
Hemostasis and related-disorders - WordPress.com · Hemostasis and related-disorders Susanna Hilda...
30
Hemostasis and related-disorders Susanna Hilda Hutajulu, MD, PhD Div Hematology and Medical Oncology Department of Internal Medicine Universitas Gadjah Mada Yogyakarta
Transcript of Hemostasis and related-disorders - WordPress.com · Hemostasis and related-disorders Susanna Hilda...

Hemostasis and related-disorders
Susanna Hilda Hutajulu, MD, PhD
Div Hematology and Medical Oncology
Department of Internal Medicine
Universitas Gadjah Mada Yogyakarta

Outline
• Normal hemostasis
• Coagulation disorders
– Hemophilia
– Disseminated intravascular coagulation
• Disorders of platelets
– Thrombocytopenia
– Thrombocytosis

Hemostasis
• Hemo= blood; sta= remain � the stoppage of bleeding.
• Primary hemostasis:
– Platelets play an important role
– Disorders include vasculopathy, quantitative thrombopathy
(trombocytopenia, thrombocytositosis), qualitative
thrombopathy
• Secondary hemostasis:
– Coagulation cascade, occurs after platelet adhesion and
aggregation
– Disorders include hemophilia and DIC
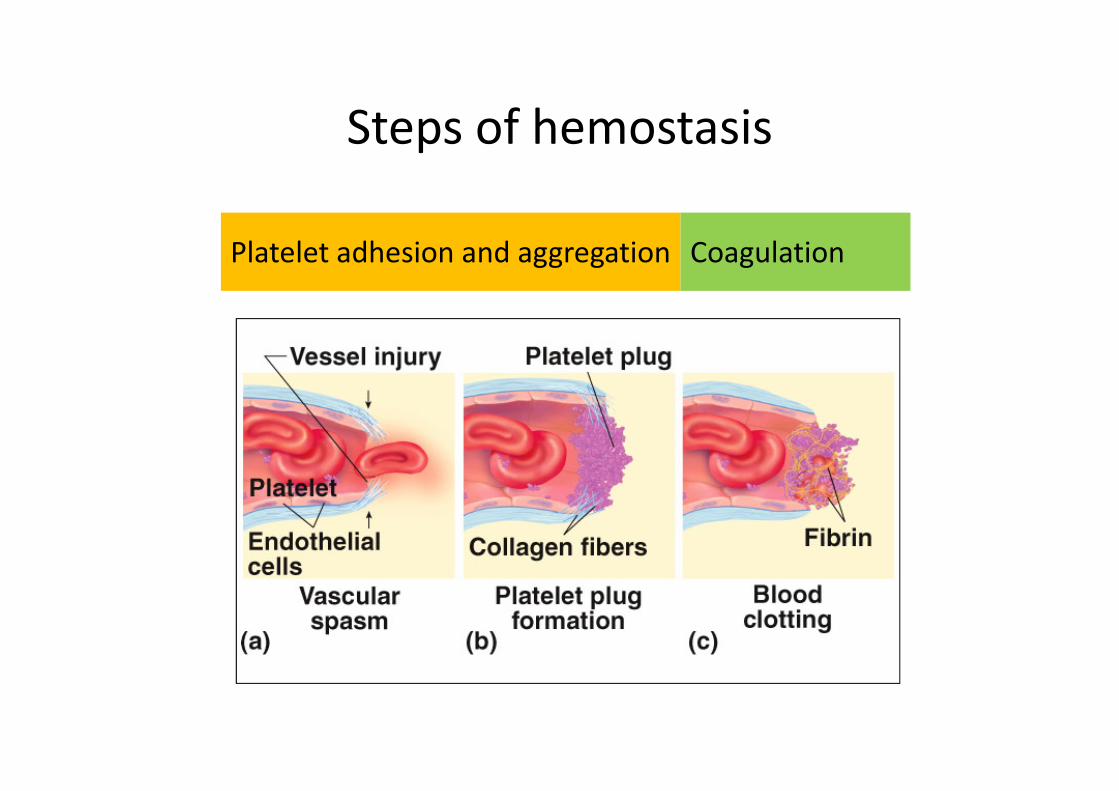
Steps of hemostasis
Platelet adhesion and aggregation Coagulation

Platelet adhesion
and aggregation
• Injured blood vessel releases ADP, which attracts platelets
• Platelets adhere to damaged subendothelial spaceand undergo activation
• Platelets come in contact with exposed collagen release: serotonin, ADP, Thromboxan A2, which accelerate vasoconstriction and causes platelet aggregation (thrombus).

Coagulation cascade
Tissue
injury
pathway
Contact
pathway
Fibrin aggregates to form a meshlike network at the site of vascular damage.

Hemophilia
• Hemophilia ~ “love of bleeding”
• Inherited deficiency of factor VIII (hemophilia A) or factor IX
(hemophilia B).
• Sex-linked recessive; almost all patients male.
• Second most common inherited clotting factor abnormality
(after von Willebrand disease).
• Incidence rate:
1/5000-10000 live male births.

Genetics
• 50% of cases of hemophilia A show an inversion mutation
in intron 1 or 22.
• Remainder genetically heterogeneous
– Nonsense/stop mutations prevent factor production
– Missense mutations may affect factor activity rather
than production
• 15-20% of cases due to new mutations.

Pathophysiology
• F VIII is a cofactor for
intrinsic Xa
• Activates Xa and thrombin

IX
X
Fibrinogen Fibrin
PT
XIa
Xa
V
VIII
XIInjury
TF
VIIaIXaVIIIa
Xa
Va
ThrombinPropagation
Initiation
•Deficiency of factor VIII affects
the propagation phase of
coagulation.
•Most likely to cause bleeding in
situations where tissue factor
exposure is relatively low.

Clinical features
• Most bleeding into joints, muscles
• Mucosal and CNS bleeding are uncommon.
• Severity is inversely proportional to factor level:
≤ 1% - severe, spontaneous bleeding or bleeding after minimal injury, occur from early infancy
1-5% - moderate, bleeding after mild injury or surgery
> 5% - mild, bleeding after significant trauma or surgery

Hemarthrosis
• The most common, painful and most physically, economically and psychologically debilitating manifestation.
• Clinically:
� Aura: tingling warm sensation
� Excruciating pain
� Affects one joint at the time
� Sites: knee (commonest), elbows, wrists and ankles.
� Edema, erythema, warmth and low of movement
� Treated early, it subsides in 6-8 hrs and disappears in 12-24 hrs.

Long-term complications of
hemarthrosis
Joint destruction Nerve damage
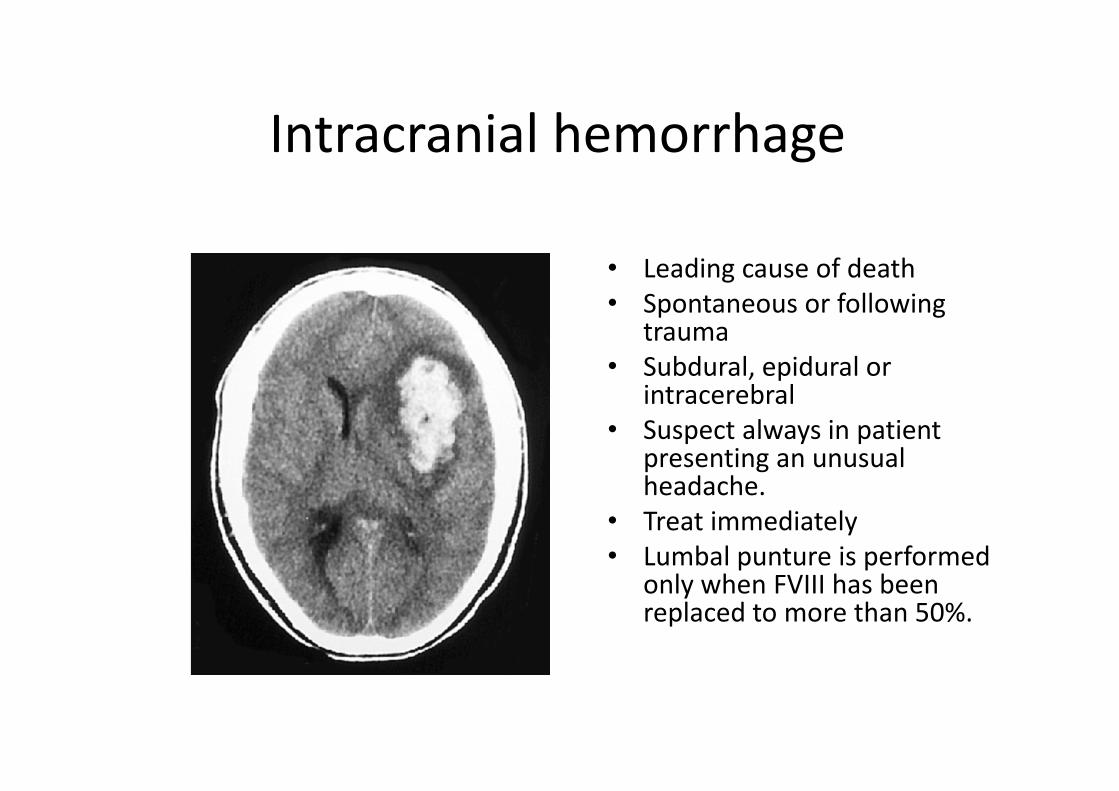
Intracranial hemorrhage
• Leading cause of death
• Spontaneous or following trauma
• Subdural, epidural or intracerebral
• Suspect always in patient presenting an unusual headache.
• Treat immediately
• Lumbal punture is performed only when FVIII has been replaced to more than 50%.

Other sites of bleeding
• Gastrointestinal Bleeding:
– 5x more common in hemophiliac patients than regular males
– associated with ingestions of NSAIDs for hemarthrosis.
• Mucous Bleeding
Epistaxis, gum bleeding
• Genitourinary Bleeding

Laboratory diagnosis
• FVIII activity, expressed in % of normal
• Prolonged aPTT, PT and BT

Treatment: Factor replacement
Replacement of F VIII is the cardinal step to prevent or reverse acute bleeding episodes.
Target level and duration of treatment depends of severity and site of bleeding.
Site of hemorrhage Desired F VIII level
Duration of treatment
(days)
Hemarthrosis 30-50 1-2
Superficial intramuscular hematoma 30-50 1-2
GI tract 50 7-10
Epistaxis 30-50 Until resolved
Oral Mucosa 30-50 Until resolved
Hematuria 30-100 Until resolved
CNS 50-100 At least 7-10 days
Retropharyngeal 50-100 At least 7-10 days
Retroperitoneal 50-100 At least 7-10 days

Treatment
other source of FVIII
� Plasma
• FFP was used as the only replacement therapy until 1960s.
• Not really much effective, only raise FVIII to 20%, by giving the patient many liters
• Side effect: volume overload (needs furosemide)
� Cryoprecipitate
• 1 unit of FFP prepared by cryoprecipitate contains 50-120 U of VIII
� Plasma Derived FVIII prepared by monoclonal antibodies.

Disseminated Intravascular Coagulation:Causes
• Infection
– Most common cause of DIC.
– The syndrome particularly is associated with gram-negative or gram-
positive sepsis
– Can be triggered by a variety of other
• Bacterial - Fungal
• Viral - Rickettsial, and protozoal microorganisms.
• Obstetrics
– The placenta and uterine contents are rich sources of tissue factor
– Other procoagulants that normally are excluded from the maternal
circulation

Coagulation cascade in sepsis

Effects of DIC
Interactions between coagulation and fibrinolytic pathways
may results in bleeding in patients with DIC

Diagnosis
• Diagnosis of severe, acute (easy)
– Prolongation of PT, aPTT and Thrombin time
• Due to consumption and inhibition of clotting factors
– Thrombocytopenia
– Fibrin degradation products
• Increased due to secondary fibrinolysis
• Assays: latex agglutination or D-dimer

Treatment
• Principals: Identify underlying cause and treat
• Asymptomatic patients with self-limited DIC
– Only lab manifestations of the coagulopathy
– No treatment
• Actively bleeding or at high risk of bleeding
– Blood component transfusion
• Platelets - improve thrombocytopenia
• Fresh-frozen plasma (FFP)
– replace all consumed coagulation factors
– correct the prolonged PT and aPTT.
• Large volumes of plasma in severe cases
– Heparin � blocks thrombin and the secondary fibrinolysis.
• Might exacerbate the bleeding tendency

• Special cases
– Profound hypofibrinogenemia
• Add cryoprecipitate
• Plasma concentrate enriched in fibrinogen
– Sepsis
• Antithrombin III concentrate - an adjunctive
measure
Treatment

Disorders of Primary HemostasisI. Platelet Disorders
A. Qualitative Platelet Disorders
1. Disorders of Platelet Adhesion
a. Bernard Soullier/Giant Plt Syndr.
b. von Willebrand Disease
2. Disorders of Platelet Aggregation
a. Glanzmann’s thrombasthenia
b. Acquired von Willebrand Disease
3. Disorders of Platelet Secretion or Release Rxs.
a. Storage Pool Diseases
1. Electron dense/delta granules deficiency
- Hermansky Pudlak, Wiskott Aldrich, Chediak Higashi, TAR
2. Alpha granules deficiency
- Gray Platelet Syndrome, Quebec platelet disorder
3. Primary granules deficiency – Hemmeler anomaly
b. Thromboxane Pathway Disorders
1. Hereditary – aspirin like defects
2. Acquired due to inhibitors of prostaglandin pathway (chronic aspirin
intake or inhibitors of thromboxane or cyclooxygenase pathway)

Disorders of Primary Hemostasis
B. Quantitative Platelet Disorders
1. Thrombocytopenia
2. Thrombocytosis

Laboratory Tests for
Primary Hemostasis
1. Bleeding Time
2. Capillary Resistance / Fragility/ Tourniquet /Rumpel Leedesor Hess Test
3. Clot Retraction Time
4. Platelet Adhesiveness Test
5. Platelet Aggregation Test
6. Platelet Count
7. Platelet Morphology and MPV

Disorders of Secondary Hemostasis
• Hereditary
– individual coagulation factor deficiencies
• Acquired
– DIC
– Liver disease
– Vitamin K deficiency
– Acquired pathologic inhibitors or the circulating
anticoagulants

Laboratory Tests for Secondary Hemostasis
1. Clotting Time – slide, Dale-Laidlaw, Lee-White, Howell
2. Plasma Recalcification Time
3. Activated Clotting Time
4. PTT / APTT / DAPTT
5. PT
6. Stypven Time
7. Thrombin Time / Thrombin Clotting Time
8. Reptilase Time
9. Substitution Test (Mixing Studies)
10. Prothrombin Consumption/Serum Prothrombin Test
11. Thromboplastin Generation Test
12. Specific Factor Assay
13. Assay of vWR:Ag and vWR:Reo – Rockett/Laurel
14. Duckert’s or Clot Solubility Test
15. Tests for Circulating Inhibitors of Coagulation

Disorders of the Fibrinolytic System
• Hemorrhagic Disorder
• Thrombotic Disorder
– Hereditary
• Deficiency in plasminogen and in the activators of
plasminogen or plasmin
– Acquired
• Primary fibrinolysis
• Secondary fibrinolysis
















![Hemostasis & Coagulation Disorders(Ringkas II) - Dicky [Compatibility Mode]](https://static.fdocuments.us/doc/165x107/577cc4cd1a28aba7119a7e52/hemostasis-coagulation-disordersringkas-ii-dicky-compatibility-mode.jpg)

