
Huntington’s disease Also called Huntington Chorea, affects men and women. 4 to 10 cases in...
75
Huntington’s disease Also called Huntington Chorea, affects men and women. 4 to 10 cases in 100,000 people (frequency between 0.004% and 0.01%) . Chorea is defined as brief, irregular, unpredictable, purposeless movements that flow from one body part to another without a rhythmic pattern. HD is a Hyperkinetic Disorder. The symptoms in HD are characterized by Excessive motor activity Involuntary movements (dyskinesias) Decreased muscle tone Behavioral and emotional alteration and cognitive decline, which may precede motor abnormalities. Importance of cognitive and neuropsychological test to identify the pre-symptomatic carriers of the disease.
-
date post
20-Dec-2015 -
Category
Documents
-
view
218 -
download
0
Transcript of Huntington’s disease Also called Huntington Chorea, affects men and women. 4 to 10 cases in...

Huntington’s disease
Also called Huntington Chorea, affects men and women. 4 to 10 cases in 100,000 people (frequency between 0.004% and 0.01%) .
Chorea is defined as brief, irregular, unpredictable, purposeless movements that flow from one body part to another without a rhythmic pattern.
HD is a Hyperkinetic Disorder. The symptoms in HD are characterized by Excessive motor activityInvoluntary movements (dyskinesias)Decreased muscle tone
Behavioral and emotional alteration and cognitive decline, which may precede motor abnormalities. Importance of cognitive and neuropsychological test to identify the pre-symptomatic carriers of the disease.
HD is a dominant autosomal inherited disease characterized by the expansion of a triplet repeat CAG, that encodes for Glutamine, in the N-terminal domain of the protein.

George Huntington (1850-1916)

Specific localization of Basal Ganglia in a coronal section

HD is caused by selective degeneration of the GABAergic neurons in the striatum, in particular in
the caudate

HD
Normal
Volumetric analysis in Huntington disease (HD)

Age of onset:
HD is thought to be a true dominant disorder, since homozygous carriers of the disease are no more severely affected than heterozygous carriers. In a normal individual, the number of CAG repeats are <36. In HD patients, age of onset when the repeats are 46 ranges between 25 and 52 years old. However, many factors influence the onset of the disease, such as:
Mitochondrial dysfunctionCell death by apoptosisLoss of neurotrophic factorsNeuron excitotoxicity
Length of the CAG repeat and onset of the disease: Inverse correlation, but not when onset is >50 years of age.

Deposition of fibrillar proteinacious material in Huntington’s disease
Huntington’s disease: a progressive neurodegenerative disease characterized by CAG repeats causing glutamine expansion motifs (polyQ) in the N-terminal region of the protein huntingtin. Onset of the disease inversely correlates with the number of CAG repeates. Huntingtin can aggregate forming intracellular inclusion bodies that contain also proteasome proteins and ubiquitin. Huntingtin aggregates contain -sheet structures similar to amyloid.

In homo sapiens, the gene for huntingtin encodes a large, cytosolic protein, comprised of 3144 aa and with a molecular weight of about 348 kDa.
NH2COOH
aa3144
PolyQ P
In normal individuals, PolyQ<36. In HD patients, PolyQ>36
aa1
Huntingtin

NH2
aa3144
PolyQ P
aa1Cleavage by caspase 2, 3, 6calpain
N-terminal fragment
C-terminal fragment
COOH
COOHNH2
Processing of PolyQ, mutant huntingtin
Cdk5, Akt
N-term htt forms aggregates

In HD, the aggregates are present in the affected brain regions, although not necessarily in the degenerating neurons
In HD, are the aggregates protecting from neurodegeneration? Are htt oligomers the
most toxic species?

Huntington’s disease: loss of function of the wild type huntingtin and gain of
toxic function of the mutant huntingtin
-Heterozygous patients are NOT different in any way from the homozygous patients: in heterozygous patients, the amount of wild-type “good” huntingtin does not make it to “buffer’ the effects of the mutant, PolyQ expanded huntingtin.
-Mutant, PolyQ expanded huntingtin can recruit wild type huntingtin into insoluble aggregates to form the inclusions.
-Neurons with larger inclusions are less compromised than those ones with diffused mutant htt (which might contain oligomers not aggregated into inclusions).

Potential mechanism of cell death in Huntington’s disease

Which is the physiological role of wild type huntingtin?
-Huntingtin is a molecule required during development, as hungtintin KO mice die at 7.5 postnatal day. Heterozygosity is enough to reach adulthood.
-Huntingtin interacts with several proteins, i.e. cytoskeletal proteins and others that are associated with membrane vesicles, suggesting a role in intracellular trafficking, membrane recycling and retrograde fast axonal transport.
-The presence of Q repeats (CAG) followed by PolyP domain associates with transcriptional regulatory proteins. Inverse correlation between number of CAG repeats and transcriptional activity. After interacting, huntingtin transports transcriptional regulatory proteins in the cytosol and within the nucleus. (BDNF).

Physiological roles of wild type huntingtin

Physiologic function of wild-type huntingtin:
Cell survival, anti-apoptotic function

Immortalized embrional striatal cells
Normal Serum withdrawal
N548Mu
N548WT
Rigamonti et al., 2000
Cell death is prevented by huntingtin wt, but is promoted by huntingtin PolyQ mutant

Rigamonti et al., 2000
Temperature-dependent apoptotic DNA fragmentation is sustained by huntingtin PolyQ mutant, but is prevented by huntingtin wt
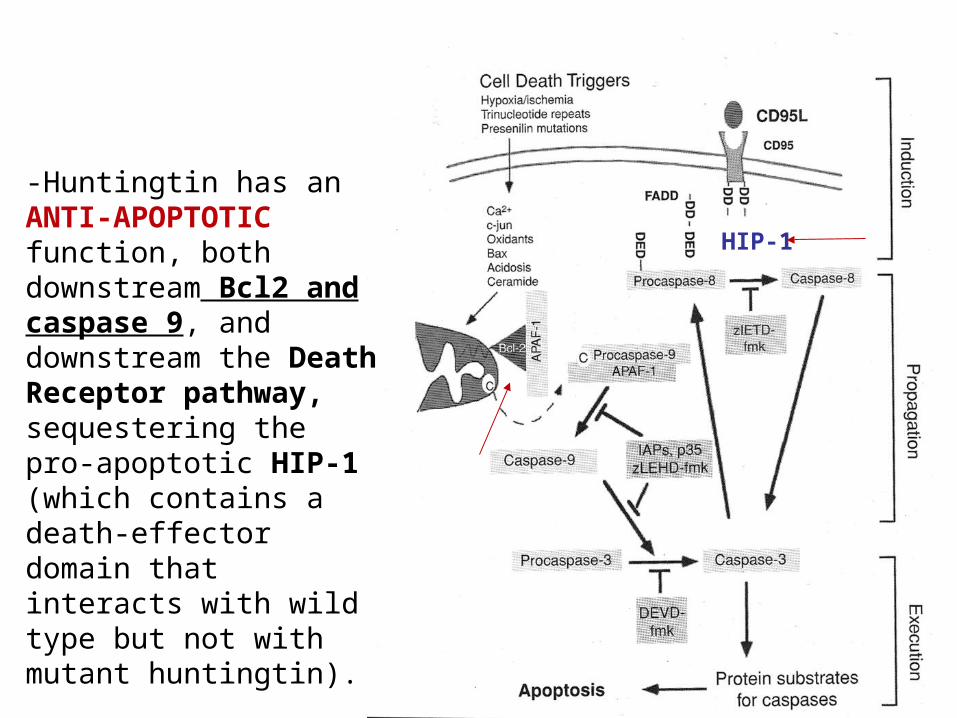
-Huntingtin has an ANTI-APOPTOTIC function, both downstream Bcl2 and caspase 9, and downstream the Death Receptor pathway, sequestering the pro-apoptotic HIP-1 (which contains a death-effector domain that interacts with wild type but not with mutant huntingtin).
HIP-1

Physiologic function of wild-type huntingtin:
modulating the expression and the transport of BDNF (Brain Derived Growth Factor) to striatal neurons

-Peculiarity of HD is that lethality occurs specifically in striatal neurons, although huntingtin is ubiquitously expressed.
-It is known that huntingtin promotes neuronal survival (and is required during development)
-BDNF has certainly a role in maintaining the “health” of striatal neurons. In facts, BDNF:
a)promotes growth and differentiation of striatal neurons
b)protects striatal neurons from excitotoxicity

Wild-type huntingtin, but not mutant PolyQ huntingtin, specifically increase the levels of secreted and cellular BDNF
in cell culture
Zuccato et al., 2002

BDNF seems likely to be produced in cortex and hippocampus, and to be subsequently transported by afferents to the striatal neurons
What happens in the cortex of HD patients?
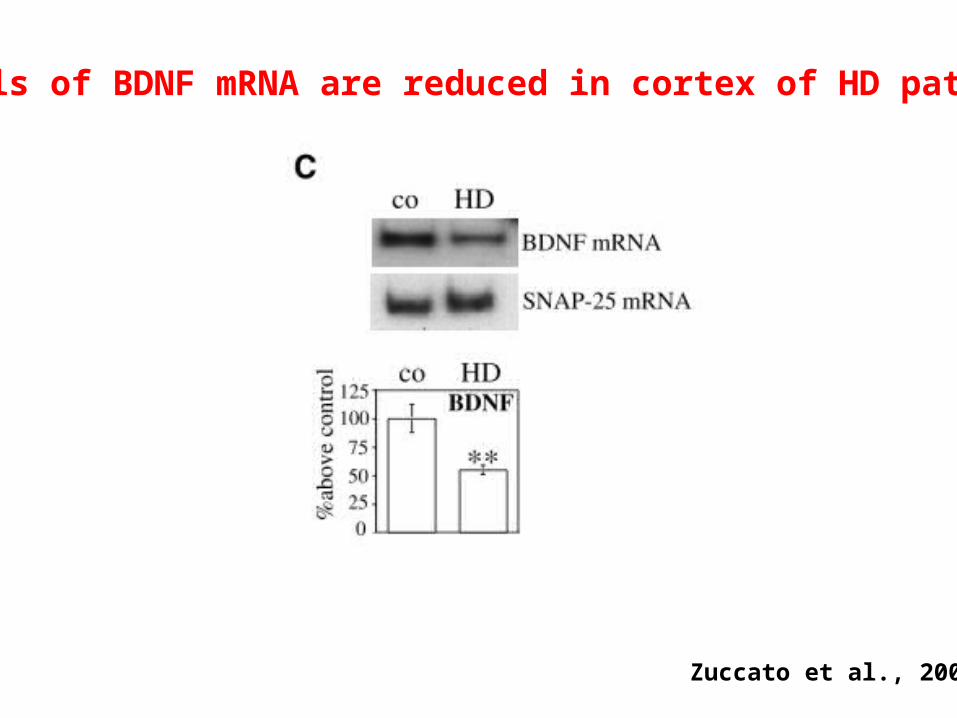
Levels of BDNF mRNA are reduced in cortex of HD patient
Zuccato et al., 2002

Pathogenic mechanisms of mutant PolyQ huntingtin:
Htt processing and formation of aggregates

NH2
aa3144
PolyQ P
aa1Cleavage by caspase 2, 3, 6calpain
N-terminal fragment
C-terminal fragment
COOH
COOHNH2
Processing of PolyQ, mutant huntingtin
Cdk5, Akt
N-term fragment forms aggregates

N-terminal fragment
NH2
Capabilities to form aggregates
nucleus cytosol
Soluble form
Inhibits Glu Uptake
Glu vesicle
Different translocation of PolyQ, mutant huntingtin N-terminal fragment
activates caspase-1 positive feedback in regulating apoptosis

How does the CAG, PolyQ expanded motif cause HD?
-It induces mitochondrial dysfunction, that leads to mitochondrial disruption of Ca++ homeostasis: trigger of cytochrome c and caspase-dependent apoptosis.
-It avoids certain post-translation modifications (like SUMOylation and palmitoylation) to occur on the protein, with consequences on the protein’s functionality.
-The N-terminal fragment translocates to the nucleus where it causes:a) reduction of the synthesis of neurotrophic factors (like BDNF
Brain-Derived Neurotrophic Factor), that are essential for the neuronal growth and life.
b) formation of intranuclear aggregates that recruit also transcription factors (CREB binding protein CBP), inducing transcriptional deregulation.

Pathogenic mechanisms of mutant PolyQ huntingtin:
Mitochondrial dysfunction

Membrane Potential: Difference in the voltage between the two sides of a membrane, caused by an influx and efflux of ions to and from a cell or neuron and an organelle. In the mitochondrion, the membrane potential is caused in particular by exchange of Ca++ ions between the mitochondrion and the cytosol.
Ca++
Ca++
PTPs: Permeability Transition Pores
PTP

Juvenile HD patient: 65 repeatsAdult HD patient: 46 repeats Panov et al., 2002
In HD patients, the membrane potential is reduced according to the number of CAG, PolyQ repeats
Physiologically, the potential drops when the PTP open

The tendency of the PTP to be open directly correlates with the number of PolyQ repeats in huntingtin mutant molecule

Panov et al., 2002
Mitochondria of HD patients have reduced Calcium levels

Huntingtin aggregates are visible on the mitochondrial membrane of PolyQ mutant huntingtin transgenic mice,
but not of wild-type huntingtin transgenic mice
Huntingtin PolyQ mutant transgenic mice
Huntingtin wild-type transgenic mice
Panov et al., 2002

Calcium homeostasis is altered
1-in mitochondria of HD patients
2- in mitochondria of transgenic mice overexpressing mutant huntingtin, but not in mice overexpressing wild type huntingtin
3-administration of synthetic PolyQ repeats to human wild-type mitochondria in vitro causes alteration of calcium homeostasis
4-huntingtin aggregates are visible on the membranes of mitochondria in mutant huntingtin mice, but not wild type mice
THUS:

Mutant PolyQ huntingtin disrupts Calcium homeostasis, probably forming aggregates on the mitochondrial membrane that cause abnormal opening of the Permeability Transition Pores.
This is an early event in the pathogenesis of HD, as the mice that show reduced membrane potential and formation of aggregates do not show the phenotype characteristic of HD animal model.
Improvement of PTP functionality could be a potential therapeutic target in HD

Proteostasis and HD
A role for the htt aggregates?

R6/2: characterization of HD phenotype in an animal model
Motor deficitMotor performance (rotarod)
Neurobehavioral PhenotypeClasping: the tendency to bring the limbs to press the stomach
Tremor
Grooming: none or obsessively repeated is shown when the animal is symptomatic. It is thought to mimic the choreiform movements displayed by HD patients
Spontaneous and locomotor activity: the capacity of the animal to cover the four quadrants of the cage with its movements.
Physical phenotype
Body weight
Coat appearance: worsens as the disease progresses. Can be also a consequence of the changes of the grooming ability/interest of the animal.
Body and tail position: hunched and rounded (body) and dragging/straub (tail) indicate improper muscle contraction


Loss of HSP70 decrease survival in HD mouse model

Loss of HSP70 exacerbates motor deficit and neuropathological phenotype in HD mouse model

Loss of HSP70 is associated with increased size of the inclusions in HD mouse model

RIPA lysates:SDS-insoluble aggregates
Formic acid lysates:SDS-resistant oligomers
Native agarose gel
Loss of HSP70 may influence the formation of oligomers

Loss of HSP70 exacerbates neuropathological deficit in HD mouse model

Chaperones mediated proteostasis does not affect the formation of aggregates in HD animal model, but probably regulate the
formation of toxic oligomers
Pharmacological regulation of proteostasis for the treatment of HD?


Inhibition of HSP90 regulates HSR in wild type mouse brain: protein levels of HSF

HSP990 increases mRNA levels of HSFs and activates HSF1

Levels of aggregates decrease by pharmacological stimulation of HSR

Increasing HSR ameliorates HD mice motor skills and reduces loss of brain volume

The effects of HSR on aggregates formation are age-dependent
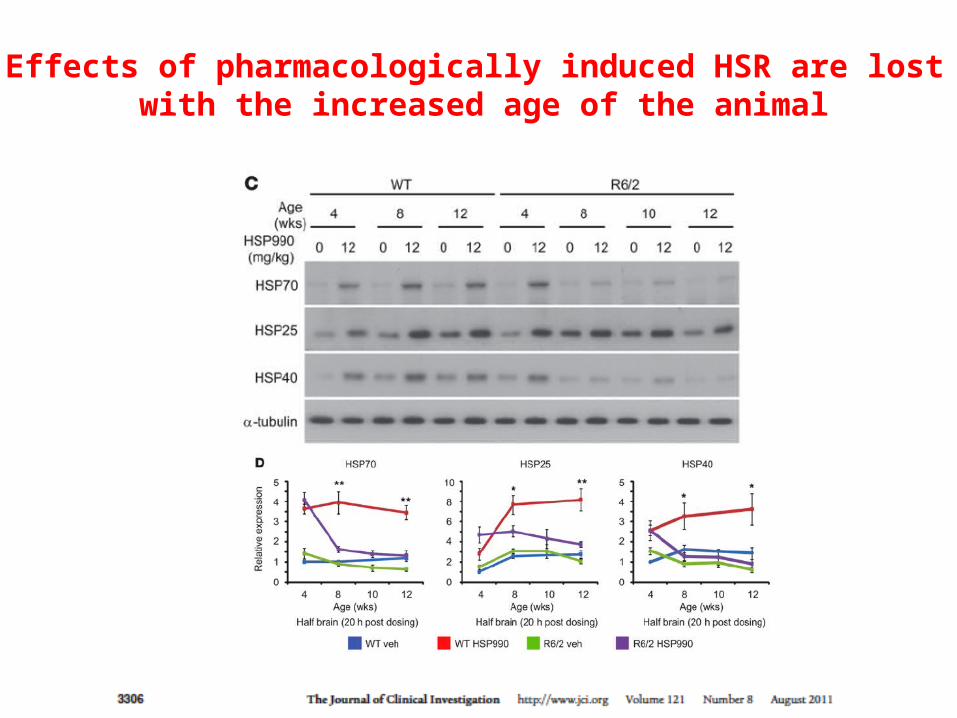
Effects of pharmacologically induced HSR are lost with the increased age of the animal

HSP990 does not improve transcription of HSFs in older animals

Pharmacological activation and nuclear localization of HSF1 are not age-dependent

Reduced HSF1 promoter binding to HS loci in 12 weeks…

…and 22 weeks old HD animals

Reduced acetylation on the target gene reduces HSF1 binding and transcriptional activity


Mutant HTT causes mitochondrial fragmentation in rat cortical neurons…
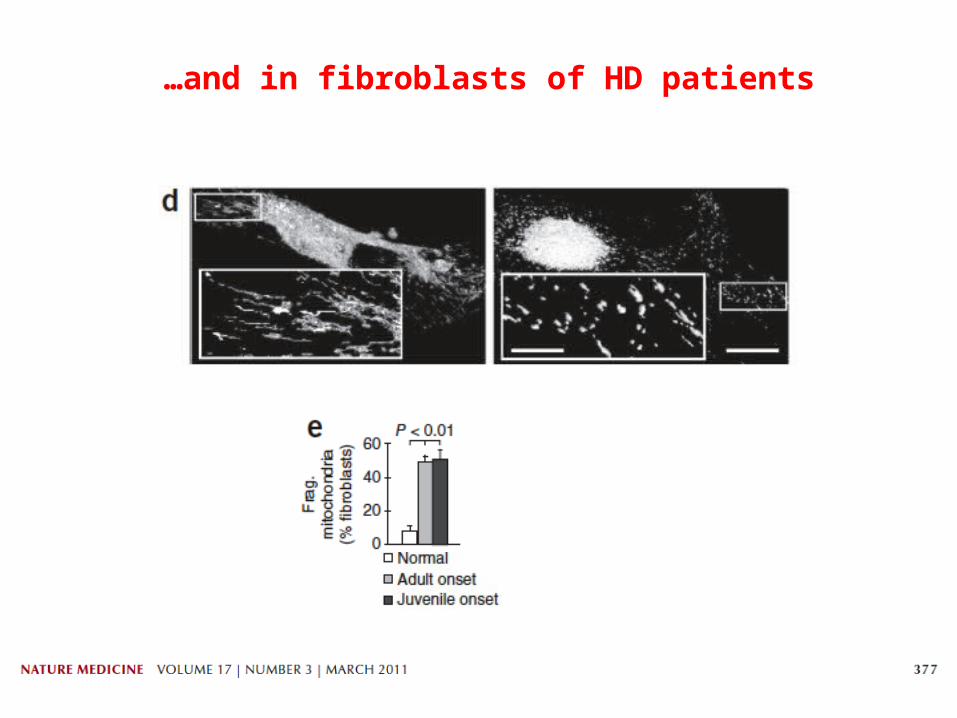
…and in fibroblasts of HD patients

PolyQ expansion associated with HD impairs mitochondrial movement

Movies

WT
YAC128
PolyQ expansion associated with HD affects mitochondrial size …

…and structure: loss of cristae’s density and presence of constrictions
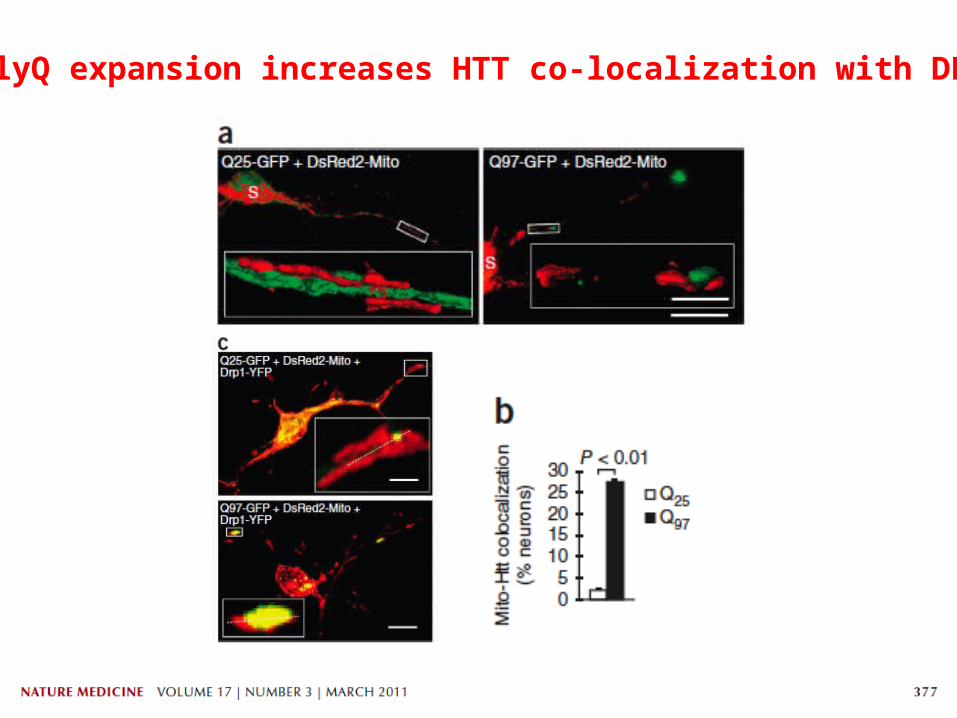
PolyQ expansion increases HTT co-localization with DRP1

Lymphoblasts Brains
Mitochondrial lysates from mouse brain
Mutant HTT associates to DRP1 in HD

nucleotides GTP GTP+httQ ext1-53
Mutant HTT has greater affinity than wt HTT for DRP1: effects on DRP1 GTPase acrivity

Inactive Drp1 mutant rescues the effects of polyQ HTT on mitochondrial fragmentation…

…and motility…

…and cell death

Re-established mitochondrial fission and fusion balance improves the toxic effects of polyQ HTT on mitochondrial motility, fragmentation and on cell death.

PolyQ HTT binds to Drp1 and disrupts the equilibrium between fission and fusion: a model for HTT toxicity in HD

LC3II accumulates in HD: autophagy is induced or is defective?

Rescuing autophagy with Rapamycin reverts the eye degenerating phenotype in htt mutant flies
Autophagy as a protective mechanism against HD

Therapy for HD-Possible presymptomatic treatment, after genetic test.-Symptomatic treatments aimed at treating:
DepressionPsychosis
ChoreaTetrabenazine, depletes DA from central neurons.
-Mutant Htt siRNA
-Caspase inhibitors (risks of higher susceptibility to develop cancer after a long-term treatment). Minocycline (antibiotic already in use in humans) used now in clinical trails for HD. In HD prevents apoptosis by inhibiting caspases and mitochondrial permeability. Possible other unknown mechanisms of action.
-Enhancing the clearance of mutant Htt, for example by autophagy.
-NMDA receptors antagonists to block glutamate-induced excitotoxicity.
-To induce wt Htt physiological function

Potential therapeutic strategies in HD


















