
IMWG updates on plasma cell dyscrasias
77
PLASMA CELL DYSCRASIAS BY EKTA JAJODIA
-
Upload
ekta-taparia -
Category
Health & Medicine
-
view
169 -
download
0
Transcript of IMWG updates on plasma cell dyscrasias

PLASMA CELL DYSCRASIAS
BY EKTA JAJODIA

B-CELL DEVELOPMENT
• B lymphocytes arise from lymphoid stem cells in bone marrow
• Initial development occurs in primary lymphoid organ (BM) from where B cells migrate to the secondary lymphoid organs (LN & spleen) and further differentiation occurs on antigenic stimulation
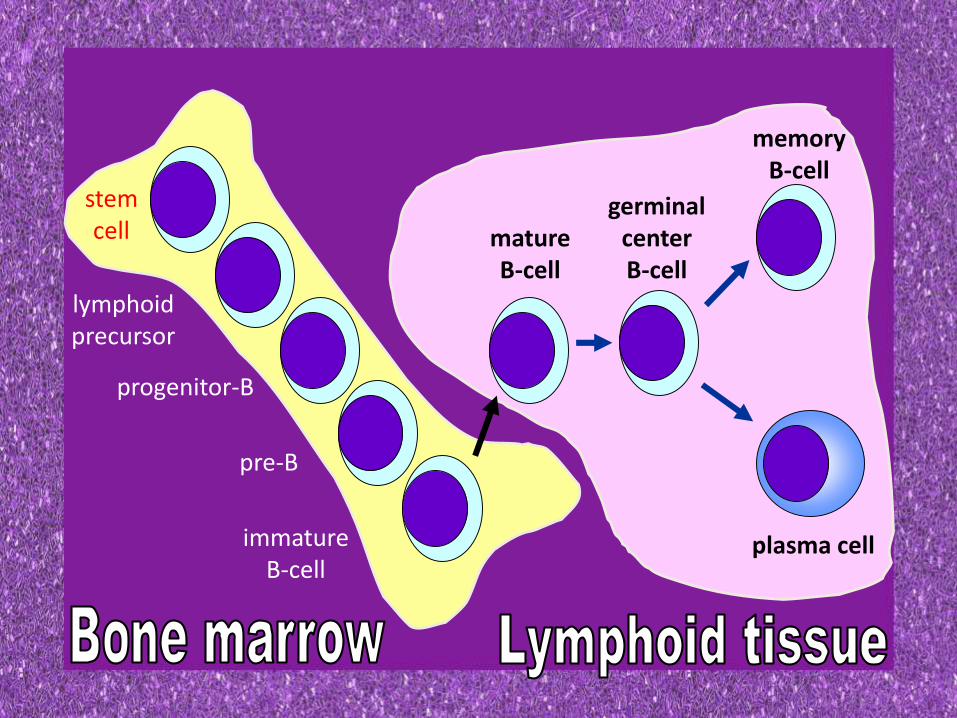
stemcell
lymphoidprecursor
progenitor-B
pre-B
immatureB-cell
matureB-cell
germinalcenterB-cell
memoryB-cell
plasma cell
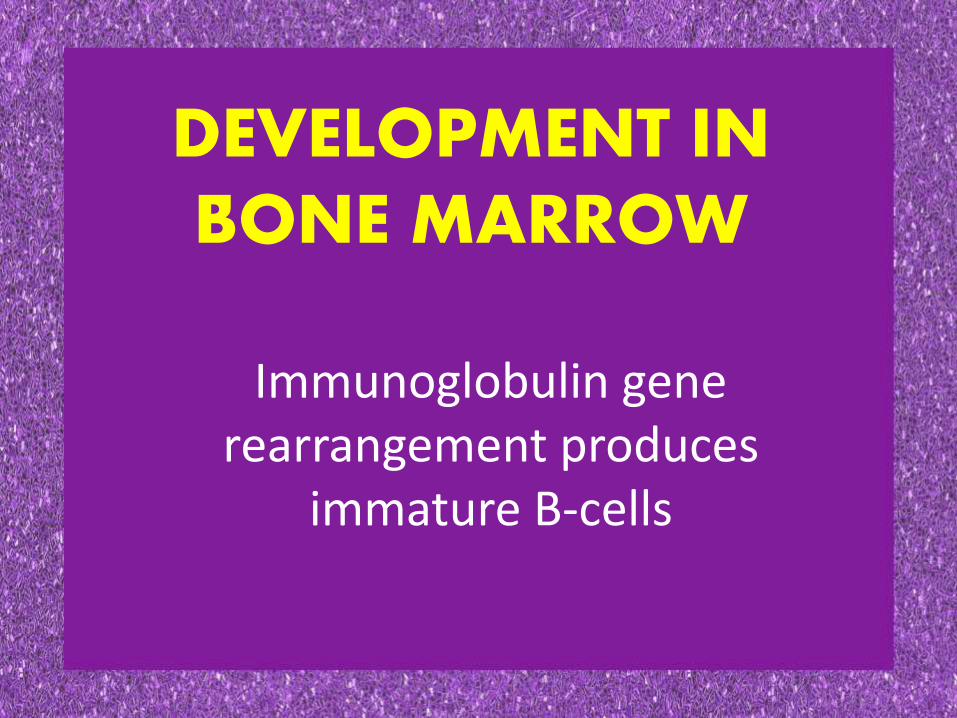
DEVELOPMENT IN BONE MARROW
Immunoglobulin gene rearrangement produces
immature B-cells

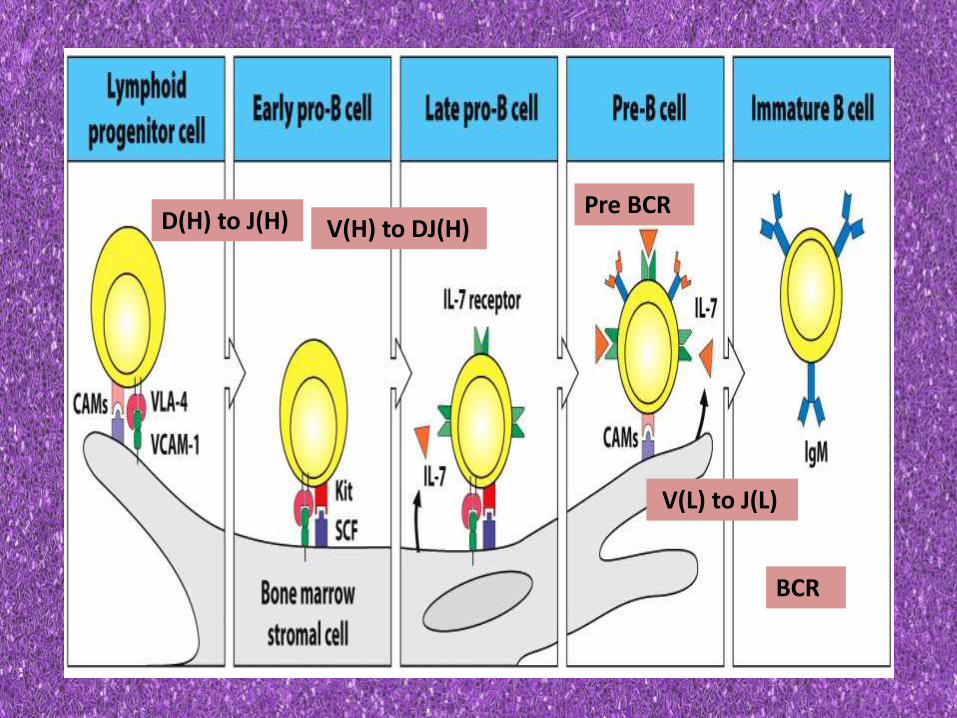
D(H) to J(H) V(H) to DJ(H)Pre BCR
V(L) to J(L)
BCR

ENZYMES REQUIRED
• TdT- (terminal deoxyribonucleotidyltransferase)
• An enzyme that catalyses heavy chain rearrangement • So active in pro-B cell and ceases to be active in pre b cell stage

• RAG-1 and RAG2 –
• An enzyme reqd for both heavy chain and light chain rearrangement
• So active in both pro B ell and pre b cell stage

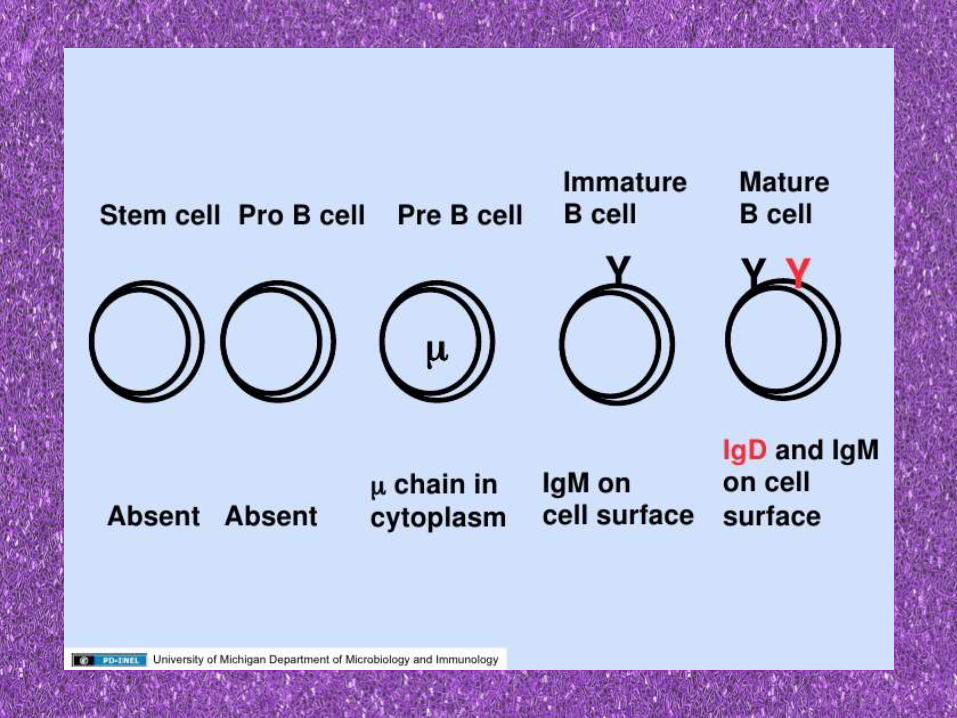
• BM phase of B cell development ends in production of an IgM bearing immature B cells

PLASMA CELL DIFFERENTIATION
Immature B cell leave BM and migrate to secondary lymphoid organs (spleen ,LN)
IgD appears on the surface
Known as naïve/virgin B cells

Encounter an antigen
Memory B cellsPlasmablasts
Plasma cells



In the spleen – plasma cells reside in red pulp
In the lymph node plasma cells reside in the medullary cords

ROLE OF BM MICROENVIRONMENT• Most important is BM stromal cells (BMSC)
• Data indicate that plasma cells cultured in presence of BMSC – survived and continued to secrete antibody for at least 3 weeks
• When cultured withour BMSC – Abs were detected only for 7 days
• This suggest that BMSC and its secreted factors are reqd for Plasma cell survival and longevity

When plasma cell VLA-4 binds to an unknown ligand on BMSC (not VCAM-1)
IL-6 mRNA is upregulated in BMSC
So increased IL-6

• IL-6 is critical for Plasma cell survival and longevity• Produced by BMSC
• Not produced by non-malignant plasma cells
• May be produced by myeloma/ malignant plasma cells

• B cell precursors have CXCR4 receptor on their surface
Binds with CXCL12 on the bone marrow stromalcells (BMSC)
This binding is responsible for retaining precursor B cells in the BM

But the immature B cells lose expression of CXCR4
Therefore they are no longer retained in BM and migrate to the secondary lymphoid organs
• Plasma cells also exp CXCR4 which interacts with BMSC CXCL12 •This is responsible for Plasma cell survival and localisation in BM


MOLECULAR AND CELLULAR EVENTS OF
PLASMA CELL DIFFERENTIATION

1. Blimp-1 (B lymphocyte induced maturation protein 1)
• Is a master regulator of plasma cell differentiation
• Cytokine activation of BCL1 – induces Blimp1 expression
• Expression of blimp- 1 results in concomitant repression of BCL6 , Pax5, c-myc, CIITA and BCR signalling markers – Spi-B and Id3
• Conversely Blimp-1 upregulates J-chain exp ( and thus Ig synthesis) and syndecan-1 protein synthesis


2. XBP-1 (X- box binding protein)
• A transcription factor essential for plasma cell differentiation
• XBP-1 is sufficient to induce generation of plasma cells
• Absence of XBP-1 does not affect Blimp-1 exp

3. SYNDECAN-1 (CD138)
• Plasma cells are best distinguished from other population by their membrane exp of syndecan-1
• Syndecan-1 binds to fibronectin, collagen and basic fibroblast growth factor
• So reqd for contact with BMSC

•Plasma cells are committed to synthesis of antibody• Their cell surface phenotype differs greatly from memory B-cells

MORPHOLOGY
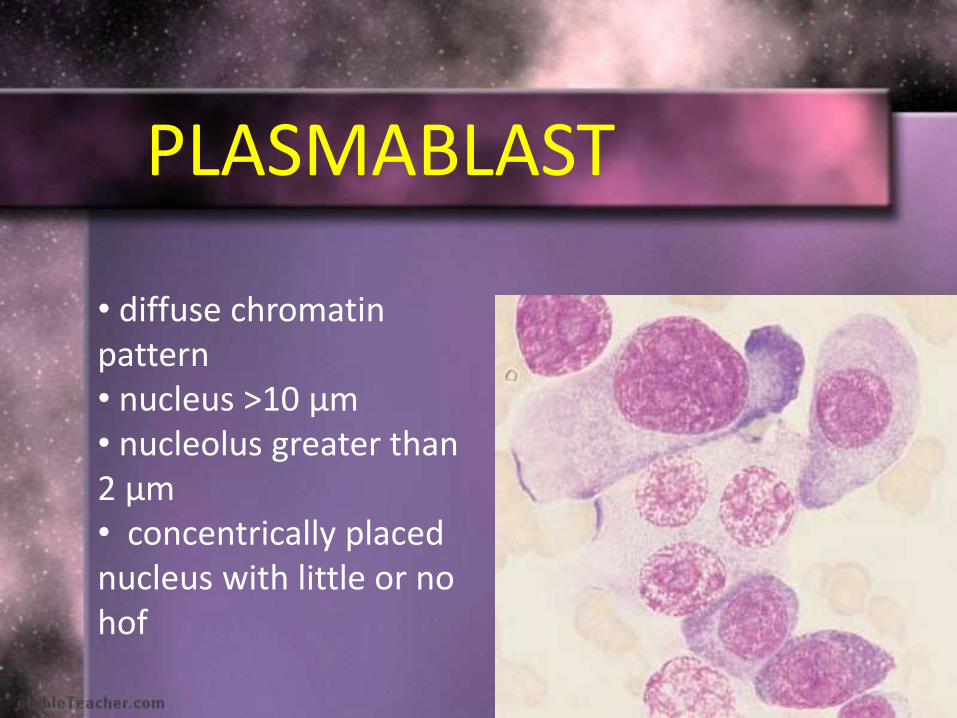
PLASMABLAST
• diffuse chromatin pattern• nucleus >10 μm • nucleolus greater than 2 μm• concentrically placed nucleus with little or no hof
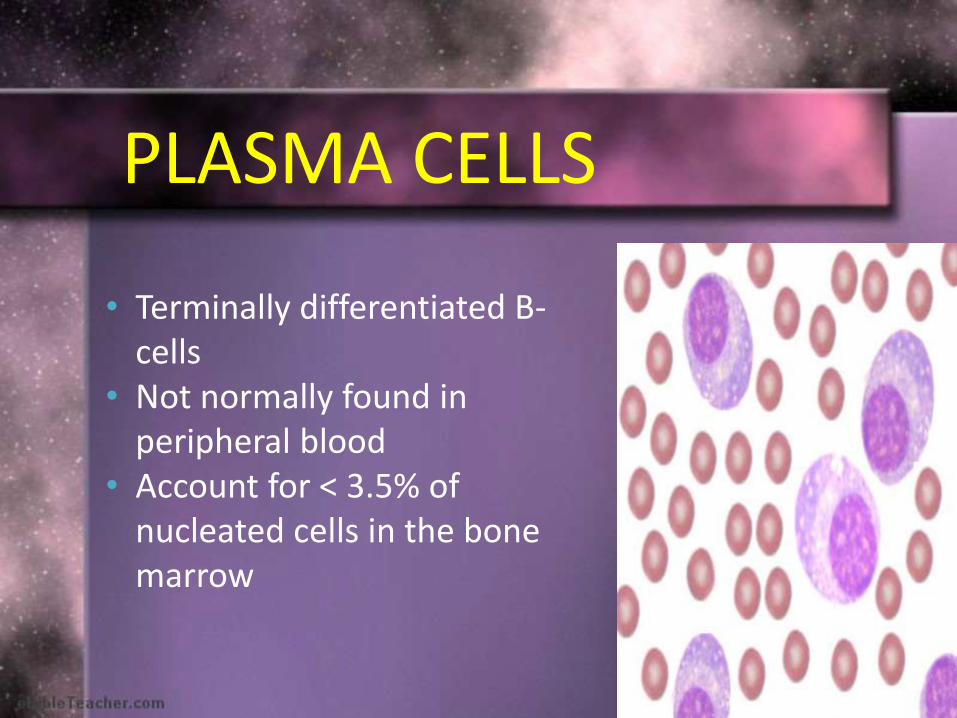
• Terminally differentiated B-cells
• Not normally found in peripheral blood
• Account for < 3.5% of nucleated cells in the bone marrow
PLASMA CELLS

• Oval cells with cart-wheel nucleus
• Cytoplasm is basophilic blue
• Nucleus is oval or round and typically placed eccentrically
• A hof is present adjacent to the nucleus contains Golgi apparatus

• Globules (2-3 μm) of accumulated immunoglobulins in the cytoplasm of plasma cells• Usually round• May be found in normal bone marrow• 1st described by William Russell
RUSSELL BODIES

MOTT CELLS/MORULA CELLS
• Plasma cells crowded with Russell bodies
• An obstruction blocks the release of Golgi secretions
• These cells can be found in any case of chronic plasmacytosis

FLAME CELLS
• Eosinophilic torn cytoplasm
•Usually associated with IgA myeloma
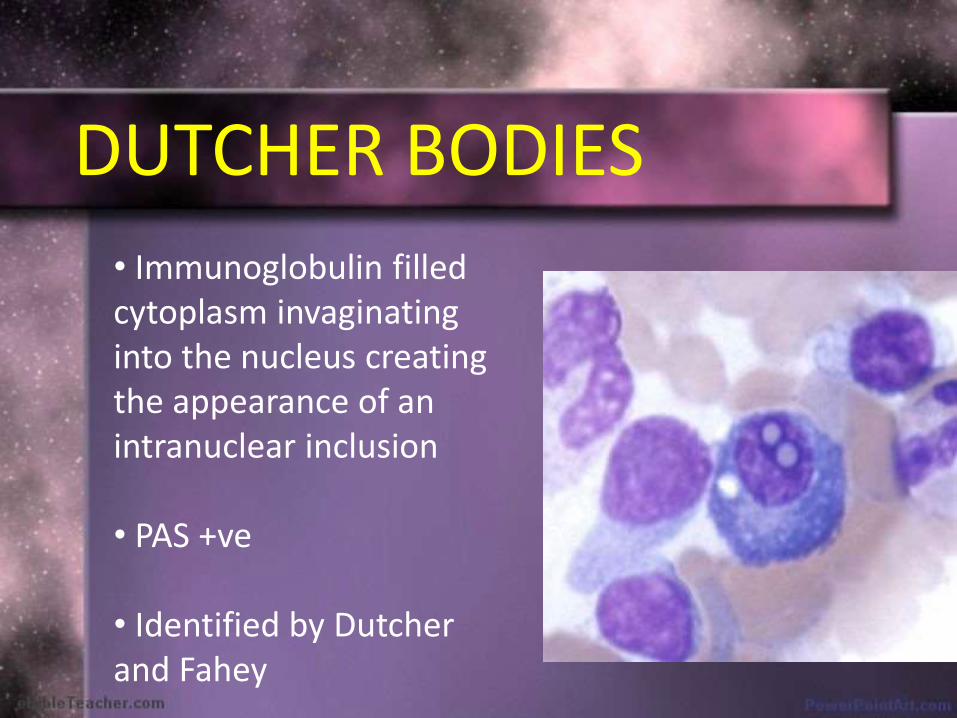
DUTCHER BODIES
• Immunoglobulin filled cytoplasm invaginatinginto the nucleus creating the appearance of an intranuclear inclusion
• PAS +ve
• Identified by Dutcherand Fahey

RHOMBOID CRYSTALLINE INCLUSIONS
intracellular extracellular IHC – lambda chain

PLASMA CELL DYSCRASIA
• Plasma cell dyscrasias are disorders of the plasma cells
• They are produced as a result of abnormal proliferation of a monoclonal population of plasma cells that may or may not secrete detectable levels of a monoclonal immunoglobulin or immunoglobulin fragment (paraprotein or M protein)

CLASSIFICATION
1. Monoclonal gammopathy of undetermined significance (MGUS)
2. Plasma cell myeloma Variants-a. Asymptomatic(smoldering myeloma)b. Non-secretory myelomac. Plasma cell leukemia
3. Plasmacytomaa. Solitary plasmacytoma of boneb. Extraosseous(extramedullary) plasmacytoma

4. Immunoglobulin deposition diseasesa. Primary amyloidosisb. Systemic light and heavy chain deposition diseases
5. Osteosclerotic myeloma (POEMS syndrome)

OTHER IMMUNOGLOBULIN SECRETING NEOPLASMS
1. Lymphoplasmacytic lymphoma/ Waldenstrommacroglobulinemia
2. Heavy chain diseases
• These are classified separately under mature B-cell neoplasms

M-component can also be detected in –
1. LYMPHOID NEOPLASMS – CLL, some T and B cell neoplasms
2. NON-LYMPHOID NEOPLASMS – CML, Breast Ca, Colon Ca
3. NON NEOPLASTIC CONDITIONS – Cirrhosis, sarcoidosis, gaucher’s disease, parasitic infection
4. AUTOIMMUNE DISEASES – RA, Myasthenia gravis, Cold agglutinin disease
5. SKIN DISEASES – Lichen myxedematous, necrobiotic xanthogranuloma

MGUS
• Monoclonal expansion of Ig secreting plasma cells but non- neoplastic
• Preneoplastic condition
• 0.5-1% / year of MGUS progress to Multiple myeloma (MM)

Non IgM MGUS
Light chain MGUS
IgM MGUS
• 80% of MM originates from this• Primary amyloidosis
• 2% of MM originates from this
• This usually evolves into waldenstrom’s or lymphoplasmacytic lymphomaRarely progress to MM
MGUS

DIAGNOSTIC CRITERIA
Non IgM MGUS / IgM MGUS –
1.Serum monoclonal protein < 30g/L
2. BM clonal plasma cells / lymphoplasmacytic cells < 10%
3. Absence of CRAB or amyloidosis

Light chain MGUS –
1. Urinary monoclonal protein <500mg/24hrs2. BM plasma cells <10%3. Abnormal FLC ratio - >1.65 (in case of increased
kappa light chain)< 0.26 (in case of
increased lambda light chain )4. No Ig heavy chain expresion on immunofixation5. Absence of CRAB or amyloidosis

1) Size and type of M –protein- M-protein of >25 g/l -IgM or IgA MGUS- greater risk of progression
2) Detection of DNA aneuploidy and subnormal levels of polyclonal Igs
Risk factors for progression of MGUS to plasma cell myeloma

• BM aspirate shows median of 3% plasma cells
• Trephine bx- no or minimal increase of plasma cells
• Plasma cells are mature looking however occasional cell may have cytoplasmic inclusion or prominent nucleoli
MORPHOLOGY

IMMUNOPHENOTYPING
2 population of plasma cells are seen
CD38 bright +,CD19+ and CD56 -
This is polyclonal with normal immunophenotype
• CD19-/CD56-• CD19-/CD56+• Weak CD38+
This is monoclonal population

PLASMACYTOMA
CLASSIFICATION-
1.Solitary plasmacytoma of bone2.Extraosseous (extramedullary)
plasmacytoma

1. Single area of bone/soft tissue destruction due to clonal plasma cells
2. Normal BM biopsy without clonal disease- ruled out by B/L iliac crest BM biopsy
3. Normal results on radiologic skeletal survey & MRI of spine, pelvis, proximal femur & humerus (except for the primary solitary lesion)
CRITERIA FOR DIAGNOSIS

4. No anemia, hypercalcemia or renal impairment attributable to myeloma
5. No or low serum or urinary level of monoclonal protein & preserved levels of uninvolved immunoglobulins (paraprotein may be seen in 50% cases but usually <2g/dl)

• Solitary plasmacytoma with minimal marrow involvement –1. One solitary bone lesion2. BM clonal plasma cells <10%
• If BM clonal plasma cells <10% with >1 bone lesion – MM
• If BM clonal plasma cells >10% with solitary plasmacytoma - MM

• Long & flat bones, axial skeleton, vertebra
• Dense homogenous PC infiltrate with varying dysplasia- irregular nuclei with dispersed chromatin, prominent nucleoli & multinucleation
• 25% cases may show amyloid deposition & foreign body reaction
SOLITARY PLASMACYTOMA OF BONE

Types:
Plasmacytic (WD)- clumped chromatin & little cytoplasmic dysplasia. Russel & dutcher bodies +ve
Plasmablastic (MD)- dispersed chromatin, prominent nucleoli & amphophilic cytoplasm
Anaplastic (PD)- nuclear immaturity & dysplasia, mitosis +ve

• Kappa or lambda light chain restriction
• Non monoclonal Ig usually normal with IgGpreponderance in bone & IgA in extramedullary
• 20% cases show B-cell markers CD 79a, CD 19,
CD 20
IMMUNOHISTOCHEMISTRY

Adverse prognostic factors
• Persistance of monoclonal protein > 1 yr after Tx• Predominance of non-IgG class immunoglobulin• Older patients• Solitary plasamcytoma >5cm
•They have a higher incidence of progression to MM• Progression to MM is more in solitary plasmacytoma of bone compared to extraosseousplasmacytoma

• Localized plasma cell neoplasms that arise in tissues other than bone
• 80% Extra osseous plasmacytomas occur in URT
• 20% of patients have a small M-protein, most commonly IgA
EXTRAOSSEOUS PLASMACYTOMA

IMMUNOGLOBULIN DEPOSITION DISEASES
1.Primary amyloidosis
2.Systemic light and heavy chain deposition disease

• Monoclonal plasma cell proliferative disorder associated with deposition of mostly light chain(usually lambda) or rarely heavy chain fragments leading to organ dysfunction
• About 10 % of patient with amyloidosis may also have MM
• Overtime a small number of patient with MGUS may eventually develop amyloidosis
PRIMARY AMYLOIDOSIS

DIAGNOSIS
• Demonstration of amyloid in abdominal subcutaneous fat pad ,bone marrow or rectum
• Grossly – waxy or porcelain like• In H&E – pink amorphous waxy appearing
substance with a characteristic cracking artifact
• Congo red stain- by light microscopy – red to pink• Under polarised light – apple green birefringence

Systemic light and heavy chain deposition diseases
CLASSIFICATION -
1.Light chain deposition disease2.Heavy chain deposition disease3.Light and heavy chain deposition disease

• Secrete abnormal light chains or heavy chains or both which deposit in tissues causing organ dysfunction • Most commonly involves – kidneys• Extrarenal deposition rarely may occur in – heart , liver and Peripheral nervous system• In LCDD – Primary defect – multiple mutations in Ig light chain variable region• In HCDD – primary defect – deletion of CH1 constant domain – causes failure to associate with heavy chain binding protein – results in premature secretion

•Do not form amyloid B-pleated sheets, bind Congo red or contain amyloid P-component
• LCD usually involves kappa light chains ; amyloidosis usually involves lambda light chains
•lambda LCD has a three times worse prognosis than kappa LCD

Morphology – tissue deposits of non-amyloid, non- fibrillary amorphous refractileeosinophilic material in glomerular and tubular basement membrane
•IF – Prominent smooth linear peritubulardeposits along the outer edge of tubular BM of mostly kappa light chains
• EM – Dense granular non-fibrillary deposits

OSTEOSCLEROTIC MYELOMA(POEMS SYNDROME)
CRITERIA FOR DIAGNOSIS
1. Polyneuropathy2. Monoclonal plasma cells (almost always
lambda)3. Any one of the 3 major criteria-
a. Sclerotic bone lesionsb. Castleman’s diseasec. Elevated levels of VEGF

4. Any one of the following 6 minor criteria-
a. Organomegaly (splenomegaly, hepatomegaly, lymphadenopathy)
b. Extravascular volume overload (edema, pleural effusion, ascitis)
c. Endocrinopathyd. Skin changes (hyperpigmentation,
hypertrichosis, glomeruloid hemangiomata, plethora)
e. Papilledemaf. Thrombocytosis/polycythemia

HEAVY CHAIN DISEASE (HCD)
1.Gamma HCD2.Mu HCD3.Alpha HCD
Patients secrete a defective heavy chain immunoglobin

1. Gamma HCD –
• aka franklin’s disease• Palatal edema (due to involvement of
waldeyer’s ring)• For diagnosis – Serum M component of IgG
type < 2g/dl• M- component present in both urine and serum
– usually IgG1 type• Truncated gamma heavy chain is produced
which lacks light chain binding sites – so cannot form a complete immunoglobulin molecule

Mu HCD –
• Produces a defective Mu heavy chain that lacks a variable region
• resembles CLL• High frequency of hepatosplenomegaly and
absence of peripheral lymphadenopathy – these features distinguish this from CLL
• Presence of vacuoles in malignant lymphocytes
• Excretion of Kappa light chains in urine

Alpha HCD –• aka seligmann’s disease/ mediterranean
lymphoma• MC HCD• Also known as IPSID ( immunoproliferative small
intestinal disease) – results in malabsorption and diarrhoea
• Lymphoplasmacytoid infiltration in the lamina propria of small intestine that secretes truncated alpha chains
• Campylobacter jejuni causative organism• Sometimes reversible by antibiotics but may
progress to DLBCL

LYMPHOPLASMACYTIC LYMPHOMA (LPL)
• Neoplasm of small B lymphocytes, plasmacytoidlymphocytes and plasma cells• Does not fulfill criteria for any other small B cell lymphoid neoplasms that may have plasmacyticdifferentiation
• WALDENSTROM MACROGLOBULINEMIA – Found in a subset of LPL and is defined as LPL with BM involvement and an IgM monoclonal component of any concentration

• Serum M component – usually 3 g/dl• Cryoglobulins present – these are Pure M components ( not mixed cryoglobulins which is seen in RA that c/o IgM/IgA complexed with IgG
• May sometimes be asociated with HCV
• Hyperviscosity syndrome (15%) : visual impairment, neurologic manifestations
• In WM – there may be loss of MAG(myelin associated glycoprotein) – a protein associated with demyelinating disease of PNS – so WM patients may develop peripheral neuropathy

What is KAHLER’S disease
• AKA Multiple myeloma
• Kahler’s disease is named after an Austrian doctor called Otto Kahler who first investigated and described MM

What is Randall disease
Monoclonal light and heavy chain deposition disease

What is Crow-Fukasesyndrome
Aka POEMS syndrome
1st described by Dr R.S Crow in 1956 and then By Dr M.Fukase in 1968

TO BE CONTINUED….


















