
Haemoglobinopathies thalassemia, prophyrias and sickle cell disease-
Upload
anastasia-sandersCategory
view
221download
1
HaemoglobinopathiesHaemoglobinopathies

HaemoglobinopathiesHaemoglobinopathies
Inherited disorders of Hb Inherited disorders of Hb Structure and/or functionStructure and/or function
ThalassaemiasThalassaemias Sickle cell disordersSickle cell disorders Unstable haemoglobinsUnstable haemoglobins

Worldwide occurrenceWorldwide occurrence 5% of world population harbor alleles for hemoglobinopathies5% of world population harbor alleles for hemoglobinopathies 300,000 children born each year with hemoglobinopathy300,000 children born each year with hemoglobinopathy 200,000 children born yearly in Africa with Sickle Cell Disease200,000 children born yearly in Africa with Sickle Cell Disease
Areas of PrevalenceAreas of Prevalence Sub-Saharan Africa Sub-Saharan Africa
S.C. trait frequency 10-40%S.C. trait frequency 10-40% S.C. disease freq </= 2%S.C. disease freq </= 2% Highest rates in Ghana,Highest rates in Ghana, Nigeria, UgandaNigeria, Uganda
http://www.nslc.wustl.edu/sicklecell/part3/biogeography.html
EpidemiologyEpidemiology

What is Sickle Cell Anemia?A serious condition in which red blood
cells can become sickle-shapedNormal red blood cells are smooth and
round. They move easily through blood vessels to carry oxygen to all parts of the body.
Sickle-shaped cells don’t move easily through blood vessels. They’re stiff and sticky and tend to form clumps and get stuck in blood vessels.
The clumps of sickle cell block blood flow in the blood vessels that lead to the limbs and organs. Blocked blood vessel can cause pain, serious infection, and organ damage.
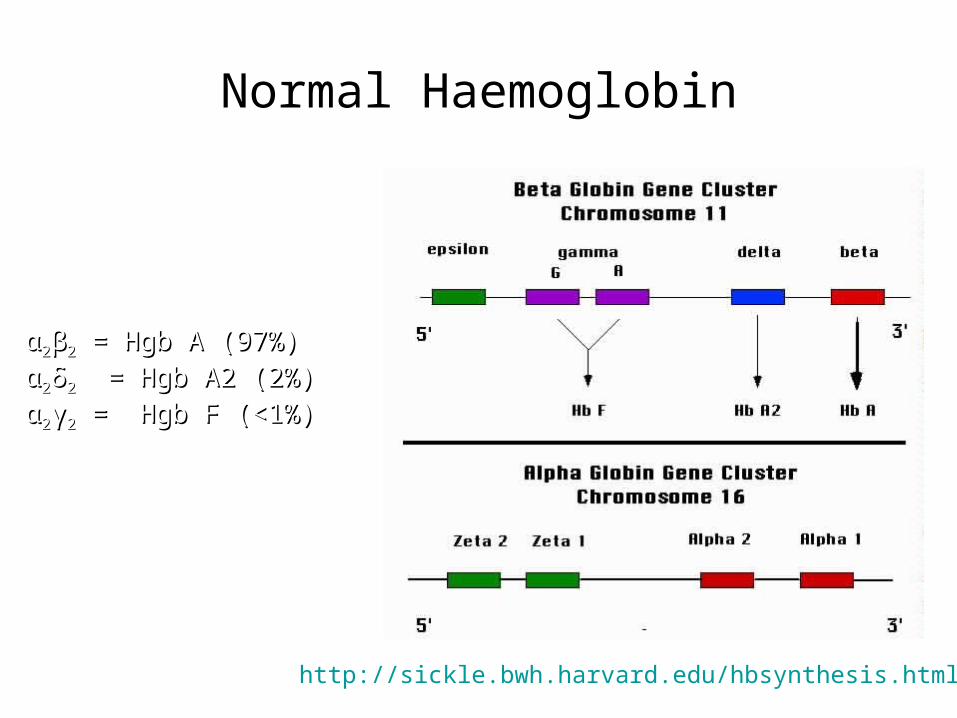
Normal Haemoglobin
Normal Adult Blood
αα22ββ22 = Hgb A (97%) = Hgb A (97%)
αα22δδ22 = Hgb A2 (2%) = Hgb A2 (2%)
αα22γγ22 = Hgb F (<1%) = Hgb F (<1%)
http://sickle.bwh.harvard.edu/hbsynthesis.html

Pathophysiology• Inheritance of mutated hemoglobin β-globin
chain• Mutation of GAG GTG at 6th codon at
chromosome 11• Glutamic acid Valine at 6th AA
– α1α2, β1β2 = normal hemoglobin
– α1α2, β1βS = heterozygote = Sickle trait
– α1α2, βSβS = homozygous recessive = Sickle cell disease
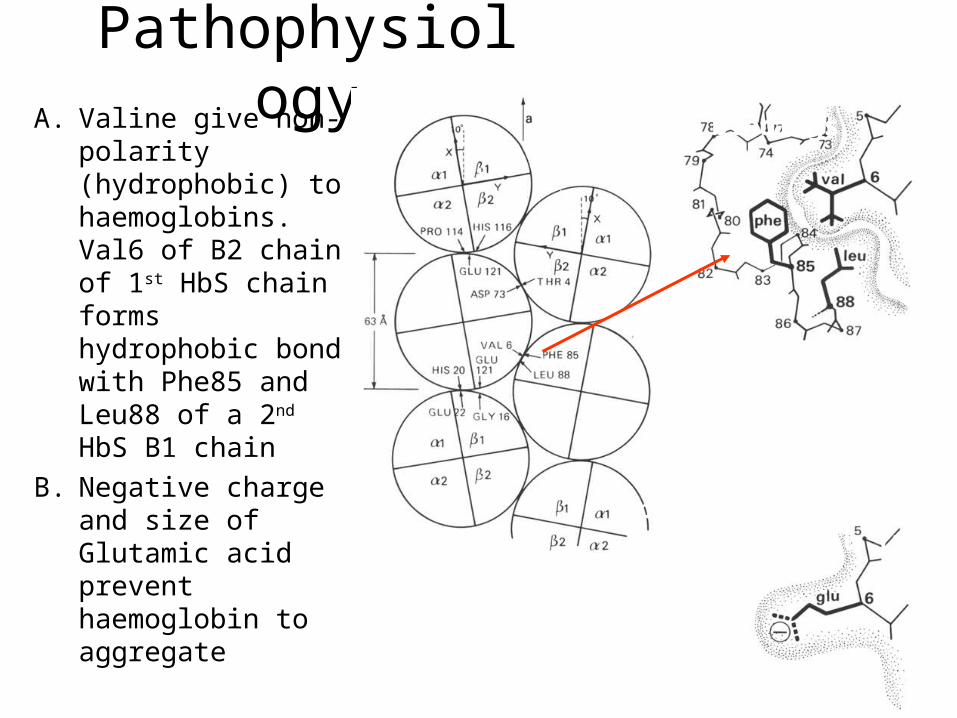
PathophysiologyA. Valine give non-
polarity (hydrophobic) to haemoglobins. Val6 of B2 chain of 1st HbS chain forms hydrophobic bond with Phe85 and Leu88 of a 2nd HbS B1 chain
B. Negative charge and size of Glutamic acid prevent haemoglobin to aggregate
B) Charge and size prevent 6 Glu from binding.
A) Haemoglobin bindings
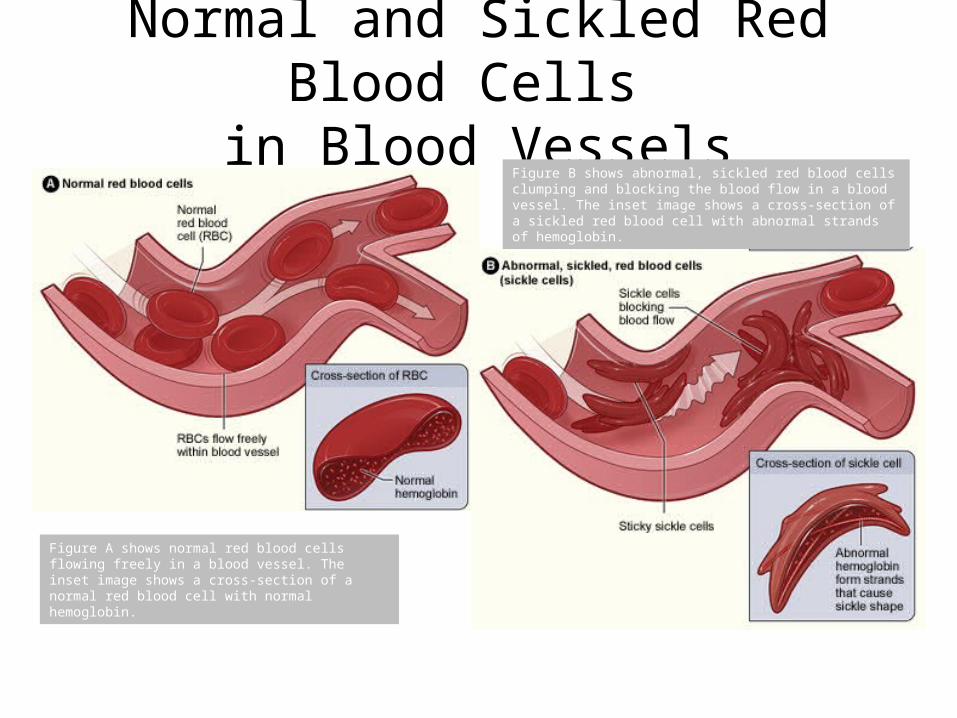
Normal and Sickled Red Blood Cells in Blood Vessels
Figure A shows normal red blood cells flowing freely in a blood vessel. The inset image shows a cross-section of a normal red blood cell with normal hemoglobin.
Figure B shows abnormal, sickled red blood cells clumping and blocking the blood flow in a blood vessel. The inset image shows a cross-section of a sickled red blood cell with abnormal strands of hemoglobin.
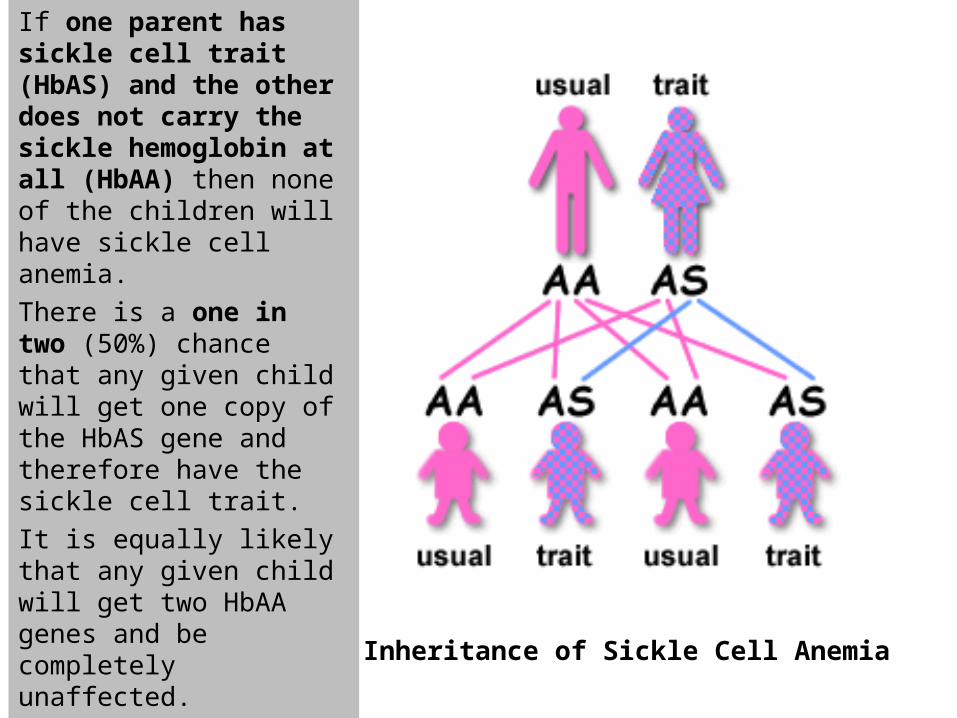
Inheritance of Sickle Cell Anemia
If one parent has sickle cell trait (HbAS) and the other does not carry the sickle hemoglobin at all (HbAA) then none of the children will have sickle cell anemia.
There is a one in two (50%) chance that any given child will get one copy of the HbAS gene and therefore have the sickle cell trait.
It is equally likely that any given child will get two HbAA genes and be completely unaffected.
Inheritance of Sickle Cell Anemia
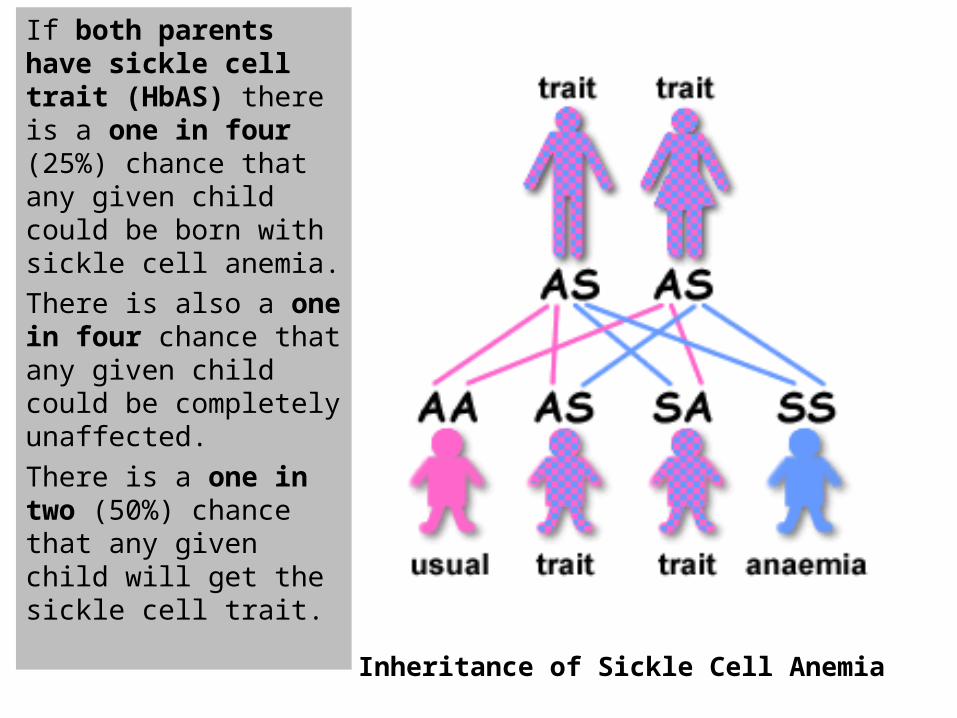
If both parents have sickle cell trait (HbAS) there is a one in four (25%) chance that any given child could be born with sickle cell anemia.
There is also a one in four chance that any given child could be completely unaffected.
There is a one in two (50%) chance that any given child will get the sickle cell trait.
Inheritance of Sickle Cell Anemia
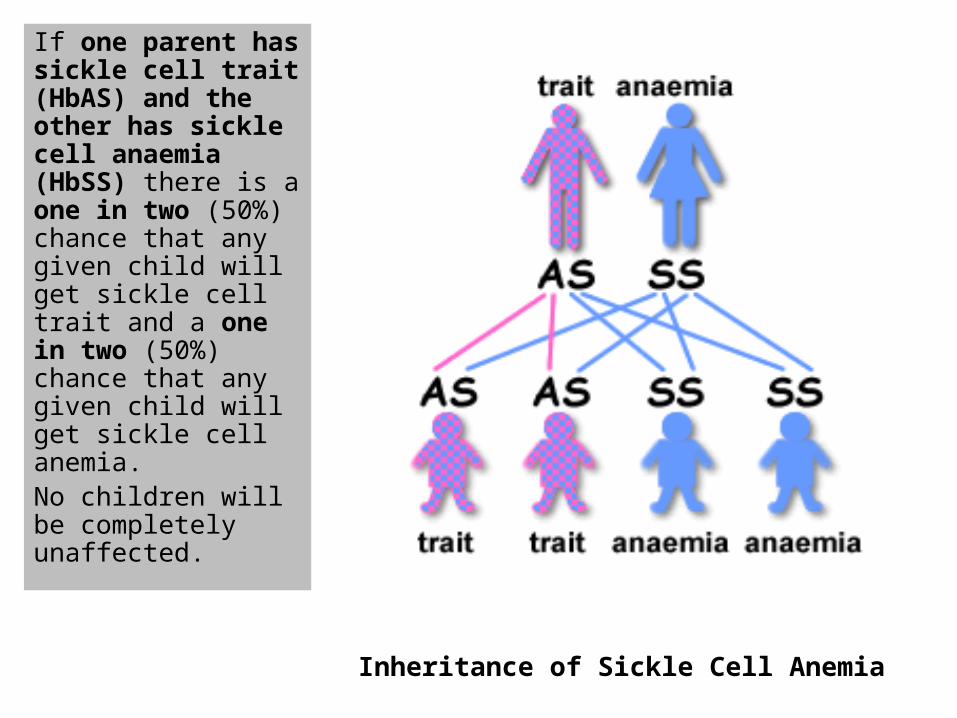
If one parent has sickle cell trait (HbAS) and the other has sickle cell anaemia (HbSS) there is a one in two (50%) chance that any given child will get sickle cell trait and a one in two (50%) chance that any given child will get sickle cell anemia. No children will be completely unaffected.
Inheritance of Sickle Cell Anemia
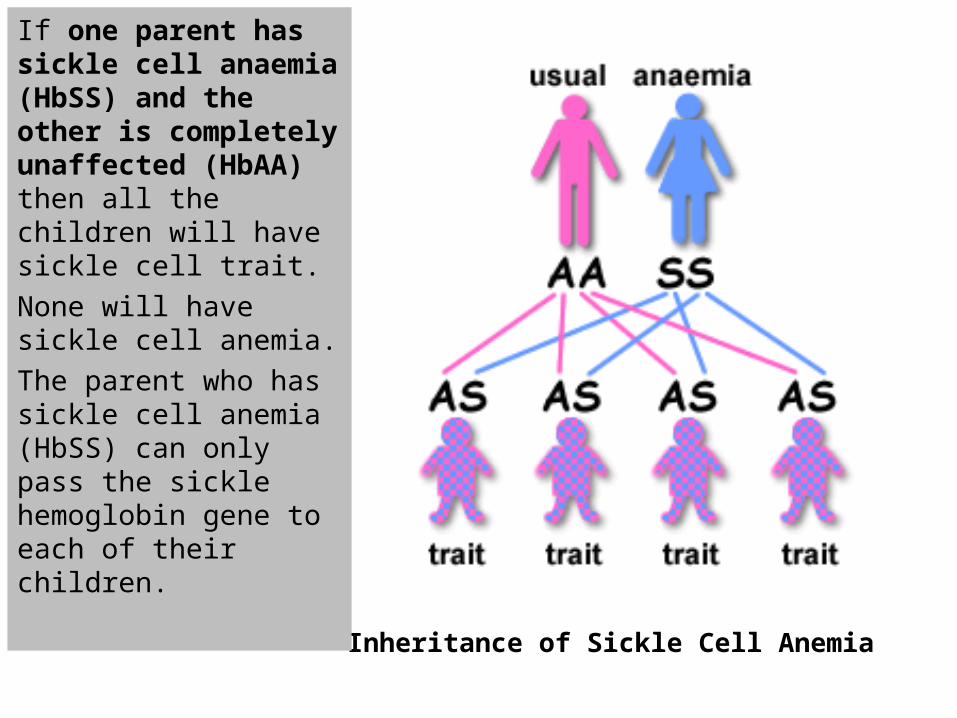
If one parent has sickle cell anaemia (HbSS) and the other is completely unaffected (HbAA) then all the children will have sickle cell trait.
None will have sickle cell anemia.
The parent who has sickle cell anemia (HbSS) can only pass the sickle hemoglobin gene to each of their children.

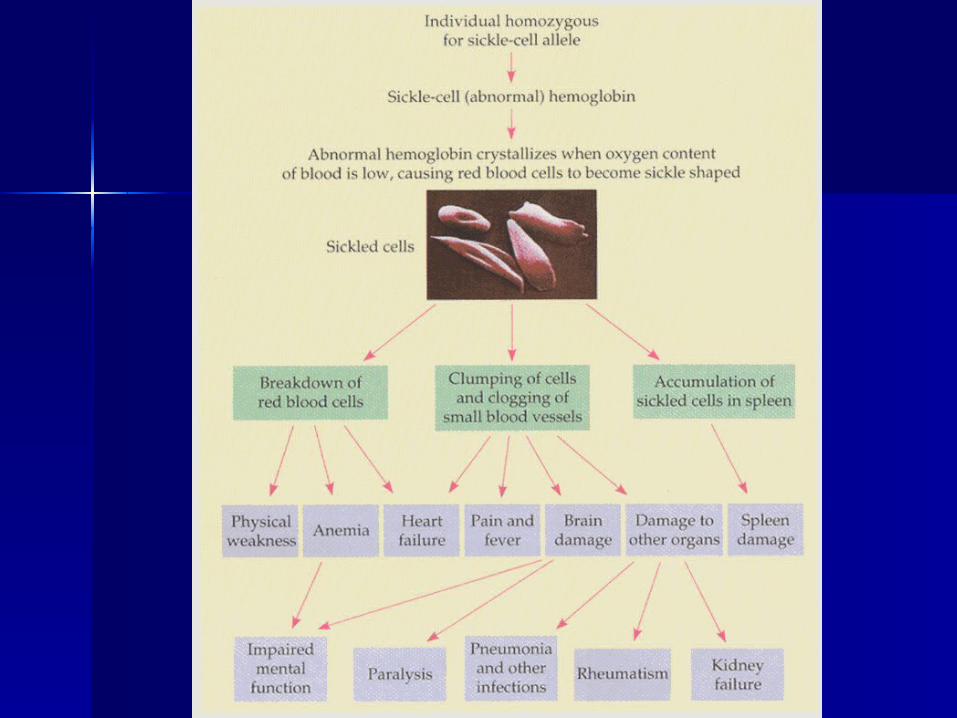
Sickle cell disease: Sickle cell disease: clinical problemsclinical problems
Anaemia (Anaemia (Hb 7-9g/dl in Hb Hb 7-9g/dl in Hb SSSS))
InfectionsInfections Painful crisesPainful crises StrokeStroke Leg ulcersLeg ulcers Visual lossVisual loss Chronic organ damageChronic organ damage
– Kidneys, lungs, joints, heartKidneys, lungs, joints, heart

Clinical problems by ageClinical problems by age
ChildrenChildren::InfectionInfection
Splenic sequestrationSplenic sequestration
PainPain
StrokeStroke
AdultsAdultsPainPain
InfectionInfection
Chest syndromeChest syndrome
Chronic organ damageChronic organ damage

Painful crisisPainful crisis Commonest problem for patientsCommonest problem for patients Pain is variable in severity and site and Pain is variable in severity and site and
may be excruciating may be excruciating Unpredictable throughout lifeUnpredictable throughout life Often precipitated by infection, physical Often precipitated by infection, physical
environment, stress, menstrual cycleenvironment, stress, menstrual cycle Associated with fear and anxietyAssociated with fear and anxiety Majority of patients manage at home Majority of patients manage at home
and only require admission for severe and only require admission for severe pain or other complicationspain or other complications
Appropriate management in the early Appropriate management in the early stages will reduce length and severity stages will reduce length and severity of crisisof crisis

Management of acute Management of acute sickle crisissickle crisis Analgesia Analgesia
– stepladder approachstepladder approach Treat associated infectionTreat associated infection FluidsFluids Monitor for acute complications Monitor for acute complications

Infections in SCDInfections in SCD Most common cause of death in Most common cause of death in
children but a major problem at all children but a major problem at all agesages
Due to splenic dysfunction from Due to splenic dysfunction from sickle damagesickle damage– occurs from a few months of ageoccurs from a few months of age– especially with certain bacteria eg especially with certain bacteria eg
pneumococcal sepsis : 400 x pneumococcal sepsis : 400 x risk risk Infection may be rapidly Infection may be rapidly
overwhelmingoverwhelming

Infection in SCD Infection in SCD
prevention:prevention:– educationeducation– Penicillin from 3/12 agePenicillin from 3/12 age– Pneumococcal, Hib, Meningococcal Pneumococcal, Hib, Meningococcal
vaccinesvaccines– travel prophylaxis : malariatravel prophylaxis : malaria
aggressive treatment of aggressive treatment of infectionsinfections

Acute sequestration Acute sequestration crisiscrisis SplenicSplenic
– mostly < 2yrsmostly < 2yrs– acute massive splenic enlargement, acute massive splenic enlargement,
Hb, shock Hb, shock– often associated with infectionoften associated with infection– significant mortalitysignificant mortality– requires emergency transfusionrequires emergency transfusion

TREATMENT FOR SCDTREATMENT FOR SCD
1 Folic acid and penicillen 1 Folic acid and penicillen administrationadministration
2 analgesics2 analgesics
3 transfusion therapy3 transfusion therapy
4 bone marrow transplant4 bone marrow transplant














![Raised Haemoglobin F (HbF) Level in Haemoglobinopathies ... · Haemoglobinopathies are the worldwide prevalent monogenic genetic disorders with variable geographic distribution [1]-[5].Although](https://static.fdocuments.us/doc/165x107/5f1b8427d7f40f077a680f2a/raised-haemoglobin-f-hbf-level-in-haemoglobinopathies-haemoglobinopathies.jpg)


