
Homogene Katalyse durch Metallkomplexe - uni-marburg.de · 3 Einleitung: Problemstellung: Wie kann...
84
1 Homogene Katalyse durch Metallkomplexe Teil I: Technische Katalyse empfohlene Literatur: B. Cornils, W.A. Herrmann: Applied Homogeneous Catalysis with Organometallic Compounds, A Comprehensive Handbook in Two Volumes, Vol 1: Applications, VCH, Weinheim 1996. G.W. Parshall, S.D. Ittel: Homogeneous Catalysis: The Applications and Chemistry of Catalysis by soluble Transition Metal Complexes, 2. Edition, Wiley, New York 1992. Ch. Elschenbroich, A. Salzer, Organometallchemie, Teubner, Stuttgart 1993. R.A. Sheldon, J.K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981. F.A. Cotton, G. Wilkinson, Advanced Inorganic Chemistry, 5. Edition, Wiley, New York 1988. Gliederung: Einleitung 1. Hydrierungen 1.1 Olefine (inkl. stereoselektive Varianten) 1.2 Transferhydrierung 1.3 Ketone (inkl. stereoselektive Varianten) 2. Isomerisierungen 2.1 Olefine via Hydrometallierung / β-H-Abstraktion an Alkylintermediaten 2.2 Olefine via 1,3-H-Shift an Allyl-Intermediaten 2.3 Allylalkohole 3. Oligomerisierungen 3.1 Olefin Oligomerisierung (Ziegler-Aufbaureaktion, SHOP) 3.2 Ethin-Tetramerisierung (Reppe) 3.3 Butadien-Di- und Trimerisierung (Wilke)
Transcript of Homogene Katalyse durch Metallkomplexe - uni-marburg.de · 3 Einleitung: Problemstellung: Wie kann...

1 Homogene Katalyse durch Metallkomplexe Teil I: Technische Katalyse empfohlene Literatur: B. Cornils, W.A. Herrmann: Applied Homogeneous Catalysis with Organometallic Compounds, A Comprehensive Handbook in Two Volumes, Vol 1: Applications, VCH, Weinheim 1996. G.W. Parshall, S.D. Ittel: Homogeneous Catalysis: The Applications and Chemistry of Catalysis by soluble Transition Metal Complexes, 2. Edition, Wiley, New York 1992. Ch. Elschenbroich, A. Salzer, Organometallchemie, Teubner, Stuttgart 1993. R.A. Sheldon, J.K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981. F.A. Cotton, G. Wilkinson, Advanced Inorganic Chemistry, 5. Edition, Wiley, New York 1988. Gliederung:
Einleitung 1. Hydrierungen 1.1 Olefine (inkl. stereoselektive Varianten) 1.2 Transferhydrierung 1.3 Ketone (inkl. stereoselektive Varianten) 2. Isomerisierungen 2.1 Olefine via Hydrometallierung / β-H-Abstraktion an Alkylintermediaten 2.2 Olefine via 1,3-H-Shift an Allyl-Intermediaten 2.3 Allylalkohole 3. Oligomerisierungen 3.1 Olefin Oligomerisierung (Ziegler-Aufbaureaktion, SHOP) 3.2 Ethin-Tetramerisierung (Reppe) 3.3 Butadien-Di- und Trimerisierung (Wilke)

24. Polymerisationen 4.1 1-Olefine (inkl. stereoselektive Varianten) 4.2 Butadien (inkl. stereoselektive Varianten) 4.3 ROMP 4.4 Epoxide und Lactone (inkl. stereoselektive Varianten) 5. Carbonylierungen 5.1 Hydrierende Carbonylierung von Olefinen (Hydroformylierung-Oxosynthese) 5.2 Protolytische Carbonylierung von Olefinen (BASF-Propionsäure-Prozeß) 5.3 Oxidative Carbonylierung von Alkoholen und Aminen 5.4 Copolymerisation von CO und Olefinen 5.5 Protolytische Carbonylierung von Alkinen 5.6 Carbonylierung von Alkoholen (Monsanto-Essigsäure-Prozess) 5.7 Carbonylierung von Nitroaromaten 5.8 Carbonylierung von Benzyl-X und Aryl-X Verbindungen in Gegenwart von Nu-H 5.9 Cn Bausteine aus Synthesegas (Fischer-Tropsch-Folgechemie) 6. HX-Addition an Olefine 6.1 Hydrocyanierung 6.2 Hydrosilylierung 6.3 Hydroaminierung 7. C-C-, C-N- und C-O-Kupplungen an Olefinen und Aromaten (nicht-oxidativ) 7.1 Heck-Reaktion und Verwandte 7.2 Cyclopropanierung (inkl. stereoselektive Varianten) 7.3 Metathese 7.4 Aminierung und Veretherung von Aryl-X Bindungen 8. Oxidationen 8.1 Olefine zu Carbonylverbindungen (Wacker-Prozess) 8.2 Olefine zu Epoxiden und Diolen (inkl. stereoselektive Varianten) 8.3 Olefine zu Allylalkoholen + α,β-ungesättigten Carbonylverbindungen + Lactonen 8.4 Aromaten-Oxidation 8.5 Aliphaten-Oxidation 8.6 Alkohol-Oxidation 8.7 Aldehyde und Ketone zu Carbonsäurederivaten 8.8 N-, und S-Oxidation 8.9 Ammonoxidation von Olefinen (allylisch / SOHIO, Diaminierung, Aziridinierung).

3Einleitung: Problemstellung: Wie kann auf Basis der leicht verfügbaren Cn-Bausteine (n = 1- 6) sowie von Wasserstoff, Sauerstoff, Stickstoff und Chlor ein ganzer Baukasten an organischen Grundprodukten ökonomisch sinnvoll und ökologisch vertretbar synthetisiert werden ? C1-Bausteine: CO, CO2, CH4, HCN C2-Bausteine: Ethen, Ethin C3-Bausteine: Propen C4-Bausteine: Butadien, 1-Buten C6-Bausteine: Benzol, Cyclohexan Wasserstoff: H2 Sauerstoff: O2, H2O2, H2O Stickstoff: N2, N2H4, NH3 Chlor: Cl2, HCl Schwefel: SO2, SO3 Diese meisten dieser Bausteine werden heterogen-katalytisch hergestellt: Technische Darstellung von H2 und CO
1. Steam-Reforming-Verfahren im Steamcracker (= Röhrenartiger "Dampfspalter" mit überhitztem Wasserdampf als Reaktandgas) liefert Spaltgas ("Synthesegas"): H2 aus Erdgas od. leichter Erdölfraktion in Gegenwart von Nickel-Katalysatoren bei 700-800°C CH4 + H2O 3 H2 + CO ∆H° = + 206 kJ/mol −CH2− + H2O 2 H2 + CO ∆H° = + 151 kJ/mol 2. Partielle Oxidation von schwerem Heizöl 2 CnH2n+2 + n O2 2n CO + 2(n+1) H2 ∆H° < 0 (exotherm) 3. Kohlevergasung (Erzeugung von "Wassergas" für synthetisches Benzin nach Fischer-Tropsch) C + H2O CO + H2 ∆H° = + 131 kJ/mol (endotherm) C + O2 CO2 ∆H° = − 349 kJ/mol (exotherm, liefert Energie) Im Anschluß an Verfahren 1 - 3: Konvertierung von CO zu CO2 im Wassergas-Gleichgewicht, Fe-Cr oder Cu-Zn Oxid Katalysatoren bei 200-400°C: CO + H2O (g) CO2 + H2 ∆H° = − 41 kJ/mol (exotherm)
Absorption von CO2 a) physikalisch: MeOH; b) reversibel chemisch: organische Amine

44. Thermisches Cracken von Erdöl (Benzingewinnung aus höheren KW) höhere Alkane CnH2n+2 ∆ → Cn-1H2n (kürzerkettiges Alkan) + H2 + C(s) Ruß höhere Alkane CnH2n+2 ∆ → 2 Cn/2Hn (2 kürzerkettige Alkene) + H2
Technische Darstellung der Basis-Olefine:
Nichtkatalytisches, thermisches Steamcracken von Erdgas (mit hohem Ethan-, Propan-, Butan-Anteil) bzw. von Naphtha (höhere KW). CnH2n+2 (Propan, Butan) ∆ → 2 CnH2n (Propen, Buten) + H2 C5H11 (Alkyl-Radikal) ∆ → C3H7 (Alkyl-Radikal) + Ethen C3H7 (Alkyl-Radikal) + C5H11 (Alkyl-Radikal) ∆ → C5H12 (Alkan) + Propen etc. Olefinanteil erhöht sich durch Unterbindung von Olefin-Folgereaktionen, d.h.
- kurze Verweilzeit im überhitzten Zustand, z.B. 900°C / 0.5 sec - Verringerung des KW-Partialdruckes durch Beimengung des (hier unreaktiven)
Wasserdampfes
Technische Darstellung von Cyanwasserstoff:
1. Ammonoxidation von Methan (Andrussow-Verfahren): CH4 + NH3 + 3/2 O2 HCN + 3 H2O ∆H° = − 473 kJ/mol drucklos, kurze Verweilzeit bei 1200°C an Pt-Netz
2. Ammondehydrierung von Methan (Degussa-BMA-Verfahren): CH4 + NH3 HCN + 3 H2 ∆H° = + 251 kJ/mol Trend: C1-Bausteine und Wasserstoff sind billiger als Cn-Bausteine; O2 ist am billigsten.

5
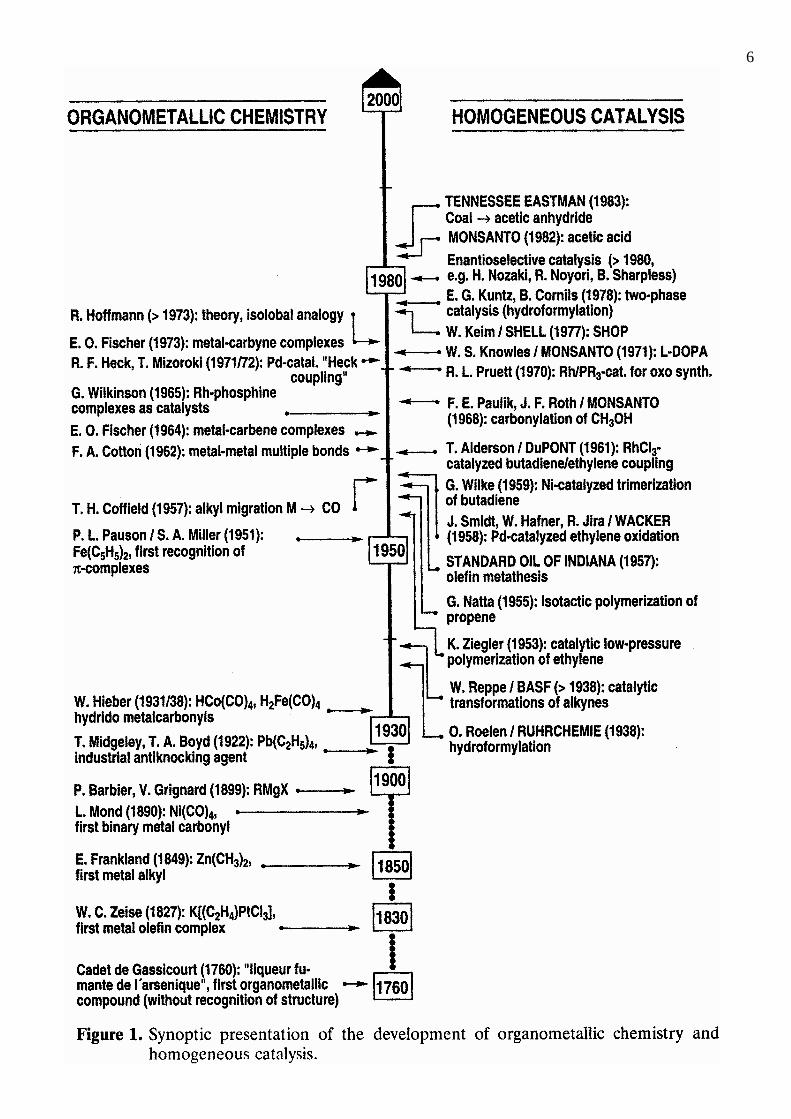
6

7

8Tabelle 1.2 Industrial Applications of Enantioselective Catalysis Chapter Reaction Product Use 2 Isomerisation of allylic amine L-Menthol Aroma and flavour chemical 3 Hydrogenation of enamides Levodopa Pharmaceutical L-Phenylalanine Food additive Hydrogenation of substituted S-Naproxena Pharmaceutical Acrylic acid (2-Arylpropion5ren) („Protene“) 6 Oxidation of allylic alcohol Dispalure Insect attractant Glycidol Intermediate Epoxidation of Indene Pharmaceuticals Syn-Dihydroxylations 9 Cyclopropanation Cilastatin Pharmaceuticals 1. Hydrierungen
1.1 Olefine (inkl. stereoselektive Varianten) Im wesentlichen 4 Klassen homogen löslicher Komplexe: 1. Rhodium(I)-Komplexe: Aktivierung von H2 durch Oxidative Addition
[Rh(PPh3)3Cl] Wilkinson-Katalysator (16VE für terminale > interne C=C-Bindungen) [Rh(PPh3)3(H)(CO)] (18VE für ausschließlich terminale Olefine) [Rh(L-L)(COD)]+ (16VE-Dienkomplexe) bzw. daraus durch Hydrierung des Diens → [Rh(L-L)(Solv)2]+ (16VE-Solventokomplexe insbes. für asymmetrische Hydrierung).
2. Platin(II)-Komplexe mit dem Trichlorostannat(II)-Liganden: Heterolytische H2-Aktivierung der Ligand (SnCl3)− ist ein schwacher σ-Donor und guter π-Acceptor (wie CO, PCl3) K2[PtCl4] + KCl + 5 SnCl2 K3[Pt(SnCl3)5] K3[Pt(SnCl3)5] + H2 K3[H-Pt(SnCl3)4] + H+SnCl3− K3[H-Pt(SnCl3)4] + Olefin K3[R-Pt(SnCl3)4] + H+SnCl3− K3[R-Pt(SnCl3)4] + H+SnCl3− K3[Pt(SnCl3)5] + RH (Protolyse) 3. Cobalt(0, I, II, III)-Komplexe: Homolytische H2-Aktivierung Homolytische Dissoziation von M-M-Bindungen oder paramagnetische Komplexzentren [Co2(CO)8] + H2 2 [H-Co(CO)4] [Co(CN)5]3− [Co2(CN)10]6− H2 → 2 [H-Co(CN)5]3−
wasserlöslich und selektiv für C=C-Bindungen in Konjugation zu C=C, C=O, C≡N, Ph. 4. Ziegler-Typ-Katalysatoren: löslicher Metallkomplex der 1. ÜM-Reihe + AlEt3, AliBu3 NiCl2, CoCl2, Co(acac)2 oder Co-2-ethylhexanoat plus AlR3 / H2 in Hexan.

9Typische mechanistische Charakteristika zum Katalyse-Mechanismus mit [Rh(PPh3)3Cl]: • terminale C=C-Bindungen rascher als sterisch anspruchsvollere, höhersubstituierte, interne
C=C-Bindungen • C=C- und C≡C-Bdg. sehr viel rascher als tolerierte Funktionalitäten −CH=O, −C≡N, −NO2 • cis-Addition von 2 H, wobei anti-Markovnikov-Orientierung bevorzugt wird
(Hydrometallierung erfolgt formal über stabileres, weniger substituiertes Carbanion). • bei Anwesenheit v. H2 und D2 kaum Scrambling (HD-Addition) → Synchrone 1,1-Addition • Mechanismus über Dissoziation des lagerbaren 16VE-Präkatalysators [Rh(PPh3)3Cl] in eine
hochgradig ungesättigte, energiereiche 14VE-Spezies: 14VE trans-[Rh(PPh3)2Cl] 16VE cis-[Rh(PPh3)2(µ-Cl)]2 (sterisch anspruchsvolle Phosphane mit großem Kegelwinkel dissoziieren leichter ab)
• → Oxidative Addition von H2 (synchron, cis über Zwischenstufe eines H2-Komplexes) zum 5-fach koordinierten 16VE-Komplex [Rh(PPh3)2Cl(H)2]
• → Koordination des Olefins zum oktaedrischen 18VE-Komplex [Rh(PPh3)2Cl(H)2(Olefin)] mit trans-ständigen Phosphanen,
• → cis-Insertion von Olefin in M-H-Bindung (H-Verschiebung anti-Markovnikov) zu einem 5-fach koord. 16VE-Hydrido-Alkylkomplex (mit diesem Kat: geschwindigkeitsbestimmender Schritt),
• → Reduktive 1,1-Eliminierung von R-H zur 14-VE-Spezies [Rh(PPh3)2Cl], die erneut H2 addiert oder sich unter Anlagerung des Phosphans zum quadr.-planaren 16-VE-Katalysatorkomplex stabilisiert.
η2-H2-Komplex als Intermediat der oxidativen Addition Mechanismus der Hydrierung mit dem Wilkinson-Katalysator [Rh(PPh3)3Cl] Andere Mechanismen mit anderen Rh(I)-Katalysatoren:
18VE-[Rh(PPh3)3(H)(CO)] (Wilkinson 1968): Dieser Komplex zeigt eine ausgesprochene Chemoselektivität gegenüber terminalen Olefinen. Dieser Präkatalysator muß zunächst in eine sterisch wenig anspruchsvolle, quadr.-planare 16VE-Spezies [Rh(PPh3)2(H)(CO)], dissoziieren, bei der im Gegensatz zu obigem Beispiel zuerst das Olefin addiert, dann H2 anlagert.

10[Rh(L-L)(Solv)2]+ (16VE-Solventokomplexe, Schrock, Osborn): • Speziell geeignet für die asymmetrische Hydrierung funktionalisierter und sterisch
anspruchsvoller Olefine. • Chelatisierendes, chirales Bisphosphan (keine 2 trans-ständigen Phosphane), vgl. (1). • Durch Koordination des Rhodiums von der re- oder si-Seite an das prochirale Olefin
entstehen 2 Distereomere (2), die über die Solventospezies im raschen Gleichgewicht miteinander stehen. si (linke Spalte) / re (rechte Spalte) = 5 / 95.
• Geschwindigkeitsbestimmender Schritt: Oxidative Addition von H2 erfolgt am in geringer Konzentration vorliegendem si-Komplex jedoch erheblich schneller als am re-Komplex, so daß nach rascher Insertion zu den diastereomeren Alkylkomplexen und reduktiver Eliminierung das R-Produkt (linke Spalte) mit ca. 95% ee überwiegt ! typische chirale Bisphosphane
L-Dopa (Therapie gegen Morbus-Parkinson) nach Monsanto 1977 (“PH”=3,4-C6H3(OH)2)

111.2 Transferhydrierung Bei der Transferhydrierung wird nicht molekularer Wasserstoff eingesetzt, sondern Wasserstoffdonatoren, die über ein vorgelagertes Gleichgewicht formal H2 (ein H+ und ein H−) bzw. Hydrido-Komplex-Zwischenstufen liefern. Die zwei wichtigsten Wasserstoffdonator-Systeme sind Isopropanol und Ameisensäure / NEt3. Erklärung: Das Prinzip der β-H-Eliminierung ist nicht allein auf Alkylmetallverbindungen
beschränkt (M = elektronenreiches Übergangsmetall, z.B. Ru, Rh).
OH + [M]-Cl [M] O
H
+ HClO
+
Isopropanol AcetonAlkoxid-Komplex
OH + [M]-Cl
H
+ HCl
Ameisensäure Formiat-Komplex
O
H O
[M] O [M]
H
[M]
H
+ CO2
[M]-Cl + H2
[M]-Cl + H2
HCl
HCl
1.3 Hydrierung von Ketonen (inkl. stereoselektive Varianten) Beobachtung von R. Noyori et al.; JACS 117 (1995) 2675: Die Turn Over Number (TON = Mol Produkt pro Mol Kat.) und die Turn Over Frequency (TOF [h-1] = Mol Produkt pro Mol Kat. pro Stunde) des für Benzophenon schlechten 16VE-Hydrierkatalysators [RuCl2(PPh3)3] nimmt enorm zu bei Anwesenheit von Ethylendiamin + KOH in Isopropanol (mögliche Erklärung s.u.) ! Enantioselektive Variante: Verwendet man ein C2-symmetrisches, chirales Bisphosphan: (S)-BINAP C2-symmetrisches, chirales Diamin: (S,S)-DPEN in Gegenwart von KOH und H2 in Isopropanol, so lassen sich prochirale, elektronenarme Ketone (Arylketone, z.B. Acetophenon + α,β-ungesättigte Ketone, z.B. vinyloge Ketone) hochenantioselektiv zu sekundären Alkoholen hydrieren (ee > 90-99 %). Beispiel:
Schema11. Asymmetric hydrogenation of unfunctionalized ketones

12Die re/si-Seiten-Stereodifferenzierung ist ein synergetischer Effekt des axial-chiralen (S)-Bisphosphans und des chiralen trans-(S,S)-Diamins an einem Koordinationszentrum (match-Fall), denn die Kombination von (S)-Bisphosphan und chiralem trans-(R,R)-Diamin oder mit dem meso-Diamin cis-(R,S)-DPEN führt zu drastischen Einbußen im ee-Wert (mismatch-Fall). Auf das chirale Diamin kann verzichtet werden, falls ein prochirales Keton vorliegt, das über eine α-Hydroxy- oder veresterten β-Carboxylfunktion am chiralen 16-VE-Solventokomplex [(S)-BINAP-RuCl2(Solv)] vorfixiert wird (chelatartige Präkoordination). Der Mechanismus dieser asymmetrischen Hydrierungen prochiraler Ketone ist im Falle der asymmetrischen Transferhydrierung elektronenarmer Arylketone mit Isopropanol als Wasserstoffdonator am weitesten aufgeklärt. vgl. R. Noyori et al.; Angew. Chem. 109 (1997) 297
• Eingesetzt wird eine 16VE-[(η6-Aren)RuCl2] Spezies (Aren = Mesitylen, Benzol, Cymol),
die im Festkörper als doppelt choroverbrücktes Dimer [(η6-Aren)(Cl)Ru(µ-Cl)2Ru(Cl)(η6-Aren)] (18VE) und in Lösung als 18VE-Solventospezies [(η6-Aren)RuCl2(Solv)] vorliegt.
• (S,S)-DPEN wird einfach N-tosyliert, wobei das R-NH(Ts) Proton eine Acidität, vergleichbar der eines Alkohols bekommt.
• Die Koordination dieses TsDPEN in Gegenwart von KOH führt zur einem Sulfonamido-amino-Chelatkomplex (Katalysatorvorstufe).
• In Gegenwart von KOH unterliegt dieser einer 1,2-Eliminierung von HCl, wobei eine katalytisch aktive Diamido-Spezies entsteht. Die nicht-tosylierte Amidofunktion stabilisiert mit ihrem hohen Ru=N-Doppelbindungsanteil das 16VE Ru-Zentrum.
• In Gegenwart von Isopropanol reagiert diese Spezies zum einem 16VE-Hydridokomplex unter Bildung von Aceton. Dies erfolgt wahrscheinlich über die Addition von iPrOH an die Ru=N-Bindung mit anschließender β-H-Eliminierung eines in geringer Gleichgewichts-
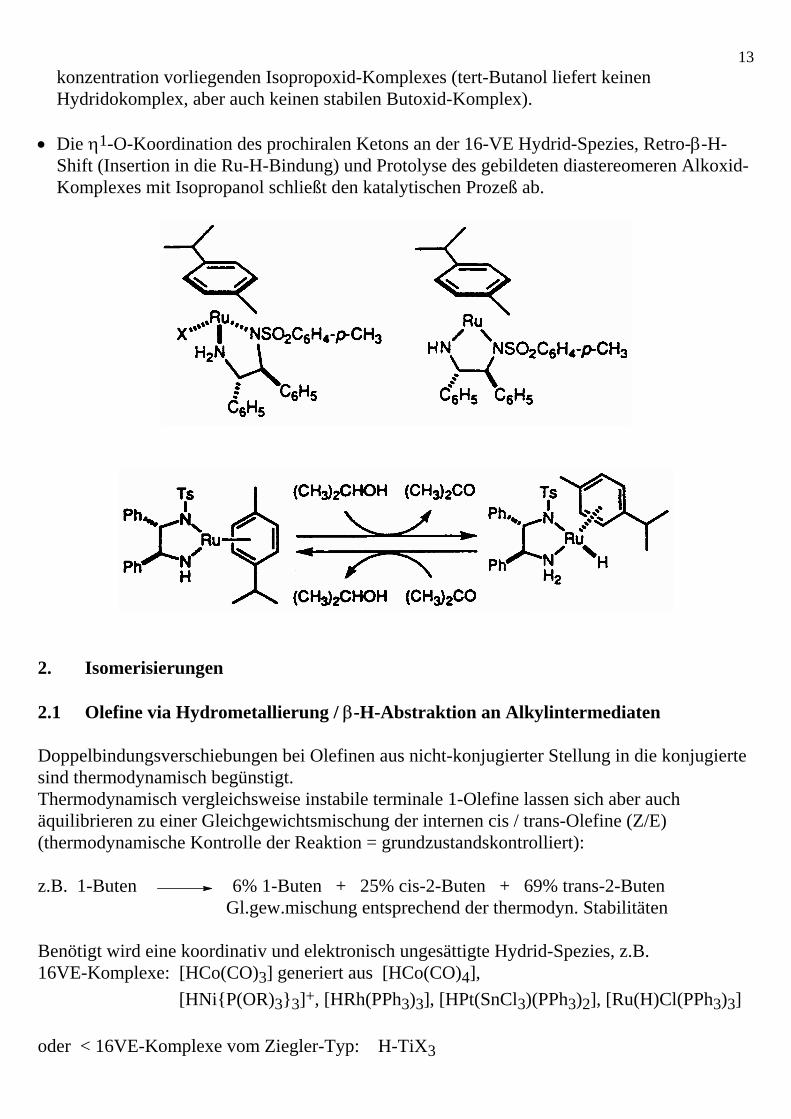
13konzentration vorliegenden Isopropoxid-Komplexes (tert-Butanol liefert keinen Hydridokomplex, aber auch keinen stabilen Butoxid-Komplex).
• Die η1-O-Koordination des prochiralen Ketons an der 16-VE Hydrid-Spezies, Retro-β-H-Shift (Insertion in die Ru-H-Bindung) und Protolyse des gebildeten diastereomeren Alkoxid-Komplexes mit Isopropanol schließt den katalytischen Prozeß ab.
2. Isomerisierungen 2.1 Olefine via Hydrometallierung / β-H-Abstraktion an Alkylintermediaten Doppelbindungsverschiebungen bei Olefinen aus nicht-konjugierter Stellung in die konjugierte sind thermodynamisch begünstigt. Thermodynamisch vergleichsweise instabile terminale 1-Olefine lassen sich aber auch äquilibrieren zu einer Gleichgewichtsmischung der internen cis / trans-Olefine (Z/E) (thermodynamische Kontrolle der Reaktion = grundzustandskontrolliert): z.B. 1-Buten 6% 1-Buten + 25% cis-2-Buten + 69% trans-2-Buten Gl.gew.mischung entsprechend der thermodyn. Stabilitäten Benötigt wird eine koordinativ und elektronisch ungesättigte Hydrid-Spezies, z.B. 16VE-Komplexe: [HCo(CO)3] generiert aus [HCo(CO)4],
[HNi{P(OR)3}3]+, [HRh(PPh3)3], [HPt(SnCl3)(PPh3)2], [Ru(H)Cl(PPh3)3] oder < 16VE-Komplexe vom Ziegler-Typ: H-TiX3

14Die Hydrometallierung zu 7 (anti-Markovnikov) ist unproduktiv in bezug auf die Isomerisierung des terminalen Olefins. Die mit geringerer Geschwindigkeit (Wahrscheinlichkeit) erfolgende Hydrometallierung zu 8 (Markovnikov) liefert dagegen ein Alkylintermediat, für das zwei Wege der β-H-Eliminierung bestehen, die zum cis- bzw. trans-Olefin führen (vgl. Newman-Projektion).
2.2 Olefine via 1,3-H-Shift an Allyl-Intermediaten Ein alternativer Mechanismus der Isomerisierung bietet sich mit Metallkomplexen, die eine besonders ausgeprägte Tendenz zur Ausbildung von η3-Allyl-Intermediaten besitzen (z.B. Ni0/2+, Pd0/2+). Schlüsselschritt ist die intramolekulare CH-Aktivierung, d.h. die oxidative Addition einer allylischen CH-Bindung an ein niedervalentes Pd(0)-Zentrum, gefolgt von einer reduktiven Eliminierung aus dem Allyl(hydrido)-Intermediat.

15Läßt man Isomerisierungen nicht bis zum thermodynamischen Gleichgewicht fortschreiten, so lassen sich mitunter Produkte der kinetischen Kontrolle, d.h. thermodynamisch instabilere Olefine (Enamine) isolieren. → Anwendung in der enantioselektiven Isomerisierung prochiraler Olefine durch bevorzugte Koordination von re- oder si-Seite, Kat: [Rh(BINAP)COD]+ClO4:
Takasago-Prozeß: 1995: 1500 jato (-)Menthol; größte industrielle Anwendung asymmetrischer Katalyse
Terpen- Duftstoff-Chemie
2.3 Allylalkohole via 1,3-OH-Shift Tert-Allylalkohole, erhalten beispielsweise durch Addition von Ethin an Ketone und nachfolgender selektiver Hydrierung oder durch 1,2-Addition an α,β-ungesättigte Ketone, erleiden in Gegenwart von Vanadinsäureestern [VO(OR)3] (R = Alkyl, Silyl) einen 1,3-OH-Shift zu den thermodynamisch stabileren prim. und sek. Allylalkoholen mit interner Doppelbindung (thermodyn. Kontrolle). Der Mechanismus entspricht einer Metallo-Variante der Claisen-Allyl-Umlagerung (konzertiert über 6-Ring, → Terpen-Chemie → Duftstoffe):

163. Oligomerisierungen 3.1 Olefin-Oligomerisierung (Ziegler-Aufbaureaktion, SHOP) Aluminiumorganyle zeichnen sich aus durch • unübertroffene elektrophile Reaktivität des monomeren "R3Al" (falls kein Etherkomplex) • sehr bereitwillige Hydro- und Carbaluminierung von Alkenen und Alkinen • kostengünstige technische Produktion (billigstes Aktivmetall in der metallorganischen
Chemie). Technische Darstellungsverfahren von Aluminiumalkylen: Trimethyl- und Triethylaluminium über das Sesquichlorid (Fa. Hüls) 4 Al + 6 MeCl 2 Me3Al2Cl3 Me4Al2Cl2 + Me2Al2Cl4 Al mit "Sesquichlorid" Et3Al aktiviert + 2 NaCl ↓ 2 Me6Al2 + 2 Al + 6 NaCl ← 6 Na auf 3 Äq. Me4Al2Cl2 (l) + 2 Na[MeAlCl3] ↓ Kostengünstiger durch Verwendung von H2 und Ethen anstatt Natrium + EtCl ist das Ziegler-Direktverfahren: Al + 1.5 H2 + 2 [Et3Al]2 80-160°C, 100-200 bar → 3/n [Et2AlH]n "Vermehrung"
3/n [Et2AlH]n + 3 Ethen 80-110°C, 1-10 bar → 3/2 [Et3Al]2 "Anlagerung" _________________________________________________________________________ 2 Al + 3 H2 + 3 Ethen [Et3Al]2 Prinzip: • Al reagiert in Gegenwart von R3Al mit H2! Al-H addiert sich an Alkene, anti-Markovnikov. • Reaktionsfähigkeit von Al wird durch Zulegieren von 0.01 - 2% Ti erhöht. • Anlagerungsschritt (Hydroaluminierung) ist reversibel und die Affinität von Al-H zu
Alkenen nimmt mit steigendem Substitutionsgrad der Doppelbindung (sterischen Anspruch) ab. Deshalb läßt sich Isobuten aus Triisobutylaluminium mit weniger anspruchsvollen Olefinen (1-Octen → Propen → Ethen) im Gleichgewicht verdrängen.
2 Al + 3 H2 + 6 H2C=CMe2 [iBu3Al]2 [iBu2Al-H]2 + H2C=CMe2
[iBu2Al-H]2 + 2 Propen [iBu2(nPr)Al]2 über Sdp. Gl.gew. verschieben Al(iBu)3 + 3 1-Octen Al(nOct)3 + 3 H2C=CMe2 iBu = −CH2−CHMe2

173.1 Oligomerisierung von Ethylen zu höheren linearen α-Olefinen: C4-C8: Co-Monomere für Polyethylen C6-C10: als primäre Alkohole → Weichmacher für PVC und im Waschprozeß C12-C20: als Sulfonate und Sulfate → anionische Tenside, bioabbaubare Detergenzien C10: als Oligomere → synthetische Hochleistungs-Schmierstoffe C10-C12: über Epoxide zu 1,2-Diolen → nichtionische Tenside 3.1.1 Oligomerisation durch die Ziegler-Aufbaureaktion:
→ Höherkettige 1-Alkene, synthetische Fettalkohole und ihre Sulfate (Tenside)
3.1.2 Oligomerisation durch den Shell Higher Olefin Process (SHOP)
→ besonders ökonomische Kombination von 4 homogen- / heterogen-katalytischen Verfahren: Oligomerisation - Isomerisierung - Metathese - Hydroformylierung
1. Oligomerisation von Ethen an einem Nickelhydrid Chelatkomplex (zur effizienteren
Katalysatorabtrennung im 2-Phasensystem 1,4-Butandiol + Ni-Kat. / α-Olefingemisch; 2 Anlagen mit zusammen 1 Mio jato Kapazität). Die eingesetzten Ni-Komplexe bestehen aus einem P∩O-Chelatliganden, der die Selektivität kontrolliert und einem organischen Anteil, der den Komplex stabilisiert (Phosphan) und unter den Reaktionsbedingungen die katalytisch aktive Hydridspezies liefert (Alkyl-, Arylligand).

18Katalysatorvorstufen und Aktivierungsschritte im SHOP Prozeß:

192. Isomerisierung der nicht brauchbaren Fraktionen der 1-Olefine C4-C6 bzw. C20-C30 zu
internen Olefinen, die für die Olefin-Metathese geeignet sind.
3. Metathese der kurz und langkettigen Olefine zu den gewünschten C10-C18 -Olefinen. 4. Isomerisierung und Hydroformylierung, gefolgt von einer Hydrierung zu Fettalkoholen
durch ein Cobaltcarbonylhydrid. Fließschema der SHOP-Anlage:

203.1.3 Katalytische Propen-Dimerisierung → Isopren + Methan → Synthesekautschuk:
2-Methylpenten ist auch das Hauptprodukt der sechs möglichen Primärprodukte der Propen-Dimerisierung durch kationische Nickelhydrid-Komplexe (Hydro- und Carbometallierung anti-Markovnikov):
3.3.4 Dimerisierung funktioneller Olefine (AlR3 ungeeignet !) Für die Gewinnung von Nylon (Polyamid 6,6) basierend auf Propen ist das folgende Verfahren ausgearbeitet worden (als Alternative für die Sequenzen Cyclohexan → Cyclohexanonoxim → Lactam bzw. Butadien + 2 HCN): Allylische Oxidation von Propen liefert Acrolein → Acrylsäure → Acrylsäureester (Acrylate) Allylische Ammonoxidation von Propen liefert Acrylnitril (SOHIO-Prozess, s.u.) Die Alkoholyse von RhCl3, RuCl3 bzw. PdCl2 liefert auf dem Wege der β-H-Eliminierung der Alkoxide die notwendige Hydridspezies: MCl3 + EtOH HCl + H-MCl2 + MeCHO alternative Generierung kationischer Hydridspezies: [(Ethen)2RhCl]2 + Lewis-Säure + Proton

21
Die linearen Dimerisierungsprodukte lassen sich hydrieren zum Adipinsäurediester und zum Hexamethylendiamin, deren Kondensation das Polyamid Nylon 6,6 liefert. 3.2 Ethin-Tetramerisierung (Reppe, BASF 1940) Katalysator: Nickelacetylid, gebildet aus Ni(CN)2 / CaC2 od. Ni(CN)2 / HC≡CH in THF absorbiert Ethin (20 bar, 70°C) unter Bildung von COT ! Bis(allyl)nickel und Ni(0)-Komplexe gehen auch: Ni(CO)4 und Ni(COD)2 in Gegenwart von 1 PPh3 / Ni ist die letzte Cycloinsertion
(Carbometallierung) gehemmt → es entsteht Benzol. Alkine: Ethin → COT, 1-Alkine → subst. COT (disubst. Alkine gehen nicht) Mechanismus: weitgehend ungeklärt, jedoch wie folgt plausibel:
Ni Ni0 +2
Ni+2
Ni+2
Ni+2
Ni+2 Ni
0
Alkin K. vs. Metallacyclopropen Carbomet.
Redukt. Elim.
COT

223.3 Butadien-Di- und Trimerisierung (Wilke) Folgende Allyl-Insertionsmechanismen und Allyl-Allyl-Kupplungen spielen als Elementarreaktionen bei Transformationen an 1,3-Dienen eine Rolle: • 1,3-Dien-Insertion in M-H (Hydrometallierung eines 1,3-Diens) zum Allyl-Komplex
Different configurations and modes of coordination of the terminally mono- substituted allyl anion (R, organyl, e.g., the growing polybutadienyl chain • 1,3-Dien-Insertion in M-C (Carbometallierung eines 1,3-Diens) zum neuen Allyl-
Kompl. Schematic representation of the “allylinsertion mechanism“ as the catalytic Principle of chain growth in the complex-catalyzed diene polymerisation • Reduktive Eliminierung eines 1,5-Diens aus einem Bis(allyl)-Komplex wichtigste Dimerisierungsprodukte:

23Trimerisierung von Butadien zu 1,5,9-Cyclododecatrien (CDDT): 2 Einstiegsalternativen: t,t,t-1,5,9-CDDT c,t,t-1,5,9-CDDT erhält man in 70-90% aus einem in situ aus TiCl4 / Al2Cl3Et3 dargest. Kat. Vergleiche:
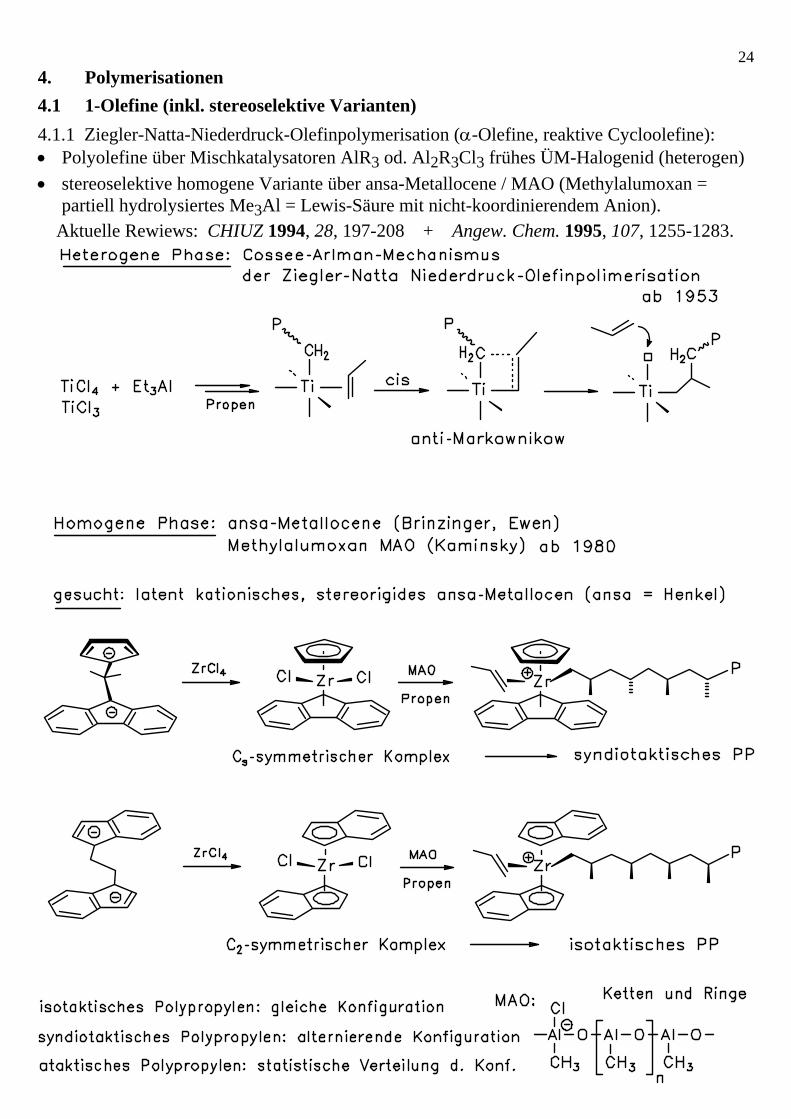
244. Polymerisationen
4.1 1-Olefine (inkl. stereoselektive Varianten)
4.1.1 Ziegler-Natta-Niederdruck-Olefinpolymerisation (α-Olefine, reaktive Cycloolefine): • Polyolefine über Mischkatalysatoren AlR3 od. Al2R3Cl3 frühes ÜM-Halogenid (heterogen) • stereoselektive homogene Variante über ansa-Metallocene / MAO (Methylalumoxan =
partiell hydrolysiertes Me3Al = Lewis-Säure mit nicht-koordinierendem Anion). Aktuelle Rewiews: CHIUZ 1994, 28, 197-208 + Angew. Chem. 1995, 107, 1255-1283.
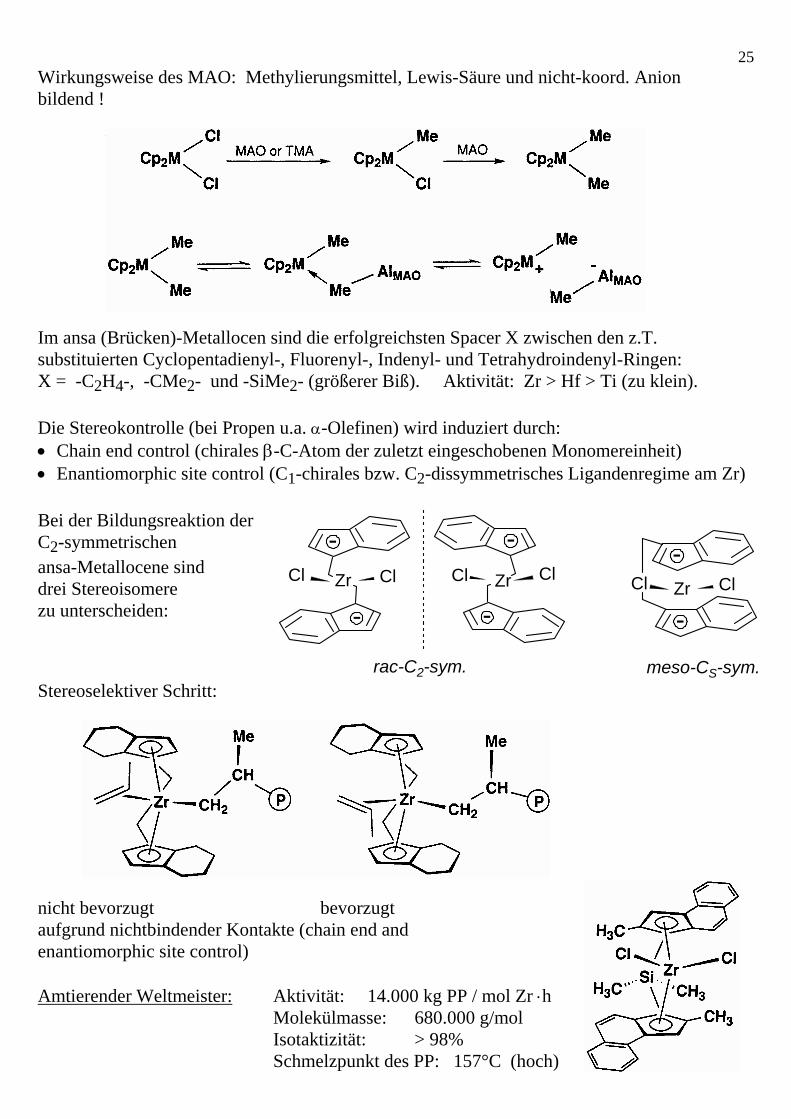
25Wirkungsweise des MAO: Methylierungsmittel, Lewis-Säure und nicht-koord. Anion bildend ! Im ansa (Brücken)-Metallocen sind die erfolgreichsten Spacer X zwischen den z.T. substituierten Cyclopentadienyl-, Fluorenyl-, Indenyl- und Tetrahydroindenyl-Ringen: X = -C2H4-, -CMe2- und -SiMe2- (größerer Biß). Aktivität: Zr > Hf > Ti (zu klein). Die Stereokontrolle (bei Propen u.a. α-Olefinen) wird induziert durch: • Chain end control (chirales β-C-Atom der zuletzt eingeschobenen Monomereinheit) • Enantiomorphic site control (C1-chirales bzw. C2-dissymmetrisches Ligandenregime am Zr) Bei der Bildungsreaktion der C2-symmetrischen ansa-Metallocene sind drei Stereoisomere zu unterscheiden:
Zr ClCl
meso-CS-sym.
-
-Zr ClCl
-
-ZrCl Cl
rac-C2-sym.
-
-
Stereoselektiver Schritt: nicht bevorzugt bevorzugt aufgrund nichtbindender Kontakte (chain end and enantiomorphic site control) Amtierender Weltmeister: Aktivität: 14.000 kg PP / mol Zr ⋅h
Molekülmasse: 680.000 g/mol Isotaktizität: > 98% Schmelzpunkt des PP: 157°C (hoch)

26Spezielle Polymere und andere Polymerisationskatalysatoren: Phillips-Katalysatoren: Cr(II) / Cr(III) auf SiO2 in Gegenwart von Al2Et3Cl3, Union Carbide 1. Imprägnieren von Silicagel mit löslichen Chromkomplexen, z.B. CrO2(OSiPh3)2 od. Cp2Cr. 2. Reduktion mit Aluminiumorganylen liefert H-Cr(III) bzw. R-Cr(III). 3. Homogener Katatysator: [Cp*Cr(CH3)Cl]2 / MAO Mitsui Petrochemical's high value amorphous polyolefin "APO";V(III) + Al2Et3Cl3,V(II) ist inakt.; Cycloolefin-Copolymere (COC) für Compact Discs, optische Linsen und Datenspeicher. CS-sym. ansa-Metallocen + MAO für Norbornen / Ethen - COC funktioniert ebenso. Dow und Exxon "constrained geometry catalysts" für die Copolymerisation von Ethylen und 1-Octen (→ Elastomere) in Gegenwart von MAO: Alternative Einführung eines nichtkoord. Anions: [M-CH3] + [PhNMe2H]+ [B(C6F5)4]− oder [M-CH3] + B(C6F5)3 Waymouth : Stereoselektiv-cyclisierende Polymerisation von 1,5-Hexadien zu Poly(methylen-1,3-cyclopentan)
M Cpoly
M Cpoly
M Cpolyn
trans-isotaktisch
Kat.: C2-sym. ansa-Metallocen mit C2-sym. Binaphthol-Liganden / MAO

26aCyclopentadienyl-Imido-Ligandanalogie
Empirischer Vergleich
der Grenzorbital-Eigenschaften
[η5-C5H5M] ⇔ [RN≡M]
Metall-d-Orbitale gleicher Symmetrie werden zu (σ- und) π-Bindungen herangezogen:
+
+ +
+
++
++
N +
+
+ +
+
+
dxz dyz dxz dyz
px py
e1
++ +++
+
pz
a1
+
+
dz2
N+
pz
σ π π π π
+
+ +
++
σ
z
xy
Anzahl der Elektronen in beiden Fragmenten gleich, wenn ....
Nb Mo+ (isoelektr.) + [C5H5]- [RN]2- (6-Elektr.-Donor)
________________________________
[C5H5 Nb]- [RN Mo]- (gleiche Elektronenzahl) Die Anzahl, Gestalt, ungefähren Energie, die Symmetrie-Eigenschaften und die Zahl der Elektronen in den Grenzorbitalen sind in folgenden Baugruppen vergleichbar:
M
Metall
M
NR
σ,2π σ,2π
'
"(n+1)"
Ligand
Gruppe "n"

26bBeispiele für Isolobalbeziehungen:
Ti V
NR
CrRN
NR
RNVTi
Cr
NR +
Jüngste Entwicklung bei Single-Site-Katalysatoren (L)MX2 für die Olefin-Polymerisation Bekannte Katalysatoren:
ClTiN
Cl
Me2Si
Me3C
Dow, Exxon, EP 420436 (1991) Waymouth et al. Chem. Rev. 98 (1998) 2587
ClTi
ClO
Nomura et al. OM 17 (1998) 2152.
N
NTi
Cl
Cl
McConville et al. JACS 118 (1996) 10008.
ClTi
NCl
tBu3P
Stephan et al. Nova Chemicals WO 00/05236.
H.W. Turner, V.J. Murphy, Exxon USP 5,851,945 (1997) OM 16 (1997) 2495
ClV
OCl
N
ClV
NCl
N
Me
Cr
N
NCl
Cl
CH3H3CCH3
CH3
CH3
H3C
V.C. Gibson > 1994 BP Chemicals, V.C. Gibson, JCS, Chem. Commun. (1998) 849 Union Carbide, Brookhart et al. JACS 120 (1998) 4049
N
M
ClCl
M = Fe, Co R = Me, iPr
NN
R
R
R
R
NAr
NAr M
ClCl
NAr
NAr M
ClCl M= Ni, Pd
Brookhart et al. > 1994
Review: Metallocen-Substitute als Single-Site Katalysatoren, Angew. Chem. 111 (1999) 449.

274.2 Butadien und Isopren (inkl. stereoselektive Varianten) Lediglich das aus einer cis-1,4-Polymerisation hervorgegangene Polyisopren besitzt als synthetischer Kautschuk die herausragenden Eigenschaften des natürlichen Kautschuks. Stereoselektivität der Isopren Polymerisation; die 1,2- und 3,4-Addition kann iso-, syndio- oder ataktisch erfolgen: Wie bei der Oligomerisation (s.o.) ist der Allyl-Insertionsmechanismus wirksam: • man geht von isolierbaren Allylkomplexen von Ni, Co, Ti, Nd als Katalysatoren aus • oder man erzeugt die Initiator-Allylspezies in situ
→ durch technische Carbolithiierung des Isoprens mit n-Buli, t-Buli: → wichtige technische Verwendung von Alkyllithium-Verbindungen in der anionischen cis-1,4-Polymerisation von 1,3-Dienen → oder durch Behandlung von Metallcarboxylaten mit Aluminiumalkylen:
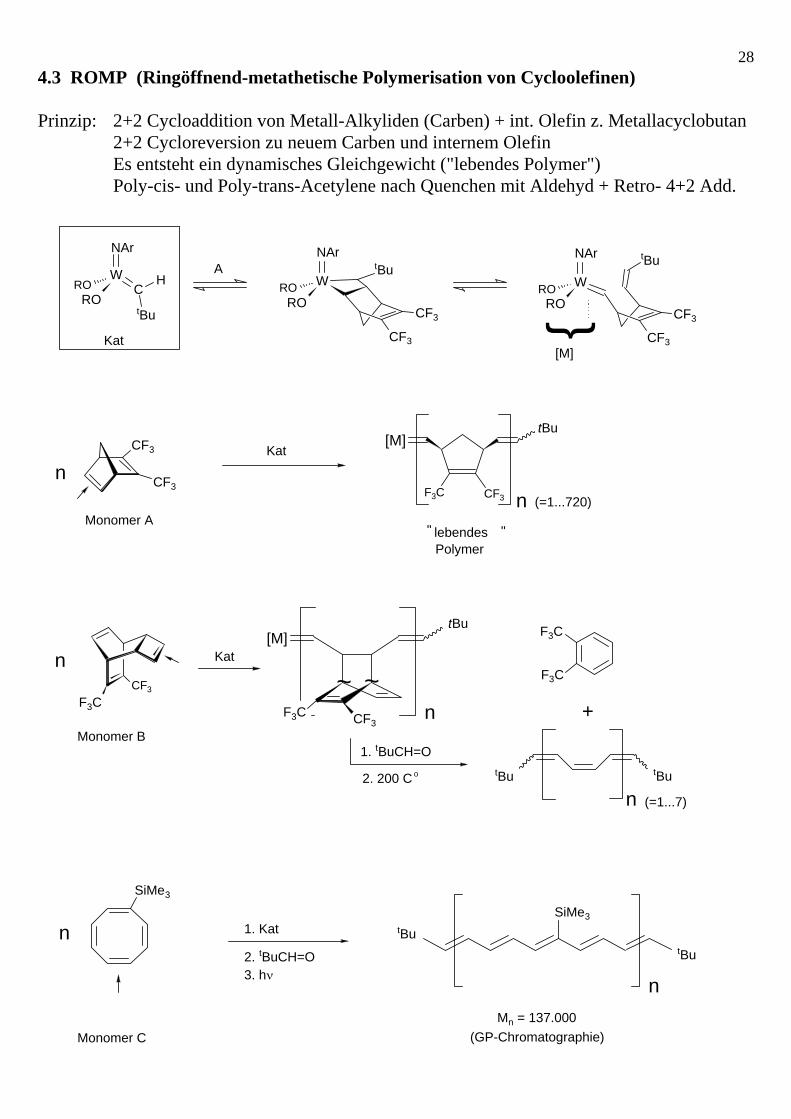
284.3 ROMP (Ringöffnend-metathetische Polymerisation von Cycloolefinen) Prinzip: 2+2 Cycloaddition von Metall-Alkyliden (Carben) + int. Olefin z. Metallacyclobutan 2+2 Cycloreversion zu neuem Carben und internem Olefin Es entsteht ein dynamisches Gleichgewicht ("lebendes Polymer") Poly-cis- und Poly-trans-Acetylene nach Quenchen mit Aldehyd + Retro- 4+2 Add.
W
NAr
tBu
CF3
CF3
CF3
CF3
NArtBu
WRO
CH
RORO RO
CF3
CF3
NAr tBuW
RORO
Monomer AlebendesPolymer
" "
CF3
F3C
[M]
n
tBu
CF3F3C
[M]
n
tBu
CF3F3C
~~
tButBu
n
n
Kat
F3C
F3C
1. tBuCH=O
2. 200 C o
+
A
n
Monomer B
(=1...720)
(=1...7)
SiMe3
Kat
n 1. Kat
2. tBuCH=O3. hν
tButBu
SiMe3
nMn = 137.000
(GP-Chromatographie)
Kat
Monomer C
{
[M]
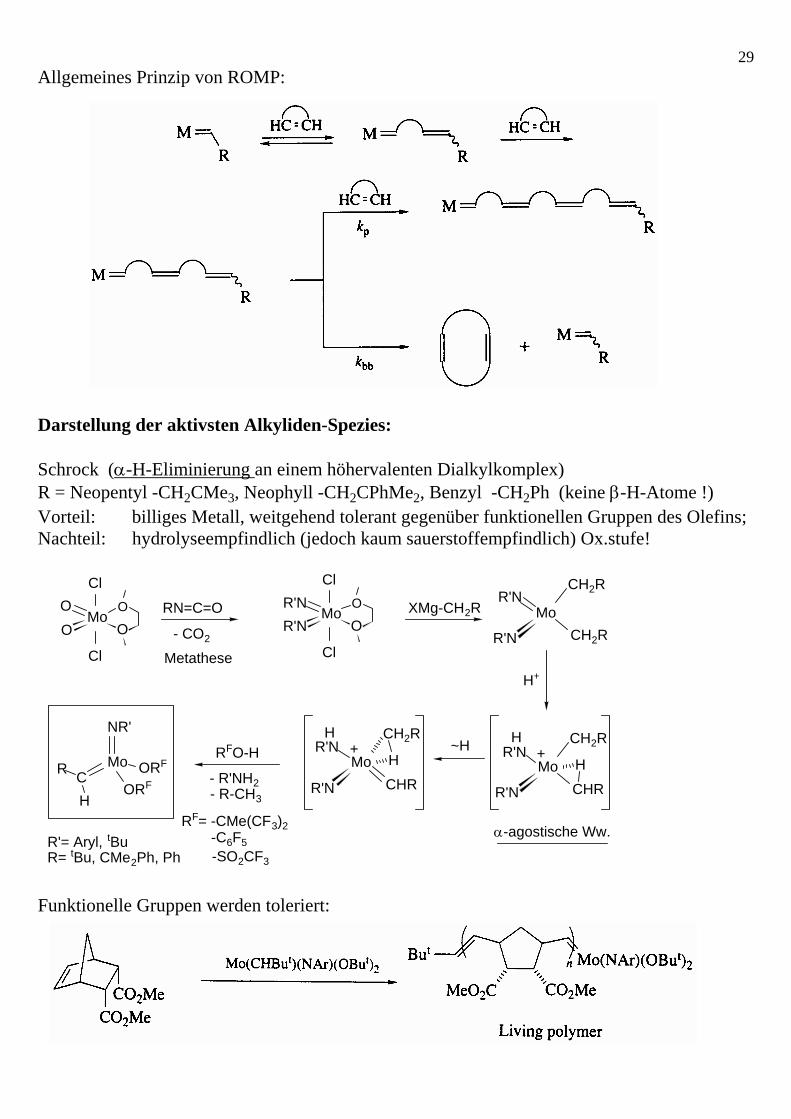
29Allgemeines Prinzip von ROMP: Darstellung der aktivsten Alkyliden-Spezies: Schrock (α-H-Eliminierung an einem höhervalenten Dialkylkomplex) R = Neopentyl -CH2CMe3, Neophyll -CH2CPhMe2, Benzyl -CH2Ph (keine β-H-Atome !) Vorteil: billiges Metall, weitgehend tolerant gegenüber funktionellen Gruppen des Olefins; Nachteil: hydrolyseempfindlich (jedoch kaum sauerstoffempfindlich) Ox.stufe!
Mo
Cl
Cl
O
OOO Mo
Cl
Cl
O
OR'NR'N
Mo
R'N
R'NCH2R
CH2R
Mo
R'N
R'NCH2R
CHR
H+
H
H+
α-agostische Ww.
Mo
R'N
R'NCH2R
CHR
H
H+
RN=C=O
- CO2
XMg-CH2R
RFO-H
RF= -CMe(CF3)2-C6F5-SO2CF3
~HMo
NR'
C ORF
ORFR
H
R= tBu, CMe2Ph, PhR'= Aryl, tBu
- R'NH2
Metathese
- R-CH3
Funktionelle Gruppen werden toleriert:

30Grubbs (aus dem Tebbe Reagens durch Zugabe von Base, z.B. Dimethylaminopyridin DMAP) Nachteil: [Ti-C] sehr hydrolyseempfindlich + intolerant gegenüber funktionellen Gruppen, "[Cp2Ti=CH2]" nur als Basen- oder Olefinaddukt (Metallacyclobutan) stabil Grubbs (aus Carben-Vorläufern und der 16-VE-Spezies RuCl2(PPh3)3 ) Vorteil: recht hydrolysestabil (jedoch mäßig sauerstoffempfindlich) Ox.stufe!,
tolerant gegenüber funktionellen Gruppen.

314.4 Polymerisation von Epoxiden u.a. cyclischen Ethern,
von Estern (MMA) und Lactonen (inkl. stereoselektive Varianten) Epoxide u.a. cyclische Ether (inkl. stereoselektive Varianten) Im rac-Propenoxid läßt sich stereoselektiv ein Enantiomer zu isotaktischem Material s,s,s ...-[-CH2-C*HMe-O-] polymerisieren, während das andere Enantiomer eine andere Kette (r,r,r ...) startet. Verwendet werden Metall-Lewis-Säuren generiert durch Reaktion von ...: ZnEt2 / H2O bzw. AlEt3 / H2O / Acetylaceton (→ Chain end control) ZnEt2 / (+)-borneol (Chain end control + enantiomorphic site control → kinetische
Racematspaltung, ein Enantiomer bleibt unreaktiv zurück !) Oligomere und Polymere von Ethylen- und Propylenoxid finden Verwendung als: Klebstoffe, Weichmacher, Tensid-Bausteine, Beschichtungsmittel etc. THF - Oligomere mit OH-terminalen Enden interessant als Polyurethan-Bausteine:
3 THF + H2O + Lewis-Säure-Kat. HO{(CH2)4-O}3H (→ + Diisocyanate) Methacrylsäure-Ester (MMA) (stereoselektive Variante, nicht-radikalische Polymerisation) Als Katalysatoren für die stereoselektive Polymerisation von Methylmethacrylat MMA (und nichtfunktionalisierten α-Olefinen) dienen metallorganische Lewis-Säuren der Lanthanide Ln: Chain-end control der α-Methylgruppe führt insbesondere bei MMA zu Bildung eines • syndiotaktischen "lebenden" Polymers, • hoher Molekülmasse Mn > 500 000 g / mol • enger Molekülmassenverteilung (Polydispersität PDI = Mw / Mn = 1.05) • das nach dem Quenchen heiß verformbar und biologisch abbaubar ist (Plexiglas).

32Mechanismus: Michael-Addition über Metall-Enolat Zwischenstufe Ringöffnende Polymerisation von Lactonen und Lactiden Anwendung finden Metall-Lewis-Säuren vom Typ:
Cp*2Sm(THF)2 Y5O(OiPr)13 Bu3Sn-OR
Y(OCH2CH2OEt)3 Y(OCH2CH2NMe2)3 TON 30 / min (1.3 M Dilactid bei 70°C) vgl. Al(OiPr)3 TON 0.8 / min
Mechanismus: Alkoxid M-OR' wird auf den O-koordinierten Ester R''COOR übertragen; metal-
lierter ortho-Ester M-O-C(OR)(OR')R'' zerfällt unter C−O Bindungsspaltung. Anwendung: Medizinische Polymere: enantiomerenreine d,d- bzw. l,l-Dilactide (dimere,
cyclische Milchsäureanhydride) lassen sich ringöffnend polymerisieren zu resorbierbarem Nahtmaterial, Knochenschrauben etc.
Ebenso: bioabbaubares Poly(ε-caprolacton), Poly(1,4-dioxanon) etc.

335. Carbonylierungen
5.1 Reduktive Carbonylierung: Olefine + CO + H2 (Hydroformylierung = Oxosynthese)
Hydroformylierung = Addition von H• und •CHO an ein Olefin. Entdeckung durch Otto Roelen (Ruhrchemie, 1938): aus Ethen und Synthesegas: Propionaldehyd Die wichtigsten "Oxoprodukte" heutzutage: aus 1-Octen, 1-Dodecen: lineare Fettalkohole (Detergentien) vgl. SHOP aus Propen und Synthesegas: n-Butylaldehyd, n-Butanol,
2-Ethylhexanol (→ PVC-Weichmacher) Gängigste Katalysatoren: Co2(CO)8, HCo(CO)3PBu3, HRh(CO)(PPh3)3, HRh(CO)2(PPh3)2 Probleme mit billigem Cobalt-Katatysator: - Hoher H2 Druck erforderlich zur Co-Co Spaltung und Kat.-Stabilisierung, - Katalysatorverluste, da HCo(CO)4 labil und leicht flüchtig ist (Abtrennung !), - Alkenverluste durch Hydrierung oder i.d.R. ungewollte, verzweigte Produkte. Trends in der Kat.-Aktivität / Selektivität • Rh >> Co, Pd > Ir, Ru >> Ni • P(OR)3 > PPh3 >> NPh3 > AsPh3 Trends in der Reaktivität der Olefine / Selektivität • terminale Olefine >> interne Olefine
(daher läßt sich terminales Olefin aus dem Isomerisierungs-Gl.gew. abfangen) • meist überwiegen lineare Oxoprodukte (anti-Markovnikov-Hydrometallierung) die verzweigten (Markovnikov).

34Mechanismus mit nicht Phosphan-modifizierten Co- u. Rh-Katalysatoren:
1. Co-Co Homolyse durch H2 unter Bildung des aciden Hydrids (hoher Druck) 2. Dissoziation von CO unter Bildung einer 16-VE Spezies 3. Koordination des Olefins (Olefin-Isomerisierung möglich) 4. Hydrometallierung (anti-Markovnikov vs. Markovnikov) zur 16-VE Spezies 5. CO Anlagerung zur 18-VE Spezies 6. 1,2-Alkyl-Wanderung auf CO (aus cis-Position!) liefert 16-VE Acylkomplex 7. Oxidative cis-Addition von H2 liefert 18-VE Dihydrido-Acyl-Komplex,
der einer reduktiven 1,1-Eliminierung von Aldehyd unterliegt. Isotopen-Markierungsexperimente belegen "1,2-Migratory Insertion" Mechanismus mit Phosphan-modifizierten Rh-Katalysatoren: • Hydrid bereits vorgebildet (thermo-
dynamisch stabiler, kinetisch labiler)
• zwei Phosphanliganden sorgen für sterischen Anspruch → anti-M. günstiger
• die katalytisch aktive, quadrat.-planare 16-VE Spezies trans- HRh(CO)(PPh3)2 bildet sich durch Dissoziation von HRh(CO)(PPh3)3 oder HRh(CO)2(PPh3)2

35Stereoselektive Varianten: Die Hydroformylierung prochiraler, interner Olefine bzw. bei Verzweigung auch terminaler Olefine (z.B. Styrol bei Markovnikov-Orientierung) liefert ein Chiralitätszentrum. Dieses läßt sich bei Verwendung chiraler Bis-Phosphin-, Phosphin-Phosphit- bzw. Bis-Phosphit-Liganden enantioselektiv (> 90% ee) aufbauen: Katalysator mit L∩L(1) und L∩L(2): (L∩L)Pt(SnCl3)2 Katalysator mit L∩L(3) bis L∩L(6): (L∩L)Rh(CO)H Verfahrenstechnische Varianten: Das Problem der Abtrennung und Wiederverwertung des teuren Rhodium-Triphenylphosphan-Komplexes aus dem gebildeten Gemisch von n- / i-Butanal (aus Propen) gelingt durch eine effiziente Zweiphasen-Katalyse (Ruhrchemie / Rhone-Poulenc Verfahren). Hierbei werden wasserlösliche Rhodiumkomplexe mit dem Liganden Triphenylphosphan-tris-sulfonat (TPPTS) eingesetzt. In einem nachgeschalteten Dekantierungstank trennt sich die wässrige Katalysatorphase von der organischen Aldehydphase. Neuste Entwicklungen: grenzflächenaktive Phosphane und axial-chirale, wasserlösliche Phosphane.
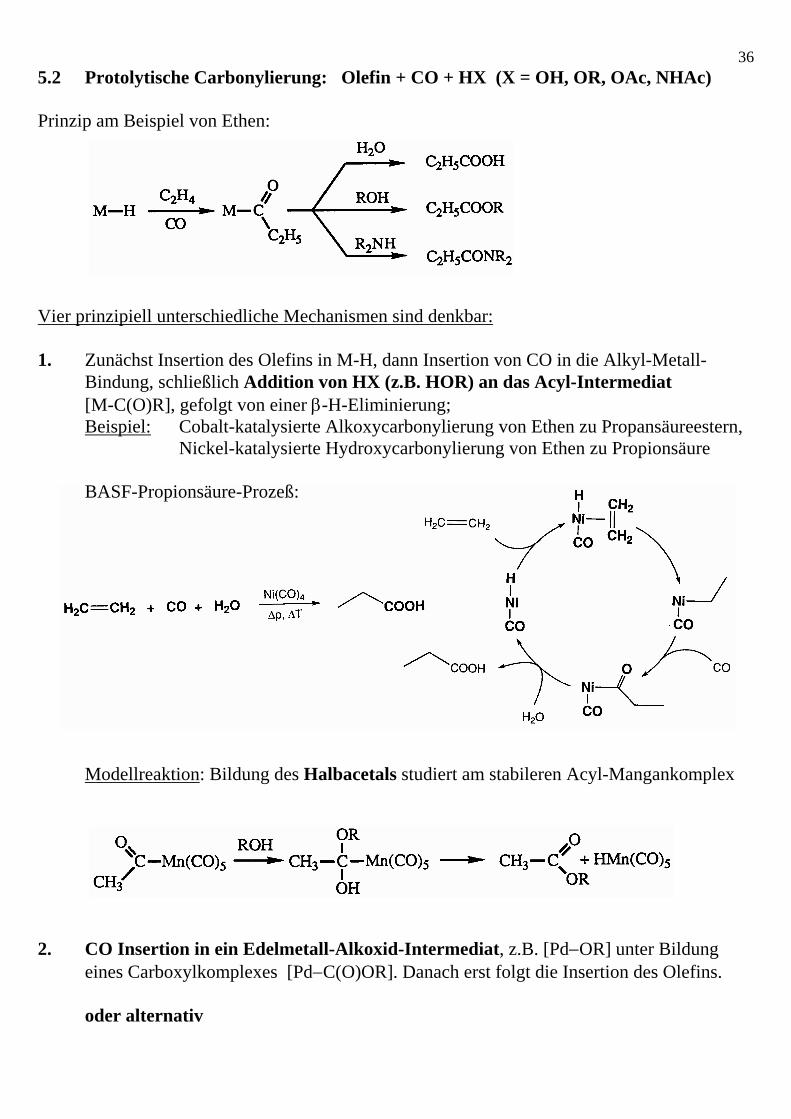
365.2 Protolytische Carbonylierung: Olefin + CO + HX (X = OH, OR, OAc, NHAc) Prinzip am Beispiel von Ethen: Vier prinzipiell unterschiedliche Mechanismen sind denkbar: 1. Zunächst Insertion des Olefins in M-H, dann Insertion von CO in die Alkyl-Metall- Bindung, schließlich Addition von HX (z.B. HOR) an das Acyl-Intermediat [M-C(O)R], gefolgt von einer β-H-Eliminierung; Beispiel: Cobalt-katalysierte Alkoxycarbonylierung von Ethen zu Propansäureestern, Nickel-katalysierte Hydroxycarbonylierung von Ethen zu Propionsäure BASF-Propionsäure-Prozeß: Modellreaktion: Bildung des Halbacetals studiert am stabileren Acyl-Mangankomplex 2. CO Insertion in ein Edelmetall-Alkoxid-Intermediat, z.B. [Pd−OR] unter Bildung
eines Carboxylkomplexes [Pd−C(O)OR]. Danach erst folgt die Insertion des Olefins. oder alternativ

373. Addition von H-OR an einen CO Liganden (Direktangriff von OR− am CO) unter Bildung eines Hydroxycarbens, z.B. Pd=C(OR)(OH) und dessen Deprotonierung zum Carboxylkomplex [Pd−C(O)OR]. Danach erst folgt die Insertion des Olefins. Beispiel: Palladium-katalysierte Alkoxycarbonylierung von Propen zu Butansäureestern S. 190 S. 189 Beispiel: Cobalt-katalysierte Methoxycarbonylierung von Butadien zu Adipinsäureestern (BASF-Verfahren) S. 193 S. 105

384. In Gegenwart von HI oder LiI als Promotor bildet sich in einem vorgelagerten
Gleichgewicht aus einem Alkohol ein Alkyliodid, das oxidativ addiert wird. Es folgt eine CO Insertion zum Acylkomplex und dessen Protolyse zum Carbonsäurederivat. Alternativ wäre die reduktive Eliminierung des Carbonsäureiodids diskutabel (vgl. auch Monsanto-Essigsäure-Verfahren der Carbonylierung von Methanol in Gegenwart von Rhodiumiodid).
Bildet sich der Alkohol durch Addition eines Carbonsäureamids an einen Aldehyd (der aus einem Olefin erzeugt wurde) so resultiert der Spezialfall der Synthese von N-Acylaminosäuren über Amidocarbonylierung von Olefinen: S. 161 Mechanistisches Prinzip: • Olefin wird zunächst zum terminalen Aldehyd hydroformyliert, • der Aldehyd addiert ein Carbonsäureamid zu einem N-Acyl-Amino-Halbacetal, • Promotoren wie LiI oder HI überführen die [R-OH] Funktion des Halbacetals in eine [R-I]
Funktion, die leichter oxidativ an das Metallzentrum addiert werden kann. • Es folgt die CO Insertion und die protolytische Abspaltung der Carbonsäure (oder des
Carbonsäureiodids, das anschließend einer Hydrolyse unterliegt).

395.3 Oxidative Carbonylierung von Alkoholen oder Aminen Alkohol + CO + O2 ergibt Carbonate bzw. Oxalsäurediester Ähnlich wie beim Wacker-Prozeß dient das System PdCl2 / CuCl2 / O2 als Katalysator: Herm. 176 Kennzeichen der Katalyse: • Ein Pd(II)-Carbonyl addiert Alkoxid (oder Alkohol) unter Bildung einer
Bis(alkoxycarbonyl)-Spezies ROOC-[Pd]-COOR, die reduktiv Oxalsäurediester eliminiert. • CuCl2 reoxidiert Pd(0) zu Pd(II). • Das gebildete Cu(I) wird durch Sauerstoff reoxidiert. • Als Nebenprodukt bilden sich Kohlensäureester (z.B. Dimethylcarbonat), generiert durch
reduktive Eliminierung an RO-[Pd]-COOR Mechanistisch weitgehend ungeklärt ist die Bildung von Dimethylcarbonat mit dem Redoxkatalysator Cu(I / II) (Enichem-Prozeß):

39a Verfeinerte Vorstellungen zum Mechanismus der Oxidativen Carbonylierung von Methanol zu Dimethylcarbonat: 2 MeOH + CO + 0.5 O2 MeO-CO-OMe + H2O
CuCl
CO
Cu(CO)Cl
O2
CuCl, H2O
MeOH
O
MeO OMe
+1
+2CuClClCu
O
O
+2
+2CuClClCu
+2H
H
O
O2
2
2
4
H2O
+2CuClClCu
+2Me
Me
O
O2
+1CuClClCu
C
+2MeO
2
O
+1CuClClCu
C
+2MeO
2
O
CO-Aktivierung
reduktiveO2 -Aktivierung
Peroxokomplex
Hydroxykomplex
Methoxykomplex
2
Methanolyse
H2O
CO2 MeOH"HCuCl"
oxidativer Transfervon MeO /(MeO -1e )
Methoxy- CarbonylIntermediat
· - -
Verwendung von DMC: • Dipolar aprotisches Lösungsmittel, biologisch abbaubar. • Benzinadditiv zur Erhöhung der Octanzahl. • Phosgenersatzstoff bei der Gewinnung von Polycarbonaten (nach Umesterung mit Phenol zu
Diphenylcarbonat). • Hochtemperaturalkylierungsmittel (ungiftig im Vergleich zu Dimethylsulfat).

40Amin + CO + O2 ergibt Carbamate bzw. Isocyanate Mechanismus noch weitgehend ungeklärt: • Oxidative Carbonylierung von Anilin in Gegenwart von Ethanol liefert Phenylcarbamat und
Wasser (Nebenprodukt: N,N'-Diphenylharnstoff). • Nach Hydroxymethylierung mit Formaldehyd entsteht ein Diurethan als
Kondensationsprodukt, das zum Methylendiisocyanat (MDI) gecrackt werden kann (→ Polyurethane).
5.4 Copolymerisation von CO und Ethen (Shell-Polyketon-Prozeß) Zufallsentdeckung eines modernen, schlagfesten Polymers (Polyketon) im Rahmen der Untersuchungen zur Methoxycarbonylierung von Ethen mit Katalysatoren vom Typ [L2PdX2] (X = OTos, OTf, schwach-koord. Anion) zum Methylpropionat Mit trans-[(Ph3P)2PdX2] (X = OTos, OTf) → Protolyt. Carbonylierung zu Methylproprionat: 336 Mit cis-[(Ph2P-(CH2)3-PPh2)PdX2] (X = OTos, OTf) → Copolymerisation von CO + Ethen:

41Zwei Insertionsmechanismen ausgehend von [L2PdX2] bzw. [L2Pd(CO)X]+ werden diskutiert: • der Methoxy-Cyclus wird initiiert durch Protolyse einer Pd-X Bindung zu [Pd-OR] • der Hydrido-Cyclus wird initiiert durch die Hiebersche Basenreaktion: [Pd-CO] + H2O →
[Pd-H] + CO2 Da das cis/trans-Verhältnis bei monodentaten Triphenylphosphan-Komplexen weit auf der Seite des trans-Produktes liegt, eine CO-Insertion aber lediglich aus der cis-Position erfolgt, wird die Acylspecies in trans-Form "konserviert". → Sie unterliegt der Protolyse mit Methanol, ehe es zu einer weiteren Insertion von Ethen (→ Bldg. von Polyketon) kommt: Der Wechsel zum bidentaten Chelatphosphan Ph2P-(CH2)3-PPh2 (dppp) führt zu einer Propagation der Insertionen mit streng alternierenden CO und Ethen-Einheiten. Der Grund: Lediglich "insertionsaktive" cis-Phosphankomplexe sind möglich.

42Initiation: • In Anwesenheit von MeOH entstehen ausgehend vom Methoxycarbonyl [Pd-COOMe]
Polyketone mit terminaler Esterfunktion. • In Anwesenheit von H2O entstehen initiiert durch die Insertionssequenz [Pd-H] → [Pd-Et] → [Pd-CO-Et] → [Pd-C2H4-CO-Et] Polyketone mit terminaler Ketofunktion.
Propagation: Streng alternierend Der Grund: Die irreversible cis-Insertion von Ethen in die Acyl-Pd-Bindung liefert ein stabiles 5-Ring-Alkyl-Chelat, dessen Keton-Donorfunktionalität zwar durch CO, nicht aber durch Ethen verdrängt werden kann. Nach CO Insertion bildet sich ein weniger stabiles 6-Ring-Acyl-Chelat, das sowohl durch CO als auch durch Ethen in der Keton-Donorfunktionalität geöffnet werden kann. Da die doppelte CO Insertion thermodynamisch ungünstig und reversibel ist, gewinnt die irreversible Ethen Insertion unter Bildung eines neuen 5-Ring-Alkyl-Chelats. Terminierung: Protolyse auf der Stufe des Alkylkomplexes mit H2O liefert terminales Keton + CO2 + [Pd-H] Protolyse auf der Stufe des Alkylkomplexes mit ROH liefert terminales Keton + [Pd-OR] Protolyse auf der Stufe des Acylkomplexes mit ROH liefert terminalen Ester + [Pd-H] d.h. die beiden Kettenenden der Polyketone können als α,ω-Ketoester, als α,ω-Diketone oder als α,ω-Diester vorliegen, wobei in Methanol als Lösungsmittel die α,ω-Ketoester überwiegen. Stereoselektive Variante: Die Copolymerisation von Propen und CO kann regioselektiv (Kopf-Schwanz) und darüber hinaus diastereoselektiv zu isotaktischen oder syndiotaktischen Polyketonen führen. 347

435.5 Carbonylierung von Propin in Gegenwart von ROH zu Methylmethacrylat (MMA) Basierend auf dem veralteten Acrylsäureprozeß von Reppe HC≡CH + CO + H2O + [Ni-H] H2C=CH-COOH ist ein modernes Verfahren für die Gewinnung von Methylmethacrylat (MMA) basierend auf der Carbonylierung von Propin entwickelt worden (Shell). Propin ist ein Produkt des Naphtha-Steamcrackers, MMA ist das Basismonomer für Plexiglas: Mechanismus: zwei mögliche Cyclen Kontrolle der Regioselektivität durch spezielles Liganddesign des P,N-koordinierenden Pyridylphosphan-Liganden: 1125

445.6 Carbonylierung von Methanol (Monsanto-Essigsäure-Prozeß und Verwandte) In Gegenwart von HI oder MeI als Promotor läßt sich das Fischer-Tropsch-Produkt Methanol carbonylieren, wobei Rhodium(I / III) carbonyliodide als Homogenkatalysatoren dienen: Me−OH + CO Me-CO-OH (Essigsäure) MeOH → Me-CO-OMe (Methylacetat) Me−OMe + CO Me-CO-OMe (Essigsester, Methylacetat) Me−OCOMe + CO Me-CO-O-COMe (Acetanhydrid, Tennessee Eastmann) Kennzeichen der Katalyse: • In einem vorgelagerten, rein organischen Gleichgewicht aktiviert der Promotor HI die
Bausteine Methanol, Dimethylether oder Methylacetat Me-OR (R = H, CH3, COOMe) unter Bildung von Methyliodid, das oxidativ an Rh(I) addiert werden kann.
• Nach CO Insertion erhält man durch Reduktive Eliminierung Acetyliodid als entscheidendes Intermediat, das dann mit protischen Reaktionspartnern zum Essigsäurederivat unter Rückbildung von HI als Promotor abreagiert.
• Me-COI + H-OH Me-COOH + HI Me-COI + H-OMe Me-COOMe + HI Me-COI + H-OCOMe Me-CO-O-COMe + HI • Alternativ zur Reduktiven Eliminierung von Acetyliodid ist die Addition von HO-R (R = H,
Me, COMe) an die Metall-Acylfunktion zu diskutieren:
[Rh]-C=O
CH3
IH+
[Rh]=C-OHCH3
I
[Rh] = [RhI2(CO)2] R = H, CH3, COCH3-
Acetyl-Komplex Hydroxycarben-
OR-[Rh]-C-OR
CH3
I OHβ-H-El.δ+
Halbacetal-
[Rh]-H
I
+H3C-COOR
Red. El.[Rh]
+
H-I

455.7 Carbonylierung von Nitroaromaten Drei exergonische Prozesse, von denen lediglich der erste technisch realisiert ist (Umweltproblematik): Selen als Redoxkatalysator (Arco) neue Umweltproblematik?, die Pyrolyse des Carbamats liefert Alkohol + Isocyanat (vgl. oben) Katalysemechanismus:

46PdCl2 als Katalysator + CuCl2, CoCl2, FeCl3 als redoxaktiver Promotor zur Regenerierung von Pd2+ PdCl2 wird in Gegenwart von Wasser und CO zu Palladiumschwarz Pd(0) reduziert. Prinzipiell das gleiche gilt für Anilinderivate in Gegenwart von CO: Mechanistische Hypothese I: Reduktive Spaltung der N-O Bindung des Ar-NO2 über Palladacyclen, gebildet durch oxidative 1,2-Addition von Ar-NO2 an [Pd(0)-CO] Mechanistische Hypothese II: Reduktive Spaltung der N-O Bindung des Ar-NO2 über Imido (Nitren)-Komplexe?

47
Ir
N
CMe3
2 CO Ir
N
CMe3
COCO
d6 Ir(III) 18 VE d6 Ir(III) 18 VE
Ir
N
CMe3
COCO
d8 Ir(I) 18 VE
Modellreaktion zur metallvermittelten Kupplung von CO und [NR] Bergman, JACS 113 (1991) 2041
Reduktive Carbonylierung von Nitroaromaten zu Urethanen
Wird die Carbonylierung in Alkoholen durchgeführt, so entstehen primär Carbamate (Urethane), die sich in einem Folgeschritt thermisch zu Isocyanaten und Alkohol wieder spalten lassen.
R-N=C=O R-NH-CO
-OR+ ROH
∆, - ROHUrethan
5.8 Carbonylierung von Benzyl-X, Allyl-X und Aryl-X in Gegenwart von Nu-H Prinzip: HX muß durch Base abgefangen werden (daher „Heck-Typ-Carbonylierung“) General Mechanism of the carbonylation of C-X.

48Beispiele: Hydroxycarbonylierung Nu-H = HO-H Alkoxycarbonylierung Nu-H = RO-H Amidocarbonylierung Nu-H = RCONH-H Doppelcarbonylierung n = 2 Prinzipielle Schritte: (2) Generierung einer koord. ungesättigten Pd(0) Spezies (auch Ni, Pt, Co) (3) Oxidative Addition von R-X meist geschwindigkeitsbestimmender Schritt (Trends entsprechend der R-X Bindungsenergien): allgemein: allyl > benzyl > phenyl = methyl > vinyl > propyl > ethyl
bei Aryl-X: C-I > C-OTf ≥ C-Br >> C-Cl darüber hinaus bei Allyl-, Benzyl-X: > OCOCF3 > OCO2Et > OCOPh > OCOMe (4) Meist einfache (selten zweifache) CO Insertion zum Acylkomplex. (5) Reduktive Eliminierung von RCO-X, das mit Nu-H zu HX + RCO-Nu reagiert. (6) Alternativ: Hydrolytische Spaltung der Acylspecies mit Nu-H unter Bildung eines [X-Pd-H]-Komplexes, der (7) in Gegenwart von Base reduktiv H-X eliminiert. Industrielle Anwendungen: Carbonylierung von Halogenaromaten (wertsteigernde Feinchemikalien-Synthese) Phenylessigsäure (→ Duftstoffe, Pestizide) aus Benzylchlorid: Zweiphasensystem: PhOPh / 40% NaOH / kationisches Tensid [Benyl-NMe2-C12H25]+
49Profene (Arylpropionsäuren → entzündungshemmende Phamaka, 2.5 Mrd. Markt) über in situ gebildete 1-Methyl-benzylchloride 5.9 Cn -Bausteine aus Synthesegas (Fischer-Tropsch-Folgechemie) Die Fischer-Tropsch-Reaktion ist eine heterogen katalysierte Hydrierung des C1-Bausteins CO zu Methanol bzw. je nach Bedingungen unter C-C-Knüpfung zu höheren Cn-Bausteinen: m CO + n H2 (Synthesegas) CxHyOz Produkte + H2O Bedingungen: 170-350 °C, Katalysatoren: Co / ThO2 / MgO oder Fe-Oxide Synthesegas kann aus Kohle gewonnen werden (Kohlevergasung), duch partielle Verbrennung von Erdöl oder aus einem Reforming-Prozeß aus Erdgas (vgl. Einleitung). Die relevanten Schritte des Wassergas-Gleichgewichtes, das den H2 Anteil relativ zu CO erhöht, lassen sich mechanistisch wie folgt deuten: 958 Mechanistic proposal fort he metal-catalyzed water-gas shift reaction; equilibria Disregarded. M: metal complex, e.g. [Fe(CO)4]

50Mechanistische Modellvorstellungen, die die Bildung von Methanol und höherer Alkane als wichtige F.-T.-Produkte erklären könnten: Modellreaktionen: erste Reduktion von CO durch H2 und erste C-C-Kupplung (Bercaw 1976) Technisches Ziel: Ethylenglycol aus F.-T.-Produkt Methanol (statt Ethylenoxid + H2O) durch „Hydroxycarbonylierung von Formaldehyd“ alternativ: durch "Hydroformylierung von Formaldehyd" gefolgt von Hydrierung
H2C=O + CO + H2 H-O-CH2-CH=O
H-O-CH2-CH=O + H2 HO-CH2-CH2-OH

516. HX-Addition an Olefine
6.1 Hydrocyanierung Adipinsäuredinitril (Adiponitril ADN, aus Butadien + HCN, DuPont-Prozeß)
.... ADN-Hydrierung → Polyamide Schritte der Katalyse: 1. Ni(0)-Phosphit-Komplex addiert oxidativ HCN zu einem Kontaktionenpaar 2. C-C-Knüpfung über Allyl-Zwischenstufen zu Penten-nitrilen 2-PN, 3-PN, 4-PN 3. Isomerisierung von 3-PN zum terminalen 4-PN 4. Erneute Insertion des reaktiveren, terminalen Olefins 4-PN in die Ni-C-Bindung eines Lewis-
Säure (A) aktivierten Ni-CN-A Komplexes (für A = R3Sn+, BPh3: "Heteroisonitril") Interessant: Elektronenreichere Ni(0)-Phosphankomplexe addieren HCN zwar rascher, doch es folgt eine Desaktivierungreaktion: H-NiL3CN + HCN H2 + L + L2Ni(CN)2 erster Schritt: Ni{P(OAr)3}4 P(OAr)3 + Ni{P(OAr)3}3 HCN → [H-Ni{P(OAr)3}3]+ CN−

52Hydrocyanierung prochiraler Ketone und Aldehyde an chiralen Lewis-Säuren: Problem: Hohe Katalysatorkonzentration (TON > 10), Katalysator i.d.R. nicht recyclebar. Beispiel 1: TMS-CN als Synthon für HCN Andere Schiffbasen-Liganden Beispiel 2: 6.2 Hydrosilylierung R3Si−H Addition (statt H−H) an Olefine,katalysiert durch d8-Pt(II) und d10-Pt(0)-Komplexe, die gewöhnlich aus H2PtCl6, also Pt(IV)-Vorstufen, in Gegenwart von Reduktionsmitteln (Phosphanen, i-Propanol) gewonnen werden. Prinzip der Katalyse: A Ein 14 VE-Komplex, gebildet aus einer quadratisch-planaren 16 VE-Vorstufe addiert Olefin. B Es folgt die Oxidative Addition der Si-H Bindung zum 18 VE-Komplex (A und B können auch in umgekehrter Reihenfolge durchlaufen werden). C Es folgt eine Hydrometallierungsschritt (1,2-H-Shift auf Olefin, Olefin-Insertion in M-H). D Reduktive Eliminierung aus dem Alkyl-Silyl-Komplex in Gegenwart von Olefin. Bei der Hydrosilylierung von C=N und C=O Bindungen wird eine 1,2-Wanderung der Silylgruppe auf das Heteroatom diskutiert.

53Anwendungen bei der Gewinnung von Polymeren und Dendrimeren: z.B. Addition von H-funktionellem Silan an Vinylsilan →
H
SiR3[Pt][Pt]
H
SiR3
+SiR3
[Pt] [Pt]SiR3
+
Oxidat. Addition Insertion Redukt. Eliminierung
SiR3[Pt] [Pt]
SiR3
+
Si-H + HC≡CH [Pt]
Mechanismus:HC≡CH
Si-H + H2C=CH-Si [Pt]Si-CH2-CH2-Si
Vernetzung H-funktioneller Polysiloxanöle zu Elastomeren
Si-CH=CH2
Gewinnung von Carbosilan-Polymeren: 6.3 Hydroaminierung Die Addition einer N-H-Bindung an Ethylen ist ein exergonischer Prozeß. Er ist kinetisch inhibiert, da keine Synchronaddition möglich ist. Das Gleichgewicht liegt aufgrund des hohen negativen Entropieterms bei hohen Temperaturen auf der Seite der Edukte: → ein industriell interessanter Fall für die Katalyse Prinzip der Katalyse: Ein Olefin insertiert in eine Metall-Stickstoff-Bindung eines Metallamids.

54Fall 1: Das Metallamid kann durch Oxidative Addition an ein 14 VE-Edelmetall-Komplexfragment gebildet werden. Nach Olefin-Insertion unterliegt der Hydridoalkyl-Komplex in Gegenwart des NH-funktionellen Amins einer reduktiven Eliminierung. Fall 2: Das Metallamid kann durch Protolyse an ein 14 VE-Lanthanid-Alkylkomplex (oder Alkalimetallamid, z.B. LiNEt2) gebildet werden. Nach Olefin-Insertion unterliegt der gebildete Alkylkomplex (Metallalkyl) in Gegenwart des NH-funktionellen Amins einer Protolyse.
18 VE [M]HI
NR2 18 VE [M]HI
NR2
14 VE [M] + H-NR2 18 VE [M]
HI
NR216 VE [M]
HINR2
: 16 VE [M]
HINR2
Ethen
H-NR2
Et-NR2
14 VE [M] = [Ir(PEt3)2Cl]
18 VE [M] 18 VE [M] NR2
14 VE [M]-R + H-NR2 16 VE [M] NR216 VE [M]
HINR2
Ethen
H-NR2
Et-NR2
14 VE [M]-R = [Cp*2Ln]-CH(SiMe3)2
RI
R-H
NR2
H-NR2
16 VE [M] NR2
Anwendungsbeispiele: insbesondere bei entropisch begünstigter Cyclisierung von α,ω-Aminoolefinen (Marks)

557. Weitere Transformationen an Olefinen (C-C-Kupplungen) 7.1 Heck-Reaktion und Verwandte Ein vinylisches H-Atom wird durch eine Aryl-, Vinyl- oder Benylgruppe (generiert aus entsprechenden Halogeniden oder Triflaten) ersetzt. Als Katalysator dient ein Pd(0)-Komplex, in situ generiert aus Pd(OAc)2 + NEt3 + PPh3 (als Oxidationsprodukt bildet sich Ph3PO). Es wird eine äquimolare Menge Base NEt3, NaOAc oder K2CO3 zum Abfangen von HX benötigt. 713 Mechanismus: 1) PdL2 (14 VE, d10) addiert oxidativ (trans → SET-Mechanismus) Ar-X, Vinyl-X oder Benzyl-X. 2) - 4) Nach Austausch von L gegen Olefin erfolgt regioselektiv (s.u.) eine syn-Insertion (Carbometallierung) gefolgt von einer syn-β-H-Eliminierung. → Die syn-Add. / syn-Elim. Sequenz führt bei 1-Olefinen zu einer trans-Geometrie, bei 1,2-disubstituierten Olefinen zu einer invertierten Olefin-Geometrie, da vor der β-H-Eliminierung eine Rotation notwendig ist:
Pd C
RR
ar Pd
RR
Ar
H HH H
PdR
R Ar
H
H
syn-Elim.Rotationsyn-Add. 5) Die reduktive HX-Eliminierung durch Base regeneriert das Pd(0)-Zentrum.

56 719 720
Spezielle Anwendungen: Konjugierte Polymere: all-trans-Poly-1,4-phenylen vinylen (M 5.000-10.000) Enantioselektive C-C-Kupplung (intermolekular) Heck-Reaktion, gefolgt Von Doppelbindungs 716 Isomerisierung Enantioselektive C-C-Kupplung (intramolekular)

577.2 Cyclopropanierung (inkl. stereoselektive Varianten) Pd(OAc)2 katalysiert die Zersetzung von Diazomethan unter syn-Addition des Methylens an das Olefin (aus trans-Olefin entsteht trans-Cyclopropan, aus cis-Olefin dagegen cis-Cyclopropan). Mechanismus nicht gesichert, jedoch plausibel über Metall-Carbenkomplex, der Olefin zum Metallacyclobutan addiert, welches anschließend reduktiv Cyclopropan eliminiert. Asymmetrische Cyclopropanierung: Erstes Beispiel einer enantioselektiven Katalyse durch Metallkomplex: Asymmetrische Addition von Ethyldiazoacetat an Styrol katalysiert durch chirale Cu(II)-Schiffbasen-Komplexe (Noyori 1966). Die katalytisch aktive Spezies ist allerdings ein in situ reduktiv gebildeter Cu(I)-Komplex. Moderne Bis-Oxazolin-Chelatliganden: → Ester und Amide der Chrysanthemsäure → Cilastatin = in-vivo-Stabilisator für Antibiotika; Pyrethroide = natürliche Insektizide. 738, 739 7.3 Metathese (vgl. auch ROMP, s.o.) Die katalytisch aktive Spezies ist ein koordinativ ungesättigter Carbenkomplex, der ein Olefin anlagern kann. Diese Alkylidenspezies bildet sich entweder aus einem Dialkylkomplex, der über eine über α-agostische Ww. (s.o.) einer intramolekularen α-H-Eliminierung von Alkan unterliegt oder sie bildet sich durch Liganddissoziation aus einem bereits vorgebildeten Alkylidenkomplex:
z.B. CO-Dissoziation aus [(OC)5W=CPh2] oder Bromidabstraktion durch GaBr3 aus [Br2(OR)2W=CHtBu].

58
M CH-R
HM CH2 R
MH
C RH
R'R'R' M CH-R
-R'-H
α-agostische Ww. reduktive Eliminierung CHAUVIN-Mechanismus der Olefin-Metathese über Metallacyclobutan-Zwischenstufe: Olefin-, Alkin- und Halogenid-Methathese- Reaktionen verlaufen i.a. über die Spaltung von 4-Ring-Zwischenstufen, die σ-Bindungsmetathese, z.B. R-Zr(IV) + H-H, dagegen über 4-Ring- Übergangszustände. Spezielle Anwendungen definierte Komplexe: [Cl2(PPh3)2Ru=CHR] und [(RO)2(ArN)Mo=CHR] in situ gebildete Spezies: Re2O7 / SiO2 / EtAlCl2 oder WCl6 / Me4Sn. Tolerant gegenüber funktionellen Gruppen; terminale und gespannte Cycloolefine bevorzugt. ROMP (s.o.) und ROM (Ring opening metathesis) RCM (Ring closing metathesis) Alkin-Metathese über Alkylidin-Komplexe: Metathetische 1-Alkin-Polymerisation über Alkyliden-Komplexe:
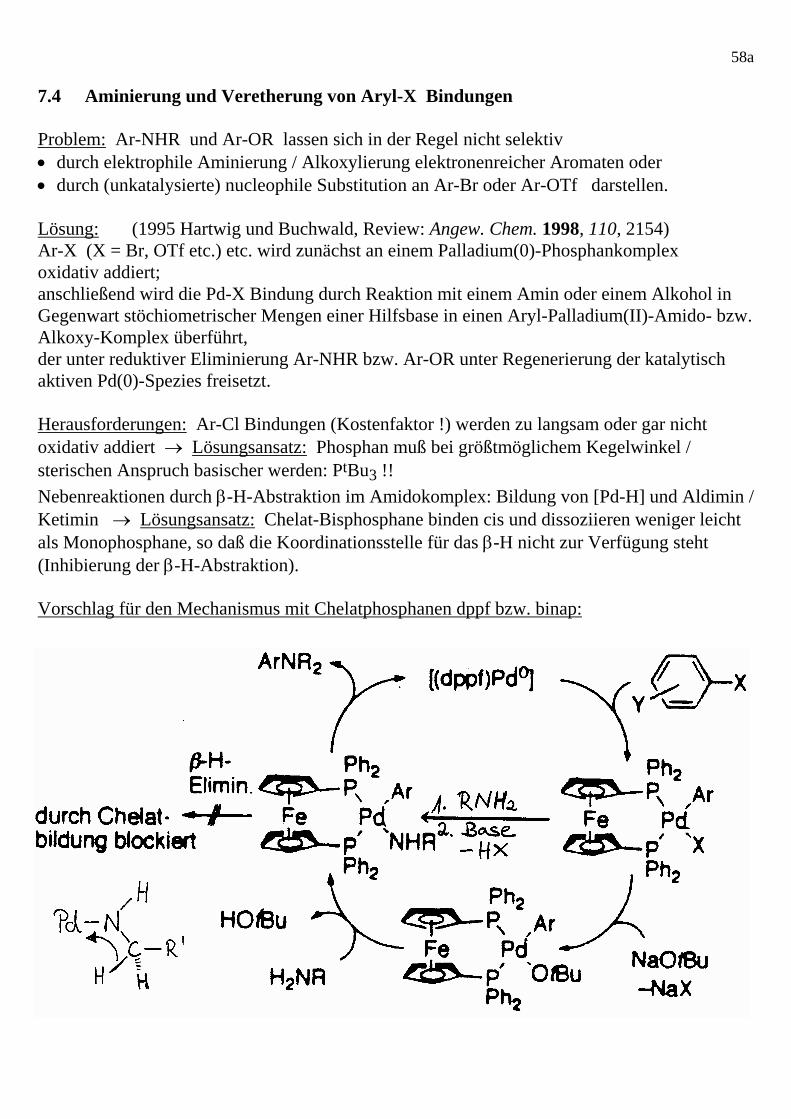
58a 7.4 Aminierung und Veretherung von Aryl-X Bindungen Problem: Ar-NHR und Ar-OR lassen sich in der Regel nicht selektiv • durch elektrophile Aminierung / Alkoxylierung elektronenreicher Aromaten oder • durch (unkatalysierte) nucleophile Substitution an Ar-Br oder Ar-OTf darstellen. Lösung: (1995 Hartwig und Buchwald, Review: Angew. Chem. 1998, 110, 2154) Ar-X (X = Br, OTf etc.) etc. wird zunächst an einem Palladium(0)-Phosphankomplex oxidativ addiert; anschließend wird die Pd-X Bindung durch Reaktion mit einem Amin oder einem Alkohol in Gegenwart stöchiometrischer Mengen einer Hilfsbase in einen Aryl-Palladium(II)-Amido- bzw. Alkoxy-Komplex überführt, der unter reduktiver Eliminierung Ar-NHR bzw. Ar-OR unter Regenerierung der katalytisch aktiven Pd(0)-Spezies freisetzt. Herausforderungen: Ar-Cl Bindungen (Kostenfaktor !) werden zu langsam oder gar nicht oxidativ addiert → Lösungsansatz: Phosphan muß bei größtmöglichem Kegelwinkel / sterischen Anspruch basischer werden: PtBu3 !! Nebenreaktionen durch β-H-Abstraktion im Amidokomplex: Bildung von [Pd-H] und Aldimin / Ketimin → Lösungsansatz: Chelat-Bisphosphane binden cis und dissoziieren weniger leicht als Monophosphane, so daß die Koordinationsstelle für das β-H nicht zur Verfügung steht (Inhibierung der β-H-Abstraktion). Vorschlag für den Mechanismus mit Chelatphosphanen dppf bzw. binap:
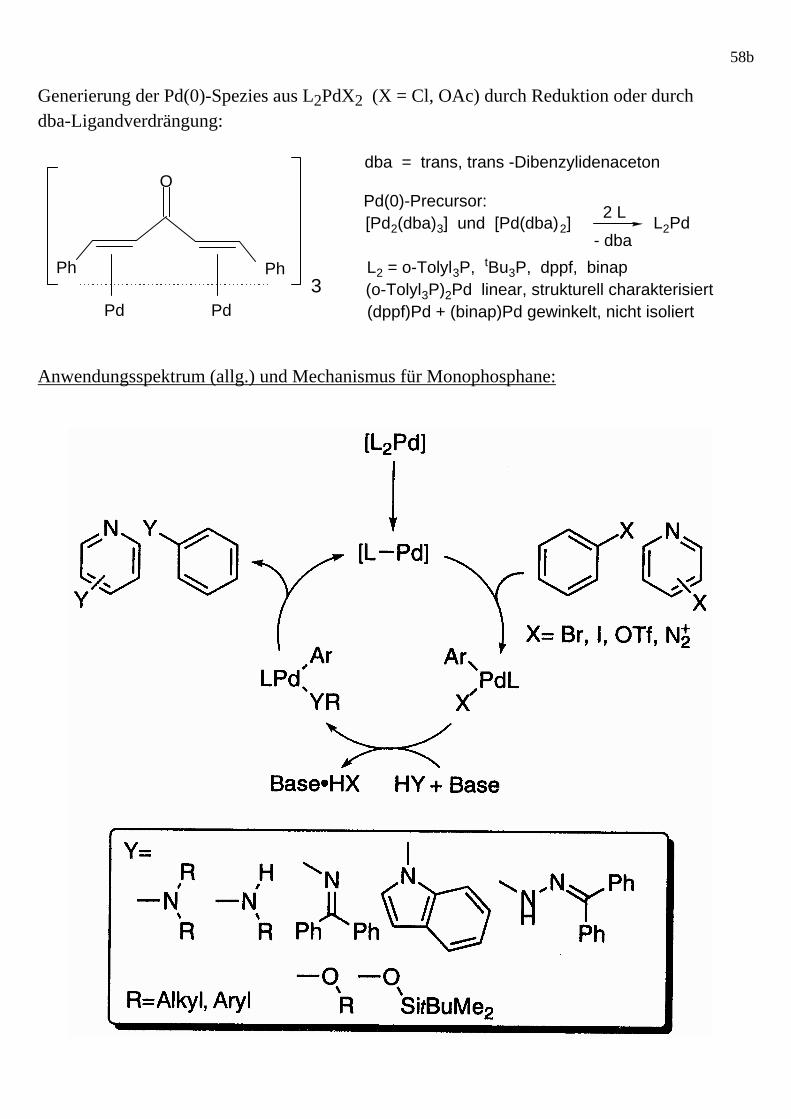
58b Generierung der Pd(0)-Spezies aus L2PdX2 (X = Cl, OAc) durch Reduktion oder durch dba-Ligandverdrängung:
Ph
O
Ph
Pd Pd3
dba = trans, trans -Dibenzylidenaceton
Pd(0)-Precursor:[Pd2(dba)3] und [Pd(dba)2] 2 L
- dbaL2Pd
L2 = o-Tolyl3P, tBu3P, dppf, binap (o-Tolyl3P)2Pd linear, strukturell charakterisiert(dppf)Pd + (binap)Pd gewinkelt, nicht isoliert
Anwendungsspektrum (allg.) und Mechanismus für Monophosphane:

598. Oxidationen 8.1 Olefine zu Carbonylverbindungen (Wacker-Prozess) und Vinylverbindungen Vinyl-X Verbindungen wurden früher durch Addition von HX an Ethin dargestellt (X = OH → Aldehyd, OR, OAc, Cl). In den 60er bis 80er Jahren setzte sich die Oxypalladierung (= Addition von Pd + OH−) von Olefinen durch. Heute überwiegt CO Technologie bei der Darstellung von > C2 Aldehyden und Ketonen. Modellversuch: Wässrige PdCl2 Lösung reagiert mit Ethen Acetaldehyd + Pd(0)-Schwarz Propen Aceton + Pd(0)-Schwarz 1- u. 2-Buten MEK (Methylethylketon) + Pd(0) 1-Olefine Methylketone Styrol Acetophenon + Pd(0) Cyclohexen Cyclohexanon Die Möglichkeit der Reoxidation von Pd(0) zu Pd(II) durch das Redoxpaar CuCl / CuCl2 / O2 läßt die Oxypalladierung katalytisch werden: 138 Anwendungsbreite: Vinylische H-Atome lassen sich durch X = Cl, OR, OAc, OH ersetzen, z.B. in Ethen: Eb 497

60Mechanismus: Kennzeichen der Katalyse: 1) - 3) Zwei Chloroliganden in [PdCl4]2− werden durch Olefin und durch H2O bzw. OH− substituiert (hohe Chloridkonzentration inhibiert die Katalyse). 4) syn-Hydroxypalladierung (Verschiebung des cis-ständigen OH− Liganden auf das Olefin) liefert eine β-Hydroxyethyl-Spezies (hierfür spricht das Geschwindigkeitsgesetz) alternativ: anti-Hydroxypalladierung (Addition von Wasser (bzw. OH− ) ohne vorherige Koordination an das Olefin) liefert eine andere β-Hydroxyethyl-Spezies (hierfür spricht die Stereochemie der Addition an DHC=CHD). Wahrscheinlich sind je nach Randbedingungen beide mechanistischen Alternativen wirksam. 5) + 6) β-H-Eliminierung liefert einen Vinylalkohol-Hydridokomplex (der Vinylalkohol = Aldehyd wird nicht abgespalten!, da hier ein H / D Austausch zu erwarten wäre). 7) + 8) Hydridverschiebung zurück auf den Vinylalkohol liefert einen α-Hydroxyethyl-Komplex, der schließlich streng intramolekular einer β-H-Eliminierung unterliegt (beim Arbeiten in D2O oder MeOD wird kein Deuterium in den Aldehyd eingebaut !).

618.2 Olefinoxidation zu Epoxiden (inkl. stereoselektive Varianten) 8.2.1 Elektronenreiche Olefine Ethenoxid (EO) und Propenoxid (PO) sind wichtige Basischemikalien (→ Polyglycole und ihre Ether, Tenside, Weichmacher, Polyester, Polyurethane etc.) Ethylenoxid wird durch Luftoxidation von Ethen am Silberkontakt (heterogen) dargestellt. Die Darstellung von Propenoxid gelingt nicht auf analoge Weise, sondern beispielsweise klassisch über das Chlorhydrin (Problem: Giftigkeit, Salzabfälle). Oxiranprozeß (Arco + Halcon): Aktivierung von Alkylhydroperoxiden Radikalische Autoxidation von Isobutan oder Ethylbenzol mittels Luftsauerstoff unter Bildung der Alkylhydroperoxide tBuOOH (TBHP) bzw. Ethylbenzolhydroperoxid (EBHP). ARCO- und HALCON-Verfahren: Alkylhydroperoxide werden durch homogen lösliche Mo(VI)- und W(VI)-Komplex unter Bildung reaktiver Metallpersäureester [M-OOR] aktiviert SHELL-Verfahren: Aktivierung von ROOH an heterogenen Katalysator TiO2 auf SiO2-Träger. Nachteil beider "gekoppelten" Verfahren: Pro Mol PO entsteht ein Mol eines Alkoholes, für den eine Verwendung gefunden werden muß. Problem gelöst: Tert-Butanol wird zu MTBE (Benzinadditiv) verethert, 1-Methylbenzylalkohol dagegen zu Styrol (Monomer) dehydratisiert.

62Aktivierung von Wasserstoffperoxid (ENICHEM) Da der Bedarf an PO stärker wächst als der von Styrol, arbeitet man z.Z. an nicht gekoppelten Verfahren, d.h. an der katalytischen Aktivierung von Wasserstoffperoxid (umweltfreundlich, nach dem Anthrachinon(AO)-Verfahren aus H2 und Luft produziert). Vielversprechende aktuelle Entwicklungen: ENICHEM-Verfahren: Titansilicalit TS-1 (Titan dotierter Zeolith). Heterogene Zweiphasen-Katalyse (fest-flüssig). Tenside als Liganden für Molybdänpersäuren in Zweiphasen-Katalyse (flüssig-flüssig, vgl. 62a). Mechanismen: Prinzip der Perhydrolyse einer Metallsäure mit M=O (Oxofunktion) über M-OOH (Hydroperoxyfunktion, Metallpersäure) zu M(η2-O2) (Metallperoxid).
Dioxirane vs. Metall-Peroxide
Percarbonsäuren vs. Metall-Persäuren
Mechanistisches Modell der Perhydrolyse einer terminalen Metall-Oxofunktion [M=O]:

63Synthese und Struktur der Katalysatoren:
L + MoO3 + 2 H2O2 [L-Mo(O)(O2)2(H2O)] + H2O (L = ONR3, OPR3, Py etc.) Me-ReO3 + 2 H2O2 [Me-Re(O)(O2)2(H2O)] + H2O
Isoelektron. Kompl.: → Struktur-Wirkung-Beziehung!
Mimoun-Komplexe (Mo, W) Herrmann-Komplex (Re)
Phasentransfer-Oxidationskatalyse:
H2O, H2O2, diols
MoO
O
OO
O
OH2
OER3
MoO
O
O
OER3O
O
MoO
O
OO
R3EO OMo
OO
O
OER3O
O kdim
MoO
O
O
OH2O
O
OH2
kdiss
O
MoO
O
OER3O
O
MoO
O
OER3O
O
OH2
H2O2 H2O
kass
MoO
O
O
OER3O
O
OER3
kass
inactive
inactive
active active
inactive
inactive
inactive
O
E+
_O
E+
_O
E+
_
interphase
MoO3 + 2 H2O2perhydrolysis aqueous phase
O transfer
interphase
olefin, epoxide
organic phase
OE(n-dodecyl)3E = N, P, As
pH 2 - 3
ksubst
tensidetype ligands
R' R'
kdiff
- H2O
kdiff
+ H2O
+ OER3molybdylmono peroxide

64Mechanistisches Prinzip des O-Transfers: (zwei Wege Gegenstand des wissenschaftl. Disputs)
MO
OCH2
MO OM
O
O
MOO
MO
OH2C
=
Sharpless: Butterfly, konzertiert, orbitalkontrolliert
Mimoun: 1,3-Dipol, schrittweise, ladungskontrolliert
Argumente: • Diamagnetische, Lewis-acide Metalloxokomplexe der Elektronenkonfiguration d0 (= höchste
Oxidationsstufe, Metallsäuren = Abkömmlinge Lewis-acider Metalloxide) besitzen keine besetzten d-Orbitale für Rückbindungen zum Olefin !
• Je Lewis-acider das Metallzentrum, desto elektrophiler der Peroxo-Sauerstoff → starke Donorliganden (auch H2O), insbesondere auch Chelatliganden, inhibieren die Katalyse.
• Je elektronenreicher das Olefin (Zahl der Alkylsubstituenten), desto reaktiver ist es → SN2 Typ Reaktion?
• Eine η2-Peroxofunktion ist ein schwächeres Oxenoid ( [O]-Überträger) als eine einseitig protonierte, alkylierte, silylierte oder metallierte Peroxofunktion. Der oxenoide Charakter wächst aufgrund der Polarisierung der ansonsten unpolaren O-O-Bindung → das LUMO (σ*O-O -Charakter) wird energetisch abgesenkt und damit leichter angreifbar durch das Substrat HOMO (Olefin π-Charakter).
• Die Epoxidation verläuft stereospezifisch (Bindungsknüpfung zu beiden C-Atomen synchron): Aus cis-Olefin entsteht cis-Epoxid.
Aktivierung der η2- Peroxyspezies [M(η2-O2)] durch Säure: Der Angriff des Olefin HOMO's erfolgt orbitalkontrolliert in das Komplex LUMO. Der Prozeß ähnelt einer SN2-Reaktion am peroxidischen O-Zentrum. Zur Erinnerung: Orbital-WW der SN2-Reaktion:
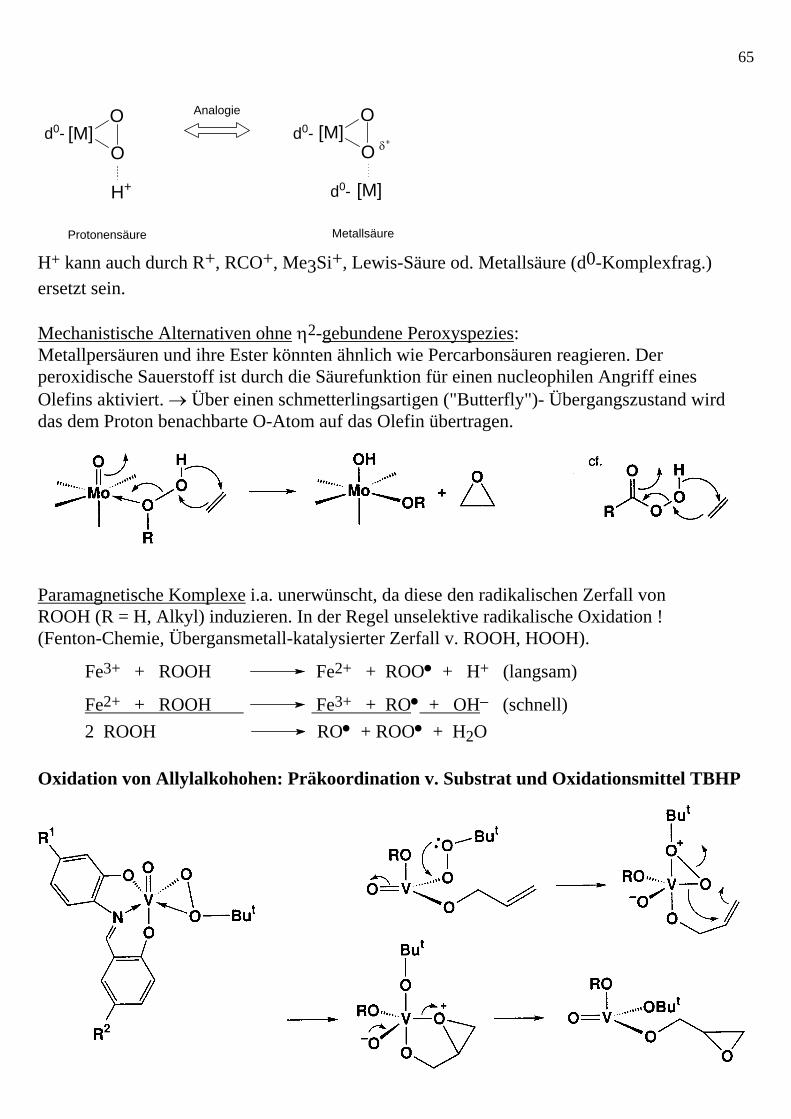
65
[M]O
O[M]
O
O
[M]H+ d0-
d0-d0-δ+
Protonensäure Metallsäure
Analogie
H+ kann auch durch R+, RCO+, Me3Si+, Lewis-Säure od. Metallsäure (d0-Komplexfrag.) ersetzt sein. Mechanistische Alternativen ohne η2-gebundene Peroxyspezies: Metallpersäuren und ihre Ester könnten ähnlich wie Percarbonsäuren reagieren. Der peroxidische Sauerstoff ist durch die Säurefunktion für einen nucleophilen Angriff eines Olefins aktiviert. → Über einen schmetterlingsartigen ("Butterfly")- Übergangszustand wird das dem Proton benachbarte O-Atom auf das Olefin übertragen. Paramagnetische Komplexe i.a. unerwünscht, da diese den radikalischen Zerfall von ROOH (R = H, Alkyl) induzieren. In der Regel unselektive radikalische Oxidation ! (Fenton-Chemie, Übergansmetall-katalysierter Zerfall v. ROOH, HOOH).
Fe3+ + ROOH Fe2+ + ROO• + H+ (langsam)
Fe2+ + ROOH Fe3+ + RO• + OH− (schnell) 2 ROOH RO• + ROO• + H2O Oxidation von Allylalkohohen: Präkoordination v. Substrat und Oxidationsmittel TBHP

668.2.2. Stereoselektive Varianten: Sharpless-Epoxidierung von Allylalkoholen: Templatartige Vorfixierung des prochiralen Allylalkohols, des Alkylhydroperoxids TBHP und des chiralen Induktorliganden S,S-Diethyltartrat an einem Ti4+ Komplexzentrum. Inhibierendes Wasser wird durch Molekularsieb gebunden: Enantioselektive Variante: Diastereoselektive Variante (kinetische Racematspaltung möglich):

67Enantiomer-Differenzierung: Das langsam reagierende R-Enantiomer (R4 = H, R5 ≠H) erleidet sterische Repulsion zwischen Tartrat und R5). Jacobsen-Katsuki-Epoxidierung
Anwendungsbreite: Insbesondere arylsubstituierte Olefinen ohne Ankerfunktion Anderes Prinzip des Oxentransfers: Epoxidation mittels C2-symmetrischer Salen-Mangan(III)-Komplexe, Hypochlorid oder PhIO als Oxidanzien, Mn(V)-Oxen-Komplexe als Oxenoid, Pyridin-N-oxid als Hilfsligand (verdrängt Chlorid).
Mn
O
Mn3+ 5+
O Cl
Mn
O4+2e
- Cl
•
•O
4+•
•
ArC=C R
O3+••
ArR
- Mn
O3+
•
••
Salen (2-) Salen (1-) Salen (2-)Mn Mn
••
R
Ar
H
H
ONC5H5
Mn5+
Salen(2-)
Cl
Mn5+
ClPy-N-Oxid
Kation leichter nucleophil angreifbar Mn stärker in die Salen-Ebene gezogen
Wirkung von Py-N-Oxid:
Salen redoxaktiv
O O**
bessere asym. Induktion
8.2.3 Elektronenarme Olefine α,β-ungesättigte (elektronenarme) Olefine (z.B. MMA) lassen sich nicht durch elektronenarme Metallpersäuren (oder Percarbonsäuren) epoxidieren! Stöchiometrisch läßt sich ein elektronenarmes Olefin durch das Nukleophil Na+[OOH]− angreifen und epoxidieren!
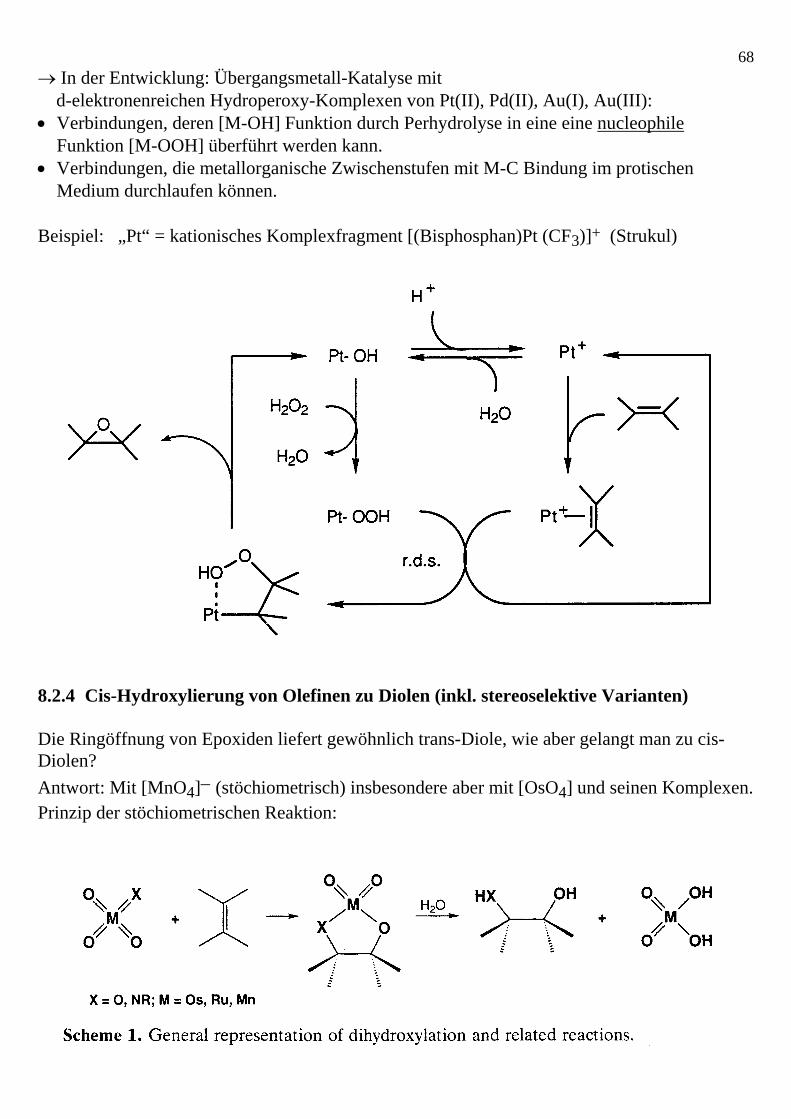
68→ In der Entwicklung: Übergangsmetall-Katalyse mit d-elektronenreichen Hydroperoxy-Komplexen von Pt(II), Pd(II), Au(I), Au(III): • Verbindungen, deren [M-OH] Funktion durch Perhydrolyse in eine eine nucleophile
Funktion [M-OOH] überführt werden kann. • Verbindungen, die metallorganische Zwischenstufen mit M-C Bindung im protischen
Medium durchlaufen können. Beispiel: „Pt“ = kationisches Komplexfragment [(Bisphosphan)Pt (CF3)]+ (Strukul) 8.2.4 Cis-Hydroxylierung von Olefinen zu Diolen (inkl. stereoselektive Varianten) Die Ringöffnung von Epoxiden liefert gewöhnlich trans-Diole, wie aber gelangt man zu cis-Diolen? Antwort: Mit [MnO4]− (stöchiometrisch) insbesondere aber mit [OsO4] und seinen Komplexen. Prinzip der stöchiometrischen Reaktion:

69Mechanistische Alternativen: konzertiert [2+3],
energetisch günstiger (Ea !)
MO
O
CH2
CH2M
O
OM
O
O
MO
Od0 d2
d0
=
schrittweise [2+2], 1,2-Uml. Asymmetrische Dihydroxylierung (AD): Chirale tert.-Amin-Liganden L: Zwei Cinchona-Alkaloid-Einheiten (DHQD, DHQ = „Alk“) verknüpft über einen N-Heterocyclus als Spacer (z.B. Phthalazin PHAL): Katalytischer Kreisprozeß: Reoxidation mit NMO (N-Methyl-morpholin-N-oxid)
homogen in Aceton-Wasser

70Katalytischer Zweiphasen-Kreisprozeß: Reoxidation mit rotem Blutlaugensalz Fe(III) 8.3 Olefine zu Allylalkoholen + α,β-ungesättigten Carbonylverbindungen Heterogene Katalyse Allylische Oxidation von Propen zum Allylalkohol, Acrolein und schließlich zur Acrylsäure an einem MoO3 / Bi2O3 Kontakt: H2C=CH-CH3 + O2 (MoO3 / Bi2O3 300-400°C) H2C=CH-CH=O + H2O Mechanismus evtl. vergleichbar mit homogener SeO2-Oxidation (M = Se, Mo):
Übertragen in die Oberflächen-Chemie des o.g. Kontakts:

71
In ähnlicher Weise wird die Oxidation (Dehydrierung) von 1-Buten zu Butadien am MoO3-Kontakt mechanistisch diskutiert: 8.4 Aromaten-Oxidation Produktion von Menadion (Vitamin K3) durch Oxidation von 2-Methylnaphthalin bislang stöchiometrisch mit CrO3 (18 kg Cr-Abfall pro kg Chinon) Katalytische Varianten: Aktivierung von H2O2 / HOAc mit Methylrheniumtrioxid (MTO), Wasser inhibiert Kat. ! Aktivierung von (Me3Si)2O2 mit Trimethylsilylperrhenat (Me3SiO-ReO3) Chemoselektivität: ca. 60 % Regioselektivität 9 : 1: Der el.reichere methylsubstituierte Ring wird bevorzugt oxidiert

72Katalytisch aktive Spezies: Perrheniumsäuren bzw. ihre O-Silylester. Kupfer-katalysierte Phenol-Oxidation mit Luft (Mitsubishi-Prozeß, 6000 Jato): Mechanismus Gegenstand intensiver Forschungsbemühungen; Prinzipielle Schritte: Cu(I) reagiert mit O2 zu Cu(II)-Peroxid-Intermediaten (thermisch instabil) und weiter zu Cu(III)-Oxid-Spezies (H-Atom-Abstraktor, Elektronen-Acceptor !)
Ln(X)CuO2
Ln(X)Cu Cu(X)Ln
O
O
Ln(X)Cu Cu(X)Ln
O
O
Ln(X)Cu Cu(X)LnO
OLn(X)Cu Cu(X)Ln
+2 +2
+2 +2
+1 +2
+2 +1
+1
2e-Transfer 2e-Transfer
Ln(X)Cu Cu(X)Ln
O
O
Ln(X)Cu Cu(X)LnO
OLn(X)Cu Cu(X)Ln
+3 +3
+2 +2
+2 +2
H-Atom-AbstraktorenPeroxid-Intermediate
Protolysevon Cu(III)Phenol
O Cu+3
O Cu• +2
O Cu+2
H•O Cu
+2H
Phenoxy-Radikal-Komplex-Intermediate
OO
O2
+ Cu(II)
OO
• - Cu(III)-OH
N
(N = 2)

738.5 Aliphaten-Oxidation n-Butan zu Methyl(ethyl)keton (MEK) / Cyclohexan zu Cyclohexanon Co(III)-katalysierte Radikalkettenreaktion (Autoxidation der KW mit Sauerstoff) AMOCO-Terephthalsäure (TPA)-Prozeß (auch „Mid-Century-Prozeß“: > 4 Mio Jato !!) Seitenketten-Oxidation von p-Xylol zu TPA mit Luft (HBr bzw. Bromid als Promotor)

74Mechanismus: 8.6 Alkohol-Oxidation Stöchiometrisch: β-H-Abstraktion an Chromsäureestern
(Chromsäureanhydrid CrO3 als Oxidans) Kupferkatalysierte Oxidation von (ungesättigten) Alkoholen mit Luftsauerstoff (Marko et. al.; Science (1996) 274, 2044; Angew. Chem. (1997) 109, 2297).

75Mechanismus: 8.7 Aldehyde und Ketone zu Carbonsäurederivaten Radikalische Wege: Die Autoxidation von Aldehyden zu Percarbonsäuren wird durch paramagnetische Metallionen katalysiert: H-Abstraktion eingeleitet z.B. durch [M(III)-X] [M(II)] + HX + Carbonylradikal RCO Carbonylradikal RCO + O2 RCO-OO RCO-OO + RCHO RCO-OOH + RCO RCO-OOH + RCHO 2 RCOOH Polare Wege: Die Baeyer-Villiger Oxidation von Ketonen wird durch die gleichen d-elektronenreichen Platin-Peroxokomplexe katalysiert, wie die Epoxidation elektronenarmer Olefine (s.o.).

768.8 N-, und S-Oxidation N-Oxidation: Katalysatoren: Oxokomplexe → Peroxokomplexe (bzw. Persäuren) von Re, Mo und W Oxidans: Wasserstoffperoxid Tert. Amine und Pyridine ergeben die entsprechenden N-Oxide Sek. Amine ergeben Nitrone Prim. Amine ergeben insbesondere für R = Aryl Produktspektrum (R = Alkyl → Nitroalkane) Modellreaktion:

77S-Oxidation: Asymmetrisch substituierte Sulfide zu chiralen Sulfoxiden (ee = 50 - 98 %) Titan-Katalysatoren: Ti(OR)4 + chirales Diol + H2O (1:2:1) Oxidans: tBuOOH (TBHP) bzw. besser Cumolhydroperoxid (CHP). C2-sym.-Diole: R,R-Diethyltartrat DET (Kagan bzw. Modena 1984),
R,R-1,2-Diphenyldiol, BINOL. Mechanismus: Vergleichbar mit Sharpless-Epoxidierung,
jedoch keine Substrat-Präkoordinierung Vanadium-Katalysatoren: V(IV)-Schiffbasen-Komplexe synthetisiert aus [V(O)(acac)2] Oxidans: H2O2 30%, gute ee-Werte insbesondere mit 14 (Bolm 1996) 8.9 Ammonoxidation von Olefinen Allylische Ammonoxidation von Propen zu Acrylnitril (SOHIO-Prozeß) Heterogene Katalyse (SOHIO-Prozeß): Allylische Oxidation von Propen in Gegenwart von Ammoniak an einem MoO3 / Bi2O3 Kontakt mit Sauerstoff (vgl. o., Acrylsäure und Acrolein): H2C=CH-CH3 + 1,5 O2 - NH3 (MoO3 / Bi2O3 300-400°C) H2C=CH-CN + 3 H2O

78Mechanistische Modellvorstellungen (SOHIO): Asymmetrische Aminohydroxylierung von Olefinen (AA) Katalysator: Wie bei Asym. Dihydroxylierung (AD); K2[OsO2(OH)4] + PHAL-Liganden Oxidans: Nitrenoide; N-Halogen-amide generiert aus Säureamiden + NaOH + tBuOCl
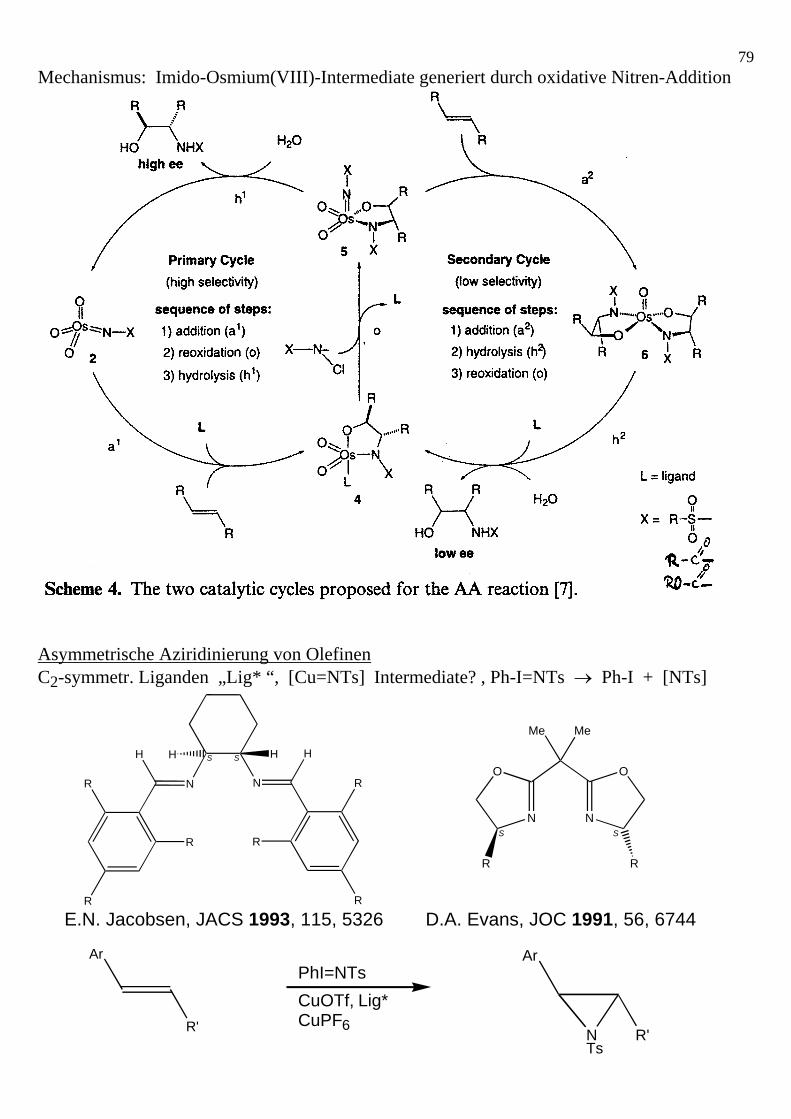
79Mechanismus: Imido-Osmium(VIII)-Intermediate generiert durch oxidative Nitren-Addition Asymmetrische Aziridinierung von Olefinen C2-symmetr. Liganden „Lig* “, [Cu=NTs] Intermediate? , Ph-I=NTs → Ph-I + [NTs]
R
R
R
R
R
R
NN
HHH HS S
O
N
R
O
N
R
MeMe
SS
E.N. Jacobsen, JACS 1993, 115, 5326 D.A. Evans, JOC 1991, 56, 6744
Ar
R'
PhI=NTsCuOTf, Lig*CuPF6
Ar
R'NTs


















