
Hypokalemic Paralysis as First Manifestation of Sjögren Syndrome ...
Upload
laxmikant-joshiCategory
view
453download
1
HYPERKALEMIC PP VS HYPOKALEMIC PERODIC PARALYSIS

OUTLINE
• Causes Of Primary Periodic paralysis
• Causes Of Secondary Periodic paralysis
• PRESENTATION OF HYPERKALEMIC PP
• COMPARISON BETWEEN Hyper PP Vs Hypo PP

• Primary Periodic paralysis
• Secondary Periodic paralysis
Causes of Primary Periodic paralysis
– Hypokalemic (CACNA1S/ SCN4A)
– Hyperkalemic (SCN4A)
– Anderson Tawil syndrome (KCNJ2)
3
Periodic paralysis

Secondary Periodic Paralysis
• Hypokalemic:– Thyrotoxic periodic
paralysis– hyperaldosteronism– RTA– villous adenoma– cocaine binge– diuretics, licorice,
steroids, ETOH
• Hyperkalemic(k>5.5)– hyporeninemic
hypoaldosteronism(DM/CKD) (ie, GFR or ≤ 20 mL/min).
– type IV renal tubular acidosis (RTA)
– AKI/Metabolic acidosis– High oral K, DRUGS– chronic heparin therapy– Rhabdomyolysis– Adrenal insufficiency
4

MACHINE

HYPERKALEMIC PERIODIC PARALYSIS
MUTATED GENE SCN4A
CHROMOSOME 17q
DEFECTIVE CHANNEL SODIUM
MODE OF INHERITENCE
AUTOSOMAL DOMINANT

Hyperkalemic Periodic Paralysis
• term hyperkalemic is misleading since patients are often normokalemic during attacks.
• Onset first decade• M : F 1:1• Attacks are brief and mild, usually lasting 30
minutes to 4 hours.• Weakness affects proximal muscles, sparing
bulbar muscles. • Attacks are precipitated by rest following exercise
and fasting.

• In a variant of this disorder, the predominant symptom is myotonia without weakness (potassium-aggravated myotonia).
• The symptoms are aggravated by cold, and myotonia makes the muscles stiff and painful.
• Clinically apparent myotonia is seen less than 20% of patients, but electrical myotonia may be found in 50-75%.

Pathophysiology
In hyperKPP, Na+ channels fail to inactivate and prolonged openings and depolarization result.
Increased extracellular K+ levels worsen the inactivation of Na+ channels

INVESTIGATIONS
• Potassium may be slightly elevated but may also be normal during an attack.
• NCV reduced motor amplitudes and In between attacks of weakness, the conduction studies are normal.
• EMG may be silent in very weak muscles. • often demonstrate myotonic discharges during
and between attacks.• Muscle biopsy shows vacuoles that are smaller,
less numerous, and more peripheral compared to the hypokalemic form or tubular aggregates.

TREATMENT
• For patients with frequent attacks, acetazolamide (125–1000 mg/d) is helpful.
• mexiletine is helpful in patients with significant myotonia.
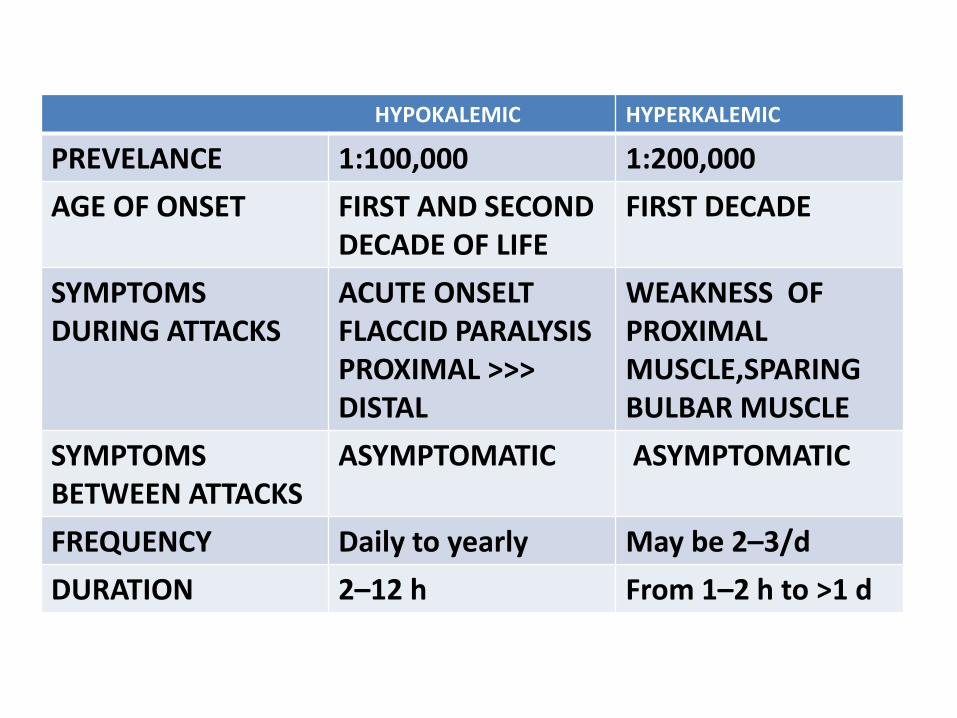
HYPOKALEMIC HYPERKALEMIC
PREVELANCE 1:100,000 1:200,000
AGE OF ONSET FIRST AND SECOND DECADE OF LIFE
FIRST DECADE
SYMPTOMS DURING ATTACKS
ACUTE ONSELT FLACCID PARALYSISPROXIMAL >>> DISTAL
WEAKNESS OF PROXIMAL MUSCLE,SPARING BULBAR MUSCLE
SYMPTOMS BETWEEN ATTACKS
ASYMPTOMATIC ASYMPTOMATIC
FREQUENCY Daily to yearly May be 2–3/d
DURATION 2–12 h From 1–2 h to >1 d

HYPOKALEMIC HYPERKALEMIC
Effect of muscle cooling
No change Increased myotonia
TRIGGERS HIGH CARBOHYDRATE,HIGH SALT,DRUGS-BETAAGONISTS, INSULINREST FOLLOWING PROLONGED EXERCISE
REST AFTER EXERCISESTRESSFATIGUEFOOD HIGH IN POTASSIUMALCOHOLINFECTION
POSTASSIUM SUPPLEMENTATION
TREATMENT PROVOCATIVE TEST
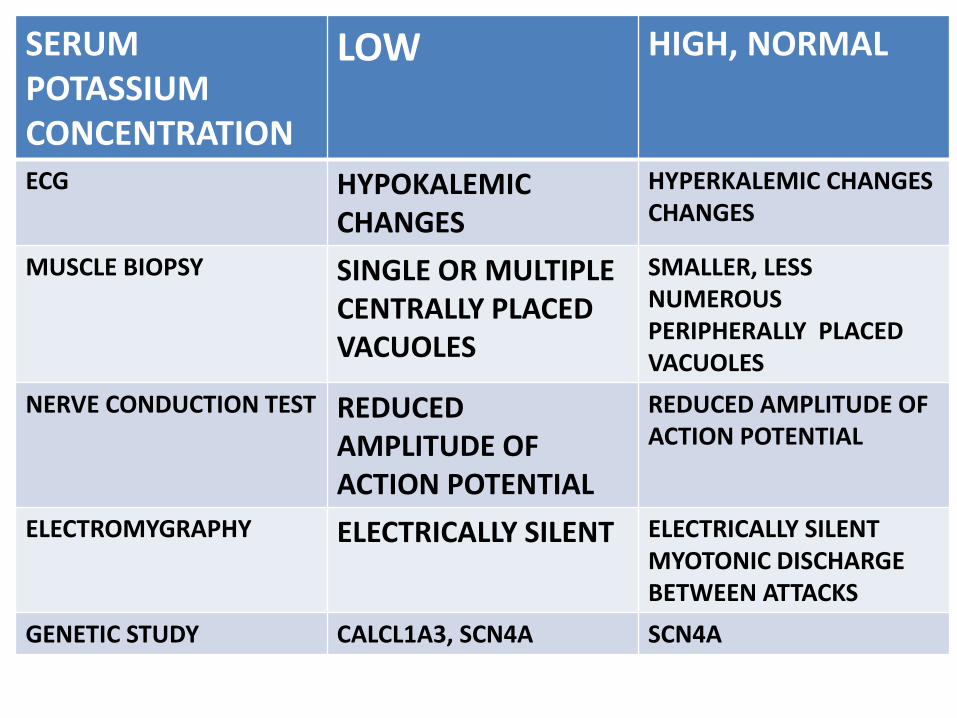
SERUM POTASSIUM CONCENTRATION
LOW HIGH, NORMAL
ECG HYPOKALEMIC CHANGES
HYPERKALEMIC CHANGES CHANGES
MUSCLE BIOPSY SINGLE OR MULTIPLE CENTRALLY PLACED VACUOLES
SMALLER, LESS NUMEROUS PERIPHERALLY PLACED VACUOLES
NERVE CONDUCTION TEST REDUCED AMPLITUDE OF ACTION POTENTIAL
REDUCED AMPLITUDE OF ACTION POTENTIAL
ELECTROMYGRAPHY ELECTRICALLY SILENT ELECTRICALLY SILENTMYOTONIC DISCHARGE BETWEEN ATTACKS
GENETIC STUDY CALCL1A3, SCN4A SCN4A
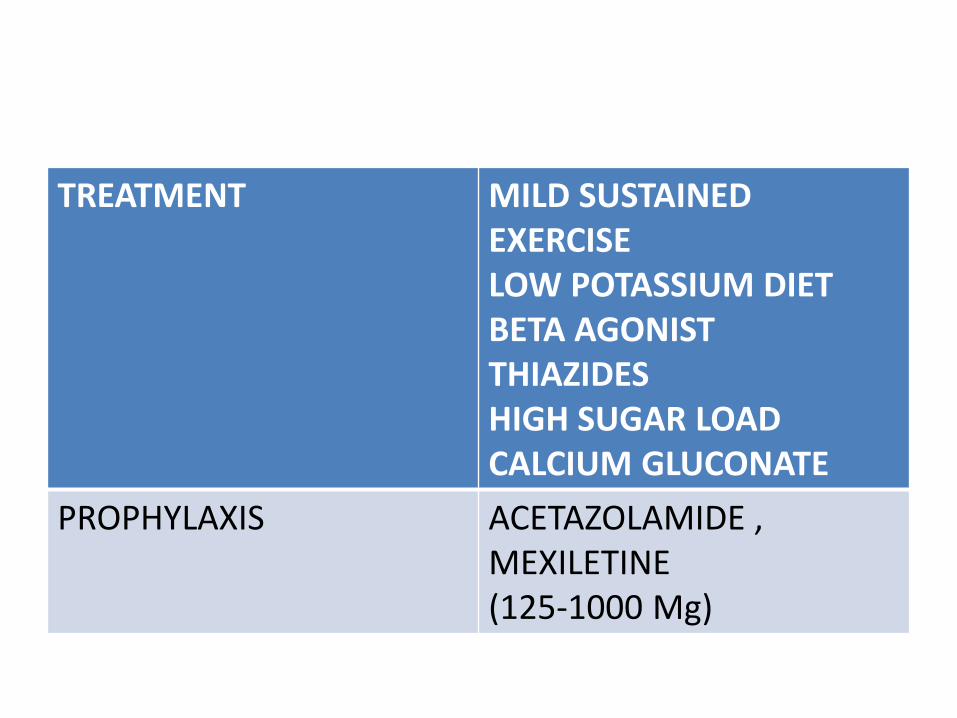
TREATMENT MILD SUSTAINED EXERCISELOW POTASSIUM DIETBETA AGONISTTHIAZIDESHIGH SUGAR LOADCALCIUM GLUCONATE
PROPHYLAXIS ACETAZOLAMIDE , MEXILETINE(125-1000 Mg)


ACUTE ONSET WEAKNESS –AIDP VS HYPOKALEMIC PERODIC PARALYSIS

OUTLINE
• DIFFERENTIAL DIAGNOSIS OF AIDP
• PRESENTATON OF AIDP
• CAUSES OF PERIODIC PARALYSIS
• PRESENTATION OF HYPOKALEMIC PP
• COMPARISON BETWEEN AIDP VsHypo PP

DIFFERENTIAL DIAGNOSIS OF AIDP
PERIPHERAL NEUROPATHIES Toxic • Vincristine• Glue sniffing• Heavy metal• Organophosphate pesticidesInfections • HIV• Diphtheria• Lyme diseaseInborn errors of metabolism • Leigh disease (Subacute Necrotizing
Encephalomyelopathy)• Tangier disease (Familial alpha-
lipoprotein deficiency)• PorphyriaCritical illness: polyneuropathy
SPINAL CORD LESIONS Acute transverse myelitisEpidural abscessTumorsPoliomyelitisVascular malformationsCord infarctionCord compression from vertebral subluxation related to congenital abnormalities or traumaAcute disseminated encephalomyelitis (ADEM)
NEUROMUSCULAR JUNCTION DISORDERS Tick paralysis, myasthenia gravis, botulism, hypercalcemiaMyopathiesPeriodic paralyses, dermatomyositis, critical illness myopathy

OTHER NAMES
• LANDRY’S ASCENDING PARALYSIS
• ACUTE INFLAMMATORY DEMYELINATING POLYNEUROPATHY (AIDP)
• ACUTE IDIOPATHIC POLYRADICULONEURITIS
• ACUTE IDIOPATHIC POLYNEURITIS
• FRENCH POLIO
• LANDRY GUILLAIN BARRE SYNDROME

AIDP
• The most common acute neuromuscular disease seen in the intensive care unit is GBS
Epidemiology
• incidence 2 /100,000/year.
• Sex
• M/F 1.1-1.7:1
• Age
• 2 months to 95 years
• Average 15-35 years
• Childhood GBS average age is 4-8 years

Antecedent events (causes)
• 60-70% cases –post infectious .
– 1-3 weeks after an acute infectious process respiratory or GIT.
– 20-30% cases –campylobacter jejuni
– Other agents –HHV (EBV,CMV)
– Mycoplasma pneumoniae
• Recent immunisation –swine influenza vaccine,olderrabies vaccine (nervous system)
• Can be seen in patients with lymphoma,HIVpositive,SLE.

Presentation• FEVER AND CONSTITUTIONAL SYMPTOMS ARE ABSENT AT THE ONSET
AND IF PRESENT ,CAST DOUBT ON DIAGNOSIS.
• Progressive weakness usually begins in the feet. at presentation, 60% have weakness in all 4 limbs. usually symmetric.
• GBS with a descending pattern of weakness seen in 14% cases.
• Paresthesias often precede the onset of weakness by 1 or more days. Often gait ataxia with distal limb paresthesias. Pain, temp relatively spared.
• At presentation half have some facial weakness
• Ophthalmoparesis see in 10-20% of patients. (around 10%.Dyck and Thomas ). Abducens palsy most common; usually bilateral.

Oropharyngeal weakness present in almost 50% of cases
>1/3 require mechanical ventilation
Areflexia: 70% at presentation and eventually in all
58 to 76% of patients have sensory NCS abnormalities (Oh et al, NEUROLOGY 2001)
Autonomic dysfunction two thirds of patients. most common is sinus tachycardia. Retention of urine 1/3 cases, GI dysmotility 15% (Semin Neurol 2008)
plateaus 50% in 2 weeks, over 90% by 4 weeks Improvement usually begins 1-4 weeks after the plateau.

IMMUNOPATHOGENESIS
• An autoimmune basis.
• Both cellular and humoral immunity involved.
• T cells activation –IL2,IL2 receptor,IL-6,TNF alpha,IFN gamma.
• All GBS results from immune responses to non self anitgnes(infectious agents,vaccines) that misdirect to host nerve tissue through a resemblance of epitope(molecular mimicry).
• Neural targets are gangliosides.
• Anti ganglioside ab –GM1 (20-50% cases of C.jejuni)
• Anti GQ1b ab - >90% MFS

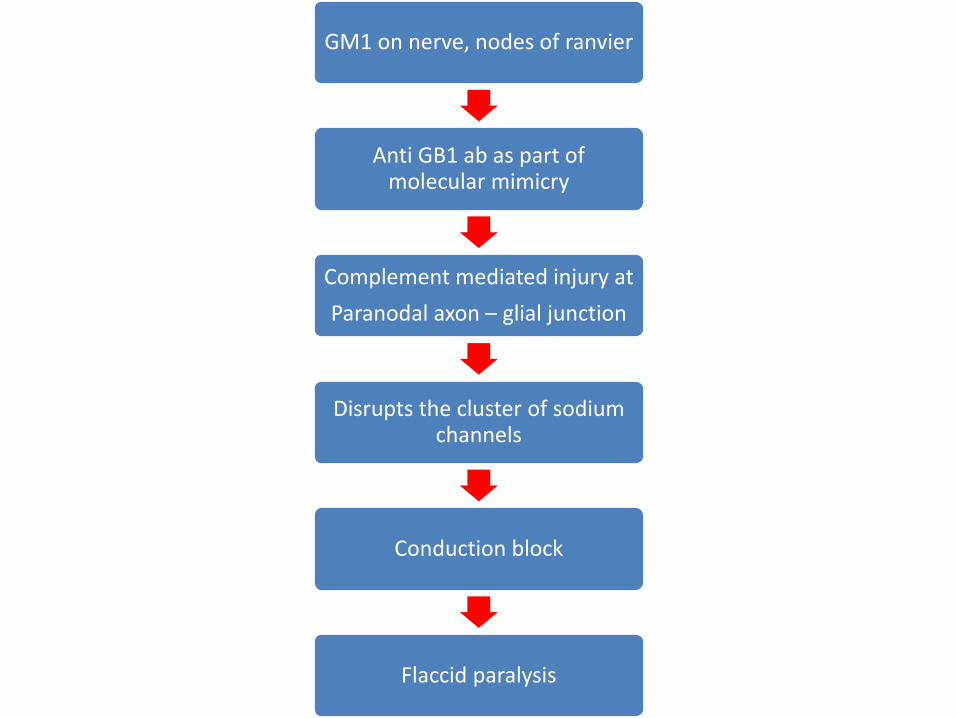
GM1 on nerve, nodes of ranvier
Anti GB1 ab as part of molecular mimicry
Complement mediated injury at
Paranodal axon – glial junction
Disrupts the cluster of sodium channels
Conduction block
Flaccid paralysis

Asbury Criteria for diagnosis
REQUIRED :• 1.progressive weakness of 2 or more limbs due to neuropathy.
• 2.areflexia
• 3.disease course < 4 weeks
• 4 exclusion of other causes (vasculitis,PAN,SLE,churg strausssyndrome,toxins,lead,botulism,diptheria,porphyria.,cauda equinalsyndrome)
SUPPORTIVE:• 1.relatively symmetric weakness
• 2.mild sensory involvement
• 3.facial nerve or other cranial nerve involvement
• 4.absence of fever
• 5.typical CSF profile(aceelualr,increase in protein level)
• 6.electrophysiologic evidence of demyelination

Variants
• MFS: most common variant, ophthalmoplegia, areflexia, and ataxia usually in adults, also common in children. Most have GQ1b Ab
• Regional variants : eg pharyngeal-cervical-brachial weakness are rare (acute progression of oropharyngeal, neck, and shoulder weakness. facial palsy, blepharoptosis, no sensory disturbance, preserved DTR in the legs, elevated CSF protein and denervation and decreased conduction velocity on EMG, reported in 1986 by Ropper )
• Pure pandysautonomia usually no weakness, many have areflexia
• AMAN China, developing countries. weakness only. little or no demyelination or inflammation, more prevalent in kids
• AMSAN

Work up
• ESR and SE are normal ,Sometimes Liver enzymes are elevated
Albuminocytologic dissociation on CSF
• protein > 55mg/dl
• cells <10 mononuclear leukocytes/ml
• 2/3 in 1st week, 82% have it by 2 weeks after symptom onset.
• no association with clinical severity.
• Some have oligoclonal bands or Myelin basic protein

• Early on, NCS often normal.
• 90% are abnormal within 3 weeks of onset.
• 3 of the 4 NCS criteria = clear primary demyelinating neuropathy (Cornblath)
• Reduced conduction velocity
• Conduction block or abnormal dispersion
• Prolonged distal latencies
• Prolonged F-waves
• Autonomic tests
• PFT: FVC < 20 mL/kg ICU; FVC< 15mL/kg or NIF<-25 Intubation
• Nerve biopsy if prolonged clinical course.

Treatment
• Initiate as soon as possible.
• Each day counts 2 weeks after the first motor symptoms –immunotherapy is no longer effective.
• IVIg IVIg-first choice,easy to administer Five daily infusions 2g/kg body weight
• Plasmapheresis - 40-50ml/kg four times a week
• Combination is not effective
• Treatment reduces need for ventilation by half.,increases full recovery at an year.
• Glucocorticoids are not effective in GBS.
• Conservative management in mild cases.

Prognosis
• total recovery in adults around 75%
• during the early stage of GBS, increasing severity in neurologic disability scores ,cranial nerve involvement, urinary incontinence, respiratory signs, and the need for ventilator support are associated with poor prognoses
• Low CMAP amplitudes (< 20% of normal) bad prognostic indicator.
• 10% may have a relapse in 1-6 weeks after completing immunomodulatory therapy
• 15% end up with significant neurological residuals
• Mortality 2-6 %

• Primary Periodic paralysis
• Secondary Periodic paralysis
Causes of Primary Periodic paralysis
– Hypokalemic (CACNA1S/ SCN4A)
– Hyperkalemic (SCN4A)
– Anderson Tawil syndrome (KCNJ2)
34
Periodic paralysis

• Hypokalemic:– Thyrotoxic periodic paralysis– hyperaldosteronism– RTA– villous adenoma– cocaine binge– diuretics, licorice, steroids, ETOH
• Hyperkalemic (k>7): – hyporenemic hypoaldosteronism (DM/CRF)– oral K, CRF, chronic heparin, rhabdomyolysis
• Normakalemic: – Guanidine, sleep paralysis, MG, TIA, conversion
35
SecondaryPeriodic Paralysis
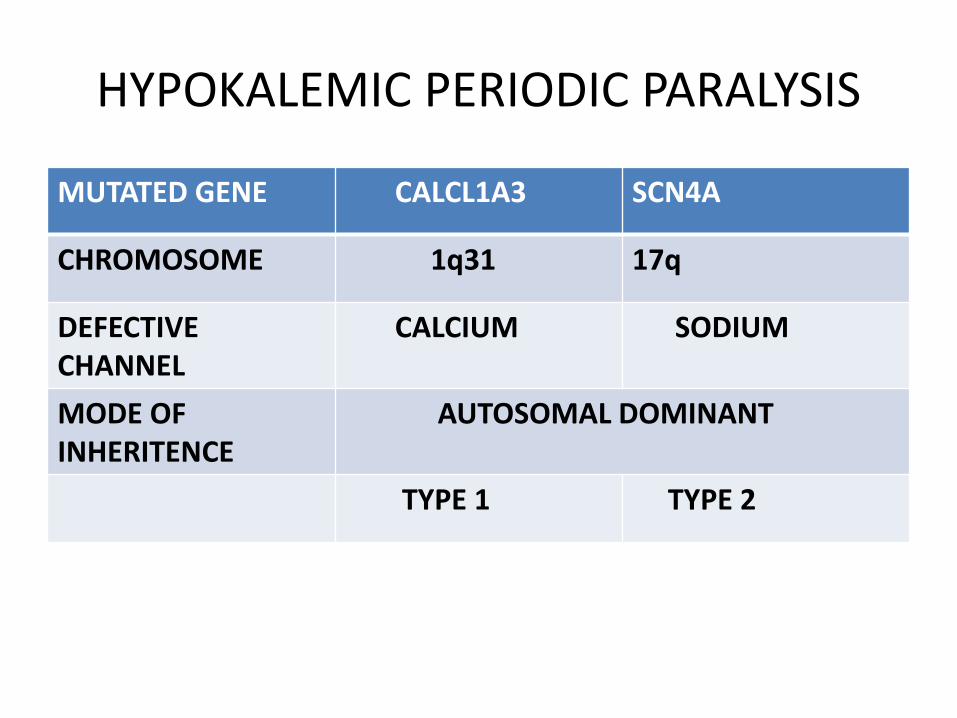
HYPOKALEMIC PERIODIC PARALYSIS
MUTATED GENE CALCL1A3 SCN4A
CHROMOSOME 1q31 17q
DEFECTIVE CHANNEL
CALCIUM SODIUM
MODE OF INHERITENCE
AUTOSOMAL DOMINANT
TYPE 1 TYPE 2

HYPOKALEMIC PERIODIC PARALYSISPREVELANCE 1:100,000, AD
AGE OF ONSET FIRST AND SECOND DECADE OF LIFE
M:F 3 or 4:1
SYMPTOMS DURING ATTACKS Occur anytime of the day; more common in morningAbsence of myotoniaProximal > distal weakness; legs > armsSparing of facial, ventilatoryand sphincter musclesLasts several hours to more than a day
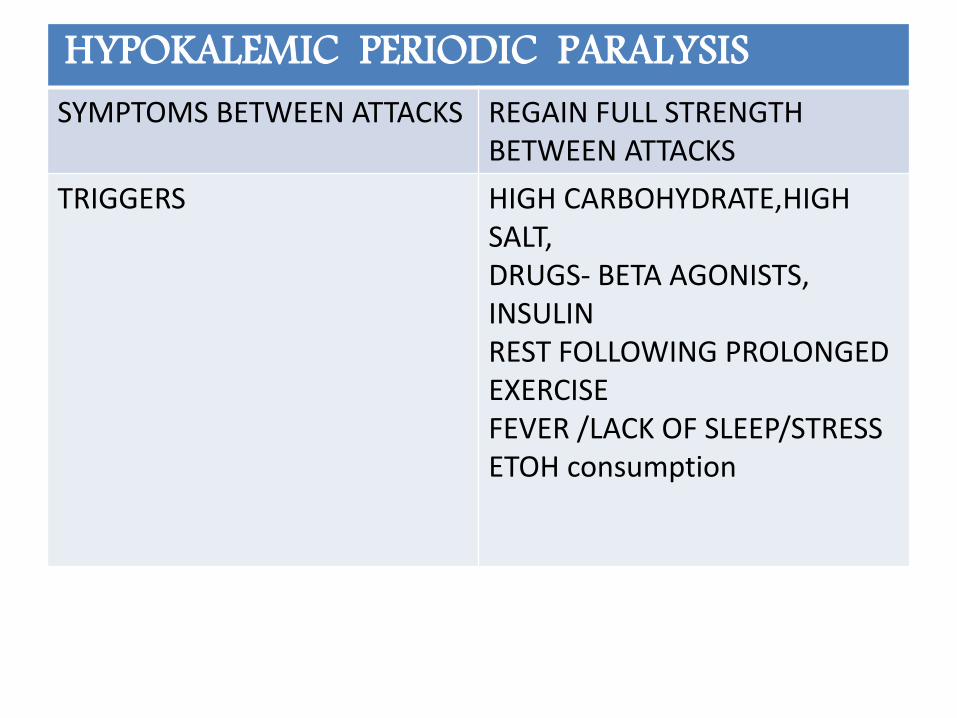
HYPOKALEMIC PERIODIC PARALYSISSYMPTOMS BETWEEN ATTACKS REGAIN FULL STRENGTH
BETWEEN ATTACKS
TRIGGERS HIGH CARBOHYDRATE,HIGH SALT,DRUGS- BETA AGONISTS, INSULINREST FOLLOWING PROLONGED EXERCISEFEVER /LACK OF SLEEP/STRESSETOH consumption

SERUM POTASSIUM CONCENTRATION
LOW
ECG U waves, flattening of T waves
MUSCLE BIOPSY SINGLE OR MULTIPLE CENTRALLY PLACED VACUOLES
NERVE CONDUCTION TEST REDUCED AMPLITUDE OF ACTION POTENTIAL
ELECTROMYGRAPHY ELECTRICALLY SILENT

TREATMENT ORAL KCL SUPPLEMENTATIONKCL VIA INFUSIONDONOT GIVE IN DEXTROSE
PROPHYLAXIS ACETAZOLAMIDE(125-1000 Mg)
PROGNOSIS USUALLY GOODRARE DEVELOPMENT OF PROXIMAL MYOPATHY
*Never forget to measure the thyroid hormones.
Hypokalemic Periodic Paralysis
41

• Frequency Of Attacks : highly variable
• Frequency decreases after age 30; may become attack free in 40s and 50s
• Permanent fixed weakness or slowly progressive weakness more common with HypoKPP1
42
PROGNOSIS

SUMMARY
• 1.acute rapidly evolving areflexic ascending motor paralysis with or without sensory disturbances.
• 2.fever is absent at the onset of weakness
• 3.bladder involvement in severe cases –transient
• 4.campylobacter jejuni 20-30 % cases
• 5.autoimmune basis ,molecular mimicry
• 6.anti GM1 ab (MC),anti GD1a,anti GQ1b (MFS)
• 7.autonomic involvement is common
• 8.facial nerve is the one most commonly affected,optic nerve.
• 9.30% require ventilatory support.

• 10.course is usually < 4 weeks
• 11 .typical CSF profile shows high protein,no pleocytosis
• 12.electrographically conduction block present
• 13.treatment as soon as possible
• 14.IVIg,plasmapheresis –both are equally good
• 15.glucocorticoids are not effective in GBS
• 16.think of miller fischer variant in presence of ophthalmoplegia,ataxia,areflexia.
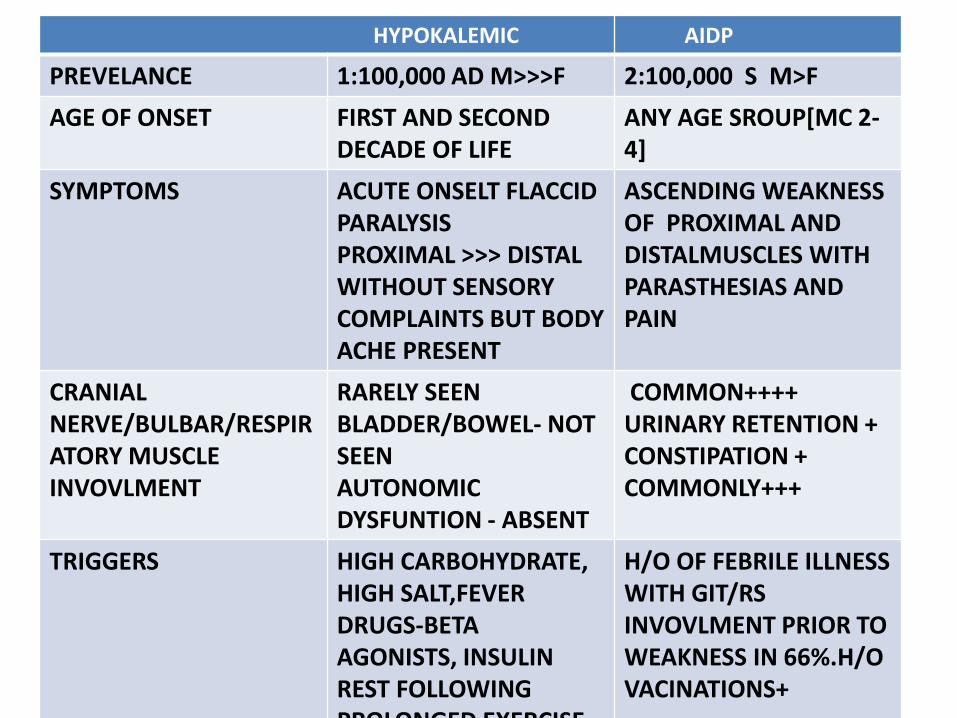
HYPOKALEMIC AIDP
PREVELANCE 1:100,000 AD M>>>F 2:100,000 S M>F
AGE OF ONSET FIRST AND SECOND DECADE OF LIFE
ANY AGE SROUP[MC 2-4]
SYMPTOMS ACUTE ONSELT FLACCID PARALYSISPROXIMAL >>> DISTAL WITHOUT SENSORY COMPLAINTS BUT BODY ACHE PRESENT
ASCENDING WEAKNESS OF PROXIMAL AND DISTALMUSCLES WITH PARASTHESIAS AND PAIN
CRANIALNERVE/BULBAR/RESPIRATORY MUSCLE INVOVLMENT
RARELY SEENBLADDER/BOWEL- NOT SEENAUTONOMIC DYSFUNTION - ABSENT
COMMON++++URINARY RETENTION +CONSTIPATION +COMMONLY+++
TRIGGERS HIGH CARBOHYDRATE,HIGH SALT,FEVERDRUGS-BETAAGONISTS, INSULINREST FOLLOWING PROLONGED EXERCISE
H/O OF FEBRILE ILLNESS WITH GIT/RSINVOVLMENT PRIOR TO WEAKNESS IN 66%.H/O VACINATIONS+
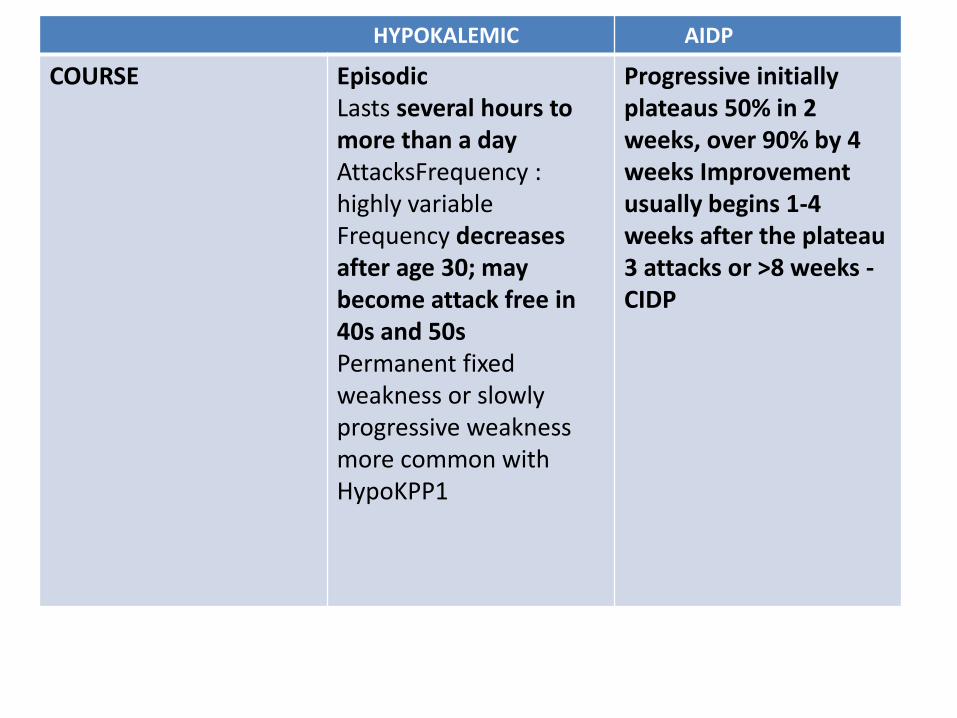
HYPOKALEMIC AIDP
COURSE EpisodicLasts several hours to more than a dayAttacksFrequency : highly variableFrequency decreases after age 30; may become attack free in 40s and 50sPermanent fixed weakness or slowly progressive weakness more common with HypoKPP1
Progressive initiallyplateaus 50% in 2 weeks, over 90% by 4 weeks Improvement usually begins 1-4 weeks after the plateau3 attacks or >8 weeks -CIDP
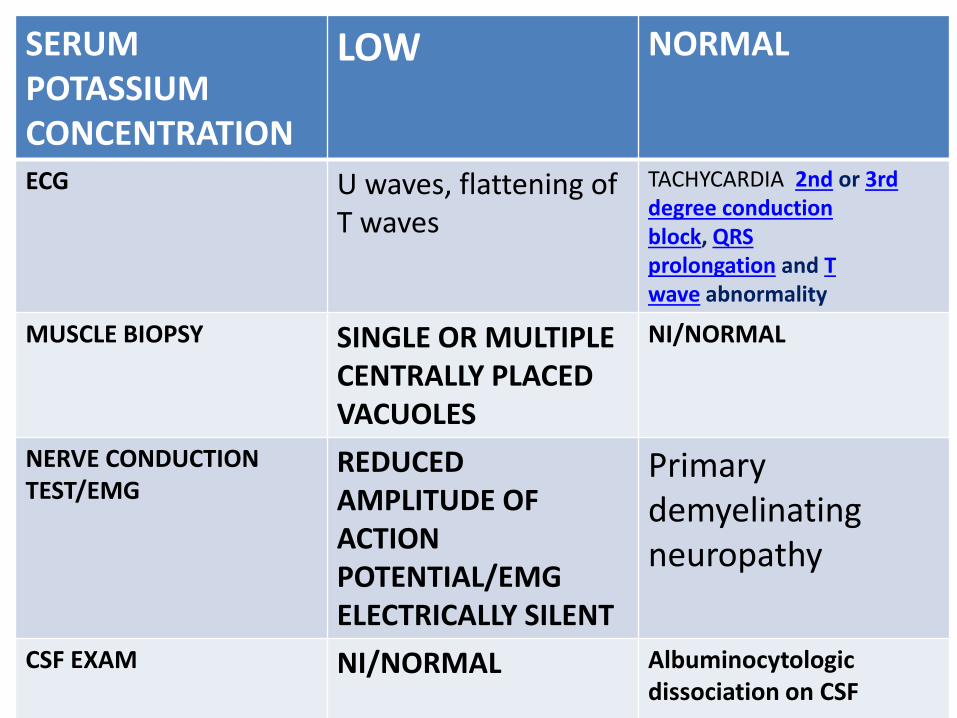
SERUM POTASSIUM CONCENTRATION
LOW NORMAL
ECG U waves, flattening of T waves
TACHYCARDIA 2nd or 3rd degree conduction block, QRS prolongation and T wave abnormality
MUSCLE BIOPSY SINGLE OR MULTIPLE CENTRALLY PLACED VACUOLES
NI/NORMAL
NERVE CONDUCTION TEST/EMG
REDUCED AMPLITUDE OF ACTION POTENTIAL/EMG ELECTRICALLY SILENT
Primary demyelinating neuropathy
CSF EXAM NI/NORMAL Albuminocytologicdissociation on CSF



















