (e.g., nifQ,nifX, nifY, and nafY).
The identification and isolation of in vivo synthesizedFeMo-co precursors, such as NifB cofactor (NifB-co) and theVK cluster† (10–12), entailed very important progress andallowed the in vitro reconstitution of the late steps of theFeMo-co biosynthetic pathway in reactions that contained onlypurified Nif proteins, providing direct biochemical evidence tosupport those steps in the current model.
Much less is known about the early steps of the FeMo-cobiosynthetic pathway, especially at the interface with the path-way for the biosynthesis of more simple [2Fe-2S] and [4Fe-4S]clusters, which involves the activities of the cysteine desulfuraseNifS and the scaffolding protein NifU (13, 14). Pioneeringstudies on nitrogenase maturation by Dean and colleagues(reviewed in ref. 13) uncovered the existence of a complexcellular machinery involved in the assembly of ubiquitous [2Fe-2S] and [4Fe-4S] clusters that, at present, is recognized to be verysimilar for a diversity of Fe-S proteins in all organisms. It isgenerally accepted that the biosynthesis of the complex FeMo-couses common [2Fe-2S] and/or [4Fe-4S] cluster as precursors, butsupport from direct biochemical evidence is currently lacking.
Genetic evidence has revealed the critical function of the nifBgene product for the in vivo accumulation of NifB-co and the VKcluster. The NifB protein has been recently purified and used toactivate apo-NifDK when added to cell-free extracts of a NifB-deficient A. vinelandii mutant strain. This NifB-dependent invitro activation of apo-NifDK required ferrous iron, sulfide,S-adenosyl methionine (SAM), molybdate, sodium dithionite(DTH), Mg-ATP, and R-homocitrate (15). It is still unknownwhether other gene products (in combination with NifB) arerequired for the synthesis of NifB-co.
All currently known proteins that are critical for FeMo-cobiosynthesis are available in purified form. This fact has given usthe unprecedented opportunity to attempt to reconstitute in vitrothe complete FeMo-co biosynthesis pathway. The present studyestablishes a minimal and composition-defined reaction mixtureshowing that FeMo-co can be synthesized in vitro from Fe, S, Mo,
Author contributions: L.C. and L.M.R. designed research; L.C., J.A.H., R.Y.I., B.S., and D.Z.performed research; L.C., J.A.H., and L.M.R. analyzed data; and L.C. and L.M.R. wrote thepaper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Abbreviations: FeMo-co, iron–molybdenum cofactor; NifB-co, NifB cofactor; DTH, sodiumdithionite; SAM, S-adenosyl methionine.
*To whom correspondence should be addressed. E-mail: [email protected].
†NifB-co, the metabolic product of NifB activity, is an isolatable [Fe-S] cluster of unknownstructure that serves as precursor to FeMo-co. NifB-co has also been proposed to be aprecursor of the iron–vanadium and iron-only cofactors of the alternative nitrogenases(29). The VK cluster is a precursor to FeMo-co that accumulates on NifEN in a �nifH mutantstrain and is thought to represent an intermediate after NifB-co in FeMo-co synthesis.
This article contains supporting information online at www.pnas.org/cgi/content/full/0703050104/DC1.
© 2007 by The National Academy of Sciences of the USA
17626–17631 � PNAS � November 6, 2007 � vol. 104 � no. 45 www.pnas.org�cgi�doi�10.1073�pnas.0703050104
and R-homocitrate with only purified NifB, NifEN, and NifHproteins. This demonstration provides direct biochemical evi-dence to confirm the current model for FeMo-co biosynthesisand apo-NifDK activation.
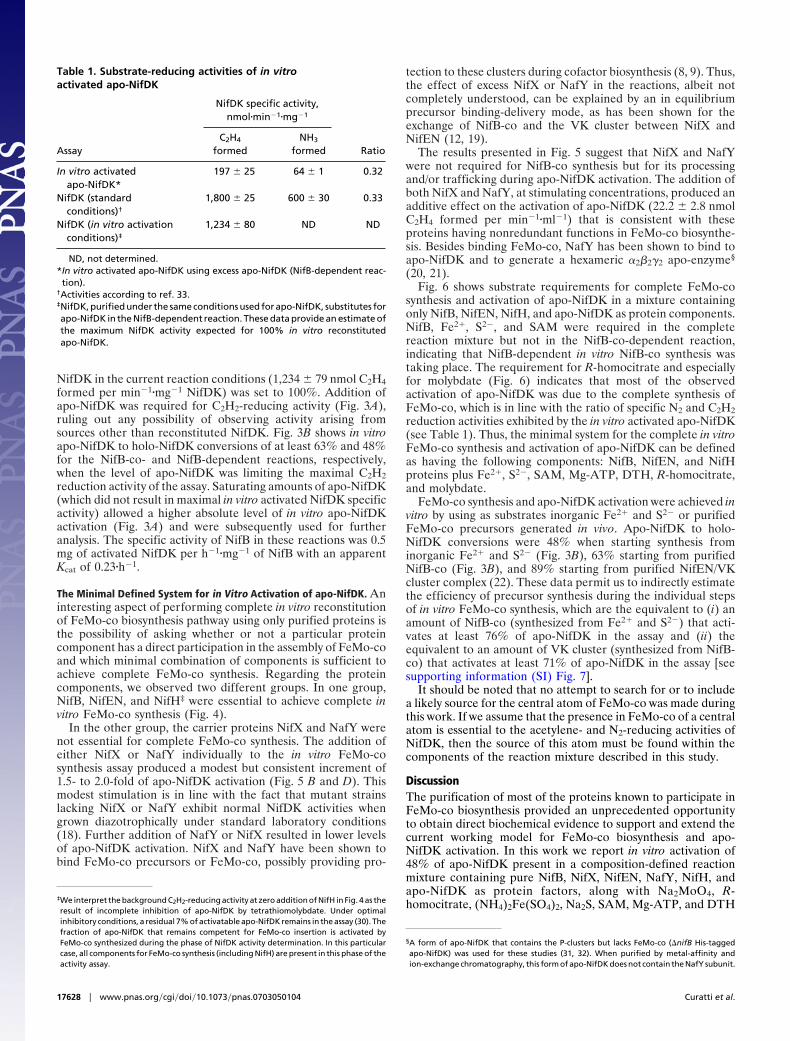
ResultsIn Vitro Activation of apo-NifDK Using Purified Nif Proteins. In theinitial set of experiments, we preincubated NifB with a 50-foldmolar excess of each (NH4)2Fe(SO4)2 (or FeSO4), Na2S, andSAM. Then, NifX, DTH, R-homocitrate, Na2MoO4, Mg-ATP,NifEN, NafY, NifH, and apo-NifDK were sequentially added.After allowing 35 min for apo-NifDK activation, we determinedthe activity of reconstituted NifDK by the acetylene reductionassay. As shown in Fig. 2C, this complete reaction resulted in theNifB- and (Fe2�, S2�, and SAM)-dependent activation of apo-NifDK. Remarkably, NifB, Fe2�, S2�, and SAM completelysubstituted for the requirement of NifB-co, the metabolic prod-uct of NifB activity, indicating that efficient synthesis of thisFeMo-co precursor had been achieved in vitro. It is apparent thatFe2�, S2�, and SAM were spontaneously assembled on NifB tobuild precursors required for NifB-co biosynthesis. This assem-bly might be assisted by NifU and NifS in vivo. It was hypoth-esized that NifB-co would accumulate on the NifB protein in amutant strain having the FeMo-co biosynthetic pathway inter-rupted at the level of NifEN (10). Although it is still possible thatthis is the case in vivo, our data indicate that NifB as purifiedfrom an A. vinelandii �nifHDK-TY-ENX strain does not containbound NifB-co. Seven independent preparations of NifB puri-
fied from a �nifHDK-TY-ENX A. vinelandii strain and twoindependent preparations of NifB purified from A. vinelandiiwild type showed minimal differences in Fe content, specificactivity, requirement of Fe2�, S2, and SAM for activity, andUV-visible spectra (data not shown).
NifDK protein activated in vitro by the NifB-dependent assaysalso presented N2 to NH3 reduction activity, the physiologicalreaction catalyzed by nitrogenase. In vitro activated NifDKexhibited a ratio of specific activities for NH3 production to C2H4production very similar to that of purified wild-type NifDKenzyme (Table 1). The fact that this ratio was remarkablydifferent from that of the alternative nitrogenases, especially theFe-only nitrogenase (ratio of 1.35) (16, 17), strongly supportsthat most of apo-NifDK activation was due to synthesis andinsertion of FeMo-co.
Addition of increasing amounts of apo-NifDK to the reactionmixture was carried out to estimate the percentage of apo-NifDKmaturated in vitro. The activities obtained were compared withthe specific activity of purified wild-type NifDK as measured inthe NifB-dependent reaction conditions. The specific activity of
Fig. 1. Current model for FeMo-co biosynthesis and insertion into apo-NifDK. The model shows in dark blue the core components of the FeMo-cobiosynthesis pathway: NifB, NifEN, and NifH (this work). Simple [Fe-S] clusters(most likely [2Fe-2S] and [4Fe-4S]), which can be chemically reconstituted onNifB, are converted into NifB-co precursor in a reaction that requires SAM andreducing conditions. Presumably, NifS and NifU would be responsible for thesynthesis of those ‘‘NifB-co precursors’’ on NifB in vivo (8, 9). NifB-co is acommon precursor for the biosynthesis of FeMo-co and the cofactors for thealternative nitrogenases, iron–vanadium cofactor and iron-only cofactor.NifEN binds NifB-co and converts it into the VK cluster. Incorporation of Moand R-homocitrate into the VK cluster and final assembly of FeMo-co requireNifH and MgATP. The resulting FeMo-co is inserted into apo-NifDK to activateit. Analogous reactions are hypothesized to occur during the maturation ofalternative nitrogenases. The model shows in light blue the other Nif/Nafproteins known to participate in FeMo-co biosynthesis and insertion intoapo-NifDK. NifS, NifU, NifQ, and NifV provide physiologically relevant formsof Fe, S, Mo, and R-homocitrate. NifX and NafY may assist NifB, NifEN, andNifDK on FeMo-co precursor trafficking among them and may serve as areservoir of precursors giving some buffering capacity to the pathway.
Fig. 2. Purification of A. vinelandii Nif/Naf proteins and in vitro activation ofapo-NifDK with purified Nif proteins. (A) SDS/PAGE analysis of purified NifB,NifEN, NifH, and apo-NifDK proteins on a 12% polyacrylamide gel. (B) SDS/PAGE analysis of purified NifX and NafY on a 14% polyacrylamide gel. (C)NifB-dependent in vitro activation of apo-NifDK. The complete reaction(NifB-dependent assay) contained NifB, NifEN, NifH, NifX, NafY, apo-NifDK,FeSO4, Na2S, SAM, Na2MoO4, R-homocitrate, Mg-ATP, and DTH. In the NifB-co-dependent assay, purified NifB-co substitutes for NifB, FeSO4, Na2S, and SAM.Data are the mean and standard deviation of two to four independent assays.
Curatti et al. PNAS � November 6, 2007 � vol. 104 � no. 45 � 17627
BIO
CHEM
ISTR
Y
NifDK in the current reaction conditions (1,234 � 79 nmol C2H4formed per min�1�mg�1 NifDK) was set to 100%. Addition ofapo-NifDK was required for C2H2-reducing activity (Fig. 3A),ruling out any possibility of observing activity arising fromsources other than reconstituted NifDK. Fig. 3B shows in vitroapo-NifDK to holo-NifDK conversions of at least 63% and 48%for the NifB-co- and NifB-dependent reactions, respectively,when the level of apo-NifDK was limiting the maximal C2H2reduction activity of the assay. Saturating amounts of apo-NifDK(which did not result in maximal in vitro activated NifDK specificactivity) allowed a higher absolute level of in vitro apo-NifDKactivation (Fig. 3A) and were subsequently used for furtheranalysis. The specific activity of NifB in these reactions was 0.5mg of activated NifDK per h�1�mg�1 of NifB with an apparentKcat of 0.23�h�1.
The Minimal Defined System for in Vitro Activation of apo-NifDK. Aninteresting aspect of performing complete in vitro reconstitutionof FeMo-co biosynthesis pathway using only purified proteins isthe possibility of asking whether or not a particular proteincomponent has a direct participation in the assembly of FeMo-coand which minimal combination of components is sufficient toachieve complete FeMo-co synthesis. Regarding the proteincomponents, we observed two different groups. In one group,NifB, NifEN, and NifH‡ were essential to achieve complete invitro FeMo-co synthesis (Fig. 4).
In the other group, the carrier proteins NifX and NafY werenot essential for complete FeMo-co synthesis. The addition ofeither NifX or NafY individually to the in vitro FeMo-cosynthesis assay produced a modest but consistent increment of1.5- to 2.0-fold of apo-NifDK activation (Fig. 5 B and D). Thismodest stimulation is in line with the fact that mutant strainslacking NifX or NafY exhibit normal NifDK activities whengrown diazotrophically under standard laboratory conditions(18). Further addition of NafY or NifX resulted in lower levelsof apo-NifDK activation. NifX and NafY have been shown tobind FeMo-co precursors or FeMo-co, possibly providing pro-
tection to these clusters during cofactor biosynthesis (8, 9). Thus,the effect of excess NifX or NafY in the reactions, albeit notcompletely understood, can be explained by an in equilibriumprecursor binding-delivery mode, as has been shown for theexchange of NifB-co and the VK cluster between NifX andNifEN (12, 19).
The results presented in Fig. 5 suggest that NifX and NafYwere not required for NifB-co synthesis but for its processingand/or trafficking during apo-NifDK activation. The addition ofboth NifX and NafY, at stimulating concentrations, produced anadditive effect on the activation of apo-NifDK (22.2 � 2.8 nmolC2H4 formed per min�1�ml�1) that is consistent with theseproteins having nonredundant functions in FeMo-co biosynthe-sis. Besides binding FeMo-co, NafY has been shown to bind toapo-NifDK and to generate a hexameric �2�2�2 apo-enzyme§
(20, 21).Fig. 6 shows substrate requirements for complete FeMo-co
synthesis and activation of apo-NifDK in a mixture containingonly NifB, NifEN, NifH, and apo-NifDK as protein components.NifB, Fe2�, S2�, and SAM were required in the completereaction mixture but not in the NifB-co-dependent reaction,indicating that NifB-dependent in vitro NifB-co synthesis wastaking place. The requirement for R-homocitrate and especiallyfor molybdate (Fig. 6) indicates that most of the observedactivation of apo-NifDK was due to the complete synthesis ofFeMo-co, which is in line with the ratio of specific N2 and C2H2reduction activities exhibited by the in vitro activated apo-NifDK(see Table 1). Thus, the minimal system for the complete in vitroFeMo-co synthesis and activation of apo-NifDK can be definedas having the following components: NifB, NifEN, and NifHproteins plus Fe2�, S2�, SAM, Mg-ATP, DTH, R-homocitrate,and molybdate.
FeMo-co synthesis and apo-NifDK activation were achieved invitro by using as substrates inorganic Fe2� and S2� or purifiedFeMo-co precursors generated in vivo. Apo-NifDK to holo-NifDK conversions were 48% when starting synthesis frominorganic Fe2� and S2� (Fig. 3B), 63% starting from purifiedNifB-co (Fig. 3B), and 89% starting from purified NifEN/VKcluster complex (22). These data permit us to indirectly estimatethe efficiency of precursor synthesis during the individual stepsof in vitro FeMo-co synthesis, which are the equivalent to (i) anamount of NifB-co (synthesized from Fe2� and S2�) that acti-vates at least 76% of apo-NifDK in the assay and (ii) theequivalent to an amount of VK cluster (synthesized from NifB-co) that activates at least 71% of apo-NifDK in the assay [seesupporting information (SI) Fig. 7].
It should be noted that no attempt to search for or to includea likely source for the central atom of FeMo-co was made duringthis work. If we assume that the presence in FeMo-co of a centralatom is essential to the acetylene- and N2-reducing activities ofNifDK, then the source of this atom must be found within thecomponents of the reaction mixture described in this study.
DiscussionThe purification of most of the proteins known to participate inFeMo-co biosynthesis provided an unprecedented opportunityto obtain direct biochemical evidence to support and extend thecurrent working model for FeMo-co biosynthesis and apo-NifDK activation. In this work we report in vitro activation of48% of apo-NifDK present in a composition-defined reactionmixture containing pure NifB, NifX, NifEN, NafY, NifH, andapo-NifDK as protein factors, along with Na2MoO4, R-homocitrate, (NH4)2Fe(SO4)2, Na2S, SAM, Mg-ATP, and DTH
‡We interpret the background C2H2-reducing activity at zero addition of NifH in Fig. 4 as theresult of incomplete inhibition of apo-NifDK by tetrathiomolybdate. Under optimalinhibitory conditions, a residual 7% of activatable apo-NifDK remains in the assay (30). Thefraction of apo-NifDK that remains competent for FeMo-co insertion is activated byFeMo-co synthesized during the phase of NifDK activity determination. In this particularcase, all components for FeMo-co synthesis (including NifH) are present in this phase of theactivity assay.
§A form of apo-NifDK that contains the P-clusters but lacks FeMo-co (�nifB His-taggedapo-NifDK) was used for these studies (31, 32). When purified by metal-affinity andion-exchange chromatography, this form of apo-NifDK does not contain the NafY subunit.
Table 1. Substrate-reducing activities of in vitroactivated apo-NifDK
NifDK specific activity,nmol�min�1�mg�1
AssayC2H4
formedNH3
formed Ratio
In vitro activatedapo-NifDK*
197 � 25 64 � 1 0.32
NifDK (standardconditions)†
1,800 � 25 600 � 30 0.33
NifDK (in vitro activationconditions)‡
1,234 � 80 ND ND
ND, not determined.*In vitro activated apo-NifDK using excess apo-NifDK (NifB-dependent reac-tion).
†Activities according to ref. 33.‡NifDK, purified under the same conditions used for apo-NifDK, substitutes forapo-NifDK in the NifB-dependent reaction. These data provide an estimate ofthe maximum NifDK activity expected for 100% in vitro reconstitutedapo-NifDK.
17628 � www.pnas.org�cgi�doi�10.1073�pnas.0703050104 Curatti et al.

(Fig. 3). Because no carry-over of in vivo-generated precursorsbound to protein factors was evident through this work, theseresults indicate that all relevant chemistry essential for theassembly of FeMo-co and its insertion into apo-NifDK takesplace among these components.
NifB, NifEN, and NifH are shown to be essential and sufficientfor the conversion of Fe2�, S2�, molybdate, and R-homocitrateinto FeMo-co in a complex reaction that also requires Mg-ATP,SAM, and DTH (Fig. 6). The essentiality of NifB, NifEN, andNifH (compared with the accessory character of NafY and NifX)was not surprising because it matches previous genetic evidenceobtained from mutating the corresponding genes in A. vinelandiiand other diazotrophs (7, 8). However, it is remarkable becauseit shows that the reconstitution in vitro of this complex metabolicpathway roughly mimics the pathway occurring in vivo.
Although the genetic–biochemical approach has been pow-erful in identifying key nif gene products and their arrangementin the pathway of FeMo-co biosynthesis, it did not rule out thepossibility that other nif or non-nif gene products were alsorequired for this trait. This report gives strong support to thecurrent genetic–biochemical model for FeMo-co biosynthesisand indicates that no yet-unidentified protein or group of
proteins has essential direct participation in FeMo-co assemblyin vitro.
The demonstration that NifB, NifEN, and NifH are sufficientfor a significant activation of apo-NifDK from Fe2�, S2�, mo-lybdate, and R-homocitrate (13% activation) strongly suggeststhat within a complex multiprotein machinery for the biosyn-thesis of FeMo-co there is a catalytic core composed of NifB,NifEN, and NifH that might be responsible for all of the criticalbiochemical transformations of Fe, S, Mo, and R-homocitratethat lead to mature FeMo-co (Fig. 1). Other proteins (minimallyNifU, NifS, NifX, NafY, NifQ, and NifV) might provide thephysiological substrates and accessory support to the catalyticcore. Because FeMo-co and its precursors are extremely unsta-ble and sensitive to oxidation (10, 18), the in vitro activation ofapo-NifDK in the absence of carrier proteins suggests that NifB,NifEN, NifH, and apo-NifDK might be in close proximity oreven forming a multiprotein complex during the synthesis ofFeMo-co. There is evidence that the A. vinelandii NifEN proteinforms a stable complex with the L127� variant of NifH (23). Itis also known that the genes encoding NifB and NifN are fusedin a single ORF in several nitrogen-fixing Clostridium strains(24). Although these observations point to the existence of amultiprotein complex composed of NifB, NifEN, and NifH,copurification experiments performed during the course of thiswork have not provided any evidence of its existence (data notshown).
The proposal of a minimal catalytic core for the assembly ofFeMo-co does not underestimate the function of the accessoryproteins. As discussed above, an efficiency of 48% in the in vitroconversion of apo-NifDK to NifDK indicates that no criticalfactor is missing. However, the estimated apparent Kcat forNifB-dependent in vitro activation of apo-NifDK (0.23�h�1) isunlikely to reflect the in vivo efficiency of the pathway. Thisstimulates new research to integrate more accessory proteinsinto the in vitro reconstituted pathway. As demonstrated here forNifX and NafY, the in vitro reconstitution of the catalytic coreof the pathway represents a platform onto which the activity ofother Nif proteins can be analyzed in vitro as a complementaryand powerful tool to confirm and refine current knowledge onFeMo-co biosynthesis.
It is expected that the availability of a minimal definedreaction for the in vitro synthesis of FeMo-co from Fe2�, S2�,
Fig. 3. Effect of apo-NifDK concentration on the specific activity of recon-stituted NifDK. Reactions were set essentially as those in Fig. 2C, but theconcentration of apo-NifDK present in the assay varied from 0 to 2.4 �M. Œ,NifB-dependent reactions; ■ , NifB-co-dependent reactions. (A) Plot of C2H2
reduction activity versus apo-NifDK concentration in the assay. (B) Specificactivity of in vitro activated NifDK versus apo-NifDK concentration in theassay, as calculated from data in A. The percentage of apo-NifDK activated inthe assay is indicated on the right axis. The specific activity of purified NifDKdetermined under the same reaction conditions (1,234 nmol C2H4 formed permin�1�mg�1) represents 100% activation.
Fig. 4. NifB, NifEN, and NifH are essential for in vitro activation of apo-NifDK. Shown is the effect of NifB, NifEN, or NifH concentration on in vitroapo-NifDK activation. The NifB-dependent assays were conducted in theabsence of NifX and NafY (see Experimental Procedures) with the followingmodifications: in A the concentration of NifB in the assay was increased from0 to 11 �M, in B the concentration of NifEN in the assay was increased from 0to 4.5 �M, and in C the concentration of NifH in the assay was increased from0 to 7.5 �M. In C, apo-NifDK activation was terminated by addition of 0.3 mM(NH4)2MoS4 before the addition of excess NifH for the determination of NifDKactivity. The scale of activity axes in B and C are the same. Representativeexperiments are shown.
Curatti et al. PNAS � November 6, 2007 � vol. 104 � no. 45 � 17629
BIO
CHEM
ISTR
Y

Mo, and R-homocitrate would enormously facilitate and focusfuture research to advance in the structural and mechanisticaspects of the pathway, especially those involved in the earlysteps of FeMo-co assembly, which are shared by the alternativenitrogenase cofactor biosynthetic pathways.
Recent progress has been made in understanding the assembly ofanother heavily studied complex [Fe-S] cluster, the H-clusterlocated at the active site of the [FeFe] hydrogenase. In vitrobiochemical complementation of Escherichia coli cell-free extractsexpressing either apo-HydA or the accessory proteins HydE, HydF,and HydG led to reconstitution of hydrogenase activity (25).Despite the structural differences between FeMo-co and the H-cluster (26), it is apparent that their biosynthetic pathways sharesome common features, including the participation of specific SAMradical proteins and nucleotide-hydrolyzing enzymes (27). Thus,new insights in one of the pathways might help advancing in theother as well. For the longer term, understanding how complex[Fe-S] are biochemically assembled not only will complement theknowledge on the ecologically and geochemically relevant reactionsthat are catalyzed by these cofactors, but also has the potential toconstitute the basis for the bio-inspired design of potent catalyst forthe industrial production of H2 and other commodities.
Experimental ProceduresIn Vitro FeMo-co Synthesis and Activation of apo-NifDK. NifB, NifEN,NifH, apo-NifDK, NifX, and NafY protein preparations were atleast 95% pure as analyzed by SDS/PAGE (Fig. 2 A and B). In thein vitro FeMo-co biosynthesis system, the addition of R-homocitrateand Na2MoO4 substitutes for NifV and NifQ activities, respectively(10). Thus, the requirements for NifQ and NifV were not tested inthis work.
The complete NifB-dependent FeMo-co synthesis and apo-NifDK activation assay contained 3.0 �M NifB, 3.0 �M NifX, 1.5�M NifEN, 1.2 �M NafY, 3.0 �M NifH, and 0.6 �M apo-NifDKin 22 mM Tris�HCl buffer (pH 7.5), 3.5% glycerol, 7.5 �MNa2MoO4, 0.175 mM R-homocitrate, 0.125 mM FeSO4, 0.125mM Na2S, 0.125 mM SAM, 3 mM Na2S2O4, and an ATP-regenerating mixture (1.23 mM ATP, 18 mM phosphocreatine,2.2 mM MgCl2, 3 mM Na2S2O4, and 40 �g of creatine phos-phokinase). For the experiments shown in Figs. 4 A and C and5 B and D, 5 mg�ml�1 BSA, 0.42 mM (NH4)2Fe(SO4)2, 0.42 mMNa2S, and 0.88 mM SAM were used with no observable changein apo-NifDK activation.
The NifB-co-dependent FeMo-co synthesis and apo-NifDK ac-tivation assay contained NifB-co (8.5 �M Fe), 1.5 �M NifEN, 3.0�M NifH, and 0.6 �M apo-NifDK in 22 mM Tris�HCl buffer (pH7.5), 3.5% glycerol, 7.5 �M Na2MoO4, 0.175 mM R-homocitrate, 3mM Na2S2O4, and an ATP-regenerating mixture (1.23 mM ATP,18 mM phosphocreatine, 2.2 mM MgCl2, and 40 �g of creatinephosphokinase). For the experiments shown in Fig. 5 A and C, 5mg�ml�1 BSA was included in the reaction mixture.
Reactions were carried out at two different scales in totalvolumes of 200 �l or 600 �l inside 3.6-ml or 9-ml stoppered vials,respectively, for 35 min at 30°C under an argon atmosphere toallow for the FeMo-co synthesis and insertion reactions. Exten-sive vacuum and gas exchange was performed to saturate themixture with either argon or nitrogen. No difference in activa-tion was observed in either case. The resulting activation ofapo-NifDK present in the reaction mixtures was routinely ana-lyzed by the acetylene reduction assay after addition of excessNifH and ATP-regenerating mixture (28).
Details. Additional methods are detailed in SI ExperimentalProcedures.
Fig. 5. Effect of NifX and NafY on the in vitro activation of apo-NifDK. Shownis the effect of NifX (A and B) or NafY (C and D) in the NifB-co-dependent (Aand C) or in the NifB-dependent (B and D) reactions. The NifB-co-dependentor NifB-dependent assays were supplemented with 0 to 30 �M NifX (A and B)or 0 to 30 �M NafY (C and D). A representative experiment is shown for eachtitration experiment.
Fig. 6. Substrate requirements for in vitro activation of apo-NifDK. FeMo-cosynthesis and apo-NifDK activation reactions (containing NifB, NifEN, NifH,and apo-NifDK as protein components) were conducted in the absence of oneor more substrates at a time. A reaction containing all substrates (completereaction) and a NifB-co-dependent reaction for FeMo-co synthesis and apo-NifDK activation were carried out as controls. Acid-washed vials were used toshow the Mo dependency of the reaction. Data are the average and standarddeviation of two to four independent determinations.
17630 � www.pnas.org�cgi�doi�10.1073�pnas.0703050104 Curatti et al.

We thank Susan O’Connor for excellent technical assistance; MaryWildermuth and Marc Strawn for assistance with high-performanceliquid chromatography; Dennis Dean (Virginia Tech, Blacksburg, VA)for providing A. vinelandii strains DJ884, DJ995, and DJ1143; Juan
Imperial for stimulating discussions; and Paul Ludden for discussionsand support. This work was supported by National Institute of GeneralMedical Sciences/National Institutes of Health Grant GM-35332 (to PaulLudden).
1. Falkowski PG (1997) Nature 387:272–275.2. Oldroyd GE (2007) Science 315:52–53.3. Igarashi RY, Seefeldt LC (2003) Crit Rev Biochem Mol Biol 38:351–384.4. Sakurai H, Masukawa H (2007) Mar Biotechnol 9:128–145.5. Chan MK, Kim J, Rees DC (1993) Science 260:792–794.6. Einsle O, Tezcan FA, Andrade SL, Schmid B, Yoshida M, Howard JB, Rees
DC (2002) Science 297:1696–1700.7. Ludden PW, Rangaraj P, Rubio LM (2004) in Catalysts for Nitrogen Fixation:
Nitrogenases, Relevant Chemical Models, and Commercial Processes, eds SmithBE, Richards RL, Newton WE (Kluwer Academic, Dordrecht, The Nether-lands), pp 219–253.
8. Dos Santos PC, Dean DR, Hu Y, Ribbe MW (2004) Chem Rev 104:1159–1174.9. Rubio LM, Ludden PW (2005) J Bacteriol 187:405–414.
10. Shah VK, Allen JR, Spangler NJ, Ludden PW (1994) J Biol Chem 269:1154–1158.
11. Hu Y, Fay AW, Ribbe MW (2005) Proc Natl Acad Sci USA 102:3236–3241.12. Hernandez JA, Igarashi RY, Soboh B, Curatti L, Dean DR, Ludden PW, Rubio
LM (2007) Mol Microbiol 63:177–192.13. Johnson DC, Dean DR, Smith AD, Johnson MK (2005) Annu Rev Biochem
74:247–281.14. Lill R, Muhlenhoff U (2006) Annu Rev Cell Dev Biol 22:457–486.15. Curatti L, Ludden PW, Rubio LM (2006) Proc Natl Acad Sci USA 103:5297–
5301.16. Schneider K, Gollan U, Drottboom M, Selsemeier-Voigt S, Muller A (1997)
Eur J Biochem 244:789–800.17. Chisnell JR, Premakumar R, Bishop PE (1988) J Bacteriol 170:27–33.
18. Shah VK, Brill WJ (1977) Proc Natl Acad Sci USA 74:3249–3253.19. Shah VK, Rangaraj P, Chatterjee R, Allen RM, Roll JT, Roberts GP, Ludden
PW (1999) J Bacteriol 181:2797–2801.20. Paustian TD, Shah VK, Roberts GP (1990) Biochemistry 29:3515–3522.21. Rubio LM, Rangaraj P, Homer MJ, Roberts GP, Ludden PW (2002) J Biol
Chem 277:14299–14305.22. Soboh B, Igarashi RY, Hernandez JA, Rubio LM (2006) J Biol Chem
281:36701–36709.23. Rangaraj P, Ryle MJ, Lanzilotta WN, Goodwin PJ, Dean DR, Shah VK,
Ludden PW (1999) J Biol Chem 274:29413–29419.24. Chen JS, Toth J, Kasap M (2001) J Ind Microbiol Biotechnol 27:281–286.25. McGlynn SE, Ruebush SS, Naumov A, Nagy LE, Dubini A, King PW,
Broderick JB, Posewitz MC, Peters JW (2007) J Biol Inorg Chem 12:443–447.26. Drennan CL, Peters JW (2003) Curr Opin Struct Biol 13:220–226.27. Posewitz MC, King PW, Smolinski SL, Zhang L, Seibert M, Ghirardi ML
(2004) J Biol Chem 279:25711–25720.28. Shah VK, Brill WJ (1973) Biochim Biophys Acta 305:445–454.29. Bishop PE, Joerger RD (1990) Annu Rev Plant Physiol Plant Mol Biol
41:109–125.30. Shah VK, Ugalde RA, Imperial J, Brill WJ (1985) J Biol Chem 260:3891–3894.31. Christiansen J, Goodwin PJ, Lanzilotta WN, Seefeldt LC, Dean DR (1998)
Biochemistry 37:12611–12623.32. Schmid B, Ribbe MW, Einsle O, Yoshida M, Thomas LM, Dean DR, Rees DC,
Burgess BK (2002) Science 296:352–356.33. Barney BM, Igarashi RY, Dos Santos PC, Dean DR, Seefeldt LC (2004) J Biol
Chem 279:53621–53624.
Curatti et al. PNAS � November 6, 2007 � vol. 104 � no. 45 � 17631
BIO
CHEM
ISTR
Y


















