
Heterogeneous nucleation in the crystallization of high ...
205
University of Massachusetts Amherst University of Massachusetts Amherst ScholarWorks@UMass Amherst ScholarWorks@UMass Amherst Doctoral Dissertations 1896 - February 2014 1-1-1974 Heterogeneous nucleation in the crystallization of high polymers Heterogeneous nucleation in the crystallization of high polymers from the melt. from the melt. Ananda Mohan Chatterjee University of Massachusetts Amherst Follow this and additional works at: https://scholarworks.umass.edu/dissertations_1 Recommended Citation Recommended Citation Chatterjee, Ananda Mohan, "Heterogeneous nucleation in the crystallization of high polymers from the melt." (1974). Doctoral Dissertations 1896 - February 2014. 598. https://doi.org/10.7275/grr1-n529 https://scholarworks.umass.edu/dissertations_1/598 This Open Access Dissertation is brought to you for free and open access by ScholarWorks@UMass Amherst. It has been accepted for inclusion in Doctoral Dissertations 1896 - February 2014 by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected].
Transcript of Heterogeneous nucleation in the crystallization of high ...

University of Massachusetts Amherst University of Massachusetts Amherst
ScholarWorks@UMass Amherst ScholarWorks@UMass Amherst
Doctoral Dissertations 1896 - February 2014
1-1-1974
Heterogeneous nucleation in the crystallization of high polymers Heterogeneous nucleation in the crystallization of high polymers
from the melt. from the melt.
Ananda Mohan Chatterjee University of Massachusetts Amherst
Follow this and additional works at: https://scholarworks.umass.edu/dissertations_1
Recommended Citation Recommended Citation Chatterjee, Ananda Mohan, "Heterogeneous nucleation in the crystallization of high polymers from the melt." (1974). Doctoral Dissertations 1896 - February 2014. 598. https://doi.org/10.7275/grr1-n529 https://scholarworks.umass.edu/dissertations_1/598
This Open Access Dissertation is brought to you for free and open access by ScholarWorks@UMass Amherst. It has been accepted for inclusion in Doctoral Dissertations 1896 - February 2014 by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected].


s
HETEROGENEOUS NUCLEATION IN THE CRYSTALLIZATION OF
HIGH POLYMERS FROM THE MELT
A Dissertation Presented
by
Ananda Mohan Chatterjee
Submitted to the Graduate School of the
University of Massachusetts in
partial fulfillment of the requirements for the degree of
DOCTOR OF PHILOSOPHY
March 1974
Major Subject: Polymer Science and Engineering

Ananda Mohan Chatterjee
All Rights Reserved

• •
111
HETEROGENEOUS NUCLEATION IN THE CRYSTALLIZATION OF
HIGH POLYMERS FROM THE MELT
A Dissertation Presented
by
Ananda Mohan Chatterjee
Approved as to style and content by:
raser P. Pri^e,^&-Chairman ofCommitte
Seymour [Newman, Co-Chairman of Committee
RichardS. Stein, Member
i
/mRoger S. Porter, HeadPolymer Science and Engineering
March 1974

IV
ACKNOWLEDGMENTi
The author acknowledges his deep gratitude to Professor Fraser
Price, Thesis Co-Director, for his most helpful advice and encouragement
during the course of this work. He also sincerely thanks Professor Seymour
Newman, Thesis Co-Director, for initiating this project and for his valuable
guidance and suggestions as the research progressed. Helpful suggestions
of Professor Richard Stein, member, Thesis Committee, are high appre-
ciated .
I am thankful for the help and friendliness of my fellow students
at the University of Massachusetts. Financial support of this work by the
Ford Motor Company in the form of a fellowship to the author is gratefully
acknowledged.. .

vii
HETEROGENEOUS NUCLEATION IN THE CRYSTALLIZATION OFHIGH POLYMERS FROM THE MELT
(March 1974)
AnandaM. Chatterjee
B . Tech. , University of CalcuttaM. Tech.
, University of CalcuttaM.A.Sc.
, University of Waterloo
Directed by: Dr. Fraser P. PriceDr. Seymour Newman
ABSTRACT
In spite of the large amount of work expended in the study of
crystallization of polymers , the nature of the surfaces responsible for
heterogeneous nucleation and the mechanism as to how foreign surfaces
induce nucleation are poorly understood. The present investigation has
been directed toward a better understanding of the behavior of surfaces
in the nucleation of polymer crystals from the melt.
Isotactic polypropylene (iPP) , poly (ethylene oxide) (PEO),
polycaprolactone (PCL) and poly (butene-1) (PB1) have been crystallized
in contact with various substrates (mostly polymeric) , whose nucleating
abilities have been studied using hot stage polarizing microscopy coupled
with still and motion photography. The substrates have been classified
into three groups, based on their observed nucleating ability. Type I
substrates (very active nucleants) are able to induce transcrystallinity
,
nucleation being strongly favored on the substrate. With Type II sub-
strates (moderately active) , nucleation is favored neither on the substrate
nor in the bulk polymer. With Type III substrates (inactive) ,nucleation

viii
is favored in the bulk. Furthermore , the evolution of nuclei on substrates
and in bulk has been followed by counting the number of spherulites
nucleated as a function of time , at different crystallization temperatures.
Nucleating abilities of surfaces have been characterized (a)
qualitatively by studying the morphologies of the growing polymer induced
by substrates and (b) quantitatively (i) by directly measuring nucleation
densities and nucleation rates on substrates and in the bulk polymer and
(ii) by indirectly evaluating the interfacial energy parameters appearing
in the theory of heterogeneous nucleation. The interfacial energy para-
meters were obtained from the experimentally measured temperature
coefficients of heterogeneous nucleation rates and of spherulitic growth
rates.
A survey of forty three substrate-crystallizing polymer pairs
indicated that similarity in chemical structure and in crystallographic
unit cell type of the substrate and the crystallizing polymer are no cer-
tain criteria for nucleating ability. The survey also indicated that close
match of the lattice parameters of the substrate and the forming polymer
crystal is not a necessary condition for strong nucleating action. It has
been concluded that crystallinity is a necessary but not sufficient condi-
tion for a substrate to be a strong nucleating agent and that the surface
energy of a substrate does not dictate its nucleating ability. Low energy
surfaces of polymers have been found to be able to induce transcrystalline
nucleation of polymer crystals
.
Contents of Ti and Al catalytic residues in iPS samples could
be reduced by fractionating the polymer and chelating Ti and Al with
acetylacetone. Use of these two methods and the use of an iPS sample of

IX
negligible Ti and Al contents showed that the high nucleating ability of
iPS samples in the crystallization of iPP, as evidenced by the generation
of transcrystallinity, is due to the polymer (iPS) itself.
Motion pictures of the development of transcrystallinity in poly-
caprolactone showed that in the early stage of this process, nuclei appear
sporadically on the substrate. The growth rate and melting temperature
of the transcrystalline polymer were found to be the same as those of
bulk-nucleated spherulites.
The substrate-induced morphology of a crystallizing polymer
has been found to be temperature dependent, changing from transcrystal-
line to spherulitic on increasing the crystallization temperature. At inter-
mediate temperatures,mixed surface morphologies are observed (trans-
crystalline plus spherulitic) .
In the crystallization of polymers in contact with substrates,
the existence of a fixed number of nucleating sites on the substrate has
been indicated. Two variations of the shape of the experimental nuclea-
tion density (on substrate and in bulk) vs. time curve have been observed,
namely (i) curve with an initial linear part, and (ii) S-shaped curve.

V
DEDICATION
To
All in my family and to my friends
Ashesh Chatterjee
and
Kenneth Hill

vi
XABLE OF CONTENTS
CHAPTER I . INTRODUCTION1
CHAPTER II. NATURE OF HETEROGENEOUS NUCLEATINGAGENTS AND THE MECHANISM OF NUCLEATINGACTION 4
CHAPTER in. TRANSCRYSTALLINITY 10
CHAPTER IV. THEORETICAL BACKGROUND 17
CHAPTER V . EXPERIMENTAL 40
CHAPTER VI. RESULTS 52
CHAPTER VII. DISCUSSION 69
CHAPTER VIII. SUMMARY AND CONCLUSIONS 83
CHAPTER IX. SUGGESTIONS FOR FURTHER RESEARCH 90
REFERENCES 94
TABLES 103
CAPTIONS FOR FIGURES 151
FIGURES 157

4
CHAPTER I
INTRODUCTION
The study of the crystallization of polymers from the melt andsolution is an area of considerable current interest
1 " 3. Such interest stems
from the fundamental scientific value as well as the technological implications
of the organization of long-chain molecules into ordered structures4 ' 8
. The
crystallization of polymers from the melt, the subject of concern in the pre-
sent study, proceeds through the "birth" enucleation) and growth of spheru-
lites, until mutual impingement of the spherulites occurs and the volume is
filled.The spherulite is the most prominent structural organization in
polymers crystallized from the melt. It is a spherical aggregate of twisted
lamellae and is recognizable by its characteristic appearance in the polarizing
microscope, where it is seen as a circular birefringent area possessing a
dark Maltese cross pattern. Growth and cessation of spherulitic growth is
accompanied and followed by "secondary crystallization" which involves
slow reordering of polymer chains within the spherulites2
.
The term "nucleation" denotes the formation of fragments of a
new phase within a pre-existing mother phase. Nucleation plays the cen-
tral role in a wide range of phenomena, e.g. condensation of vapors, boiling,
devitrification of glasses, crystallization of metals and polymers, atmosphericin 11
cloud formation , etc. .
Two types of nucleation are distinguished, namely "heterogeneous"
and "homogeneous", depending on whether a foreign surface facilitates the
12 13birth of the new phase or not '. Homogeneous nucleation occurs as a

result of random fluctuation of order in the supercooled (or supersaturated)
phase, while heterogeneous nucleation is induced by foreign surfaces, e.g.
the surfaces of the container, particles of impurities, dust or additives pre-
14sent in the specimen
. These foreign nucleating agents decrease the free
energy barrier to the formation of the new phase. Thus, heterogeneous
nucleation normally takes place at supercoolings lower than that required
for homogeneous nucleation to occur °'. Nucleating agents, by introducing
increased number of crystallizing centers into play, consequently enhance
the rate of phase transformation.
The importance of the phenomenon of nucleation in polymer crys-
tallization should be clear from the fact that not only it is responsible for
the initiation of the phase transformation (primary nucleation) , but addi-
tionally for the subsequent growth of the primary polymer crystals16 " 18
.
Thus, after the formation of the primary nuclei, further crystal growth
proceeds through secondary nucleation on the developed crystal faces. The
number of primary nuclei determines the ultimate size of the final morpho-
logical units (spherulites) ; this, together with the internal spherulite order,
plays important roles in determining the physical and mechanical properties
2 19of the resulting bulk polymer '
In spite of the large amount of research effort expended on studies
of polymer crystallization, the nature of the nucleating agents and mechanism
through which foreign surfaces promote nucleation are largely unknown.
The present work has been directed toward a better understanding of the
behavior of foreign surfaces in the nucleation of polymer crystals from the
melt. Detailing the objectives of the present study will be postponed until
the last section of Chapter III.

3
From this point onwards, the phrase "crystallization of polymers-
should be taken ip mean crystallization from the melt, unless crystallization
from solution is specifically mentioned.

CHAPTER II
NATURE OF HETEROGENEOUS NUCLEATING AGENTSAND THE MECHANISM OF NUCLEATING ACTION
From the results of a number of studies, one can ascertain the
efficiency of various additives in promoting nucleation of the crystallization
of polymers20 40
. These additives have been mostly low molecular weight
organic and inorganic substances. Exact correlations between the chemical
composition, crystal structure and physical properties of the nucleating
agents with their nucleating ability have not yet been clearly established,
though attempts have been made in this direction. In this chapter, the
chemical and physical characteristics of nucleating agents in polymer
crystallization, based on reports in the. literature , will be summarized.
Where appropriate , mention will also be made of the mechanism of the
nucleating action, as offered by different investigators.
The techniques used to measure nucleating efficiency include
21 28microscopic measurement of spherulite size '
, differential thermal
24 25 28analysis (DTA) ' , depolarization of plane polarized light , etc.
24Beck and Ledbetter used the DTA technique to measure the
effectiveness of additives as nucleants, in the crystallization of polypropy-
lene (PP) . This technique is based upon the ability of an active nucleant
to induce crystallization at a lower supercooling, compared to the base
polymer. The low molecular weight nucleants used by these authors varied
greatly in activity, demonstrating the specificity of heterogeneous nuclea-
tion processes. Among the more efficient nucleants reported were alkali
metal or aluminum salts of aromatic or alicyclic carboxylic acids. Salts of

aliphatic mono- and dibasic acids or arylalkyl acids were found to be "inter-
mediate" in activity. The relative nucleating ability within these classes of
compounds was found to change considerably by varying the organic sub-
stituents, metal cations, etc. Inorganic compounds used by Beck and
Ledbetter (alums, silica, Ti0 2> CaO, MgO, carbon black, clays) were
classified as "poor" nucleating agents for PP crystallization.
25Beck undertook the evaluation of the nucleating ability of a
further series of compounds (mostly organocarboxylic acid salts) in the
crystallization of PP, using the DTA technique. The nucleating efficiencies
were discussed in terms of the chemical structures of the additives. It was
suggested that a model nucleating agent for PP might be envisioned as one
consisting of an organic group (solubilizing portion) and a polar group
(insolubilizing portion) . Organocarboxylic acid salts represent such a
model. In fact, sodium benzoate and basic aluminum dibenzoate were
among the best nucleants for PP.
25Beck reached the following conclusions for the crystallization
ofPP:
(i) Aromatic groups impart higher nucleating ability than ali-
phatic groups.
(ii) Salts of carboxylic acids are more effective nucleants than
the free acids.
(iii) Sodium benzoate is a more effective nucleant than the
benzoates of a large number of other metals; however, no correlation was
found between nucleating ability and the atomic weight, atomic number,
atomic radius or valence of the cations.

(iv) Substituents in the aromatic ring generally reduce the
more
nucleanng ability; however, subshtuents in the para position areeffective than the corresponding ortho or meta substituents.
(v) A tertiary butyl group, especially in the para position
often increases the activity of the intermediate or poor nucleants, but notthat of a good nucleant.
(vi) Among aliphatic mono- and dicarboxylates and alicyclic
carboxylates, those containing four to seven chain or ring carbon atomsare the most effective.
(vii) Among fused-ring aromatic carboxylates, those having the
polar group in the 2 -position are the most effective.
The effect of polymer molecular weight on the response of PP to
heterogeneous nucleation of its crystallization by basic aluminum dibenzoate
non-existent26(an effective nucleant) was found to be small or practically
Binsbergen's study of a large number of additives in polyolefin
crystallization27
' 28led to the conclusion that nucleating agents for poly-
olefins are crystalline solids of melting point higher than that of the polymer,
insoluble in the polymer melt, having no chemical reactivity toward the
crystallizing polymer, and consisting of both hydrocarbon groups (good
"solvents" for the polymer) and either polar groups or a condensed aromatic
structure which renders the nucleant insoluble in the polymer melt. He
further concluded that nucleating ability is strongly dependent on the degree
and sometimes on the method of dispersion of the nucleant in the polymer.
The conclusions of Kargin, et al.29 " 37
, who studied the nucleating
ability of a number of foreign substances in the crystallization of isotactic

polypropylene (iPP) and isotactic polystyrene (IPS), are in agreement with
those of Binsbergen. Their work also shows the role of nucleating agents
in modifying the supermolecular structure of polymers, and the influence
of the latter,in turn
, on the mechanical behavior of the polymers.
In a study of the mechanism of the action of the nucleating agents,
33Kargin, et al.,considered the role of wettability, lattice match and nucle-
ant dimension in heterogeneous nucleation. First, wettability of a foreign
surface by the polymer was found to improve nucleating action (no mention
was made as to whether the wettability by the crystalline embryo or by
the polymer melt was the key factor) . Thus, hydrophilic inorganic oxides
exert nucleating action in hydrophilic polymers, but not, as expected, in
iPS; also, organic substances like indigo, alizarin, quinacridine and 1,5-
anthraquinone are active nucleating agents for iPS . It was also found that
hydrophilic cotton fibers do not function as nucleants for iPS, but become
active in this respect when their surfaces are rendered hydrophobic.
Secondly, a match of lattice type and lattice parameters between
the substrate and crystallizing iPS was not found to be necessary for strong
nucleating action.
Thirdly, the effect of the dimensions of the nucleating agent on
the nucleating action was explored, using the system iPS-indigo. It was
found that there is no upper limit to the dimension of indigo crystals , for
functioning as an active nucleant at any given temperature; but, a lower
limit does exist. The lower critical size of the nucleant depends on the
temperature of polymer crystallization and diminishes as the supercooling
is increased; thus, as the crystallization temperature gets lower, smaller
and smaller particles become capable of functioning as nucleants. According

8
.° Kargin. et a, 33. the^ size q{^ ^^^^
perature in the same way as does the critical size of the pollerAnother study hy Kargin, et I." lndicated that when nucl(jating
agents in the form of sohd particles are incorporated in a polymer, stresses
arise in the layer of polymer surrounding the particles, and a, the sites of
stresses micro-ordered segments are formed, which promote the initiation ofthe crystallizahon of the polymer
. The magnitude of the induced stress
depends on the size of the particle, the nature of the polymer and the
nucleating substance and on their interaction. This concept is consistent
with the observation, made by these authors as well as by Rybnikar41
.
that gas or vapor bubbles can act as efficient nucleating centers in PP.38Rybnikar studied the nucleating efficiency of six additives
(phthalic acid, benzoic acid, aluminum phthalate, sodium salicylate, carbon
black and the pigment Irgalilechtbrilant blau) in the crystallization of eight
commercial types of iPP. The results indicate that most of the nucleating
agents do not exhibit a universal nucleating ability for all the samples of
iPP examined; in other words, a particular additive shows different
nucleating ability for different samples of polypropylenes . It was suggested
that the real heterogeneities might be other substances originally present
in the polymer, possibly remainders of the catalytic system or occasional
impurities. The additives were thought to activate the originally present
heterogeneities (e.g. by improving the wetting), thus enhancing their
nucleating effect.
The above concept is consistent with the observation of
Rybnikar 38and Binsbergen 42
that the number of heterogeneous nuclei
cannot be indefinitely increased by increasing the concentration of additives.

9
in the cases studied, a. about 1% additive concentration, the number of crys-
tallization centers reached a maximum
.
Heterogeneous nucleating agents (e.g. indigotin, anthraquinone)
were necessary to induce crystallization in poly(2,6-dimethyl phenyleneoxide) which by itself has practically no tendency tc crystallize
40. Also,
the organic dyes indigo and alizarin were found to increase the crystalli-
zadon rate of the very slowly crystallizing IPS by acting as centers of
heterogeneous nucleation31
.
Several studies of the kinetics of crystallization have been madeusing added nucleating agents in polymers 19 ' 21
•42 • 43
. Tne nucieants
reduced the induction period for crystallization and enhanced the rate
of crystallization without changing the shape of the crystallinity vs. time
isotherms.
Two interesting aspects of the physical features of nucleants,
namely the influence of the substrate structure, particularly orientation44
,
and the enhancement of the nucleating ability of a substrate by morphologi-45
cal imprinting, observed by Hobbs, come within the scope of this chapter,
but will be mentioned in the following chapter, as they involve transcrystal-
linity.

10
CHAPTER III
TRANSCRYSTALLINITY
Formation
On cooling a polymer melt to a temperature below the melting
point such that it is in contact with a surface of high nucleating ability
toward the polymer, a large number of spherulites are nucleated on the
surface. These spherulites, due to lateral crowding , cannot grow with
spherical symmetry and are forced to grow in the direction perpendicular
to the substrate surface. The resulting columnar morphology, consisting
of elongated spherulites, has been termed "transcrystalline"46
' 47.
The formation of a transcrystalline region is indicative, there-
fore, of a high nucleating ability of the substrate in the crystallization of a
particular polymer. Transcrystallinity has been observed in polyamides 46 ,48
polyurethanes46
, polyethylene49 " 53
, polypropylene52
'54,55 'Present study
^
and in polycaprolactone (present study) . The formation of the transcrystal-
line region also depends upon the conditions of crystallization, e.g. crystal-
lization temperature, cooling rate, etc.54
The effect of internal transcrystal-
linity (within the bulk of a polymer) is observed if sufficient number of
adjacent nuclei are formed in the interior of the polymer, giving rise to a
"row structure"; examples found are polyamides56,57
and polyethylene53
57aand certain fiber-filled systems
It has been suggested that transcrystallinity results from a
temperature gradient between the nucleating surface and the interior of
48 49the polymer under which the polymer is crystallizing ' . Subsequent
experiments with polypropylene showed that a temperature gradient by

, as
11
itself is ineffective for the development of transcrystallinity and that poly-
propylene can transcrystallize in the virtual absence of a temperature54
gradient .
Transcrystallinity was also claimed to be induced only by high
energy surfaces like metals, metal oxides and alkali halides; low energy
surfaces (like polymers) being apparently unable to induce transcrystal-
lization '
58•
59. However
, studies by Fitchmun and Newman 53'54
well as the present study, show that transcrystallinity can be generated
by low energy surfaces as well.
The degree of penetration of the transcrystalline region into the
bulk of the crystallizing polymer depends on the conditions of crystalliza-
tion, e.g. temperature, and the relative nucleating ability of the substrate
and the adventitious particles present in the polymer sample. Thus, the
presence of bulk-nucleated spherulites in the vicinity of the transcrystal-
line region limits the depth of penetration of the transcrystalline layer to
a depth occupied by spherulites formed in the bulk. Transcrystalline
layers of depth 50-100u are often observed.
Structure
The structure of the transcrystalline region in polyethylene has
been revealed by x-ray crystallographic studies of Keller56
, Eby49
, and
6 0Gieniewski and Moore . The results indicate that the transcrystalline
region consists of symmetrically packed oriented lamellas, with their
longest axes (b) perpendicular to the substrate surface and with the a and
c axes oriented randomly in the plane perpendicular to the b axis.

ers
.
ease
12
Properties
In this section.the important physical and mechanical properties
of the transcrystalline region will be briefly reviewed to show how controlof the morphology of the surface region of polymers through heterogeneous
nucleadon enables one to modify the physical and mechanical behavior of
polymers.
Kwei, et al.51
, reported that the dynamic Young's modulii of
transcrystalline PE and PP are higher than those of the bulk polym
They showed that the modulii of molded films of these polymers deer
with increasing film thickness.
Matsuoka, et al.61
, reported that in the temperature range of
-160 to 120°C, transcrystalline PE exhibits a higher in-phase modulus, a
higher loss modulus and a higher loss tangent (in tensile mode) than bulk
PE.
62Kargin,et al
. ,showed that a transcrystalline surface layer
imparts greater abrasion resistance, greater impact strength and lower
C02permeability to polypropylene films.
Conflicting reports exist in the literature regarding the density,
heat of fusion and crystallinity of the transcrystalline region as compared
to the bulk polymer crystallized under the same conditions. Hara and63
Schonhorn observed higher values of the above three properties for
transcrystalline PE, compared to bulk PE. An opposite conclusion, namely
lower values for the transcrystalline region, was drawn earlier byfil
Matsuoka, et al.

Wettability of the transcrystalline surface has been found to be
different from that of conventional PE; thus, the critical surface tension of
wetting of gold-nucleated PE (transcrystalline) has been observed to be
considerably in excess of the value for ordinary PE 52, and the difference
has been accounted for on the basis of a modified Fowkes-Young equation
for the wettability of polymer surfaces. Transcrystalline PE was found by
Schonhorn and Ryan 64to be amenable to conventional adhesion bonding and
to form a strong adhesive joint, e.g. with aluminum, in contrast to the poor
adhesion with conventional PE. Thus, transcrystallinity offers a route to
improved adhesion of polymers to other surfaces.
Rationalization of the observed physical and mechanical properties
of the transcrystalline region in terms of structural features like lamellar
orientation and packing has not yet appeared in the literature.
Columnar Crystals in Metals
The phenomenon of transcrystallinity is not restricted to polymers
and has been observed in the crystallization of metals65-67
. In fact, the
term "transcrystallinity" was adapted from metallography 46. Casting of I
metals involves a liquid to solid phase transformation and if the nucleation
density is much higher at the mold wall (chilled) than in the bulk of the
metal, long columnar grains extend perpendicular to the mold surface
inward, in the direction of the greatest temperature gradient during freezing
The columnar crystals are not formed if the mold is preheated to a tempera-
ture approaching the freezing point of the metal. The conclusion must be
drawn, therefore, that it is the increased nucleation density at the lower
temperature of crystallization that is responsible for the generation of the
layer of columnar crystals in contact with the mold surface.

14
Northcotfs investigations into the structure of cast metal
ingots68" 70
showed that the columnar crystals are often dendritic and that
they have a preferred orientation; thus, for cubic materials the [100] direc-
tion is parallel to the growth direction.
Substrate Structure and Morphological Imprinting
Two noteworthy features of the action of nucleating agents involvin
transcrystallinity will now be pointed out. First, Hobbs44
observed that the
ability of graphite fibers to nucleate the crystallization of isotactic polypro-
pylene (iPP) is dependent on the substrate structure, particularly the
orientation. Thus, iPP transcrystallizes in contact with Morganite I, but
not in contact with Morganite II. Morganite II is composed of very small
(about 25& graphitic nuclei with a high degree of disorientation, while
Morganite I is made up of much larger (>100#) graphitic planes which are
highly oriented along the fiber axis. According to Hobbs, the observed
difference in nucleating ability between Morganites I arid II is due to the
difference in the size and orientation of their constituent graphite planes.
Secondly, in a study of the crystallization of iPP on morphological
45templates
,Hobbs showed that by using a surface replication technique,
one can transform a non-active surface (like amorphous carbon) into a
surface which can readily induce transcrystallization . A 300$ thick layer
of carbon was deposited by evaporation from a carbon electrode under high
vacuum onto iPP films . These iPP films , on subsequent melting and crystal-
lization, showed development of transcrystallinity. The increase in nuclea-
tion density was shown to result directly from the structural information
contained in the carbon replicas and neither from molecular bonding between

15
the polypropylene surface and the carbon replica nor from the unmelted seed
crystals adhering to the carbon overlayer.
Objectives of the Present Study
Surfaces responsible for heterogeneous nucleation are, in general,
poorly characterized. The first objective of the present study was to charac-
terize surfaces (a) qualitatively by examining the morphologies of the growing
polymer induced by substrates and (b) quantitatively by directly measuring
nucleation densities and nucleation rates on substrates, and by indirectly
evaluating the interfacial energy parameters appearing in the theory of
heterogeneous nucleation. The interfacial energy parameters were intended
to be experimentally determined from the temperature coefficients of hetero-
geneous nucleation rates and of spherulitic growth rates.
The second objective of this study was to generate appropriate
experimental data to throw light on the role played by the following factors
in the heterogeneous nucleation of polymer crystals:
(i) similarity in chemical structure of the substrate and the
crystallizing polymer,
(ii) similarity in unit cell type and lattice parameters of the
substrate and the crystallizing polymer,
(iii) crystallinity of the substrate , and
(iv) surface energy of the substrate.
In view of the claim in the literature that low energy substrates
52 58 59are ineffective in inducing transcrystallinity in polymers ' ' and
53 54evidence to the contrary '
, it was felt desirable to investigate the pos-
sibility of finding further examples of transcrystalline nucleation induced
by polymer substrates.

Finally, it was also intended (i) to investigate the temperature
dependence of the morphology of a growing polymer induced by a substrate,
and (ii) to compare the growth rate and melting temperature of the trans-
crystalline region with those of bulk-nucleated spherulites.

17
CHAPTER IV
THEORETICAL BACKGROUND
Much of our present understanding of phase stability and phasetransformation rests on the classical work of Gibbs
71. Theories of nuclea-
te were subsequently developed by Volmer and Weber, Becker and Coring,Turnbull, and others for low molecular weight substances72 ' 73
. Theseconcepts formed the basis for the development of the theories treating
nucleation in polymer crystallization; Gornick and Hoffman 74and, more
1
8
recently, Price have reviewed the field. The airn of this chapter is to
introduce the concepts of the classical nucleation theory and develop the
equations for the kinetics of homogeneous and heterogeneous nucleation in
polymer crystallization from the melt. The theory of the nucleation control
of spherulitic growth will complete the chapter
.
Homogeneous Nucleation - General
Homogeneous nucleation occurs as a result of random fluctuations
of order in the supercooled phase, in the absence of foreign surfaces. These
nuclei are clusters of molecules (or atoms) and appear at random throughout
the mother phase.
Let us consider the formation of a spherical embryo of a daughter
phase p in a surrounding medium of mother phase a , at constant temperatur
and pressure. The Gibbs free energy of formation of the embryo of radiu
r can be written as
e
AG = - (4/3)7tr3AG + 4tt r
2a (1)

where AGy (=G/ - Gya
) is the difference of Gibbs free energy per unit
volume between the a and p phases and o is the interfacial energy between
the embryo and the mother phase. From this point onwards, free energies
should be taken as Gibbs free energies, unless mentioned otherwise.
When p is the more stable phase, the relationship between AGand r is shown in Figure 1
.The maximum value of AG (designated AG*)
occurs at a critical size r* and represents the free energy of formation of
the "critical-sized nucleus." Such a nucleus can grow in size with a
decrease, rather than an increase, in free energy.
Differentiation of Equation 1 yields
dAG „ 2= -4tc r AG
y+ 8tt ra
Setting this derivative equal to zero, one obtains for the critical
nucleus:
-4tc (r*)2 AG
y= 8tt r*a
or
r* = 2o/AGv (2)

19
Substituting this into Equation 1
AG* =16
3 (AG )
2 (3)
The spherical shape of the nucleus is a good assumption in many
cases, but not in the case of polymers, where a cylindrical nucleus is con-
sidered to be more appropriate15
. For a cylindrical nucleus of radius r and
length 1 and characterized by two interfacial energies, namely, c for the
s
side and o&for the end (Figure 2) , we may write
2 9AG = - itr 1AG„ + 2tc r lo + 2tt r ov s e
This equation, on partial differentiation, yields
= - 2tc r 1 AG + 2tt 1 a + 4tt r aor v s e
and'
8AG 2 A „-aT = - n r AG +27iraol V s

20
Setting the partial derivatives equal to zero and solving, one
obtains for the critical nucleus
r = 2 o /AGs v (5)
J * = 2°e/AG
v (6)
Substitution of these two values into Equation 4 gives the free
energy of formation of the critical nucleus:
AG* = 87C os
2ae/(AG
v )
2
(7)
This equation is similar in form to Equation 3 for a spherical nucleus. The
insensitivity of such formulation to embryo geometry is worth noting.
75Volmer and Weber assumed that nuclei form by a series of
bimolecular reactions of the type
V a ai + 1
(8)

21
With specific reference to condensation from the vapor, they further assumedthat the concentration of critical-sued nuclei was that characteristic of
equilibrium and that the back flux of supercritical nuclei could be neglected.
Their equation for the rate of nucleation (I) per unit volume is
I = a A nv (9)
Here a is the collision frequency of single molecules with nuclei per unit
area of the nuclei, A* is the area of the critical nucleus, and ny*
is the
number of critical nuclei per unit volume, given by76
nv* = n
v°exp (-AG*/kT) .
'•
(10)
In this equation ny
is the number of unassociated molecules per unit volume,
k is Boltzmann's constant, and T is the absolute temperature.
The back flux in Equation 8 and the decrease in embryo population
caused by growth of the nuclei were subsequently taken into account by
77Becker and Doring who calculated the steady-state concentration of
critical nuclei. Their equation for the nucleation rate is

22
I = a A n B (11)
Here B is the Becker-Doring non-equilibrium factor, given by
B =(3o/kT)
1/2
2tt (r )
2(12)
where v is the molecular volume of phase p (embryo) and r* and c havebeen defined in the beginning of this section for a spherical nucleus.
For nucleation in condensed phases, the free energy of activation
for motion across the daughter phase-mother phase interface should be
included in the expression for nucleation rate. This was done by Becker78
and later more quantitatively by Turnbull and Fisher79
, who combined the
absolute reaction rate theory 80 with the Becker-Doring approach and derived
the following equation for the steady state nucleation rate:
I = IQexp (-AG /kT) exp (-AG /kT)
(13)
where AGtis the activation energy for transport across the embryo-mother
phase interface. Equation 13 is referred to as the Turnbull-Fisher equation.

23
For most nucleation problems of interest, I may be taken to
order-of-magnitude accuracy as
I s n nO V o
where nQ
is the molecular jump frequency. The term I is dependent on
molecular parameters, but is essentially independent of temperature. At
comparatively small supercoolings , the second exponential in Equation 13
that is the transport term, exerts negligible influence on the nucleation
rate; the term becomes significant at large supercoolings.*
The quantity AG depends on AGv
(Equation 3); AGv
> in turn
is proportional to the supercooling AT. Hence the Turnbull-Fisher equation
predicts that under conditions where the transport term is negligible, the
homogeneous nucleation rate should vary exponentially with the super-
o-i
cooling. Experimentally, this variation is very sharp. Thus, Turnbull
observed the nucleation rate of crystals in liquid mercury droplets to vary
by four orders of magnitude in a temperature interval of 3°C
.
*The term AG in the Turnbull-Fisher equation can be expressed
in terms of the supercooling and the interfacial energies characterizing the
nucleation process. This allows one to determine the dependence of the
nucleation rate on these parameters.
With the above-mentioned aim in mind, let us start with a model
for a small crystal of a low molecular weight substance in contact with the

melt from which it formed (Figure 3) . The crystal dimensions are a, b,
and c, respectively. Let oebe the interfacial energy of the two AB faces
and a be the interfacial energy of all the other faces and AGy
the bulk free
energy of fusion per unit volume. The free energy of formation of the
above-mentioned crystal, relative to the melt, is given by
AG = - abc AGy
+ 2 ab ag
+ 2a (be + ac) (1/
This equation represents a hyper-surface whose positions of extrema are
determined by equating to zero the partial derivatives of AG with respect
to a, b, and c and solving for the critical values, namely a , b' and c
which define the dimensions of the critical nucleus. Thus, Equation 14
yields
9AG-~— = - be AG + 2 b o + 2acoa v e
9AG _
9b- ac AG + 2 a a + 2ac
v e
and
= - ab AG + 2ab + 2aadc v

For the critical nucleus:
25
a* = b* = 4a/AGv
C*=4
°e/AG
v (16)
Substitution of these values in Equation 14 gives the free energy of formation
of the critical nucleus:
* 2 9AG = 32 o oe/(AG
v)
z
(17)
Heterogeneous Nucleation - General
The nucleation of most phase transformations takes place hetero-
geneously on impurity particles, container walls or structural imperfec-
tions14
'
81a'
81b. Intentionally added nucleating agents also catalyze the
nucleation process. A formal description of heterogeneous nucleation will
1 *3
be presented in this section, following Turnbull .
Let us consider the formation of a spherical cap of the daughter
phase p from the mother phase a, on a flat substrate s (Figure 4); the

substrate-nucleus contact angle is 0 . The net free energy of formation in a
of unit area of the p- s interface is given by
Wc= on -a
s ps as (18)
where oqs
and are, respectively, the a-s and p-s interfacial energies.
The magnitude of Wgdetermines whether or not the substrate catalyzes the
nucleation of p. The substrate will be a preferred (relative to the bulk of
a) p nucleation site, provided Wg
is less than oap , the a-p interfacial energy
The greater are the intermolecular attractive forces between p and s, the
less is Wg
.If the substrate interacts more strongly with p than with a,
W will be negative (since on < a )fa ps as"
The relation of Wgwith the contact angle 0 is obtained by a hori-
zontal force balance in Figure 4:
o a cos0 + on = aap ps as
or
cos0 = (a - o„ )/oas °ps J/aap (19)

Using Equation 18, we have
27
cos© = - w /as «P (20)
Volmer 82showed that AG
g*, the free energy of formation of a
critical nucleus heterogeneously, is given by
AG * -167tO
a33f(0)
S3 (AG
y+ E)
2 (21)
= AG* f (0)(22)
where
f (0) = (2 + cos0) (1 - cos0)2
4 (23)
Here E is the elastic strain energy per unit volume and AG* is the thermo-
dynamic barrier for homogeneous nucleation in the bulk of a (see, for
example, Equation 3 for a spherical nucleus)
.
The magnitude of f (0) lies between zero and unity, depending
on Wg
. The limiting values are:

28
f (0) = 0 when - W > as ^ ap
and
f (0) = 1 when W = os ap
Equations 22 and 23 predict that the thermodynamic barrier to
nucleation on a substrate should decrease with decreasing contact angle 6,and approach zero as 0 -> 0.
Following the Turnbull-Fisher 79treatment for homogeneous
nucleation, Turnbull 83 ' 84derived the following equation for the rate of
heterogeneous nucleation (per unit area of substrate)
:
Is= K
sexp(-AG
s"/kT) exp(-AG
t/kT)
where
gfc
Ks= n n
sf (a
ap/kT)1/2
(2V/ 9*)
1/3[f(0)] 1/6
(24)
Here n is the number of molecules in the nucleus in contact with a, V is
the volume per molecule of liquid, ng
is the number of molecules of a in
contact with unit area of the substrate, and f is the fundamental jump fre-
quency. According to the absolute rate theory, f = kT/h, where h is
Planck's constant.

29
It should be noted that Equation 24 is of the same form as the
Turnbull-Fisher equation, i.e. Equation 13.
The general action of a nucleating substrate is to reduce the
free energy barrier for nucleation to occur. When a nucleus forms on a
substrate, in addition to the creation of the nucleus-mother phase interface,
some relatively high energy substrate-mother phase interface is replaced
by a lower energy substrate-nucleus interface.
The subject of retention of embryos in cavities of foreign bodies,
under conditions where such embryos would be unstable on a flat surface
,
was treated in detail by Turnbull83
for conical as well as cylindrical cavities
In the latter case, embryos will be retained in cavities of radii less than r ,
c
which is given by
r = 2a n cos0/AGc ccp v
Here 0 is the angle shown in Figure 5 and AGv
is the motivating free energy
for a •* p transformation.
If a system is heated above the melting temperature (T ) by an
amount AT+
to eliminate embryos in cavities of radii greater than r , andc
then cooled below T , embryos retained in cavities of radii less than rm Jc
will grow to the mouth of the cavities. They will not, however, serve as
nuclei unless the radius at the cavity opening equals or exceeds r , the

30
critical size. Hence a linear relation between AT+and AT~ (undercooling)
is anticipated, until the range of undercooling is reached where normal
heterogeneous nucleation on the substrate is possible.
Nucleation in Polymer Crystallization.
Extension of the concepts discussed above to the nucleation of
polymer crystals will be presented in this section, following the treatment18
of Price.The treatment for crystallization from the melt, which will be
presented, is essentially valid for crystallization from dilute solution also,
excepting for minor corrections like replacing the polymer melting tempera-
ture by the solution temperature, etc. The validity stems from the fact
that the heats of solution are small compared to heats of crystallization and
that the entropies of solution can be easily calculated.
Homogeneous Nucleation in Polymers . For homogeneous
nucleation in polymer crystallization , a model for the nucleus depicted in
Figure 6 will be used. The embryo is built up by the successive addition
of adjacent segments of the polymer chain . The length c is called the
"fold length" or "segment length". The AB surfaces are called "fold
surfaces" and since most folds occur in the AC plane, this plane will be
called the "fold plane" . According to the treatment given, the size and
free energy of formation of the critical nucleus are given by Equations 15
through 17.
To illustrate the insensitivity of the aforementioned formulations
to embryo geometry, it should be pointed out that if a right circular cylinder
had been assumed, the diameter would be given by Equation 15, the length

would be given by Equation 16, and the constant 32 in Equation 17 wouldbe replaced by 8tt .
The temperature dependence of AG* resides in the quantity
AGv .
If AHyand AS
yare, respectively, the enthalpy and entropy of
fusion of the polymer per unit volume
,
AGv
= AHv
- T ASv
At the melting temperature (T ) , we may write
0 = AH - T As flJ{nv m v 126)
or
T = Ah /As , 97 ,m v v (27)
Substituting this equation into Equation 25
AG = AH - T AH /Tv v v' m
= AH (1 - T/T )v m J
= AHvAT/Tm (28)

32
where AT (* Tm - T) is the supercooling.
Combining Equation 28 with Equations 13 and 17, one obtains
the rate of homogeneous nucleation (folded chain nuclei):
I = Iqexp (-AG
t/kT) exp
2 9-32 o o T
e mT (AT)
2k (AH )
2 (29)
Experimental studies of homogeneous nucleation in the crystalli-
zation of polyethylene 85' 86
and polypropylene87
have been performed, using
the "droplet" technique to annul the effect of foreign heterogeneities. Before
the crystallization, the polymer samples were divided into sufficiently large
number of parts so that the heterogeneities, of finite number, resided in only
a small fraction of the parts. Supercoolings of about 60°C and 100°C were
necessary for polyethylene and polypropylene, respectively, for homogeneous
nucleation to occur.
Heterogeneous Nucleation in Polymers . Referring to the model
of Figure 6, let one AC face of the crystal be in contact with a foreign surface.
Also, let a be the substrate-crystal interfacial energy and a be themsubstrate-melt interfacial energy . The free energy of formation of the
crystal will be given by
AG - - abc AG + 2 ab o + 2 be a + ac AaO V c (30)

33
where
Aa = a + a - ac m (31)
es
The term oerepresents the interfacial energy of the fold surfaces (AB)
,
while o represents the interfacial energy of all the other fac
The situation just described is referred to as "non-coherent"
nucleation, which is characterized by the condition Aa £0. The expressions
for the size and free energy of formation of the critical nucleus will be
derived now.
From Equation 30,
-^-= - be AG +2 bo +c Ao
3AGW = " ac AGy
+ 2 a ae+ 2 c a
9AG-j^r = - ab AG
v+ 2 bo + a Aa
Equating the partial derivatives to zero, and solving, one obtains the size
of the critical nucleus:

34
a = 4o/AGv (32)
*
b s 2Ao/AGv (33)
c = 4o /AGe v (34)
Substitution of Equations 32 through 34 in Equation 30 gives the free energy of
formation of the critical nucleus:
AGg
16 aoeAo/ (AG
y ) (35)
Using Equation 28, and substituting Equation 35 into Equation 24, one obtains
the rate of heterogeneous nucleation per unit area of substrate (folded chain
nuclei)
:
Ig= K
seXp (-AG /kT) exp
-16 oa Ao Te m
T (AT) k(AHv )
2 (36)
or

35
16 aa Aa Tlog I = log K - AG./ (2.3 kT) e "
"mb St „ 9 r (37)
2.3 T(ATr k (AH )
2
This equation describes the dependence of the rate of heterogeneous nuclea-
tion on supercooling.
When the substrate is absent, am= 0 and o = a
c. Under this con-
dition.Equation 32 reduces to Aa = 2a, and Equation 36 reduces to Equation 29,
the case for homogeneous nucleation.
Comparison of Equations 29 and 36 shows that if Aa < 2a at a given
temperature, Ig
will exceed I; that is, heterogeneous nucleation will be
favored. According to Equation 31, Ao < 2a when (a - a ) < a. It should
be noted that this condition is identical to that due to Turnbull for the
spherical cap model of heterogeneous nucleation, namely W < a (sees ap
Equation 18) . The quantity aR is the same as a , a is the same as a ,P° c as mand o^p is the same as a
.
Heterogeneous nucleation processes are always accompanied by
a transient in the number of nuclei. However, the interaction between the
substrate and the polymer crystal may be strong or weak.
When the substrate-polymer crystal interaction is strong, the
wetting of the substrate by the crystal is good, and a has a low value.c
The rate of heterogeneous nucleation will be high in such cases, at tempera-
tures well above the range of homogeneous nucleation. If there exists a
finite, but very large, number of heterogeneities, on cooling the melt all
the crystals will appear close to zero time and will seem to develop from a

«xed number „ nuclei . The transiem in the number of ^ thjs ^of very short duration and large magnitude.
When the substrate-polyrner crystal interaction is weak, o is
large and Ao may approach 2o. In such a case, the substrate affects the
nucleation rate only slightly and crystals may appear sporadically i„ ,imeand space. This heterogeneous nucleation process mimics the homogeneousprocess and has been called "pseudohomogeneous nucleate 18 ' 88
. How-ever, the number of heterogeneities being finite, if the crystals do no, growso rapidly as to fill the vo.ume ,00 soon, eventually the number of growingcenters will become fixed (this cannot happen in homogeneous nucleation) .
In this case the transient in the number of nuclei can become quite long in
duration.
Figure 6a schematically illustrates the nucleation rate curves for
homogeneous, heterogeneous and pseudohomogeneous nucleation, the third
being a special case of the second.
Nucleation Control of Spherulite Growth
The very large temperature dependence of spherulite growth rate
at relatively small supercoolings led to the idea that the growth rate of
spherulites is nucleation-controlled16 '
17. The idea is based on an earlier
concept due to Volmer 89that the addition of a new layer of crystalline
material to an already completed crystal face is determined by the rate of
appearance upon that face of small crystal patches, which are extensions of
the underlying parent crystal and have been called "coherent growth nuclei".
The problem of crystal growth then reduces to one of describing the forma-
tion of coherent growth nuclei9,15
.

37
Figure 7 is a schematic representation of a coherent growth
nucleus of dimensions a,b
, and 1 on the tip of a crystal at the spherulite
boundary. The free energy of formation of such a nucleus from the melt is
given by
AG = - abl AG + 2 blv a + 2 ab
e (38)
Here AGy
is the bulk free energy of fusion per unit volume and o and o aree
'
respectively, the interfacial energies of the bl and ab faces. No term involving
the al faces appears because building the nucleus destroys as much such a
face as it produces. Setting dAG/da = 0 and 3AG/91 - 0, and solving, one
obtains for the critical nucleus:
a* = 2o/AGv (39)
1 =2a/AG (40)
Substituting Equations 39 and 40 into 38,

AG = 4b 00 /AGe v
38
(41)
Using Equation 28, this yields
* 4b oo TAG = e m
AHy
(AT) (42)
The growth rate (G) of the polymer spherulite is proportional to I,
the rate of formation of coherent growth nuclei on the tips of the crystals at
the spherulite boundary. Using the Turnbull-Fisher equation, one can
write
G = zl = zIq
eXp (-AGt/kT) exp (-AG /kT) (43)
where z is the proportionality constant. If z!Q
= GQ , combination of Equation
42 and 43 yields
G = Gqexp(-AG
t/kT) exp
- 4b oo Te m
T ( AT) k (AH )v(44)

The term Gq
is a constant depending on molecular parameters, but
essentially independent of temperature. Equation 44 describes the
temperature dependence of the growth rate of polymer spherulites.
Taking logarithms
,
i n ,4b oa T
log G = log G - AG./ (2.3 kT) - —.
e m0 1 2.3 k (AH ) T (AT) (45 ^
According to this equation, it is possible to calculate ooefrom
the temperature coefficient of spherulitic growth rates. Also, one can
obtain the quantity aoeAo from the temperature coefficient of heterogeneous
nucleation rates according to Equation 37. By dividing oa Aobyaae J
e'one gets Ao (= a + o
q- aj . The quantity o is independent of the sub-
strate; (om - CM,the difference between the. substrate-melt and
substrate-crystal interfacial energies, reflects the nucleating ability of
substrates. A higher value of (om" aj indicates a better heterogeneous
nucleating agent since, relative to the melt, less energy is expended in
creating a unit area of substrate-crystal interface. This approach has
been used in the present study to characterize the nucleating efficiency
of substrates
.

CHAPTER V
EXPERIMENTAL
5 . 1 Materials
Table I describes the materials used in the present investiga-
tion.
Melting points of the samples of iPP, poly (ethylene oxide),(PEO)
,
poly(butene-l) (PB1) and polycaprolactone (PCL) were determined by
heating the sample in the hot stage of a polarizing microscope at 0.2°C/min.
and observing the spherulites under crossed polars. The melting point
was taken as the temperature at which the birefringence of the sample dis-
appeared .
"Ash",Ti and Al contents were analyzed at the Microanalysis
Laboratory at the Department of Chemistry, University of Massachusetts.
For the determination of "ash" content, the sample was burned in oxygen
at 1000°C; the residue consists usually of metallic oxides. Mass of this
residue, as a percentage of the initial mass of the sample, is reported as
"ash content"
.
Ti and Al contents of isotactic polystyrene samples were deter-
mined by atomic absorption spectroscopy.
Molecular weights of PEO were obtained by Gel Permeation
Chromatography, through the courtesy of Dr. Robert Ulrich90
.
The intrinsic viscosities of two fractions of isotactic polystyrene
(entry 5d in Table I) were determined following the conventional proce-
91dure . Viscosity average molecular weights were calculated from the
intrinsic viscosities, using the Mark-Houwink equation. Details are
included in this chapter.

The remainder of the characterization data shown in Table I
was supplied by the vendors.
5.2 Equipment
Figure 8 gives an overall view of the equipment constructed forthe present study. The essential parts of this photomicrographs equip-ment, which is supported on a drill press stand (a,
, are the following:
(i) A Zeiss Polarizing Microscope (b) , equipped with a vertical
eyepiece (inside c) and a binocular eyepiece (d) .
(ii) A Mettler FP2 Hot Stage (e), placed on the microscope
stage. Temperature programming is accomplished using the control unit
(f).
(iii) A Prontor-Press shutter (g) (Geiss America, Inc.),
fitted on top of the vertical eyepiece. The shutter is provided with a cable
release and can give exposure times down to 1/125 second.
(iv) A light-tight black wooden box (h) tapering downwards,
at the bottom of which the shutter is held in place with screws. This box
is also fitted on the outside with a viewing telescope (i) facing the observer
and a solenoid (j) on the observer's right.
(v) A photographing device at the top. In Figure 8, a custom
70 mm. camera (k) is seen. In its place a Polaroid Film Holder or a motion
picture camera can also be used.
At the bottom of the wooden box is a 45° mirror which , when
vertical, allows light to reach the photographic film just above the top of
the wooden box. When the mirror is at 45°, the light coming through the
vertical eyepiece of the microscope, after passing through the shutter, is

reflected by the mirror and reaches the observer's eye through the tele-
scope (i).
This arrangement allows one to focus the image at the film plane
by focusing it on the ground glass in the telescope for magnifications of 80,
160, 200 and 400 X. The telescope in its present state suffers from the
following two disadvantages: (i) focusing at the ground glass of the tele-
scope does not insure focusing at the film plane for all magnifications, and
(ii) the telescope allows one to see only a small part of the field of view of
the microscope (as seen through the eyepiece) .
Focusing can alternatively be effected by placing an acrylic
sheet (size 6" x 4.75" x 0.25") with one face ground at the film plane
(that is, on top of the wooden box) and viewing through a comparator.
The custom 70 mm. camera (k) seen at the top is linked with
an Intervalometer (1) with which the exposure time and the time between
exposures can be controlled. For the present study, a Polaroid Film Holder
was used for photographing the crystallization process on a microscope
slide inside the hot stage. Motion pictures of transcrystalline nucleation
and growth processes were also taken using a Bolex 16 mm movie camera.
/ At the bottom right corner in Figure 8 can be seen an arrange-
ment (m) consisting of solid state rectifiers for converting the 110 volt
alternating current (A.C.) from the main to direct current (D.C.) which
drives the solenoid activating the mirror. The D.C. source is schematically
represented in Figure 9. The light dimmer in Figure 9 provides a contin-
uously variable wattage (zero to 600 watts) . A side view of each silicon
rectifier (6 ampere, 200 PIV , stud mount) , represented by a triangle in
Figure 9, is shown in Figure 10.

The D.C. source is used in conjuncUon with the Intervalometerto operate the solenoid. For taking Polaroid pictures, neither the Inter-
valometer nor the D.C. source is needed. In this case, the 45° mirror is
kept vertical (no obstruction to light passage), the exposure time being
controlled by the cable-release shutter (g) .
5.3 Procedure
The polymers were crystallized in contact with different sub-strates inside the hot stage mentioned in the preceding section. Both the
substrate and the crystallizing polymer were used in the form of films of
thickness 2 to 6 mils, prepared as described below.
5.3.1 Sample Preparation
5.3.1.1 Substrates. Films of polymers to be used subsequently as sub-
strates were prepared by pressing the polymer between two aluminum foils
in a Carver press at 15°C above the melting temperature and 2500-5000 psi
pressure, followed by quenching with cold water (15°C) . The films were
stored in a desiccator.*
The polymer substrates in many cases were initially quenched
to below their glass transition temperatures, while being made into films.
Crystallinity developed in the crystallizable polymer substrates (e.g.
,
isotactic polystyrene, nylons 6, 66, and 610) while the polymer whose
crystal nucleation was being studied was being melted and crystallized
in contact with the substrate.
The nylon 6, 66, and 610 substrates were heated in an oven
(105°C) overnight before use, in order to remove any residual moisture.
Since the glass transition temperatures of these nylons lie in the range

44
50 - 75 C.annealing at !05°C is expected to increase the crystallinity of
the nylons. The degrees of crystallinity of the substrates were „o, measuredAluminum and gold substrates were used as received, in the form of foils.^^W^Sl^. Films of the polymer ,o be crystalli2ed
prepared following the same method as used for the substrates. and stored
in a desiccator
.
5.3.2 Crystallization
Freshly-cut surfaces of the substrate films were used for
studying their nucleating ability. The substrate films were cut
perpendicular to the film plane with a clean pair of scissors. The sample
mounting is shown in Figure 11. Two films of the polymer to be crystallized
and one cut substrate film were assembled on a microscope slide and covered
with a glass cover slip. The microscope slide was then mounted inside the
Mettler FP2 Hot Stage. The crystallizing polymer was in contact with the
substrate at the AB and CD interfaces (Figure 11) , while in the regions
AEFB and CGHD, bulk crystallization of the polymer took place.
For crystallization,the temperature of the hot stage was first
raised to a point (T^ above the melting temperature (TJ of the polymer
to be crystallized (but below the melting temperature of the substrate),
held at Tj for a few minutes, and then the temperature dropped rapidly to
the crystallization temperature. The exact temperatures and times are shown
in Tables II through Vin. The morphology of the crystallizing polymer
induced by the substrate was then observed directly under the microscope
and photographed.
Calibration, using pure compounds of known melting points,
namely para-nitro toluene, biphenyl, naphthalene, and benzoic acid,
showed that the indicated temperature was accurate within ±0.5°C.

5.3.3 Nucleation Densities
The number of spherulites nucleated on the substrate surface
and in bulk were counted directly under the microscope, during isothermal
crystallization. Nucleation densities were calculated from these numbers bydetermining the area of the substrate and the volume of the bulk from the
measured sample thickness and the diameter of the field of view of the microscope.
Figure 12 shows the experimental arrangement for counting the
number of nuclei. The circle ACBD represents the field of view of the
microscope. The substrate was placed so that its cut surface (AB) was
along a diameter of the field of view. Figure 12 shows five hemispherical
spherulites nucleated on the substrate and four spherulites nucleated in
the bulk. As the polymer sample is sandwiched between two glass sur-
faces (Figure 11) ,the number of nuclei counted in the bulk includes those
originating from the glass surfaces.
The diameter of the field of view of the microscope was measured
with the help of a stage micrometer.
The total thickness of the assembly (glass slide plus polymer
sample plus cover slip) in Figure 11 was measured after the crystallization
with a micrometer. Subtraction of the thickness of the glass slide plus the
cover slip from the total thickness gave the thickness of the polymer sample.
5.3.4 Nucleation Rates
Rates of nucleation on the substrate and in bulk were measured
by counting the number of nuclei as a function of time , as crystallization
progressed isothermally, and determining the slopes of nucleation density
vs. time plots.
The temperature dependence of the nucleation rates was studied
by performing crystallization experiments at different temperatures, keeping

46
to the same portion of the sample (substrate and bulk, under observabonwithout disturbance between experiments. This technique ehminatcd thevariation of the nucleation arising from one area eiement of the substrate to
another at the same temperature. Variations in the volume elements in thebulk were also thereby minimized.
5.3.5 Growth Rates
Linear growth rates of the transcrystalline region and the radial
growth rates of the spherulites in the bulk were measured at the same crys-tallization temperature by direct observation of the growth process underthe microscope, using a micrometer eyepiece. The scale in the micrometer
eyepiece was calibrated using a stage micrometer. Growth rates were
obtained from the slopes of linear distance-time plots.
5.3.6 Ti and Al Imp uritie s in Isotactic Polystyrene Samgles
In the present investigation, it was observed that samples of
isotactic polystyrene (iPS) are able to induce transcrystallinity in isotactic
polypropylene (iPP) as well as in polycaprolactone. A series of experiments
was performed to ascertain whether the high nucleating ability of the isotactic
polystyrene substrate was due to the polymer itself or to the catalyst resi-
dues (Ti and Al compounds) in the sample of iPS, which was synthesized
by Ziegler-Natta polymerization93
. The iPS sample was subjected to (i)
centrifugation,
(ii) treatment with a chelating agent, and (iii) fractionation,
as described below.
5.3.6.1 Centrifugation. In anticipation of the lower solubility and higher
density of the metal-containing impurities, compared to the polymer, the
following experiment was performed. A sample of iPS (3 g .) was taken in
toluene (125 ml.) and stirred until partial solution of the polymer resulted.

47
The supernatant was centrifuged at 12000 rpm for 6 . 5 hours, usjng ,^
centrifuge. From the centrifugate IPS was recovered oy precipitaUon in
methanol; iPS was then filtered, washed with methanol and dried undervacuum
This technique did not reduce the ash content of iPS (initial
2.07%, after centrifugation 2.17%)
5-3.6.2 Use of a Chelat^A^^Both Ti and Al are
known to form organo-soluble chelates with acetylacetone (AA)94
' 95and it
was possible to remove these metals, though not completely, from the iPS
sample by this method.
Acetylacetone (7 ml.) was suspended in distilled water (40 ml.)
and ammonium hydroxide added dropwise with stirring until a clear solution
resulted. Then 0.5 g of the iPS sample was added and the suspension was
stirred at 29°C for 5 hours with a magnetic stirrer. The solid polymer was
then filtered and washed successively with benzene, carbon tetrachloride,
methanol and acetone, to dissolve and remove Ti- and Al-acetylacetonates.
Finally, acetone was removed from the sample by drying under vacuum.
Ash contents as well as Ti and Al contents of the initial iPS and
AA-treated iPS samples were determined. The difference in the nucleating
abilities of these two iPS samples in the crystallization of iPP was then
investigated by crystallizing iPP in contact with the iPS substrates and
observing the polypropylene morphologies induced by the iPS substrates.
The results are included in the following chapter.
5.3.6.3 Fractionation of Isotactic Polystyrene . The sample of iPS
(2g.) was taken in 200 ml. toluene and dissolved by heating at 100°C and

48
stirring „ith a magnetic^ A^ ^^^ ^theBM
.
four fractions of iPS were iso.ated, usln s the foUowing tech.nique.
Methanol was added dropwise with stirring to the solution ofiPS. When turbidity developed, the nature was warmed gently unti. theturbidity disappeared and then allowed to cool slowly to room temperatureThe precipitated first fraction was separated by decanting the supernatantFrom the supernatant three more fractions were collected using the sameprocedure. The precipitated fractions were washed with methanol andthen dried under vacuum to remove methanol.
The first fraction was found to have ash content twenty times
higher than that of the fourth fraction. The nucleating abilities of these
two fractions of iPS in the crystallization of iPP were compared by observingthe polypropylene morphologies induced by the iPS substrates.
5.3.7 Determination of Intrinsic Viscosities
Intrinsic viscosities of the two fractions of iPS in toluene at 30°C
were determined following the conventional technique2
. An Ubbelohde-type
capillary viscometer was used for flow time measurements, so that successive
dilution of the polymer solution within the viscometer could be made.
Intrinsic viscosity is defined as

49
where c is the concentration of the polymer solution; nsp
is the specific
viscosity defined as
and r, and r,
oare the viscosities of the polymer solution and the solvent, res-
pectively
.
For negligible kinetic energy contributions,
n/ri0 B t/t
o
where t and tQare the viscometric flow times for the polymer solution and
the solvent, respectively. Plots of both n /c vs. c and (In r, )/c vs. csp
were extrapolated to zero concentration to obtain intrinsic viscosities.
Experimentally-determined values of the intrinsic viscosities
were converted to viscosity average molecular weights (M ) using the
, * 96,97equation '
[nj = 9.3 x 10~ 5 M °' 72

50^^^^^^^^The term "surface morphology „m be used t0 refer tQ ^ mor _
phology of the crystallizing polymer in contact with the substrate. Twotypes of surface morphologies should be distinguished: "spherulitic"
(SPH) and "transcrystalline" (TC) . It should be noted that although a
transcrystalline layer is also spherulitic in nature, being composed of
very closely-spaced spherulites. the term "spherulitic surface morphology"
will be used to refer to a situation where the spherulites nucleated on the
substrate are isolated from one another, as shown in Figure 12. With con-
tinued growth, the spherulites shown in Figure 12 will eventually meet
one another.
"Transcrystalline surface morphology", on the other hand, is
characterized by intense lateral crowding of the spherulites nucleated by
the substrate and an easily recognizable columnar layer growing toward
the melt. It is clear that a substrate giving rise to a transcrystalline sur-
face morphology is of higher nucleating ability than a substrate giving
rise to spherulitic surface morphology.
In this study the substrates have been classified into the
following three groups, in order of decreasing nucleating ability.
Type I. Very Active Substrates. The very large number of
nuclei generated on such a substrate gives rise to a transcrystalline
surface morphology. Nucleation is overwhelmingly favored at the sub-
strate, compared to the bulk.
Type II. Moderately Active Substrates. This group is charac-
terized by surface morphologies of the following four types, in order of

51
decreasing nucleating ability of the substrate: (i, TC (mostly, + SPH,(ii) TC + SPH, (iii) SPH (mostly) TC, (iv) SPH.
Subdivisions (i, through (iv, represent cases of mixed surfacemorphology; examples can be seen in Chapter VI. Case (i) represents a
situation where more than half of the substrate gives rise to transcrystal-
lini.y and the rest gives rise to spherulitic surface morphology. Case (ii)
represents a substrate, one half of which gives rise to transcrystallinity andthe other half to spherulitic surface morphology. When the dominant surface
morphology is spherulitic, case (iii) results. Finally, case (iv) refers to
a completely spherulitic surface morphology
.
Type III. Inactive Substrates. These substrates generate very
few or no spherulites at all. In this case spherulites are nucleated prefer-
entially in the bulk and those close enough to the substrate grow and meet
the substrate. At the end of crystallization, the substrate is in contact with
bulk-nucleated spherulites.

52
CHAPTER VI
RESULTS
The Spectrum of Nucleating Ability of Substrates
The morphologies characterizing the three types of substrates
referred to at the end of the preceding chapter will be illustrated. Figure 13
shows an example of a Type I substrate, namely iPS which has induced trans-
crystallinity in iPP. Nucleation is strongly favored at the substrate. Figure 14
shows a Type II substrate,namely Kel-F or poly (chlorotrifluoroethylene)
,
which is only moderately active in the crystallization of iPP. Nucleation
in this case is seen to have occurred at the substrate surface as well as
in the bulk, without any preference for either. An example of a Type III
substrate is shown in Figure 15. The nylon 610 substrate is inactive as
a nucleating agent in the crystallization of PEO . Spherulites are nucleated
preferentially in the bulk of PEO.
In Tables II through VIII are listed the surface morphologies
observed and surface types (I, II or III) in the crystallization of iPP, PB1,
PEO and PCL in contact with the indicated substrates at different tempera-
tures. Four different samples of iPP were used, as indicated in Tables
II through V. For iPS substrate , data are available for different samples,
different fractions from the same sample as well as for an acetylacetone-
treated sample (of reduced Ti and Al content) . Each of these iPS sub-
strates was able to induce transcrystalline nucleation of iPP, as can be seen
from Tables II through V
.
Effect of Crystallization Temperature on Morphology Induced by Substrates
The surface morphology induced in a crystallizing polymer

53
by a substrate was observed to depend on the crystallization temperature.
Examination of a particular substrate-crystallizing polymer pair in TablesII through V and VEI shows invariably that as the crystallization temperature
is raised, the surface morphology changes from transcrystalline to spheru-
litic. At intermediate temperatures, mixed surface morphologies (trans-
crystalline plus spherulitic) are observed.
Figure 16 illustrates the reduced nucleating ability of iPS sub-
strate with increasing temperature in the crystallization of iPP. The surface
morphology is transcrystalline at 125°C (Figure 16 i) and at 130°C (Figure
16 ii);
it is half transcrystalline and half spherulitic at 133°C (Figure
16 iii),while at 135°C (Figure 16 iv) , the surface morphology is completely
spherulitic.
Surface Morphology
Examination of Tables II through V shows that iPP (monoclinic
unit cell) can transcrystallize in contact with iPS (rhombohedral), nylon 6
(monoclinic),nylons 66 and 610 (both triclinic)
, Penton (orthorhombic)
and PET (triclinic) but Kel-F (hexagonal), PPO or poly (2 ,6-dimethyl
phenylene oxide), atactic polystyrene and Al substrates have lower
nucleating ability towards iPP .
Figures 17 through 20 show the transcrystalline surface mor-
phology of iPP crystallizing at 125°C in contact with nylons 6, 66 and 610,
and Penton, respectively. Spherulitic surface morphology of iPP crystal-
lizing at the same temperature in contact with PPO is shown by Figure 21.
Table VIII shows that PCL (orthorhombic) can transcrystallize
at 50°C in contact with high density polyethylene (orthorhombic), nylons
6, 66, and 610, iPS and polyoxymethylene (hexagonal) . However, iPP,

54
Penton, Kel-F, low density polyethylene, atactic PS, PET, PB1 (rhombo-
hedral),gold and aluminum substrates could not induce transcrystallinity
at 50°C. At 45°C, iPP, Penton and Kel-F were able to induce transcrystal-
linity, but atactic PS failed to do so.
Figures 22 through 24 are photomicrographs showing trans-
crystallization of PCL at 50°C in contact with nylons 6 and 66, and poly-
oxymethylene,respectively. Spherulitic surface morphology generated in
contact with gold at 50°C is shown in Figure 25.
Figures 26 through 28 show the moderate activity of three crys-
talline substrates, namely PET, PB1 and Kel-F, respectively, in the nuclea
tion of PCL crystals at 50°C . In each case, nucleation is favored neither
on the substrate nor in bulk, and the substrate should be classified as
Type II.
None of the substrates used in the crystallization of PEO and
PB1 was found to induce transcrystallinity in these polymers, as can be
seen from Tables VI and VII. For PEO, Figure 29 illustrates a moderately
active (Type II) substrate (iPP), while Figure 30 illustrates an inactive
(Type III) substrate (nylon 6) . Nylon 66 (Figure 31) behaves similar to
nylon 6 ,but since occasionally one or two PEO spherulites were found to
be nucleated on nylon 66 substrate , the latter has been classified as
Type II substrate in Table VII.
It is also worth noting that in the crystallization of iPP (Tables
II through V) , the substrate activity was either of Type I or Type II, no
example of Type in being found. The same is true for the crystallization
of PCL (Table VHI) . In the case of PB1 (Table VI) , all the substrates used

55
were found to be moderately active (Tvoe III in *y U ype II). in the crystallization of PEO
the substrates used were either of Type II or TvnP tttiype 11 or Type III, no example of trans-crystallinity being found.
In the crystallization of iPP in contact with ips
(Table II) at 125°C, it can be seen that increasing the holding time of the
transcrystallinity was generated in each case
.
Crystallinity of the Substrate
Figure 32 shows the higher nucleating ability of iPS, compared
to atactic PS.
in the crystallization of iPP . Isotactic polystyrene was foundto induce transcrystallinity within 2 minutes at 125°C (Figure 32 i) . Atthe same temperature, atactic PS showed only moderate activity at the most(Figure 32 ii)
. Transcrystallinity was found not to develop from the atactic
PS substrate even on reducing the crystallization temperature to 115°c
(Table II).Below this temperature, the bulk nucleation densities in com-
mercial isotactic polypropylenes were found to be high enough to interfere
with the study of morphological feature on substrates.
The higher nucleating ability of iPS compared to atactic PS wasalso found to be true in the crystallization of PCL. Thus at 50°C, while
iPS could induce transcrystallinity (Figure 33 i), atactic PS could not
(Figure 33 ii)
.
Thirdly, at 50°C PCL was found to transcrystallize in contact
with high density polyethylene (Figure 34 i) , but low density polyethylene
gave rise to spherulitic surface morphology (Figure 34 ii) . These results
indicate higher nucleating ability of substrates with higher crystallinity.

56
Inspection of Tables II through VIII reveals that all the substrateswhich are able to induce transcrystallinity are crystalline. On the other
hand, there are many crystalline substrates which are either moderately
active or inactive nucleants. This suggests that crystallinUy is a necessarybut not sufficient condition for a substrate to be a strong nucleating agent.
The author speculates that the requirement of crystallinity in
a substrate for strong nucleating action is probably tied with the fact that
the newly emerging phase is crystalline. In other words, crystallinity of
the substrate may not be a necessary condition for strong nucleaUon action
in the case of heterogeneous nucleaUon of vapors from a liquid or vice
versa.
Figure 32 i shows a feature of the development of crystallinity
in iPS, previously quenched from the melt to about 15°C. Birefringent
striations (e.g. ab) on the iPS film at about 45° to the iPS-iPP interface
were observed to develop while the iPS was being annealed. The annealing
of iPS took place unavoidably while iPP was being melted and crystallized.
The striations can also be seen in Figures 13 and 16 (ii - iv) . Examination
of Figures 16 (iii) and 32 (i) reveals that the striations are not seen at the
lower part of the photomicrographs, that is, away from the iPS-iPP inter-
face. This suggests that the striations may result from the shearing action
due to the cutting of the iPS film with a pair of scissors.
Ti and Al Impurities in Isotactic Polystyrene
Table IX shows the reduction of Ti and Al contents of
iPS sample by treatment with acetylacetone (AA) . Ti content was
reduced to less than half of its initial value , while Al content went
down to one-seventh of the initial value. Comparison

of the nucleating ability of untreated and AA-treated iPS samples in the
crystallization of iPP is shown in Table X. Experiments were performed
with three samples of iPP, at indicated temperatures. It can be seen that
the AA-treated sample did not lose its original nucleating ability. For all
the three samples of iPP, identical surface morphologies resulted in con-
tact with iPS substrates of markedly different Ti and Al content.
It should also be noted in Table X that at 135°C, for the same
sample of substrate, the surface morphology varies, depending on the
sample of the crystallizing polymer (iPP); this is true for both untreated
and AA-treated iPS substrates. The crystallization temperature (135°C)
is within the range where the transition from transcrystalline to spherulitic
surface morphology takes place.
Table XI shows the ash contents of the fractions of iPS . As suc-
cessive fractions were precipitated, their ash contents decreased pro-
gressively. Measurement of the intrinsic viscosities of the iPS fractions
in toluene at 30°C yielded the following values: 3.55 dl/g. for Fraction I
and 1.75 dl/g. for Fraction IV. The calculated viscosity average molecular
weights corresponding to these intrinsic viscosities are 2.24 x 106and
g0.85 x 10 , respectively.
Table XII shows the nucleating ability of the first and fourth
fractions of iPS in the crystallization of iPP (two samples) . The two frac-
tions of iPS differ twenty-fold in ash content, as can be seen from Table XI.
At 130°C, the two substrates had similar nucleating ability. However, at
135°C, Fraction IV had a lower nucleating ability than Fraction I, as indica-
ted by the surface morphologies. This difference might be due to a lower

58
degree *cry™, of Fraction w . Cl„nity^ ^ ^*S substrate while iPP „ being nielted and ^^Unity could result from the lower molecular weight of Fraction IV (highernumber of chain ends) .
Finally, iPP was crystallized in contact with a sample of iPS(Dow EP 1340-128) which by atomic absorption spectroscopy showed < 1
Ti and < 1 ppm Al and which showed ash content of 0.025%. Even at
were
ppm
thesenegligible metal contents, transcrystailinity was generated, as shown in
Table II, Table III and Figure 13.
The results mentioned in this section lead to the conclusion that
the high nucleating ability of iPS substrate in the crystallization of iPP is
due to the polymer (iPS) itself.
Nucleation Densities
Quantitative measures of the nucleating ability of substrates
obtained by counting the number of spherulites nucleated on the substrate
and in bulk and calculating the nucleation densities, namely ng(number
of nuclei/cm 2) for substrate and n
fe(number of nuclei/cm 3
) for bulk.
The experimentally-determined nucleation densities are shown in
Tables XIII through XV, for the crystallization of iPP, PB1 and PEO,
respectively. The substrates have been listed in order of increasing
ns/n
brati0
'The temperatures of crystallization and the times of
measurement have been indicated in the tables . Zero time was taken as
the time when the hot stage reached the crystallization temperature. After
cooling from the melting temperature, the crystallization temperature was
reached in two to four minutes . The times of observation in Tables Xin

59
active at the particular temperature.
The ns/n
bratios listed in Tables XIII through XV are repre-
sented as heights of the bars in Figures 35 through 37. Referring to theIPS and nylon 610 substrates in Table XIII, ,he exact number of nucleicould not be counted by visual observation due to the overcrowding of
spherulites in the transcrystalline region. The minimum number of
nuclei that could be counted is shown. I, follows that the real heights
corresponding to the last two substrates in Figure 35 are larger than the
indicated heights.
The selective nature of heterogeneous nucleahon processes is
evident from the results of Tables II through VIII and XIII through XV . Forexample, Kel-F and iPS have comparable nucleating ability in the crystal-
lization of FBI,but when IFF is the crystallizing polymer, iPS is a much
better nucleant than Kel-F.
Examination of the nucleation density data in Tables XIII through
XV indicate that the nucleation density in bulk (nb
) is of the order of
1010/cm
3.
For PEG at 53°C the order is 109
. For spherulitic surface
morphology, the nucleation density on the substrate (n ), is of the order
5 2s
of 10 /cm and the n^ ratio is of the order of 1QS^ fo^
transcrystallinity is generated, ng
is in excess of 106/cm
2(probably of
the order of 106
- 107/cm
2) .
Figure 38 shows the evolution of nuclei in the early stage of
the development of transcrystallinity in PCL, the substrate being high
density polyethylene. The photographs were printed from motion picture

60
negatives filmed at 2 frames/ sec and 1 frame/ 6 sec. Only four or five
spherulites can be seen to be nucleated at 9 minutes (Figure 38 i) at ADinterface. With the passage of time more spherulites are nucleated on the
substrate (Figures 38 ii to iv) . These photographs show that in the early
stage of the formation of a transcrystalline layer, nuclei appear sporadi-
cally over the substrate. However
, heterogeneous, and not homogeneous
,
nucleation process is involved since Figure 38 shows the virtual absence
of nucleation in the bulk PCL (region AED) .
After counting the number of nuclei by analyzing the film in a
viewer, the nucleation density corresponding to Figure 38 (iv) was
determined to be 6.7 x 105/cm
2. This may be taken to be a stage where
approximately half of the substrate is occupied by spherulites. Considering
the fact that these spherulites were growing, it was assumed that at complete
coverage of the substrate by spherulites, half as many more nuclei will
appear on the substrate. This assumption leads to a nucleation density
(for transcrystalline formation) of 1.5 x 6.7 x 105/cm
2or 10^/cm
2, at the
point of total occupancy of the substrate by spherulites.
To check the reproducibility of the numbers of nuclei, the
experiment of crystallization and counting the number of nuclei was
repeated four times with attention to the same portion of the sample , with-
out any disturbance between crystallizations. The result is shown in
Table XVI. The data indicate that the numbers counted in bulk vary on
repeating the crystallization. The coefficient of variation, defined as (s/m) X
100, was 21% in this case. Here s is the standard deviation and m is the mean.
The number of nuclei on the substrate is too small to derive any useful

61
conclusion. However,the inactive nature of nylon 66 as a nucleant for PEG
crystals is evident.
The bulk nucleation densities in each of Tables XIII through XV at
a particular temperature and time represent nucleation densities in different
volume elements of the same sample of the crystallizing polymer. The mean,
standard deviation and the coefficient of variation for each case are shown
in Table XVII. A comparison of the sixth and last lines (PEO at 51°C) of
the table shows that the coefficient of variation for different volume elements
is greater than that calculated after repeated crystallizations within the
same volume element. It should also be noted that the coefficient of varia-
tion is higher at higher crystallization temperatures; this probably is due
to the smaller number of nuclei counted at higher crystallization temperatures
The variation of nucleation density on substrate (given sample)
from one sample of a crystallizing polymer to another is shown by Tables
XIII (part i) and XVIII. For these two tables, the same substrate sample
was used, but the iPP samples were different, namely Pro-fax X 12886-38
(Hercules, Inc.) for Table XIII and Shell 5520 for Table XVIII. A comparison
of the two tables shows that for each of the three substrates, namely Al
,
atactic PS and Kel-F , the ngand n
g/n
bvalues obtained with the Pro-fax
iPP sample are different from the values for the Shell iPP sample. The
ratios of ng/n
bvalues (Pro-fax to Shell) for the three substrates are shown
in Table XIX. Their pronounced deviation from unity in both directions
shows that ng/n
bratios for a given substrate sample vary from one sample
of a crystallizing polymer to another. It is evident that the ranking of sub-
strates based on ng/n
bratios depends on the particular sample of the crystal-
lizing polymer used.

62
Nucleation Rates
Figure 39 shows typical curves of nucleation density on sub-
strate vs. time of crystallization at three crystallization temperatures. Theinhibition period increases as the supercooling decreases. The surface
morphology at 126°C was transcrystalline and after 3 minutes the number of
nuclei were too many to count accurately by direct observation. At the other
two temperatures, the number of nuclei could be followed until the substrate
area was completely occupied.
It can be seen in Figure 39 that the number of nuclei on the sub-
strate eventually becomes fixed and the growth of these fixed numbers of
spherulites fills the total available substrate area. The number of growing
centers was observed to become fixed before the available area on the sub-
strate was totally occupied. In other words , the horizontal part of the
curve is true up to the complete coverage of the substrate area. A fixed
number of active nucleating sites on substrate surfaces is thus indicated.
The shape of the curve is general; iPP , PB1 and PEO crystallizing in con-
tact with various substrates showed this behavior.
At higher crystallization temperatures the nucleation density
decreases. The height of the plateau in Figure 39 gives the total number
of nuclei active at a particular temperature. The difference between the
plateau heights gives the additional number of nuclei that become active
for a given drop in the crystallization temperature. Thus, according to
Figure 39, 30% more nuclei became active on the substrate when the crys-
tallization temperature was dropped from 128° to 127°C.
Curves of similar shape were obtained when the nucleation den-
sity in bulk was plotted against time of crystallization, as shown by

63
Figure 40. Here too, the number of growing centers, after initial risebeco.es constant before the total volume is filled with spherulites. Thus» bulk also there exists a fixed number of active heterogeneities which areresponsible for nucleation at a given temperature.
For both substrate and bulk, two cases have been observed in
connection with the nucleation density-time curves, namely (i, the initial
part of the curve is linear (e.g. Figure 40, , and (ii, the curve is S-shaped(e.g. Figure 41) . Nucleation rates have been obtained for the first casefrom the slope of the initial linear part, and for the second case from the
maximum slope of the curve.
Experimentally determined nucleation rates on substrate (I )
ssand in bulk (I
sb ) as well as1^ ratios are listed in Tables XX and XXI,with PB! and PEO, respectively, as the crystallizing polymer. The I /I
ratios for different substrates have been represented as the heights of the
bars in Figures 42 and 43.
To study the reproducibility of the bulk nucleation rates, the
crystallization of PEG was performed four times, with the same portion of
the sample under observation. The plots of nucleation density vs. time for
the four runs are shown in Figure 41 . It can be seen that the curves are
not exactly reproduced on repeating the experiment, though the same shape
is obtained each time. However, no definite trend is noticeable in these
curves. The nucleation rates have been calculated from the maximum slopes
of the S-shaped curves in Figure 41 and shown in Table XXII. Their mean,
standard deviation and coefficient of variation have also been included.

64
The different bulk nuclea(ion raies jn TaMes ^sent variation from one volume ,tament t0 another h ^ bu[k Qf^^P-^r sample. The values of the mfian
, standard deviatjon coefflcjen[of variation are entered in Tabie XX,„ . The large coeffjclent Qf
for PEO is probably due to the smaller number of spherulites counted, thenature of heterogeneities in the PEO samole and th» o „fcsampie, and the S- shaped nucleationrate curves which introduce arrnr i«ntrociuce error m the measurement of the slope of thenucleation density vs. time curves.
In one instance, the reproducibility of the nucleation rate onsubstrate was checked. The determinabon of nucleation rate was repeatedwith the same pair of samples but different portions in contact. The agree-ment, as shown in Table XXfV, is good. However, whether this goodagreement will be observed consistently can only be ascertained by fur-ther repetition of the experiment.
Finally, it should be noted that the nucleation rates on substrates
studied are of the order of 104
- 105/min/cm 2
and the bulk nucleation rates
are of the order of 109
- 1010/min/cm 3
.
Temperature Coefficient of Spherulitic Growth Rates
Spherulitic growth rate (G) data for poly (butene-1) at different
temperatures of crystallization are shown in Table XXV. The plot of log Gvs. 1/CTAT) is shown in Figure 44. The calculated least square slope of
the straight line is - 0.622 X 105 V. According to Equation 45 (Chapter IV)
for spherulitic growth rate, the slope is equal to - 4b oa T /(2.3k AH ) .e m v
The melting temperature (TJ of PB1 was determined to be
134. 3°C, the method having been described in Chapter 5. From the litera-98
ture,the heat of fusion of poly (butene-1) was taken as AH =
v

65
35.7 cal/g. . 1.42 X 10yerg/cm 3
. Using these data and assuming b = 5A,
the interfacial energy product ooqwas calculated to be 243 erg
2/cm
4.
Temperature Coefficients of Heterogeneous Nucleation Rates
Figure 45 shows the number of nuclei counted on iPS substrate as
a function of time in the crystallization of PB1 at different temperatures. In
each case PB1 was premelted at 140°C for 4 minutes. The decreasing inhi-
bition period and increasing nucleation density with increasing supercooling,
as well as the fixed number of active nucleating sites on the substrate, as
indicated by the levelling off of the curves, have already been pointed out.
The number of nuclei on the substrate became constant before the available
area on the substrate was occupied totally by spherulites.
The nucleation rates were calculated from the slopes (N ) of the» s
plots in Figure 45. The data are shown in Table XXVI. The quantity log
Ngwas plotted against 1/[T(AT)
2] (Figure 46) and the least square
straight line was drawn through the points. According to Equation 37 for
heterogeneous nucleation (Chapter IV) , the slope of the straight line is
equalto- 16 aaeAo Tm
2/ [2. 3 k (AH
y )
2]. To make this clear , it should
be noted that Nghas the unit # nuclei/min, while I has the unit #
s2
nuclei/min/cm and if a = area of the substrate,
I = N /as s

66
Differentiating,
d log I d log N1
s
r1
J (AT)2 d
T (AT)2
since a = constant, by experimental setup.
Using the previously-mentioned values of Tm and AHy
for poly-
(butene-1),the triple product oo
qAc for iPS substrate was calculated to be
531 erg3/cm 6
.
Similar experiments, namely measurement of nucleation rates
of PB1 at different crystallization temperatures, were performed using iPP as
the substrate. The data are shown in Table XXVII and Figures 47 and 48.
Plots of nucleation density vs. time for the bulk crystallization
of PB1 at different temperatures are shown in Figure 49. The existence of
a fixed number (decreasing with increasing crystallization temperature) of
active heterogeneities in the bulk polymer, and the increasing inhibition
period and decreasing nucleation rate with increasing crystallization tem-
perature are in agreement with the results of Sharpies" for the crystalli-
zation of poly (decamethylene terephthalate) . The temperature dependence
of the nucleation rates of PB1 is shown in Table XXVHI and Figure 50. In
this case the value of oa Aa was calculated to be 283 erg3/cm
6.

67
The calculated values of the interfacial energy parameters are
summarized in Table XXIX. The value of Ac was obtained by dividing 00*0by aa .J e
The quantity (om - 0(J in Table XXIX represents the difference
between the substrate-melt and substrate-crystal interfacial energies and
hence is a measure of the nucleating ability of substrates; larger values of
(om - oj indicate better nucleating ability. Table XXIX shows that iPS and
iPP substrates have practically the same nucleating ability toward PB1. This
is in agreement with the surface morphology studies (Table VI) . Both sub-
strates showed moderate activity (Type II) in the nucleation PB1 crystals.
Table XXIX also shows that the nucleating ability of the surfaces
of the foreign particles present in the PB1 sample is comparable to those of
iPS and iPP substrates.
Transcrystalline Growth Rates
Transcrystalline layers of iPP and PCL were found to grow with
a constant linear velocity. Comparison of the linear growth rate of the trans-
crystalline region with the radial growth rate of bulk-nucleated spherulites
is shown in Table XXX. For iPP, the values of the radial growth rates of
spherulites are in good agreement with those reported in the literature100
.
For PCL, no growth rate data at 50°C was available for comparison at this
writing
.
Table XXX indicates that the linear growth rate of the transcrys-
talline region is the same as the radial growth rate of the spherulites. Exam-
ination of the last two lines of this table also shows that the transcrystal-
line growth rate is independent of the substrate responsible for the trans-
crystalline nucleation.

68
Melting Temperature
The meltag temperatures of the transcrystalline polymer-d that of the bulh-nucleated spherulites were determined by directobservation of the disappearanoe of the birefringence of these tworegions, under the same field of view of a polarizing microscope, a,
a beating rate of 0.2°C/min. The two melting temperatures were foundto be identical, namely 171. 8°C for iPP and 64.2°C for PCL.
*

69
CHAPTER VII
DISCUSSION
Examination of the forty three substrate-crystallizing polymer
pairs during the present study has confirmed the selectivity of heterogeneous
nucleation processes. However
, the necessary and sufficient conditions for
effective heterogeneous nucleation are not yet definitely established. The
present study represents progress towards this goal.
It has been noted during the present investigation, particularly
in the crystallization of PEO and PB1, that a number of particles in the melt
visible under the microscope gave rise to no spherulites at all. This indi-
cates that the mere presence of a foreign surface in a supercooled melt is not
enough for nucleation to occur.
No definite correlation has been found between nucleating ability
and similarity of chemical structure of the substrate and the crystaUizing
polymer. In fact, examples have been found for all the four classes, shown
in Table XXXI. The conclusion must be drawn that similarity in chemical
structure is no certain criterion for strong nucleating ability.
No definite correlation was found between strong nucleating action
and similarity of crystallographic unit cell type of the substrate and the
crystallizing polymer. Again, examples found for each of the four classes
are shown in Table XXXII. Similarity of unit cell type evidently is not a
necessary condition for strong nucleating action.
The role of crystallographic unit cell parameters in heterogeneous
nucleation will now be discussed. In this study, strong nucleating action
has been observed in both cases, namely with and without similarity in

70
the unit cell parameters of the substrate and the crystallizing polymer. Someexamples will follow. The crystal structures of PCL and PE are very similar.
Both crystallize with orthorhombic unit cells, the dimensions (in % of which
have been reported to be as follows: 101
c
(fiber axis)
PE 7.400 4.930 2.534
PCL 7.496 4.974 17.279
During the present study, PCL has been found to transcrystallize
in contact with high density polyethylene (Figure 34 i); however, low den-
sity polyethylene showed a lower nucleating ability (Figure 34 ii) . Decreased
crystallite size or increased lattice distortion in low density polyethylene may
be responsible for this result.
The other substrates which have been observed to induce trans-
crystallinity in PCL are nylons 6, 66 and 610, isotacuc polystyrene and poly-
oxymethylene . However , comparison of the unit cell parameters of these
polymers102
with those of PCL 101shows no particular similarity.
In the case of iPP, transcrystallinity has been found to be induced
by iPS, nylons 6 , 66 and 610, PET and Penton substrates. The crystallo-
graphic b and c axes of the unit cells of iPP and iPS are very similar and
1 fi?both polymers form 3-fold helices in the crystalline state . However, no
significant similarity exists in the unit cell parameters of iPP and any of the
1 02other five polymer substrates mentioned above

71
The evidence of strong nucleating action without any close match of
lattice parameters is an exception to the theory of Turnbull and Vonnegut 103,
according to which nucleating efficiency should increase with increasing
closeness of match between the lattice parameters of the substrate and the
forming crystal. Exceptions to the Turnbull-Vonnegut theory have been
found earlier also, e.g. Beck35
noted that although sodium formate has a
better match of lattice parameters with PP than does lithium benzoate, the
latter is a much better nucleating agent. One must conclude that although
there are known examples where the Turnbull-Vonnegut theory is obeyed 103,
match of lattice parameters is not a necessary condition for strong nucleating
action.
The three nylons (6, 66 and 610) have been found in this study to
behave similarly in the nucleation of crystals of iPP. This is also true for the
crystallization of PCL and PEO . Thus , each of the three nylons can induce
transcrystallinity in iPP and PCL, but is inactive in the case of PEO. The
pronounced difference in the nucleating ability of a particular substrate,
from one crystallizing polymer to another, should be noted.
Transcrystalline nucleation and growth of polymers on polymer
substrates have been reported earlier, e.g. PE in contact with Mylar53
and iPP in contact with Mylar and TFE-Teflon54
. The present study has
brought to light further examples of this phenomenon, with iPP and PCL
crystallizing in contact with crystalline polymer substrates. To the author's
knowledge, no known example of transcrystallinity induced by an amor-
phous substrate exists.

72
It should be noted that well-developed transcrystalline morphologyis observed only in a certain range of crystallization temperature. At highertemperatures, transcrystallinity is not seen because of the reduced nuclea-te density on the substrate. On the other hand, at temperatures belowthe range, although a very high nucleation density initially exists on the
substrate, a large number of spherulites are also nucleated in the bulk of
the crystallizing polymer. These spherulites meet the transcrystalline
region very early in its development. Thus the volume is filled quickly anda well-developed transcrystalline morphology is not seen. The convenient
ranges of crystallization temperature for observing transcrystallinity have
been found to be: 120-130°C for iPP and 48-52°C for PCL
.
The results of the present investigation, as well as those of Fitchmun
and Newman 53' 54
,indicate that low energy surfaces of polymers are capable
of inducing transcrystallinity, contrary to claims in the literature that only
high energy surfaces can induce transcrystallinity52
' 58 ' 59. in fact, during
the present study, PCL has been found to transcrystallize in contact with a
number of low energy surfaces at 50°C (Table VIII), while gold and aluminum
(high energy surfaces) were unable to induce transcrystallinity (Figures 22
through 25, 33 i and 34 i) . Also , Table II reveals that Al could not induce
transcrystallinity in iPP at 125°C, while a number of crystalline polymers
were able to do so.
The results mentioned above lead one to believe that surface
energy of a substrate does not dictate its nucleating ability. This thesis
is supported by the results of a study by Koutsky, Walton and Baer on the
heterogeneous nucleation of PE from the melt on cleaved surfaces of alkali

73
halide singie Wtth»< Changing the surface energy ^ hajidesubstrates by a factor of more than four ^ ^ resuu ^^ signiacantchange in th. t, - ^ values for toe ^ ^ ^ ^^of 5 - 7 erg/cm . As indicated earlier
, a Mgher value of _ o^ indjcatesa better heterogeneous nucleating agent.
The melting temperature of the transcrystalline region has beenfound in this study to be identical with that of the bulk-nucleated spheruiitesfor both iPP and PCL as the orystailizing polymer. This finding is not in
agreement with that of Matsuoka Pt a i61 uiatsuoka
,et al
. ,who reported that transcrystalline
PE has a melting point 5°C lower than that of spherulitic bulk PE. It shouldbe noted that in the present study the melting process was directly observedunder the polarizing microscope at a heating rate of 0.2°C/min
( with the
transcrystalline region and the bulk-nucleated spheruiites occupying the
same field of view. The data of Matsuoka, et al., are based on differential
scanning calorimeter thermograms obtained at the heating rate of 10°C/min.
The change of the surface morphology from transcrystalline to
spherulitic on increasing the crystallization temperature (Figure 16) is due
to the reduced rate of nucleation at smaller supercooling, as predicted by
the theory of heterogeneous nucleation.
At intermediate temperatures (e.g. 133°C for Figure 16 iii),
mixed (transcrystalline plus spherulitic) surface morphologies have been
observed. Examples can be seen in Tables II through V and VIII. The sub-
strate-crystallizing polymer interface (ab) in Figure 16 iii is 2.015 mm long.
The left half of the interface shows spherulitic morphology while the right
half shows transcrystallinity. Evidently, the right half has a stronger

74
nucleaang effect. Any explanation for this observabon should take into
account the fact that mixed surface morphologies are observed only at the
intermediate temperatures of crystallizadon; above this range one observes
spherulitic surface morphology, while below this range, transcrystallinity
is seen.
One speculation will be made in order to explain the existence
of mixed surface morphologies, namely that the right half of the substrate
(Figure 16 iii) is more wettable by iPP crystals, compared to the left half,
thereby giving rise to a higher nucleation density in the right half. The
wettability differences do not result in a morphological difference at lower
temperatures (when transcrystallinity is generated) or at higher tempera-
ture (when spherulitic surface morphology results) . In other words,
probably there exists a difference in wettability between the right and
left sides of the substrate at all temperatures, but only at intermediate tem-
peratures this difference gives rise to different morphologies in different
regions of the substrate.
Apart from transcrystallinity, there exists a phenomenon involving
another type of oriented overgrowth on substrates, namely epitaxial crystal-
lization105 115
. However, it should be noted that while transcrystallinity
is defined by its characteristic morphology, for epitaxy a definite orienta-
tion relation exists between the crystalline substrate and the forming
crystal, namely that the planes and directions in the two crystals in which
the atomic arrangement is most similar are parallel103
. High polymers
(e.g. PE, polyamides, iPS, Penton) have been grown epitaxially from

75
solution onto selected faces of cleaved alkali halide single crystals107
' 109 '
110,113-115 _ .
.Oriented rod-like crystallites of the growing polymer on
the substrate are usually observed. The macromolecular chains lie
parallel to the substrate surface. Koutsky, et al.
104, observed that rod-
like PE crystallites form on the (001) face of alkali halide single crystals
when PE melt is nucleated on them .
Epitaxy of low molecular weight substances on oriented polymer
substrates was observed by Richards 116 and Willems108
. Richards found
that paraffin wax crystals grow epitaxially from solution onto cold-drawn
PE and polyethylene sebacate, but not onto cold-drawn PET, nylon 66,
nylon 610, natural rubber or gutta percha substrates. The chain molecules
in the substrates were oriented parallel to the direction of drawing. With
the PE substrate, the paraffin wax molecules were found to be oriented
parallel to the PE molecules.
108Willems observed the epitaxy of pentachlorophenol (PCP) and
hexaethylbenzene on the surface of a strip of cold-drawn PE and the epitaxy
of PCP, pentabromophenol, pentachloroaniline and anthraquinone on the
surface of cold-drawn strips of nylon 66.
117Takahashi, et al. , reported the epitaxial crystallization of
polymers from the melt on uniaxially drawn and annealed polymer substrates.
This study involved the crystallization of polymers from the melt on the
surface of other polymers and, therefore, among the studies mentioned,
bears the greatest resemblance to the present study. The results of
Takahashi, et al. , and those obtained during the present study are in
excellent agreement. Thus , Takahashi, et al. , reported that PCL grows

76
epitaxially on HOPE, nylon 6 and POM substrates, but not on LDPE or PP
According to the present study (Table VIII), PCL transcrystallizes at 50°C
in contact with HDPE,nylon 6 and POM, but LDPE induces a spherulitic
surface morphology and iPP induces a mixed surface morphology.
Based on the tendency of crystallographic planes of closest
atomic packing to appear on the surface (as a stable crystal habit),
Takahashi, et al., assumed that the (110) planes of PE are exposed
locally on the substrate and are responsible for the nucleation of PCL
crystals.
In the present study the polymer substrates used were cut
surfaces from pressed films (undrawn) . The orientation of the chain
molecules in the substrate was not determined. In the absence of the
necessary information, no particular crystal plane of the substrate can
be singled out to be responsible for generating transcrystallinity . It
certainly is desirable to clarify whether only particular crystal planes of
the substrates are responsible for transcrystalline nucleation or all the
crystallographic faces of the substrates are capable of inducing trans-
crystallinity .
The nucleation density vs. time curves for crystallization in
contact with a substrate (Figures 39, 45, 47) and for crystallization of
the bulk polymer (Figures 40, 49) exemplify pseudohomogeneous nucleation
in those cases where the transient in the number of nuclei is relatively long
in duration. The levelling off of the number of spherulites before the
available area (or volume) is filled indicates that homogeneous nucleation
is not involved. However, due to weak substrate-polymer crystal

77
interaction, spherulites are nucleated sporadically in hme on the substrate(and in bulk)
,thus minicking the process of homogeneous nucleation.
It has already been mentioned that the shape of the nucleation
density vs. time curve can have two variations, namely (i) with an initial
linear part (Figure 40) , and (ii) S-shaped curve (Figure 41) . When the
embryo size distribution adjusts itself to that corresponding to the crystal-
lization temperature at a rate greater than or equal to that at which the
crystallization temperature (T) is reached, curves of type (i) result. Onthe other hand, in cases where the hot stage has reached the crystalliza-
tion temperature, but the embryo size distribution has not yet adjusted
itself to the state corresponding to T, S-shaped curves result. This is
because even though the crystallization temperature is reached, the system
is still adjusting itself from a state of higher temperature to one of lower
temperature, and a lower nucleation rate is observed in the early stage,
giving rise to the foot of the S-shaped curve.
Tables XXVI through XXVIII reveal that for the crystallization
of PB1 in contact with substrates (iPS, iPP) and for the bulk crystallization
of this polymer,the heterogeneous nucleation rates increase by a factor
of approximately 5 for a 10°C increase in the supercooling. Such tempera-
ture coefficients are to be contrasted with the much stronger dependence
of homogeneous nucleation rate on supercooling. For PE, the homogeneous
nucleation rate changes by a factor of 10 for a degree change in super-
1
8
cooling.
For normal alkane liquids, Turnbull and Cormia118
reported
increase of homogeneous nucleation rate by a factor of 5000 to 8000 per
degree decrease in temperature.

78
Examination of the temperature dependence of spherulitic growthrates of FBI (Table XXV) indicates a (5 to 10) fold increase in the growthrate for 10°C increase in the supercooling. This dependence is comparableto the temperature coefficient of the heterogeneous nucleation rates of FBIand is much weaker than the temperature dependence of homogeneous
nucleation rates. This result is not surprising since spherulitic growth is
believed to be controlled by coherent nucleation on already-formed crystal
faces, and not governed by homogeneous nucleation processes.
Measurements of the temperature coefficients of the heterogeneous
nucleation rates have permitted the calculation of Ao and (a - a ) values
for substrates in the crystallization of PB1 (Table XXIX) . The values of
(om - oc ) can be used to compare the nucleating ability of the indicated
substrates. Furthermore, the substrate-melt interfacial energy(0 ) can
be obtained from measurements of the contact angle of a drop of the melt on
the substrate. This will enable one to obtain absolute values of a , thec
substrate-crystal interfacial energy. To the author's knowledge, values
of ochave neither been directly measured nor indirectly calculated so far.
Glicksman and Childs119
also obtained values of (c - a ) fromm c
experimental studies of the heterogeneous nucleation of metallic tin from the
melt on inorganic substrates, namely metals, metallic oxides, carbides
and sulfides. They used the spherical cap model of the crystalline phase
nucleated on a plane substrate and experimentally measured the critical
supercooling necessary to induce a pre-estimated sensible nucleation rate.
Their calculated values of (a - a ) were in the range of 35-58 erg/cm2
.
The values indicated that in the crystallization of tin , metallic substrates
are better nucleants than metallic oxides, carbides or sulfides.

The nucleation density data (Tables yttt aUables XI" through XIX) are applicable for one temperature and one ^ ^
obtained from the temperatoe ^rates, the effects of both time and temperature are taken^^ ^
terizing the nucleating ability of =:iih«t,- a .„.my substrates compared to either nucleationdensities or nucleation rates by themselves.
in the present study the rates of heterogeneous nucleation havebeen measured by counting the number of spherulites every minute. Moreaccurate nucleation rates will be obtained by counting the number of
spheruHtes at smaller intervals of time. e.g. 10 Seconds. from motionPicture frames. This technique will enable one to investigate in detail
«he reproducibility of the nucleation density vs. time curves on repeatingthe crystallization experiment. Such a study by Abbs with the isotachc
polypropylene-isotactic polystyrene system is in progress now 120.
The present study has indicated the existence of a fixed numberof active nucleating sites on substrate surfaces. The work of Zettlemoyer
and his coworkers 121 indicated isolated nucleating sites on the surface of
silver iodide crystals which have been used as nucleating agents for ice
formation. They showed that silver iodide surface is largely hydrophobic,
but has hydrophihc sites on it. These hydrophilic sites are responsible
for the nucleation of ice crystals.
The existence of localized active nucleating sites on a substrate
thus appears to be a general phenomenon. The distinguishing feature of

80
these active sites is only a matter of speculation at this writing. It is also
unknown whether the nature of the active sites for polymer crystallization
is the same as or different from that of the active sites responsible for the
nucleation of low molecular weight substances. During the present study
direct light microscopic observations did not reveal any distinguishable
physical feature characterizing the active sites. Four speculations as to
the nature of the active nucleating sites will be offered now.
First, steps and dislocations on the substrate surface may be
responsible for nucleation. Secondly, the active sites may have cracks or
fissures inside which crystalline residues can survive at temperatures suf-
ficient to melt the polymer on the rest of the substrate, in accordance with a
theory due to Turnbull83
. On supercooling the melt, the crystalline resi-
dues act as seeds for the nucleation of spherulites. Thirdly, the active
sites may be envisioned to have more wettability (by the crystalline
embryo) compared to the inactive sites. In other words, (a - a ) valuesHi C
differ from one site on the substrate to another. Sites characterized by
(om "°c ) values lower than som e critical value are inactive. Finally, the
active sites on polymer substrates may have chain conformations which are
conducive to nucleation. Results of the present study do not favor any of
the four speculations.
This chapter will be concluded with some remarks on the poly-
morphism of poly (butene-1), 1,4-trans polybutadiene , and nylons 6, 66
and 610.
Three crystalline polymorphs of PB1 have been reported98
'122-127
,
namely forms I (rhombohedral) , II (tetragonal) , and III (orthorhombic),

81
with molting points of 135.5°, 124°, and 306. 5°C, respectively 127. On
crystallization from the melt, form II is formed first; it slowly transforms
into form I which is the more stable form124
' 126 - 127 - 128 o• torm I may be
taken to be responsible for the behavior of FBI substrate in the crystalli-
zation of PCL , shown in Table VIII
.
According to Natta and Corradini129
, a first order crystal-
crystal transition takes place in 1 , 4-trans polybutadiene (TPB) at 67°C.
The high temperature form is characterized by a packing mode in which
the macromolecules have increased freedom of rotation about the chain
axes, compared to the low temperature form. A density decrease of more
than 9% accompanies this phase change.
While studying the nucleating ability of TPB in the crystalliza-
tion of PEO (Table VII), the pair of polymers was first heated at 80°C for
5 minutes to melt the PEO, and then the temperature was dropped rapidly
to 51°C, in order to crystallize PEO. The low temperature form of TPB can,
therefore, be taken to be responsible for the behavior shown in Table VII.
According to Holmes, et al.130
, most samples of nylon G contain,
in addition to the normal crystalline form (a) , varying amount of another
crystal form (p) . These two forms differ in the side-by-side packing of
131hydrogen-bonded sheets . The p form is unstable and the transforma-
tion p -» a occurs almost completely by immersing nylon 6 in boiling water
1 30for 5 or 6 hours
. During the present study, the nylon 6 samples used
as substrates were heated in an oven at 105°C overnight to remove moisture
Based on these facts, the nylon 6 substrates used may be taken to contain
the a crystalline form almost completely.

82
According to Bonn and Garnor 132, fibers of nylon 66 also con-
tain two different crystalline forms: a and (3. These two forms involve
different packings of geometrically similar molecules 132•1 33
. Most
nylon 66 specimens contain predominantly the a form. These remarks
apply for nylon 610 also41,132
.
The above is a summary of information from the literature which
is relevant as to which crystalline polymorphs of the polymers used were
responsible for the results obtained in the present study. It should be
kept in mind that the substrates used were in the form of unoriented films
which were spherulitic for crystallizable polymers. Spherulitic specimens
are known to contain amorphous material also. The degrees of crystal-
linity of the substrates were not determined. The degree of crystallinity
of the substrate was increasing due to annealing, as the other polymer was
being melted and crystallized.

83
CHAPTER VIII
SUMMARY AND CONCLUSIONS
Summary
1.
Heterogeneous nucleation of isotactic polypropylene (iPP)
,
poly(butene-l) (FBI),poly (ethylene oxide) (PEO)
, and polycaprolaetone
(PCL) crystals have been studied using hot stage polarizing microscopy
coupled with still and motion photography. The polymers have been crys-
tallized from the melt in contact with various substrates (mostly polymeric) .
Attention has been paid to the morphologies of the growing polymer induced
by substrates. The morphologies have been used to characterize and com-
pare the nucleating ability of substrates qualitatively. The evolution of
spherulites on the substrate (as well as in the bulk polymer) has been
directly observed and studied in order to characterize the nucleating
ability of substrates quantitatively.
2. Heterogeneous nucleation in polymer crystallization is a
selective process. A given substrate has different nucleating abilities
for different crystallizing polymers. Also, different substrates have dif-
ferent nucleating abilities for the same crystallizing polymer.
3. The mere presence of a foreign surface in a supercooled
melt is not enough for nucleation to occur.
4. Similarity in chemical structure of the substrate and the
crystallizing polymer is no certain criterion for nucleating ability.
5. Similarity of crystallographic unit cell type of the substrate
and the crystallizing polymer cannot be correlated with nucleating ability.

Close match of the lattice parameters of the substrate and the crystallizing
polymer is not a necessary condition for strong nucleating action
.
6.
Crystallinity is a necessary but not sufficient condition for a
substrate to be a strong nucleating agent.
7. Surface energy of a substrate does not dictate its nucleating
ability. Low energy surfaces of polymers are capable of inducing trans-
crystalline nucleation of polymer crystals.
8. Substrates can be classified into the following three groups,
based on their nucleating ability: highly active (Type I), moderately
active (Type II),and inactive (Type III) . Substrates of Type I are able to
induce transcrystalhnity. In this case nucleation is favored on the sub-
strate. With a Type II substrate, nucleation occurs on the substrate as
well as in the bulk polymer, without any preference for either. With a
Type III substrate, nucleation is favored in the bulk polymer.
9. The morphology of a crystallizing polymer induced by a sub-
strate is temperature dependent. The morphology changes from transcrys-
talline to spherulitic on increasing the crystallization temperature. The
reduced nucleation density at the higher temperature is the result of lower
nucleation rate at smaller supercooling, in accordance with the theory of
heterogeneous nucleation.
10. At intermediate temperatures, that is in the temperature
range where the transition from transcrystalline to spherulitic surface
morphology takes place, one observes mixed surface morphology (trans-
crystalline plus spherulitic) . Three cases of the latter are observed,
namely (i) transcrystalhnity. predominating, (ii) transcrystalline and

85
spherulitic surface morphology in equal proportions, and m spherulWcsurface morphology predominating
.
11.
Well-developed transcrystalline morphology is ohserved onlyin a certain range of crystallization temperatures. The range for iPP is
120-130°C, and for PCL it is 48-52°C.
12. In the early stage of the development of transcrystallinity in
polycaprolactcne.sporadic nucleation on the substrate ,high density poly-
ethylene) has been observed.
13. The linear growth rates of the transcrystalline region in iPP
and PCL are equal to the radial growth rates of bulk-nucleated spherulites.
The transcrystalline growth rate is independent of the substrate which is
responsible for the transcrystalline nucleation.
14. The melting temperatures of transcrystalline iPP and PCL
are identical with those of the bulk-nucleated spherulites.
15. The high nucleating ability of isotactic polystyrene samples
in the crystallization of isotactic polypropylene, as evidenced by the genera-
tion of transcrystallinity, is due to the polymer (iPS) itself and not due to
the Ti and Al impurities in the sample of iPS . Ti and Al contents (residues
of Ziegler-Natta catalysts) of iPS samples can be reduced by two methods:
(a) by fractionating the polymer, and (b) by chelating Ti and Al with
acetylacetone
.
16. Nucleation densities on substrate and in bulk have been
determined by counting the number of spherulites nucleated under the
polarizing microscope. The nucleation density on the substrate (n ) is
5 2S
of the order of 10 /cm . In cases where transcrystallinity is generated,

86
n is of the order of 106
- i n7 /rm2 TK „ , .eroiAU 10/cm •
The nucleation density in the bulkpolymer (n, ) is of the order of m 10 /™ 3 *h )
me oraer ot 10 /cm, for commercial iPP, PB l and PEO
samples. These data were obtained for the crystallization of iPP (i 2 50c/
3 min, 129°C/14 min),PBl (85.5°C/9 min, 88°C/9 min) and PEO (50°C/
3 min, 51°C/5 min
, 53°C/34 min) .
17.
Bulk nucleation densities vary from one volume element of
the crystallizing polymer to another. The volume elements under observa-
tion were of the order of l(f9cm 3
. Coefficients of variation of 20-40% are
typical. The variation from one volume element to another is larger than
that within a given volume element with the crystallization experiment
being repeated.
18. Nucleating ability of substrates can be compared using the
ns/n
bratio as an index. However, for a given substrate, n /jl ratios
s bvary from one sample of a crystallizing polymer to another.
19. Repetition of the crystallization of PEO with the same volume
element under observation does not produce identical nucleation density
(bulk) vs. time curves. However, no definite trend showing progressive
deactivation of nuclei was noticeable with this polymer, the number of
spherulites being counted every minute.
20. In the crystallization of polymers in contact with substrates,
there exists a fixed number of nucleating sites on the substrate. Similarly,
in the crystallization of the bulk polymer, a fixed number of heterogeneities
are responsible for nucleation at each temperature.
21. Pseudohomogeneous nucleation has been observed on sub-
strate and in bulk, in the crystallization of iPP, PBl and PEO.

87
22. The shape of the experimental nucleation density vs. time
curve has two variations, namely (i) curve with an initial linear part, and(ii) S-shaped curve.
23. Experimental heterogeneous nucleation rates of PB1 at
85-88°C are of the order of 105/min/cm 2
on substrates and 109-10 10/min/cm 3
in bulk. The coefficient of variation of bulk nucleation rates has been calcu-
lated to be 12-16%. For PEO crystallization at 51°C, measured heterogeneous
nucleation rates are of similar orders of magnitude, while the coefficient of'
variation of bulk nucleation rates has been calculated to be 56%.
24. The heterogeneous nucleation rates in the crystallization of
PB1 (on iPS and iPP substrates and in bulk) increase by a factor of about 5
for a 10°C increase in supercooling. The spherulitic growth rates of PB1
have comparable temperature coefficients. These temperature coefficients
are much lower than those of homogeneous nucleation rates.
25. Experimentally-measured temperature coefficients of hetero-
geneous nucleation rates of PB1 yielded values of the interfacial energy
parameter aaeAa. The temperature coefficients of spherulitic growth
rates yielded the value of ca • By division, values of Aa (= o + a - a )e cmwere obtained for different substrates. The values of Ao, in turn, yielded
values of (aQ
- om ) . The quantity aQ
is the substrate -crystal interfacial
energy, while om is the substrate-melt interfacial energy. Higher values
of (am - oQ
) indicate a better heterogeneous nucleating agent. Absolute
values of o may be obtainable by further measurements of o by contactu m J
angle techniques.

88
Conclusions
1.
Morphologies of crystallizing polymers induced by substratescan be used to characterize the nucleating ability of substrates qualitatively
The morphologies are temperature dependent.
2. Experimentally-determined nucleation densities, nucleation
rates and the interfacial energy parameters appearing in the theory of
heterogeneous nucleation can be used to characterize the nucleating ability
of substrates quantitatively. The third approach is preferred, since the
effect of both time and temperature are taken into account
.
3. Similarity in chemical structure and in crystallographic unit
cell type of the substrate and the crystallizing polymer are no certain cri-
teria for nucleating ability. Close match of the lattice parameters of the
substrate and the forming crystal is not a necessary condition for strong
nucleating action.
4. The surface energy of a substrate does not dictate its nuclea-
ting ability. Low energy surfaces of polymers are capable of inducing
transcrystalline nucleation of polymer crystals.
5. Crystallinity is a necessary but not sufficient condition for a
substrate to be a strong nucleating agent.
6. In the crystallization of polymers in contact with substrates,
there exists a fixed number of nucleating sites on the substrate. When
these nucleating centers become active successively during crystallization,
pseudohomogeneous nucleation on the substrate obtains.
7. The high nucleating ability of isotactic polystyrene samples
in the crystallization of isotactic polypropylene, as evidenced by the genera-
tion of transcrystallinity, is due to the polymer (iPS) itself. Titanium and

89
alundnum contents (residues of Zie gler-Natta catalysts) of ips ^be reduce, by two methods: W by fracUonatog fte^ ^ ^chelating Ti and Al with acetylacetone.
8. The growth rate and the melting temperature of transcrystal-hne polymer are identical with those of bulk-nucleated spherulites.

90
CHAPTER IX
SUGGESTIONS FOR FURTHER RESEARCH
In the present study the number of nuclei was counted by visual
observation of spherulites. When transcrystallinity is generated this tech-
nique is insufficient due to the crowding of spherulites on the substrate.
Motion pictures of the transcrystalline nucleation process will permit more
accurate determination of nuclcabon densities in the transcrystalline region
as well as their time dependence.
Molecular weight of the crystallizing polymer does not enter
directly as a parameter in the theories of heterogeneous nucleation of
polymer crystallization. Furthermore, the theories treat the substrate as
a plane surface and no distinction is made as to whether the substrate is
polymeric or not. It seems desirable to generate experimental data on the
effect, if any, of molecular weight of the crystallizing polymer and of the
substrate on the nucleating action.
One study of the effect of molecular weight of the crystallizing
polymer on the nucleating effect of a low molecular weight substance has
26been reported. A hundred-fold change in the molecular weight of
crystallizing iPP resulted in a negligibly small change in the nucleating
ability of basic aluminum dibenzoate . The effect of molecular weight of the
crystallizing polymer on the nucleating effect of a polymer substrate should
also be investigated.

91
The present study has indicated that probably the molecular weightof a polymer substrate does influence its nucleating ability. The molecular
weight of the substrate can affect its crystallization kinetics from the
quenched state and hence the degree of crystallinity of the substrate.
Further research in this direction is recommended.
The role of the adsorption of the chain molecules in the melt on
substrates in nucleation phenomena should be clarified. It is felt that for a
better understanding of the heterogeneous nucleation processes, it should
be inquired whether the nature of the adsorbed layer can be related to
the activity of the nucleating surface , and how far the conformation of
adsorbed molecules is conducive to nucleation.
From this study the crystallinity of the substrate has emerged as
an important factor in the heterogeneous nucleation of polymer crystals.
The dependence of the nucleating ability of polymer substrates on their
degree of crystallinity should be quantitatively studied. Also, the effect
of the nucleation density (number of spherulites per cm 2) of the substrate
and the orientation of the chain molecules on the substrate on the nucleatin
ability of the substrate should be investigated. The effect of orientation can
be studied by using stretched polymer films as substrates. The possibility
of epitaxy in such cases should also be investigated.
The bulk nucleation densities reported in the present study include
the contribution from nuclei generated by the glass surfaces. It is recommen-
ded that the effect of the glass surfaces be determined by performing crystal-
lization experiments at different thicknesses of the crystallizing polymer.
ao

92
During the course of repeated crystallization on a substrate in
this study, it seemed that spherulites reappeared at the same positions.
This point should be clarified.
On repeating the crystallization of PEO, identical nucleation
density (bulk) vs. time curves were not produced (Figure 41) . The first
crystallization produced the highest slope and the coefficient of variation
of the nucleation rates was 63% (Table XXII) . It is suggested that repeated
crystallization experiments be performed with other polymers in order to
clarify whether any definite trend exists or not. Nucleation both on sub-
strates and in bulk should be studied.
From measurements of temperature coefficients of heterogeneous
nucleation rates of PB1,values of (om
- oq
) have been obtained. It is recom-
mended that experiments on wettability of substrates by PB1 melts be per-
formed so that values of am can be calculated. In this way, absolute values
of acwill be obtained. Such data have not been reported so far, to the
author's knowledge.
The present study has shown the ability of polymer substrates
to induce transcrystallinity in other polymers. One can envision the
development of composite fibers using the technique of growing one polymer
onto another, e.g. iPP onto PET or nylons 6, 66, or 610. Such fibers will
have contributions from the properties of both polymers. The feasibility
of the process and the usefulness of the final materials should be investigated.
Heterogeneous nucleation by the dispersed phase of polyblends is
expected to influence their crystallization kinetics, morphology and physical
properties. Quantitative study of this area is recommended. Finally, a

93
systematic study of the role of contact surfaces in the manufacture of polymerfilms will help in controlling and improving film properties.
A comparative study of the nucleating ability of substrates with trans-
crystalline and spherulitic morphologies, respectively, will throw light on
the role of morphology of a substrate on its nucleating ability. Also, during
the present study, cut surfaces of polymer films and metals were used as
substrates. Fracture surfaces should also be studied as nucleating sub-
strates.
Corners and cavities in crystals are high energy sites and are con-
sidered to have significant nucleating potential134
. A study of the role of
crystal defects on the nucleating ability of substrates is recommended, with
the hope that a better understanding of the dependence of the nucleating
ability of substrates on their physical features will evolve.

94
REFERENCES
1. L. Mandelkern, CRYSTALLIZATION OF POLYMERS. McGraw-Hill,
New York, 1964.
2. A. Sharpies. INTRODUCTION TO POLYMER CRYSTALLIZATION
,
Edward Arnold, London, 1966.
3.
P.
H.
Ceil,POLYMER SINGLE CRYSTALS
, Interscience. New York
,
1963.
4. L. Mandelkern, Chem . Rev. 56. 903 (1956).
5. F. P. Price, Ann. N. Y. Acad. Sci. 83, 20 (1959).
6. H. D. Keith in PHYSICS AND CHEMISTRY OF THE ORGANIC SOLID
STATE, Vol. I, D. Fox, M. M. Labes and A. Weissberger, Eds.
,
Interscience, New York, 1963, Chapter 8.
7. A. Keller, Reports on Progress in Physics 31, Part 2, 623 (1968) .
8.
J.
M.McKelvey
, POLYMER PROCESSING. John Wiley
, New York,
1962, Chapter 5.
9. F. P. Price in ENCYCLOPEDIA OF POLYMER SCIENCE AND TECH-
NOLOGY, Vol. 8, H. F. Mark, N. G. Gaylord and N. M. Bikales,
Eds., John Wiley, New York, 1968, p. 63.
10.
NUCLEATION PHENOMENA, American Chemical Society Publications
,
Washington, D.C.. 1966 (papers presented at ACS Symposium on
"Nucleation Phenomena," Washington, D.C. , June, 1965, Alan S.
Michaels, Symposium Chairman; also published in Ind. Eng. Chem.
57, 58 (1965-66))

95
11. A. C. Zettlemoyer,Ed.
, NUCLEATION, Marcel Dekker, New York,
1969.
12. D. Turnbull in THERMODYNAMICS IN PHYSICAL METALLURGY,
American Society for Metals, Cleveland, Ohio, 1949, pp. 282-306.
13. D. Turnbull, Solid State Physics 3, 225 (1956) .
14.
J.H
.Hollomon in THERMODYNAMICS IN PHYSICAL METALLURGY
,
American Society for Metals, Cleveland, Ohio, 1949, pp. 161-177.
15. L. Mandelkern, CRYSTALLIZATION OF POLYMERS. McGraw-Hill,
New York, 1964, Chapter 8.
16. P. J. Flory and A. D. Mclntyre, J. Polymer Sci . 18, 592 (1955) .
17. B. B. Burnett and W. F. McDevit, J. Appl. Phys. 28, 1101 (1957).
18. F. P. Price in NUCLEATION, A. C. Zettlemoyer, Ed., Marcel
Dekker, New York, 1969, Chapter 8.
19. W. A. Kargin, T. I. Sogolova , N. Ya. Rapoport and I. I. Kurbanova,
J. Polymer Sci. Part C, No. 16, 1609 (1967) .
20. A. G. M. Last, J. Polymer Sci. 39, 543 (1959) .
21. M. Inoue, J. Polymer Sci. Al, 2013 (1963) .
22. C.J. Kuhre, M. Wales and M. E. Doyle, SPE Journal 20, 1113
(1964) .
23. C. G. Vonk, Kolloid Z. u. z. Polymere 206 , 121 (1965).
24. H. N. Beck and H. D. Ledbetter , J. Appl. Polymer Sci. 9, 2131
(1965) .
25. H. N. Beck, J. Appl. Polymer Sci. 11, 673 (1967) .
26. H. N. Beck, J. Polymer Sci. Part A-2, 4. 631 (1966).
27. F. L. Binsbergen. NUCLEATION IN THE CRYSTALLIZATION OF
POLYOLEFINS, Ph.D. Thesis, Groningen, The Netherlands, 1969.

96
28. F. L. Binsbergen, Polymer 11, 253 (1970) .
29. V. A. Kargin. T. I. Sogolova and N. Ya. Rapoport-Molodtsova
,
Vysokomol. Soyed. 6. 2090 (1964); English Translation: PolymerSci. USSR 6, 2318 (1964)
.
30. V. A. Kargin. T. I. Sogolova and T. K. Shaposhnikova. Dokl.
Akad. NaukSSSRISO, 1156 (1964); English Translation: Dokl.
Phys. Chem. 156, 612 (1964).
31. V. A. Kargin, T.I. Sogolova and N. Ya. Rapoport-Molodtsova,
Dokl. Phys. Chem. 156, 644 (1964) .
32.
V.
A.
Kargin,T
.1
. Sogolova and I . I . Kurbanova, Dokl . Phys
.
Chem. 162, 467 (1965) .
33. V. A. Kargin, T. I. Sogolova and N. Ya . Rapoport, Dokl. Phys.
Chem. 163, 600 (1965) .
34. V. A. Kargin, T.I. Sogolova and T. K. Shaposhnikova, Polymer
Sci. USSR 7, 250 (1965) .
35. V. A. Kargin, T. I. Sogolova and T. K. Shaposhnikova, Polymer
Sci. USSR 7, 423 (1965) .
36. V. A. Kargin, T.I. Sogolova and N. Ya. Rapoport-Molodtsova,
Polymer Sci. USSR 7, 631 (1965) .
37. V. A. Kargin, T.I. Sogolova and I. I. Kurbanova, Polymer Sci.
USSR 7, 2308 (1965) .
38. F. Rybnikar, J. Appl. Polymer Sci. 13, 827 (1969).
39. A. Yim and L. E. St. Pierre, Polymer Letters 8, 241 (1970) .
40. A. Packter and K. A. Sharif, Polymer Letters 9, 435 (1971) .
41. F. Rybnikar, Polymer 10, 747 (1969).

97
42. F. L. Binsbergen, NUCLEATION IN THE CRYSTALLIZATION OFPOLYOLEFINS
,Ph.D. Thesis, Groningen
, The Netherlands,
1969, Chapter V.
43. K. Hara and H. Schonhorn, J. Appl. Polymer Sci. 16, 1103 (1972)
44. S. Y. Hobbs, Nature Physical Science 234 (44), 12 (1971).
45. S. Y. Hobbs, Nature Physical Science 239 (89), 28 (1972).
46. E. Jenckel, E. Teege and W. Hinrichs, Kolloid-Z 129, 19 (1952) .
47. A. Sharpies, INTRODUCTION TO POLYMER CRYSTALLIZATION,
Edward Arnold, London, 1966, p. 21.
48. R. J. Barriault and L. F. Gronholz , J. Polymer Sci. 18, 393
(1955) .
49. B. K. Eby, J. Appl. Phys . 35, 2720 (1964) .
50. H. Schonhorn, Polymer Letters 2, 465 (1964).
51. T. K. Kwei, H. Schonhorn and H . L. Frisch , J. Appl. Phys. 38,
2512 (1967) .
52. H. Schonhorn, Macromol. 1, 145 (1968).
53. D. R. Fitchmun and S. Newman, Polymer Letters 7, 301 (1969) .
54.
D.R
.Fitchmun and S . Newman , J . Polymer Sci . , Part A-2 , 8
,
1545 (1970)
.
J. R. Shaner and R. D. Corneliussen , J. Polymer Sci. Part A-2,
10, 1611 (1972)
.
56. A. Keller, J. Polymer Sci. L5, 31 (1955).
57. A. Keller, J. Polymer Sci. 21, 363 (1956).
57a. K. H. Yang and J. L. Kardos. Bull. Am. Phys. Soc. , Series II,
17. No. 3, 249 (1972); Paper No. AL2 presented at the American
Physical Society meeting, Atlantic City, New Jersey, March, 1972.
55

98
58. H. Schonhorn, Polymer Letters 5, 919 (i 96 7) .
59.
J.
P.
Luongo and H. Schonhorn
, J . Polymer Sci . Part A-2, 6
,
1649 (1968) .
60. C. Gieniewski and R. S . Moore, Macromol. 2, 385 (1969) .
61. S. Matsuoka, J. H. Daane, H. E. Bair and T. K. Kwei, Polymer
Letters 6 , 87 (1968) .
62. V. A. Kargin, T. I. Sogolova and T . K . Shaposhnikova, Dokl.
Phys. Chem. 180, 406 (1968).
63. K. Hara and H. Schonhorn, J. Appl. Phys. 42, 4549 (1971) .
64. H. Schonhorn and F. W. Ryan, J. Polymer Sci. Part A- 2, 6, 231
(1968) .
65.
C.S
.Barrett
,STRUCTURE OF METALS
, McGraw-Hill, New York
,
1952, p. 510.
66. J. W. Christian, THE THEORY OF TRANSFORMATIONS IN METALS
AND ALLOYS, Pergamon Press
, Oxford , 1965, p . 582
.
67. J. Nutting and R. G. Baker, THE MICROSTRUCTURE OF METALS,
The Institute of Metals, London, 1965, pp. 4-7.
68. L. Northcott, J. Inst. Met. 62, 101 (1938) .
69. L. Northcott, Ibid., 65, 173, 205 (1939) .
70. L. Northcott, Ibid., 72, 283 (1946) .
71. J. W. Gibbs, SCIENTIFIC PAPERS, Vol. I, Dover, New York,
1961.
72. D. R. UhlmannandB. Chalmers, Ind. Eng. Chem. 57 (9) , 18 (1965)
73. W.J. Dunning in NUCLEATION, A. C. Zettlemoyer , Ed. , Marcel
Dekker, New York, 1969, Chapter 1.

99
74. F. GornickandJ. D. Hoffman, fed. Eng. Chem. 58, 41 (1966).
75. M. Volmer and A. Weber, Z. Physik. Chem. 119, 277 (1925).
76. J. C. Fisher, J. H. Hollomon and D . Turnbull, J. Appl. Phys. 19,
775 (1948) .
77. R. Becker and W. Doring, Ann. Phys. 24, 719 (1935).
78. R. Becker, Ann. Phys. 32, 128 (1938) .
79. D. Turnbull and J. C. Fisher, J. Chem. Phys. 17, 71 (1949) .
80. S. Glasstone, K. J. Laidler and H . Eyring, THE THEORY OF RATE
PROCESSES, McGraw-Hill, New York, 1941.
81. D. Turnbull, J. Appl. Phys. 20, 817 (1949) .
81a. F. P. Price, J. Am. Chem. Soc. 74, 311 (1952) .
81b. W. J. Barnes, W. G. Luetzel and F . P. Price, J. Phys. Chem. 65,
1742 (1961) .
82. M. Volmer, Z. Elektrochem. 35_, 555 (1929) .
83. D. Turnbull, J. Chem. Phys. 18, 198 (1950).
84. D. Turnbull, J. Chem. Phys. 20^, 411 (1952) .
85. R. L. Cormia, F. P. Price and D. Turnbull, J. Chem. Phys. 37,
1333 (1962) .
86.
F. Gornick ,
G.S . Ross and L.J. Frolen , J . Polymer Sci . , Part C ,
18, 79 (1967) .
87. J. R. Burns and D. Turnbull, J. Appl. Phys. 37, 4021 (1966) .
88-. J. D. Hoffman and J.I. Lauritzen, Jr. , J. Research NBS, 65A ,
297 (1961) .
89. M. Volmer, Z. Physik. Chem. 102, 267 (1922).

100
90. R. D. Ulrich, CRYSTALLIZATION BEHAVIOR OF SHEARED POLY-
ETHYLENE OXIDE MELTS, Ph.D. Thesis
, University of Massachusetts,
1972.
91. F. W. Billmeyer, Jr., TEXTBOOK OF POLYMER SCIENCE, Wiley-
Interscience. New York. 1971, Chapter 3, p. 84.
92. J. Brandrup and E. H. Immergut, Eds., POLYMER HANDBOOK,
Interscience, New York, p. 111-74.
93. R. W. Lenz, ORGANIC CHEMISTRY OF SYNTHETIC HIGH POLYMERS,
Interscience, New York, 1967, Chapter 15, p. 579.
94. F. A. Cotton and G. Wilkinson, ADVANCED INORGANIC CHEMISTRY,
Interscience, New York, 1966, pp. 444, 808.
95. W. C. Fernelius, Ed. , INORGANIC SYNTHESES, Vol. II, McGraw-
Hill, New York, 1946, pp. 25, 119.
96. J. Brandrup and E. H. Immergut, Eds. , POLYMER HANDBOOK,
Interscience, New York, 1966, p. IV-13.
97. L. Trossarelli, E. Campi and G. Saini, J. Polymer Sci. 35, 205 (1959)
98. J. Brandrup and E. H. Immergut, Eds. , POLYMER HANDBOOK,
Interscience, New York, 1966, p. Ill- 3
.
99. A. Sharpies, Polymer 3, 250 (1962) .
100. J. Brandrup and E. H. Immergut, Eds. , POLYMER HANDBOOK,
Interscience, New York, 1966, p. III-100.
101. H. Bittiger, R. H. Marchessault and W . D. Niegisch, Act. Crystal.
B26 , 1923 (1970) .
102. J. Brandrup and E. H. Immergut, Eds. , POLYMER HANDBOOK,
Interscience, New York, 1966, Chapter III.

101
103. D. Turnbull and B.Vonnegut, Ind. Eng. Chem. 44, i 292 (1952).
104. J. A. Koutsky, A. G. Walton and E . Baer, Polymer Letters 5, 185
(1967) .
105. J. H.vander Merwe, Disc. Faraday Soc . 5, 201 (1949).
106. J. Willems, Experientia 13, 276 (1957) .
107. J. Willems and I. Willems, Experientia 13. 465 (1957) .
108. J. Willems, Disc. Faraday Soc. 25, in (1958)
109. E. W. Fischer, Disc. Faraday Soc. 25, 204 (1958) .
110. E. W. Fischer, Kolloid-Z. 159, 108 (1958) .
111. J. Willems, Experientia 15, 175 (1959) .
112. J. Willems, Experientia 17, 344 (1961).
113. J. A. Koutsky, A. G. Walton and E . Baer, J. Polymer Sci., PartA-2,
4, 611 (1966)
.
114. J. A. Koutsky, A. G. Walton and E. Baer, Polymer Letters 5, 177
(1967) .
115. K. A. Mauritz, E. Baer and A. J. Hopfinger, J. Polymer Sci.,
Polymer Phys. Ed. 11, 2185 (1973) .
116. R. B. Richards, J. Polymer Sci. 6, 397 (1951) .
117. T. Takahashi, M. Inamura and I. Tsujimoto, Polymer Letters 8, 651
(1970) .
118. D. Turnbull and R. L. Cormia, J. Chem. Phys. 34, 820 (1961) .
119: M. E. Glicksman and W.J. Childs, Acta Metallurgica 10, 925 (1962) .
120. B.J. Abbs, M.S. Thesis, in preparation, University of Massachu-
setts, 1974.
121. N. Tcheurekdjian , A. C. Zettlemoyer and J.J. Chessick, J. Phys.
Chem. 68, 773 (1964) .

102
122. G. Natta, J. Polymer Sci. 16, 143 (1955) .
123. G. Natta, P. Pino, P. Corradini, F. Danusso, E . Mantica,
G. Mazzanti and G. Moraglio, J. Am. Chem. Soc. 77, i 708 (19 55 ) .
124. G. Natta, P. Corradini and I. W. Bassi, Makromol. Chem.2J_, 240
(1956) .
125. F. Danusso and G. Gianotti, Makromol. Chem. 61, 139 (1963) .
126. R. L. Miller and V. F. Holland, Polymer Letters 2, 519 (1964) .
127.
R.L
.Miller in CRYSTALLINE OLEFIN POLYMERS
, Part I
,
R.
A.
V.Raff and K
.W . Doak , Eds . , Interscience
, New York
,
H 1965, Chapter 12, p. 577.
128. A. Turner-Jones, Polymer Letters 1, 455 (1963)
129. G. Natta and P. Corradini, J. Polymer Sci. 39, 29 (1959).
130. D. R. Holmes, C. W. BunnandD. J. Smith, J. Polymer Sci. 17,
159 (1955) .
131. D. C. Vogelsong, J. Polymer Sci. , Part A, 1, 1055 (1963).
132. C. W. Bunn and E. V. Garner, Proc. Roy. Soc. A, 189, 39 (1947)
133. P. Meares, POLYMERS: STRUCTURE AND BULK PROPERTIES,
VanNostrand, London, 1967, p. 108.
134. J. W. Christian, THE THEORY OF TRANSFORMATIONS IN METALS
AND ALLOYS, Pergamon Press , Oxford, 1965, pp. 405-415.

103
TABLE I
Material
MATERIALS USED
Source Characterization Data1. Isotactic —
Polypropylene(iPP)
(a) Shell 5520 Shell Chemical Isotactic content: -95%Company
M.Pt. : 170. 3°C
Ash Content : 1.6 5%
Molecular Weight:
Mn
M\
Mw
v
426,00
337,000
281,000
(b) Pro-fax 6323 HerculesInc.
Additives (stabilizers)::
(i) thioester : 0.15% (wt)(dimyristyl-thiodipropion-ate)
(ii) hindered phenol: 0.1% (wt)(3,5-di-t-buty1-4-methyl-phenol )
(iii) calcium stearate : 0 . 1%(wt)
Isotactic content: 96-97
M.Pt. : 171. 8°C
Ash content : 3.57%
Intrinsic viscosity in de-calin at 135°C:1.8 dl/g.
Mv 209,000
(Cont.
)

104
Material
(c) Profax 6509
(d) Pro-faxX 12886-38
(e) PR 84919
2. Poly (ethyleneoxide) (PEO)
Carbowax 20-M
3. Poly (butene-1
)
(PB1)
(a) Petrotex14988-1(for all theentries inTable VI ex-cept thelast two)
(b) Vestolen BT6000(for the lasttwo entriesin Table VI)
TABLE I
(cont.
)
Source Characterization Data
HerculesInc
.
HerculesInc
.
HerculesInc.
Union CarbideCorp.
Petrotex Co
Hiils Co.
Isotactic content: 96-97%
Isotactic content: 96%
Ash content: 0.76%
Mw
"7 X 10
Melt index: 0.35
M.Pt. 61.3°C
Molecular Weight
Mn 26 ,600
Mw : 395,000
Ash content: 0.90%
M.Pt. : 134. 3°C
(Cont.
)

105
Material
4. Polycaprolac-tone (PCL)
[<CH2
)5-C-0-]
n
TABLE I
(cont.
)
Source Characterization Data
PCL- 700
5. IsotacticPolystyrene
(iPS)
(a) iPS-MPL
(b) iPS-MPL-U-1
(c) iPS-MPL-AA-1
Union CarbideCorp
.
Monomer Poly-mer Labora-tories
, BordenChemical Co.
This sample wasrecovered fromthe centrifugatewhen a toluenesolution of iPS-MPL was centri-fuged (details insection 5.3.6.1of Ch. v)
This sample was ob-tained when iPS-MPLwas treated withacetylacetone toreduce Ti and Alcontent (details insection 5.3.6.2 ofCh. v)
M.Pt.
Mw
64.2°C
40,000
Chemical analysisTiAlAsh
Ash
0.60%0. 36%2. 07%
2.17%
Chemical AnalysisTiAlAsh
0.26%0.05%1.24%
(Cont.
)

106
fd
-Pcd
QcoH-Pfd
N•H
CD
-POfd
(d
x:u
IS
<D
o >. c•H p <DCO -H pC CO H•H 0 0M u 4J-P 03
C •H CH > •H
«—
<
r-—
«
co
<•
u
uooco
-P
OrHXCN
CM
lolo
00
CN
OHXLO00
i
o
LO
CO
LOCO
•PC0)
-p
coo
o•HPofd
-P
0CO
co
•Ho COo >1o rHrd
LO CiH fd
LOrHfd
«
•
0•HgCD
u
G LOfi Q.CN
HHOV V
"H rH CO
Eh r< <
-PC0u
HwrJ
<
-Pcou
<d
uu
oCO
4-1
o
u
o
-p
<D
cn
CD
4-)
0)
rH -P
13
fd o o0) CD
-p o.CO CO
u-H
H
O•H-PO I
fd co
r-3
03
co >
ftH
CO MHCD CO rH O
O fd roH -P •
-P CD vofd u ^ •
fd w roco UCD M-i > i LOH rHtt-p a) c2 co > ofd fd -h -hCO rH -p -P
>1cfd
e0u
fd
ah
u
0a
fd
ouo•HU-Pua)
w
a)
c<D
to
Hna)
rd«-<
H >
fa fal l
& &I I
CO COft ft•H -H
oQI
CO
ftH
00CNrH
I
oro
ftw
>1—x: <d
-p rdCD -H6 XH O*V
I 0)
^> C- CD
CN rH
rH CD
o &
L£>
ro
u o
7T1
u
uoft
ft

107
TABLE I
(Cont.
)
Material Source Characterization Data
Poly [3,3-bis(choromethyl)oxacyclobutane]
CH-C11
2
(0-CHo-C-CHo -)/-I 2 n
CH2C1
•Penton" HerculesInc
.
8. Polyoxymethy-lene (POM)
(CH9-0)
B 5849 DuPont Co.
9. Poly (chlorotri-f luoroethylene)
(CCIF-CFJ2 n
'Kel-F
'
KF-6060 3M Co. M . Pt . . 2 12°CDegree of Polymerization: 1600
10. Low density (Molecular weight: 1,85,600)Polyethy-lene (LDPE)
Monsanto 8011 Monsanto Density: 0.925 g./cm3
Co.
11. High densityPolyethylene
(HDPE)
Marlex TR-885 Phillips Density: 0.965 g./cm 3
Petro- m : <110,000leum Co. _w
M /M : <7w n
Ash: 0.085%(Cont.
)

108
TABLE I
(cont.
)
Material Source Characterization Data
13. Atacticpolystyrene
14 . Nylon 6
15. Nylon 66Zytel 101
16. Nylon 610Zytel 38
16. Aluminum
Monsanto Co.
DuPont Co.
DuPont Co.
DuPont Co.
'Reynolds Wrap'Foil Due to the known ten-
dency of pure Al tooxidize rapidly in air,the surface layer canbe taken to be asA1 2°3
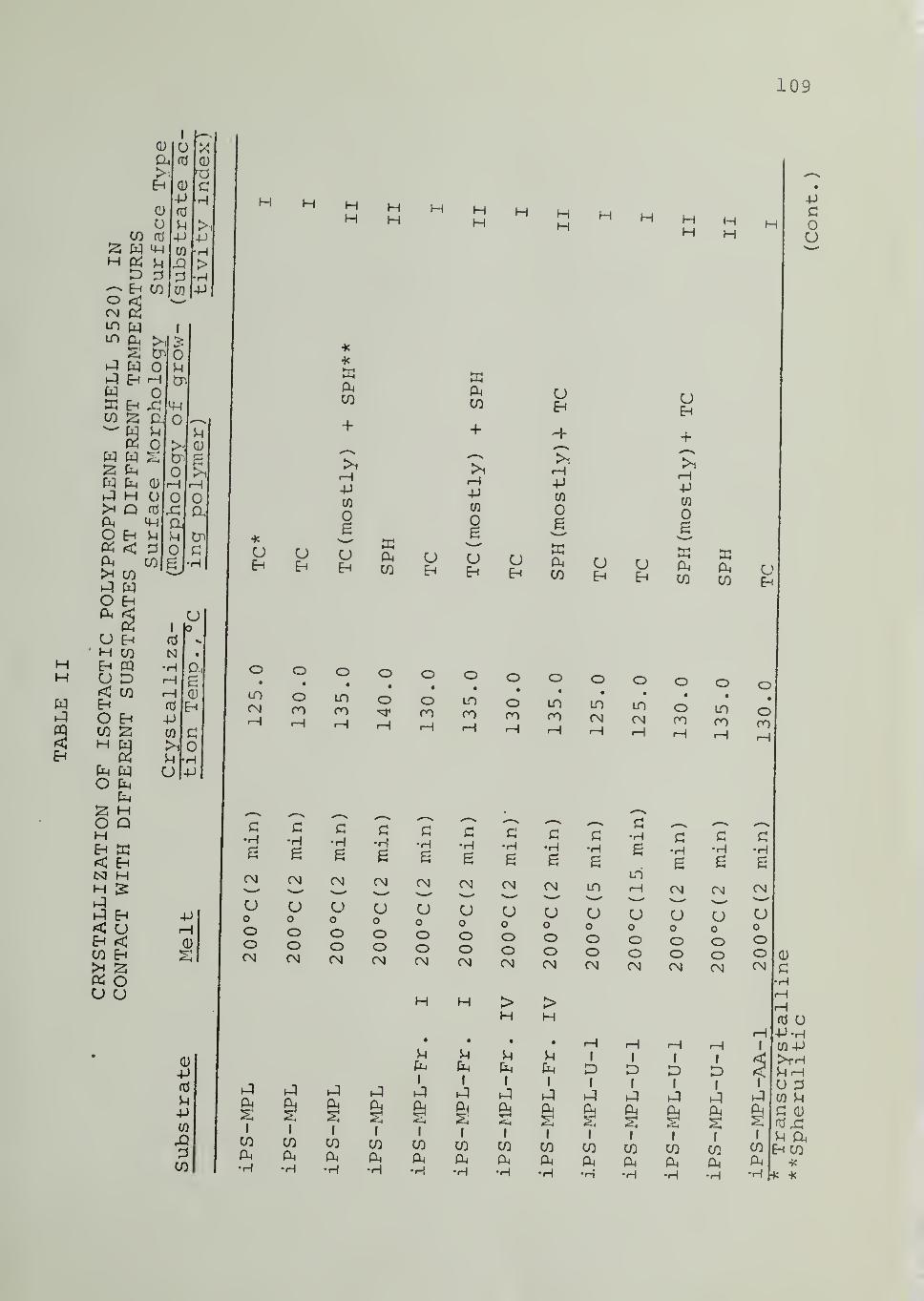
109
HHWi-h*
COZ WH f*J
D— EhOCMin win ftSMEh
1
oCD
>• X)Eh CD c
-P Ha td
0 u >1fd +j -P
CO •H5H >p p rH
CO P
WKCO
CT>
Eh
w wS EhW Phf—h
1 H>h Q
2 ^
COw
>h
O
U EhH COEh CQU D<C coEh
O EhCO £H
OJGQiuo
0)
ofd
muPCO
1
0rH
4-4
0Jh
i CD
tr g0rH rH
0 0a
a&
0 ce •H
Q
Cm WO Ph
OHEh B< EhN HH £
EhCO>H
rd
-Pto
Ua
u10
eCU
Eh
CcH+J
Eh
U<Eh
0)
sK OU U
<D+J
cd
M
CO
&CO
uEh
LOCN
l-H
CU
I
COp-f
•H
H
UEh
OoCO
I
COft•H
H
*KftCO
+
•PCO
O
u
inro
a a•H -H -H
e 6
CN CN CN
U u U0 o oO o Oo o OCN CN CN
ft
I
COft•H
H
CO
O
nI
COft•H
H H H
ULH
Oro
UfaI
KH1
&I
COft•H
ftco
-p
CO
IuEh
UEh
UPmI
hi
SI
COCU•H
MfaI
•J
&I
COCU•H
HH
H H HH
H
UEh
rH-PW0
CUCO
CJEh
UEh
UEh
H+J
CO
0Js
K Kfa ftCO CO
q c a c•H •H H •H -H Hg g g g 6 gCN CN CN CN CN mu u U u u Uo o o 0 o oO o O o O OO o o o o OCN CN CN CN CN CN
H H > >H H
ufa
I
ftS
I
coft*H
rHI
DI
COft
I
D!
fts
I
coft•H
I
DI
^\
&I
COft•H
I
D
ft
I
coft•H
Eh
ot
o•
o•
o•
O O o oLOrorH
oroH
mrorH
inCNrH
•
inCNrH
•
oroH
135.
•
ororH
•H C ag -H •H •H
g g ginrH CN CN CN
u u U UO 0 0 0
o o o oo o o oCN CN CN CN
-p
cou
-P-HCO -P
< JM rHO pCO VH
C 0)
WHO)CU *HI* *
I

110
i
r \KJ
?sd
)
rrtl>
Eh *
H<D uU 4-> >fd co
Mh •HU p >w •H
co 4->
W -P^ c
ou
Eh —9
l
*>»>
CP oofHoxi '4-1
Cu o ^ ^
u U0 > Q)
S tp io0) rH rHa 0 0fd X! am a
u3 0 ca] H
ui ofd
N•H DtrH gH ofd Eh
CO C0rl
U -P
2
0)
4->
fd
5h
4->
CO
hpCO
HH
S3ftCO
+
4J
CO
o
u
H
S3
CO
H
uEh
o o o• • •
o inro CMH rH rH
H
UEh
HH
HH
H
CO
S3
ftCO
UEh
UEh
H
uEh
o•
o•
O•
O o Oo in in
•
m•
m•
ro CM rH CM CM CMrH rH fH iH rH rH
rH rH1 1 00 CO
% 3CN CN CO COrH rH Cm Cm
1 1 1 1
\A £ o £ o a Oft Cm 0 ^ 0 •H H
s Q ro Q ro 4-> 4J1 1 1 rH 1 H O UCO CO CO CO fd fd
Cm ft ft Cm Cm Cm +J 4->
•H •H H W "H W < <
oH
OrHKO
corH
>12
CD
(1) 4-»
C fd
Q) rHiH fd
Xi 4-»
p x:
>i <D
rH M0 CD
Cm Eh
HH
CmCO
-PCO
0£
UEh
Ooro
0) -P
a fd
<D HH fd
>i£x: -p
-p £0) Cm>i a)
rH U0 <3)
Cm Eh
4J
H 0H u
KCmCO
LD<NrH
C C Is 2 n•He
He
•Hg IUX lllX TUI mi mi mi: 1LLX
.
[TUI
CN CM CM CM CM CN in in CN CM CN
U0
Uo
uo
Uo
u0
U0
uo
uo
u0
u0
U0O O o O o O o O o o Oo o o O o o o o o o oCN CM CM CM CM CM CM CM CM CM CM
PhI
rHCD
«

Ill
rrt A
> 0)f~*
Hin i jHUai»u
ij j I
i_i<-<
•"1•-J rr»w "H
+-11
l
Lj
J-nO 1
—_i
U_l
u
Li Li
o* . 0
rH
0O 0ftf a. a,MhU 0P eco H
HHW JCQ O< UEh —
-P
5h
U
Ua
-PrHCD
s
H H
CO COuEh
o o o• • •
o in inm CN egrH rH rH
c c C•H •H •H
E ein CN CN
U u uo 0 oO o oo oCN CN rH
CD4J
<d
u
cn i
rHPCO
co
c
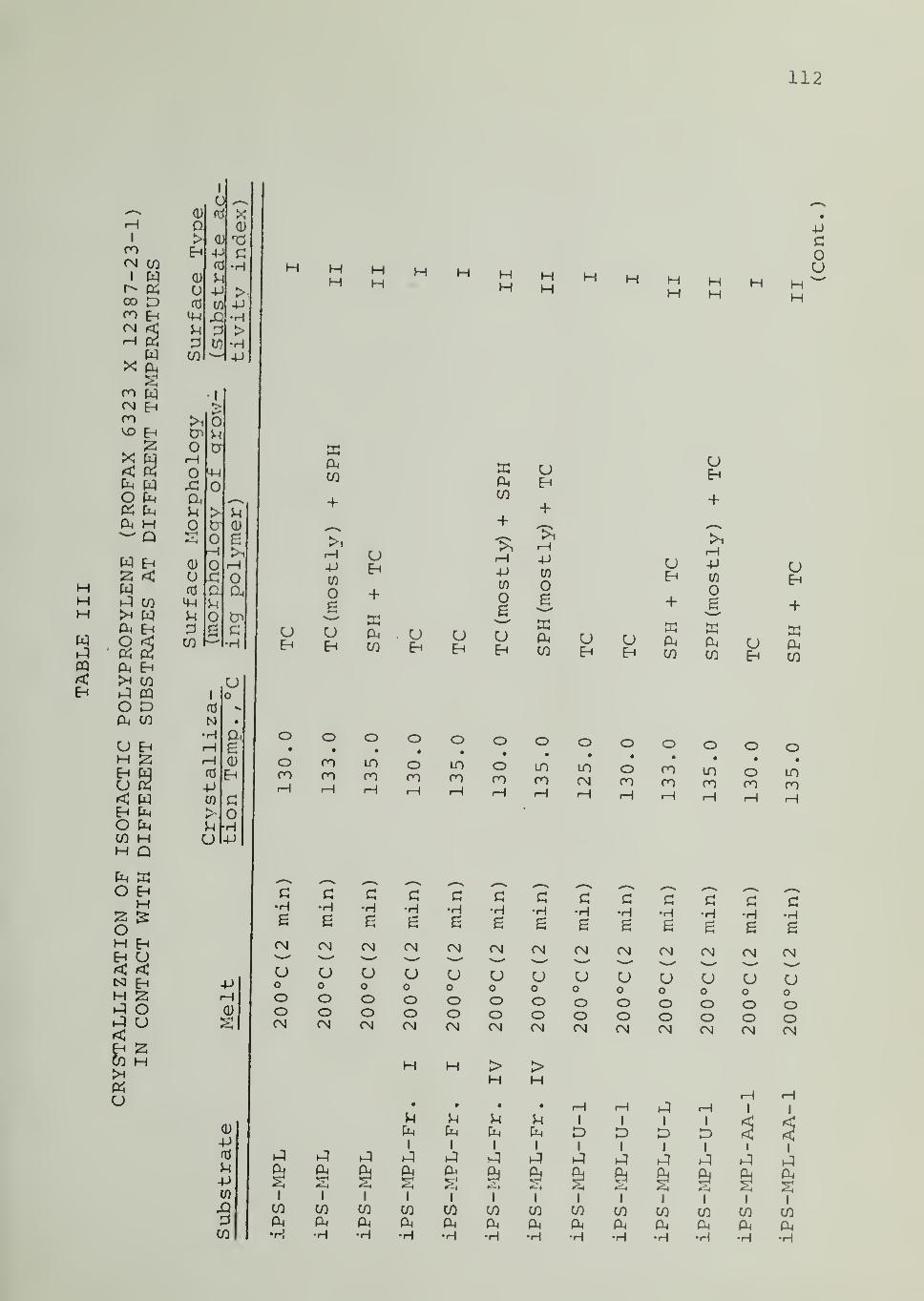
112
HHwrH*
gE-t
I
ro<N CO
I
oom D
Eh
WX ft
r-:
tn wcm Ehrovr> Eh
X<ft WO ftft ftft hw Q
Eh
COWEh
WW
ftoft
ft Eh>h COi-h* ftO D04 CO
EhUHEh
u<C WEh ftO ftCO HH Qft ffi
O EhH
2OEh
<NH
<,
tV) H
u
Eh
Eh
Ou
o
p <D
> (D
Eh P cid H
(U Sh
a -P > t
rd CO -Pm pQ •H*h 3 >p V> •Hco -P
>101orHo.c
Sh
o
i
:-
ouQ
O
>
<d
ufd
4-1
upCO
o
u0
U0}
|Ho
cH
td
-pCO
Jh
U
uo
Oke
Eh
C0H4-'
0)
-prd
u-pCO
pCO
uEn
I
CO
H
PnCO
-p
o
uEh
HrH H H H
H H
CO
uEh
UEh
UEh
+
COuEh
urH
-PCO
o£
uEh
-Pto
O
S3PnCO
ejEh
UEh
UEh
EC
CO
-p
CO
o6
PnCO
H
UEh
o•
o•
O•
O•
O•
O•
O O O o o oororH
rororH
if)
rorH
OrorH
mroH
ororH
•
IDrorH
•
LD<NrH
130. 133.
•
mrorH
*
oroH
1-3
2i
COa.•H
i
COPn•H
H H >H
>H
rH• • • • rH rH rH 1
U >H !h u 1 1 1
Ch Ch ft D D D D1 1 1 I 1 1 1 1
hi
MP g MP &*-*
&*u
MP
l r I 1 l 1 1 1 1
CO CO CO CO CO CO CO CO CO111 a. Pn Pn 0^ Oh (U-H -H •H •H •H -H •H •H •H
•p
cou
H
uEh
a.CO
inro
C C C C C c C•He
H6
•H6 mi
•HE TUI
•
TUI
•
mi TUI
•
mi :tui
*
[TUI mi]
CN CM CN CN CN CN CN CM CM
uo
u0
uo
uo
CJo
U0
Uo
uo
U0
uo
Uo
U0
u0o o o o O O O o O o O O oo o o o o o O o O o o o oCM (N CN CN CN CN CN CM <M CM CM CM CN
I
&4
sI
CO
a,•H
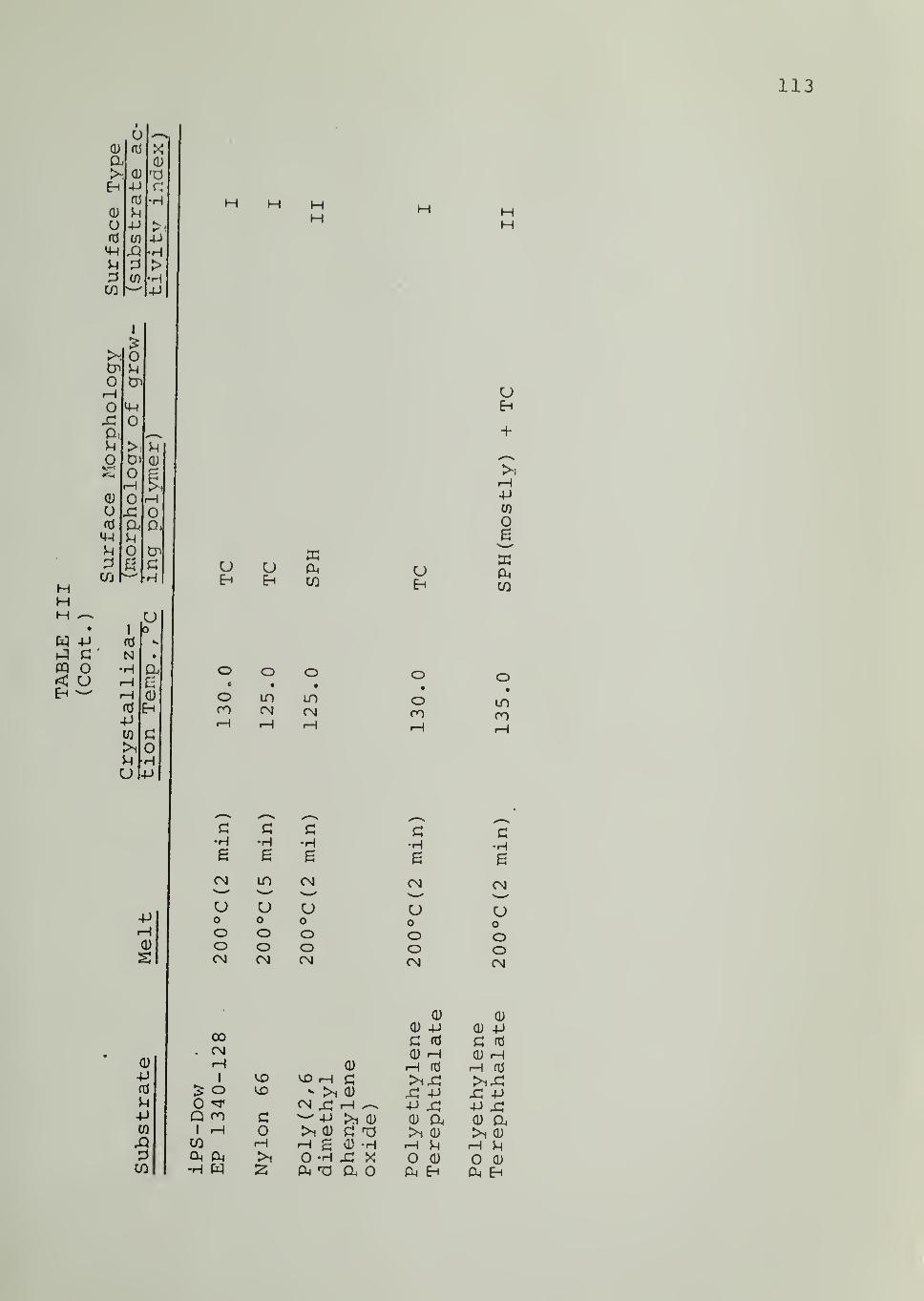
113
HH
W -P3 CCQ 0< uEn —
i
V./
(1)
> CD
t" 4J e* -I
fd •HCD
o -P K* !
fd -Po
Lj
p rn - r- I
rn
otn Lj
o W 1
rHoX!
> Li
0 (1)
^ .^—
H
orH
o 0ofd
u o tn
C/j H
M|
fd
N •
Hr—
1
Gr—
1
0)
fd HPCfi £>i 0M Hu P
-p
H(D
g
CD
-Pfd
u-p
CO
pQ
CO
uEh
OQI
CO
00<N
OCO
P4
COrH>12;
S3
CO
c C•H -H
6 e e
CN CN
u U uo 0 0o O oo o oCN (N CN
CD
CN xi iH ^W -P >i 0)
>i CD C!H g 0) -H0 -H £ X
H
uEh
o o o O• • • •o in m o
CO CM CN COrH rH rH H
HH
UEh
-PWo£
CMC/3
Omco
a•H •He e
CN CN
u u0 oo oo oCN CN
CD CD
CD -P CD -Pc fd c fd
CD rH CD rHrH fd rH fd
>iXi£ p x: -p•p £ -p x:a) a cd a,>i <D >i CD
rH ^ H U0 CD 0 CD
04 EH Eh
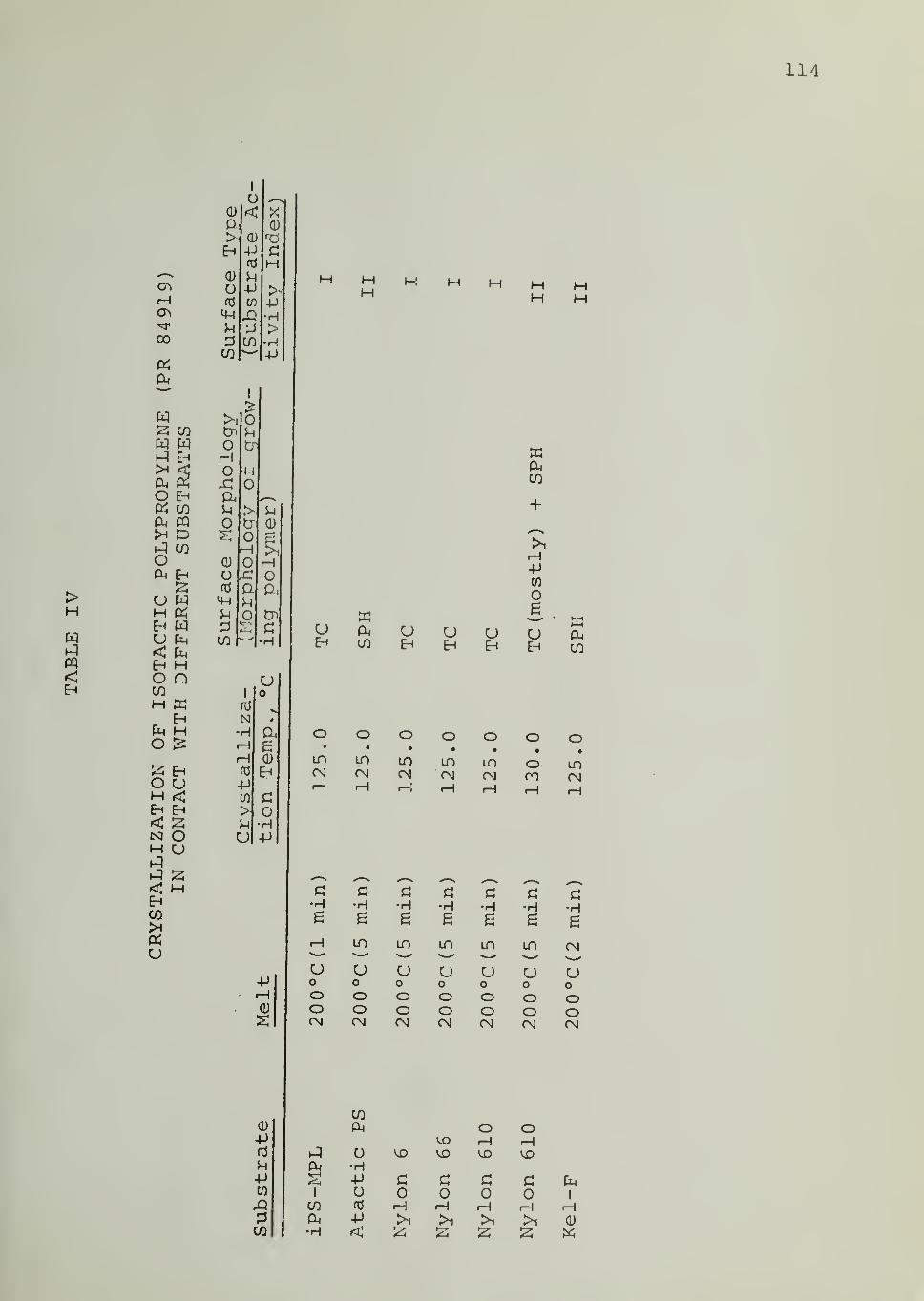
114
wft
<Eh
i
o<U
D (D
>1 'OEh
atCH
<D jh
O -p
H rd co 4J
m Xi HM >
CO CO -HCO
ft
wIS COw w*A Eh>< <;ft ftO Ehft COft ft>h Dt-P COOft Eh2
U WH ftEh ftU ft< ftEh HO QCOH ft
rEh
ft Ho 5S3 EhO UH <Eh Eh
< ZN OH Uhi
< HEhCO
su
>w
tw Li
0HO 44
0a> 7i
0 0)
0i,iH
<u 0 rHu 0aj Dim>H 0 tn
IB -4 CCO H
U1 0rd
NH a
iH e—
1
a)
rd Eh
CO CJ
> 0U H
-P
s
0)+J
td
u
CO
co
Eh
I
CO
H
CMCO
H
CO
ft
o•H+>
Otd
+J
uEh
uEh
CO
2
CorH>12
OrHvr>
OrH
Z
HH
CO
>irH4J
CO
O
uEh
orH
COrH>12
WftCO
o O o O O O OLO in in in LO O mCM CM CM <N CO CMrH rH rH rH rH rH rH
a c fi a c•H •H -H •H •H -H •He E g 6 6 e erH in m m in in CM
U u U U U U uO 0 o o o 0 oO o O O O O OO o o O O o OCN CM CM CM CM CM CM
ft
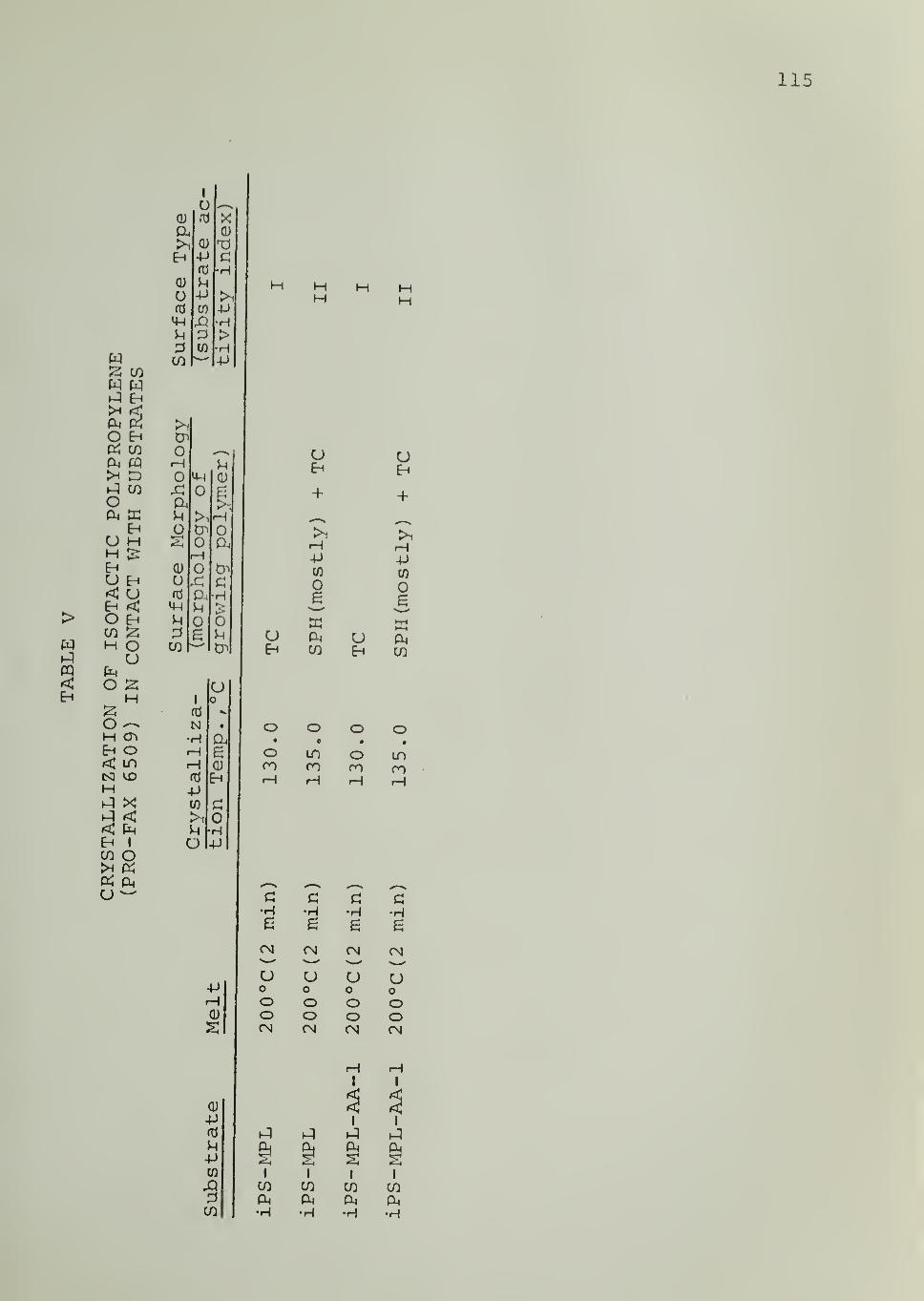
115
wl-H
<Eh
W53 COW Wi-h
o
Eh
sEh
& COP-i CQ
DCO
>H
oEh
U HH &EhU<EhOCO
O
Eh
UEh
Ou
HO —H ChEh O< LOCS3 *X)
Ha <:
Eh I
CO o>H «u —
1
oCD XOu CD
>i CD
Eh C
CD uU -Pfd •P
H>
CO -HCO
>i
0rH M0 m CD
o
>, rH"
0 00rH
CD 0 &U A cfd •H4-1 u »>
U 0 0P gco
I
fd
NHrHrHfd
-Pw
Uo
aEh
0
O
0)
a)
-pfd
u-PCO
.Q
w
uEh
H
UEn
-PW
1EC
ftco
uEh
H
uEh
rH-PCO
O6
ftCO
o O O o• « • •
o LO o LOro CO m COrH rH rH rH
a fi c•H H •H •HB s e gCN CN CM
U u u L)0 0 0 oO o o OO o o OCM CM CM CM
H rH1 1
1
sI
hJft
g 21 1 i 1
CO CO CO COft ft ft ft•H •H •H •H
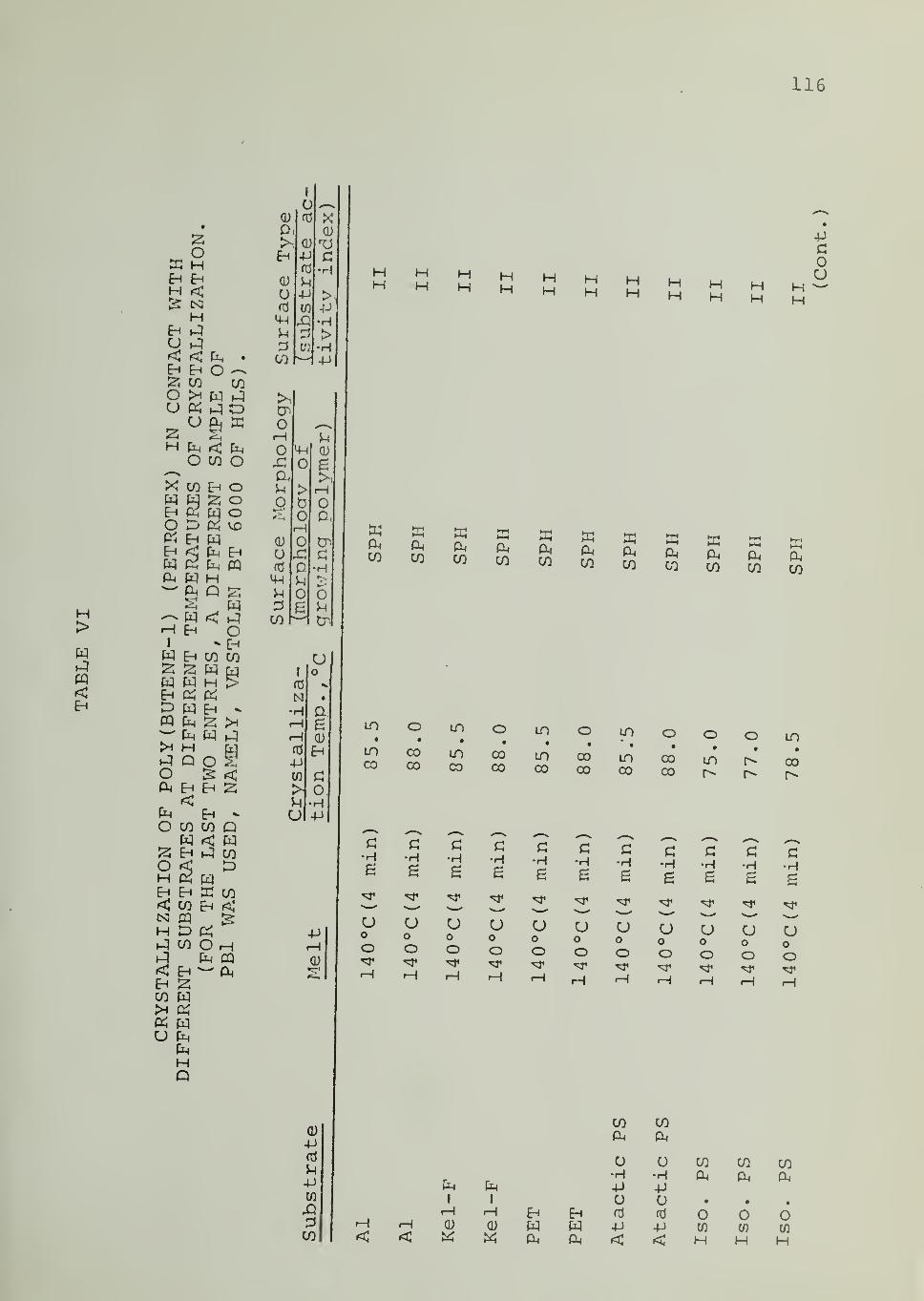
116
EhH
EhU<Eh
O
oHEh<:toHhi
*cEhCO>H
ftow
cohi
U Pj ffi
Ho co
fto
Eh ooolb
ftft Eh
ft cq
X COW WEh ftO Oft EhEhWft WH— ftSWEh
I
H
W ft Hft ftW Eh •
ft 53 >h
ft W i-lH ftQ
Eh
Dft
Eh
Whio
- Ehco m
>H
hioft
ft
Eh
<
OEh
Eh -
o in in aW <C ft
oHEh
<
Eh M
EhCO
to ftH D
ftftEh
CODCO
hi
Eh S3CO W>h ftft ftU ft
ftHQ
ftw Oh
ft ftEh ^ ft
I ) ^CD n:
P 0)
<L > vEh +- l c
•HCD ^O 4- >>
+J4~l HJh >D w *H00 4-)
>-.
bi0rH0
0 g>(
>H > rH0 0
0 arH
CD 0o x:
Q •H4-1 Sq
h- .,
h- '
U 0 03 gCO tnl
U1 0
OJ
N •
H P:iH grH CD
id Eh
CO c> 0,
jH •H
CD
CD
-P<d
-p
co
CO
HH H
H H H
coftCO
ft ftCO CO
KftCO
He
uoo
C•H6
uoo
Uo
O
•Hg
UoO
CH
UoO
C-Hg
Uo
OrH
HH H
H
C0u
K 33 S3 H-t
ft ft ft ftM-l
ftCO CO CO CO CO
ftCO
LO•
O•
LO•
o•
LO o LO o oLOCO
COCO
LOCO
0000
•
LO00
•
CO00
•
LOCO
•
CO00
•
LO
LO
CO
c c•H •Hg g g g g
u U u u Uo o o 0 0o O o o OrH rH H H
< <
CO CO
ft ft
0 u CO CO CO
ft ft-H •H ft ft ft4J 4-»
1 i O O • • •
rH rH Eh Eh td rd 0 0 0a) CD W W CO CO CO
ft ft < H H H

117
H
WPQ
<En
COU
I
o
a,>.Eh -P C
•HCD
0 •P >icd co -P4-1 JQ •HH >
co -HCO -P
>OiH M0 0)
0 ia*h >,0 0g lo dCD 0 Cn0 Xtd •Hmsh 0 0
e uco
id
-p
w>iUu
uo
Eh
coH
CD
s
-prd
5h
•PCO
co
HH H
H HH
H H HH
EC 5304
CO CO CO
K H! 53Ch 0<CO CO CO CO
04CO
c•H -He e e
U u uo 0 0O O OH rH H
C•H •H rl6 6 e e
CN
U U u uo 0 0 0O O o OH H H
CO CO CO CO CO04 04 04 04 04
• • • • •
0 0 0 0 00) CO CO CO COH H H H H
00ro
I
<x>
0000CMfHX
04 X
4H• o0 UCO 04
00roI
L£>
00COCNfHX
a* x04 Ol
• MHo oCO Jh
H &
00ro
I
COCOCNiHXX
MH• 0c uCO OfH —
'
HH
5304CO
o*04
oCNLOLO
04
• <D
0 £CO COH ^
HH
5304CO
o•
o•
LO•
o•
O•
ro•
LO o o oo00
ro00
LO00 CO
00CO
o00
•
LO00
•
0000
•
oCO 100,
c C•H -H
e e e eLO LO
u u O uo o 0 0o o LO LO
H rH H rH
oCNLOLO
04
04
• 0)
o x;CO CO

118
H>
a<Eh
EhU
COWEh
DHsin
WQ PhH OX fn
OU
H
2uoo
W oo
Eh
<>h
ffi
EhW O>H H
EhO r^0< W
COWEh
PhO
OEh
< EhN COh pqA D
EhCO
CO
KEh
i
rd
& <u
<D
Eh -Prd H
0)i)
frt +*nM»—
'
»-* rn
rn
1
*>
nhn
o* i
no
&4H0 tn CD
s: 0H
o 0 rHo 0fd
4-1
op £ Cco H
fd
-PCO
u
uo
gCD
oH4J
<D
-P<d
-Pco
XI
co
HH
OOO rHLO If)
H HH H
o a o o•H •H -H •H-P 4-> -P P•H •H •H •H
rH rH rHP P p
CD CD CD CD
x:a
CO CO CO CO
o
in
0C0)
rH>1.c-p
a;
e>ixo>1
Eh Eh Eh rHW W H 0P4 04 0<
H
t3CD
-p
rd
(D
O rH•H O-P P"H CrH |
P ^M rHCD P
CO
H
0•H-P•HrHp
CD
a.CO
HHH H
CD
c(D
rH
PCD
i0>1r—
I
0On
HH
(1) QJ
Pfd ffl
<u QJ
O rH 0 rH o•H O •H U •H-P P -P 3 -pH C •H C •HM I H 1 rH
rl H r( rH0) P (1)
-aa— a-—CO CO CO
o•
O•
o«
o o orH m o
•
rH• •
oLO in in m in m
HH
U-H-P•HrHpUCD
CO
orHin
4J
C0u
H
0H-P•HHPUCD
n3d)
Pcd
<D
rHup
I
HP
CO
oCOin
w w WCM CM rH (d fd
rH CH>i >1 >i <D 0 0+J P -P x:•H •H •H CO oo COW W W ^ ma C C 1 l
(1) cu KD04 00 04 oo
• • 00 • 000 0 CN 0 CN
0 0 0 CO CO <H CO rHrH H H X H X
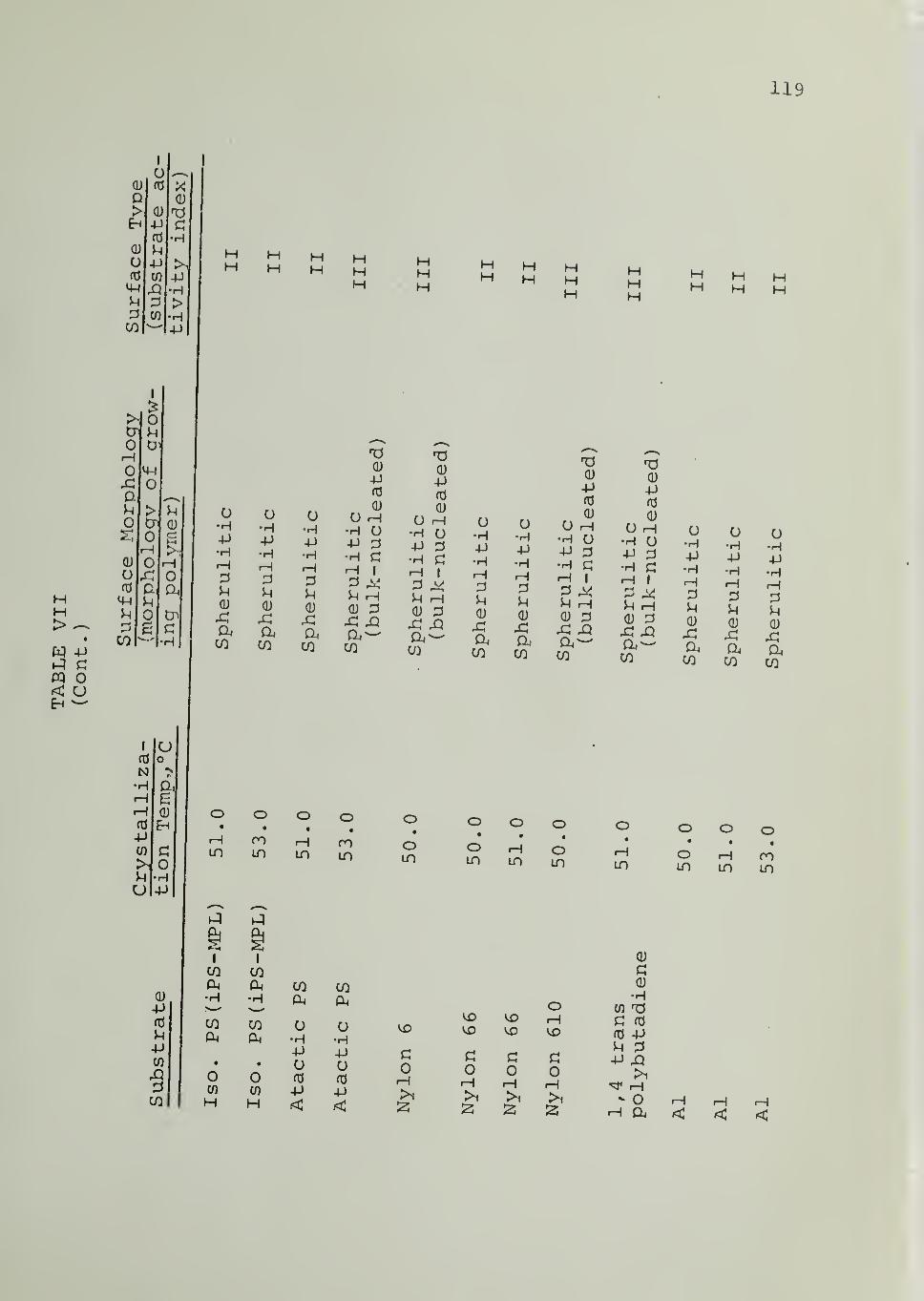
119
HH
co-
rf;
Eh
4J
Cou
i
CJ
1 u v0 GJ
> (D
Eh -P C05 -H
<D U0 -P >trd CO +Jm XI •H
3 >3 w •HCO
> 0tr Jh
0 triH0 <*H
-C 0Q-i
U >O trS3 0
iH0) 00 x:rd c<H n^ 03 £CO
oa,
S3
H
H HH
o u 0•H H H-P -P •P•H •H HiH H iH3 3 3M Sh UOJ 0) 0)X! X! XJ
a.CO CO CO
HHH
<D
+Jid
to
O rH•H O-P 3•H C•H I
3U <HCJ 3XJ X)
CO
HH
Ptd
O rH•H O•P 3H CtH I
3 *M rH<D 3XJ Xi£WCO
H
O•H-P•HiH3
<D
CO
HH
HHH
00)
Pcd
a o i-h
•H H O4J -P 3•H *H CH rH |
3 3 ^H H
a; <D 3x; X}
cu—CO CO
HH
HH
a(D
4J
rd
a>
O H 0•H O H-P 3 P•h a HrH 1 rH3 * 3rM rH M0) 3 0)X3 XI x:a,—CO CO
H
OH4-)
HrH3U0)
x;
CO
HH
O•HP•HH3
0)
X!aCO
i
cd
N•HrHfH03
-PCO
u
uo
G.E0)
Eh
CoH-P
o o o o orH ro rH CO oin m in m m
o•
o•
o oo rH
•
o•
Hm in in m
o o o• • •
o rH COm m m
-Prd
UPCO
XI3CO
I
COPui
-H
CO04
OCO
H
I
CO
r|
COft
oen
COft
o•H-Pard
-P
COft
a•H-purd
-P
<
c0rH
CorH
corH
orH
c0rH>12
o•H
CO T3C rd
rd -PJh 3-P jQ
>i^ rH- 0
rH arH

120
/
HHH>W
<EH
COwEh
sEh
COCQDCO
«EhH&Eh
Urtl
EhS!OU
1
o<L) (0 X
<D
> CD
Eh -p C•H
0)
O -P05 CO -PM-l £) •HIH >P CO •HCO -P
HooiHO
uos
o03
4-1
CO
0
>
OHa
MOg
u
oQ
H
I
td
NH
rd
-PCO
>i
U
Uo
«
i
eoEh
OH-P
-PiH
uEh
CD
C•H
03
-PCO
>
OCO
c03
Eh
oin
e
LO
uoO00
wft
>1-p•H
a) CO
-p c03 CD
U 13-PCO X!Xi Cn
•HCO
H H
uuEh Eh
>i >i4-> +J•H -hCO 0)
C CCD CD
tn tj>H -H
H H H H H H
0•H-P•HrH
0xiQaCO Eh Eh Eh
UEh
UEh
o•
o•
O•
O•
O•
O•
o O O O oLO o
LOoLO
LO oLO
oLO
•
oLO
•
oLO
•
oLO
•
LO•
oLO
c c
e s C G a C
in to• •
•H •H -H •rl •Hg E e e g
ro ro LO CN CN CD
U U u U u U uo o o 0 0 o 0o o o o o O o00 CO CO 00 00 00 00
w wft ft W
ft
>1-P•HCO
acu
0
c co orH rH
CorH
2
VO
corH>iZ
orH
corH
I
COft-H
COa,
oCO
H
CO CO
uH-p
a03
-p
o•H-pu03
•p
<
-p
cou
EC wEh CO CO Eh
in
C fl C C•H •H H -H Hfi g g g g
<N m LO LO
U U u U Uo 0 0 0 0O o O O o00 00 CO 00 CO
COCO
I
V£>
00COCNrHX
ft Xft oJ
<w• 00 MCO 04H ^-

121
HHH> ~
ffl o< uEh —
1
oCD
a>< CD
Ehat
<D
O -p >.rd CO -P<H H
>3 CO HCO P
:h oDO
o.C
OS
uidm
tn
O
>CPoHoX!
o
B
MOQ,
H
u1 0
03
N * OH a •
rH E orH CD LOrd Eh-PCO C>i Ou •Hu +»
CD
-Prd
U-Pco
,Q
CO
HH
HiCO
+
•He
in
Uo
o00
00ro
I
v£>
0000CMiHXX
ft rd
ft, M-l
• 00 MCO ftH
H H H H
Eh Eh
C CH -He g
S3ftCO
CHe
mm ro
u u uo o oo o o00 00 00
CD CD
c c0) CD
rH iH>i >i
-P -PCD CD
6>iX ><
o o>* >rH rHC 0ft ft
I
CD
CCD
-P
P
>1
HH
H
S3 as Kft ft ftCO CO CO
uEh
HftCO
uEh
(JEh
o o O O O o• « • • •
lo o o o o oLO LO LD LO LO
rH
<oft
o
CD
CD -P
C fd
CD rHrH rd
x: -p-p .acd a>i CD
rH U0 CD
ft H
HH
H
ftCO
ftCO
o O o o o• • • • •
IT) o LO o oLO LO LO
C c S3 C fi c•H •H •H -H •H •H -HE 0 E E E E 6LO LO LO LO LO LO LO LO
u u u U U u U UO 0 o 0 0 0 0 0o o o O O o O O00 00 00 00 00 00 CO CO
C C0 0 CO+J -p 1 1 CO
S3 c rH H rd
CD (D CD CD rHft ft U

122
TABLE IX
REDUCTION OF TITANIUM AND ALUMINUM CONTENT*? op tc™POLYSTYRENE SAMPLE BY TREATMENT S»^1S^tSS?^2) C
CodeAl °-
Untreated sample of iPS
AA-treated iPS
iPS-MPL
iPS-MPL-AA-1
2 -07 0.60 0.36
1.24 <0.26 0.05

123
Xwft<Eh
CO CmW S
COS Cm
CO w
wzw
CMoft ftft
I
I
I
COft-H
W>HEhCOI*
O
1-1
Oft u
XEhCO!x
oU CmHEh
T3
OP
C
+J
wAP
tmcoo
o
CU
OS
0id
Jh
P
i
ji
§SiI
COa*•H
+J
W>1
ua
P
cH
U Eh UH O <Eh CO EhU H O T3<C CO Q)
Eh Q H OO W PCO Eh Q TJ 4JH rtj W cW Eh
ft ft <H
in
>1 "4-1
0Z 5 Eh 0 4JO 1 rH fd aH EC W 0 u 0Eh Eh 2 XJ -p •Hflj h o tt co -PN 5 Eh Sh XI fd
H pq 0 Nhh Eh (J s CO •HKH U < H< < 3 (-H* HEh Eh {H 0 fd
CO 2 Eh aJ •P>• O W 4-1
1 CO
ft U U CO >iU < P Cm UZ CO •H uh a
a0H+Jfd
NH uH 0Hfd
-PCO
>1 ea)
u Eh
rHrH OQ) CNX! inco m
fd CO H4-1 co co
|
O fN (n nU CO rH CNHi X |
Xfd
O oM inCm
rH O0) CN
CO LO
X r-fd coM-i co coO CN CN CO^ ro rH CNCm X J
rH
I
Xfd
O oP-i
UEh
UPh
uEh
U UrH
UrH
urH
I
-P Ko co
5 +
u SirH H
I
P Mco (14
O COg
U >,rH H
uEh
CO
I
-P uCO EhO5 +
ta —Cm >iCO H
I
P Kco pnO CO
5 +
Eh H
UfH
CO
I
-pCO
o
CO
uEh
+
S3CmCO
aCmCO
o O O O o• • • • •
m o CO in oCN CO CO COrH rH rH rH rH

Fraction
TABLE XI
FRACTIONATION OF ISOTACTIC POLYSTYRENE(1PS-MPL) OF ASH CONTENT 2?07%
Ash %
11.80
6. 92
1.52
0.51
0.34
Insoluble in Toluene
1st Fraction
2nd Fraction
3rd Fraction
4th Fraction

125
Ht—
I
Xwhi
aEn
Ua EhO U<
oEh
S3OU
w
En
<H H
COXu
EhCO
co OW 04
8 EhCO U
wS3 <NEh w Eh
OCO
COwu ooHH"
o
w Hzhi o>H
fH CO2
Pi oA H
K hi04 O« Pmos uHW Eho uPh EhPi OD COCO H
Eh
U
Ph
oEh
EhHS:
rda)
o
cH>
0iHOx:auo
CD
Ufd
<4H
Sh
CO
CD
-P
•H
dp
CO
4
>
0
o•H+Jrj
N•H
id
CO
>u(J
rH OCD <NXl LOco m
ro rHr\J I
ro rocm
I
rd coMh ro0 <NU rH
CD
>-P
cCD •Hu
rHo\o rH OCN CD CM
H rC m• CO LO
> MH0
0 • •
rH XI co CO 0x: < -HQ -PU fd ro rHO H N CN 1
s •H ro roC rH <£> CN
d) 0 rH 1
o •H fd X r-fd -P -P cd 00MH o CO Md rou fd >^ 0 CN
5h SH U rHCO o 0, X
u1 0rd
N •
•HrH erH 0)
td Eh-PCO
> 0u •Hu -P
d)
rd
-PCO
>iiH
OCO
c«d
!H
-p
0)
•HrHrH
o c rd
•H fd -P-P CO
•H >;rH >i u3 rH 0u -p CO
CD CO c0 fd
uCO -p
CD (DC Cf—i-H *HrH rH•H -a rHfd c u o c fd
-P fd -H -h fd -PCO -P P CO
>\^ -H -H ^ >,u >jrH rH >i HO rH 3 3d UCO -P SH 5H -P CO
CO CD CD CO C0 XI X! 0 fd
U 6 ^ 6 rH
-P w CO CO^— -P
CD CD
C c•H •HrH rHrH rHfd fd c a-P -p fd -hCO CO -P>1!H >H >irHO Ud 3CO CO -P rH
c C CO 0fd fd o x:Sh u g a-P -P ^
^ CO
CD CD
C-H -HrH rHrH rHfd fd
•P -PCO CO
>1 >iu uu uCO CO
cfd fd
u u-p -p
oro
inro

126
HHHXW3<:Eh
Eh
oHEh
<toH
<EhCO Q>h
Ph
uwEh
H
Dm
co
EhH
Eh
<Eh
OU
COWEh
EhCO
^1DU CQ
W Pn DEh W CO
gm
Eh -
CO 00
D I
COCO
*Z ooO CM
>H
EhHCO
wa
xX<cPh
o
oHEh
<
U PhD O
a*Cm•H
E3
JQ U
•Hwc
QC0•H-Prtf
(U
rHU
2
CO
E oo rH\ 1
•H OrH
a XrH PP A
c
CN
a)
(-i
u LO•p \ 1
aj •H oCD rH
-P rHCO a X
Ps CO
co c
*2
a) 1
rHo co
e •H C/)
CO Eh
CD rH-P P
PQP0u
Q)
H +J
Q) 03
rH >-l
0 -PP CO
i3 pQPCO
<D
•P03
4-)
CO
X)PCO
o inCO CM
LOO
CN
O
CN
CMOO
00LOoo
oCO
CN
OO
«
o
CN
oo
oo
oo
oCMOo
CO
g•He
CO
• ».
uoLOCMrH
<1
Eh H•H W <D
LO
Cocj
rHCO
•
o•
OO
•
OLO
•
LO oo O o
LO CO• •
CN
•
A
•
00LOA
CNCN
•
CM
oH
00 orH
CN00
oCM
LOCO
O00
OO
CTt LO-1
CNrH
CMrH
r-CN
•
A
*
oCO
A
COHoo
o O o o O
CN H orH H CM
rH CM 00 00rH H CN
A A
COCLj o
rHyo *x> O
•HG G -P fi
0 0 U 0rH rH id CO H>. >1 -P >iZ z < •H Z

in
HHHX —
PQ O<: uEh —
co
eo\•H<D
rH-p U*H rH0) 3e 03 =«=
CD
QCN
C e0 <D U i
•H -P-P fd •Hfd M CD
<D -P rHrH 0O
CO
3"
CD 1 ;drH
O U)
e H U)
fd 0)
CO Eh G
CD rH-P 3C pq30U
0)
H •p
0) rd
iH uU 4J
0)i
£1
=&=
0)
4Jfd
u-pw
CO
HE
U0
P
o rH CN o00 r- CN
CN CN
00
co
co
P4I
rHCD
HCN
CN
LO in CN rH roro m rH rH •
• • • • inCN LO ro
CN
OCN
ro
CN CNin CO
CN
roA
O00
A
CNCN
Oo
roA
rH CN 00CO CN O
O CN H rH rH
oCN
A
rH O 00 ro rH m CNKD CN CN CO CM CN CN LOO O o o O O O OO O o o O o O OO O o o O o o O
00CO
<
CO m in mrH rH CN
A A
CO
OrH
o VO•H-P C GO 0 0 0fd H rH En H CO-P >i >1 W >1 Oi< 2 ft 2 •H

>HXw3«En
25O •
H COEh W<< EhtS3
H
<EhCO CO
LOo
Eh
COPQD
Eh
Zwa w
DPQ
fafa
EhH
Eh
U<:Eh
ZOU
QS3
w zEh H
Eh Xco wPQD
Eh
OCO &
EhZ WO A>hEh —CO
WI
Wz
Q WEh
ZoHEh
m>H
.-H
w oJ fauDZ
fa
o
>i+J
•HCO
c
Q
3pq
eu\•HCU
HU
Z
CNc E0 o•H -p \4-> rd Hrd 0)
<D -P rHrH CO OO
CO
CD 1
CO
H CO
0CO »—
.
0) iH4J PC CQ
0U
0)
H -P<D rd
iH MU +J
COi h
CO
0)
+Jrd
•PCO
XIPCO
LOIo
CO
LOo 00 orH•
CO• •
00 CO•
CO• •
ro
O CO rH 00LO
•
o•
LO•
00 ro
CN ro CN•
CM•
CN•
rH
rH O00 H
• • •
CM LO rH
LO LOCNro oo
co
C•He
uoLO
•
LOCO
-P
<
CN
LO
CO
oLO
LOLO
LO CO ro CN rHro r> ro LOo o o o o oo o o o o Oo o o o o O
COLO
rH orH rH
CO
o-H
fa -P1 O
CU rH Eh (d COCU <D W -p CU•H 04 < •H

LDO
X —W -P^ cm o< uEh —
LOo o CMH ^* in CO
H H rH H CN
e oo rHN . 1
•H o0) HrHU XP rH
•H »-v ACO m -U qCa)
Q(N
C E0 (1) u m-H -P \ i
-P rd *H oid M <D H<u -P rHiH 0) O Xo & 33 3 2: CO
Z CO =**= C
'HCD
Hu CO
e •H to
rd ^1 0)
CO Eh ft
<U rH+J
C PQ30U
0) •
-H 4J CO
Q) rd fiiH U •HO +J 63 co
en
uo0000
prd -PU+J
CO
•H3 •HCO
VD 00 H 00ID
• •
00 CO O rH
CN rH•
H•
•H•
CN•
iH
rH in 0000 00 r- <T\ 00 00
CN CM CN <N CNJ
l> CN 00 Hco If) oo o O o o rHo o o o o Oo o o o o O
mo in
in
CN
o00
CO ro in
COft
u•H-P
I CJ
rH CO E-i rd ftCD ft W P ft« -H ft <! •H
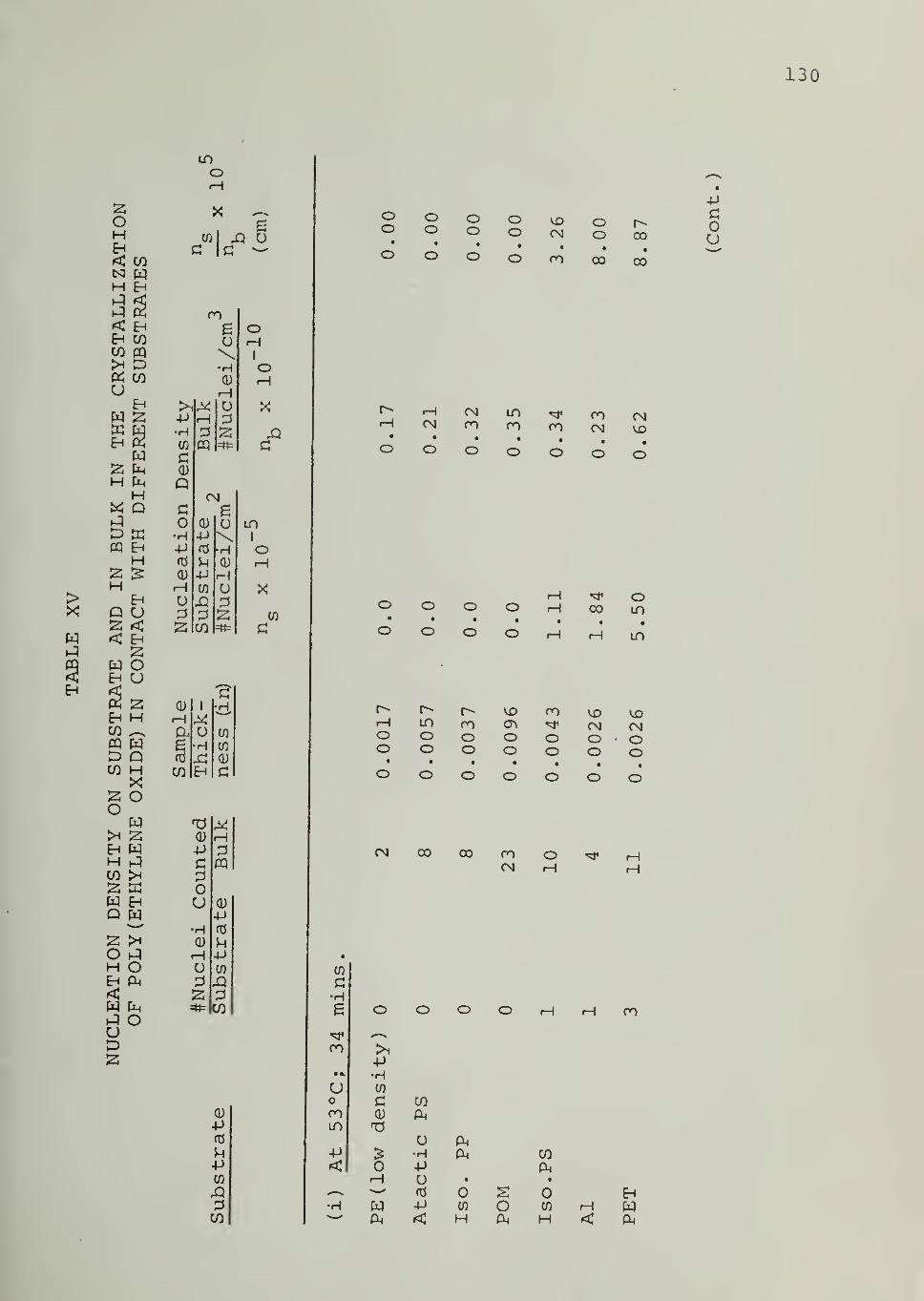
130
wl-H
2oHEh< WN WH Eh
<Eh
>h DU
Eh
WKEh
Eh
2
W
« QD ECCQ EhH
HEh
Q U2 <
Eh
aW OuEh
Eh H
DW
o>H
Eh
QHXoww
cn2 EC
W Eh
Q W>H
Oft
S3oHEh
<O
uD2
IT)
o
X
Wl jQ 0
6o\•H<D
•HU
4J f—
1
fi 2:=**=
cCD
QCM
C £>—
i
0 (U O•H -P \-P fd Hrd <U
(U -p HiH w oU XI353 co
CD i HiH
O CO
§•H w,C
CO Eh c
d) rH-PC PQ
0u 0)
•PH fd
CD
iH -p
O3
4J
td
+>
W§CO
orH
I
X
IDI
oH
oo
•
oo
•
oo
*
oo CN
oo r>
CO
o o o•
o•
co•
00•
00
-p
cou
rH CN If) CO CNrH
•
CN•
CO•
CO•
CO CN
o O o o•
ot
O•
o
c•HB
CO
Uo
COin
-P<:
rH oo o o o rH 00 mo o o o rH H LO
r- vo CO VDrH LT) CO a> CN CNO o o o O O • Oo o o o O o oo o o o O o o
CN 00 00 CO O rHCN H •H
o o o o H H CO
p•HWCa)
o
w
CO
a•H-PO«J
-P
«5
oa)
So04
CO
o EH
w

W -Pa cPQ O< uEh —
C0•H-Pid
0)
rHUP2
CD
H
CO
CM
eu
-Pfd H•P rHCO O
3CO
1
Mo cn
•H w0)
Eh C
*V MQ) rH-PC CQ
0u 0)
-P•H fd
<u
rH -PO CO
£13
=**= CO
LT)
IOrH
X
-Pfd
u-P0)
XI
CO
0)
oo rH rH
LO
o rH rH rH rH CN OrH
.0018 .0033 .0028 ,0024 ,00640057 0027
o o o o o o o
CMCM CM CN
CNCM
COCO
•
co O rH rHc•H6
in
>i-P
o •H0 CO
rH C COm OJ
-P 0 04
<g
•H ft-P
rH O •
-H fd 0•H w -P CO
a* < H
CN
COCO
CO
CO
9
0CO o wH

W -P^ cffl o<< uEh ~
mo
Ig
c Is -
-prlCO
CD
Q
3
go\•HCD
O3
oHI
p:
0 CD r
\
\_/
-H -P \-P Cd •H(d u CD
CD -p rHiH 0) oO XI 3
CO
CD 1 •H,iH
u CO
g •H CO
c* CD
CO EH c
0) fH-P
C
0u
CD
H -PCD ctf
H HO -P
CO
rQ
0]
LDI
O
X
0)
CD
-P03
UPCO
,Q
CO
00 roCN o
• • •
CN ro
CN 00O• • •
00 rH CN
ro iH00 ro 00
• • •
LT) CNrH
00 00CN H CNO O OO o o
• • •
o o o
00 Hm CM
• CNCO
c•Hg
ro
U po •HO CO
LO CCD
4-»
§H rH•H•H H W
ft
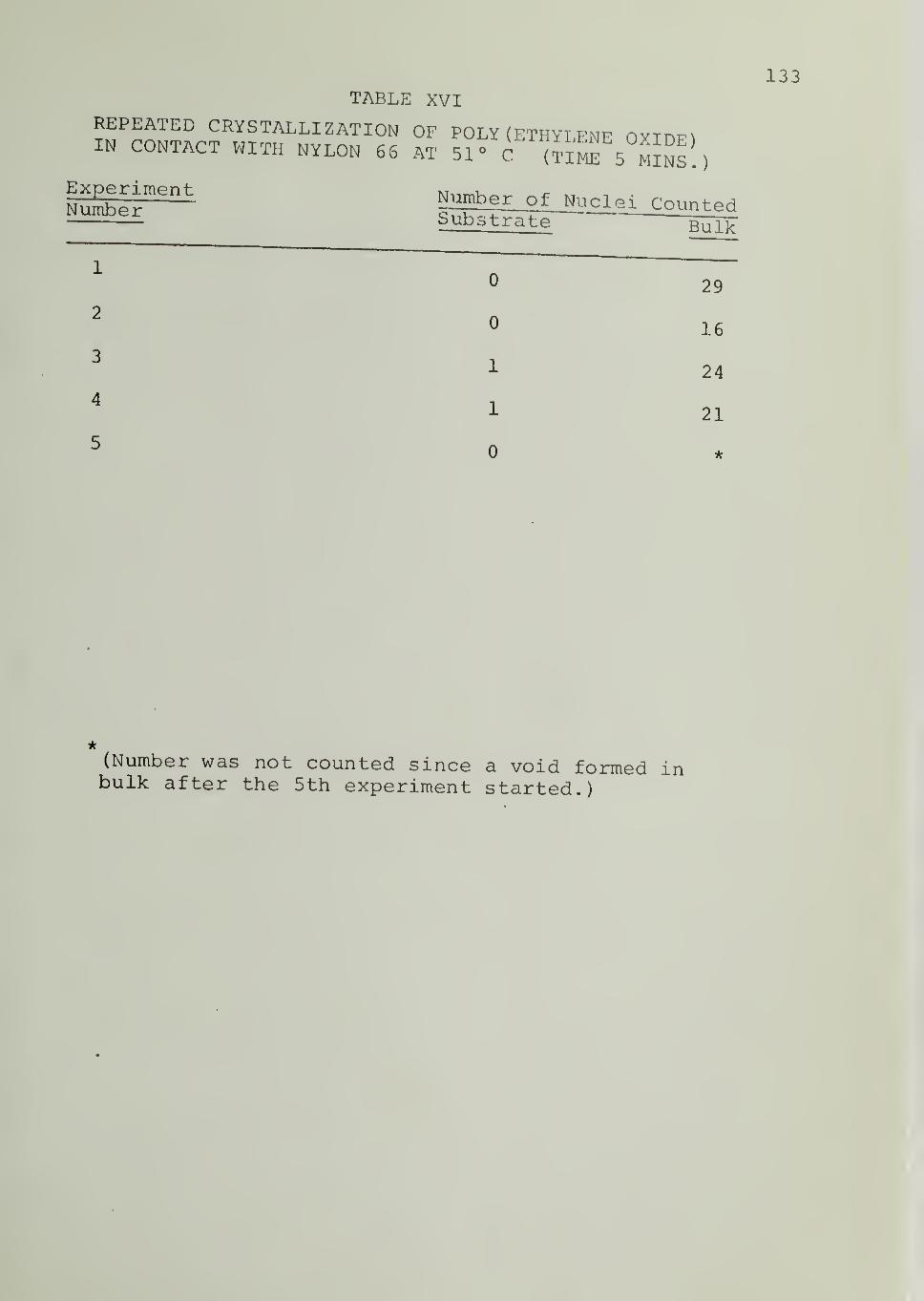
TABLE XVIREPEATED CRYSTALLIZATION OF POLY (ETHYLENE OXTnFlIN CONTACT WITH NYLON 66 AT 51° C (SSY52S!
)
gSberment »yer of^
Substratej^JXk"
10 29
2
3
4
50
(Number was not counted since a void formed inbulk after the 5th experiment started.)
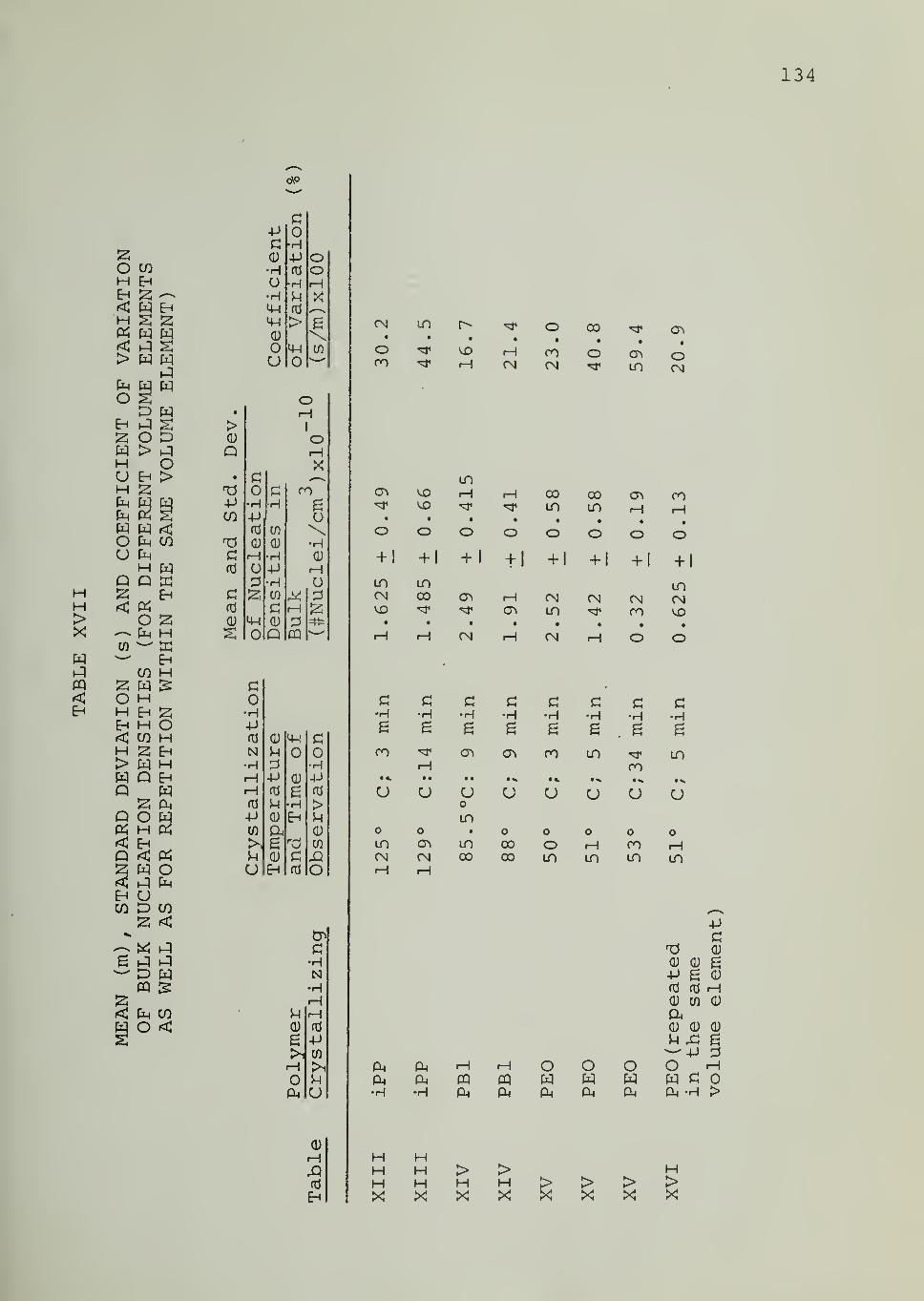
134
HH
EH
o
ou
S3
53O
H>WQQ2<:QS3<CEhCO
PO>Eh
S3Cm
hw w
ftoft
uD53
Eh
22
WEh
PSW
, £ .
« w w< ^ s> w w
HwspH"o>
Eh
<
ftOEh
53WHU
Cm COftH ft
Q Q ffi
S3Eh
CO HW SH
H Eh 53Eh H O< CO H
53HQ Eh
ftft53
OEh<; ft
W Oft
CO
6 J Klw P WPQ 5
< ft COcq o <:
>
Q
•0+J
CO
C
CftJ
0)
S
-Pc<D
•HO•HMHm0)
ou
coH-P(d
H>H
>
o
OOHX
g
w
xc0 c•H H-P0J to
0) 0)
H HO +>
P •HCO
C rH<4H 0 30 Q m
co
go
•H0)
rHO
=tt=
co*HPfd
N•HiHrHfd
-Pto
uu
CD
0
fd
aCD
EH
CJH c0 0
•H0) Pe fd
>
CDrCJ CO
cfd O
CHNH
CD
g5H w
oft
(0
Sh
U
CD
rH
Eh
CM r- O 00 <T\
O H CO o Oco H CM (N LO CN
rH H CO CO CO<o LO LO rH rH
O o o o O O o o+ | + 1 + 1 tl + 1 + 1 + 1 +LO LO LOCM 00 <y\ rH CN CM CM CM
cn LO ro V£>
H rH CN rH CM H O O
C c a a C C G•H •H •H •H •H •H •He g g g e g . 6 gco co LO LO
iH CO
U u u0
U U u U uLO
0 o • 0 o 0 0 0LO LO 00 O H CO HCN CN 00 00 LO LO LO LO
04 ft rH rH o O oP-J ft PQ PQ H w w•H •H 0^ p^
HH
HH > >
H H H HX X X X
>X
>X
+J
a) <u g4J g cu
is <rj i—
i
<d w a)
aQJ <U CD
U SI
ow cft -H
g
H0>
>X
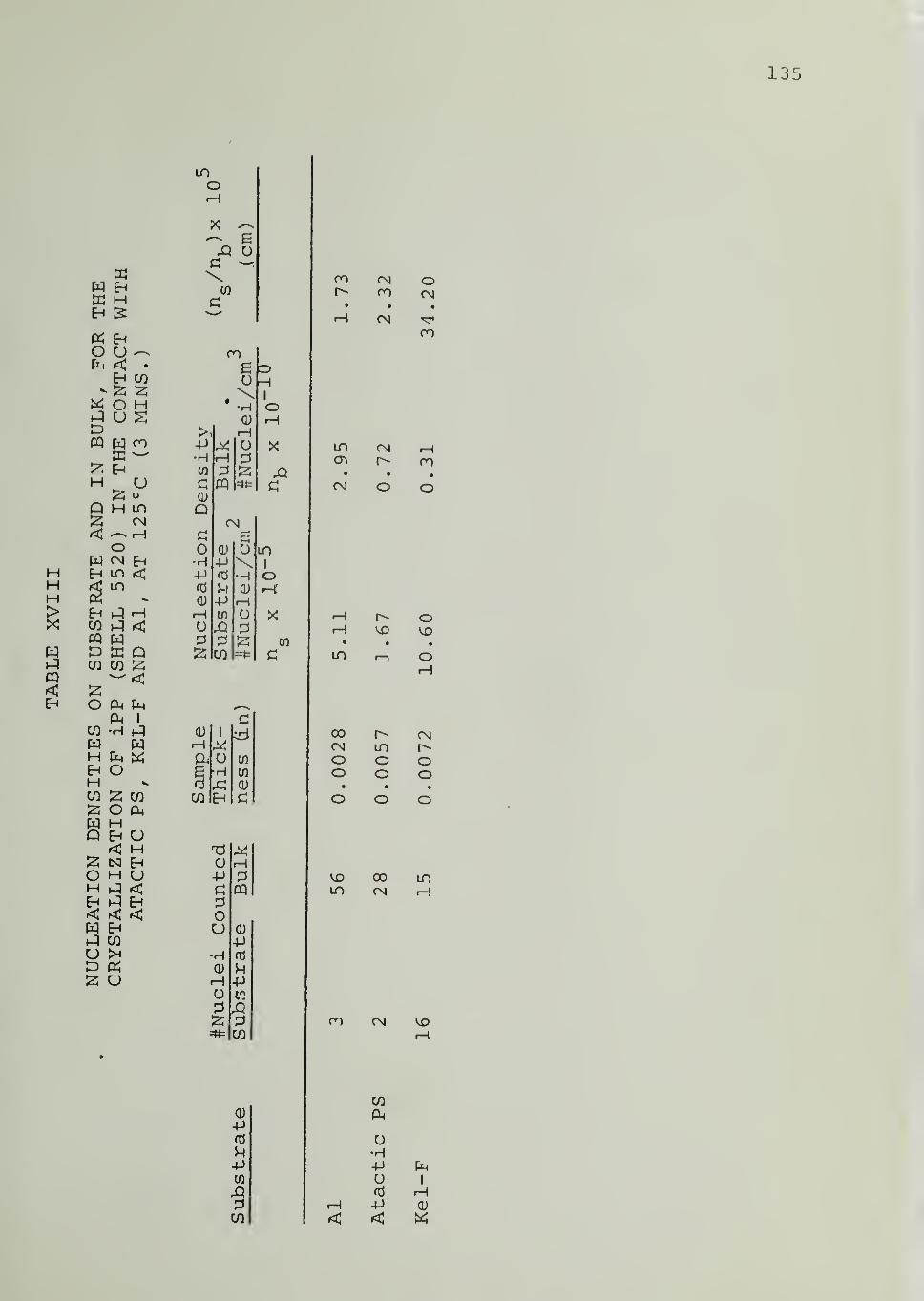
135
in
HH
3PQ
<
KW Ehffi HEh £
Eh
O U£ rtj
Eh- 55« O HJUSDW pq n55 EhH U
55 oQHin2 CM
CO55
oEh
sEhCO« WD WCO CO
Eh
LD
Qa
o cu hfin I
HCOWH m kEh OCO
55WQ
OHEh<:wuD
55 COO CUHEh U<C HCO EhH UJ EH
EhCO>H
U
oiH
XE0
C\<z
CO
-PHCO
C(1)
Q
o•H•P<fl
0)
rHOP
CD
CO
•\•HCU
1 rH0
rH pPCP =tt=
CNBo
-P \-H
r-l CD
P rHco O
PpCO
I
o co
H CO
XJ CD
C
T3rH
-p Pm
0u CD
-P
H «5
rH -PU K
XIPco
CD
-P(d
-PCO
XIP0)
X
io
0)
CO CN Oro CN
« • •
rH CNco
LO CN rHro
* • •
CN o o
rH OrH VD
• * •
LD rH OrH
00 CN(NO o oo o o
• • •
o o o
00 inLO CN rH
ro CN
COft
oH4J
Otd
p<
faI
rH0)

136
/
XHX
w
i<EH
CO
in
c\
O
\CO
CO
(U
id
-P03
CO
00LO
• • •
o COIT
I
CM
o•H+J
ofd
p
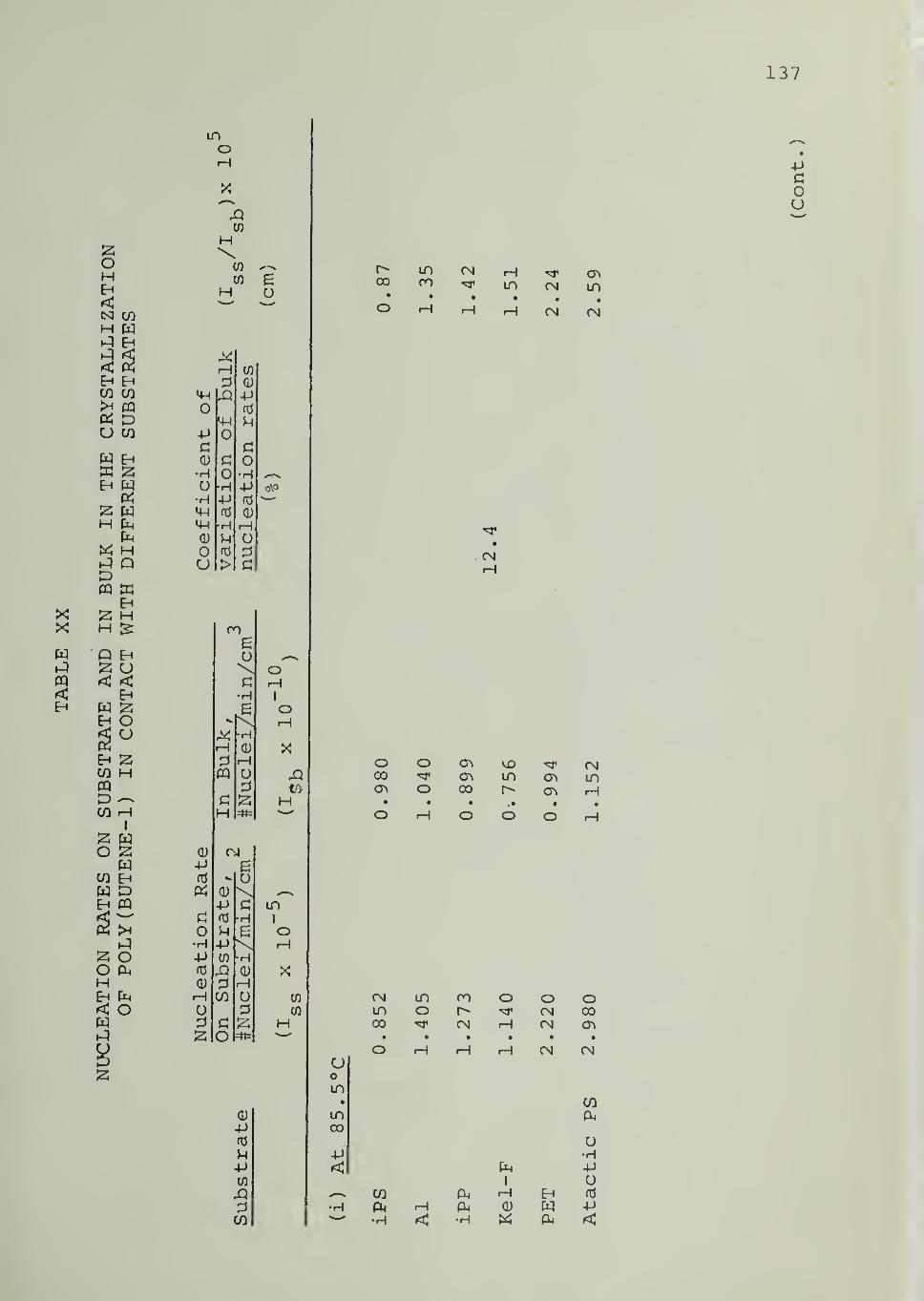
137
Xw•J
a
§HEh
<N3 COH W^ Bh
En E-«
CO CO>h W« DU CO
En
S5
WWEh
HX H5m x
HEhH
Q EH
UEh
Eh
EhCO«DCO
S3OCOwEh
2
Ou
I
wwEhD
2 OO Ch
wH
S3
faO
ino
Mv.
cn
CO eH u
rH cn
34J
0 rd4-1
p 0c cd) £ 0•H 0 HU H -p*H -P to
U-l <d 0M-i rHCD U0 <d
U > c
CD
•Prd
co•rH
-P
CD
rHo3
eoo
c rH•H 1o\ rHH
rH <D XrH
PQ 03 to
C HH =»=
CN
Q)
4J
rd
M4J
V)
S2
3CO
2IO
su
cMs
HCD
HU3
inlo
X
CD
-Prd
-p
w.Q3CO
PCou
in CN rHCO CO in CN in
o rH rH rH CN CN
CN
o o CN00 o\ ID LOo 00 rH
o rH o O o rH
CO CN in ro O O OCO in o r- CN 00
H 00 CN rH CN <J\
o rH H CN CN
uom
•
inoo
-P
<COft
1
ft rHft CD W
ft
COft
u•H-PUfd
p
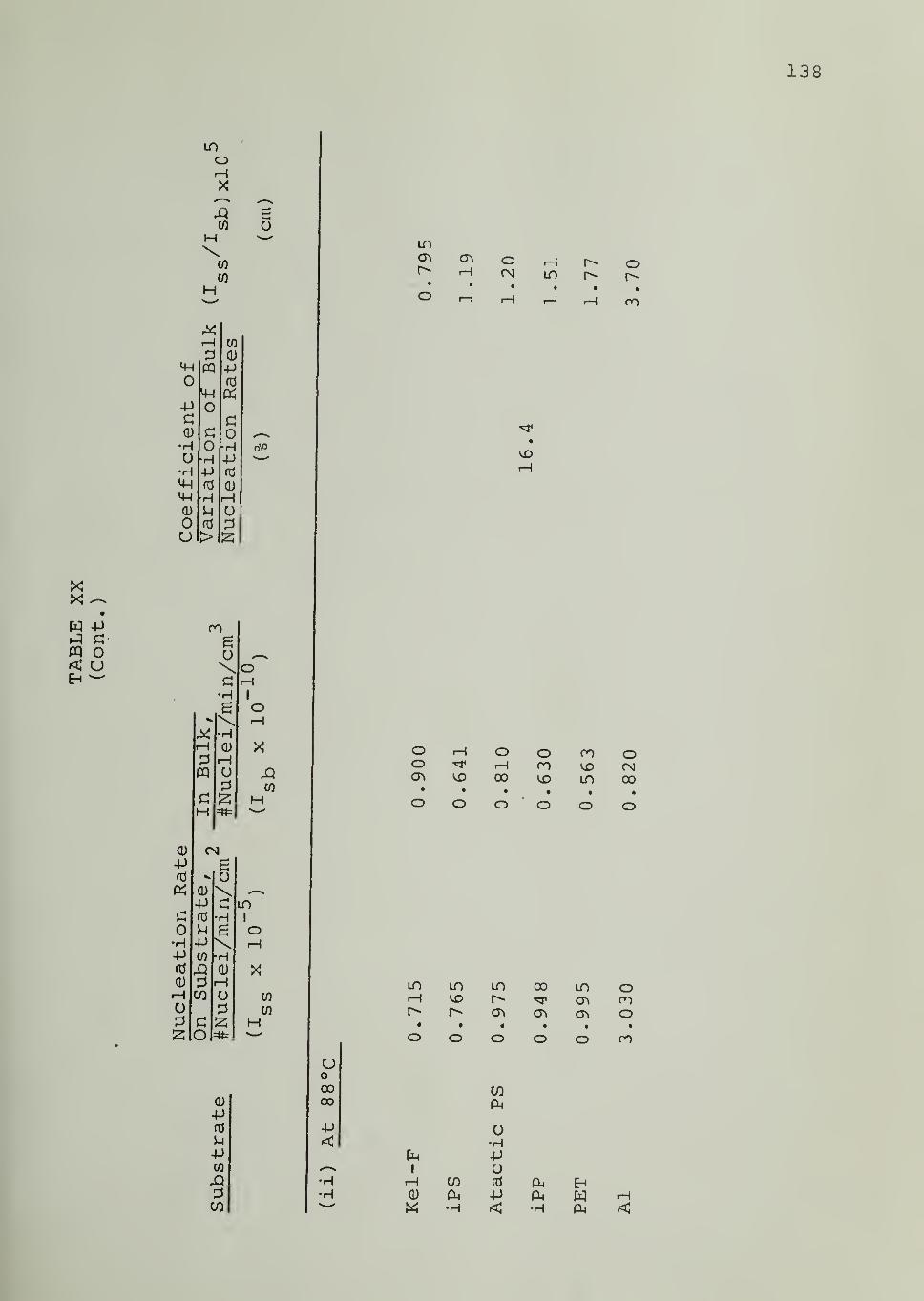
138
LOoX
X
mH\
CO
cn
H
LOcn o H O
•
iH LO r-
o rH rH rH m
rH CO
3 <U
CQ P0 (0
4-1 «-P 0
CCD G 0•H O HO H•H Pm 05 a)
•H rH<D U o0u >
rH
W -P
a cm o< u
-Pcd
05
CO•H-Pto
rHo
2
apq
cH
g0\c•Hg\•H<D
rHO0
OrH
co
a)
+Jrj
rH
CO
en
ao
H(U
H0
=tt=
ini
o
CO
CO
CD+J
fd
u-p
CO
.Q
CO
o rH O o co oo rH VD<J\ 00 m 00
o O o o o o
uo0000
p
HH
in LO LO 00 LO orH cor- r- <Ti oo o o o O
CO
ft
0-H-P
l OrH CO rd Ph Eh
CM P Oh W rH•H •H ft
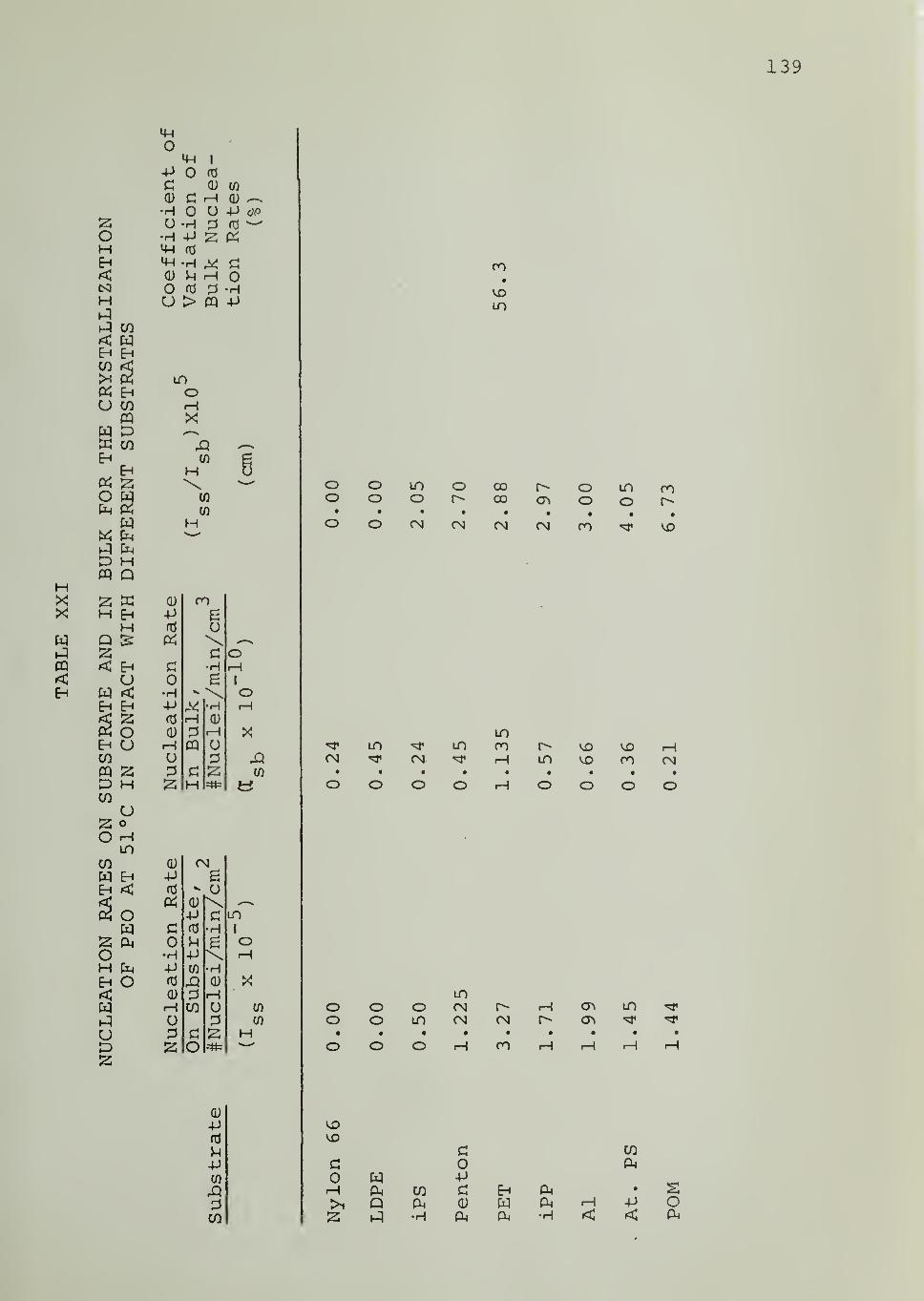
HXXw
aEh
oH
COHi-h" co< wEh EhCOX« Ehu co
W Dffi coEh
Eh
OfaW
D
CO
zO
HQ
UorH
mo
UH+> oc0)
HU
coH
•H -PMH n3
*H -H ^
I
fd
—I CL> -—
^
U -P oV°
£-4
oH
u > mo•H-p
LOO
H
2 K 0) COH £h 4J
H Id uQ S: «
c o< Eh C H rH
u 0 e 1
w <; •H \ oEh Eh *P •H rH< Z td 1—
1
0)
ft O 0) rH XEh U rH CQ Uen U
NuA
C wP H !3 H
CO 0) Csl
W Eh -P eEh < rcJ u
KK O -P c LO
W C •H 1
0 SH £ oO •H -P \ rHH Pm -P 01 •H
fd rQ CDg°a) rH
w iH CfJ o <
a <
u Cp Oz
+J
id
u-p
en
X!PCO
CO
LO
o o in o 00 O LO coo o o 00 o oo o CN CM CM CM CO
inLO LO CO H
CM CM H m CO CM
o O o o H o o o O
mo o o CM r** H ino o in CM CM r-
o o o H CO rH rH rH
C CO
0 ft
0 W +J
iH ft EH ft •
>1 Q ft W ft rH -P
z •H ft ft •H <aoft

140
HHXXw
E-<
o
in
Eh
Wa.
a
o
wwEh
oHswuD
DPQ
*Ho
oo
0) c•H ou •H X•H -PM-l 05
H0-) U \0 COu >
noXI
(0
d Hcfd <Hp 0CO
CO
qfd
0 oH iH
e; -P 1
fd o•H H>
(D X:: Q
(Dm-P afd oPS
c oC •H H0 B 1
O-P »H rHfd <U
0) rH XrH Oo XJM ^. CO
r
'
ti H
o>
11
u
orHOrH
X
orH cH\
0) Hu
rHCO ufd
:•;
y,II
CNCM
•
O+ 1
inCO
#
o
LO co oCN CO o o
rH co (N• • * •
o o o o
o
CO
PS
rH CN• • • •
orH
• • • •
rH CN ro
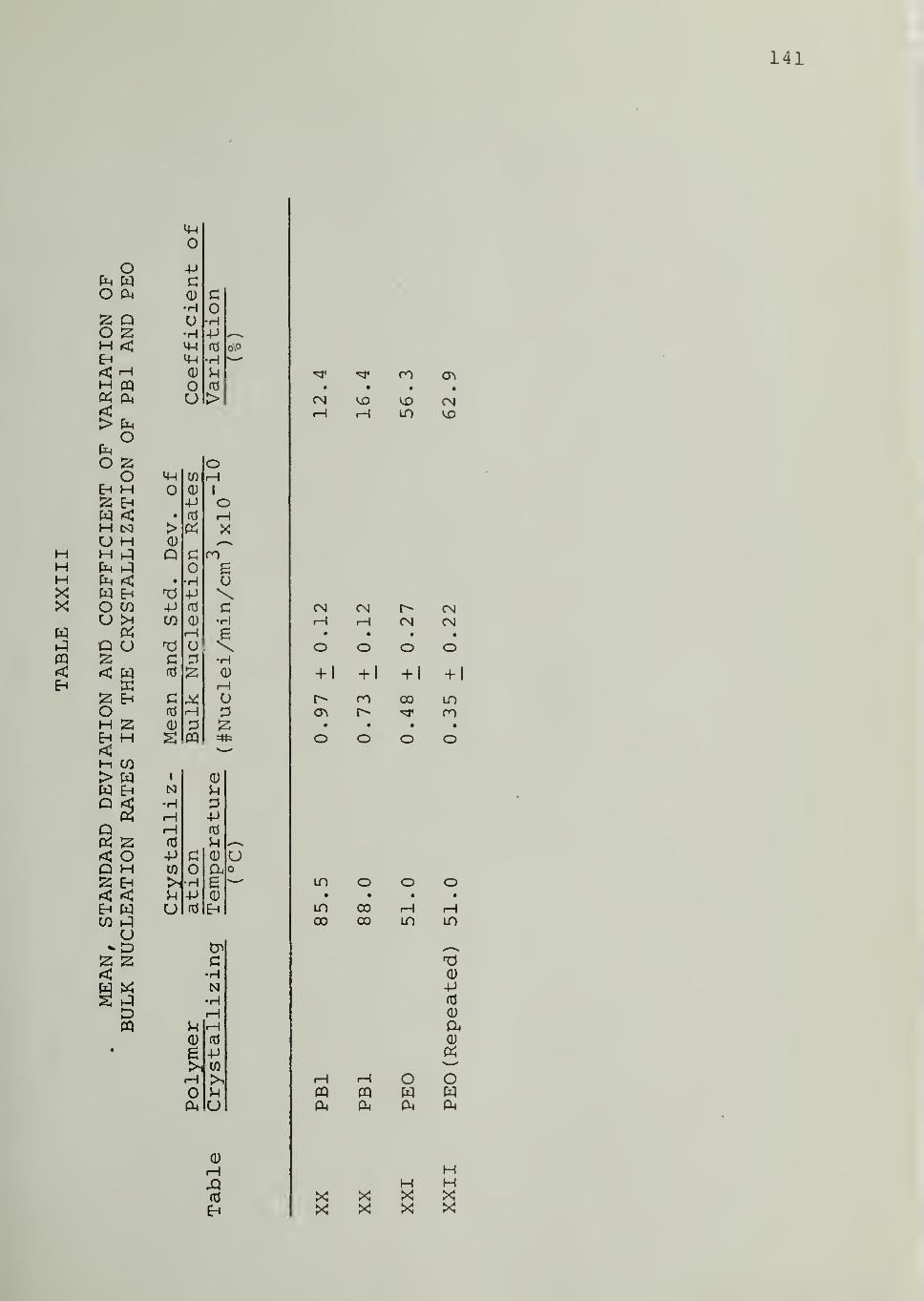
141
HHHXXwKH-
3Eh
0
o -Pw c
O CM CD c•H 0
2 Q O •HO £ •H -PH < M-l fd dpEh *H •H<: iH d) >H
H CQ 0 fd
« CM u >3>O
fnO 2 oO 03 rH
Eh H 0 <D 1
2 Eh •P OW < • fd rHH CO > « XU HH J Q c coPh ^ oPm rij • •H OW Eh -p V\O to -P fd cU » tf) cu •H
rH sQ U u v,s.
G •H<C W fd <D
rH2 Eh c M 0O fd rH 3H £ 3 53EH H £ m =**=
<Hi CO
> W i 0W Eh N rH
Q < •H 3rH -P
Q rH fd
PS 2 fd Jh
< O -P c CD UQ H CO 0 CI. o
2 EH >1 •H e< < u -P CD
Eh W u fd EhCOU
•» DS5 £ c
•H
a « N•H
D rHrH rH
ocu
fa
u
H
Eh
ro• • • #
(NH rH m VO
CM r- CNrH H CM
• • • •
O O o o+ 1 + 1 + 1 + 1
r- ro 00 inro
• • • •
o o o o
in o o o•
in•
00•
rH
•
H00 00 in in
+>fd
QJ
a>
rH rH O oPQ H w
ft Cu ft CM
HHH
X X X XX X X

142
>HXX
ATE
STR
rrtCMD uCO 0
LOco •
CM LOrl
Eh
oCO iHw mEh
ooHEh o< HW Eh
<:u IS3
D H
PhO Eh
COX >H
EhH UH wm KH EhuDQ HO«
-Pid
u-p
wpQ
CO
co -
0) g-P u«S \« CHC £ in0 \ i
•H -H oU (D rHffl rHCD O XH 3O 2 CO
w2 =«=
oo
CMIDCO
O
4J
xw CN
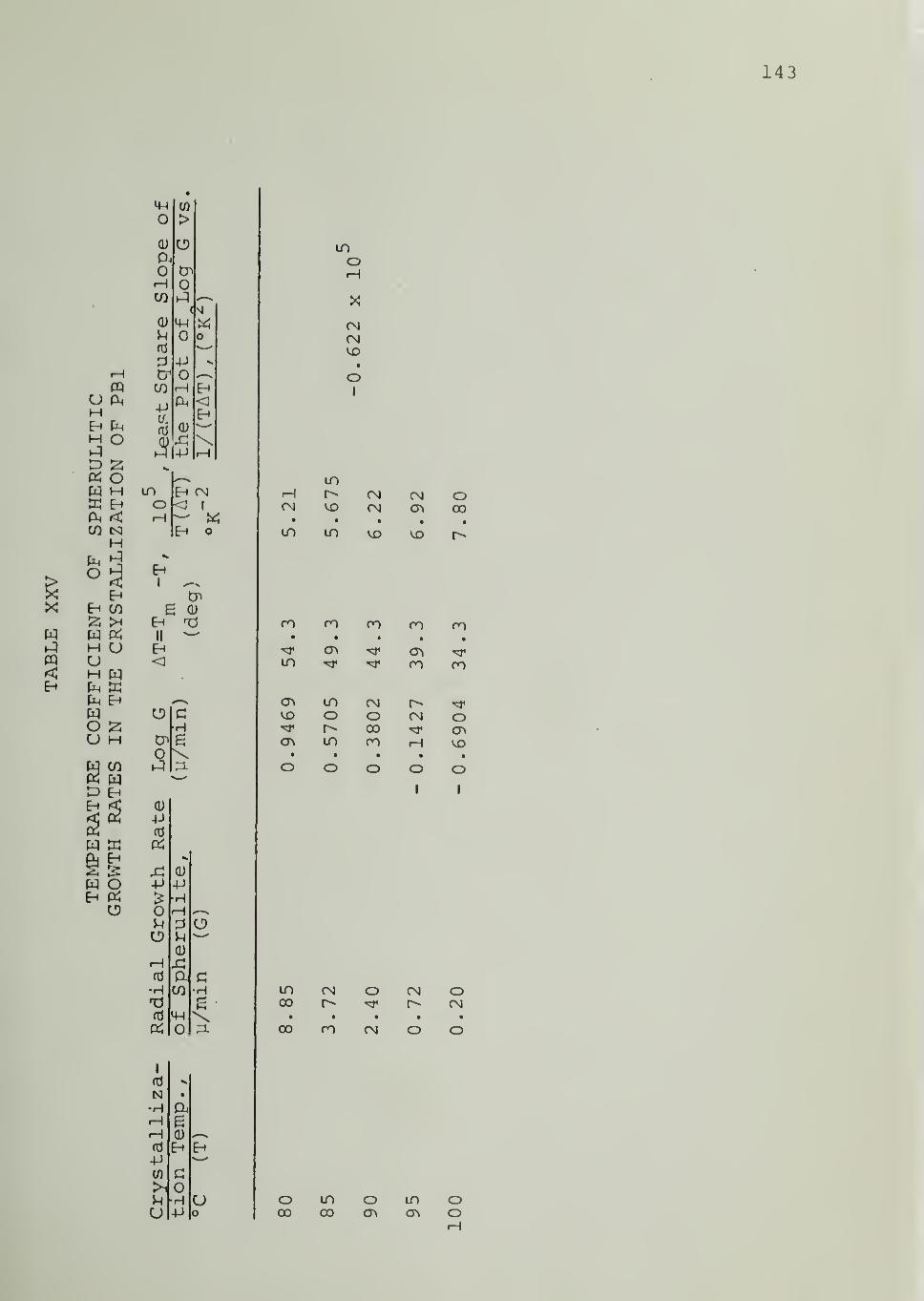
143
Xw
<Eh
mMEh Eh
O
Eh
2
2O
5W HEC Eh
Ph <<CO tS3
Hp4O
l-H
Eh
CO>H
W KH UUHhWOU
wECEh
W CO3 wD Eh
W K§ &W OEh
o
p,oiHCO
0
(0
D1
CO
-P
3
>
o
0
-Po
ft
CD
C-P
Eh<Eh
LT)
OEh CN
\ o
Eh
6 OEh TiIIw
Eh
<
o c
CP S0»J
-p
«<L)
-P
H0 i—
1
u Ou M
0)
rHtd a C-H H
g\id "4-1
« 0 P.
i
NHrH erH Q)
fd Eh Eh-Pcn a
0H u-M 0
ino
X
cnCN
oI
LDiH CN CN oCN vr> CM c\ 00
in in
m CO ro m
m co
in CNo O CN or* 00 CT\
a\ m ro iH
o o o o oi 1
in CN o CN o00 CN
00 CO (N o O
o00
inoo
o m oo

H
xw
En
HKEhH
Eh
uEh
Ou
H
2;oHEh
<WhH"
uD
Cm
O
Eh
WH 2U OH H
Eh
W tsi
Ou
o
DEh
aEhCO
Uw
EhEh
COWEh
CD
XI-P
0
<u •
Qj <0
0 >rHCO CO
0)
>H
id
CO UH
0-Pto
id
0)
O co
C\J
-P
orH
to
o Eh<
O(J
o•HPid
<D
rHo
CD
id
Jh
-Pt/>
CO
I
NH
03
4J
CO
>iuu
CD
orH(/)
'OCD
SH
3HCO
id
CD
£
(A
4J
id
0
03
Eh
GOHP o
Eh
<]
COI
o
U
X
COCN
•
OI
in orH rH <£>
00 00 o rHrH H
in o oCO 00 <^ OCN *p r*» o
o o Oo o o o o o
1
in 00 O O OCM r- CN O 00
in CM rH rH o
in•
oo ooo
CO00 00

145
HsXwm<
(±H
EnH
Eh
UEh
ZW O^ UUD ZZ H
O
ZoHEh
U
DEh
SW
wEh
OEh
zwH Zu oHEh
<W tsi
O
EhCO>H
«uwKEh
COWEh
S
CD
-p
4-1
0
<D •
Di CO
0 >iHCO
m<D r-M 0 o01 0
*».
D"1 X
co 4h <N
0 a\-P Eh rHCO -P < CN(0 c rH •
CD oHi Eh 1
oCN
Eh<
l
o
O
o-H 0-P rHfd CO CO
0)
rHU ©3 Jh C
HCO B
d) nJ
-P CD •Hrd 6 <y
sh rH+j OCO CD
XI -Prd
to =«=
1
«J
N•HrH ri
rH CD
cd Eh Eh
CO C>i 0u H Uu P 0
CN o o O oCO rH Ch
o H rH CMrH rH rH rH
O O O OVD O rH O orH a\ O O oIs- CO CO o oO o O o o
o O O OCN O o o
• • • • •
lO CN CN rH
o00
CM00 00 00
0000

146
HH
X
3Eh
CO
H
OMEh
WU «D £=4
DCQ
O
EnO
OH HZ Eh
w <JH IS]
U HHEnEn <Cwou
§DEh
$
wEh
Eh
CO
&u
KEh
<U
•P
mo
Q)
aorHCO
u
D1
CO
+J
CO
cu
CO
>
tno
4Ho
oHft
o
CM
<
oiH
H rn< I
o
t7>
Oh1
XLO<NiH
•
OI
ro rH cn O ro
O iH rH CN inH rH iH rH i—
i
rH
rH O m H ro iniH CN o m CNO VD rom ro CN CN o 00
iH iH H iH H o
Id
0rHw S3
C0•H 0)
-P C•H
a) CO grH 05 \o •H
£ (D
rH
O0)
rH -p
pq =8=
i
td
N•H ftrH grH 0)
(d
-p
en
>i 0u
U •P o
o o ro CN H 00
m o 00 00 rHro CN H rH rH
<N00 oo CO
0000
o CN

147
CM
Q<
I
WZwEh
D
eo\U
U
i
CM
CHE
CO
to
ft ^ft O*H ft
6o
5h
CD
inII
D
faCO Oft
*
e CN
•H eo 1
OH II
It \tnCO Eh o
TE ZA D+ er
HH Ds
E-tLL
X C/3 <X
UB ST
w w Csl
m fa u CMS3 O 6E-i
HE0\
U EhO u
w D CD
H H bUH Wfa wfa H co
WIT
o W gD U
H W < \Eh O CP<3 O 0)
W « D 0)
W t>
u EhD W2 K
p4J
+J
w
(N ^3*
00 00• • •
CM CN co
00rH o rH
• • •
<N CN rH
CO
CN
rH in COCO 00in CN
COft•H
ftft•H
CO
CD
I -H0 -PM -H
* a) a)
rH +> C3 0 0BE &

148
Xgwa<Eh
Dw mH 2HH H
EhCO>H
fauCO
S3
1Eh
faOwEh
KEh
sO faU
Pi51w
faO
oCO
COw
Dfa
WafaCO
faowEh
Eh
O
HQ
WS3Eh
33H EhPi h< &
o oU Hu
0)
-P-HrH
HCD
HCO
•
c•He\ ne
li
rHCO fd
CD -P-P co
rd >cr
C-p c 0
CD fd •H0 C U tn
•H <D
15 «
c0-H-Pfd
NH
fd
-p
to
U
Uo
a,>i B
CD
Eh
CD
-Pfd
UPco
CO
tnCHNH
Oft
fd
-P
>i CO
in
u
Hon ro CN
• • • •
o o
00H CM CN
• • • •
o O
o O O O• • • •
LO O o(N CN LOH
CO CO sft oH •H ft
cn in
•H Cw x: a)
fa — T3
ft ftft ft u U•H -H ft ft

149
HXXXwam
wEh
co
EhCO>h
ftUEh
COft ftDCO
wS3Eh
WS
Oft
ft ftO O
DEh
oHEH
U ft
5 ^ft uEh DCO St-H WU Eh
swftufto
Eh
ft
>h
Os
0*
H HS (S3
H HCO Jw <:ftEh
EhCO>H
ft fto uft ft
ft
9 ehft
Q
CP
u
tn
QCO\u<u
6
iH0fa
C•HN•H
CO
tHrH
oHa co
>iMU
w
crc•H-P03 C
c <D 00 rH HSH 0 4J
OCO 53 <
CD
rH
ua rd
•H o rH
e p Hn ep •H
u CO co
co
tn
fd
rHU
COfa•H\fafa•H
S 0^ COO 04 fafa *H -H
OW CQ CQ04 fa 04
CO
CD O
CO CO
a) cu
vo
cfd
VO(d VD
04 fa
CO
(1)
o
VO
cfd
ID
rH rH -
<C < vo
v v CO rHfa fa c <I I o
0) Q)
O Eh CO rHfa W fa CQfa fa -H 04
fa rH O Jfa CQ fa CJ•H O* 04 fa
o53
O2
PQ

150
HXXX
wCQ
<Eh
CO
<W EhEh CO
« «Eh CJCOP3 «D WCO S
>h
W ^S3 OEh CU
Cm PhO O
o5
EhCOX
wS3Eh
Cm
OW
§
0)
Pfd
M-PCO
ja
CO\u<u
i
c•HN•HrH
to iHCD fd
rH -P
D- CO
>>
X UK
ngO •H
-PH fd c(S3 CD 0H iH H
o -p
0<Eh
p a)
u a >iD Eh
wa) H
Eh B Hfd a)
Q CO u
CO
CO
fd
rH
u
CO
CD
orH
04 J04 U•H CL
—-P (D
•H -p
C fd
-H UrH -PrH CO
rd xiP 3CO CO
>1rH HOCO <D
c a. ufd >, i
Vh Eh I
Eh — I
orH
CO
GorH>1r~
*
VD
c CO
0 C4J 0C H(D >i
C
oEH s HW o VD
G G0 0
CO H Ho. >i >i•H •H G G\ \ \ \04 C o04 u w H•H Pn
02
(D
-Pfd
Sh
-PCO
CO
0) H l
> H I
•H I-POOO a n3fd >-, i
C Eh I
H w I
CO
CD 02
U

151
CAPTIONS FOR FIGURES
Gibbs free energy (AG) as a function of the radius (r) of a spherical
nucleus.
A cylindrical nucleus of radius r and length 1 , with interfacial
energies o and a •
e s
Model for a small crystallite of low molecular weight substance.
Heterogeneous nucleation of a spherical cap of phase p from
phase a on a flat substrate s (0 is the contact angle) .
Retention of embryo in cylindrical cavity.
Model for the nucleus in polymer crystallization with regular folding
and adjacent re-entry
.
Nucleation rate curves (schematic) for homogeneous (A) , hetero-
geneous (B) , and pseudohomogeneous (C) nucleation.
Model of growth nucleus on spherulite boundary, illustrating
nucleation control of spherulite growth.
Overall view of photomicrographic equipment: (a) drill press stand,
(b) Zeiss polarizing microscope, (c) eyepiece to shutter connector,
(d) binocular eyepiece, (e) hot stage for sample mounting, (f)
temperature control unit for the hot stage, (g) shutter with cable
release, (h) wooden box,
(i) viewing telescope , (j) solenoid,
(k) camera, (1) intervalometer , and (m) A.C.-D.C. converter for
solenoid
.
Schematic diagram of the direct current source for the solenoid.
Side view of rectifier.

152
11. Arrangement for sample mounting before crystallization (top view) .
(ABDC: substrate; ABFE, CDHG: polymer to be melted and crystal-
lized)
12. Experimental arrangement (top view) for counting the number of
nuclei on the substrate (area AB) and in bulk (volume ABCA) .
13. Light micrograph showing transcrystalline surface morphology of
iPP crystallizing in contact with iPS (ab) (substrate type I) at 130°C
(time 15 min. ) .
14. Spherulitic surface morphology of iPP crystallizing in contact with
Kel-F or poly (chlorotrifluoroethylene) (substrate type II) at 125°C
(time 9 min. ) . (abed: Kel-F overlaid with iPP
.
)
15. Nylon 610 as an inactive substrate (type III) in the crystallization of
PEO at 50°C (time 2 min.)
16. Effect of crystallization temperature (T) on the surface morphology
of iPP crystallizing in contact with iPS: (i) T = 125°C (time 4 min.)
;
ab: iPS overlaid with iPP; ac: transcrystalline region; (ii) T =
130°C (time 15 min.) ; ab: iPS; ac: iPS overlaid with iPP; ad:
otranscrystalline region, (iii) T = 133 C (time 42 min .) ; abca: iPS;
abd: iPS overlaid with iPP . (iv) T = 135°C (time 84 min. ) .
17. Transcrystalline surface morphology of crystallizing iPP generated
by nylon 6 substrate at 125°C (time 4 min.) .
18. * Transcrystallinity of iPP crystallizing in contact with nylon 66 at
125°C (time 3 min.) . (abc: nylon 66)
19. Transcrystallinity of iPP crystallizing in contact with nylon 610 at
125°C (time 4 min.)

153
Transcrystallinity of iPP crystallizing in contact with Penton or
poly[3,3-bis(chloromethyl) oxacyclobutane] at 125°C (time 12 min.)
Spherulitic surface morphology of iPP generated in contact with
poly(2,6-dimethyl phenylene oxide) or PPO at 125°C (time 4 min.) .
(ac: PPO overlaid with iPP)
Transcrystallinity of PCL generated in contact with nylon 6 at 50°C
(time 2 hr. 57 min.) . (abc: nylon 6)
Transcrystalline morphology of PCL induced by nylon 66 substrate
at 50°C (time 3 hr. 22 min.) . (abed: nylon 66)
Transcrystalline morphology of PCL generated in contact with poly-
oxymethylene (POM) at 50°C (time 2 hr. 43 min.). (ab: POM-PCL
interface)
Spherulitic surface morphology of PCL crystallizing in contact with
gold (abed) at 50°C (time 2 hr. 42 min.) .
Spherulitic surface morphology of PCL crystallizing in contact with
polyethylene terephthalate (PET) at 50°C (time 2 hr. 42 min.) .
(abc: PET overlaid with PCL)
Spherulitic surface morphology of PCL crystallizing in contact with
poly(butene-l) (PB1) at 50°C (time 2 hr. 52 min.) . (abc: PB1
overlaid with PCL)
Spherulitic surface morphology of PCL crystallizing in contact with
Kel-F at 50°C (time 2 hr. 35 min.) . (abc: Kel-F overlaid with PCL)
Isotactic polypropylene as a moderately active substrate (type II)
in the crystallization of PEO at 50°C (time 1 min. )
.

30. Nylon 6 as an inactive substrate (type III) in the crystallization of
PEO at 50°C (time 1 min.) .
31. Nylon 66 as an inactive substrate in the crystallization of PEO at
50°C (time 1 min. ) .
32. Crystallization of iPP at 125°C in contact with: (i) iPS (time 2 min.)
:
transcrystalline surface morphology, (ab: striation on iPS film)
(ii) atactic PS (time 5 min.) : spherulitic surface morphology,
(ab: atactic PS-iPP interface; abc: atactic PS overlaid with iPP)
33. Crystallization of PCL at 50°C in contact with: (i) iPS (time 1 hr.
53 min.): transcrystalline surface morphology, (ii) atactic PS
(timelhr. 15 min.): spherulitic surface morphology . (ab:
atactic PS overlaid with PCL; cad: atactic PS-PCL interface) .
34.. Crystallization of PCL at 50°C in contact with: (i) High density
polyethylene (HDPE) (time 2 hr. 50 min.): transcrystalline surface
morphology (abc: HDPE) . (ii) Low density polyethylene (LDPE)
(time 2 hr. 50 min.): spherulitic surface morphology, (abed:
LDPE)
.
35. Ratio of nucleation densities (substrate to bulk) in the crystallization
of iPP in contact with indicated substrates at 125°C (3 min.) and
129°C (14 min.) ; data of Table XIII. (Actual heights for iPS and
nylon 610 substrates are greater than the indicated heights.)
36. Ratio of nucleation densities (substrate to bulk) in the crystallization
of PB1 in contact with indicated substrates at 85.5°C (9 min.) and
88°C (9 min.); data of Table XIV
.

155
37. Ratio of nucleation densities (substrate to bulk) in the crystallization
of PEO in contact with indicated substrates at 50°C (3 min.), 51°C
(5 min. ) and 53°C (34 min
. ) ; data of Table XV .
38. Appearance of spherulites on substrate (high density polyethylene)
in the early stage of the formation of transcrystallinity in PCL at 50°C
at (i) 9 min., (ii) 14 min. , (iii) 19 min. , and (iv) 24 min. (ABCD:
HDPE; AED: supercooled PCL) .
39. Nucleation density on substrate as a function of time in the crystal-
lization of iPP in contact with nylon 66 at three crystallization tem-
peratures.
40. Nucleation density in bulk as a function of time in the crystallization
of PB1 at 85.5°C.
41. Bulk nucleation density as a function of time for repeated crystalliza-
tion of PEO at 51°C.
42. Ratio of nucleation rates (substrate to bulk) in the crystallization of
PB1 at 85.5°C and 88°C in contact with indicated substrates; data of
Table XX.
43. Ratio of nucleation rates (substrate to bulk) in the crystallization of
PEO at 51°C in contact with indicated substrates; data of Table XXI.
44. Dependence of the growth rate (G) of PB1 spherulites on supercooling
(AT) . (T = crystallization temperature; straight line drawn by
least square technique.)
45. Nucleation density on substrate as a function of time in the crystal-
lization of PB1 in contact with iPS , at indicated temperatures.

156
46. Heterogeneous nucleation rate (on iPS substrate) as a function of
supercooling in the crystallization of PB1;*N represents the slope
measured from Figure 45. (Straight line drawn by least square
technique.
)
47. Nucleation density on substrate (iPP) as a function of time in the
crystallization of PB1 at indicated temperatures.
48. Heterogeneous nucleation rate (on iPP substrate) as a function of
supercooling in the crystallization of PB1; N represents the slopes
measured from Figure 47. (Straight line drawn by least square
technique.
)
49. Bulk nucleation density as a function of time in the crystallization
of PB1 at indicated temperatures.
50. Heterogeneous nucleation rate (bulk) as a function of supercooling
in the crystallization of PB1; represents the slope measured from
Figure 49. (Straight line drawn by least square technique.)
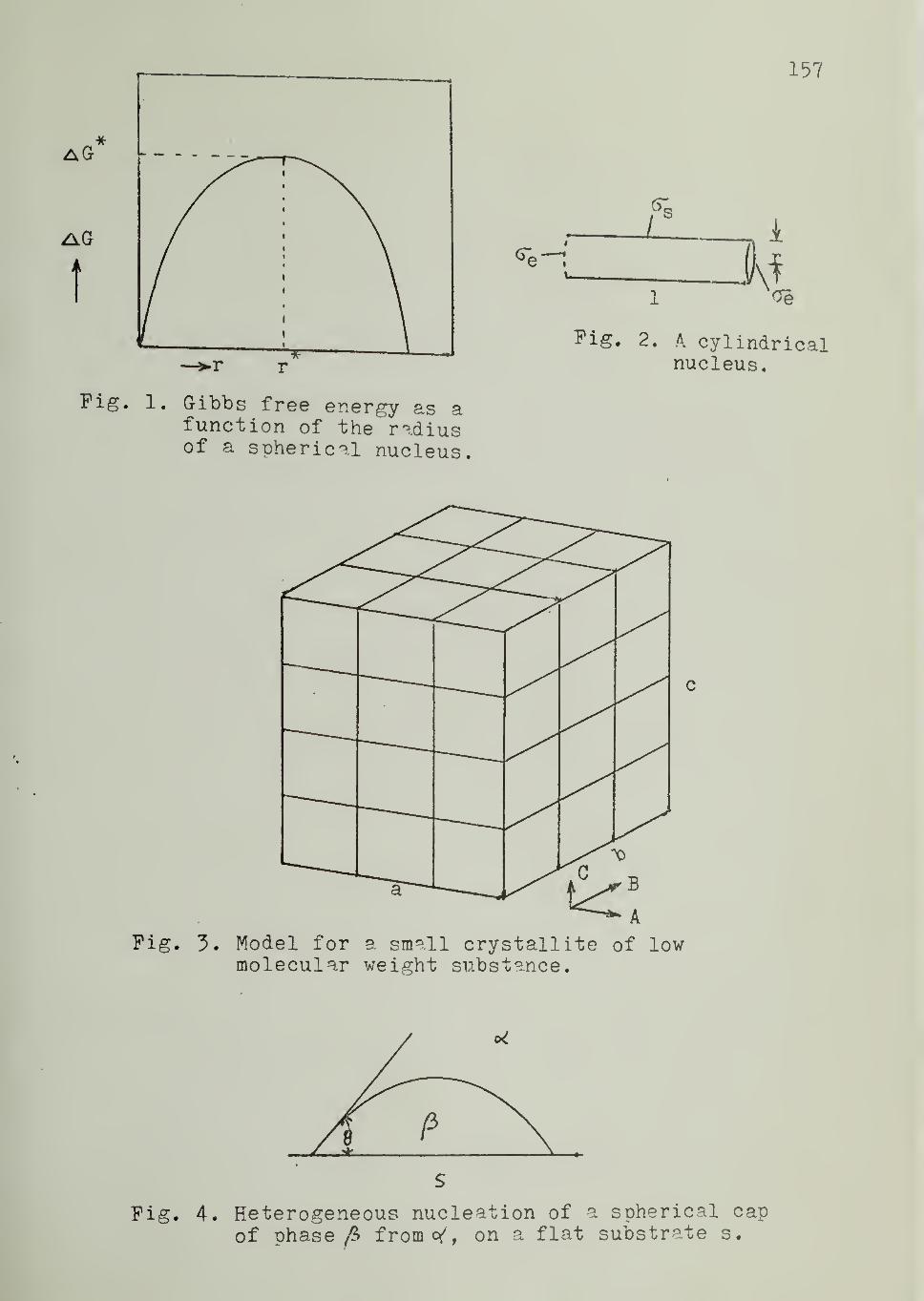
AG
ZlG
t
157
Fig. 1. Gibbs free energy as afunction of the radiusof a spherical nucleus.
Fig. 2. A cylindricalnucleus.
Fig. 3. Model for a small crystallite of lowmolecular weight substance.
Fig. 4. Heterogeneous nucleation of a spherical cap
of phase from on a flat substrate s.
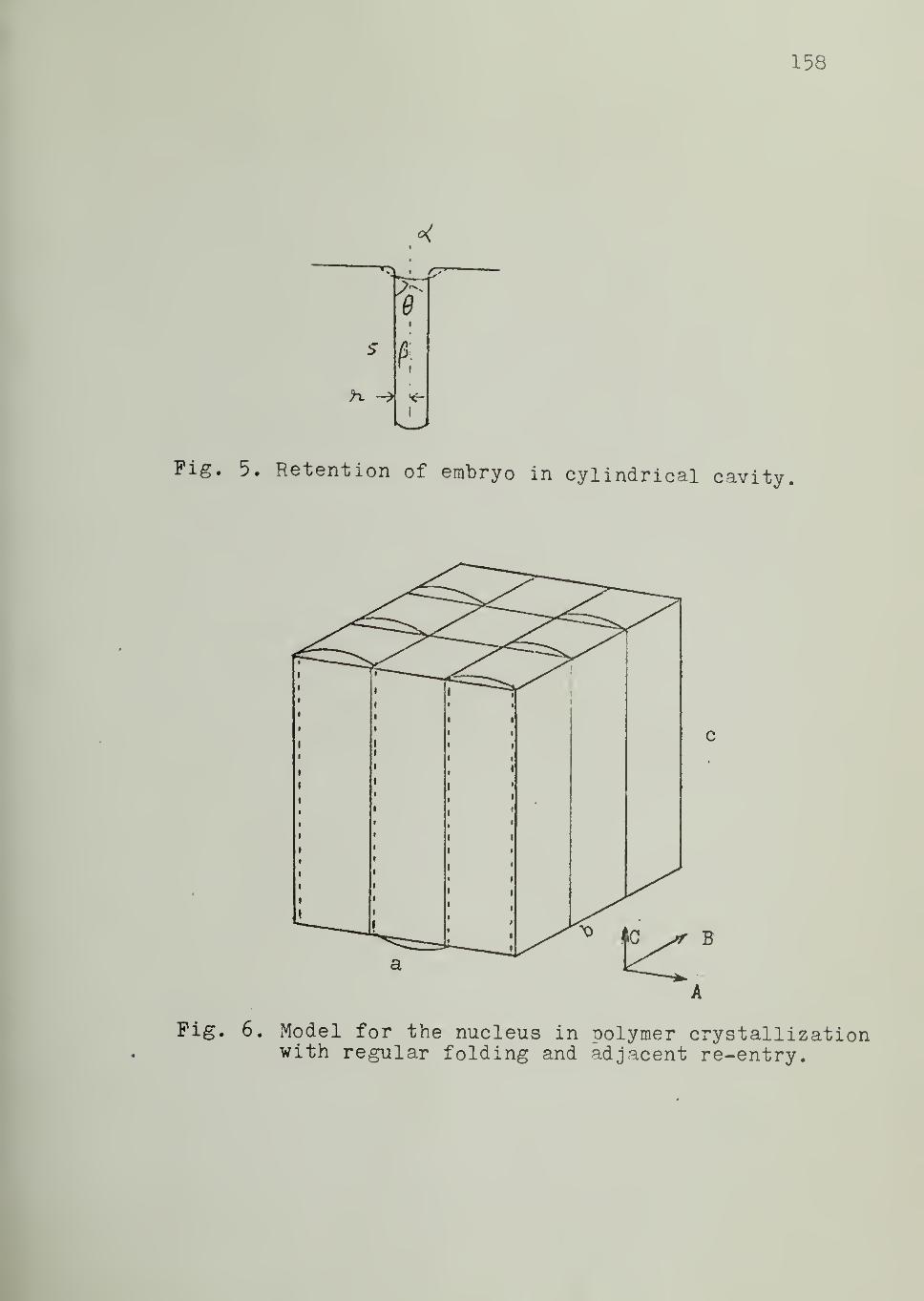
158
j<
fs
0
Fig. 5. Retention of embryo in cylindrical cavity
Pig. 6. Model for the nucleus in polymer crystallizationwith regular folding and adjacent re-entry.
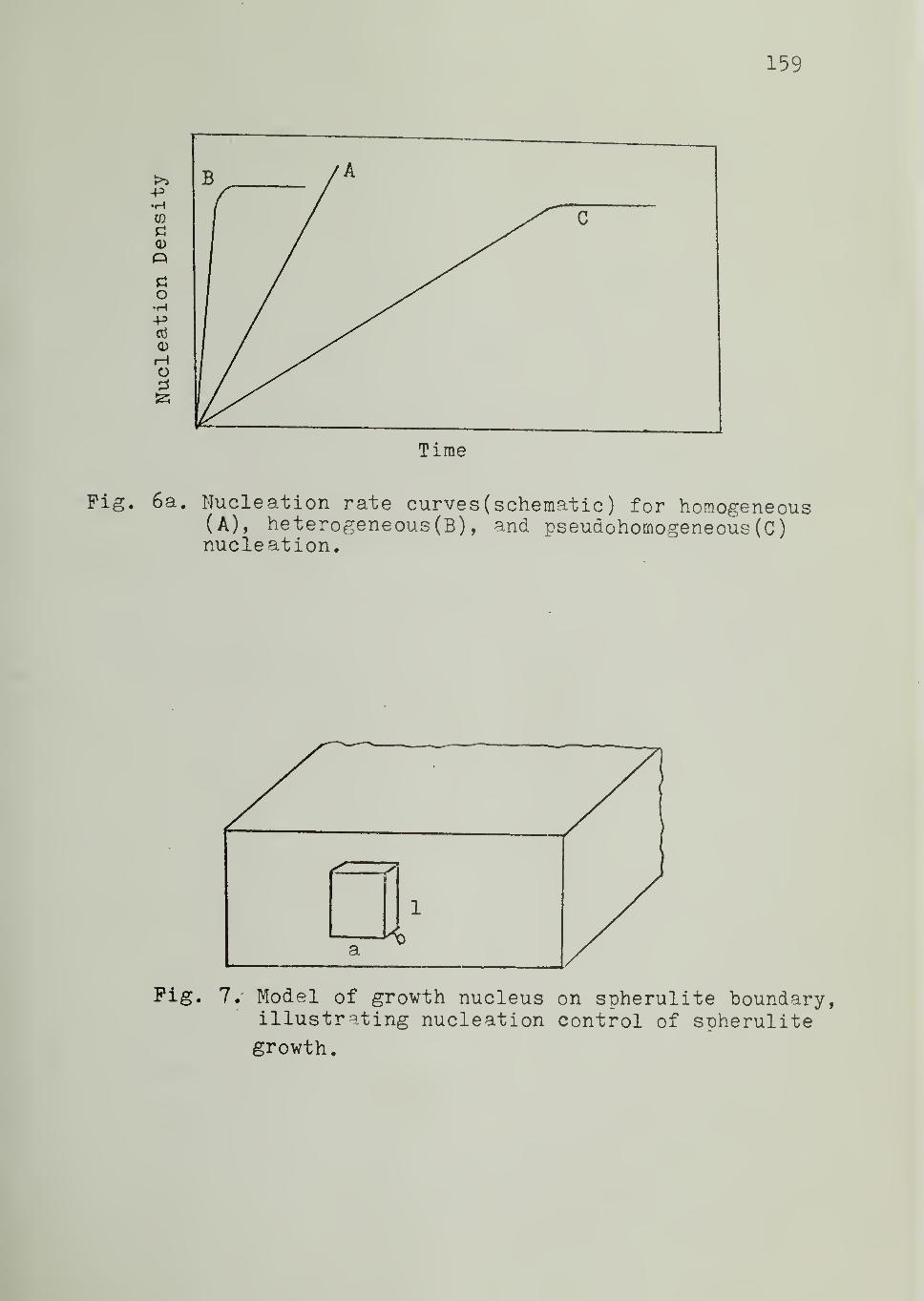
Time
Pig. 6a. Nucleation rate curves (schematic) for homogeneous(A), heterogeneous (B) , and pseudohomogeneous (C
)
nucleation.
Pig. 7. Model of growth nucleus on spherulite boundary,illustrating nucleation control of spherulite
growth.

160
Figure 8

161
Light Dimmer
Maleplug
Rectifiers (to solenoid)
Figure 9
o
:
Anode
Cathode
Figure 10

162
microscope slide
glass cover slip
polymer to be crystallized
substrate
Figure 11
Crystallizingpolymer
Substrate
Figure 12

163
Figure 13
Figure 14

164
Figure 15

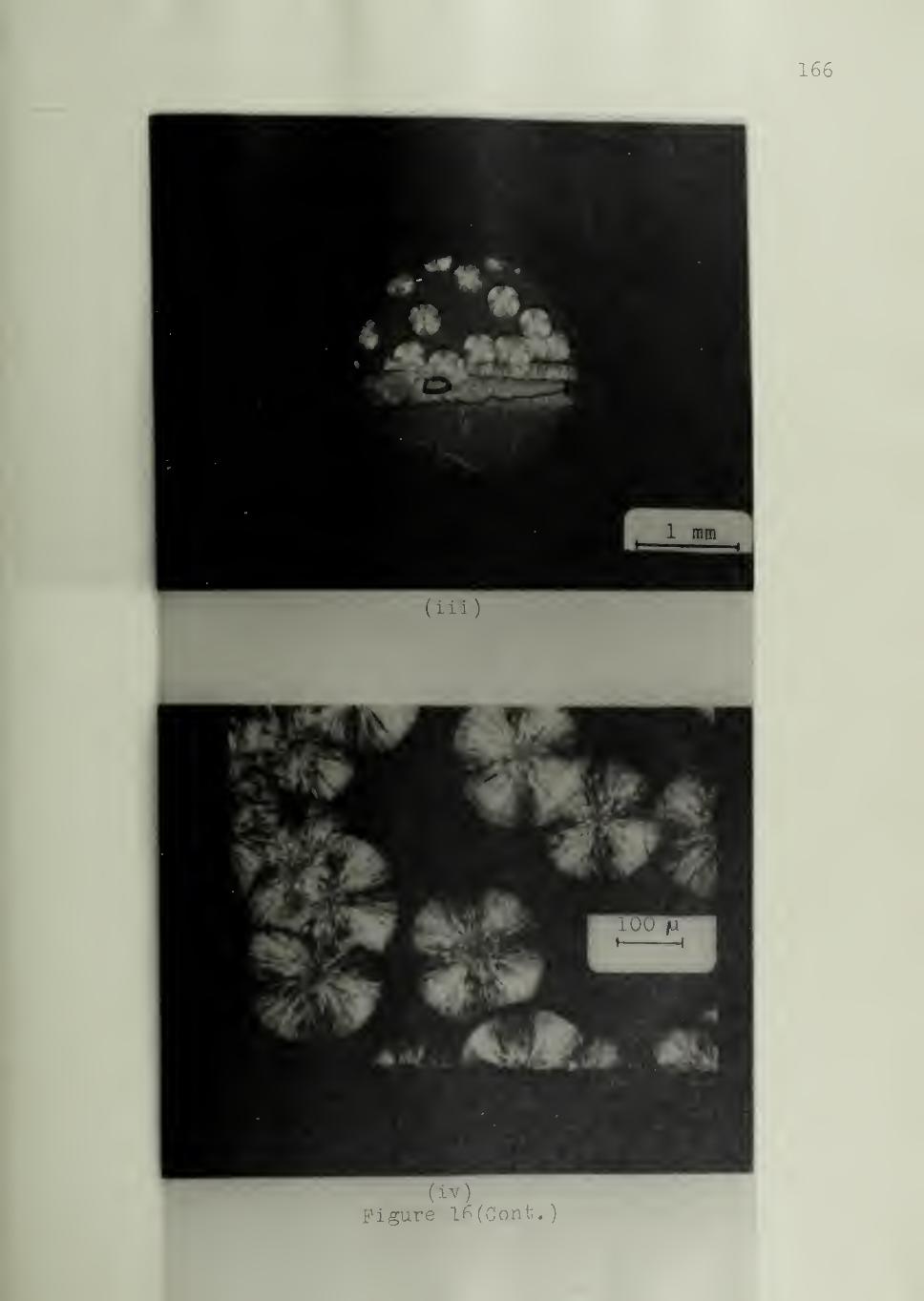
166
Figure l6(Cont.)
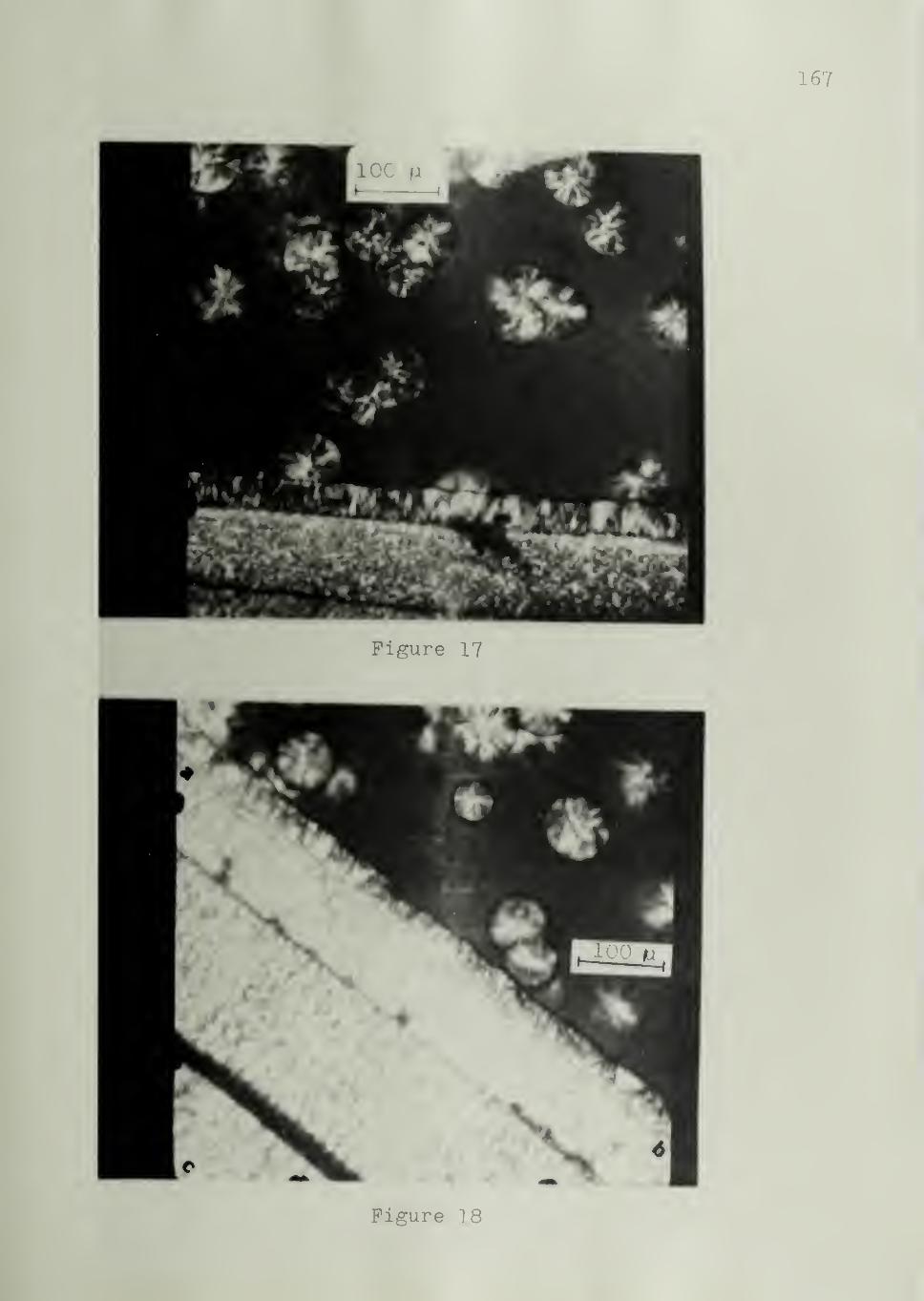
Figure 17
Figure 18

Figure 20

169

170
Figure 23
Figure 26

173
Figure 29
Figure 30

(ii)Figure 32
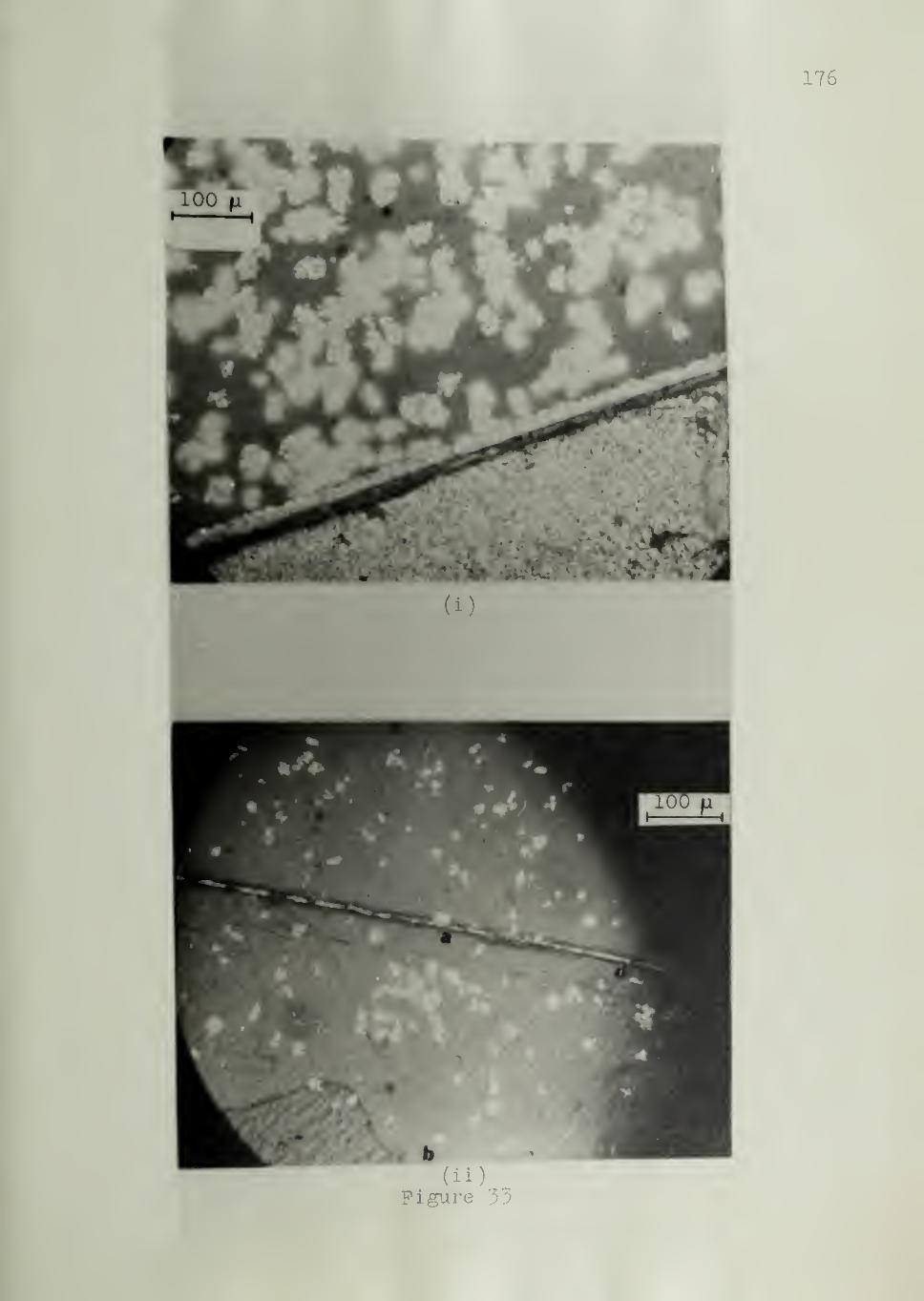
176
Figure 33
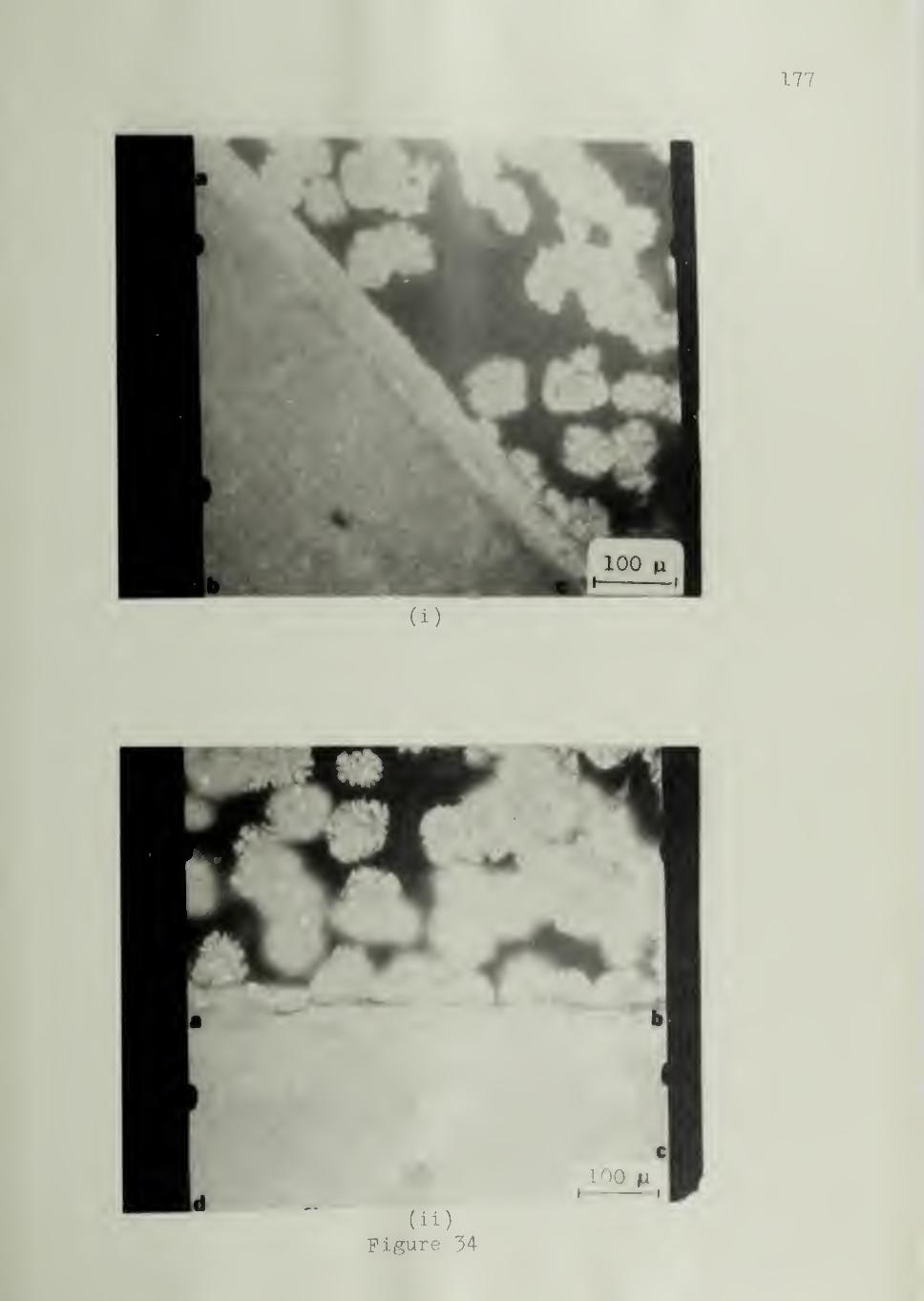
Figure 34

178
cr
mZDCD
03
tin1
gGl X (
qLi/
Su)

179
4
inCL•H
u
•
cf_J •H0 E
CO cn
CJ •
0 cLH
• ECO cn
LTl CM D
UJ34
.01 X (
qu/Su)
uHCC
L:J
C_
CDCL
LUh-ct QJ
cr Ph
h-1 tn cn
_J QD •H
UJcn
CL•H

180
Li J
EX
c
L3 Ec
in
t> E
lo in
- cCJ >HB Ea
LO
LPl
LO
LO
in D_
CJ
CJ tnct CL
UCLQ
WUJI
—
cTcrh-
m
m03
Z3
CP•H
COC\J
Ja
•013 .01 X (V u)
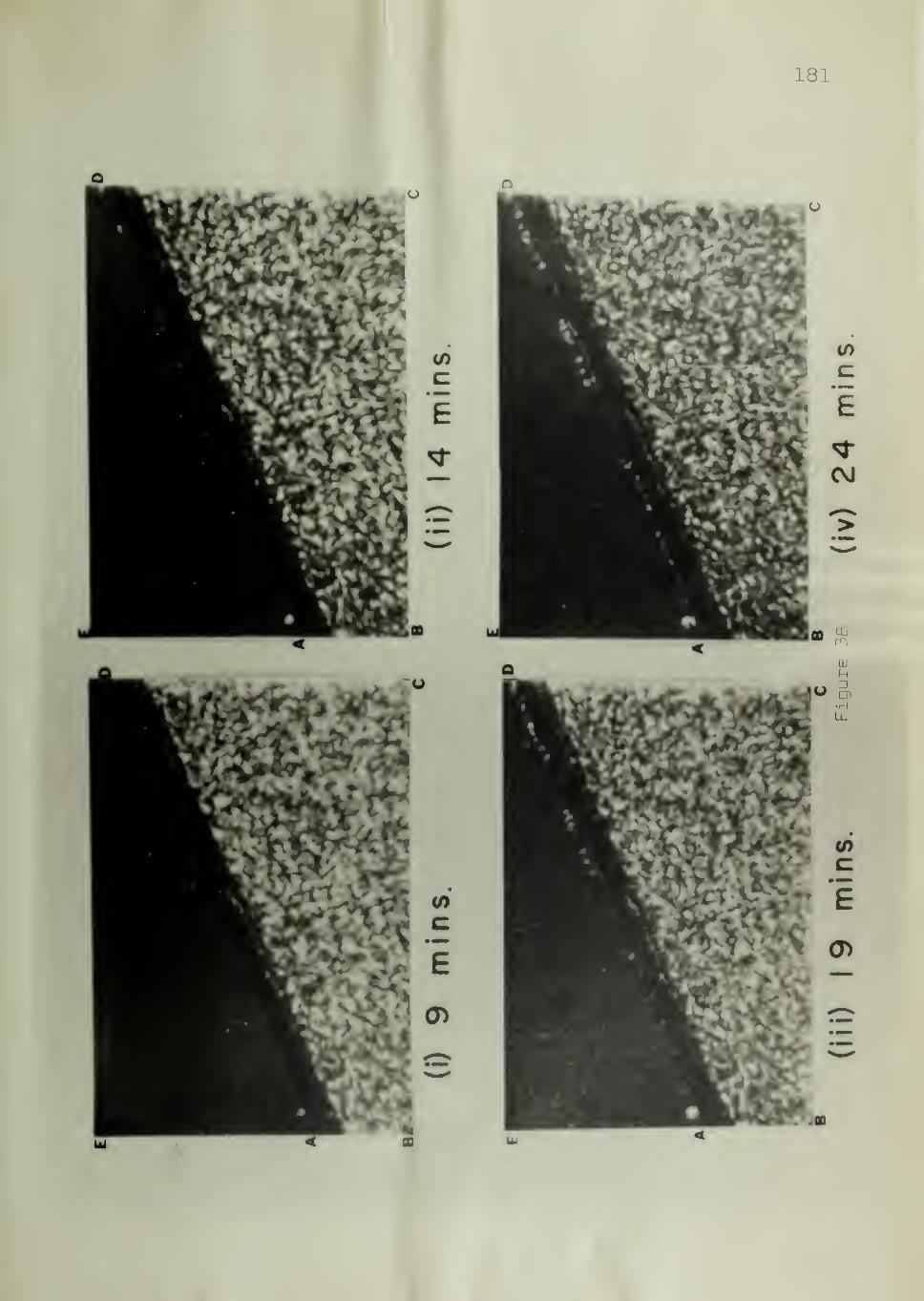
181

132
CRYSTALLIZATION TIKE, WIN.
Figure 39
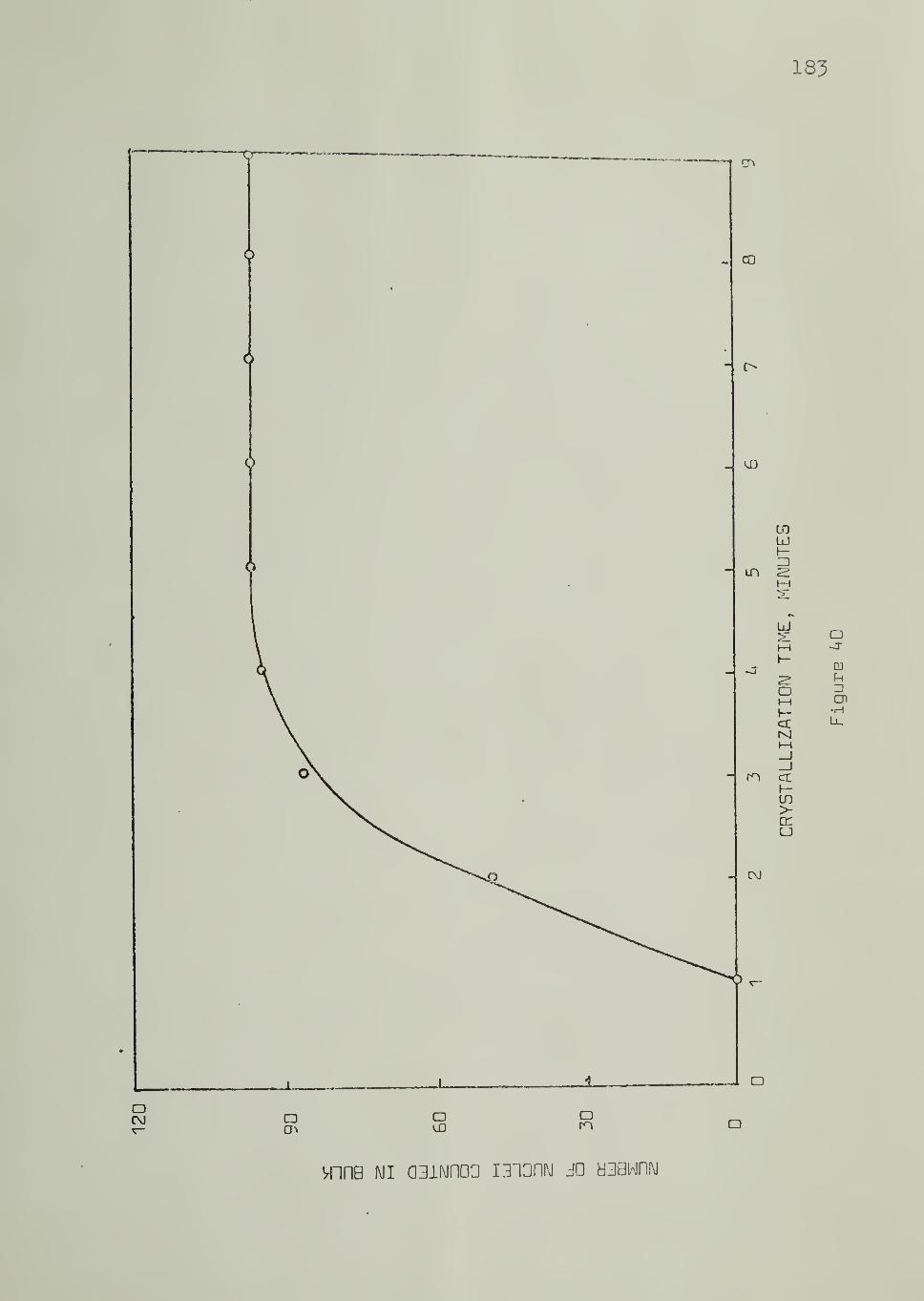
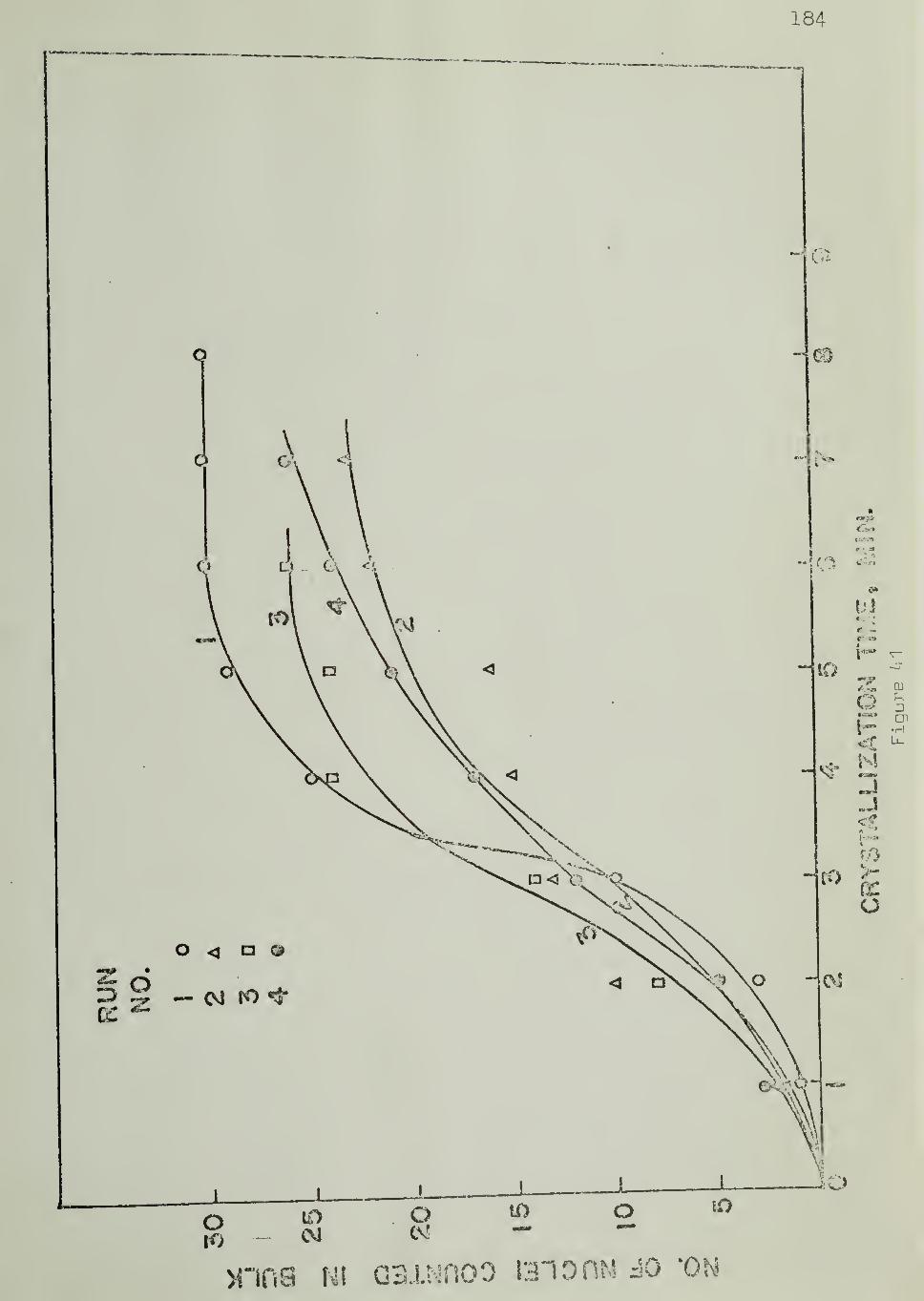
184
>riria ni oaiNnoo m om do -oh

185
u incl ELh-'
Q_
Lu1
_J lq CMUJ LU j-
:c i—ct QJ
cr Ph
f— =!
CTm •HZD Lu
a in
LTJ
UJ3qs ,rs
v
01 X ( 1/ I)

186
aa.
uMCJ in
CLCL
LUa.
tnLdh-(.r
cr
LQcn
ID
QJ
cn
CL
cn£L•H
uCLO
a> 03
C\J
as ,ss .
01 X ( 1/ I)
a

187


189
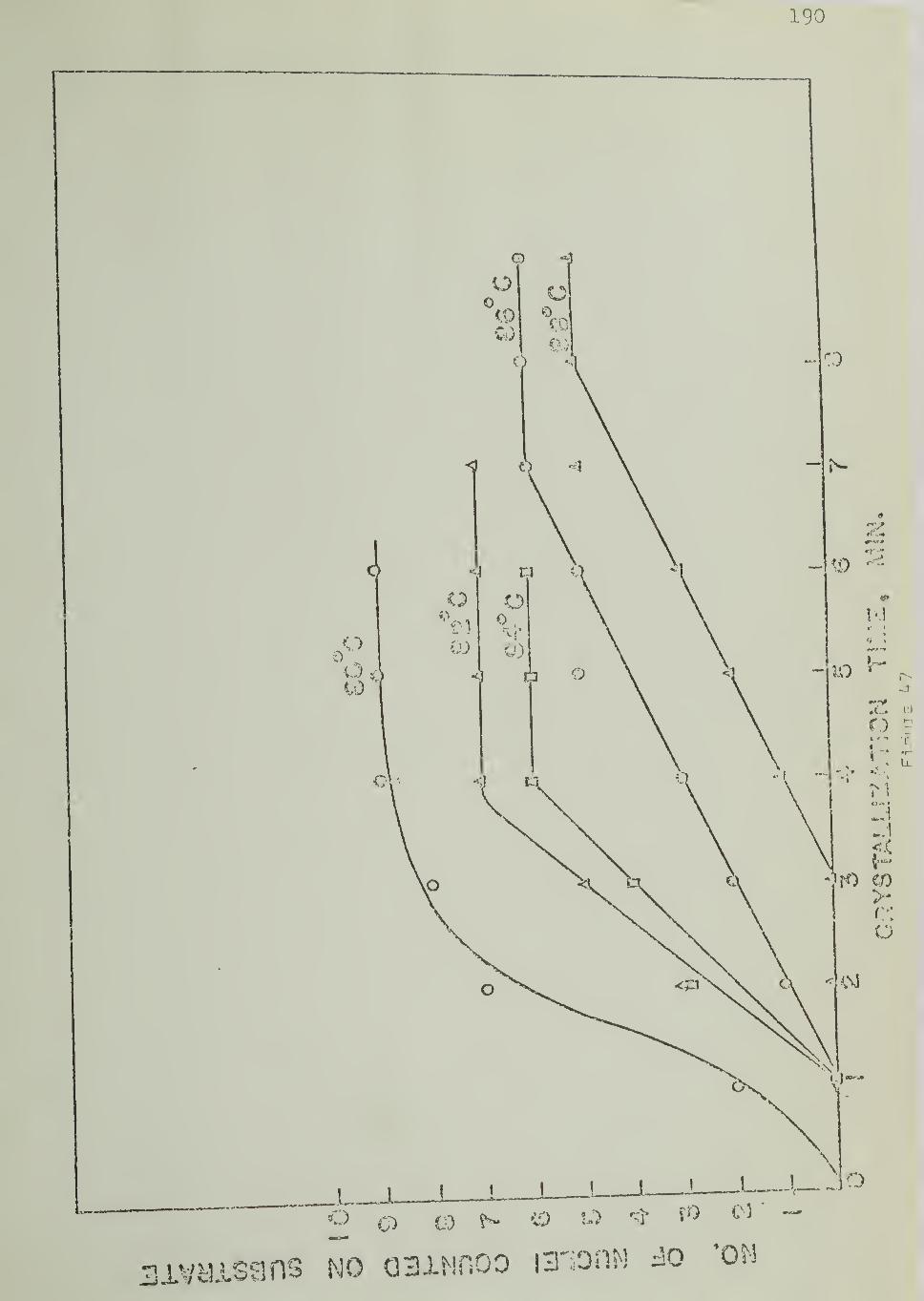
190

191
03
CP
C\J
UIUJ/TaXDHLJft
[\IGai

192

193



















