
Highly selective inhibition of IMPDH2 provides the basis ... · effects on neuroinflammation by...
9
Highly selective inhibition of IMPDH2 provides the basis of antineuroinflammation therapy Li-Xi Liao a,1 , Xiao-Min Song a,1 , Li-Chao Wang a,b,1 , Hai-Ning Lv a , Jin-Feng Chen a , Dan Liu c , Ge Fu a , Ming-Bo Zhao a , Yong Jiang a , Ke-Wu Zeng a,2 , and Peng-Fei Tu a,2 a State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China; b State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing 210009, China; and c Proteomics Laboratory, Medical and Healthy Analytical Center, Peking University Health Science Center, Beijing 100191, China Edited by Jerrold Meinwald, Cornell University, Ithaca, NY, and approved June 7, 2017 (received for review April 28, 2017) Inosine monophosphate dehydrogenase (IMPDH) of human is an attractive target for immunosuppressive agents. Currently, small- molecule inhibitors do not show good selectivity for different IMPDH isoforms (IMPDH1 and IMPDH2), resulting in some adverse effects, which limit their use. Herein, we used a small-molecule probe specifically targeting IMPDH2 and identified Cysteine residue 140 (Cys140) as a selective druggable site. On covalently binding to Cys140, the probe exerts an allosteric regulation to block the catalytic pocket of IMPDH2 and further induces IMPDH2 inactivation, leading to an effective suppression of neuroinflammatory responses. How- ever, the probe does not covalently bind to IMPDH1. Taken together, our study shows Cys140 as a druggable site for selectively inhibiting IMPDH2, which provides great potential for development of therapy agents for autoimmune and neuroinflammatory diseases with less unfavorable tolerability profile. IMP dehydrogenase-2 | druggable site | covalent binding | allosteric regulation | immunosuppression I nosine monophosphate dehydrogenase (IMPDH) is a major rate-limiting enzyme involved in guanosine and deoxyguanosine biosynthesis and widely expressed in immunocytes (1). There exist two IMPDH isoforms (IMPDH1 and IMPDH2), which are encoded by distinct genes (2, 3). Many inflammation-relevant diseases have been specially characterized by the high expression of isoform II of IMPDH (IMPDH2) in rapidly proliferating immunocytes, rather than the “housekeeping” type I isoform (IMPDH1) in nor- mal human leukocytes and lymphocytes (4, 5). Therefore, selective targeting of IMPDH2 with small molecules is an attractive topic for development of antiinflammation agents with low side effects. Both IMPDH isoforms contain two major domains: the cata- lytic domain for substrate interaction and the Bateman domain, which is not required for catalytic activity but exerts an important allosteric regulation effect on IMPDH activity by communicating with the catalytic domain (6, 7). By influencing catalytic domain activity, the Bateman domain can regulate IMPDH function and further blocks the downstream-of-inflammation signaling path- ways (8, 9). Currently, IMPDH inhibitors are divided into two major categories. One kind of inhibitor, including 6-chloropurine riboside ribavirin and mizoribine, targets the binding pocket of the natural substrate, inosine monophosphate (IMP). Another kind of inhibitor (e.g., mycophenolic acid and thiazole-4-carboxamide adenine dinucleotide) targets the site of the cofactor, NAD + / NADH, which usually leads to low selectivity or even side effects in clinical trials, such as diarrhea and leukopenia (10, 11). More- over, a third ligand has been speculated to bind to a possible site far from the IMP and NAD + pockets as an allosteric inhibitor. However, an allosteric site for designing selective IMPDH2 inhibitors has been largely unexplored. Natural small molecules remain promising drug sources (12, 13). In the present study, we report that a natural small-molecule probe, sappanone A (SA, Fig.1A), demonstrated significant inhibitory effects on neuroinflammation by directly targeting the conserved cysteine residue 140 (Cys140) in the noncatalytic Bateman domain of IMPDH2. Interestingly, SA selectively targets and inactivates IMPDH2 but not IMPDH1. The selectivity is explained by differ- ential substituent groups of amino acids in two IMPDH isoforms. The thiol in cysteine 140 of IMPDH2 can lead to irreversible co- valent binding via the Michael addition to the α,β-unsaturated car- bonyl in SA. However, the corresponding amino acid in IMPDH1 is serine with a weaker nucleophilic group hydroxyl, resulting in a weakened covalent binding effect of SA. The selective modification of SA on IMPDH2 caused an allosteric effect on its catalytic domain to narrow the substrate combination space in the catalytic pocket, which led to a suppression of IMPDH2 activity and IMPDH2- dependent neuroinflammatory response without obvious hemato- logical side effects. These findings indicate that cysteine 140 is a druggable binding site for selectively targeting IMPDH2. Small molecules binding to cysteine 140 of IMPDH2 can exert an effective antineuroin- flammation therapy in clinical trials with fewer side effects. Results IMPDH2 Is Selectively Targeted by SA. First, we found that sap- panone A (SA) was a potent inhibitor of microglial activation. As shown in SI Appendix, Fig. S1 A and B, SA significantly suppressed the releases of NO, TNF-α, IL-6, and PGE 2 and decreased the gene expressions of TNF-α, IL-6, IL-1β, MCP-1, iNOS, and COX-2 in BV-2 cells. Similarly, SA significantly suppressed the production of NO, TNF-α, and IL-6 in primary microglia (SI Appendix, Fig. S2). Significance Inosine monophosphate dehydrogenase (IMPDH) is an attractive target for immunosuppressive agents. Currently, small-molecule inhibitors do not show good selectivity for different IMPDH iso- forms (IMPDH1 and IMPDH2), resulting in some adverse effects, which limit their use. Here, we identified Cys140 as an isoform- selective druggable binding site for IMPDH2 inhibition but not for IMPDH1. We found small-molecule sappanone A directly co- valently targets Cys140 in IMPDH2 to block its activity, resulting in neuroinflammatory inhibition with less side effects than pan- IMPDH inhibitor. In summary, our findings reveal Cys140 is a druggable binding site for selectively inhibiting IMPDH2 for neu- roinflammatory diseases with less unfavorable tolerability profile. Author contributions: K.-W.Z. and P.-F.T. designed research; L.-X.L., X.-M.S., L.-C.W., H.-N.L., J.-F.C., and K.-W.Z. performed research; H.-N.L., J.-F.C., D.L., G.F., M.-B.Z., and Y.J. contributed new reagents/analytic tools; L.-X.L., X.-M.S., L.-C.W., J.-F.C., D.L., G.F., and K.-W.Z. analyzed data; and X.-M.S., L.-C.W., K.-W.Z.., and P.-F.T. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. Freely available online through the PNAS open access option. 1 L.-X.L., X.-M.S., and L.-C.W. contributed equally to this work. 2 To whom correspondence may be addressed. Email: [email protected] or pengfeitu@ bjmu.edu.cn. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1706778114/-/DCSupplemental. E5986–E5994 | PNAS | Published online July 3, 2017 www.pnas.org/cgi/doi/10.1073/pnas.1706778114
-
Upload
nguyendung -
Category
Documents
-
view
215 -
download
0
Transcript of Highly selective inhibition of IMPDH2 provides the basis ... · effects on neuroinflammation by...

Highly selective inhibition of IMPDH2 provides thebasis of antineuroinflammation therapyLi-Xi Liaoa,1, Xiao-Min Songa,1, Li-Chao Wanga,b,1, Hai-Ning Lva, Jin-Feng Chena, Dan Liuc, Ge Fua, Ming-Bo Zhaoa,Yong Jianga, Ke-Wu Zenga,2, and Peng-Fei Tua,2
aState Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China; bState Key Laboratoryof Natural Medicines, China Pharmaceutical University, Nanjing 210009, China; and cProteomics Laboratory, Medical and Healthy Analytical Center, PekingUniversity Health Science Center, Beijing 100191, China
Edited by Jerrold Meinwald, Cornell University, Ithaca, NY, and approved June 7, 2017 (received for review April 28, 2017)
Inosine monophosphate dehydrogenase (IMPDH) of human is anattractive target for immunosuppressive agents. Currently, small-molecule inhibitors do not show good selectivity for different IMPDHisoforms (IMPDH1 and IMPDH2), resulting in some adverse effects,which limit their use. Herein, we used a small-molecule probespecifically targeting IMPDH2 and identified Cysteine residue 140(Cys140) as a selective druggable site. On covalently binding toCys140, the probe exerts an allosteric regulation to block the catalyticpocket of IMPDH2 and further induces IMPDH2 inactivation, leadingto an effective suppression of neuroinflammatory responses. How-ever, the probe does not covalently bind to IMPDH1. Taken together,our study shows Cys140 as a druggable site for selectively inhibitingIMPDH2, which provides great potential for development of therapyagents for autoimmune and neuroinflammatory diseases with lessunfavorable tolerability profile.
IMP dehydrogenase-2 | druggable site | covalent binding | allostericregulation | immunosuppression
Inosine monophosphate dehydrogenase (IMPDH) is a majorrate-limiting enzyme involved in guanosine and deoxyguanosine
biosynthesis and widely expressed in immunocytes (1). There existtwo IMPDH isoforms (IMPDH1 and IMPDH2), which are encodedby distinct genes (2, 3). Many inflammation-relevant diseaseshave been specially characterized by the high expression of isoformII of IMPDH (IMPDH2) in rapidly proliferating immunocytes,rather than the “housekeeping” type I isoform (IMPDH1) in nor-mal human leukocytes and lymphocytes (4, 5). Therefore, selectivetargeting of IMPDH2 with small molecules is an attractive topic fordevelopment of antiinflammation agents with low side effects.Both IMPDH isoforms contain two major domains: the cata-
lytic domain for substrate interaction and the Bateman domain,which is not required for catalytic activity but exerts an importantallosteric regulation effect on IMPDH activity by communicatingwith the catalytic domain (6, 7). By influencing catalytic domainactivity, the Bateman domain can regulate IMPDH function andfurther blocks the downstream-of-inflammation signaling path-ways (8, 9). Currently, IMPDH inhibitors are divided into twomajor categories. One kind of inhibitor, including 6-chloropurineriboside ribavirin and mizoribine, targets the binding pocket ofthe natural substrate, inosine monophosphate (IMP). Another kindof inhibitor (e.g., mycophenolic acid and thiazole-4-carboxamideadenine dinucleotide) targets the site of the cofactor, NAD+/NADH, which usually leads to low selectivity or even side effects inclinical trials, such as diarrhea and leukopenia (10, 11). More-over, a third ligand has been speculated to bind to a possible sitefar from the IMP and NAD+ pockets as an allosteric inhibitor.However, an allosteric site for designing selective IMPDH2 inhibitorshas been largely unexplored.Natural small molecules remain promising drug sources (12, 13).
In the present study, we report that a natural small-molecule probe,sappanone A (SA, Fig.1A), demonstrated significant inhibitoryeffects on neuroinflammation by directly targeting the conservedcysteine residue 140 (Cys140) in the noncatalytic Bateman domain
of IMPDH2. Interestingly, SA selectively targets and inactivatesIMPDH2 but not IMPDH1. The selectivity is explained by differ-ential substituent groups of amino acids in two IMPDH isoforms.The thiol in cysteine 140 of IMPDH2 can lead to irreversible co-valent binding via the Michael addition to the α,β-unsaturated car-bonyl in SA. However, the corresponding amino acid in IMPDH1 isserine with a weaker nucleophilic group hydroxyl, resulting in aweakened covalent binding effect of SA. The selective modificationof SA on IMPDH2 caused an allosteric effect on its catalytic domainto narrow the substrate combination space in the catalytic pocket,which led to a suppression of IMPDH2 activity and IMPDH2-dependent neuroinflammatory response without obvious hemato-logical side effects.These findings indicate that cysteine 140 is a druggable binding
site for selectively targeting IMPDH2. Small molecules bindingto cysteine 140 of IMPDH2 can exert an effective antineuroin-flammation therapy in clinical trials with fewer side effects.
ResultsIMPDH2 Is Selectively Targeted by SA. First, we found that sap-panone A (SA) was a potent inhibitor of microglial activation.As shown in SI Appendix, Fig. S1 A and B, SA significantlysuppressed the releases of NO, TNF-α, IL-6, and PGE2 anddecreased the gene expressions of TNF-α, IL-6, IL-1β, MCP-1,iNOS, and COX-2 in BV-2 cells. Similarly, SA significantlysuppressed the production of NO, TNF-α, and IL-6 in primarymicroglia (SI Appendix, Fig. S2).
Significance
Inosine monophosphate dehydrogenase (IMPDH) is an attractivetarget for immunosuppressive agents. Currently, small-moleculeinhibitors do not show good selectivity for different IMPDH iso-forms (IMPDH1 and IMPDH2), resulting in some adverse effects,which limit their use. Here, we identified Cys140 as an isoform-selective druggable binding site for IMPDH2 inhibition but not forIMPDH1. We found small-molecule sappanone A directly co-valently targets Cys140 in IMPDH2 to block its activity, resulting inneuroinflammatory inhibition with less side effects than pan-IMPDH inhibitor. In summary, our findings reveal Cys140 is adruggable binding site for selectively inhibiting IMPDH2 for neu-roinflammatory diseases with less unfavorable tolerability profile.
Author contributions: K.-W.Z. and P.-F.T. designed research; L.-X.L., X.-M.S., L.-C.W.,H.-N.L., J.-F.C., and K.-W.Z. performed research; H.-N.L., J.-F.C., D.L., G.F., M.-B.Z., andY.J. contributed new reagents/analytic tools; L.-X.L., X.-M.S., L.-C.W., J.-F.C., D.L., G.F.,and K.-W.Z. analyzed data; and X.-M.S., L.-C.W., K.-W.Z.., and P.-F.T. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Freely available online through the PNAS open access option.1L.-X.L., X.-M.S., and L.-C.W. contributed equally to this work.2To whom correspondence may be addressed. Email: [email protected] or [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1706778114/-/DCSupplemental.
E5986–E5994 | PNAS | Published online July 3, 2017 www.pnas.org/cgi/doi/10.1073/pnas.1706778114
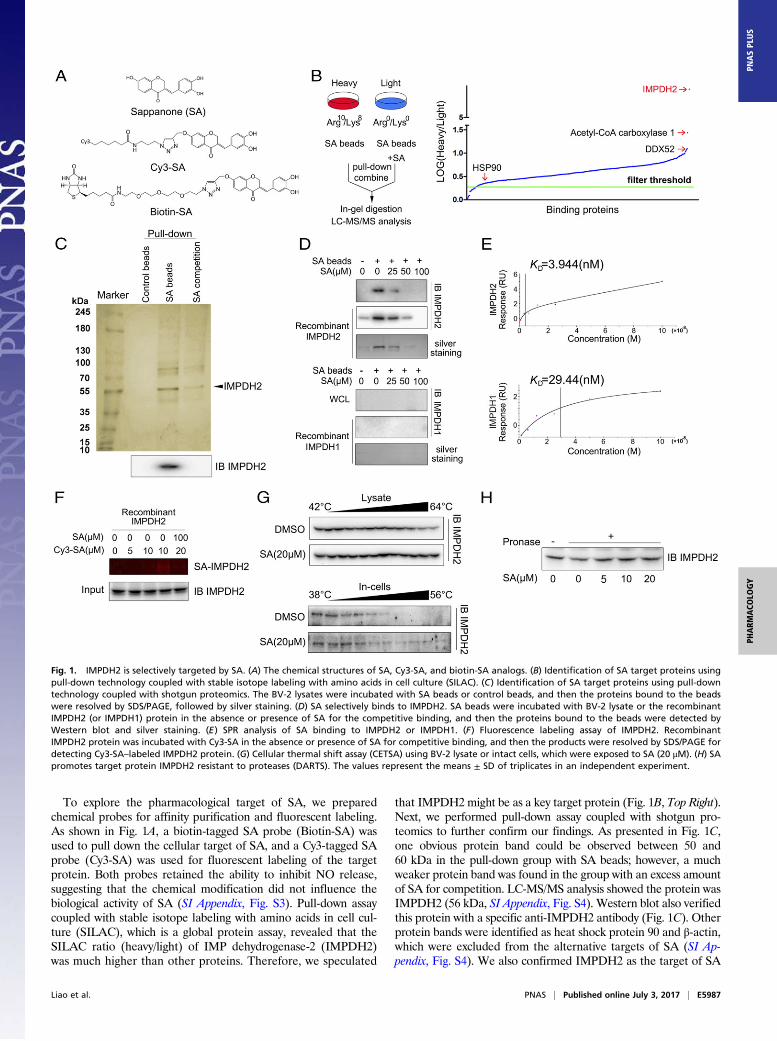
To explore the pharmacological target of SA, we preparedchemical probes for affinity purification and fluorescent labeling.As shown in Fig. 1A, a biotin-tagged SA probe (Biotin-SA) wasused to pull down the cellular target of SA, and a Cy3-tagged SAprobe (Cy3-SA) was used for fluorescent labeling of the targetprotein. Both probes retained the ability to inhibit NO release,suggesting that the chemical modification did not influence thebiological activity of SA (SI Appendix, Fig. S3). Pull-down assaycoupled with stable isotope labeling with amino acids in cell cul-ture (SILAC), which is a global protein assay, revealed that theSILAC ratio (heavy/light) of IMP dehydrogenase-2 (IMPDH2)was much higher than other proteins. Therefore, we speculated
that IMPDH2 might be as a key target protein (Fig. 1B, Top Right).Next, we performed pull-down assay coupled with shotgun pro-teomics to further confirm our findings. As presented in Fig. 1C,one obvious protein band could be observed between 50 and60 kDa in the pull-down group with SA beads; however, a muchweaker protein band was found in the group with an excess amountof SA for competition. LC-MS/MS analysis showed the protein wasIMPDH2 (56 kDa, SI Appendix, Fig. S4). Western blot also verifiedthis protein with a specific anti-IMPDH2 antibody (Fig. 1C). Otherprotein bands were identified as heat shock protein 90 and β-actin,which were excluded from the alternative targets of SA (SI Ap-pendix, Fig. S4). We also confirmed IMPDH2 as the target of SA
Fig. 1. IMPDH2 is selectively targeted by SA. (A) The chemical structures of SA, Cy3-SA, and biotin-SA analogs. (B) Identification of SA target proteins usingpull-down technology coupled with stable isotope labeling with amino acids in cell culture (SILAC). (C) Identification of SA target proteins using pull-downtechnology coupled with shotgun proteomics. The BV-2 lysates were incubated with SA beads or control beads, and then the proteins bound to the beadswere resolved by SDS/PAGE, followed by silver staining. (D) SA selectively binds to IMPDH2. SA beads were incubated with BV-2 lysate or the recombinantIMPDH2 (or IMPDH1) protein in the absence or presence of SA for the competitive binding, and then the proteins bound to the beads were detected byWestern blot and silver staining. (E) SPR analysis of SA binding to IMPDH2 or IMPDH1. (F) Fluorescence labeling assay of IMPDH2. RecombinantIMPDH2 protein was incubated with Cy3-SA in the absence or presence of SA for competitive binding, and then the products were resolved by SDS/PAGE fordetecting Cy3-SA–labeled IMPDH2 protein. (G) Cellular thermal shift assay (CETSA) using BV-2 lysate or intact cells, which were exposed to SA (20 μM). (H) SApromotes target protein IMPDH2 resistant to proteases (DARTS). The values represent the means ± SD of triplicates in an independent experiment.
Liao et al. PNAS | Published online July 3, 2017 | E5987
PHARM
ACO
LOGY
PNASPL
US
in primary cultured microglial lysates and the tissue lysate of LPS-injected mouse brains (SI Appendix, Figs. S5 and S6).To verify the interaction of SA with IMPDH2, we incubated
BV-2 cell lysates or recombinant IMPDH2 protein with SAbeads in the absence or the presence of an excess amount of SAfor competitive binding. As shown in Fig. 1D, IMPDH2 wasobviously pulled down by SA beads, which were detected byWestern blot and silver staining. Moreover, an excess amount ofSA effectively blocked the binding of IMPDH2 to SA beads.Meanwhile, we did not detect obvious binding of SA with thetype I isoform (IMPDH1) (Fig. 1D). SPR analysis revealed thatthe target affinity [KD (equilibrium dissociation constant) value]of SA binding to IMPDH2 was 3.944 nM, almost 10 times lowerthan the KD of SA binding to IMPDH1 (29.44 nM, Fig. 1E).Fluorescent labeling assay also showed a specific fluorescentband around 56 kDa for Cy3-SA–labeled IMPDH2 protein(Fig. 1F).
Small-molecule inhibitors can perturb protein function and in-crease the protein stability via forming a ligand–protein complex(14). Thus, we attempted to investigate whether SA could bind toIMPDH2 protein and increase its stability in intact cells or lysateusing two target engagement assays (15, 16). From cellular thermalshift assay (CETSA), we found that SA treatment efficiently pro-tected IMPDH2 protein from temperature-dependent degradation(Fig. 1G). Second, DARTS assay was used to monitor target en-gagement based on SA-induced stabilization of IMPDH2 protein.Our data also demonstrated a concentration-dependent reducedproteolysis with the incubation of SA (Fig. 1H).
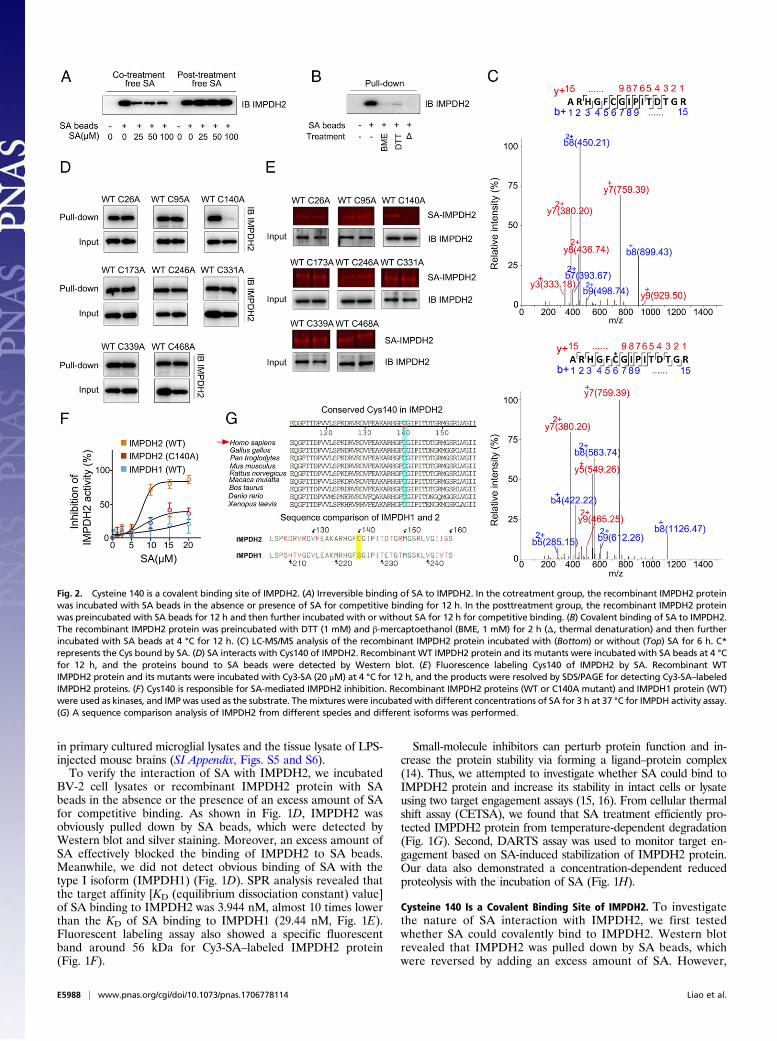
Cysteine 140 Is a Covalent Binding Site of IMPDH2. To investigatethe nature of SA interaction with IMPDH2, we first testedwhether SA could covalently bind to IMPDH2. Western blotrevealed that IMPDH2 was pulled down by SA beads, whichwere reversed by adding an excess amount of SA. However,
Fig. 2. Cysteine 140 is a covalent binding site of IMPDH2. (A) Irreversible binding of SA to IMPDH2. In the cotreatment group, the recombinant IMPDH2 proteinwas incubated with SA beads in the absence or presence of SA for competitive binding for 12 h. In the posttreatment group, the recombinant IMPDH2 proteinwas preincubated with SA beads for 12 h and then further incubated with or without SA for 12 h for competitive binding. (B) Covalent binding of SA to IMPDH2.The recombinant IMPDH2 protein was preincubated with DTT (1 mM) and β-mercaptoethanol (BME, 1 mM) for 2 h (Δ, thermal denaturation) and then furtherincubated with SA beads at 4 °C for 12 h. (C) LC-MS/MS analysis of the recombinant IMPDH2 protein incubated with (Bottom) or without (Top) SA for 6 h. C*represents the Cys bound by SA. (D) SA interacts with Cys140 of IMPDH2. Recombinant WT IMPDH2 protein and its mutants were incubated with SA beads at 4 °Cfor 12 h, and the proteins bound to SA beads were detected by Western blot. (E) Fluorescence labeling Cys140 of IMPDH2 by SA. Recombinant WTIMPDH2 protein and its mutants were incubated with Cy3-SA (20 μM) at 4 °C for 12 h, and the products were resolved by SDS/PAGE for detecting Cy3-SA–labeledIMPDH2 proteins. (F) Cys140 is responsible for SA-mediated IMPDH2 inhibition. Recombinant IMPDH2 proteins (WT or C140A mutant) and IMPDH1 protein (WT)were used as kinases, and IMPwas used as the substrate. Themixtures were incubatedwith different concentrations of SA for 3 h at 37 °C for IMPDH activity assay.(G) A sequence comparison analysis of IMPDH2 from different species and different isoforms was performed.
E5988 | www.pnas.org/cgi/doi/10.1073/pnas.1706778114 Liao et al.
when IMPDH2 was preincubated with SA beads, posttreatmentof an excess amount of SA could not prevent IMPDH2 bindingto SA beads (Fig. 2A), indicating a covalent bond formationbetween SA and IMPDH2 protein. Because SA contains anα,β-unsaturated carbonyl group, which has a potential to reactcovalently with the thiols of cysteine on IMPDH2 (17), IMPDH2was incubated with SA beads in the absence or the presence ofβ-mercaptoethanol (BME)/DTT for competitive binding to IMPDH2via thiols (18). As shown in Fig. 2B, BME or DTT completelyabolished IMPDH2 binding to SA beads, suggesting that SAmight covalently bind to the thiols of cysteine. Additionally,the SA-glutathione (GSH, a thiol donor) complex formed viaMichael addition was also accurately confirmed using LC-pMRManalysis (SI Appendix, Fig. S7).Next, we used BLAST analysis with full-length IMPDH2 protein
sequence and found eight conserved cysteine residues (SI Appen-dix, Fig. S8). To determine which cysteine residue was attacked bySA, we incubated IMPDH2 protein with or without SA, followedby LC-MS/MS analysis. Tryptic peptides containing cysteine wereevaluated, and Fig. 2C presents a peptide with a calculated mass of1884.86 Da, which is 284.07 Da larger than the Cysteine140(Cys140)-containing peptide ARHGFCGIPITDTGR, which has acalculated mass of 1600.79 Da. The mass difference of 284.07 Daexactly matches the molecular weight of an SA molecule. MS/MSspectrometry of this peptide revealed that a 284.07 Da mass shiftoccurred starting from the b5 to the b6 fragment ions, in-dicating that the Cys140 residue was covalently modified bySA. This finding was also confirmed by synthetic peptides de-rived from human IMPDH2 containing Cys140 (PeptideC140,ARHGFCGIPIT-His) incubated with SA (SI Appendix, Fig.
S9). Then, we mutated each cysteine residue of IMPDH2 intoalanine. Pull-down assay with recombinant cysteine-mutatedIMPDH2 proteins further supported that SA covalently mod-ifies Cys140 but not the other cysteines of IMPDH2 (Fig. 2D).Additionally, these observations were supported by fluorescentlabeling experiments, which showed that only Cys140-mutatedIMPDH2 (C140A) could not be labeled by Cy3-SA (Fig. 2E).We further investigated whether Cys140 mutation could impactthe inhibitory effect of SA on IMPDH2. As shown in Fig. 2F,SA significantly inhibited WT IMPDH2 activity, which wasmarkedly suppressed in Cys140-mutated IMPDH2. Meanwhile,SA did not show obvious inhibitory effect in WT IMPDH1 proteinactivity. A sequence comparison in Fig. 2G suggests that theCys140 residue in IMPDH2 is conserved among various species.Interestingly, IMPDH1 does not possess a cysteine (Cys140) at thecorresponding site, but a serine instead (Fig. 2G). Because hy-droxyl in serine is a weaker nucleophilic group than the thiol incysteine, this could explain why SA tends to preferably bind toIMPDH2 over IMPDH1.
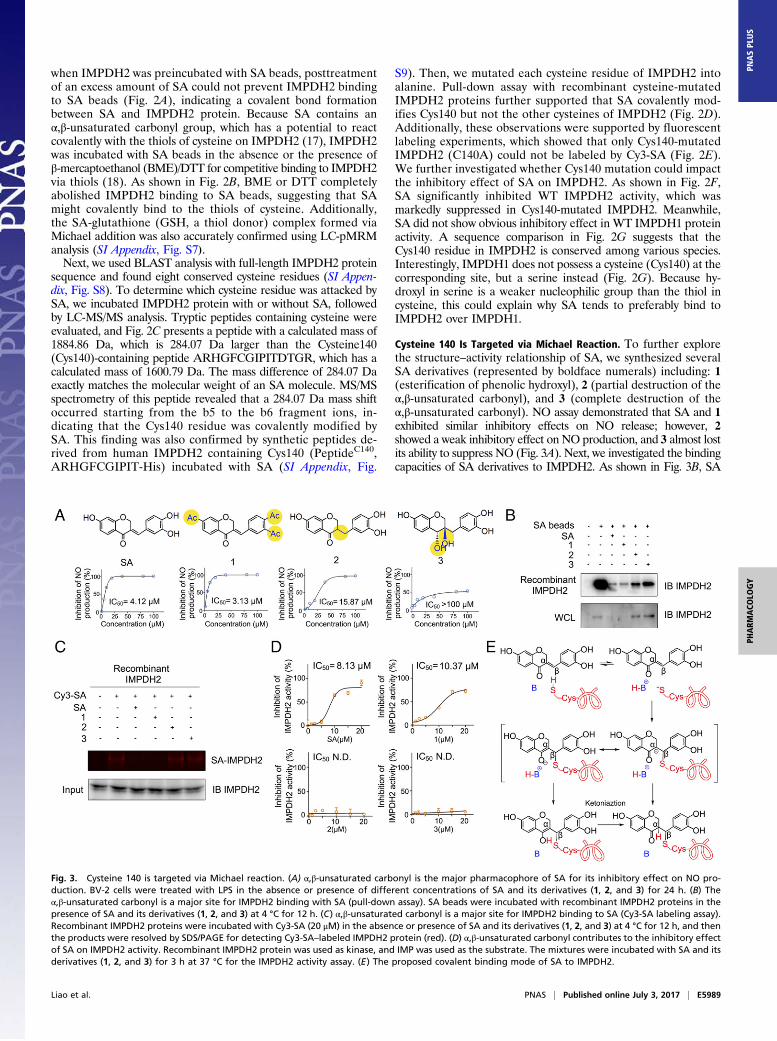
Cysteine 140 Is Targeted via Michael Reaction. To further explorethe structure–activity relationship of SA, we synthesized severalSA derivatives (represented by boldface numerals) including: 1(esterification of phenolic hydroxyl), 2 (partial destruction of theα,β-unsaturated carbonyl), and 3 (complete destruction of theα,β-unsaturated carbonyl). NO assay demonstrated that SA and 1exhibited similar inhibitory effects on NO release; however, 2showed a weak inhibitory effect on NO production, and 3 almost lostits ability to suppress NO (Fig. 3A). Next, we investigated the bindingcapacities of SA derivatives to IMPDH2. As shown in Fig. 3B, SA
Fig. 3. Cysteine 140 is targeted via Michael reaction. (A) α,β-unsaturated carbonyl is the major pharmacophore of SA for its inhibitory effect on NO pro-duction. BV-2 cells were treated with LPS in the absence or presence of different concentrations of SA and its derivatives (1, 2, and 3) for 24 h. (B) Theα,β-unsaturated carbonyl is a major site for IMPDH2 binding with SA (pull-down assay). SA beads were incubated with recombinant IMPDH2 proteins in thepresence of SA and its derivatives (1, 2, and 3) at 4 °C for 12 h. (C) α,β-unsaturated carbonyl is a major site for IMPDH2 binding to SA (Cy3-SA labeling assay).Recombinant IMPDH2 proteins were incubated with Cy3-SA (20 μM) in the absence or presence of SA and its derivatives (1, 2, and 3) at 4 °C for 12 h, and thenthe products were resolved by SDS/PAGE for detecting Cy3-SA–labeled IMPDH2 protein (red). (D) α,β-unsaturated carbonyl contributes to the inhibitory effectof SA on IMPDH2 activity. Recombinant IMPDH2 protein was used as kinase, and IMP was used as the substrate. The mixtures were incubated with SA and itsderivatives (1, 2, and 3) for 3 h at 37 °C for the IMPDH2 activity assay. (E) The proposed covalent binding mode of SA to IMPDH2.
Liao et al. PNAS | Published online July 3, 2017 | E5989
PHARM
ACO
LOGY
PNASPL
US

beads effectively pulled down IMPDH2 protein, which was markedlyreversed by an excess amount of SA and 1, but not 2 or 3. Addi-tionally, a fluorescent labeling experiment showed that Cy3-SA–labeled IMPDH2 protein bands were significantly decreased byadding an excess amount of SA or 1, but not 2 or 3 (Fig. 3C). Toverify the functional significance of the α,β-unsaturated carbonylin SA, we performed in vitro kinase assay (19, 20). As shown inFig. 3D, both SA and 1 markedly inhibited IMPDH2 activity;however, 2 and 3 did not demonstrate any inhibitory effects onIMPDH2 activity. Overall, we demonstrated that SA directly targetsand inactivates IMPDH2 protein via theMichael addition of thiol incysteine to the α,β-unsaturated carbonyl (Fig. 3E).
Cysteine 140 Is an Allosteric Regulatory Site of IMPDH2. IMPDH2 proteinhas a two-domain structure: (i) a catalytic domain (amino acidresidues 2–92 and 224–492) forming the core of the activeenzyme; and (ii) a regulatory Bateman domain (amino acidresidues 93–223) (Fig. 4A) (7, 9). Molecular dynamics simulationanalysis reveals that SA is deeply embedded in the cleft of theIMPDH2 Bateman domain and further promotes its bending tocatalytic domain (“head-lowering” conformation). Additionally, asshown in Fig. 4B, a strong hydrogen-bonding interaction exists be-tween SA and the residues of IMPDH2, including Thr225, Arg224,and Arg226. Such noncovalent interactions can serve as an initial site-recognition step when SA binds to IMPDH2 and hence raises theprobability of the covalent reaction. Upon SA binding to Cys140, aprotein loop region containing 20 amino acids (amino acid residues322–342) in catalytic domain moves into the substrate IMP-bindingpocket (Fig. 4C), leading to the inactivation of IMPDH2. Moreover,
the loop region interacts with the IMP-binding pocket via hydrogenbonds and enhances the conformational stabilization (Fig. 4D). Thevariations of hydrogen bond length in simulated movement locuswere shown in SI Appendix, Fig. S10. Additionally, the interactionsurface of the loop region contains several hydrophobic amino acids,tending to interact with the hydrophobic surface of the IMP catalyticpocket (SI Appendix, Fig. S11). These observations were also con-firmed by pull-down assay using IMP-coupled beads. We foundIMPDH2 protein could bind to IMP-coupled beads and was inhibitedby adding an excess amount of SA (Fig. 4E).IMPDH2 protein functions as a tetramer by clustering four
monomers (7, 21). We tried to evaluate the effect of SA onIMPDH2 clustering by observing the colocalization of GFP-taggedIMPDH2 (green) and mCherry-tagged IMPDH2 (red). As shown inFig. 4F, the overlap of green and red fluorescence (yellow) wasobvious in control cells; however, SA treatment markedly suppressedthe overlap of green and red fluorescence (Fig. 4F). Moreover,nondenaturing gel electrophoresis and cross-linked whole-cell ex-tracts also showed that IMPDH2 tetramers were decreased by SAtreatment (Fig. 4G).
NF-κB and p38 MAPK Pathways Contribute to IMPDH2-DependentNeuroinflammation. GTP is a key cellular metabolite of IMPDH2(22). Fig. 5A shows that SA markedly reduced GTP level inBV-2 cells by about 50%. We next sought to elucidate whether thefunction of IMPDH2 is required for SA to inhibit microglialactivation. As shown in Fig. 5B, blockage of IMPDH2 geneexpression using a specific IMPDH2 siRNA significantly reversedSA-mediated inhibition of NO production. Moreover, we found
Fig. 4. Cysteine 140 is an allosteric regulatory site of IMPDH2. (A) A representative global view of the SA-mediated allosteric effect of IMPDH2 protein.IMPDH2 protein alone (head-raising) was shown by the yellow ribbon model, SA-IMPDH2 complex (head-lowering) was shown by the green ribbon model.(B) Hydrogen bonds between SA and the binding pocket in the Bateman domain were identified and indicated by red lines. (C) A representative view of theconformation change of the catalytic pocket in IMPDH2 protein. The catalytic pocket is shown by gray (IMPDH2 alone) or green color (SA-IMPDH2 complex).IMP is indicated by red color. (D) The hydrogen bonds forming between loop region and neighboring amino acid residues in catalytic pocket. (E) SA inhibitsIMPDH2 binding to IMP. Recombinant IMPDH2 protein was incubated with IMP-bound beads (IMP beads) in the absence or presence of SA for competitivebinding at 4 °C for 12 h. (F) The effect of SA on IMPDH2 clustering was investigated by dual-label fluorescence analysis (bar = 50 μm). The colocalization ratioof IMPDH2 clusters was calculated. (G) The clusters of IMPDH2 tetramer were detected by nondenaturing gel electrophoresis and cross-linked whole-cellextracts. The values represent the means ± SD of triplicates in an independent experiment. **P < 0.01, compared with the control group.
E5990 | www.pnas.org/cgi/doi/10.1073/pnas.1706778114 Liao et al.
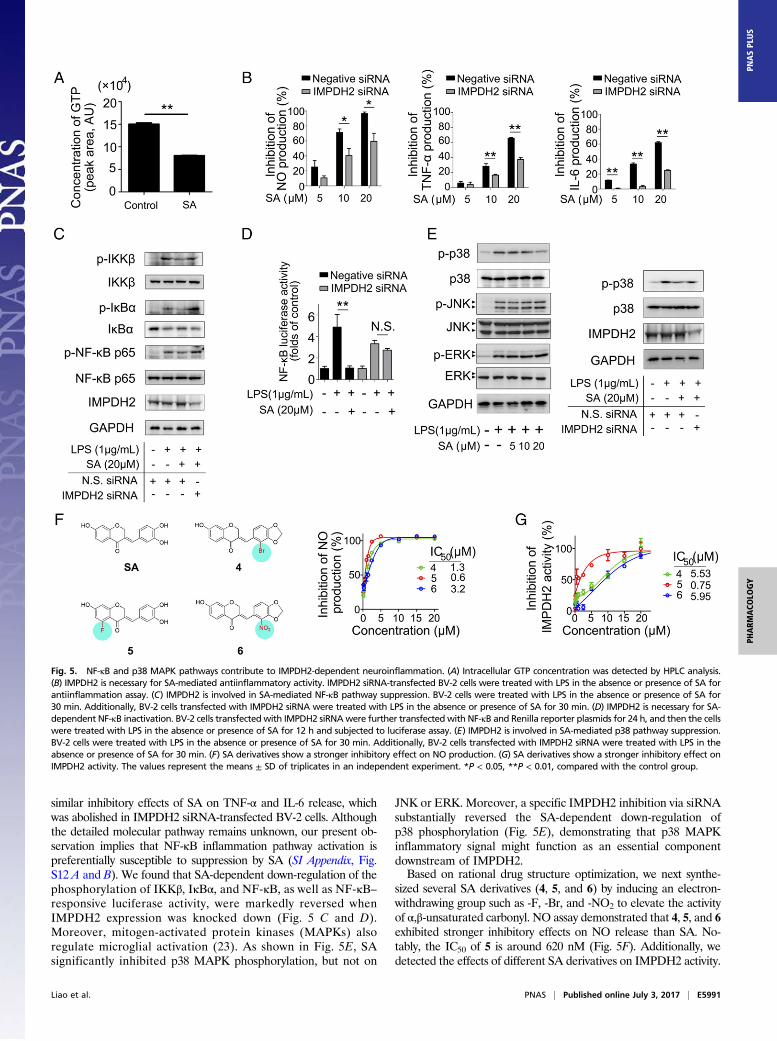
similar inhibitory effects of SA on TNF-α and IL-6 release, whichwas abolished in IMPDH2 siRNA-transfected BV-2 cells. Althoughthe detailed molecular pathway remains unknown, our present ob-servation implies that NF-κB inflammation pathway activation ispreferentially susceptible to suppression by SA (SI Appendix, Fig.S12 A and B). We found that SA-dependent down-regulation of thephosphorylation of IKKβ, IκBα, and NF-κB, as well as NF-κB–responsive luciferase activity, were markedly reversed whenIMPDH2 expression was knocked down (Fig. 5 C and D).Moreover, mitogen-activated protein kinases (MAPKs) alsoregulate microglial activation (23). As shown in Fig. 5E, SAsignificantly inhibited p38 MAPK phosphorylation, but not on
JNK or ERK. Moreover, a specific IMPDH2 inhibition via siRNAsubstantially reversed the SA-dependent down-regulation ofp38 phosphorylation (Fig. 5E), demonstrating that p38 MAPKinflammatory signal might function as an essential componentdownstream of IMPDH2.Based on rational drug structure optimization, we next synthe-
sized several SA derivatives (4, 5, and 6) by inducing an electron-withdrawing group such as -F, -Br, and -NO2 to elevate the activityof α,β-unsaturated carbonyl. NO assay demonstrated that 4, 5, and 6exhibited stronger inhibitory effects on NO release than SA. No-tably, the IC50 of 5 is around 620 nM (Fig. 5F). Additionally, wedetected the effects of different SA derivatives on IMPDH2 activity.
Fig. 5. NF-κB and p38 MAPK pathways contribute to IMPDH2-dependent neuroinflammation. (A) Intracellular GTP concentration was detected by HPLC analysis.(B) IMPDH2 is necessary for SA-mediated antiinflammatory activity. IMPDH2 siRNA-transfected BV-2 cells were treated with LPS in the absence or presence of SA forantiinflammation assay. (C) IMPDH2 is involved in SA-mediated NF-κB pathway suppression. BV-2 cells were treated with LPS in the absence or presence of SA for30 min. Additionally, BV-2 cells transfected with IMPDH2 siRNA were treated with LPS in the absence or presence of SA for 30 min. (D) IMPDH2 is necessary for SA-dependent NF-κB inactivation. BV-2 cells transfected with IMPDH2 siRNAwere further transfected with NF-κB and Renilla reporter plasmids for 24 h, and then the cellswere treated with LPS in the absence or presence of SA for 12 h and subjected to luciferase assay. (E) IMPDH2 is involved in SA-mediated p38 pathway suppression.BV-2 cells were treated with LPS in the absence or presence of SA for 30 min. Additionally, BV-2 cells transfected with IMPDH2 siRNA were treated with LPS in theabsence or presence of SA for 30 min. (F) SA derivatives show a stronger inhibitory effect on NO production. (G) SA derivatives show a stronger inhibitory effect onIMPDH2 activity. The values represent the means ± SD of triplicates in an independent experiment. *P < 0.05, **P < 0.01, compared with the control group.
Liao et al. PNAS | Published online July 3, 2017 | E5991
PHARM
ACO
LOGY
PNASPL
US
As expected, 4, 5, and 6 significantly suppressed IMPDH2 activity,and the IC50 of 5 is around 750 nM (Fig. 5G).
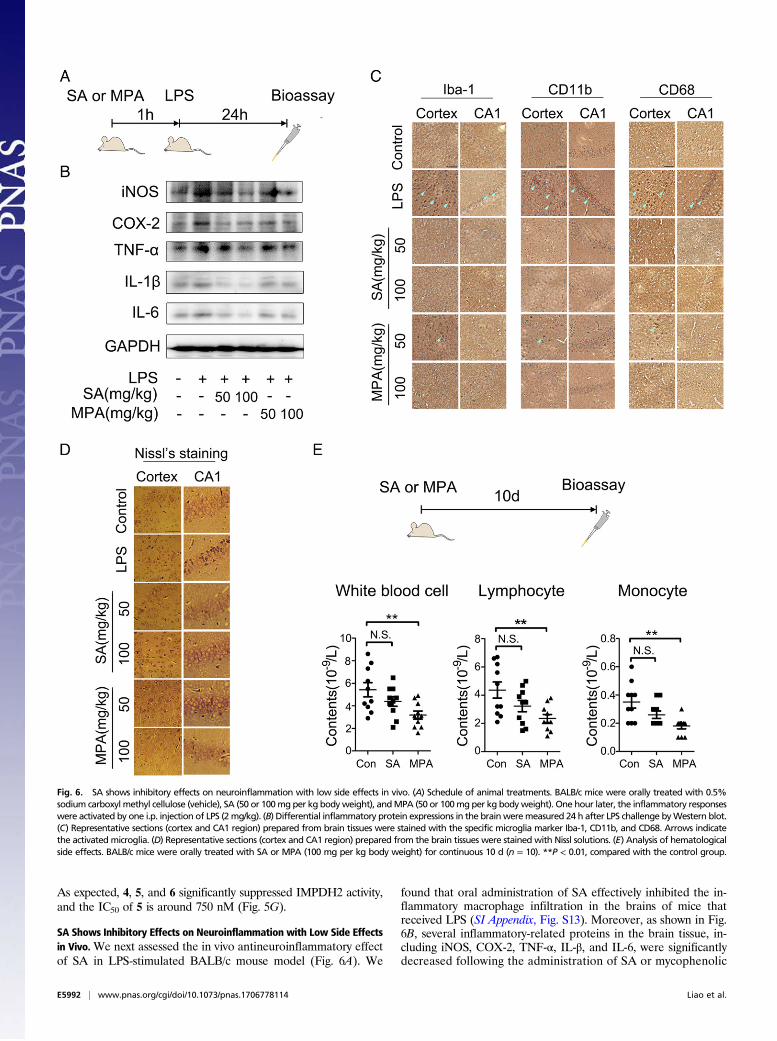
SA Shows Inhibitory Effects on Neuroinflammation with Low Side Effectsin Vivo.We next assessed the in vivo antineuroinflammatory effectof SA in LPS-stimulated BALB/c mouse model (Fig. 6A). We
found that oral administration of SA effectively inhibited the in-flammatory macrophage infiltration in the brains of mice thatreceived LPS (SI Appendix, Fig. S13). Moreover, as shown in Fig.6B, several inflammatory-related proteins in the brain tissue, in-cluding iNOS, COX-2, TNF-α, IL-β, and IL-6, were significantlydecreased following the administration of SA or mycophenolic
Fig. 6. SA shows inhibitory effects on neuroinflammation with low side effects in vivo. (A) Schedule of animal treatments. BALB/c mice were orally treated with 0.5%sodium carboxyl methyl cellulose (vehicle), SA (50 or 100mgper kg bodyweight), andMPA (50 or 100mgper kg bodyweight). One hour later, the inflammatory responseswere activated by one i.p. injection of LPS (2mg/kg). (B) Differential inflammatory protein expressions in the brainweremeasured 24 h after LPS challenge byWestern blot.(C) Representative sections (cortex and CA1 region) prepared from brain tissues were stained with the specific microglia marker Iba-1, CD11b, and CD68. Arrows indicatethe activated microglia. (D) Representative sections (cortex and CA1 region) prepared from the brain tissues were stained with Nissl solutions. (E) Analysis of hematologicalside effects. BALB/c mice were orally treated with SA or MPA (100 mg per kg body weight) for continuous 10 d (n = 10). **P < 0.01, compared with the control group.
E5992 | www.pnas.org/cgi/doi/10.1073/pnas.1706778114 Liao et al.

acid (MPA), which is a non-isoform-selective pan-IMPDH in-hibitor. Furthermore, immunohistochemical staining showedthat activated microglia (indicated by specific Iba-1, CD11b,and CD68 antibodies) in the cortex and hippocampal CA1 re-gions were effectively inhibited by SA or MPA (Fig. 6C).Meanwhile, SA or MPA effectively protected neurons againstmicroglia-mediated neuroinflammatory injuries by Nissl’s stainingassay (Fig. 6D). It is noteworthy that SA did not show signifi-cant hematological side effects; however, the same dose of MPAcaused significant decrease in several hemogram indexes, includ-ing whole-blood-cell counts, lymphocyte counts, and monocytecounts, suggesting that specific IMPDH2 inhibitor SA can inhibitneuroinflammatory responses with high drug safety and less sideeffects (Fig. 6E).
DiscussionOver the past decade, IMPDH has been viewed as an attractivedrug target for the chemotherapy for autoimmune disorders, andIMPDH inhibitors appeared to act as effective immunosup-pressive agents in clinical trials (24). Thus, there has been aconcerted effort to identify small-molecule inhibitors of IMPDHfor inflammation-related diseases (5). However, current small-molecule inhibitors do not show good selectivity for differentIMPDH isoforms (IMPDH1, IMPDH2), resulting in some ad-verse effects, which limit their clinical use.The druggable target identification is extremely important for
seeking therapy drugs (25–27). To explore the direct cell target ofSA (28), we designed a small-molecular probe based on SA struc-ture and found it selectively targets IMPDH2, but not IMPDH1.Notably, IMPDH2 contains a critical cysteine residue (Cys331) in itscatalytic domain that was targeted by several current inhibitors (29,30). Interestingly, SA is selective toward Cys140 in regulatoryBateman domain but not Cys331 in catalytic domain. Hitherto,Cys140 has not been reported as a druggable site for IMPDH2inhibition. Notably, SA showed selectivity to IMPDH2 rather thanIMPDH1. We assume that the molecular geometry of SA and thechemical environment surrounding the binding pocket might be thekey factors. The covalent binding site of SA in IMPDH2 is the thiolof Cys140, which supplies a structural specificity for the recognitionand binding of SA. However, the corresponding site in IMPDH1 isserine, which contains a hydroxyl group and is reactionless to SA.Molecular dynamics (MD) stimulation analysis indicated that the
covalent binding of SA to Cys140 induced an allosteric effect onIMPDH2 by promoting the Bateman domain to bend to the cata-lytic domain. To our surprise, we found that the IMP-binding site inthe catalytic domain was also affected by the SA-induced allostericeffect. In the SA-IMPDH2 complex, the IMP-binding site was oc-cupied by the neighboring loop region and caused a dysfunction ofsubstrate processing as well as IMPDH2 inactivation. We foundthat SA-induced serpentine flow mainly passes from the Batemandomain to the IMPDH domain and arrives in the catalytic domain.Notably, Ile461, Leu235, Ser237, and Ala236 play important roles inserpentine flow passing (SI Appendix, Fig. S14). Thus, we speculatedthat IMPDH2 activity might be subject to distinct regulation by SA
in the Bateman domain and further contribute to conformationalchanges of the catalytic domain. Collectively, these observationssuggest a physiologically important role in the regulatory regionoutside of the catalytic site of IMPDH2. To our knowledge, SArepresents the first small-molecule allosteric inhibitor that blocksIMPDH2 by directly targeting Cys140 residue in the regulatoryBateman domain.Notably, S. Lee et al. have reported that SA could inhibit in-
flammation response on murine periphery macrophages viaNrf2 and NF-κB pathways (31); however, the direct target of SA islargely unexplored. Here, we showed SA directly targeted Cys140 inIMPDH2 to block IKKβ kinase activity, leading to an effectivesuppression of the NF-κB pathway. Moreover, we revealed that SAshowed an inhibitory effect on IMPDH2-mediated guanine nucle-otide biosynthesis, which was important for DNA or RNA synthesis.This could cause the blockage of various inflammation-associatedgene expressions. Thus, we speculate that SA might also inhibit theexpression of heme oxygenase (HO)-1, which was revealed in S. Leeet al.’s work (31) by direct inhibition of IMPDH2. Interestingly, SAwas also found to inhibit cellular tyrosinase activity via repressingtyrosinase gene expression in mouse B16 melanoma cells (28). Thiscould be explained by SA-mediated IMPDH2 inhibition and re-sultant guanine nucleotide biosynthesis stagnation, which is impor-tant for the tyrosinase genetic transcription process.In summary, we discovered Cys140 as a covalent allosteric reg-
ulatory site for selective IMPDH2 inhibition. The small moleculestargeting Cys140, such as SA, can induce an allosteric effect oncatalytic pocket and suppress IMPDH2 activity, leading to antiin-flammation and immunosuppressive action. Therefore, Cys140 mayrepresent a promising drug-binding site of IMPDH2 inhibitors toaccelerate clinical drug development for neuroinflammation withlow side effects.
MethodsCell Survival Assay. The cell survival assay was performed using the MTTmethod. The detailed protocol is found in SI Appendix, SI Methods.
Identification of SA Target Proteins. Identification of SA target proteins wasbased on pull-down technology coupled with SILAC and shotgun protemicsanalysis. The methods are found in SI Appendix, SI Methods.
Determination of the SA-Binding Site on IMPDH2. The SA-binding site onIMPDH2was detected using LC-MS/MS analysis on LTQ-Orbitrap. The detailedprotocol is found in SI Appendix, SI Methods.
Molecular Dynamics Simulation. The force field parameters for inhibitor co-valently bonded to Cys140 residue of IMPDH2 protein were generated by theGeneral AMBERForce Field (GAFF) andRestrainedElectrostatic Potential (RESP).The detailed methodologies for MD are provided in SI Appendix, SI Methods.
All other methods, including cell culture, chemical synthesis, target identi-fication, enzyme activity, gene or protein expression, animal experiments, datacollection, and so forth, are described in detail in SI Appendix, SI Methods.
ACKNOWLEDGMENTS. This work was supported by National Key TechnologyR & D Program “New Drug Innovation” of China Grant 2012ZX09301002-002-002 and Natural Science Foundation of China Grants 81303253 and 30873072.
1. Zimmermann AG, Gu JJ, Laliberté J, Mitchell BS (1998) Inosine-5′-monophosphatedehydrogenase: Regulation of expression and role in cellular proliferation and Tlymphocyte activation. Prog Nucleic Acid Res Mol Biol 61:181–209.
2. Collart FR, Huberman E (1988) Cloning and sequence analysis of the human and Chinesehamster inosine-5′-monophosphate dehydrogenase cDNAs. J Biol Chem 263:15769–15772.
3. Natsumeda Y, et al. (1990) Two distinct cDNAs for human IMP dehydrogenase. J BiolChem 265:5292–5295.
4. Bremer S, et al. (2008) Expression of IMPDH1 and IMPDH2 after transplantation andinitiation of immunosuppression. Transplantation 85:55–61.
5. Jonsson CA, Carlsten H (2003) Mycophenolic acid inhibits inosine 5′-monophosphatedehydrogenase and suppresses immunoglobulin and cytokine production of B cells.Int Immunopharmacol 3:31–37.
6. Sintchak MD, et al. (1996) Structure and mechanism of inosine monophosphate de-hydrogenase in complex with the immunosuppressant mycophenolic acid. Cell 85:921–930.
7. Zhang R, et al. (1999) Characteristics and crystal structure of bacterial inosine-5′-monophosphate dehydrogenase. Biochemistry 38:4691–4700.
8. Scott JW, et al. (2004) CBS domains form energy-sensing modules whose binding ofadenosine ligands is disrupted by disease mutations. J Clin Invest 113:274–284.
9. Thomas EC, et al. (2012) Different characteristics and nucleotide binding properties ofinosine monophosphate dehydrogenase (IMPDH) isoforms. PLoS One 7:e51096.
10. Sobiak J, Kami�nska J, Głyda M, Duda G, Chrzanowska M (2013) Effect of mycophe-nolate mofetil on hematological side effects incidence in renal transplant recipients.Clin Transplant 27:E407–E414.
11. Staatz CE, Tett SE (2014) Pharmacology and toxicology of mycophenolate in organtransplant recipients: An update. Arch Toxicol 88:1351–1389.
12. Hung TM, et al. (2010) Homoisoflavonoid derivatives from the roots of Ophiopogon ja-ponicus and their in vitro anti-inflammation activity. Bioorg Med Chem Lett 20:2412–2416.
13. Waller CP, Thumser AE, Langat MK, Crouch NR, Mulholland DA (2013) COX-2 in-hibitory activity of homoisoflavanones and xanthones from the bulbs of the Southern
Liao et al. PNAS | Published online July 3, 2017 | E5993
PHARM
ACO
LOGY
PNASPL
US

African Ledebouria socialis and Ledebouria ovatifolia (Hyacinthaceae: Hyacinthoi-deae). Phytochemistry 95:284–290.
14. Chang J, Kim Y, Kwon HJ (2016) Advances in identification and validation of proteintargets of natural products without chemical modification. Nat Prod Rep 33:719–730.
15. Jafari R, et al. (2014) The cellular thermal shift assay for evaluating drug target in-teractions in cells. Nat Protoc 9:2100–2122.
16. Pai MY, et al. (2015) Drug affinity responsive target stability (DARTS) for small-molecule target identification. Methods Mol Biol 1263:287–298.
17. Hartman RF, Rose SD (2006) Kinetics and mechanism of the addition of nucleophilesto alpha,beta-unsaturated thiol esters. J Org Chem 71:6342–6350.
18. Yu X, He L, Cao P, Yu Q (2015) Eriocalyxin B inhibits STAT3 signaling by covalentlytargeting STAT3 and blocking phosphorylation and activation of STAT3. PLoS One 10:e0128406.
19. Dunkern T, et al. (2012) Virtual and experimental high-throughput screening (HTS) insearch of novel inosine 5′-monophosphate dehydrogenase II (IMPDH II) inhibitors.J Comput Aided Mol Des 26:1277–1292.
20. Dunkern T, et al. (2014) Design, synthesis and biological evaluation of novel inosine5′-monophosphate dehydrogenase (IMPDH) inhibitors. J Enzyme Inhib Med Chem 29:408–419.
21. Gunter JH, et al. (2008) Characterisation of inosine monophosphate dehydrogenaseexpression during retinal development: differences between variants and isoforms.Int J Biochem Cell Biol 40:1716–1728.
22. Zhang Q, et al. (2014) The role of IMP dehydrogenase 2 in Inauhzin-induced ribo-somal stress. eLife 3:e03077.
23. Kyriakis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signaltransduction pathways activated by stress and inflammation. Physiol Rev 81:807–869.
24. Braun-Sand SB, Peetz M (2010) Inosine monophosphate dehydrogenase as a targetfor antiviral, anticancer, antimicrobial and immunosuppressive therapeutics. FutureMed Chem 2:81–92.
25. Farha MA, Brown ED (2016) Strategies for target identification of antimicrobialnatural products. Nat Prod Rep 33:668–680.
26. Fetz V, Prochnow H, Brönstrup M, Sasse F (2016) Target identification by imageanalysis. Nat Prod Rep 33:655–667.
27. Wang J, et al. (2016) Target identification of natural and traditional medicines withquantitative chemical proteomics approaches. Pharmacol Ther 162:10–22.
28. Chang TS, Chao SY, Ding HY (2012) Melanogenesis inhibition by homoisoflavavonesappanone A from Caesalpinia sappan. Int J Mol Sci 13:10359–10367.
29. Antonino LC, Straub K, Wu JC (1994) Probing the active site of human IMP de-hydrogenase using halogenated purine riboside 5′-monophosphates and covalentmodification reagents. Biochemistry 33:1760–1765.
30. Mizushina Y, et al. (2007) Inhibitory action of polyunsaturated fatty acids on IMPdehydrogenase. Biochimie 89:581–590.
31. Lee S, et al. (2015) Sappanone A exhibits anti-inflammatory effects via modulation ofNrf2 and NF-κB. Int Immunopharmacol 28:328–336.
E5994 | www.pnas.org/cgi/doi/10.1073/pnas.1706778114 Liao et al.


















