
Highly ordered mesoporous carbon nanomatrix as a new approach to improve the oral absorption of the...
9
Highly ordered mesoporous carbon nanomatrix as a new approach to improve the oral absorption of the water-insoluble drug, simvastatin Yanzhuo Zhang a,⇑ , Huan Wang b , Cunqiang Gao c , Xue Li b , Lusi Li c a Department of Pharmaceutics, Xuzhou Medical College, 209 Tongshan Road, Quanshan District, Xuzhou, Jiangsu Province 221004, China b Department of Pharmaceutics, School of Pharmacy, Shenyang Pharmaceutical University, 103 Wenhua Road, Shenhe District, Shenyang, Liaoning Province 110016, China c Jiangsu Hengrui Medicine Co. Ltd., 11 Changjiang Road, Economic and Technological Development Zone, Lianyungang, Jiangsu Province 222047, China article info Article history: Received 23 April 2013 Received in revised form 27 May 2013 Accepted 30 May 2013 Available online 18 June 2013 Keywords: Nanomatrix Mesoporous carbon Water-insoluble drugs Solubility Crystalline state Bioavailability abstract Three different kinds of highly ordered mesoporous carbon (HMC) matrices with different morphologies (hexagonal, spherical and fibrous), particle sizes (700 nm, 400–900 nm and 1–4 lm) and pore diameters were compared as drug carriers for a model drug, simvastatin (SIM). The physicochemical properties of the SIM-loaded composites were studied using field emission scanning electron microscopy (FESEM), specific surface area analysis, differential scanning calorimetry (DSC), wide-angle X-ray scattering (WAXS), HPLC, solubility measurement and dissolution testing. Furthermore, the oral bioavailability of SIM-loaded SHMC (spherical HMC nanomatrix) in beagle dogs was compared with that of the reference formulation (Zocor Ò ). The results obtained showed that SIM molecules are encapsulated in a noncrystal- line state due to geometric confinement in the nanopores of HMC. In vitro dissolution testing showed that the dissolution rate of SIM released from monodispersed SHMC was significantly faster compared with that of crystalline SIM and other SIM-loaded composites. In addition, in vivo bioavailability study demon- strated that the relative bioavailability of SIM and SIM b-hydroxy acid (an active metabolite of SIM) for SIM-loaded SHMC formulation was 138.42% and 163.55%, respectively. In conclusion, monodispersed SHMC appear to be a more promising candidate as a new oral drug delivery vehicle providing a rapid drug release and enhanced oral bioavailability. Ó 2013 Elsevier B.V. All rights reserved. 1. Introduction The poor aqueous solubility and insufficient dissolution of many active pharmaceutical ingredients are a major technical problem, especially for pharmaceutical researchers involved in for- mulation development (Blagden et al., 2007). It is estimated that 40% of new chemical entities are poorly soluble or insoluble in water, and up to 50% of orally administered drugs have formula- tion problems related to low bioavailability (Lipinski et al., 1997). In order for a drug to be absorbed into the systemic circulation fol- lowing oral administration, it must be dissolved in gastrointestinal fluids (Hörter and Dressman, 2001). For many hydrophobic active pharmaceutical ingredients that cross the gastrointestinal mucosa easily, the drug levels achieved will be dictated by the time re- quired for the dosage form to release its contents, and for the drug to dissolve (Huanga and Tong, 2004; Manly et al., 2007). Hence, improving the saturation solubility and dissolution rate of water- insoluble drugs is very important and presents a major challenge to formulation scientists seeking to obtain complete absorption of active pharmaceutical ingredients (Vasconcelos et al., 2007). Over the years, the major formulation tools employed to in- crease the dissolution rate and saturation solubility include: the use of solid dispersions (Vasconcelos et al., 2007), nanosizing (Kesi- soglou et al., 2007), inclusion complexes (Sadighi et al., 2012), and salt forms of active pharmaceutical ingredients (Bastin et al., 2000). Another, alternative technique is to prepare pharmaceutical agents in an amorphous form. The amorphous form is a high-energy state that exhibits increased saturation solubility and dissolution rate and, thus, enhanced oral bioavailability (Manly et al., 2007; Vasconcelos et al., 2007). One strategy that is increasingly popular as a means of stabilizing amorphous drugs is adsorption in the nanopores of mesoporous materials (Ambrogi et al., 2008; Barbé et al., 2004; Charnay et al., 2004; Heikkilä et al., 2007; Thomas et al., 2009). Although a variety of chemically different mesoporous matrices have been reported, mesoporous silica is the most widely investigated in the field of drug delivery (Kim et al., 2011; Tan et al., 2011; Jaganathan and Godin, 2012; Tang et al., 2012; Wani et al., 2012; Zhang et al., 2012). In recent years, there has been sig- nificant interest in the development of mesoporous carbon materials with a uniform pore structure. Highly ordered mesopor- ous carbon (HMC) can be synthesized by the nanocasting 0928-0987/$ - see front matter Ó 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.ejps.2013.05.031 ⇑ Corresponding author. Address: Xuzhou Medical College, P.O. Box 62, 209 Tongshan Road, Quanshan District, Xuzhou, Jiangsu Province 221004, China. Tel./fax: +86 516 83262116. E-mail address: [email protected] (Y. Zhang). European Journal of Pharmaceutical Sciences 49 (2013) 864–872 Contents lists available at SciVerse ScienceDirect European Journal of Pharmaceutical Sciences journal homepage: www.elsevier.com/locate/ejps
Transcript of Highly ordered mesoporous carbon nanomatrix as a new approach to improve the oral absorption of the...

European Journal of Pharmaceutical Sciences 49 (2013) 864–872
Contents lists available at SciVerse ScienceDirect
European Journal of Pharmaceutical Sciences
journal homepage: www.elsevier .com/ locate /e jps
Highly ordered mesoporous carbon nanomatrix as a new approach toimprove the oral absorption of the water-insoluble drug, simvastatin
0928-0987/$ - see front matter � 2013 Elsevier B.V. All rights reserved.http://dx.doi.org/10.1016/j.ejps.2013.05.031
⇑ Corresponding author. Address: Xuzhou Medical College, P.O. Box 62, 209Tongshan Road, Quanshan District, Xuzhou, Jiangsu Province 221004, China.Tel./fax: +86 516 83262116.
E-mail address: [email protected] (Y. Zhang).
Yanzhuo Zhang a,⇑, Huan Wang b, Cunqiang Gao c, Xue Li b, Lusi Li c
a Department of Pharmaceutics, Xuzhou Medical College, 209 Tongshan Road, Quanshan District, Xuzhou, Jiangsu Province 221004, Chinab Department of Pharmaceutics, School of Pharmacy, Shenyang Pharmaceutical University, 103 Wenhua Road, Shenhe District, Shenyang, Liaoning Province 110016, Chinac Jiangsu Hengrui Medicine Co. Ltd., 11 Changjiang Road, Economic and Technological Development Zone, Lianyungang, Jiangsu Province 222047, China
a r t i c l e i n f o
Article history:Received 23 April 2013Received in revised form 27 May 2013Accepted 30 May 2013Available online 18 June 2013
Keywords:NanomatrixMesoporous carbonWater-insoluble drugsSolubilityCrystalline stateBioavailability
a b s t r a c t
Three different kinds of highly ordered mesoporous carbon (HMC) matrices with different morphologies(hexagonal, spherical and fibrous), particle sizes (700 nm, 400–900 nm and 1–4 lm) and pore diameterswere compared as drug carriers for a model drug, simvastatin (SIM). The physicochemical properties ofthe SIM-loaded composites were studied using field emission scanning electron microscopy (FESEM),specific surface area analysis, differential scanning calorimetry (DSC), wide-angle X-ray scattering(WAXS), HPLC, solubility measurement and dissolution testing. Furthermore, the oral bioavailability ofSIM-loaded SHMC (spherical HMC nanomatrix) in beagle dogs was compared with that of the referenceformulation (Zocor�). The results obtained showed that SIM molecules are encapsulated in a noncrystal-line state due to geometric confinement in the nanopores of HMC. In vitro dissolution testing showed thatthe dissolution rate of SIM released from monodispersed SHMC was significantly faster compared withthat of crystalline SIM and other SIM-loaded composites. In addition, in vivo bioavailability study demon-strated that the relative bioavailability of SIM and SIM b-hydroxy acid (an active metabolite of SIM) forSIM-loaded SHMC formulation was 138.42% and 163.55%, respectively. In conclusion, monodispersedSHMC appear to be a more promising candidate as a new oral drug delivery vehicle providing a rapid drugrelease and enhanced oral bioavailability.
� 2013 Elsevier B.V. All rights reserved.
1. Introduction
The poor aqueous solubility and insufficient dissolution ofmany active pharmaceutical ingredients are a major technicalproblem, especially for pharmaceutical researchers involved in for-mulation development (Blagden et al., 2007). It is estimated that40% of new chemical entities are poorly soluble or insoluble inwater, and up to 50% of orally administered drugs have formula-tion problems related to low bioavailability (Lipinski et al., 1997).In order for a drug to be absorbed into the systemic circulation fol-lowing oral administration, it must be dissolved in gastrointestinalfluids (Hörter and Dressman, 2001). For many hydrophobic activepharmaceutical ingredients that cross the gastrointestinal mucosaeasily, the drug levels achieved will be dictated by the time re-quired for the dosage form to release its contents, and for the drugto dissolve (Huanga and Tong, 2004; Manly et al., 2007). Hence,improving the saturation solubility and dissolution rate of water-insoluble drugs is very important and presents a major challenge
to formulation scientists seeking to obtain complete absorptionof active pharmaceutical ingredients (Vasconcelos et al., 2007).
Over the years, the major formulation tools employed to in-crease the dissolution rate and saturation solubility include: theuse of solid dispersions (Vasconcelos et al., 2007), nanosizing (Kesi-soglou et al., 2007), inclusion complexes (Sadighi et al., 2012), andsalt forms of active pharmaceutical ingredients (Bastin et al., 2000).Another, alternative technique is to prepare pharmaceutical agentsin an amorphous form. The amorphous form is a high-energy statethat exhibits increased saturation solubility and dissolution rateand, thus, enhanced oral bioavailability (Manly et al., 2007;Vasconcelos et al., 2007). One strategy that is increasingly popularas a means of stabilizing amorphous drugs is adsorption in thenanopores of mesoporous materials (Ambrogi et al., 2008; Barbéet al., 2004; Charnay et al., 2004; Heikkilä et al., 2007; Thomaset al., 2009). Although a variety of chemically different mesoporousmatrices have been reported, mesoporous silica is the most widelyinvestigated in the field of drug delivery (Kim et al., 2011; Tanet al., 2011; Jaganathan and Godin, 2012; Tang et al., 2012; Waniet al., 2012; Zhang et al., 2012). In recent years, there has been sig-nificant interest in the development of mesoporous carbonmaterials with a uniform pore structure. Highly ordered mesopor-ous carbon (HMC) can be synthesized by the nanocasting

Y. Zhang et al. / European Journal of Pharmaceutical Sciences 49 (2013) 864–872 865
technique using highly ordered mesoporous silica (HMS) as a tem-plate (Liu et al., 2006, 2008). HMC has a larger pore volume andhigher specific surface area compared with other mesoporousmaterials (zeolite, silica, aluminum oxide, etc.) (Liang et al.,2008; Vashist et al., 2011) and, so, it should have a higher drugloading capacity. In addition to the nanocavities of HMC being ableto change the crystalline state of a drug to an amorphous one(Wani et al., 2012), the nanocavities also effectively restrict drugrecrystallization and significantly reduce the particle size of theamorphous drug (Qian and Bogner, 2011). All these propertiesdemonstrate the potential and advantages of using HMC in oraldrug delivery systems. However, few in vitro studies have beenconducted using oral drug delivery systems involving HMC carriers(Liang et al., 2008; Li et al., 2011; Vashist et al., 2011; Wang et al.,2011) and, to the best of our knowledge, there are no reports of theuse of HMC nanomatrix for oral drug delivery in vivo.
Based on the above considerations, we have designed threekinds of HMC matrices as oral drug carriers and loaded a modelwater-insoluble drug into the nanopore channels. SIM is a choles-terol-lowering agent which is widely used to treat hypercholester-olemia (Illingworth and Tobert, 2000). SIM has a poor solubility(6.3 lg/ml, pH 1–7, at 25 �C) and is, thus, a good model drug forformulation in the amorphous state to enhance its dissolution rate(Graeser et al., 2008). The purpose of the present study was to de-velop and investigate the properties and mechanism of enhance-ment of dissolution and oral bioavailability of the SIM-loadedHMC. To achieve this aim, the effects of different morphologies,particle sizes and pore diameters of HMC on the drug loadingand release behaviors of the model drug SIM were systematicallystudied using FESEM, transmission electron microscopy (TEM),specific surface area analysis, DSC, WAXS, HPLC, solubility mea-surement and dissolution testing. Furthermore, pharmacokineticprofiling of SIM after oral administration of SIM-loaded SHMC orthe commercial product Zocor� in beagle dogs was made withuse of ultraperformance liquid chromatography equipped withelectrospray ionization mass spectrometry (UPLC/ESI-MS). The re-sults obtained showed that monodispersed SHMC nanomatrix is apromising carrier able to improve the dissolution rate and subse-quently the oral absorption of the water-insoluble drug SIM.
2. Materials and methods
2.1. Chemicals
SIM (USP grade, P98%), Lovastatin (LOV, P99%) and SIMAammonium salt were kindly donated from Hisun Pharma (Zhe-jiang, China). Guaranteed-grade sucrose, tetraethyl orthosilicate,HPLC-grade acetonitrile and ethyl acetate were supplied fromKemiou (Tianjin, China). Pluronic 123 was obtained from BASF(Ludwigshafen, Germany). SIM tablets (Zocor�) were obtainedfrom Merck Sharp & Dohme (Hangzhou, China). Lactose monohy-drate and pregelatinized starch were donated from Roquette(Jiangsu, China). Avicel� PH microcrystalline cellulose (MCC) andcroscarmellose sodium were supplied from FMC BioPolymer (Phil-adelphia, PA, USA). All other reagents were of reagent grade andwere used as purchased without further purification.
2.2. Preparation of HMS nanomatrix and micromatrix
A series of HMS matrices were synthesized according to proce-dures described in the literature with some modifications, usingtetraethyl orthosilicate as a silicate source. For the synthesis ofhexagonal HMS (HHMS) nanomatrix (Wang et al., 2009a,b), 2.0 gglycerol and 2.0 g Pluronic 123 were dissolved in 76 ml 2.5 MHCl solution at ambient temperature. Then 4.2 g tetraethyl
orthosilicate was added to the Pluronic 123 solution. The stirringwas allowed to continue for 10 min before switching to static syn-thesis conditions at 38 �C. After 24 h, the resulting gel mixture wastaken to a Teflon-coated stainless-steel autoclave and kept forapproximately 20 h at 80 �C. The resulting precipitate was recov-ered, washed, and dried. Finally, the solid product was calcinedin air at 600 �C for 5 h. Spherical HMS (SHMS) nanomatrix (Liuet al., 2009) were synthesized in the same way as HHMS, exceptfor the use of 0.33 g cetrimonium bromide as a co-template and76 ml 1.5 M HCl solution. To prepare fibrous HMS (FHMS) microm-atrix (Zhao et al., 1998), 2.0 g Pluronic 123 was dissolved in 76 ml1.75 M HCl solution. Subsequently, 4.2 g tetraethyl orthosilicatewas added to this solution with stirring at 38 �C for 24 h and thesynthesized material was filtered, washed, and calcined at 600 �Cfor 5 h.
2.3. Preparation of HMC nanomatrix and micromatrix
Three different kinds of HMC matrices with different morphol-ogies and particle sizes were replicated from the HMS templatesusing sucrose as the carbon precursor (Lee et al., 2008). In a typicalexperiment, a precursor solution, containing 0.26 g boric acid,0.14 g H2SO4, 1.25 g sucrose and 4.0 ml purified water was allowedto infiltrate the nanocavities of the respective HMS template (1.0 gHHMS, SHMS and FHMS, respectively). Following this, the mixturewas dried at 100 �C for 6 h in a DGT-10 drying oven (Haier, China),then the temperature was increased to 160 �C and held there forapproximately 6 h. The infiltration and drying procedure were re-peated again with an additional 3.68 g precursor solution, beforethe silica/sucrose composite was carbonized at 900 �C for 3 h undera nitrogen purge of 60 ml/min. The silica template was removed bytreatment with 10% HF solution for 24 h. Finally, the respectiveHMC product (HHMC, SHMC and FHMC, respectively) was filtered,washed with ethanol, and dried in a vacuum oven. SHMC-2 wassynthesized in the same way as SHMC but without adding boricacid. In vitro cytotoxicity of HMC on Caco-2 cells was assessed bythe MTT reduction assay. The results of MTT assay indicated thatthe prepared HMC exhibited a very low cytotoxicity (specific datacan be found in Fig. S1 in Supplementary Data).
2.4. Loading SIM into the porous materials
A solvent immersion/evaporation procedure was used to loadthe porous materials with SIM. In a typical experiment, 0.6 gHMC (HHMC, SHMC, SHMC-2 and FHMC, respectively) was addedto 20 ml 0.34 M SIM solution. Ethanol was used as the loading sol-vent. Then, the particle suspensions were ultrasonicated for a fewminutes and kept gentle stirring on a HJ-A6 magnetic stir plate(Runhua, China) for 24 h to achieve maximum adsorption in thenanocavities of HMC. The adsorption procedure was protectedfrom the light. Subsequently, the SIM-loaded particles were sepa-rated from the drug solution by filtration under vacuum. The moistpowder obtained was dried at 40 �C under reduced pressure(10�3 bar) for 6 h to remove any residual ethanol from the nano-cavities of HMC. The drug-loaded samples were referred to asSIM-HHMC, SIM-SHMC, SIM-SHMC-2 and SIM-FHMC, respectively.The same process was repeated for HHMS, SHMS and FHMS. Inaddition, the SIM-SHMC tablets were prepared by a process of di-rect compression with physical blending of SIM-SHMC (20 mg, ex-pressed as SIM equivalents), lactose monohydrate, MCC (PH-102),pregelatinized starch, MCC (PH-200), croscarmellose sodium andmagnesium stearate (in a mass ratio of 20/142/24/26/39.5/9.2/4.5).

866 Y. Zhang et al. / European Journal of Pharmaceutical Sciences 49 (2013) 864–872
2.5. Particle size and morphology analysis
The particle size and surface morphology of the prepared nano-porous silica and carbon samples were characterized using aJSM-6301F field emission SEM instrument (JEOL, Japan). Prior toimaging, the samples were mounted onto aluminum stages usingdouble-sided carbon tape and sputter coated using a K550 sputtercoater (Emitech, UK) to render them electrically conductive. TEMmicrographs were obtained using a JEM-2100F TEM instrument(JEOL, Japan) operated at a voltage of 200 kV.
2.6. Specific surface area and pore size analysis
An ASAP 2020 rapid surface area and pore size analyzer(Micromeritics, USA) was used to measure the pore characteristicsof the nanoporous silica and carbon samples. The nitrogen adsorp-tion–desorption isotherms were recorded at �196 �C. The preparednanoporous silica and carbon matrices were degassed at 300 �C for6 h prior to measurements, while the SIM-loaded composites weredegassed at 40 �C for 6 h. The specific surface area (SBET) wascalculated using the Brunauer–Emmett–Teller (BET) equation inthe relative pressure range between 0.05 and 0.2 Pore size distribu-tions were determined from adsorption branches of isothermsusing the Barrett–Joyner–Halenda (BJH) method.
2.7. Solid-state characterization by DSC and WAXS
The physical state of the pure crystalline SIM and the SIM-loaded composites was examined by DSC and WAXS analysis.The thermographs of each powder were obtained using a Q1000DSC instrument (TA Instruments, USA) equipped with a refriger-ated cooling system. Calibration of the DSC instrument was carriedout using indium as a standard. The powder (about 5 mg) wasaccurately weighed, placed in an aluminum pan. Testing was per-formed at temperatures increasing from 40 to 200 �C at a rate of10 �C/min under a nitrogen purge. Furthermore, WAXS measure-ments were carried out with a SMART X2S powder diffractometer(Bruker, Germany) using Cu Ka radiation at a voltage of 30 kV anda current of 50 mA. Data were obtained from 4� to 40� (diffractionangle 2h) at a step size of 0.02� and scanning speed of 4�/min.
2.8. In vitro quantification of SIM
The drug loading capacity and samples from the in vitro drugdissolution and solubility studies were assayed using a HPLC meth-od. Briefly, chromatography was carried out using an Agilent 1100HPLC system (Agilent, USA) consisting of two G1312A pumps, aG1316A column oven and a G1314B UV–VIS detector set at238 nm. The separation of SIM was acquired on a 200 � 4.6 mmZorbax C18 analytical column (Agilent, USA) at 40 �C, eluting withacetonitrile and 25 mM sodium dihydrogen phosphate solution(70/30%, v/v) at a flow rate of 1.0 ml/min. SIM was determined overthe concentration range 0.5–40 lg/ml. Parameters validated in-cluded precision (intra-day and inter-day) and accuracy. Both theintra- and inter-day relative standard deviations (RSD) of qualitycontrol (QC) standards were less than 4% over the selected range.The high accuracy of the method was confirmed with recovery val-ues of 98–102%.
2.9. Drug loading evaluation
The drug loading of the SIM-loaded samples was determined bysuspending an accurately weighed amount of SIM-loaded HMC(about 20 mg) in 250 ml acetonitrile. These suspensions were son-icated for 10 min and subsequently put in a magnetic stir plate for12 h. Following this, the nanoporous silica or carbon matrix was
separated from the SIM solution by centrifugation at 9,000 rpmfor 10 min. The supernatant layer was taken, filtered through a0.22 lm PTFE syringe filter (Millipore, USA) and then the drugconcentration was determined by HPLC. The drug loading wascalculated using the following Eq. (1):
Drug loading ð%Þ ¼ ðweight of SIM in composite=weight of compositeÞ�100 ð1Þ
2.10. Solubility study
The equilibrium solubility of SIM in phosphate buffer solution(PBS, pH 6.8) was determined by adding an excess amount ofraw SIM and SIM-loaded composites to different Eppendorf tubesand adding 4 ml freshly prepared PBS. The tubes were vortex-mixed and then placed in a constant temperature shaking waterbath maintained at 37 ± 1 �C for 48 h. Suitable aliquots were centri-fuged at 9000 rpm for 10 min in a 5702RH temperature-controlledcentrifuge (Eppendorf, Germany) at 37 ± 1 �C. The supernatantlayer was taken, filtered and then the drug concentration wasdetermined by HPLC as described above. The measurements wereperformed in triplicate for each formulation.
2.11. In vitro drug dissolution study
The in vitro drug dissolution study was conducted using a USP IIdissolution apparatus (AT7 Smart offline, Sotax, Switzerland) at arotation speed of 50 rpm. The SIM-loaded composites (equivalentto 10 mg SIM) and the pure crystalline SIM (10 mg) were placedin dissolution vessels containing 900 ml PBS (pH 6.8, 37 ± 0.5 �C).Then, 5 ml supernatant was withdrawn at each pre-determinedtime point and replaced with 5 ml fresh dissolution medium. Thewithdrawn samples were filtered and then the SIM concentrationwas determined by HPLC as described above. A similar procedurewas carried out to determine the dissolution rate of the commer-cial tablet and the SIM-SHMC tablet. To provide sink conditions,0.3% (w/v) sodium dodecyl sulfate was added to the dissolutionmedium.
2.12. In vivo experiments
2.12.1. Drug administration and blood sample analysisMale beagle dogs were housed in the animal facility of the Gen-
eral Hospital of Nanjing Military Region, (Nanjing, China) with freeaccess to water and food. All the animal experiments compliedwith the Animal Care and Use institutional guidelines and approvalwas granted by the Animal Ethics Committee of Nanjing Univer-sity. Six dogs (body weight 10–15 kg) were randomly allocated totwo treatment groups and given the reference formulation (Zo-cor�) and the SIM-SHMC formulation according to a two-periodcrossover experimental design. The washout period between twoconsecutive treatments was 1 week. The dogs were fasted for overa day to dosing and a dose of each formulation (20 mg, expressedas SIM equivalents) was given by gavage. Blood samples (about2 ml) were collected from the jugular vein pre-dosing and 0.25,0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12 and 24 h post-dosing. Immedi-ately after blood collection, samples were centrifuged at 4000 rpmfor 15 min and the plasma obtained was then transferred to freshheparinized tubes and stored with photo protection at �20 �C forfurther analysis.
The SIM and SIMA concentrations in plasma were determinedby a validated UPLC/ESI-MS method after liquid–liquid extraction.To 200 ll plasma, 25 ll internal standard solution (LOV, 200 lg/mlin methanol) was added and vortexed. After adding 3 ml ethyl ace-tate, the mixture was vortexed for 4 min using a TM-2F vortex

Y. Zhang et al. / European Journal of Pharmaceutical Sciences 49 (2013) 864–872 867
mixer (ASONE, Japan). Following centrifugation at 12,000 rpm for10 min, the upper organic phase was transferred into an Eppendorftube and evaporated to dryness at 40 �C under a gentle nitrogenpurge. The residue was reconstituted in 200 ll methanol, vortexedand a 5 ll aliquot was injected into the UPLC/ESI-MS system for thequantification of SIM and SIMA.
The chromatographic separation was performed on an AcquityUPLCTM system using a 2.1 � 50 mm Acquity UPLC™ BEH C18
column (Waters, USA). The mobile phase consisted of solvent A(acetonitrile) and solvent B (10 mM ammonium acetate containing0.1% formic acid) in a gradient system: 20–80% A (0–0.8 min), 80%A (0.8–2.7 min), 80–20% A (2.7–3 min), and 20% A (3–3.5 min). Theflow rate was 0.25 ml/min. A Micromass quattro micro™ API triplequadrupole tandem mass spectrometer (Waters, UK) interfacedwith the UPLC via an electrospray ionization (ESI) source was usedfor the mass detection and analysis. The precursor/product iontransitions were monitored at m/z 405.2 ? 285.2 for SIM, m/z419.2 ? 285.1 for SIMA and m/z 437.3 ? 303.2 for LOV (internalstandard), respectively. The mass spectrometer operating condi-tions were optimized as follows: capillary 0.2 kV, cone 30 V andextractor 20 V for SIM and SIMA, capillary 0.2 kV, cone 25 V andextractor 10 V for LOV. The desolvation temperature and thesource temperature were set at 400 �C and 110 �C, respectively.Calibration curves (0.5–200 ng/ml, seven different concentrations)and QC samples (0.5, 20 and 200 ng/ml) for both SIM and SIMAwere prepared freshly for each analysis. The linear regression coef-ficients of the calibration curves were not less than 0.99. The accu-racy and the precision of the QC samples ranged from 92.4% to108.1% of the nominal values, with a CV (coefficient of variation)of 5.3–8.6%, respectively. The lower limit of quantification valuesof SIM and SIMA were both 0.5 ng/ml. The extraction recoveriesof SIM and SIMA from the QC samples were 75.4 ± 2.2 and69.0 ± 3.1%, respectively.
2.12.2. Pharmacokinetic analysisThe pharmacokinetic parameters of SIM and SIMA were ob-
tained by non-compartmental analysis (NCA) using Drug and Sta-tistics software (version 2.0, Mathematical PharmacologyProfessional Committee, China). The maximum plasma concentra-tion (Cmax) and the time to reach the Cmax (tmax) were read directlyfrom the individual subject plasma concentration–time profiles.The area under the plasma concentration–time curve from zeroto 24 h (AUC0?24) was calculated using the classical linear trape-zoidal method and used to estimate the relative bioavailability(Frel.) of the test formulation with reference to that of the commer-cial tablet (Zocor�), according to Eq. (2):
Frel: ð%Þ ¼ ½AUC0!tðtestÞ=AUC0!tðreferenceÞ� � 100 ð2Þ
The experimental data were analyzed using the statisticalpackage for social sciences (SPSS version 15.0) software. Student’st-tests were performed to evaluate the significance of differencesbetween the two formulations. For all analysis, p < 0.05 was usedas the criterion for statistical significance.
3. Results and discussion
3.1. Morphology and porous structure of the prepared samples
The FESEM and TEM images reveal important information aboutthe morphology, particle size and porous structure, respectively.The FESEM micrographs (Fig. 1A–C) shows that the three HMSmatrices have distinctively different morphologies and particlesizes. The HHMS and SHMS matrices exhibit a hexagonal morphol-ogy (with a mean particle size of about 700 nm) and a sphericalmorphology (with a diameter of 400–900 nm), respectively. It
can be seen that FHMS is aggregated with a fibrous macrostructureconsisting of many tube-like structures (with a length of 1–4 lmand a diameter of about 300 nm). Pore structures of the preparedHMS samples can be seen in the TEM images. As an example, theTEM image of SHMS (Fig. 1D) shows cylindrical pores (with anaverage nanopore diameter of 6–8 nm) arranged in an orderedform and in parallel with each other (Fig. 1D inset). The FESEMmicrographs shown in Fig. 2 reveal the morphology of the respec-tive HMC product (HHMC, SHMC and FHMC, respectively), which issimilar to the one observed for the HMS template. It was found thatthe particles of FHMC are agglomerated or connected (Fig. 2C),which is similar to the case of FHMS (Fig. 1C). The TEM image ofSHMC (Fig. 2D) viewed perpendicular to the long axis shows cylin-drical arrays of the carbon matrix and uniform nanopores. In addi-tion, the morphology of the model drug SIM was non-uniform witha diameter ranging from 5 to 30 lm (Fig. 3A). As seen from the FES-EM image (Fig. 3B), the morphology of the SIM-loaded SHMC wasthe same as blank SHMC (Fig. 2B), and no separated particles ofdrug can be observed. This indicated that the model drug wasincorporated into the nanopores of SHMC or adsorbed uniformlyon the surface of SHMC.
3.2. Nitrogen adsorption study
The nitrogen adsorption–desorption isotherms of the preparedHMS and HMC matrices were typical type IV isotherms accordingto the International Union of Pure and Applied Chemistry classifi-cation, characteristic of mesoporous materials. The nitrogenadsorption–desorption isotherms of SHMS, HMC and SIM-loadedSHMC are presented in Fig. 4. The presence of a sharp capillary con-densation step at a higher relative pressure and H1 hysteresis loopindicate that all the HMC matrices have a uniform pore structurewith large mesopores. The major parameters including the BETspecific surface area (SBET), total pore volume (Vt), BJH pore diam-eter (wBJH) and drug loading values for each porous materials de-rived from the nitrogen adsorption–desorption experiments aresummarized in Table 1. The corresponding pore size distributiondata shows that the nanopores are uniform and have a narrow poresize distribution, centered at about 7.2, 9.3, 5.6 and 7.8 nm forHHMC, SHMC, SHMC-2 and FHMC, respectively. It is particularlynoteworthy that all the HMC matrices have a high SBET and Vt, indi-cating their potential application as a host for bonding or storingmore drug molecules in the drug storage/release system (Salonenet al., 2005; Yang et al., 2008). As an example, the SBET of SHMCis 1638.5 m2/g, which is almost 1.96-fold larger than that of thecorresponding SHMS template. Furthermore, SBET, Vt and wBJH werereduced apparently after SIM loading, confirming that SIM wasincorporated into HMC pore channels. It is interesting that themaximum drug loading of the prepared HMC was approximately40%. In contrast, the maximum drug loading of the preparedHMS was no more than 30%. The difference in the maximum drugloading between HMC and HMS could be attributed to Vt and SBET.It is well known that the relatively high Vt and SBET of mesoporousmaterial enable it to achieve a high drug loading efficiency(Wang, 2009).
3.3. Physicochemical characterization
The thermal behaviors of SIM, physical mixture and SIM-loadedHMC samples were recorded and signals due to melting could beobserved. Fig. 5 shows the DSC thermograms of both raw SIMand physical mixture (the mass ratio of SIM: SHMC was 1:4) withthe corresponding melting transition of crystalline SIM at 138.1 �C.However, no endothermic peak of SIM was identified in the DSCcurves obtained from each SIM-loaded HMC. The absence of phasetransitions owing to SIM in the DSC analysis is evidence that the

Fig. 1. FESEM photographs of (A) HHMS, (B) SHMS and (C) FHMS. TEM photograph of (D) SHMS.
Fig. 2. FESEM photographs of (A) HHMC, (B) SHMC and (C) FHMC. TEM photograph of (D) SHMC.
868 Y. Zhang et al. / European Journal of Pharmaceutical Sciences 49 (2013) 864–872

Fig. 3. FESEM photographs (A) crystalline SIM and (B) SIM-SHMC.
Fig. 4. Nitrogen adsorption/desorption isotherms of (a) SHMS, (b) HHMC, (c) SHMC,(d) FHMC, (e) SHMC-2 and (f) SIM-SHMC. The isotherms of (b), (c), (d) and (e) wereoffset vertically by 200, 600, 1000 and 1400 (cm3/g STP), respectively.
Table 1Results of sample characterizations (n = 3).
Sample SBET (m2/g) Vt (cm3/g) wBJH (nm) Drug loading (%)
HHMS 822.1 ± 6.4 1.07 ± 0.09 6.4 ± 1.7 22.6 ± 1.9SHMS 837.4 ± 9.5 1.05 ± 0.04 6.2 ± 1.2 24.6 ± 1.5FHMS 910.1 ± 10.3 1.01 ± 0.06 5.9 ± 1.4 25.1 ± 1.3HHMC 1550.2 ± 27.8 1.62 ± 0.14 7.2 ± 2.3 37.6 ± 2.3SHMC 1638.5 ± 24.3 1.79 ± 0.17 9.3 ± 2.6 39.5 ± 2.2FHMC 1703.1 ± 20.2 1.83 ± 0.22 7.8 ± 1.4 34.6 ± 3.4SHMC-2 1667.9 ± 31.7 1.81 ± 0.15 5.6 ± 1.1 40.1 ± 2.0SIM-HHMC 177.2 ± 19.6 0.22 ± 0.09 2.9 ± 0.6SIM-SHMC 156.3 ± 27.5 0.27 ± 0.06 3.1 ± 0.4SIM-FHMC 131.0 ± 24.9 0.19 ± 0.10 1.4 ± 0.7SIM-SHMC-2 149.6 ± 38.2 0.23 ± 0.05 1.7 ± 0.6
Fig. 5. DSC thermograms of (a) crystalline SIM, (b) physical mixture, (c) SIM-HHMC,(d) SIM-SHMC and (e) SIM-FHMC.
Fig. 6. WAXS patterns of (a) crystalline SIM, (b) SHMC, (c) physical mixture, (d)SIM-HHMC, (e) SIM-SHMC and (f) SIM-FHMC.
Y. Zhang et al. / European Journal of Pharmaceutical Sciences 49 (2013) 864–872 869
encapsulated drug SIM was molecularly well dispersed in the HMCmatrices, and complete drug amorphization was attained. TheWAXS patterns of the SIM-loaded HMC samples were also recordedto determine whether a crystalline drug phase could be detected.
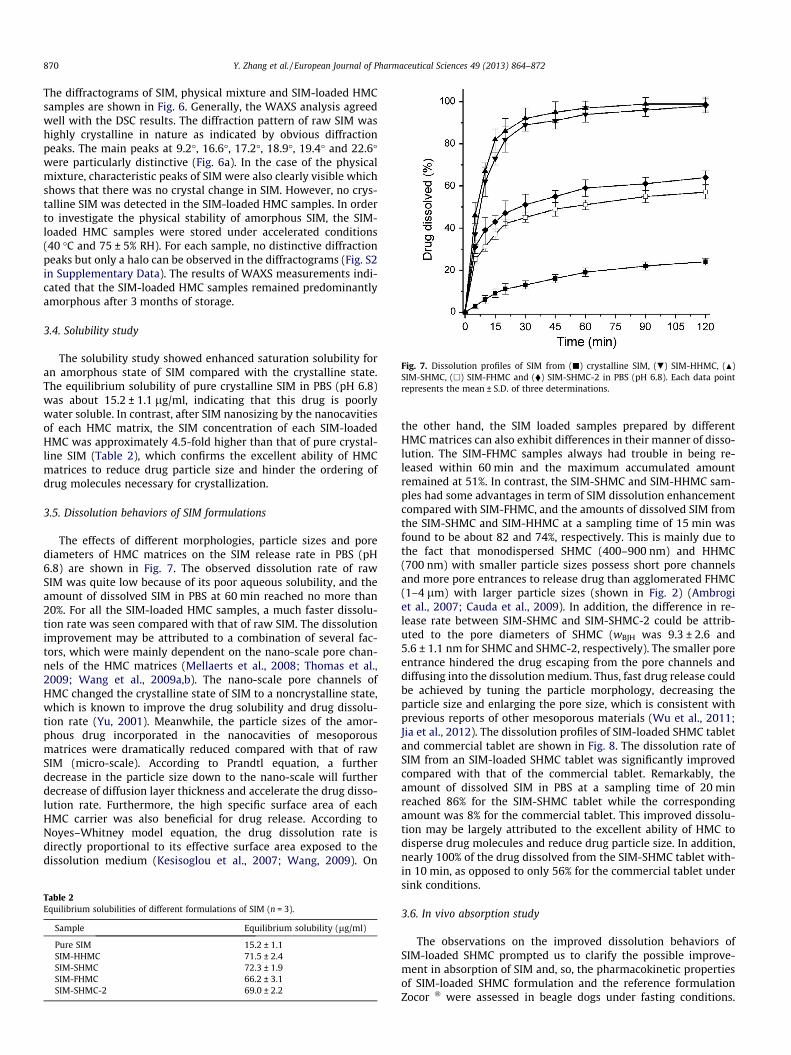
Fig. 7. Dissolution profiles of SIM from (j) crystalline SIM, (.) SIM-HHMC, (N)SIM-SHMC, (h) SIM-FHMC and (�) SIM-SHMC-2 in PBS (pH 6.8). Each data pointrepresents the mean ± S.D. of three determinations.
870 Y. Zhang et al. / European Journal of Pharmaceutical Sciences 49 (2013) 864–872
The diffractograms of SIM, physical mixture and SIM-loaded HMCsamples are shown in Fig. 6. Generally, the WAXS analysis agreedwell with the DSC results. The diffraction pattern of raw SIM washighly crystalline in nature as indicated by obvious diffractionpeaks. The main peaks at 9.2�, 16.6�, 17.2�, 18.9�, 19.4� and 22.6�were particularly distinctive (Fig. 6a). In the case of the physicalmixture, characteristic peaks of SIM were also clearly visible whichshows that there was no crystal change in SIM. However, no crys-talline SIM was detected in the SIM-loaded HMC samples. In orderto investigate the physical stability of amorphous SIM, the SIM-loaded HMC samples were stored under accelerated conditions(40 �C and 75 ± 5% RH). For each sample, no distinctive diffractionpeaks but only a halo can be observed in the diffractograms (Fig. S2in Supplementary Data). The results of WAXS measurements indi-cated that the SIM-loaded HMC samples remained predominantlyamorphous after 3 months of storage.
3.4. Solubility study
The solubility study showed enhanced saturation solubility foran amorphous state of SIM compared with the crystalline state.The equilibrium solubility of pure crystalline SIM in PBS (pH 6.8)was about 15.2 ± 1.1 lg/ml, indicating that this drug is poorlywater soluble. In contrast, after SIM nanosizing by the nanocavitiesof each HMC matrix, the SIM concentration of each SIM-loadedHMC was approximately 4.5-fold higher than that of pure crystal-line SIM (Table 2), which confirms the excellent ability of HMCmatrices to reduce drug particle size and hinder the ordering ofdrug molecules necessary for crystallization.
3.5. Dissolution behaviors of SIM formulations
The effects of different morphologies, particle sizes and porediameters of HMC matrices on the SIM release rate in PBS (pH6.8) are shown in Fig. 7. The observed dissolution rate of rawSIM was quite low because of its poor aqueous solubility, and theamount of dissolved SIM in PBS at 60 min reached no more than20%. For all the SIM-loaded HMC samples, a much faster dissolu-tion rate was seen compared with that of raw SIM. The dissolutionimprovement may be attributed to a combination of several fac-tors, which were mainly dependent on the nano-scale pore chan-nels of the HMC matrices (Mellaerts et al., 2008; Thomas et al.,2009; Wang et al., 2009a,b). The nano-scale pore channels ofHMC changed the crystalline state of SIM to a noncrystalline state,which is known to improve the drug solubility and drug dissolu-tion rate (Yu, 2001). Meanwhile, the particle sizes of the amor-phous drug incorporated in the nanocavities of mesoporousmatrices were dramatically reduced compared with that of rawSIM (micro-scale). According to Prandtl equation, a furtherdecrease in the particle size down to the nano-scale will furtherdecrease of diffusion layer thickness and accelerate the drug disso-lution rate. Furthermore, the high specific surface area of eachHMC carrier was also beneficial for drug release. According toNoyes–Whitney model equation, the drug dissolution rate isdirectly proportional to its effective surface area exposed to thedissolution medium (Kesisoglou et al., 2007; Wang, 2009). On
Table 2Equilibrium solubilities of different formulations of SIM (n = 3).
Sample Equilibrium solubility (lg/ml)
Pure SIM 15.2 ± 1.1SIM-HHMC 71.5 ± 2.4SIM-SHMC 72.3 ± 1.9SIM-FHMC 66.2 ± 3.1SIM-SHMC-2 69.0 ± 2.2
the other hand, the SIM loaded samples prepared by differentHMC matrices can also exhibit differences in their manner of disso-lution. The SIM-FHMC samples always had trouble in being re-leased within 60 min and the maximum accumulated amountremained at 51%. In contrast, the SIM-SHMC and SIM-HHMC sam-ples had some advantages in term of SIM dissolution enhancementcompared with SIM-FHMC, and the amounts of dissolved SIM fromthe SIM-SHMC and SIM-HHMC at a sampling time of 15 min wasfound to be about 82 and 74%, respectively. This is mainly due tothe fact that monodispersed SHMC (400–900 nm) and HHMC(700 nm) with smaller particle sizes possess short pore channelsand more pore entrances to release drug than agglomerated FHMC(1–4 lm) with larger particle sizes (shown in Fig. 2) (Ambrogiet al., 2007; Cauda et al., 2009). In addition, the difference in re-lease rate between SIM-SHMC and SIM-SHMC-2 could be attrib-uted to the pore diameters of SHMC (wBJH was 9.3 ± 2.6 and5.6 ± 1.1 nm for SHMC and SHMC-2, respectively). The smaller poreentrance hindered the drug escaping from the pore channels anddiffusing into the dissolution medium. Thus, fast drug release couldbe achieved by tuning the particle morphology, decreasing theparticle size and enlarging the pore size, which is consistent withprevious reports of other mesoporous materials (Wu et al., 2011;Jia et al., 2012). The dissolution profiles of SIM-loaded SHMC tabletand commercial tablet are shown in Fig. 8. The dissolution rate ofSIM from an SIM-loaded SHMC tablet was significantly improvedcompared with that of the commercial tablet. Remarkably, theamount of dissolved SIM in PBS at a sampling time of 20 minreached 86% for the SIM-SHMC tablet while the correspondingamount was 8% for the commercial tablet. This improved dissolu-tion may be largely attributed to the excellent ability of HMC todisperse drug molecules and reduce drug particle size. In addition,nearly 100% of the drug dissolved from the SIM-SHMC tablet with-in 10 min, as opposed to only 56% for the commercial tablet undersink conditions.
3.6. In vivo absorption study
The observations on the improved dissolution behaviors ofSIM-loaded SHMC prompted us to clarify the possible improve-ment in absorption of SIM and, so, the pharmacokinetic propertiesof SIM-loaded SHMC formulation and the reference formulationZocor � were assessed in beagle dogs under fasting conditions.

Fig. 8. Dissolution profiles of SIM from (.) SIM-SHMC tablet and (�) commercialtablet in PBS (pH 6.8); Dissolution profiles of SIM from (N) SIM-SHMC tablet and (j)commercial tablet in PBS (containing sodium dodecyl sulfate, pH 6.8). Each datapoint represents the mean ± S.D. of three determinations.
Fig. 9. Plasma concentration–time profiles of SIM following an oral dose of (j) acommercial tablet and (h) a SIM-SHMC tablet in beagle dogs. Each data pointrepresents the mean ± SD (n = 6).
Fig. 10. Plasma concentration–time profiles of SIMA following an oral dose of (j) acommercial tablet and (h) a SIM-SHMC tablet in beagle dogs. Each data pointrepresents the mean ± SD (n = 6).
Table 3Pharmacokinetic parameters of SIM after oral administration of SIM formulations tobeagle dogs (n = 6).
Parameter SIM-SHMC Control formulation
tmax (h) 1.08 ± 0.20* 1.58 ± 0.37Cmax (ng/ml) 106.62 ± 24.50* 57.30 ± 19.06AUC0?24h (ng h/ml) 289.95 ± 73.98* 210.06 ± 51.72Relative bioavailability (%) 138.42 ± 26.73
* P < 0.05 compared with control formulation.
Y. Zhang et al. / European Journal of Pharmaceutical Sciences 49 (2013) 864–872 871
After oral administration, SIM (inactive lactone) is known to berapidly hydrolyzed to its corresponding b-hydroxy acid (SIMA),which is a potent inhibitor of hydroxymethylglutaryl coenzyme A(HMG-CoA) reductase (Illingworth and Tobert, 2000). As a result,both SIM and SIMA in dog plasma were determined to evaluatethe in vivo pharmacokinetic behavior. The mean plasma concentra-tions versus time curves of SIM and SIMA following a single dose ofeach formulation are shown in Figs. 9 and 10, respectively. It couldbe observed that there are clear differences between the plasmaconcentration versus time curves following the administration ofboth types of tablets, indicating a clear difference in the rate andextent of SIM absorption. The major pharmacokinetic parametersof SIM and SIMA obtained by the non-compartmental analysisare summarized in Tables 3 and 4, respectively. It is worth notingthat the SIM-loaded SHMC formulation exhibited a significantly in-creased AUC0?24h and Cmax compared with the reference formula-tion (p < 0.05). Meanwhile, more rapid absorption was observed forthe SIM-loaded SHMC formulation compared with the reference
formulation (p < 0.05). Compared with the commercial tablet, therelative bioavailability judged from the AUC0?24h of SIM and SIMAwas found to be 138.42% and 163.55%, respectively (p < 0.05). Thepharmacokinetic parameters demonstrated a significant increasein the oral absorption of SIM when SHMC was used as a drug car-rier. SIM in the non-crystalline state had a limited particle size re-stricted by the nano-size pore channels and the highly dispersivedrug particles owing to the significantly increased surface areawhich all contributed to the faster dissolution rate of SIM fromthe SIM/carbon complex in gastrointestinal (GI) fluid. As a result,the oral absorption of the water-insoluble drug SIM was increased.
4. Conclusions
In this study, three different kinds of highly HMC with differentmorphologies, particle sizes and pore diameters were prepared toexplore the feasibility of using them as vehicles for water-insolubledrugs. The prepared HMC have a high drug loading (34.6–40.1%),higher than that of the HMS template. Solubility measurementshowed that SIM incorporation into the prepared HMC resultedin an approximately 4.5-fold increase in aqueous solubility in com-parison with raw SIM. In vitro dissolution testing showed that thedissolution rate of SIM released from monodispersed SHMCnanomatrix was significantly faster compared with that of crystal-line SIM and other SIM-loaded composites. In addition, a signifi-cantly shorter tmax and higher Cmax and larger AUC0?24h wereobtained from the SIM-loaded SHMC formulation compared withthe marketed tablet. Thus, monodispersed SHMC nanomatrix ap-pear to be a more promising candidate as a new oral drug deliveryvehicle providing a rapid drug release and enhanced oralbioavailability.

Table 4Pharmacokinetic parameters of SIMA after oral administration of SIM formulations tobeagle dogs (n = 6).
Parameter SIM-SHMC Control formulation
tmax (h) 1.42 ± 0.38* 1.93 ± 0.22Cmax (ng/ml) 63.19 ± 17.80* 30.10 ± 15.26AUC0?24h (ng h/ml) 168.23 ± 47.42* 104.56 ± 36.50Relative bioavailability (%) 163.55 ± 14.59
* P < 0.05 compared with control formulation.
872 Y. Zhang et al. / European Journal of Pharmaceutical Sciences 49 (2013) 864–872
Acknowledgements
Financial support for this work was provided by the NationalBasic Research Program of China (973 Program) (No.2009CB930300). We would like to thank Dr. David Jack for correct-ing language.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.ejps.2013.05.031.
References
Ambrogi, V., Perioli, L., Marmottini, F., Giovagnoli, S., Esposito, M., Rossi, C., 2007.Improvement of dissolution rate of piroxicam by inclusion into MCM-41mesoporous silicate. Eur. J. Pharm. Sci. 32, 216–222.
Ambrogi, V., Perioli, L., Marmottini, F., Accorsi, O., Paganoa, C., Riccia, M., Rossi, C.,2008. Role of mesoporous silicates on carbamazepine dissolution rateenhancement. Micropor. Mesopor. Mater. 113, 445–452.
Barbé, C., Bartlett, J., Kong, L., Finnie, K., Lin, H.Q., Larkin, M., Calleja, S., Bush, A.,Calleja, G., 2004. Silica particles: a novel drug-delivery system. Adv. Mater. 16,1949–1966.
Bastin, R.J., Bowker, M.J., Slater, B.J., 2000. Salt selection and optimisationprocedures for pharmaceutical new chemical entities. Org. Proc. Res. Dev. 4,427–435.
Blagden, N., Matas, M., Gavan, P.T., York, P., 2007. Crystal engineering of activepharmaceutical ingredients to improve solubility and dissolution rates. Adv.Drug Deliv. Rev. 59, 617–630.
Cauda, V., Muhlstein, L., Onida, B., Bein, T., 2009. Tuning drug uptake and releaserates through different morphologies and pore diameters of confinedmesoporous silica. Micropor. Mesopor. Mater. 118, 435–443.
Charnay, C., Bégu, S., Tourné-Péteilh, C., Nicole, L., Lerner, D.A., Devoisselle, J.M.,2004. Inclusion of ibuprofen in mesoporous templated silica: drug loading andrelease property. Eur. J. Pharm. Biopharm. 57, 533–540.
Graeser, K.A., Strachan, C.J., Patterson, J.E., Gordon, K.C., Rades, T., 2008.Physicochemical properties and stability of two differently preparedamorphous forms of simvastatin. Cryst. Growth Des. 8, 128–135.
Heikkilä, T., Salonen, J., Tuura, J., Hamdy, M.S., Mul, G., Kumar, N., Salmi, T., Murzin,D.Y., Laitinen, L., Kaukonen, A.M., Hirvonen, J., Lehto, V.P., 2007. Mesoporoussilica material TUD-1 as a drug delivery system. Int. J. Pharm. 331, 133–138.
Hörter, D., Dressman, J.B., 2001. Influence of physicochemical properties ondissolution of drugs in the gastrointestinal tract. Adv. Drug Deliv. Rev. 46, 75–87.
Huanga, L.F., Tong, W.Q.T., 2004. Impact of solid state properties on developabilityassessment of drug candidates. Adv. Drug Del. Rev. 56, 321–334.
Illingworth, D.R., Tobert, J.A., 2000. A review of clinical trials comparing HMG-CoAreductase inhibitors. Clin. Ther. 16, 1004–1010.
Jaganathan, H., Godin, B., 2012. Biocompatibility assessment of Si-based nano- andmicro-particles. Adv. Drug Deliv. Rev. 64, 1800–1819.
Jia, L., Shen, J., Li, Z., Zhang, D., Zhang, Q., Duan, C., Liu, G., Zheng, D., Liu, Y., Tian, X.,2012. Successfully tailoring the pore size of mesoporous silica nanoparticlesexploitation of delivery systems for poorly water-soluble drugs. Int. J. Pharm.439, 81–91.
Kesisoglou, F., Panmai, S., Wu, Y., 2007. Nanosizing–Oral formulation developmentand biopharmaceutical evaluation. Adv. Drug Del. Rev. 59, 631–644.
Kim, T.W., Slowing, I.I., Chung, P.W., Lin, V.S.Y., 2011. Ordered mesoporous polymer-silica hybrid nanoparticles as vehicles for the intracellular controlled release ofmacromolecules. ACS Nano. 5, 360–366.
Lee, H.I., Kim, J.H., You, D.J., Lee, J.E., Kim, J.M., Ahn, W.S., Pak, C., Joo, S.H., Chang, H.,Seung, D., 2008. Rational synthesis pathway for ordered mesoporous carbonwith controllable 30- to 100-angstrom pores. Adv. Mater. 20, 757–762.
Li, Y., Wang, T., Wang, J., Jiang, T., Cheng, G., Wang, S., 2011. Functional andunmodified MWNTs for delivery of the water-insoluble drug carvedilol – adrug-loading mechanism. Appl. Surf. Sci. 257, 5663–5670.
Liang, C., Li, Z., Dai, S., 2008. Mesoporous carbon materials: synthesis andmodification. Angew. Chem. 47, 3696–3717.
Lipinski, C.A., Lombardo, F., Dominy, B.W., Feeney, P.J., 1997. Experimental andcomputational approaches to estimate solubility and permeability in drugdiscovery and development setting. Adv. Drug Deliv. Rev. 23, 3–25.
Liu, R., Shi, Y., Wan, Y., Meng, Y., Zhang, F., Gu, D., Chen, Z., Tu, B., Zhao, D., 2006.Triconstituent co-assembly to ordered mesostructured polymer-silica andcarbon-silica nanocomposites and large-pore mesoporous carbons with highsurface areas. J. Am. Chem. Soc. 128, 11652–11662.
Liu, R., Wang, X., Zhao, X., Feng, P., 2008. Sulfonated ordered mesoporous carbon forcatalytic preparation of biodiesel. Carbon 46, 1664–1669.
Liu, X., Li, L., Du, Y., Guo, Z., Ong, T.T., Chen, Y., Ng, S.C., Yang, Y., 2009. Synthesis oflarge pore-diameter SBA-15 mesostructured spherical silica and its applicationin ultra-high-performance liquid chromatography. J. Chromatogr. A 1216,7767–7773.
Manly, C.J., Louise-May, S., Hammer, J.D., 2007. The impact of informatics andcomputational chemistry on synthesis and screening. Drug Discov. Today 6,1101–1110.
Mellaerts, R., Mols, R., Jammaer, J.A.G., Aerts, C.A., Annaert, P., Humbeeck, J.V.,Mooter, G.V., Augustijns, P., Martens, J.A., 2008. Increasing the oralbioavailability of the poorly water soluble drug itraconazole with orderedmesoporous silica. Eur. J. Pharm. Biopharm. 69, 223–230.
Qian, K.K., Bogner, R.H., 2011. Application of mesoporous silicon dioxide and silicatein oral amorphous drug delivery systems. J. Pharm. Sci. 101, 444–463.
Sadighi, A., Ostad, S.N., Rezayat, S.M., Foroutan, M., Faramarzie, M.A., Dorkoosh, F.A.,2012. Mathematical modelling of the transport of hydroxypropyl-b-cyclodextrin inclusion complexes of ranitidine hydrochloride and furosemideloaded chitosan nanoparticles across a Caco-2 cell monolayer. Int. J. Pharm. 17,479–488.
Salonen, J., Laitinen, L., Kaukonen, A.M., Tuura, J., Björkqvist, M., Heikkilä, T., Vähä-Heikkilä, K., Hirvonen, J., Lehto, V.P., 2005. Mesoporous silicon microparticlesfor oral drug delivery: loading and release of five model drugs. J. Control.Release 108, 362–374.
Tan, A., Davey, A.K., Prestidge, C.A., 2011. Silica-lipid hybrid (SLH) versus non-lipidformulations for optimising the dose-dependent oral absorption of celecoxib.Pharm. Res. 28, 2273–2287.
Tang, F., Li, L., Chen, D., 2012. Mesoporous silica nanoparticles: synthesis,biocompatibility and drug delivery. Adv. Mater. 24, 1504–1534.
Thomas, M.J.K., Slipper, I., Walunj, A., Jain, A., Favretto, M.E., Kallinteri, P.,Douroumis, D., 2009. Inclusion of poorly soluble drugs in highly orderedmesoporous silica nanoparticles. Int. J. Pharm. 387, 272–277.
Vasconcelos, T., Sarmento, B., Costa, P., 2007. Solid dispersions as strategy toimprove oral bioavailability of poor water soluble drugs. Drug Discov. Today 12,1068–1075.
Vashist, S.K., Zheng, D., Pastorin, G., Al-Rubeaan, K., Luongf, J.H.T., Sheua, F.S., 2011.Delivery of drugs and biomolecules using carbon nanotubes. Carbon 49, 4077–4097.
Wang, S., 2009. Ordered mesoporous materials for drug delivery. Micropor.Mesopor. Mater. 117, 1–9.
Wang, F., Hui, H., Barnes, J., Barnett, C., Prestidge, C.A., 2009a. Oxidized mesoporoussilicon microparticles for improved oral delivery of poorly soluble drugs. Mol.Pharm. 7, 227–236.
Wang, Y., Zhang, F., Wang, Y., Ren, J., Li, C., Liu, X., Guo, Y., Lu, G., 2009b. Synthesis oflength controllable mesoporous SBA-15 rods. Mater. Chem. Phys. 115, 649–655.
Wang, X., Liu, P., Tian, Y., 2011. Ordered mesoporous carbons for ibuprofen drugloading and release behavior. Micropor. Mesopor. Mater. 142, 366–385.
Wani, A., Muthuswamy, E., Savithra, G.H.L., Mao, G., Brock, S., Oupicky, D., 2012.Surface functionalization of mesoporous silica nanoparticles controls loadingand release behavior of mitoxantrone. Pharm. Res. 29, 2407–2418.
Wu, C., Wang, Z., Zhi, Z., Jiang, T., Zhang, J., Wang, S., 2011. Development ofbiodegradable porous starch foam for improving oral delivery of poorly watersoluble drugs. Int. J. Pharm. 403, 162–169.
Yang, P., Quan, Z., Lu, L., Huang, S., Lin, J., 2008. Luminescence functionalization ofmesoporous silica with different morphologies and applications as drugdelivery systems. Biomaterials 29, 692–702.
Yu, L., 2001. Amorphous pharmaceutical solids: preparation, characterization andstabilization. Adv. Drug Deliv. Rev. 48, 27–42.
Zhang, Y., Wang, J., Bai, X., Jiang, T., Zhang, Q., Wang, S., 2012. Mesoporous silicananoparticles for increasing the oral bioavailability and permeation of poorlywater soluble drugs. Mol. Pharm. 9, 505–513.
Zhao, D., Feng, J., Huo, Q., Melosh, N., Fredrickson, G.H., Chmelka, B.F., Stucky, G.D.,1998. Triblock copolymer syntheses of mesoporous silica with periodic 50–300angstrom pores. Science 279, 548–552.


















