
High Performance Liquid Chromatography HPLC · HPLC heute kurze Säulen, 2 - 5 mm ID, Stationäre...
38
High Performance Liquid Chromatography HPLC Skript zur Vorlesung „Grundlagen der Analytik“ im MSc-Studiengang „Analytik“ Prof. C. Vogt, Leibniz Universität Hannover 3.2.2007
Transcript of High Performance Liquid Chromatography HPLC · HPLC heute kurze Säulen, 2 - 5 mm ID, Stationäre...

High Performance Liquid Chromatography
HPLCSkript zur Vorlesung „Grundlagen der Analytik“
im MSc-Studiengang „Analytik“
Prof. C. Vogt, Leibniz Universität Hannover 3.2.2007

Gliederung
Kurzportrait der Methode
Stationäre Phasen
Mobile Phasen
RetentionsmechanismenNormalphasen-Chromatographie (NP)Umkehrphasen-Chromatographie (RP)
GeräteaufbauVorbereitung des EluentenPumpenInjektorSäuleDetektoren

HPLC heute
kurze Säulen, 2 - 5 mm ID, Stationäre Phase mit sehr kleinem Teilchendurchmesser (3 - 10µm) große Auswahl an stationären Phasenrelativ hohe Eingangsdrücke (bis 400 bar) reproduzierbare Injektion geringer Probemengengeeignete Durchfluss-Detektoren mit sehr kleinen Detektorzellvoluminahoher Automatisierungsgradhohe Auflösung
Moderne Flüssigchromatographie ist gekennzeichnet durch
Partikelgröße 5-10 µmInnendurchmesser 1-5 mmLänge 5-25 cm
Moleküle der mobilen Phase können sowohl mit der stationären Phase, als auch mit den Probemolekülen in Wechselwirkung treten – nicht inert !, kann als variable Einflussgröße für den Trennprozess genutzt werden

Vergleich HPLC - GC
Probenmoleküle haben in Flüssigkeiten kleinere Diffusionskoeffizienten als in Gasen (Nachteil, weil Austausch der Probenmoleküle zwischen mobiler und stationärer Phase in LC viel langsamer erfolgt)
Viskosität von Flüssigkeiten ist größer als die von Gasen (Nachteil –nur kleinere Fließgeschwindigkeiten möglich).
Flüssigkeiten lassen sich im Gegensatz zu Gasen praktisch nicht komprimieren (Vorteil – Kontrollparameter entfällt)
Signale der Analyten sind in LC breiter → Trennstufenzahlen und Auflösung sind geringer (Ursachen siehe van Deemter Gleichung)

Stationäre Phasen
Bei Verwendung als Trägermaterial wird durch chemische Reaktionen die Oberfläche des Silikagels modifiziert → Hydroxy-Gruppen werden durch Alkyl-, Phenyl-, Amino-, Cyano-Gruppen u.s.w. ersetzt→ dadurch gezielte Einstellung chromatographischer Eigenschaften möglich
Sehr feines Silikagel oder anderes Trägermaterial mit möglichst einheitlicher Korngröße entweder direkt als adsorbierende stationäre Phase (Adsorbtions-chromatographie) oder als Trägermaterial (Verteilungschromatographie)
Säulen mit kleinerem Durchmesser benötigen bei gleicher Fließgeschwindigkeit der mobilen Phase weniger Lösungsmittel → Mikro-HPLC
Höhere Trennstufenzahl erreichbar durch kleinere Teilchen der stationären PhaseKleinere Teilchen lassen mehr Austauschschritte zwischen stationärer und mobiler Phase zu

Stationäre Phasen
Irregulär geformteSilica-Partikel
SiO2 (Kieselgel, Kieselgur)Al2O3ZrO2TiO2PolymereGraphitierter KohlenstoffMagnesiumsilikat (Florisil)
Organischer Polymermonolith UNO SspherischeSilica-Partikel
Organischer Polymermonolith CIM DiskMonolith auf Silica-Basis (Chromolith)

Stationäre PhasenEinteilung nach Polarität der stationären PhaseA stationäre Phase ist polarer als die mobile Phase
→ Normalphase NP (normal-phase), -OH, -NH2
B stationäre Phase ist unpolarer als die mobile Phase→ Umkehrphase RP (reversed-phase), C8, C18, Phenyl
70 - 80% aller flüssigkeitschromato-graphischen Trennungen werden heute in "reversed-phase" Technik ausgeführt
OH
OH
OH
OH
OH
OHOH
OHOH
OH
OH
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
Normal-Phasen-Chromatographie
Umkehr-Phasen-Chromatographie
Polare stationäre Phase Unpolare stationäre Phase
UnpolarerEluent
PolarerEluent

Stationäre Phasen – Bonded phase
C-18
C8
C1
PFP
Oxy-Phenyl
C18, C8, C5, C1, Phenyl, CN, NH2
Schirmen polare Silica-OF vom Analyt-molekül ab
Starke polare WW an der Silica-OFwerden durch schwache Dispersions-kräfte ersetzt
ca. 21 Å Längeall-trans-KonformationMolekülvolumen ca.700 Å3
max. Bindungsdichte2.5 Ketten/nm2
Oder 4.1 µmol/m2 aufflacher OF

Stationäre Phasen – end-capping
nicht umgesetzte Silanolgruppen
mit TMS umgesetzte Silanolgruppen
Sekundäre Umsetzung der verbliebenen Silanolgruppen (end-capping) mit Trimethylchlorosilan

Stationäre Phasen – SchichtdickeAnordnung der gebundenen Phase auf der
Silica-Oberfläche
Idealfall
Realfall
Anzahl Kohlenstoffatome in Kette
Experimentell ermitteltTheoretisches Maximum
Dic
ke [Å
]

Stationäre Phasen
Trennung hängt von Menge und Typ der Adsorptionsplätze bzw. WW-Plätze an der Oberfläche der stationären Phase ab → große Oberfläche wichtig
Parameter des Phasenmaterials, die Retention entscheidend beeinflussen
Partikelgröße → 3 – 10 µm
Partikelgrößenverteilung → so gering wie möglich, meist ≤ d ± 10%
Porendurchmesser → 70 – 300 Å
Oberfläche → 50 – 250 m2/g
Dichte der gebundenen Phase (Anzahl Adsorptionsplätze je Einheitsfläche) → 1 – 5 Plätze je nm2

Retentionsmechanismen
sterischer Anspruch eines Analyten und seine Fähigkeit, mit den Hydroxylfunktio-nen des Kieselgels oder des Al2O3 Wasserstoffbrückenbindungen einzugehen, sind zusätzlich entscheidend für das Maß der Adsorption auf der stationären Phase
NP-Phasen → polare Dipol-Dipol-WW und sterische Effekte dominieren
Competition-Modell → gesamte Oberfläche der stationären Phase ist mit einer Monoschicht aus Solvensmolekülen belegt → Retention der Analytenerfolgt durch kompetitive Verdrängung der LM-Moleküle von der Adsorbens-oberfläche
Zwei Modelle für Mechanismus
Solvent-Interaction-Modell → Bildung einer primären und einer sekundären Schicht aus Eluensmolekülen → ZSS dieser Doppelschicht hängt vom Anteil der polaren Komponente in der mobilen Phase ab → Retentionsvorgang erfolgt durch Wechselwirkung der Analyten (Assoziation oder Verdrängung) mit der zweiten Schicht der Solvensmoleküle → Substanzmoleküle haben somit keinen Kontakt mit der Oberfläche der stationären Phase

RetentionsmechanismenRP-PhasenTrennmechanismus nicht allein durch einfache polare WW beschreibbar, da Kräfte, die zwischen den Analytmolekülen und der unpolaren stationären Phase auftreten nicht ausreichen, um das Maß der Retention auf RP-Materialien zu erklären → Kombination aus Adsorption und Verteilung
Solvophobe Theorie → unpolare stationäre Phase verhält sich wie Feststoff →Retention der Analyten aufgrund Adsorption über solvophobe Effekte an der OF der Umkehrphase → Adsorption steigt mit zunehmender OF-Spannung des Eluenten → hierdurch vermindern die Moleküle ihre Kontaktfläche mit der mobilen Phase → Retention erfolgt deshalb primär aufgrund solvophober WW mit der mobilen Phase und nicht aufgrund polarer WW mit der stationären Phase
Verteilungsmodell → chemisch modifizierte OF des Trägermaterials ähneltflüssiger stationärer Phase, in der Analyte vollständig von den an die Hydroxyl-gruppen gebundenen Alkylketten umgeben sind → keine Isotropie - durchAnbindung an die Kieselgeloberfläche ist eindeutige Vorzugsrichtung gegeben
mit zunehmender Alkylkettenlänge des RP-Materials steigt Verteilungscharakter der Trennung, mit abnehmender Kettenlänge dominiert ein Adsorptionsmechanismus
Zwei Modelle für Mechanismus

Eigenschaften der mobilen Phase
MischbarkeitEinphasiges System muss sich bildenPolare und unpolare Lösungsmittel lassen sich gar nicht, oder nur in bestimmten Verhältnissen mischen (Bsp. Methanol und Hexan)
SiedepunktSiedepunkte < 100°C, damit leichter durch Destillation reinigbarSiedepunkte < 40°C ungünstig, da beim Ansaugen durch die Pumpe Verdampfen auftreten kann (Gasblasenbildung).
Detektor-Kompatibilität (z.B. UV-Transparenz für UV-Detektor oder elektrochemische Inertheit für Amperometrischen Detektor)
PolaritätLöslichkeit der Probenbestandteile
Reinheit, Viskosität, Preis, …

Mobile Phase
LM MW Siede-punkt
Brechungs-index RI
UV Viskosität µ
[°C] [nm] [cP] [Debye]
Acetonitril 41 82 1.341 195 0.358 3.37Dioxan 88 101 1.421 215 1.26 0.45Ethanol 46 78 1.359 205 1.19 1.68Methanol 32 65 1.326 205 0.584 1.66Isopropanol 60 82 1.375 205 2.39 1.68Tetrahydrofuran 72 66 1.404 215 2.20 1.70Wasser 18 100 1.333 185 1.00 1.84
NP-Chromatographie → nicht-polare mobile PhasenRP-Chromatographie → polare Eluenten (Gemische aus Wasser und polaren
organischen LM)
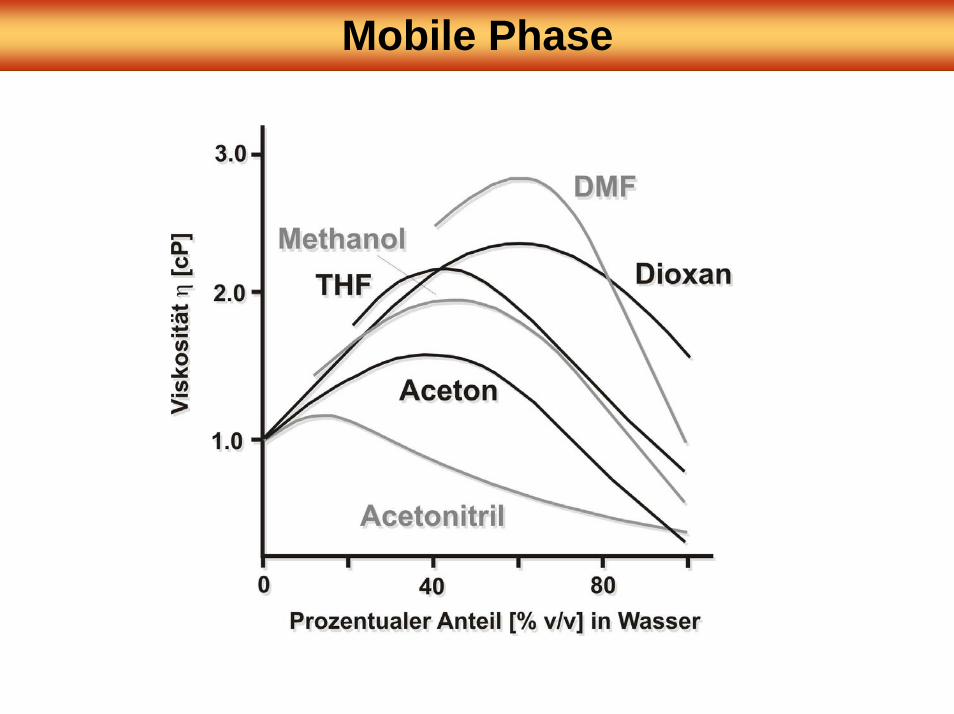
Mobile Phase

Normalphasen-Chromatographie
Pentan/Hexan < Cyclohexan < Toluen < Trichlormethan < Dichlormethan< Tetrahydrofuran < Dioxan < Acetonitril < Ethanol < Methanol << Wasser
gesättigte Kohlenwasserstoffe < Olefine < aromatische Kohlenwasserstoffe = halogenierte Kohlenwasserstoffe < organische Sulfide < Ether < Nitroverbin-dungen < Ester = Aldehyde = Ketone < Alkohole = Amine < Sulfone < Amide< Carbonsäuren
Stationäre Phasenporöse anorganische Adsorbenzien, wie Kieselgel SiO2 oder Al2O3mit polaren Hydroxylgruppen an der OberflächeMobile PhasenPentan, Hexan, Tetrahydrofuren, Dichlormethan, Methanol, Acetonitril
Elutrope Reihe → Lösungsmittel werden nach steigender Elutionskraft geordnetJe größer die Elutionskraft ist desto kleiner wird der Verteilungskoeffizient und desto schneller wandern die Analyten durch das chromatographische Bett Reihe jeweils abhängig von Polarität der stationären Phase
NP-Chromatographie besonders geeignet für Analyte, die sich in unpolarenLM sehr gut lösen → Trennung von Isomeren und von Substanzklassen, deren Elutionsreihenfolge wie folgt ist (geordnet nach steigender Retentionszeit)

Umkehrphasen-Chromatographie
elutrope Reihe und Elutionsreihenfolge der getrennten Verbindungsklassen zeigen bei Umkehrphasen inversen Verlauf verglichen mit NP-Chromatographie
Methode zur Trennung neutraler Verbindungen, die sich in Wasser oder anderen polaren LM (z.B.Acetonitril oder Methanol) lösen
Pharmazeutische IndustrieSteroide, Vitamine, Beta-Blocker und viele andere Wirkstoffe
UmweltanalytikPestiziden
BiochemieBiopolymere, wie Proteine, Peptide, Nukleinsäuren oder Oligosaccharide
Stationäre Phasenporöse anorganische Adsorbenzien auf Kieselgelbasis SiO2 mit chemisch modifizierten Hydroxylgruppen → Alkylketten unterschiedlicher LängeMobile PhasenWasser, Methanol, Acetonitril, Tetrahydrofuran

Elutionstechniken
Gradienten Elutionzwei Elutionsmittel werden stufenlos gemischtAnwendung, wenn Verbindungen der Probe große Unterschiede in derPolarität aufweisen
Isokratische Elutioneine Verbindung oder konstante Mischung mehrerer Verbindungen als mobile Phase genutzt
Elutionskraft der mobilen Phase wird so gesteigert, dass die Konzentration der stärkeren Eluentien während der chromatographischen Trennung erhöht wird
Änderung der Elutionskraft der mobilen Phase während der Laufzeit des Chromatogramms → schärfere peaks und kürzere Laufzeiten v.a. bei komplexen Gemischen von stark unterschiedlich polaren Substanzen
Gradientenerzeugunghochdruckseitig (nach Pumpe) oder niederdruckseitig (vor Pumpe)

Aufbau einer HPLCEntgaste, filtrierte mobile Phase
Förderung durch PumpenPulsationsdämpfung
Mischung der Eluentkomponenten
Injektion der Probe (in mit mobiler Phase mischbarem LM)
Reinigung auf Vorsäule
Trennung auf chromatographischer Säule(eventuell themostatierbar)
Derivatisierung
Detektion
Fraktionssammlung
Datenverarbeitung

Vorbereitung des Eluenten
Filtrieren zum Entfernen feinster Partikel → verbleiben auf stationärer Phaseund ändern deren Eigenschaften mit der Zeit
Ausgasen gelöster Gase (N2, O2) im Bereich der Pumpe oder der Mischkammer bei Kompression möglich → Flussschwankungen → Verschlechterung derReproduzierbarkeit
Ausgasen gelöster Gase (N2, O2) im Bereich des Detektors beim Entspannen→ Gasblasen in Detektorzelle → unruhige Grundlinien oder Spikes
Bei Einsatz eines Fluoreszenzsdetektors kann Sauerstoff als Quencher wirken→ Unkontrollierte Verringerung des Analytsignals
EntgasungstechnikenUltraschall off-lineVakuum-Entgasung on-lineHelium-Entgasung on-line

Pumpen für die Förderung des Eluenten
große Flusskonstanzgroßer Bereich an Flussratenschneller Eluentwechselbeständig gegenüber mobiler Phasewartungsfreundlich
Anforderungen
Schädigungen durchhohe Chloridkonzentrationenstark komplexierende Puffer-SubstanzenChlorierte KW + UV → HClstarke AlkalienHohe Salzfrachten

Pumpen für die Förderung des Eluenten
Pumpenköpfe → korrosionsbeständige EdelstähleKolben → SaphirEin- und Auslassventile → Rubin- oder Saphirkugeln
Korrosionsbeständig(er) → Polyetheretherketon PEEK, Teflon oder Titan
direkt nach Benutzung (vor Abschalten) gründlich mit inertemLösungsmittel spülen !
In Praxis Einsatz von Doppelkolbenpumpe → nockenwellengesteuertePumpenköpfe pumpen zeitversetzt, dadurch nahezu vollkommene Pulsationsdämpfung und sehr konstante Fließgeschwindigkeit
Maximaldruck 400 bar
Fließgeschwindigkeiten 0.1 – 10 mL/min

Einfluss der Fließrate
0.3 mL/min 1.2 mL/min 3.6 mL/min
Trennung von Phenylthiohydanthoin-Derivaten der Aminosäuren Asn, Asp, Glu, Ala, His, Pro, Ile und Phe
Säule: 3.2 mm x 25 cm, Stationäre Phase: LiChrosorb SI 60, 5 µmMobile Phase: tert. Butylmethylether, Detektion: UV 254 nm
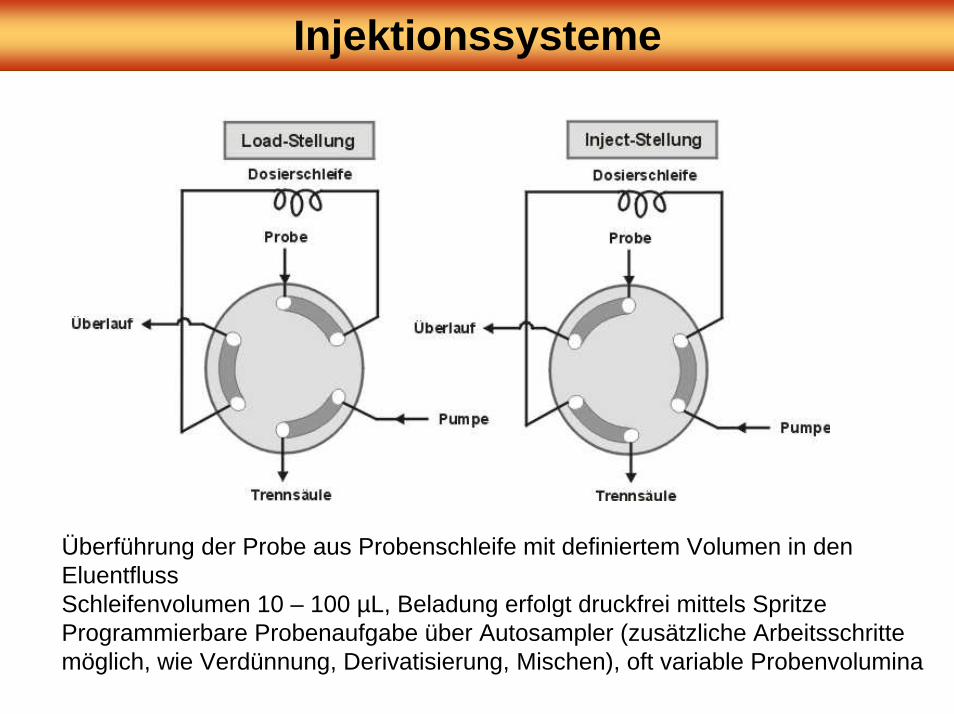
Injektionssysteme
Überführung der Probe aus Probenschleife mit definiertem Volumen in den EluentflussSchleifenvolumen 10 – 100 µL, Beladung erfolgt druckfrei mittels SpritzeProgrammierbare Probenaufgabe über Autosampler (zusätzliche Arbeitsschritte möglich, wie Verdünnung, Derivatisierung, Mischen), oft variable Probenvolumina

Trennsäule
Druck- und korrosionsbeständiger Chrom-Nickel-Molybdän-StahlL = 125 – 300 mmiD = 2 – 5 mm für analytische TrennungeniD = 10 – 25 mm für semipräparative Trennungen
Trockenpack- oder Druckfiltrationsmethode (engl. dry-fill procedure, wet-fillprocedure oder slurry-packing procedure)
kleine Teilchen haben großen Oberflächenenergie-Masse-Verhältnisse→ neigen zur Zusammenballung → nur mit Lm wird kompakteschromatographisches Bett in der Säule erhalten

Detektionssysteme
Wichtig in der HPLCVolumen der Messzelle muss so klein als irgend möglich gehalten werden, damit keine Rückvermischung schon getrennter Substanzen erfolgen kann (Peakverbreiterung)
Universelle und selektive DetektorenDetektoren, die die Änderung einer physikalischen Größe der gesamten mobilen Phase mit Analyt messen (Brechungsindex) oder die selektiv nur die Eigenschaften der gelösten Stoffe messen
Wichtigste Detektionssysteme für HPLCUV/Vis, Fluoreszenz, RI, MS, EC (Leitfähigkeit), Lichtstreuung
Wichtigste Kriterien zur Beurteilung eines DetektorsNachweisgrenze, Linearer Messbereich, Selektivität, Empfindlichkeit, Robustheit, Preis

Detektionssysteme
Detektor NWG (ng) (mol/L)
UV-Absorption 0.1 – 1 10-6 – 10-7
Brechungsindex 100 – 1000 10-4 – 10-5
Elektrochemie 0.01 – 1 10-6 – 10-9
Fluoreszenz 0.001 - 0.01 10-6 - 10-8
Leitfähigkeit 0.5 – 1 10-5 – 10-9
Massenspektrometrie 0.1 – 1 10-6 – 10-8

UV-Vis-Detektor
Quecksilber- oder Deuteriumlampen als Strahlungsquellen
Quecksilberdampflampe erzeugtLinienspektrum - 254 nm Deuteriumlampe erzeugt kontinuierliches Spektrum zwischen 190 und 380 nm
Im UV-Detektor durchstrahlt die Strahlungs-quelle eine Quarzzelle, die vom Eluentenpermanent durchströmt wird → dabei wird die Änderung der Absorption von einer Photozelle gemessen und aufgezeichnet
Festwellenlängendetektor
am häufigsten eingesetztbreites Einsatzgebiet, da die meisten organischen Verbindungen UV-Licht absorbierenDetektor außerdem relativ unempfindlich gegen Fluss-und TemperaturschwankungenFlächen abh. von Konzentration und Extinktionskoeffizienten

UV-Vis-Detektor
1 Deuterium-Lampe2 Spiegel3 Probenzelle4 Gitter5 Diodenarray6 Verstärker
Detektorzelle
Absorption wird mit Reihe von Photodioden, von denen jede bei einer anderen Wellenlänge misst, in größerem Bereich gemessen (Spektrum)Polychromatisches Licht von der UV-Strahlenquelle durch Messzelle → einzelne Wellenlängen werden unterschiedlich absorbiert → Licht in Polychromator →spektrale ZerlegungInformationen zur Peakreinheit, Identifizierung von Substanzen, Spektren-Subtraktion, Spektrenbibliotheken

Fluoreszenz-Detektor
1 Xenon-Lampe oder Laser2 Monochromator (Filter oder
Gitter)3 Fließzelle
hohe Selektivität und ausgezeichnete Nachweisempfindlichkeit
Anwendung aber beschränkt auf fluoreszierende Verbindungen (Alternative → Derivatisierung)
UV-Strahlungsquelle → Filter oder Monochromatoren → eine Wellenlänge als Anregungswellenlänge auswählenEnergieabgabe im Bereich längerer Wellenlängen als LichtPhotomultiplier rechtwinklig zur Strahlenquelle angeordnet → nur durchFluoreszenz ausgestrahltes Licht erreicht den Photomultiplier
Intensität abhängig von Konzentration, Extinktionskoeffizient und Quanten-ausbeute des Fluorophors

Fluoreszenzdetektion
Beispiel: Trennung von (+)-FLEC-D/L-Carnitin-Diastereomeren
RP 18/Ultraspere ODS240 mm x 4.6 mm20 µL Injektionsvolumen
Fluoreszenzdetektionλex 260 nm / λem 310 nm
Eluent: 50 mM Triethanol-Aminophosphat / ACNpH 2.60
A 71.5 : 28.5B 75.0 : 25.0

Brechungsindex-Detektor
Brechungsindex (RI)-Detektor Deflektion eines Lichtstrahls ändert sich, wenn sich ZSS der Probe inMesszelle relativ zu der in der Ver-gleichszelle ändert
DeflektionstypDetektor
Vorteilerobust, wenig anfällig gegen Ver-Schmutzungen, Messung über voll-Ständigen RI-BereichNachteilrelativ geringe Empfindlichkeit
ReflektionstypFresnel-Prinzip – Lichtstrahl wird Von Flüssigkeits-Glas-Interface in dieMesszelle hinein reflektiertVorteile – sehr empfindlich, geeignetfür Miniaturisierung, preiswertNachteil – extrem empfindlich Gegenüber ∆p und ∆T

GliederungBeispiel: Trennung physiologisch relevanter γ-Betaine
ohne Derivatisierung
Säule: LiChrosper 100 NH2250 mm x 4 mm, 5µm PartikelEluent: 50 mM NaH2PO4,0.5 mL/min25°C, 20 µL Injektionsvolumen
A RI-Detektion von je 1 µg Substanz je Komponente
B UV-Detektion bei 205 nm von 0.1 µg Substanz je Komponente
C RI-Detektion von je 0.1 µg Substanz je Komponente

Lichtstreudetektor
System besteht aus drei Teilen: Zerstäuber, Drift-Röhre und Lichtstreu-Zelle
(1) Probe aus Trennstrecke wird mit Zerstäuber (N2) in Aerosol umgewandelt
(2) LM wird verdampft (∆T) in Driftröhre
(3) Gebildete Tröpfchen streuenLaserlicht am Ende der Driftröhre
(4) Gestreutes Licht wird im 90°-Winkel oder bei mehreren Winkelndetektiert

LichtstreudetektorProbleme
a) Verdampfung in der Driftröhre für größere Tröpfchen schwieriger – nicht verdampftes LM erhöht das Basislinienrauschen
b) Flüchtige Verbindungen im Aerosol verdampfen in der Driftröhre und werden nicht mehr detektiert.
c) Intensität des Signals erhöht sich mit 6. Potenz der Molekülgröße – ein Teilchen mit der 10fachen Größe in Bezug auf ein anderes erzeugt ein Signal, das 106 Mio mal größer ist (Gefahr von Fehlinter-pretationen hoch)Nachweisgrenze bei Partikeldurchmessern von ca. 1/20 der verwendeten Lichtwellenlänge Detektion von Partikeln mit wenigen nm Größe (1-10 nm) – entspricht MW von wenigen Tausend Dalton
d) Staub kann erheblich störenf) optimale Gasflussrate muss für jede Trennmethode ermittelt werden, bei der das beste S/N-Verhältnis vorliegt

Beispiel 1
Trennung von Alkylphenolpolyethoxylaten in technischen GemischenSäule: NH2 (250 x 4 mm); Detektion: UV 277 nm; Fluß: 1 ml/min;
Eluent: Gradiententechnik von 100 % n-Hexan/2-Propanol 9:1 zu 100 % 2-Propanol/Wasser 9:1 (1 % pro min)
Probe: Präwozell N - Produkte, n: mittlere Anzahl der Ethoxylatgruppen
n = 2-100
0 20 40 60 80
0,00,20,40,60,81,0
Präwozell N20n=20
Nonylphenolpolyethoxylate
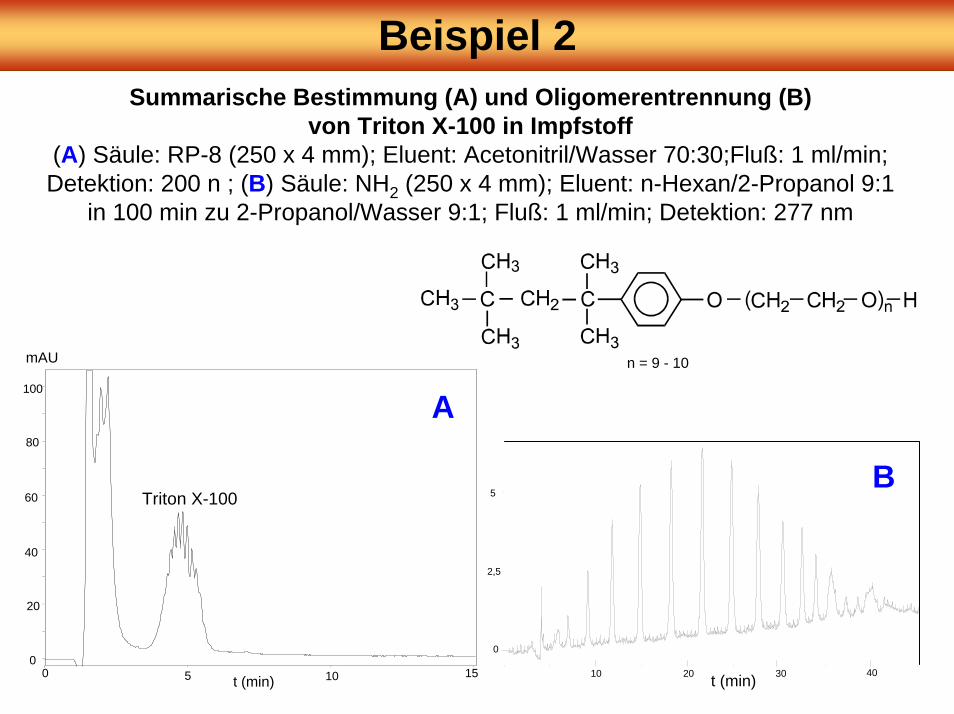
Beispiel 2Summarische Bestimmung (A) und Oligomerentrennung (B)
von Triton X-100 in Impfstoff(A) Säule: RP-8 (250 x 4 mm); Eluent: Acetonitril/Wasser 70:30;Fluß: 1 ml/min; Detektion: 200 n ; (B) Säule: NH2 (250 x 4 mm); Eluent: n-Hexan/2-Propanol 9:1
in 100 min zu 2-Propanol/Wasser 9:1; Fluß: 1 ml/min; Detektion: 277 nm
0 5 10 150
20
40
60
80
100
t (min)
mAU
Triton X-100
A
n = 9 - 10
5
2,5
0
10 20 30 40
B
t (min)
A
B


















