
Hémoglobinopathies - raft.g2hp.netraft.g2hp.net/files/2015/07/Hémoglobinopathies_F_2015-1.pdf ·...
53
Prof. Alain Gervaix Hémoglobinopathies
Transcript of Hémoglobinopathies - raft.g2hp.netraft.g2hp.net/files/2015/07/Hémoglobinopathies_F_2015-1.pdf ·...

Prof. Alain Gervaix
Hémoglobinopathies

Hémoglobinopathies
Définition: o Les hémoglobinopathies sont des maladies
héréditaires du sang qui compromettent le transport de l’oxygène dans l’organisme
o Elles se divisent en deux grandes catégories:
o La drépanocytose
o Les thalassémies

Hémoglobinopathies
o L'hémoglobine est une protéine dont la principale fonction est le transport de l’oxygène dans l'organisme humain
o L'hémoglobine se trouve essentiellement à l'intérieur des globules rouges du sang, ce qui leur confère leur couleur rouge.

o L'hémoglobine humaine est constituée de quatre chaînes identiques o deux chaînes α o deux chaînes β
o Chacune de ces chaînes est associée à un groupement prosthétique : l'hème.
o Le nom d'hémoglobine provient de deux mots : hème et globine. On la symbolise par « Hb ».
Hémoglobinopathies

Hémoglobinopathies
L’hème
o On trouve au cœur de la molécule un cycle hétérogène porphyrique, l'hème, qui contient un ion fer.
o Cet ion fer est le site de fixation de l'oxygène.

Hémoglobinopathies
Les gènes alpha et béta de la globine sont codés respectivement sur les
chromosomes 16 et 11

Hémoglobinopathies
o Comme de nombreuses protéines, les chaines d'hémoglobine peuvent présenter diverses mutations qui n'ont le plus souvent aucune incidence clinique.
o Plus de 500 hémoglobines anormales ont été répertoriées

Hémoglobinopathies
Mutations sur le gène codant pour la ß-globine (chromosome 11)
o Substitution d’un acide glutamique pour une valine
HbS
Responsable de la drépanocytose

La DREPANOCYTOSE
(également appelée hémoglobinose
S, sicklémie ou anémie à cellules
falciformes), est une maladie
héréditaire, autosomale
récessive, qui se caractérise
par une altération de
l'hémoglobine
Drépanocytose homozygote Hb S/S
La drépanocytose

La DREPANOCYTOSE
Il existe plusieurs syndromes drépanocytaires selon le type de mutation
HbS/S: propriétés de l’hémoglobine modifiées
(hémoglobinopathie qualitative)

13%
La DREPANOCYTOSE
Epidémiologie:
o > 50 millions de personnes atteintes dans le monde o >250’000 enfants présentant une forme grave
naissent chaque année o Afrique sub-saharienne, moyen-orient, Inde, Brésil

Hémoglobinopathies
Distribution de la drépanocytose Distribution de la malaria
La drépanocytose hétérozygote semble protéger contre les formes graves de la malaria

La DREPANOCYTOSE
o Les globules rouges de personnes homozygotes, HbS/S, ne contiennent pratiquement que de l'HbS.
o Or, ces molécules ont la propriété de se polymériser lorsqu'elles sont désoxygénées, donnant lieu à la formation de fibres qui déforment le globule et lui donnent un aspect en faucille.
Physio-pathologie

La DREPANOCYTOSE
HbS
Stress o Hypoxie o Froid o Déshydratation o Infection
polymérisation
falciformation

Patho-physiologie
Falciformation
Diminution de la résistance mécanique et de l’élasticité des GR
o Anémie hémolytique
o Crise vaso-occlusive douloureuse
o Endommagement des organes
La DREPANOCYTOSE

La DREPANOCYTOSE
Manifestations de la maladie
Les crises « vaso-occlusives », sont causées par l’obstruction des capilaires par les cellules en “faucille” et se manifestent par des douleurs vives et brutales dans certaines parties du corps. Elles peuvent, à la longue, entraîner la destruction de certains organes ou parties d’organes (p.ex la rate)
o Episodes douloureux aigus en raison des crises vaso-occlusives

Ces douleurs peuvent se produire dans toutes les parties du corps
ü Dos ü Thorax ü Extrémités ü Abdomen
étant les parties les plus souvent atteintes.
Les crises douloureuses, qui durent généralement trois à dix jours, sont difficilement prévisibles.
La DREPANOCYTOSE
o Episodes douloureux aigus

Ce syndrome concerne exclusivement l’enfant, avant l’âge de deux ans. Le(s) pied(s) et/ou les main(s) deviennent chauds, gonflés, et les mouvements sont douloureux. Cela peut être la première manifestation de la maladie chez les jeunes enfants, associée ou non à de la fièvre.
Syndrome pied-main ou dactylite
La DREPANOCYTOSE

La DREPANOCYTOSE
o Anémie hémolytique
L’anémie désigne un manque d’hémoglobine (ou de globules rouges) et se traduit par une fatigue excessive et une sensation de faiblesse. Lorsque l’anémie est assez sévère, le malade peut avoir des difficultés à respirer (essoufflement) et une accélération des battements du coeur (tachycardie). Les personnes atteintes sont anémiques en permanence mais s’y adaptent généralement assez bien. Parfois, les seuls signes visibles sont la fatigabilité et une couleur jaune des yeux ou de la peau, (ictère), et une coloration foncée des urines.

Aggravation de l’anémie (SÉQUESTRATION SPLÉNIQUE) Le fonctionnement intensif de la rate est une manifestation qui se retrouve surtout chez l’enfant. o Les manifestations de la séquestration splénique sont : - des douleurs abdominales ; - une augmentation très soudaine du volume de la rate
(splénomégalie) - une pâleur marquée et, de manière générale, une aggravation de toutes les manifestations de l’anémie.
o La séquestration splénique est brusque et importante. Elle peut mettre la vie en danger, surtout chez les enfants de moins de sept ans.
o Si le malade ou son entourage pense reconnaître les signes d’une séquestration splénique, il est conseillé de conduire le malade en urgence à l’hôpital
La DREPANOCYTOSE

Aggravation de l’anémie (ANEMIE APLASTIQUE)
La DREPANOCYTOSE
Chute brutale de l’hémoglobine , non régénérative
(absence de réticulocytose)
Les crises aplasiques se caractérisent par des manifestations telles que de la fièvre, des maux de tête (céphalées), des douleurs abdominales, une perte d’appétit ou des vomissements. Ces manifestations sont transitoires. Ces crises peuvent être liées à une infection par le parvovirus B19 ou à un manque en acide folique (vitamine B9) qui doit être prise régulièrement par les personnes drépanocytaires.

La DREPANOCYTOSE
o Le syndrome thoracique aïgu
La radiographie des poumons montre la présence anormale de tâches blanches (infiltrats pulmonaires). C’est une complication grave et le malade et/ou son entourage doivent en connaître les signes car ce syndrome doit être traité en urgence. Chez l’enfant il est souvent dû ou associé à une infection des poumons
Ce syndrome se manifeste par une fièvre, une gêne ou des difficultés respiratoires (dyspnée), une respiration rapide, une toux, et des douleurs dans la poitrine.

Les manifestations sont très variables, et peuvent être transitoires (on parle alors d’accidents ischémiques transitoires ou AIT) : pertes de sensibilité ou de force dans un bras, une jambe, la moitié du visage, paralysie d’un côté du corps ou d’un membre (hémiplégie), maux de tête (céphalées), difficultés soudaines à parler (aphasie), troubles de l’équilibre, convulsions, parfois coma. Des maux de tête violents ou des difficultés d’apprentissage soudaines peuvent être des signes d’alerte.
La DREPANOCYTOSE
o ACCIDENT VASCULAIRE CEREBRAL

Ces AVC concernent le plus souvent les enfants surtout entre quatre et six ans. Souvent, les symptômes apparaissent et disparaissent brutalement mais le risque qu’ils se reproduisent est élevé. L’enfant peut s’en sortir indemne mais, dans de nombreux cas, l’AVC provoque des dommages au cerveau laissant des séquelles motrices et/ou intellectuelles.
La DREPANOCYTOSE
o ACCIDENT VASCULAIRE CEREBRAL
OHENE-FREMPONG et al. Blood, Vol 91, 1998: pp 288-294

La DREPANOCYTOSE
o SUSCEPTIBILITE ACCRUE AUX INFECTIONS
o Les enfants (parfois même dès l’âge de trois mois), et dans une moindre mesure les adultes, sont très sensibles aux infections bactériennes qui peuvent se développer de manière fulgurante et doivent donc être traitées rapidement.
o Due à la dysfonction splénique
o Les personnes sont plus spécialement sensibles aux pneumonies, à la grippe, mais aussi aux hépatites, aux méningites, aux infections urinaires, aux septicémies et aux infections ostéo-articulaires
o Bactéries: S pneumoniae, H inflruenzae type B, M pneumoniae, Salmonella sp …

La DREPANOCYTOSE
o SUSCEPTIBILITE ACCRUE AUX INFECTIONS
Le risque d’infection est maximal chez les enfants de moins de 5 ans, mais il perdure toute la vie. Chez l’enfant, il est très important de prévenir les foyers infectieux chroniques (au niveau des dents, des amygdales, des os, de la vésicule, en s’assurant d’une bonne hygiène (brossage de dents suffisant etc), de maintenir à jour leurs vaccinations, et de s’assurer qu’ils prennent l’antibiotique qui leur est prescrit (pénicilline) tous les jours, sans oubli au minimum jusqu’à l’âge de 5 ans

La DREPANOCYTOSE
o Complications chroniques de la drépanocytose
o Atteinte cardiaque, rénale, dermatologique
o HTAP
o Retinopathie
o Priapisme
o Lithiase biliaire
o …etc

La DREPANOCYTOSE
Curr Probl Pediatr Adolesc Health Care, 2006

Le diagnostic est posé: o En examinant la forme des GR
o En analysant l’hémoglobine
(électrophorèse)
o Par des tests génétiques.
La DREPANOCYTOSE
o Comment poser le diagnostic de drépanocytose

La DREPANOCYTOSE
o TRAITEMENT
Il n’existe pas de traitement curatif de cette maladie génétique, hormis une transplantation de cellules souches hématopoiétiques Traitement symptômatique: Crise vaso-occlusive: Antalgiques (paracétamol, AINS, morphine) Anémie aiguë: Transfusion, acide folique Infection: Antibiotiques ü Prévention: Vaccination,
pénicilline quotidiennement (min 5 ans)

Parallèlement au traitement médicamenteux, des mesures simples doivent être prises pour apaiser le malade en cas de crise :
o repos au chaud o boire beaucoup pour bien s’hydrater o calme (l’entourage familial doit essayer autant
que possible de maintenir une atmosphère calme autour du malade)
Souvent, la mise en place d’une oxygénothérapie est proposée pendant l’hospitalisation
La DREPANOCYTOSE

La DREPANOCYTOSE
o Traitement de fond
Hydroxyurée o Augmentation de la
production HbF
o Diminution de l’adhésion à l’endothélium
o Modulation de l’inflammation

La production « forcée » de l’ hémoglobine F foetale permet de diminuer l’agglomération de l’hémoglobine S. Chez les personnes qui répondent bien au traitement, la fréquence des crises douloureuses et des hospitalisations diminuent Le besoin de transfusion et le risque de survenue d’un syndrome thoracique aigu sont également diminués Ce traitement a nettement amélioré la qualité de vie des personnes atteintes d’une forme sévère ou moyennement sévère (ayant plus de trois crises douloureuses par an nécessitant une hospitalisation, une anémie sévère ou un antécédent de syndrome thoracique aigu) Cependant, il n’agit pas sur les infections pulmonaires ou osseuses et ne met pas non plus à l’abri des accidents vasculaires cérébraux et des atteintes osseuses secondaires.
La DREPANOCYTOSE

La DREPANOCYTOSE
Références
www.orpha.net/ http://www.uptodate.com/contents/overview-of-the-clinical-manifestations-of-sickle-cell-disease?source=related_link
Diallo DA, Guindo A. “Sickle cell disease in sub-Saharan Africa: stakes and strategies for control of the disease” Curr Opin Hematol. 2014 May;21(3):210-4
Bégué P, Castello-Herbreteau B. “Severe infections in children with sickle cell disease: clinical aspects and prevention”. Arch Pediatr. 2001 Sep;8 Suppl 4:732s-741s

Les thalassémies
Les THALASSEMIES
Défaut génétique de production de la chaîne alpha ou béta de la
globine (Hémoglobinopathie quantitative)

Les thalassémies sont des
maladies héréditaires,
autosomales récessives, qui
se caractérisent par un
défaut de production
l'hémoglobine.
o Béta-thalassémie
o Alpha-thalassémie
Les thalassémies

Les thalassémies
La bêta-thalassémie atteint surtout les personnes originaires du pourtour méditerranéen, du Moyen-Orient, d’Asie (Chine, Inde, Viêt-Nam, Thaïlande) et d’Afrique noire. Elle atteint autant les femmes que les hommes.

Les ß-thalassémies
ß-thalassémie Selon que la production de chaînes ß de la globine est absente ou seulement réduite, on distingue:
o La ß-thalassémie majeure (anémie de Cooley)
o La ß-thalassémie intermédiaire
o La ß-thalassémie mineure

Les ß-thalassémies
La ß-thalassémie majeure
Les premiers signes de ß-thalassémie majeure ne se révèlent qu’après l’âge de 6 mois, car le sang du nouveau-né contient toujours beaucoup d’hémoglobine foetale HbF ( alpha2 / gamma2), sous une forme souvent très hétérogène selon les individus:
o Anémie hémolytique sévève o Paleur
o Irritabilité
o Hépatomégalie
o Splénomégalie
o Ictère
o Retard de développement
Les ß-thalassémies
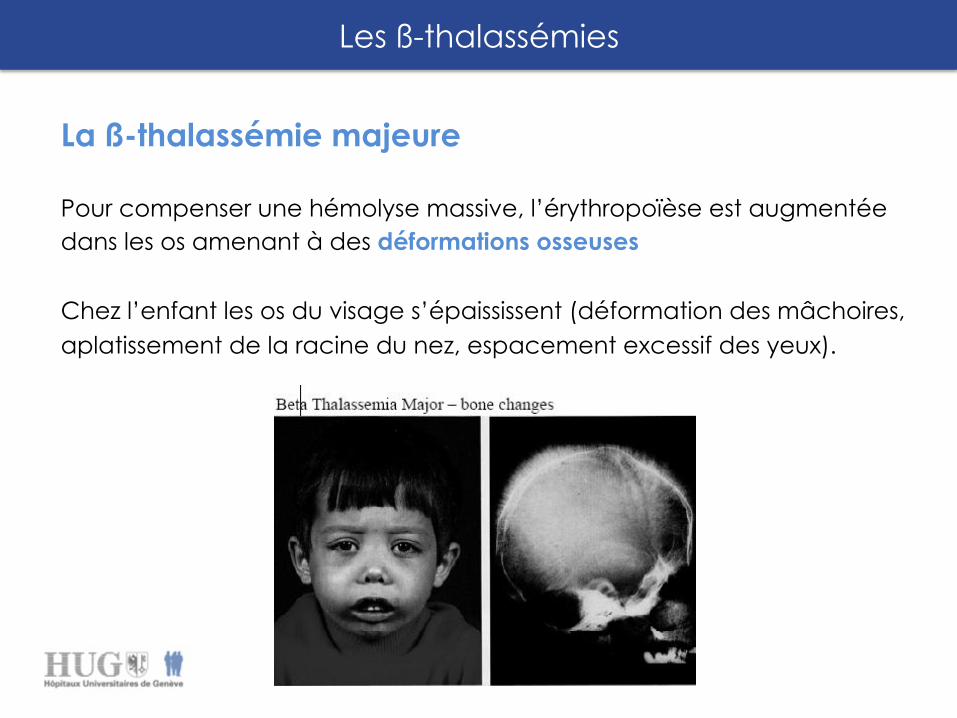
La ß-thalassémie majeure
Pour compenser une hémolyse massive, l’érythropoïèse est augmentée dans les os amenant à des déformations osseuses Chez l’enfant les os du visage s’épaississent (déformation des mâchoires, aplatissement de la racine du nez, espacement excessif des yeux).

Les ß-thalassémies
Complications secondaires due à une surcharge en fer consécutive à l’hémolyse/transfusion continue
o Anomalies endocrines et métaboliques ü Hypogonadisme 40-55% ü Retard de croissance 33%
ü Diabète 6-13% ü Hypothyroidisme 10%
La ß-thalassémie majeure
o Complications cardiaques ü Insuffisance cardiaque (hémosidérose)
ü Arythmies
o Lithiase biliaire

Les ß-thalassémies
La ß-thalassémie majeure
Diagnostic:
o Suspicion clinique (signes, symptômes, origine…)
o Frottis sanguin (anémie hypochrome, microcytaire)
Biochimie: Hémolyse (bili libre , LDH , Haptoglobine ) Fer sérique , ferritine
o Electrophorèse de l’hémoglobine ( HbA2 3.5-8%
HbF 1-2% )

Les ß-thalassémies
La ß-thalassémie majeure
Traitement:
o Transfusion sanguine
o (Splénectomie )
o Acide folique
o Chélateur du fer

Les ß-thalassémies
La ß-thalassémie intermédiaire
Dans la bêta-thalassémie intermédiaire, les deux gènes bêta sont altérés, mais ils permettent tout de même la fabrication d’hémoglobine en quantité réduite. Les symptômes sont donc beaucoup moins importants que dans l’anémie de Cooley.
o Anémie microcytaire hypochrome plus tardive ( 1-2 ans) mois profonde (>7.5 g/dl)
o Ictère, splénomégalie
o Anomalies osseuses (+/-)
o Pas de retard de croissance

Les ß-thalassémies
La ß-thalassémie mineure
o La bêta-thalassémie mineure est due à la mutation d’un seul des deux gènes bêta.
o Généralement, cette forme n’a pas de conséquence sur la santé, puisque l’autre gène est capable de compenser l’anomalie et de fabriquer suffisamment de chaînes bêta pour produire un taux d’hémoglobine normal ou proche de la normale
o Microcytose

L’alpha-thalassémie est très répandue à travers le monde. Elle affecte surtout les populations originaires d’Asie (Cambodge, Laos, Birmanie, Thaïlande notamment), dans ses formes intermédiaires ou graves, d’Afrique équatoriale, et du bassin méditerranéen dans ses formes mineures.
Les alpha-thalassémies
Les alpha-thalassémies

Les alpha-thalassémies
Les alpha-thalassémies
Sur le chromosome 16, 2 gènes codent pour les chaînes alpha de la globine

Les alpha-thalassémies
Chr 16p Chr 16m αα / αα
αα / -α
-α / -α
-- / αα
-- / -α
-- / --
Sujet normal
α thal silencieuse
α thal mineure ou
α thal majeure (Hémoglobinose H)
Hydrops fetalis
Les alpha-thalassémies

Les alpha-thalassémies
Manifestations cliniques

o L’anémie (microcytaire hypochrome) est le principal symptôme
o Celle-ci est présente dès la naissance mais elle n’est parfois diagnostiquée qu’à l’âge adulte lorsqu’elle est modérée.
o Elle est variable d’une personne à l’autre, et au cours du temps.
Les alpha-thalassémies
Hémoglobinose H

Les alpha-thalassémies
Hémoglobinose H (Hb 4ß)
Signes cliniques
o Paleur
o Fatigue
o Ictère
o Hépato-splénomégalie
o Lithiase biliaire

Les alpha-thalassémies
Diagnostic
http://www.uptodate.com/
Frottis sanguin Electrophorèse de l’hémoglobine

Hémoglobinopathies
Merci pour votre attention !


















