HIV-1: Cofactors provide the entry keys
3
Dispatch 1051 HIV-1: Cofactors provide the entry keys Dave Wilkinson The identification of the cofactors required for HIV-1 entry into cells promises to provide new insights into viral transmission and pathogenesis, and opens new avenues for AIDS therapy and prophylaxis. Address: Chester Beatty Laboratories, Institute of Cancer Research, 237 Fulham Road, London SW3 6JB UK. Current Biology 1996, Vol 6 No 9:1051–1053 © Current Biology Ltd ISSN 0960-9822 The entry pathway of the human immunodeficiency virus type-1 (HIV-1) can be divided into three major events: binding, activation and fusion. The viral envelope protein is essential for each of these stages; it consists of a surface subunit, gp120, and a transmembrane subunit, gp41, which form an oligomeric complex in the virion membrane [1]. The gp120 subunit contains a binding domain specific for the human CD4 molecule, the cellular receptor for HIV-1. The gp41 subunit contains a fusogenic hydropho- bic peptide at its amino terminus, which is essential for fusion of the viral and cellular membranes. The primary cell types infected by HIV — helper T cells, macrophages and dendritic cells — all express CD4, which is thus a fun- damental determinant of both the species and cellular tropism of HIV-1. The CD4–gp120 interaction is thought to activate the fusion activity of the envelope oligomer by inducing a conformation shift [2,3] that exposes new func- tional domains, such as the third variable region (V3) of gp120. The consequent fusion of the viral and plasma membranes releases the viral core into the cell to initiate replication. The CD4–gp120 interaction is thus important for the first two major steps in the HIV entry pathway; it provides a high affinity binding receptor and is involved in activating the fusion activity of the viral envelope. Soon after CD4 was identified as the cellular receptor for HIV-1, it became clear that expression of CD4 alone is insufficient for envelope-mediated fusion to occur. One test of this is to transfect a cell line with the CD4 gene, and see if it confers the ability to fuse with cells express- ing the HIV-1 gp120 protein. A small number of human cell lines, and the vast majority of non-human cell lines, fail this test [4,5]; the human cell lines that fail are classed as ‘restrictive’, as opposed to the ‘permissive’ cell lines that pass the test when they express CD4. The restrictive CD4 + cells were also found to resist infection by cell-free virus particles, as a result of a block at the level of virus entry [6–8]. The fusion defect of the restric- tive CD4 + cells can, however, be complemented by prior fusion with permissive CD4 – cells, indicating that the block to fusion is the result of the lack of a cofactor in some cell types [9–11]. The expression of different fusion cofactors by various cell types has been shown to be a major determinant of HIV-1 tropism [12]. The tropism of different HIV-1 strains is primarily distinguished on the basis of their ability to infect monocyte-derived macrophages as opposed to established T-cell lines. Macrophage-tropic viruses are thought to be fundamental for the transmission and pathogenesis of HIV-1; for example, macrophage- tropic viruses have been shown to predominate early in infection, possibly as a result of selection during transmis- sion when the initial cellular targets of an infection are tissue macrophages or dendritic cells. Macrophages are also believed to facilitate the dissemination of virus to various tissues within an infected individual, and thereby to contribute to the pathological course of the infection. On the virus side, important determinants of tropism have been mapped to the V3 region of gp120 [13]. Until recently, the cellular determinants of tropism were not known, but several different fusion cofactors have now been identified and established as the cellular basis for the specificity of the fusion reaction [14–20]. The first fusion cofactor to be identified was specific for virus strains adapted to T-cell lines [14], and proved to be a member of the seven transmembrane G-protein-coupled receptor superfamily. Several laboratories investigating that superfamily had previously isolated cDNAs for the same receptor, but had been unable to determine its ligand. Because of its role as a fusion cofactor, the protein has been dubbed ‘fusin’, but this may be a temporary appellation pending identification of its natural ligand. Sequence analysis shows that fusin has ~35 % sequence similarity to interleukin 8 (IL-8) receptor-b, and so is likely to be the receptor for an as yet undefined chemokine. As expected, fusin was found to be required for cell fusion mediated by the envelope protein of laboratory-adapted isolates. In addition, expression of fusin on non-permissive cell types allowed infection by cell free T-cell-tropic viruses, but not by macrophage-tropic isolates [14]. The discovery that fusin is related to chemokine recep- tors provided the catalyst for a dramatic convergence between studies of HIV-1 tropism and cell entry and investigations into the nature of antiviral factors released by CD8 (cytotoxic) T cells. Before the discovery of fusin, three b-chemokines — RANTES, MIP-1a and MIP-1b — had been shown to have anti-viral activity [15]. Follow- ing this lead, the receptors for other chemokines were
-
Upload
dave-wilkinson -
Category
Documents
-
view
215 -
download
1
Transcript of HIV-1: Cofactors provide the entry keys

Dispatch 1051
HIV-1: Cofactors provide the entry keysDave Wilkinson
The identification of the cofactors required for HIV-1entry into cells promises to provide new insights intoviral transmission and pathogenesis, and opens newavenues for AIDS therapy and prophylaxis.
Address: Chester Beatty Laboratories, Institute of Cancer Research,237 Fulham Road, London SW3 6JB UK.
Current Biology 1996, Vol 6 No 9:1051–1053
© Current Biology Ltd ISSN 0960-9822
The entry pathway of the human immunodeficiency virustype-1 (HIV-1) can be divided into three major events:binding, activation and fusion. The viral envelope proteinis essential for each of these stages; it consists of a surfacesubunit, gp120, and a transmembrane subunit, gp41,which form an oligomeric complex in the virion membrane[1]. The gp120 subunit contains a binding domain specificfor the human CD4 molecule, the cellular receptor forHIV-1. The gp41 subunit contains a fusogenic hydropho-bic peptide at its amino terminus, which is essential forfusion of the viral and cellular membranes. The primarycell types infected by HIV — helper T cells, macrophagesand dendritic cells — all express CD4, which is thus a fun-damental determinant of both the species and cellulartropism of HIV-1. The CD4–gp120 interaction is thoughtto activate the fusion activity of the envelope oligomer byinducing a conformation shift [2,3] that exposes new func-tional domains, such as the third variable region (V3) ofgp120. The consequent fusion of the viral and plasmamembranes releases the viral core into the cell to initiatereplication. The CD4–gp120 interaction is thus importantfor the first two major steps in the HIV entry pathway; itprovides a high affinity binding receptor and is involved inactivating the fusion activity of the viral envelope.
Soon after CD4 was identified as the cellular receptor forHIV-1, it became clear that expression of CD4 alone isinsufficient for envelope-mediated fusion to occur. Onetest of this is to transfect a cell line with the CD4 gene,and see if it confers the ability to fuse with cells express-ing the HIV-1 gp120 protein. A small number of humancell lines, and the vast majority of non-human cell lines,fail this test [4,5]; the human cell lines that fail areclassed as ‘restrictive’, as opposed to the ‘permissive’ celllines that pass the test when they express CD4. Therestrictive CD4+ cells were also found to resist infectionby cell-free virus particles, as a result of a block at thelevel of virus entry [6–8]. The fusion defect of the restric-tive CD4+ cells can, however, be complemented by priorfusion with permissive CD4– cells, indicating that the
block to fusion is the result of the lack of a cofactor insome cell types [9–11].
The expression of different fusion cofactors by variouscell types has been shown to be a major determinant ofHIV-1 tropism [12]. The tropism of different HIV-1strains is primarily distinguished on the basis of theirability to infect monocyte-derived macrophages asopposed to established T-cell lines. Macrophage-tropicviruses are thought to be fundamental for the transmissionand pathogenesis of HIV-1; for example, macrophage-tropic viruses have been shown to predominate early ininfection, possibly as a result of selection during transmis-sion when the initial cellular targets of an infection aretissue macrophages or dendritic cells. Macrophages arealso believed to facilitate the dissemination of virus tovarious tissues within an infected individual, and therebyto contribute to the pathological course of the infection.On the virus side, important determinants of tropism havebeen mapped to the V3 region of gp120 [13]. Untilrecently, the cellular determinants of tropism were notknown, but several different fusion cofactors have nowbeen identified and established as the cellular basis forthe specificity of the fusion reaction [14–20].
The first fusion cofactor to be identified was specific forvirus strains adapted to T-cell lines [14], and proved to bea member of the seven transmembrane G-protein-coupledreceptor superfamily. Several laboratories investigatingthat superfamily had previously isolated cDNAs for thesame receptor, but had been unable to determine itsligand. Because of its role as a fusion cofactor, the proteinhas been dubbed ‘fusin’, but this may be a temporaryappellation pending identification of its natural ligand.Sequence analysis shows that fusin has ~35 % sequencesimilarity to interleukin 8 (IL-8) receptor-b, and so is likelyto be the receptor for an as yet undefined chemokine. Asexpected, fusin was found to be required for cell fusionmediated by the envelope protein of laboratory-adaptedisolates. In addition, expression of fusin on non-permissivecell types allowed infection by cell free T-cell-tropicviruses, but not by macrophage-tropic isolates [14].
The discovery that fusin is related to chemokine recep-tors provided the catalyst for a dramatic convergencebetween studies of HIV-1 tropism and cell entry andinvestigations into the nature of antiviral factors releasedby CD8 (cytotoxic) T cells. Before the discovery of fusin,three b-chemokines — RANTES, MIP-1a and MIP-1b— had been shown to have anti-viral activity [15]. Follow-ing this lead, the receptors for other chemokines were

screened for their ability to act as cofactors for viral entry.Thus, CC-CK5, a b-chemokine receptor specific forRANTES, MIP-1a and MIP-1b, was shown to be a fusioncofactor for macrophage-tropic viral isolates [16–18]. Fur-thermore, the same three b-chemokines were found toblock envelope-induced fusion mediated by CC-CKR5but not by fusin. Additional chemokine receptors, CC-CKR2b and CC-CKR3, can also be used as cofactors bysome HIV-1 isolates [19,20] and by HIV-2 (Mark Marsh,personal communication).
Although the potential role that these additional cofactorsplay in vivo during an infection remains to be determined,compelling evidence for the importance of CC-CKR5 invivo has been reported very recently [21,22]. Individualspossessing HIV-1-resistant CD4 T cells have been identi-fied because they have remained seronegative for HIV-1despite having a high risk of infection [23]. The geneticbasis for their resistance to HIV-1 has now been shown tobe due to the disruption of both of their CKR5 alleles by a short deletion [21]. Even individuals heterozygous forthe mutation in CKR5 appear to be partially resistant toinfection [22], due either to an inhibitory effect of themutation or decreased expression of functional CC-CKR5. The finding that a mutation in CKR5 renders indi-viduals resistant to infection underscores the importanceof the interaction of macrophage-tropic viruses with theCC-CKR5 cofactor during HIV-1 transmission. Conse-quently, the development of agents that block the inter-action with CC-CKR5 is a new and promising approach toHIV-1 prophylaxis
The finding that various subsets of fusion cofactors are usedby different viral isolates provides a satisfying explanation
for the complex tropism patterns exhibited by HIV-1.Indeed, the established tropisms of many viral strains indi-cate that more fusion cofactors remain to be identified.The identification of new cofactors will also be facilitatedby the development of neutralizing monoclonal antibodiesagainst existing cofactors; for example, a neutralizing mon-oclonal antibody against fusin blocks HIV-1 infection ofsome T-cell lines but not others, suggesting that someT-cell lines may express additional, undefined cofactors(Jim Hoxie and Paul Clapham, personal communication).
The mechanism by which cofactors facilitate fusion willnow be the focus of considerable investigation, for under-standing the exact role played by chemokine receptorsduring virus entry will assist their exploitation as potentialtherapeutic targets. As predicted from tropism studies, thegp120 V3 loop is an important determinant of cofactorusage [19], and consequently also of susceptability to viralsuppression by chemokines [23]. It is interesting to notethat other retroviruses, such as murine leukaemia virus(MLV), use cellular receptors that, like chemokine recep-tors, have small extracellular domains [1]. In the case ofHIV-1, therefore, it might be that binding to a long rod-like molecule, such as CD4, provides an efficient receptorfor adsorption, but that binding to a cofactor close to thelipid bilayer is required for the subsequent events leadingto membrane fusion. One challenge now will be to under-stand how the conformational changes induced by consec-utive gp120 binding to its two receptors trigger gp41 tomediate the final fusion event (Fig. 1).
An additional and provocative possibility is that binding bygp120 V3 region to the fusion cofactor transduces a signalto the target cell via the G-protein-coupled pathway. A
1052 Current Biology 1996, Vol 6 No 9
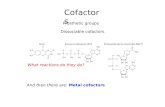
Figure 1
HIV-1
Virus entry
gp41gp120
CD4
Fusion cofactor
Fusion peptide
V3
CD4 binding Activation Insertion of fusion peptide Membrane fusion
Host cell © 1996 Current Biology
The HIV-1 cell-entry pathway. Specific binding of CD4 by gp120triggers a cascade of interactions that result in virus entry. CD4binding induces a conformational shift in gp120 which is followedby binding to a specific cofactor, determined by the V3 region ofgp120. Only after binding to the cofactor does the fusion of viraland cellular membranes occur, mediated by insertion into the cell
membrane of the fusogenic peptide at the amino terminus ofgp41. The precise mechanism by which cofactor binding togp120 leads to activation of gp41 is not known but, as gp120 isthought to occlude the fusion peptide in gp41, it is likely to involveadditional conformational changes that lead to the dissociation ofgp41 and gp120.

stimulatory signal generated during entry could possiblyplay a role in the infection of non-proliferating cells suchas macrophages. Another possibility is that a signal trans-duced during entry could induce newly infected dendriticcells to migrate, or express cell-surface molecules enhanc-ing their retention by proximal lymph nodes. A more thor-ough understanding of the mechanism of fusion is alsorequired to explain the differences between cell–cellfusion and cell-free infection that have been described.For example, macrophages express fusin and also supportsyncytia formation with cells infected with T-cell-tropicHIV-1 isolates, but entry of the same viral isolates isblocked in macrophages [24].
Understanding the entry process has importantimplications for our understanding of several key aspectsof HIV-1 transmission and pathogenesis. It has alsoopened up new avenues for therapeutic intervention andprophylaxis. Several uninfected high-risk individuals havebeen shown to have elevated levels of b-chemokines, sug-gesting that b-chemokines may provide protection againstinfection [23]. In light of this finding, the induction of pro-tracted b-chemokine production could become an aim ofvaccine design. The development of new small animalmodels for HIV-1 infection is another goal made feasibleby the identification of fusion cofactors. But it remains tobe seen whether creating transgenic animals expressing aselected few cofactors will produce a satisfactory model fora virus infection which in the natural host probablyinvolves many cofactors. Nevertheless, the recent discov-eries of crucial cofactors for HIV-1 entry into cells clearlypromise to open many more doors to an increased under-standing of HIV biology.
AcknowledgementsThe author is supported by MRC Canada with an AIDS-fellowship, and isgrateful to Matthias Dittmar and Paul Clapham for stimulating discussionsand critical reading of the manuscript. The author is also grateful to MarkMarsh, Him Hoxie and Paul Clapham for permitting mention of their unpub-lished data.
References1. Weiss RA: Cellular receptors and viral glycoproteins involved in
retrovirus entry. In The Retroviridae, vol. 2. Edited by Levy JA. NewYork, USA: Plenum Press; 1993:1–108
2. Bachelder RE, Bilancieri J, Lin W, Letvin NL: A human recombinantFab identifies a human immunodeficiency virus type 1-inducedconformational change in cell surface-expressed CD4. J Virol1995, 69:5734–5742.
3. Sattentau QJ, Moore JP, Vignaux F, Traincard F, Poignard P:Conformational changes induced in the envelope glycoproteins ofthe human and simian immunodeficiency viruses by solublereceptor binding. J Virol 1993, 67:7383–7393.
4. Ashorn PA, Berger EA, Moss B: Human immunodeficiency virusenvelope glycoprotein/CD4-mediated fusion of nonprimate cellswith human cells. J Virol 1990, 674:2149–2156.
5. Dragic T, Charneau P, Clavel F, Alizon M: Complementation ofmurine cells for human immunodeficiency virus envelope/CD4-mediated fusion in human/murine heterokaryons. J Virol 1992,66:4794–4802.
6. Chesebro B, Buller R, Portis J, Wehrly K: Failure of humanimmunodeficiency virus entry and infection in CD4-positivehuman brain and skin cells. J Virol 1990, 64:215–221.
7. Clapham PR, Blanc D, Weiss RA: Specific cell surfacerequirements for the infection of CD4-positive cells by humanimmunodeficiency virus types 1 and 2 and by simianimmunodeficiency virus. Virology 1991, 181:703–715.
8. Harrington RD, Geballe AP: Cofactor requirement for humanimmunodeficiency virus type 1 entry into a CD4-expressinghuman cell line. J Virol 1993, 67:5939–5947.
9. Broder CC, Dimitrov DS, Blumenthal R, Berger EA: The block to HIV-1 envelope glycoprotein-mediated membrane fusion in animalcells expressing human CD4 can be overcome by a human cellcomponent(s). Virology 1993, 193:483–491.
10. Dragic T, Picard L, Alizon M: Proteinase-resistant factors in humanerythrocyte membranes mediate CD4-dependent fusion with cellsexpressing human immunodeficiency virus type 1 envelopeglycoproteins. J Virol 1995, 69:1013–1018.
11. Weiner DB, Huebner K, Williams WV, Greene MI: Human genesother than CD4 facilitate HIV-1 infection of murine cells.Pathobiology 1991, 59:361–371.
12. Broder CC, Berger E: Fusogenic selectivity of the envelopeglycoprotein is a major determinant of human immunodeficiencyvirus type 1 tropism for CD4+ T-cell lines vs. primarymacrophages. Proc Natl Acad Sci 1995, 92:9004–9008.
13. Hwang SS, Boyle TJ, Lyerly HK, Cullen BR: Identification of theenvelope V3 as the primary determinant of cell tropism in HIV-1.Science 1991, 253:71–74.
14. Feng Y, Broder CC, Kennedy PE, Berger EA: HIV-1 entry cofactor:functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272:872–877.
15. Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P:Identification of RANTES, MIP-1a, and MIP-1b as the major HIV-suppressive factors produced by CD8 T cells. Science 1995,270:1811–1815.
16. Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, MurphyPM, Berger EA: CC CKR5: A RANTES, MIP-1a, MIP-1b receptor asa fusion cofactor for macrophage-tropic HIV-1. Science 1996,272:1955–1962.
17. Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di MarzioP, Marmon A, Sutton R, Hill CM, et al.: Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381:661–666.
18. Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA,Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA: HIV-1 entryinto CD4+ cells is mediated by the chemokine receptor CC-CKR5.Nature 1996, 381:667–673.
19. Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L,Mackay CR, LaRosa G, Newman W, et al.: The b-chemokinereceptors CCR3 and CCR5 facilitate infection by primary isolates.Cell 1996, 85:1135–1148.
20. Doranz GJ, Rucker J, Ye Y, Smyth RJ, Samson M, Peiper S,Parmentier M, Collman RG, Soms RW: A dual-tropic primary HIV-1isolate that uses fusin and the b-chemokine receptors CKR-5,CKR-3 and CKR-2b as fusion cofactors. Cell 1996, 85:1149–1158.
21. Liu R, Paxton WA, Choe S, Ceredini D, Martin SR, Horuk R,MacDonald ME, Stuhlmann H, Koup RA, Landau NR Homozygousdefect in HIV-1 coreceptor accounts for resistance of somemultiply-exposed individuals to HIV-1 infection. Cell 1996,86:367–377.
22. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber C-M,Saragosti S, Lapoumeroulle C, Cognaux J, Forcelle C et al.:Resistance to HIV-1 infection in caucasian individuals bearingmutant alleles of the CCR-5 chemokine resistance gene. Nature1996, 382:722–725.
23. Paxton WA, Martin SR, Tse D, O’Brien TR, Skurnick J, VanDevanterNL, Padian N, Braun JF, Kotler DP, Wolinsky SM, Koup RA: Relativeresistance to HIV-1 infection of CD4 lymphocytes from personswho remain uninfected despite multiple high-risk sexualexposures. Nat Med 1996, 2:412–417.
24. Simmons G, McKnight A, Takeuchi Y, Hoshino H, Clapham PR: Cell-to-cell fusion, but not virus entry in macrophages by T-cell linetropic HIV-1 strains: a V3 loop determined restricion. Virology1995, 209:696–700.
Dispatch 1053