Hua YIN | May 2014 1 |1 | Quality Workshop Copenhagen – May 2014 Specifications Hua YIN.
46
Hua YIN | May 2014 1 | Quality Workshop Copenhagen – May 2014 Specifications Hua YIN
-
Upload
anissa-marsh -
Category
Documents
-
view
226 -
download
5
Transcript of Hua YIN | May 2014 1 |1 | Quality Workshop Copenhagen – May 2014 Specifications Hua YIN.

Hua YIN | May 20141 |
Quality WorkshopCopenhagen – May 2014
Quality WorkshopCopenhagen – May 2014
Specifications
Hua YIN

Hua YIN | May 20142 |
OutlineOutline
Specification
Example specification exercise
Compendial Vs Non-compendial (In-house) standards
Deficiencies
Review tips

Hua YIN | May 20143 |
Information SourceInformation Source
ICH Q6A Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: Chemical substances
ICH Q3A - Q3D: Impurities
Ph Int, USP, BP, EP, JP (recognized standards in PQP)
PQP Generic Guideline – Quality Part
Other regulatory guidelines
Assessing specifications: Lynda Paleshnuik, PQP Quality Workshop, Copenhagen – January 2012

Hua YIN | May 20144 |
SpecificationsSpecifications
A list of tests, reference to analytical procedures, and acceptance criteria (numerical limits, ranges or other criteria), to which an API or FPP should conform
Confirm the quality, rather than fully characterize the API or the FPP
Should focus on those characteristics found to be useful in ensuring the safety and efficacy of the drug substance and drug product

Hua YIN | May 20145 |
SpecificationsUniversal tests/criteria
SpecificationsUniversal tests/criteria
For both API and FPP
Description /appearance
Identification (IR, HPLC/UV diode array, etc.)
Assay
Impurities

Hua YIN | May 20146 |
Specificationsspecific tests/criteria
Specificationsspecific tests/criteria
API
BCS low solubility APIs (DSV > 250ml)
Polymorphism should be investigated. If polymorphism is a factor, a test is required in specs --ICH Q6 decision tree #4
PSD limits (d10, d50, d90) are required in the API specs, based on the results of the lot used in biostudies. --ICH Q6 decision tree #3
Must be representative of the biolot characteristics.

Hua YIN | May 20147 |
Specificationsspecific tests/criteria
Specificationsspecific tests/criteria
API
Physico-chemical properties: pH, melting point
Water content (KF preferred)
Inorganic impurities, Heavy metals
Microbial limits

Hua YIN | May 20148 |
Specificationsspecific tests/criteria
Specificationsspecific tests/criteria
FPP
Performance tests: e.g. dissolution, disintegration (where applicable)
Uniformity of dosage units: mass or content uniformity, fill volume
Physical tests: e.g. LOD/water content, pH, friability, hardness, particle size
Content of preservatives
Microbial contamination
Sterility, bacterial endotoxins, particulate matter (parenteral)

Hua YIN | May 20149 |
Specification Parameters - Description Specification Parameters - Description
Description: should be detailed (colour, shape, coating, markings (score, ink, embossing))
A complete description is required to facilitate the visual identification of spurious/falsely-labelled/falsified/counterfeit (SFFC) medicines.
It is also important to distinguishing
the look of different strengths

Hua YIN | May 201410 |
Specification Parameters - DescriptionSpecification Parameters - Description
FPPs for which taste is critical (especially paediatrics): dispersible, sublingual/buccal, soluble, effervescent, and chewable tablets, and powders and granules for dispersion/solution or to be used as sprinkles.
Palatability (including taste) should be demonstrated as part of pharmaceutical development. Taste may exceptionally be part of specs if an artificial taster is available. (Ideally would be in shelf-life specs).

Hua YIN | May 201411 |
Specification Parameters - IdentificationSpecification Parameters - Identification
Identification should be specific to the API, i.e. should be able to discriminate between compounds of closely related structure (Q6A)—IR spectroscopy for API, not always applicable to FPP
A single HPLC retention time is not regarded as being specific. Two chromatographic procedures, where the separation is based on different principles (HPLC + TLC) , or combination of tests into a single procedure is generally acceptable, such as HPLC/UV diode array, HPLC/MS, or GC/MS. (Q6A)

Hua YIN | May 201412 |
Specification Parameters - AssaySpecification Parameters - Assay
Assay: should be stability indicating. May be non-specific, e.g. titration, but need to achieve overall specificity, e.g. non-specific assay plus suitable related substances test.
The assay in the release specifications of FPP is ± 5% of the label claim (i.e. 95.0-105.0%)..

Hua YIN | May 201413 |
Specification Parameters - Related substancesSpecification Parameters - Related substances
Related compounds: need limits on specified (identified and unidentified *), unspecified (individual unknowns), and total related compounds.
*specified unidentified = structurally unidentified. It is identified in the specs by means of e.g. RRT (relative retention time).
Limits must be suitably justified/qualified.
ICH Q3A, Q3B Thresholds for Reporting, Identification, and qualification
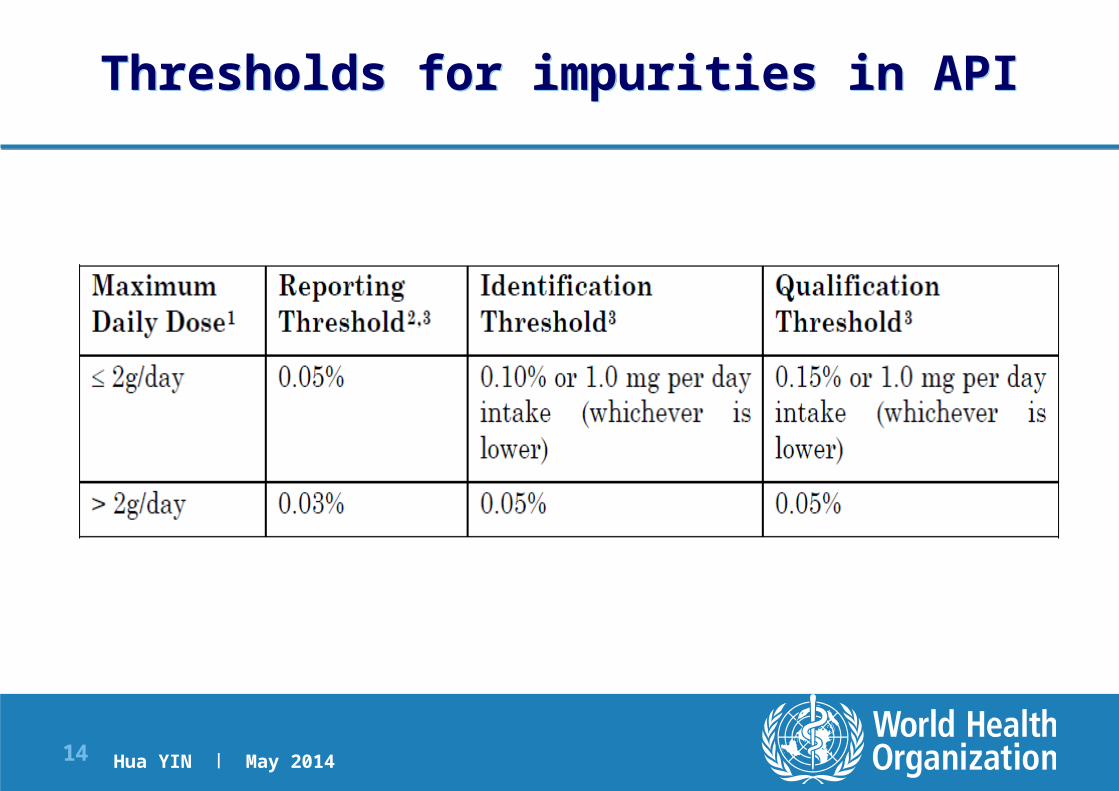
Hua YIN | May 201414 |
Thresholds for impurities in APIThresholds for impurities in API

Hua YIN | May 201415 |
Thresholds for degradation products in FPPThresholds for degradation products in FPP

Hua YIN | May 201416 |
Thresholds for degradation products in FPPThresholds for degradation products in FPP

Hua YIN | May 201417 |
Specification Parameters - Related substancesSpecification Parameters - Related substances
Any impurity
> reporting threshold should be reported
> Identification Thresholds (IT) should be Specified
Unspecified (individual unknowns) ≤ Identification Thresholds (IT)
> Qualification Thresholds (QT) should be qualified

Hua YIN | May 201418 |
Specification Parameters - Related substancesSpecification Parameters - Related substances
Qualification of limits: adopt limit QT, or qualify:
Level present in a product used in safety and/or clinical studies (ICH Q3)
If a limit refers to a significant metabolite, it is considered qualified (confirm it is a metabolite, e.g. WHOPAR, EPAR, SmPC).
Literature i.e. Pharmacopoeial (ORC) limits for specified related compounds are considered qualified; an unspecified/unknown limit in a monograph is not qualified.
Limit is similar to levels found in unstressed innovator product

Hua YIN | May 201419 |
Zidovudine 300mg tablets, Maximum daily dose is 600mg.
Int.Ph: Imp A, B, C, D are identified. Imp C is a degradant
Monograph limits of zidovudine tablets: imp C ≤ 3.0%; any other imp ≤ 1.0%; total ≤ 4.0%
The applicant claims the same method and limits as Int.Ph. Is it acceptable?
Note: RT= 0.1% ; IT= 0.2%; QT= 0.2%
Case study 1Case study 1

Hua YIN | May 201420 |
Case study 1 cont'dCase study 1 cont'd
imp C ≤ 3.0% √ qualified
any other imp ≤ 1.0%; × A, B, D ≤ 1.0%, but any other unknown individual should be ≤ 0.2%
total ≤ 4.0% √ may be tightened as per the real results of primary batches

Hua YIN | May 201421 |
Specification Parameters - DissolutionSpecification Parameters - Dissolution
Dissolution is considered product-specific. The method and limits should be appropriate for the proposed product.
Dissolution specs at release and shelf-life should be identical. It is useful to have the parameters (medium, apparatus, speed) in specs.
Some accepted specifications can be checked e.g. FDA site “Dissolution Methods for Drug Products” www.accessdata.fda.gov/scripts/cder/dissolution/
Surfactant use should be exceptional and appropriate.—not exceed 2% normally

Hua YIN | May 201422 |
Specification Parameters - DissolutionSpecification Parameters - Dissolution
The limits should be expressed as “Q”, which also implies three stage testing as per harmonized texts. Ideally the stages are expressed in the specs (i.e. S1 all individual values Q+5%)
There should be a discussion if the time is > 45 minutes.
For products containing water insoluble APIs, it is recommended to have a two tire dissolution limit. For example Artemether dissolution: NLT 40% in 1 hour and NLT 60% at the 3rd hour
It should be ascertained that the dissolution limits are not overly wide, compared to the actual results (even for an ORC limit).
ORC: officially recognized compendial

Hua YIN | May 201423 |
Specification Parameters - DissolutionSpecification Parameters - Dissolution
ICH Q6A decision trees #7 can be used to assess the proposed dissolution criteria, however:
For considering accepting DT in place of dissolution: all the considerations should be carefully assessed: highly soluble and very rapidly dissolving, plus significant supporting development data – including
when DT is more discriminating or
has a demonstrated relationship to dissolution, robustness of the formulation/manufacturing process have been demonstrated wrt DT, etc.

Hua YIN | May 201424 |
Specification Parameters - DissolutionSpecification Parameters - Dissolution
Additional considerations are required when there is a BCS-based biowaiver:
BCS Class 1 (e.g. emtricitabine, stavudine, zidovudine, levofloxacin, ofloxacin): The test and comparator products must be at least rapidly dissolving. (NLT 85% in 30 minutes)
BCS Class 3 (e.g. abacavir sulfate, lamivudine, ethambutol, isoniazid, pyrazinamide):The test and comparator products must be very rapidly dissolving (NLT 85% in 15 minutes).
Dissolution limits must meet the biowaiver requirements.

Hua YIN | May 201425 |
Case study 2Case study 2
Ethambutol hydrochloride 400mg tablets. The applicant claimed USP standard for the product.
Bioequivalent of the product is accepted as per BCS class 3 based biowavier
The applicant set the dissolution limits as below: NLT 80% (Q) in 45min at release NLT 75% (Q) in 45min at shelf life, which is in line with the
requirement of USP monograph.
Is it acceptable? What limits should be applied?

Hua YIN | May 201426 |
Case study 2 cont'dCase study 2 cont'd
Answer: not acceptable
The dissolution limits at release and shelf life should be the same.
A limit of NLT 80% (Q) in 15min should be set for both release and shelf life as for the BCS class 3 biowaiver.

Hua YIN | May 201427 |
Specification ParametersUniformity of Dosage UnitsSpecification Parameters
Uniformity of Dosage Units
PQP requirements are harmonized with the Ph Int:
Content uniformity: a test and limit for content uniformity is required for each API present in the FPP at < 5 mg or < 5% of the weight of the dosage unit
Mass uniformity: for the API(s) present at 5 mg and 5% of the weight of the dosage unit, a test and limit for weight variation may be established in lieu of content uniformity testing;

Hua YIN | May 201428 |
Specification ParametersUniformity of dosage units – scored tablets
Specification ParametersUniformity of dosage units – scored tablets
Uniformity of split portions: content uniformity requirement applies to tablets containing < 5mg or 5% of API per portion. -– on a one-time basis and does not need to be added to the specifications.
For example, 10mg tabletssplit into 2 portions, each
portion contains 5mg --
mass uniformityinto 4 portions, each protion
contains 2.5mg – content uniformity

Hua YIN | May 201429 |
Fixed-dose combination FPPsFixed-dose combination FPPs
distinguish each API in the presence of the other API(s) , acceptance criteria for degradation products should be established
with reference to the API they are derived from. if an impurity results from a chemical reaction between two or
more APIs, its acceptance limits should be calculated with reference to the worst case (the API with the smaller area under the curve).
when one API is present at less than 5 mg or less than 5% of the weight of the dosage unit, a test and limit for content uniformity is required for this API in the FPP

Hua YIN | May 201430 |
Specification(s)Specification(s)
Monographs of Ph. Int., USP, BP are acceptable + additional in-house controls, e.g. Specific impurity related to the route of synthesis Individual unspecified impurity Residual solvents Particle size distribution, polymorphism form Those standard for the type of dosage form (e.g. friability, tablet hardness,
uniformity of dosage units, viscosity) Microbial limits ID and assay of preservatives
If non-pharmacopoeial monograph, note for guidance ICHQ6A applicable
For FPP : two set of specifications, i.e. Release and Shelf life are required

Hua YIN | May 201431 |
When compendial standard is claimed When compendial standard is claimed
Generally, the monograph tests and limits should be adopted.
The monograph is the minimum standard; the authority can impose additional requirements (e.g. ICH IT for individual unspecified related compounds)
May use in-house methods
All monograph tests/limits need not be included in specifications (may use in-house methods, provide justification for not including certain parameters).
But the product must comply with the monograph, if tested (see equivalency testing).

Hua YIN | May 201432 |
When in-house standard is claimed When in-house standard is claimed
Generally, the tightest available monograph limits for assay and purity should be adopted or justified.
The available monographs can still be used as a general guideline for requirements

Hua YIN | May 201433 |
When compendial methods are claimed…(purity and assay methods)
When compendial methods are claimed…(purity and assay methods)
Ensure the same compendial method is adopted. If not:
Check the deviations against published accepted deviations: USP <621> Chromatography EP 2.2.46 Chromatographic separation techniques
Note that there are specific SST limits for EP and USP methods. If these methods are adopted, the SST requirements should apply.

Hua YIN | May 201434 |
Validation, verification, equivalencyValidation, verification, equivalency
Validation: full validation required for in-house methods, generally specificity, linearity, accuracy, repeatability, intermediate precision, plus for purity: LOD/LOQ--ICH Q2
Verification: required for most compendial methods
Equivalency: required for an in-house method, when compendial standard is claimed
Refer to : Assessing specifications: Lynda Paleshnuik, PQP Quality Workshop, Copenhagen – January 2012

Hua YIN | May 201435 |
Case Study 3Case Study 3
pH of mobile phase: 3.3 Vs 2.2 (USP)= non USP method
Ratio of the mobile phase 86:14 Vs 88:12 (USP) = USP method
<621>: NMT 0.2 pH units, NMT 10% absolute change in MP%
Detector wavelength 254nm Vs 277nm (USP) = non USP method
Dissolution test uses apparatus II Vs USP apparatus I = non USP method
Non pharmacopoeial method → full validation required
Non pharmacopoeial method → equivalent to be demonstrated if pharmacopeial standard is claimed

Hua YIN | May 201436 |
Unnecessary tests in specificationsUnnecessary tests in specifications
In-process tests: used for the purpose of adjusting process parameters within an operating range; e.g. hardness, friability (see exception for chewable tabs, Zinc tabs), individual tablet weight
Impurities from the API synthesis which are not degradants; not normally controlled in FPP testing, and not included in the total impurities limit. Note that the enantiomer should be controlled. (Q6A)
Shelf-life specifications do not need to include tests that are not stability-indicating, such as identity and content uniformity.
Redundant tests: e.g. MU when CU is required and included

Hua YIN | May 201437 | 37
Periodic /Skip TestingPeriodic /Skip Testing
Specified tests on pre-selected batches and /or at predetermined intervals. e.g.
Polymorphic form (e.g API)
Genotoxic impurity (e.g. API)
Residual solvents (e.g. solid dosage form)
Microbiological testing (e.g. solid dosage form)
Dissolution (high water soluble, ICH decision tree #7)-- carefully justified
Justified by supportive results for five production batches.

Hua YIN | May 201438 |
Periodic /Skip Testing Periodic /Skip Testing

Hua YIN | May 201439 |
Case study 4Case study 4
The supplier's specification of Zidovudine includes the control of : Methyl methane sulfonate content (by GC-MS): NMT 2.5ppm Methyl-4-toluene sulfonate content (by HPLC) : NMT 2.5ppm Sum of Methyl methane sulfonate and Methyl-4-toluene sulfonate
content : NMT 2.5ppm to be performed every 10th individual batch and first individual batch of
every campaign:
The FPP manufacturer's specification of Zidovudine does not contain the above test. Is it acceptable?

Hua YIN | May 201440 |
Case study 4 cont'dCase study 4 cont'd
Answer: no
The FPP manufacturer is requested to include that the same control of above genotoxic impurities in the specification of Zidovudine (include the same skip testing is acceptable).

Hua YIN | May 201441 |
Common DeficienciesCommon Deficiencies
• The specification should include a reference number, version, date, and appropriate standard
• The specification should be dated and signed by authorized personnel
• Any differences between release and shelf-life tests and acceptance criteria should be clearly indicated and justified.
• If an officially recognized compendial standard exists and an in-house method is used, compliance to compendial requirements should be demonstrated unless otherwise justified.

Hua YIN | May 201442 |
Common Deficiencies Cont’dCommon Deficiencies Cont’d
The specification should include all standard drug product tests and limits for that dosage form
E.g. solid orals: description, identity, uniformity of dosage units (CU or MU), assay, related compounds, dissolution, microbial limits...

Hua YIN | May 201443 |
Review of API specification Review of API specification
monograph
CEP/ API-PQ/ APIMF
FPP manufacturer’
s API specification
Generic Guidance
ImpurityMDD
RoS Specific tests
ICH Q6AICH Q3
User specific
tests
QIS

Hua YIN | May 201444 |
Review of FPP specification Review of FPP specification
FPP manufacturer’s Signed and dated specification
Compare the summary in QIScomment
Review as per •Compendial monograph •generic guide•ICH Q6A, Q3B
Review the•Specific dosage form•specific test as per the manufacturing process

Hua YIN | May 201445 |
Summary and Key Advice Summary and Key Advice
Control of drug product and establishment of specifications key areas in the marketing dossier
Specifications should have been developed based on the results of the primary batches, primarily the biolot.
A comparison of the specs to the biolot results should result in questions or further considerations when the specs are much wider than, or are not representative of, the results.
Major concerns around impurities/degradation products and safety for the patient
Confirm the sameness to compendial methods if compendial standard is claimed, when applicable
Review QIS and confirm with the submitted dossier

Hua YIN | May 201446 | 46
Any questions?Any questions?