
Guest editors Rein Ulijn and Dek Woolfson · Guest editors Rein Ulijn and Dek Woolfson ... aim is...
17
This article was published as part of the Peptide- and protein-based materials themed issue Guest editors Rein Ulijn and Dek Woolfson Please take a look at the issue 9 2010 table of contents to access other reviews in this themed issue Published on 02 August 2010. Downloaded by University of Bristol on 12/10/2015 14:30:42. View Article Online / Journal Homepage / Table of Contents for this issue
Transcript of Guest editors Rein Ulijn and Dek Woolfson · Guest editors Rein Ulijn and Dek Woolfson ... aim is...

This article was published as part of the
Peptide- and protein-based materials themed issue
Guest editors Rein Ulijn and Dek Woolfson
Please take a look at the issue 9 2010 table of contents to access other reviews in this themed issue
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online / Journal Homepage / Table of Contents for this issue

3464 Chem. Soc. Rev., 2010, 39, 3464–3479 This journal is c The Royal Society of Chemistry 2010
More than just bare scaffolds: towards multi-component and decorated
fibrous biomaterialsw
Derek N. Woolfson*ab and Zahra N. Mahmouda
Received 4th July 2010
DOI: 10.1039/c0cs00032a
We are entering a new phase in biomaterials research in which rational design is being used to
produce functionalised materials tailored to specific applications. As is evident from this Themed
Issue, there are now a number of distinct types of designed, self-assembling, fibrous biomaterials.
Many of these are ripe for development and application for example as scaffolds for 3D cell
culture and tissue engineering, and in templating inorganic materials. Whilst a number of groups
are making headway towards such applications, there is a general challenge to translate a wealth
of excellent basic research into materials with a genuine future in real-life applications.
Amongst other contemporary aspects of this evolving research area, a key issue is that of
decorating or functionalising what are mostly bare scaffolds. There are a number of hurdles to
overcome to achieve effective and controlled labelling of the scaffolds, for instance: maintaining
biocompatibility, i.e., by minimising covalent chemistry, or using milder bioconjugation methods;
attaining specified levels of decoration, and, in particular, high and stoichiometric labelling;
introducing orthogonality, such that two or more functions can be appended to the same scaffold;
and, in relevant cases, maintaining the possibility for recombinant peptide/protein production.
In this critical review, we present an overview of the different approaches to tackling these
challenges largely for self-assembled, peptide-based fibrous systems. We review the field as it
stands by placing work within general routes to fibre functionalisation; give worked examples on
our own specific system, the SAFs; and explore the potential for future developments in the area.
Our feeling is that by tackling the challenges of designing multi-component and functional
biomaterials, as a community we stand to learn a great deal about self-assembling biomolecular
systems more broadly, as well as, hopefully, delivering new materials that will be truly useful in
biotechnology and biomedical applications (107 references).
1. Introduction
1.1 Rationale for and objectives of this review
This review does not focus on any one class of biomaterial
per se—there are excellent articles on these throughout this
a School of Chemistry, University of Bristol, Cantock’s Close, Bristol,UK BS8 1TS. E-mail: [email protected]
bDepartment of Biochemistry, University of Bristol, Medical School,University Walk, Bristol, UK BS8 1TD
w Part of the peptide- and protein-based materials themed issue.
Derek N. Woolfson
Dek Woolfson received hisPhD from the University ofCambridge, UK, working withProf Dudley Williams FRSand Dr Phil Evans onproblems in peptide andprotein folding. He did shortpost-doctoral stints with ProfJanet Thornton FRS(UC London), and Prof TomAlber (UC Berkeley), wherehe honed skills in bio-informatics and biophysics tostudy sequence-to-structurerelationships in proteins. Forthe past 15 years his indepen-
dent group at the University of Sussex, and, since 2005, at theUniversity of Bristol, has focussed on protein design and itsapplication to bionanotechnology and synthetic biology. Hisgroup takes a multidisciplinary approach to its work.
Zahra N. Mahmoud
Zahra Mahmoud received herPhD from the University ofHull, where she was alsoinvolved in the develop-ment of a second-generationtracheo-oesophageal fistulaspeech valve. She currentlyworks as a research associatewith Professor Dek Woolfsonat the University of Bristol;her research interests includedesign and functionalisationof self-assembling coiled-coilbiomaterials.
CRITICAL REVIEW www.rsc.org/csr | Chemical Society Reviews
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

This journal is c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 3464–3479 3465
Themed Issue—though the ideas and concepts introduced will
be illustrated by reference to specific examples. Rather, our
aim is to layout, and to begin to address, what we see as a
major challenge in biomaterials research today. Of course,
there are multiple challenges in the general area: namely, the
further introduction of truly rational approaches to the design
of such materials; understanding, controlling and exploiting
their dynamics; and the development of methods for
producing biomaterials on large enough scales to be useful
in real-life applications. However, the problem that we focus
on here is the reliable addition of function to what are largely
at present structural materials, or bare scaffolds.
This is challenging for a number of reasons: first, as
supramolecular constructs, we have scant understanding of
the atomic structures of many of these materials, which
frustrates the processes of engineering and rational redesign;
second, and what might seem contradictory to the first point,
many of the scaffolds are so well folded, ordered and even
partly crystalline that they effectively prohibit the inclusion of
the altered or atypical components that might be used to carry
function; thirdly, many of the materials are non-covalent
assemblies and attempts to modify them post-assembly fail
because of the underlying fragility of the systems, or
the forcing conditions needed for some bioconjugations.
Nonetheless, researchers are beginning to succeed in
assembling multi-component systems, or decorating pre-
assembled materials, giving considerable hope to addressing
the challenge we have laid out.
1.2 Types of material and how these impinge on approaches to
functionalisation
Mainly, we consider three types of peptide and protein fibrous
materials here: (a) those based on amyloid-like structures,
which can be formed from natural proteins, naturally derived
peptides, or peptides of de novo design; (b) those based on
a-helical assemblies, which usually employ peptides of de novo
design; and (c) engineered peptide-synthetic hybrids, in
particular peptide amphiphiles (PAs). This is primarily because
these self-assembling systems have been developed sufficiently
to allow their decoration and functionalisation. Other structures
such as designed collagen-like assemblies and silks will be
introduced as appropriate in later sections of this review.
(a) Amyloid-like structures. The term amyloid has a specific
meaning: it refers to extracellular, insoluble, fibrous, protein
aggregates associated with diseases such as Alzheimer’s.
However, as many peptides and proteins—both natural and
engineered, and implicated in disease or not—can be induced
to form similar assemblies, including similar underlying
secondary and quaternary structures,1 the more-general term
‘‘amyloid-like structure’’ is often preferred. In this generic
structure, small model peptides, fragments of natural proteins,
or even whole proteins, undergo conformational switches
to largely b-structured states in which multiple b-strandshydrogen bond to form extended sheets, these pair to form
the generic cross-b-structured fibrils. These may or may not
bundle to form thicker fibres. In these structures, the b-strandsrun perpendicular to the long axis of the fibrils and fibres,
Fig. 1a.1
Sequence-to-structure relationships in peptides that form
amyloid-like structures are emerging, but the field is still in its
infancy in this respect. This is somewhat different from the
a-helical assemblies described next, where such relationships
tend to be better defined partly because of the better under-
standing that we have for helical assemblies from work
on free-standing and coiled-coil a-helical systems.4 None-
theless, in terms of rational peptide design, rules of thumb
for engineering peptides that form amyloid-like structures
include: using a high proportion of b-structure-favouringresidues; aromatic side chains; and employing sequence
patterns in which hydrophobic (H) and polar (P) residues
alternate, (HP)n.
(b) a-Helical assemblies. In amyloid-like structures, inter-
chain backbone hydrogen bonding plays a significant role
in the self-assembly process. For a-helical supramolecular
assemblies this is not, indeed cannot, be the case. This is
because the majority of the backbone hydrogen bonding of
the a-helix is tied up locally within the helix. This has
advantages and disadvantages for design: one advantage is
that potentially promiscuous backbone-backbone interactions
are avoided, or at least reduced, which increases the level
of control possible compared with that for amyloid-like
assemblies; a disadvantage might be stated in the question,
then how are inter-chain interactions and higher-order assemblies
achieved? The answer to this comes in part from clever protein
designs, and partly through serendipity. The design aspect is
that sequence-to-structure relationships for natural helix–helix
assemblies are used, and slightly altered to drive new modes of
helix–helix interaction that promote supramolecular assembly;
essentially, a-helical peptides can be designed as building
blocks, or tectons, for assemblies comprising many millions
of organised peptides.5 The serendipity is that, when reduced
to practice, rather than making long thin fibrils, most of these
designs form long thick fibres. It turns out that this makes
them more interesting, robust and potentially more useful than
initially envisaged.
Several groups have now succeeded in making a-helix-based fibrous systems,6–11 which have been reviewed by us
elsewhere.12,13 However, the necessary details and differences
between these systems are described in section 4 of this review.
These designs rely on programming polypeptide sequences to
form amphipathic helices that associate—usually as dimers,
trimers and pentamers. As such, the designs tend to be based
on the well-understood a-helical coiled-coil motif,4 and have
underlying heptad repeats, (HPPHPPP)n. However, with a
couple of notable exceptions,10,11 unlike in natural coiled-coil
assemblies, where the bundles are discrete or blunt ended, in
the fibrous designs the helices are programmed to pack in
slipped arrangements. This leaves overhangs, or sticky ends,
which, in turn, assemble end-to-end to drive fibrillogenesis.
This concept is illustrated in Fig. 1b.
(c) Peptide amphiphiles (PAs). Hartgerink and Stupp’s
concept for the peptide amphiphiles (PAs),3 presents a very
different approach to engineering soft materials: here
self-assembling and functional units are combined in one
molecule from the outset. In the original PA design,3 a
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

3466 Chem. Soc. Rev., 2010, 39, 3464–3479 This journal is c The Royal Society of Chemistry 2010
16-carbon alkyl chain is conjugated to the N-terminus of a
polar ter-functional linear peptide with the sequence,
Cys4-Gly3-SerP-Arg-Gly-Asp, Fig. 1c. The alkyl chain, and
the overall amphipathicity of these molecules drives assembly
into rod-like, fibrous micelles, in which the alkyl chains are
buried and the C-termini of the peptides are exposed; these
structures are stabilised by a corona of interchain disulfide
bridges between the cysteine block; and the four C-terminal
amino acids provide water solubility and function. Stupp’s
team has confirmed these concepts and basic aspects of
the design and assembly through a series of biophysics,
microscopy and rheology studies over the past decade.14,15
Each of the general classes of material a–c has advantages
and drawbacks for decoration, which can limit or even dictate
the optimal route(s) taken to functionalising the systems. For
instance, the amyloid-like materials tend to have one or both
of their N- or C-termini free. Thus, the functional moieties, be
they small molecules, other peptides or whole peptides, can
simply be appended to the amyloidogenic polypeptides prior
to assembly. Similarly, the design principles for the PAs
specifically considers the need to incorporate functional motifs
with the molecular design. Clearly in both cases a and c, this
carries many advantages, but the possible lack of control over
assembly can be an issue; often these systems employ a single
self-assembly unit, and are subject to spontaneous assembly,
or thwarted by nucleation kinetics. As outlined in section 4,
the decoration of at least some of the a-helical systems is less
straightforward; as the termini are not necessarily free for
conjugation for example. Nonetheless, the experience and
knowledge that the community has in design and engineering
a-helical systems when fully brought to bear in this area of
materials could have a major impact.
2. Decoration of self-assembled, fibrous
biomaterials
Broadly, there are two general approaches to making multi-
component and decorated materials: top-down assembly,
using a variety of patterning techniques; and bottom-up
assembly, exploiting controlled self-assembly of monomeric
units. Top-down processes are used extensively for the
fabrication of photoptical devices, nanowires and so on. More
recently, this approach has been used to pattern surfaces for
biological applications.16 However, the approach presents
limits for incorporating features at the nanoscale. This review
focuses on routes in the bottom-up design of both straight-
forward and complex (multi)functional biomaterials via the
self-assembly of peptide, and peptide-containing systems.
Several routes have been taken here, which can be divided as
follows. Firstly, there is the construction of monomeric units
that contain both the instructions for self-assembly, and a
functional moiety; we refer to these as Route 1, or co-assembly
approaches. Secondly, in Route 2, or post-assembly approaches,
bare scaffolds are assembled from more-basic building blocks,
and then decorated either through covalent (Route 2a), or
non-covalent modification (Route 2b). Thirdly, in Route 3, or
templating approaches, pre-assembled fibrous materials are
used as templates to draw down the assembly or condensation
of inorganic or polymeric materials. These routes are
illustrated in Fig. 2.
Fig. 1 Types of peptide-based self-assembling fibrous biomaterials. (a) Amyloid-like assemblies. Small peptides form b-strands that hydrogen bond
into b-sheets and then assemble to form cross-b-structured fibrils. Reproduced in part from ref. 2, by permission of the National Academy of
Sciences, copyright r 2002. (b) a-Helical assemblies. Amphipathic a-helices can be designed to interact offset and form extended coiled-coil fibrils
that bundle into long fibres. (c) Peptide amphiphiles. Here the information needed for both self-assembly and functionalisation are encoded within
the same hybrid molecule. Reproduced in part from ref. 3, with permission of Science copyright r 2001. (d) Collagen-like assemblies. Stretches of
ProHypGly sequence form polyproline-type helices that associate into right-handed triple helices. These bundle further to form collagen-like fibres.
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

This journal is c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 3464–3479 3467
We find these classifications useful, but we recognise that
they are broad and cover a wide range of different materials,
chemistry and work from many groups. As a result, they can
be split further. For instance, in the co-assembly approach for
example, building blocks for assembly and function can be
combined in the initial pre-assembled construct, as in
Stupp’s peptide amphiphiles,15 or Barker’s amyloid-forming
protein-cytochrome fusions.18 In the non-covalent approach,
functionalised building blocks can be incorporated into the
fabric of the assembling material if it is permissive, as
exemplified by Yu’s work on collagen functionalisation;63 or
through the design or selection of new binding motifs as we
demonstrate for our own Self-Assembled peptide Fibres
(SAFs).19 The final approach of templating is somewhat of a
special case, which we split according to the types of materials
that are deposited on the scaffold.
Finally, we give a short history cataloguing our attempts to
decorate the SAFs, which have met with different levels of
success; and following this we describe the work of others in
this area who work on similar systems comprising a-helicalbuilding blocks.
2.1 Route 1: co-assembly of covalently linked structural and
functional moieties
The first approach to generating peptide- or protein-based
fibres that display a functional moiety is simply to construct
a single molecule containing both the self-assembling and
the functional components in question. There are three
ways in which this might be envisaged: (1a) covalent attach-
ment of the moiety to polypeptide during peptide synthesis;
(1b) if the functional moiety is itself a peptide or a
protein, gene fusions can be designed and expressed recombi-
nantly; and (1c) hybrid peptide systems in which the peptide
parts act as the self-assembling unit, the functional moiety,
or both.
In all of these cases, functionalisation is covalent and
performed prior to assembly. Of course, this mitigates some
problems associated with incompatibility of bioconjugation
chemistry and self-assembly. However, it places restrictions on
the systems too: namely, and obviously, that the resulting
constructs must remain competent for self-assembly; and,
conversely, that the self-assembly process must not preclude
or otherwise hamper the accessibility or activity of the
attached functionality. For self-assembling peptides and
proteins, this usually means having an available N- or
C-terminus free for covalent chemistry; though, side-chain
modifications are also possible. Indeed, the latter are becoming
more-widely used with wider access to modern peptide chemistry,
and the possibility of incorporating non-proteinogenic amino
acids into proteins via new recombinant methods.
Route 1a: peptide-small molecule constructs. Modern solid-
phase peptide synthesis (SPPS) is opening a wide range of
possibilities for bringing function to synthetic peptides and
even small proteins.20 Peptides of up to B50 amino acids can
now be made routinely by SPPS. In these methods, the peptide
chain is made by a series of catalysed condensation reactions
between a resin-bound polypeptide with a free N-terminal
amino group, and an amino acid in solution with a protected
N-terminus and an activated C-terminal carboxylic acid.
During these steps, the side chains of the growing polypeptide
chain and the incoming amino acid are protected with
chemistry that is orthogonal to that used to free the
N-terminus and effect the condensation reactions. In this
way, the peptide is synthesised from its C- to its N-terminus,
and, at the end of the process, a complete, side-chain-protected
peptide is attached to the solid support. At this stage, the
N-terminus can be liberated separately—as can certain
side-chain protecting groups—to allow a variety of functional
groups to be added through straightforward acid-amine
conjugation or alternative chemistries. Finally, the (remaining)
side-chain protection can be removed, and the peptide cleaved
from the resin simultaneously to produce the free peptide
ready for self-assembly.
There are numerous examples of this chemistry being
applied to add fluorophores, biotin (for later capture by
streptavidins), small peptides and similar moieties to the
N-termini of peptides, but here we focus on functionalising
self-assembling peptides. This particular area has been
reviewed by Channon and MacPhee recently,21 but we offer
Fig. 2 Routes to decorating self-assembling systems. (a) Co-assembly
of covalently linked structural and functional moieties. (b) A func-
tional protein can be (genetically) fused to a self-assembling peptide/
protein. (c) Post-assembly covalent decoration of scaffolds using
techniques such as click chemistry. (d) Structural mimics or (e) non-
covalent interactions can be exploited for functionalisation of
pre-formed scaffolds. Reproduced in part from ref. 17 with permission
of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright r
2010. Peptides/protein fibres can be used to template hollow tubes (f),
and conducting nanowires (g).
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

3468 Chem. Soc. Rev., 2010, 39, 3464–3479 This journal is c The Royal Society of Chemistry 2010
up a few illustrative examples from the primary literature.
Most of the examples centre on using amyloid-like assemblies.
For example, Channon et al. describe the assembly of
modified peptides based on the 105–115 fragment from
transthyretin (referred to as TTR), which forms amyloid-like
assemblies. In this case, they add a fluorophore to the
N-terminus of the peptide to give F-TTR. Assembly is driven
solely by the peptide sequence, with the fluorophore still
retaining its functionality, but not contributing to the
assembly process; i.e., F-TTR assembles to 7 nm-wide fibrils
as observed by transmission electron microscopy (TEM),
which show meridional and equatorial reflections at 4.7 and
10 A, respectively, in X-ray diffraction indicative of cross-bstructure.22 The fluorophores in these assemblies exhibit new
chiro-optical properties, which, the authors suggest, may find
use or provide inspiration in the development of opto-
electronic materials for photovoltaics or synthetic photo-
synthesis. Indeed, towards the second goal, the team has
recently demonstrated the co-assembly of two independent
luminescent moieties.23 In these cases the ‘‘conjugations’’ are
straightforward, because the fluorophores are added to the
N-terminus after peptide synthesis; indeed, in the first
example, it is simply the Fmoc protecting group retained from
peptide synthesis.
N.b. The development of shorter, Fmoc and similarly
N-terminally conjugated peptides, is a rapidly growing sub-
field being explored and reviewed well by Gazit, Xu, Ulijn,
Adams, Zhang and others.24,25 Though we note in these cases
the N-terminal aromatics and fluorophores usually do play a
significant role in the assembly of these smaller peptides
compared with the TTR-based systems.
Also somewhat related to the example from Channon et al.,
though strictly it comes under Route 2b in our categorisation,
Welland and co-workers describe how concentrated amyloid-
like fibres can be cast into ordered gels, that bind and orient
fluorophores non-covalently.26 One advantage here is the use
of relatively cheap, commercially available starting materials:
the protein used is hen-egg lysozyme, and the fluorophore is
Thioflavin T.
Extending this type of study in yet another direction, Gras
et al. describe the construction of bio-active peptides based on
TTR.27 In this case, the authors make C-terminal extensions
with a Gly-Gly spacer followed by the integrin-binding peptide
Arg-Gly-Asp (RGD). The resulting peptides still form
amyloid-like assemblies that display the expected dye-binding
tinctorial properties, fibre morphology by electron micro-
scopy, and cross-b-structure in X-ray diffraction. Further-
more, dansyl fluorophore appendages, also at the C-termini,
are used to demonstrate the exposure and availability of the
C-terminus, and that blends of the parent TTR1 and
TTR1-RGD can be made; both are confirmed by immunogold
labelling using an anti-dansyl fluorophore antibody and
visualisation by electron microscopy. The RGD-decorated
fibres are used in cell studies with fibroblasts to demonstrate
their use as substrates to support cell adhesion in tissue culture.
Gras’s work adds to a growing body of studies on designed
and natural peptides that form amyloid-like assemblies, and
particularly hydrogels, being applied in the burgeoning area of
3D cell culture and tissue engineering. On the design front for
example, research groups like those of Zhang, Pochan &
Schneider, and Aggeli are contributing here: Zhang’s group
has added RGD-based peptides to his model amyloid-like
gel-forming system, RADA16;28,29 though the MAX series
of peptides from the Pochan and Schneider groups have not
been engineered in this way, they do support 3D cell growth,30
and interestingly show antimicrobial activity;31 and Aggeli and
co-workers have considered the requirements for expanding
the field to real-life applications, including the need to move to
recombinant production in place of peptide synthesis.32,33
Regarding other naturally derived peptides—in particular
those not from proteins normally associated with amylo-
idogenic peptides or amyloidoses—Ohga et al. demonstrate
that certain proteolytic fragments of laminin (a glycoprotein
component of basal lamina) form amyloid-like fibres.34,35
These can be extended with N-terminal RGD-containing
sequences, and retain the ability to form amyloid-like
structures. The resulting functionalised materials support cell
growth, and neurite-like outgrowth from a model rat phaeo-
chromocytoma (PC12) cell line in culture. The second
property is linked to the sequence motif Ile-Lys-Val-Ala-Val
(IKVAV), which some of the peptides also contain making
these assemblies multifunctional.
Route 1b: recombinant peptide-protein fusions. Recombinant
DNA technology and, with it, the ability to clone, mutate and
fuse genes for proteins, and then express these in tractable host
cells is well established.33 Nonetheless, the increased efficiency
and reduced cost of making synthetic DNA, together with the
non-specialist kit or standardised approach to cloning and
expressing such pieces, are influencing the development of the
new field of synthetic biology. In essence, the aim here is to
design de novo, or to engineer existing biological components,
and to put these together in ways to make new and interesting
systems and devices, which, hopefully, will have some new and
useful properties.36 In these respects, the design and engineering
of functional peptide- and protein-based materials, parti-
cularly when translated into recombinant production, can
be considered a legitimate aspect of synthetic biology.5
However, it is one thing to sketch out ideas for basic bio-
molecular components, it is another to start fusing them
together in predictable, and useful ways. Nonetheless, several
studies offer hope and inspiration here.
For example, using the yeast protein Ure2p, Baxa et al.
demonstrate that functional proteins can be piggybacked onto
a self-assembly framework to give functional fibres.37 The
N-terminal ‘‘prion’’ domain—either residues 1–65 or 1–80,
depending on the construct used—of Ure2p protein is natively
unfolded, but can assemble to form b-structured amyloid-like
fibrils. Baxa and co-workers fused these regions to four
proteins—three enzymes, barnase, carbonic anhydrase,
glutathione S-transferase, and the green fluorescent protein.
In all cases the soluble fusion proteins show unfolded Ure2p
domains, but assemble to b-structured fibrils. Moreover, and
though to different degrees, the appended natural proteins are
all active. Similarly, Barker et al. have fused the gene for
cytochrome b562 to the amyloidogenic SH3 dimer, which is
capable of independently forming fibrils.18 Addition of
cytochrome b562 does not interfere with fibril formation, and
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online
This journal is c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 3464–3479 3469
cytochrome b562 when displayed on the fibre retains functionality
by binding metalloporphyrins, though only half of the
potential binding sites are occupied.
An area where recombinant production of materials has a
distinct advantage is for large, repetitive and otherwise
intractable fibrous proteins such as silks.38,39 The rules relating
to sequence-to-structure in these systems are only just being
unravelled. Nonetheless, they have some superb materials
properties ripe for exploitation as engineered biomaterials.
The sequences are modular and this has been exploited, by the
Kaplan and Scheibel groups for example, to produce and
express synthetic genes. In terms of decoration, Kaplan’s
group in particular has shown that these can then be used to
introduce single and multiple cell-adhesion motifs near the
termini of fibres to generate bioactive tissue-engineering
scaffolds.40,41 In this general area, others are turning their
attention to other elastomers such as arthropod resilins,42
though at this stage this work has focused on producing
recombinant model proteins, and to our knowledge, these
have yet to be decorated.
As noted by Kyle and co-authors,33 the move towards
recombinant production of self-assembled systems, either as
independent moieties or functionalised as above, will become
increasingly important if the field is to advance and fulfil
expectations towards real applications. Though it is not with-
out difficulties—amongst these is the overproduction of small,
often ephemeral peptides; and the possible formation of
cytotoxic oligomers or matured fibres—there is encourage-
ment that the community is headed in this direction.43
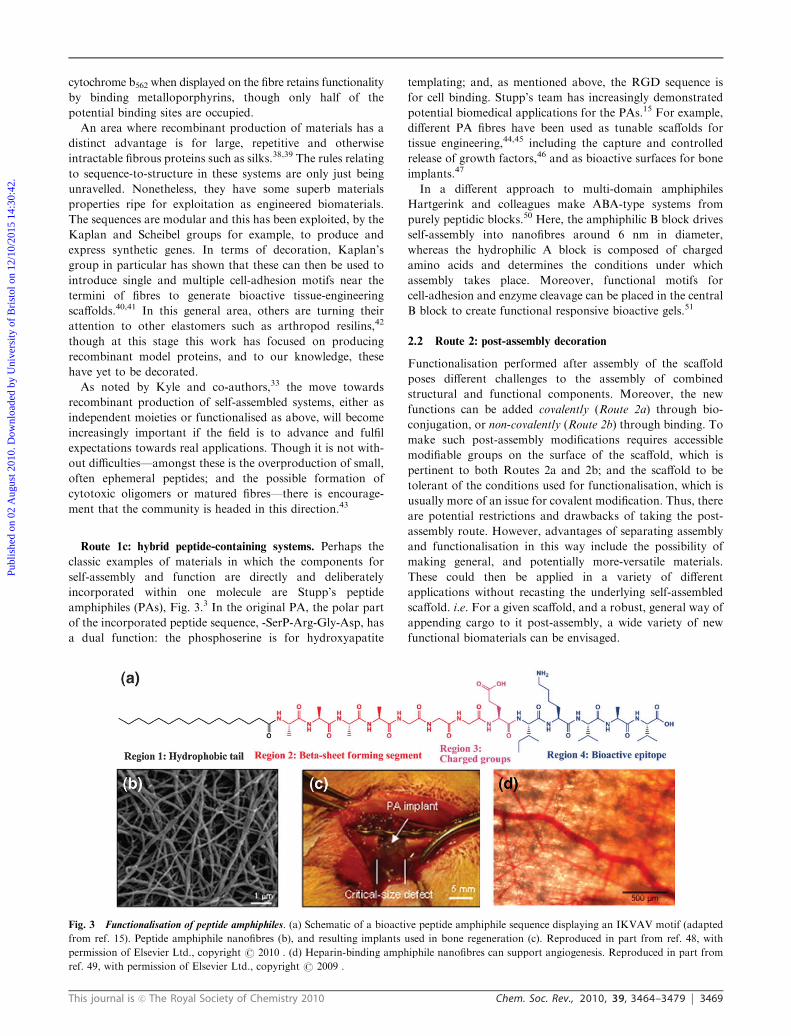
Route 1c: hybrid peptide-containing systems. Perhaps the
classic examples of materials in which the components for
self-assembly and function are directly and deliberately
incorporated within one molecule are Stupp’s peptide
amphiphiles (PAs), Fig. 3.3 In the original PA, the polar part
of the incorporated peptide sequence, -SerP-Arg-Gly-Asp, has
a dual function: the phosphoserine is for hydroxyapatite
templating; and, as mentioned above, the RGD sequence is
for cell binding. Stupp’s team has increasingly demonstrated
potential biomedical applications for the PAs.15 For example,
different PA fibres have been used as tunable scaffolds for
tissue engineering,44,45 including the capture and controlled
release of growth factors,46 and as bioactive surfaces for bone
implants.47
In a different approach to multi-domain amphiphiles
Hartgerink and colleagues make ABA-type systems from
purely peptidic blocks.50 Here, the amphiphilic B block drives
self-assembly into nanofibres around 6 nm in diameter,
whereas the hydrophilic A block is composed of charged
amino acids and determines the conditions under which
assembly takes place. Moreover, functional motifs for
cell-adhesion and enzyme cleavage can be placed in the central
B block to create functional responsive bioactive gels.51
2.2 Route 2: post-assembly decoration
Functionalisation performed after assembly of the scaffold
poses different challenges to the assembly of combined
structural and functional components. Moreover, the new
functions can be added covalently (Route 2a) through bio-
conjugation, or non-covalently (Route 2b) through binding. To
make such post-assembly modifications requires accessible
modifiable groups on the surface of the scaffold, which is
pertinent to both Routes 2a and 2b; and the scaffold to be
tolerant of the conditions used for functionalisation, which is
usually more of an issue for covalent modification. Thus, there
are potential restrictions and drawbacks of taking the post-
assembly route. However, advantages of separating assembly
and functionalisation in this way include the possibility of
making general, and potentially more-versatile materials.
These could then be applied in a variety of different
applications without recasting the underlying self-assembled
scaffold. i.e. For a given scaffold, and a robust, general way of
appending cargo to it post-assembly, a wide variety of new
functional biomaterials can be envisaged.
Fig. 3 Functionalisation of peptide amphiphiles. (a) Schematic of a bioactive peptide amphiphile sequence displaying an IKVAV motif (adapted
from ref. 15). Peptide amphiphile nanofibres (b), and resulting implants used in bone regeneration (c). Reproduced in part from ref. 48, with
permission of Elsevier Ltd., copyright r 2010 . (d) Heparin-binding amphiphile nanofibres can support angiogenesis. Reproduced in part from
ref. 49, with permission of Elsevier Ltd., copyright r 2009 .
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

3470 Chem. Soc. Rev., 2010, 39, 3464–3479 This journal is c The Royal Society of Chemistry 2010
Route 2a: post-assembly covalent modification. There are few
examples in this subtopic because of the tension between the
conditions needed to make covalent modifications, and those
required to maintain the self-assembled materials. However,
we suspect that click chemistry will transform this particular
area, facilitating new routes to functionalised self-assembled
peptide and proteins materials. Indeed, a search of Web
of Science combining the terms ‘‘click chemistry’’ and
‘‘self-assembly’’ and protein/peptide returned less than 20
papers, most of which were from the last year (2009–2010).
Thus, it seems to be an emerging and promising area.
However, most of the papers we found would be best placed
in the previous section, as they involve the conjugation of
peptides and, in one case proteins,52 to polymers to make
diblock and triblock systems that subsequently self-
assemble.53–55 Though a fascinating area for research, this
field of peptide-polymer hybrids is broad with a large literature
that is beyond the scope of this review. Therefore, we refer the
reader to a number of excellent recent reviews, which expand
this and the previous sections generally.56–58
We examine the area of click chemistry as applied to purely
peptide-based self-assembled systems with reference to our
own work in the area in section 4. Click chemistry
encompasses several types of reactions, of which the 1,3-dipolar
Huisgen cycloaddition is the most popular because of ease of
synthesis of the intermediates, high orthogonality, relatively
fast reactions kinetics and good yields. However, copper-based
click chemistry has not been widely explored for functionalisa-
tion of biomaterials because of concerns over the toxicity of
copper. However, we and others have shown that copper
can be successfully removed from gels after functionalisation;
additionally copper-free click is increasingly becoming accessible.59
As an example for this section one recent paper is parti-
cularly noteworthy even if a little off the main topic of the
review. DeForest et al. describe an elegant modular hydrogel
system that supports cell growth, Fig. 4.60 The components
comprise: a ‘‘hub’’ with four poly(ethylene glycol) (PEG) arms
each terminated by an azide functional group; a synthetic
peptide ‘‘linker’’ incorporating multiple features including:
cyclooctyne-containing terminal residues, a central alkene-
containing side chain, and a protease cleavage site; and
thiol-containing peptides that harbour fluorophores or other
protease sites to provide ‘‘functional appendages’’. Two ortho-
gonal click reactions are employed in the assembly process:
firstly, a copper-free variant of a 1,3-dipolar Huisgen cyclo-
addition between azides and alkynes is used to bring together
hubs and linkers to produce a highly cross-linked material,
which forms a physical hydrogel in aqueous buffer. Secondly,
photocatalysed thiol-ene reactions are used to tag the alkene-
containing hydrogel framework with the thiol-containing
functionalised peptides. Impressively, this can be done in a
top-down manner to allow cytocompatible photopatterning
with tens of micron resolution. In this way, the authors
prepare a number of hydrogels that can be visualised,
patterned, used to support cell growth and enzymatically
degraded.
Route 2b: post-assembly non-covalent binding. This route is
attractive for the same reasons that biology employs weak
non-covalent interactions—usually combinations of hydrogen
bonds, van der Waals’ forces, and salt bridges—to assemble
functional complexes: essentially, such forces allow for
control, specificity and reversibility; and they present possibi-
lities for building up complex (multi-component) systems. In
biomaterials design, they offer another potential advantage of
mild, biocompatible conditions for decoration. Balanced
against this, it is often difficult to rationally design specific
binders for a substrate (even if its surface structure is known);
and any binding will be reversible to some extent, and the
dynamics of cargo binding, release and, therefore, longevity on
the substrate may present problems.
Non-covalent decoration via hydrophobic interactions.
Peptides that form well-ordered supramolecular structures
are good candidates for non-covalent decoration, and make
even the notion of rationally designing binding peptides
Fig. 4 Functionalisation of polymer networks using copper-free click chemistry. A polyethylene glycol hub with terminal azides (a) is cross-linked to
cyclooctyne-bearing functional groups (b) to produce a physical hydrogel (c) with regularly arranged functional groups (R2; green). Reproduced in
part from ref. 60, with permission of Macmillan Publishers Limited, copyright r 2009.
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

This journal is c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 3464–3479 3471
possible. For example, Mihara’s group have reported a
peptide design, FI, that self-assembles to form straight, highly
ordered, tubular amyloid-like structures.61,62 Interestingly, FI
first assembles into protofibrils 5–17 nm in diameter, these
align side-by-side to form ribbons, which twist to form straight
fibres 80–130 nm in width andB10 mm in length. This packing
gives striations in negative-stain TEM—indicative of a high
level of order—and it is proposed that it exposes some of the
hydrophobic residues within the fibre. On this basis, the group
has designed anchor molecules to interact with these hydro-
phobic regions non-covalently.17 These consist of biotin linked
to a hydrophobic dodecyl group with or without a spacer unit.
Assembly of F1 is not perturbed by either anchor, and
subsequent incubation with gold-conjugated antibody shows
that the anchors were successfully recruited to the surface of
the fibres. To expand this style of non-covalent anchoring, the
dodecyl component can be replaced with Ile-Ile or Phe-Phe
dipeptide units to similar effect. Moreover, these dipeptide
binders can be extended via b-alanine linkers with imino-
diacetate moieties for Ni-chelation and subsequent capture
of hexahistidine-tagged proteins. Using these constructs, the
group present some striking images of nanoparticles arranged
with regular periodicity along the edges of fibres, Fig. 5.17
Non-covalent decoration using structural mimics. Yu and
co-workers describe how peptides that mimic collagen
sequence and secondary structure, so-called collagen-mimetic
peptides (CMPs), can be incorporated into the triple-helical
structure of collagen during the assembly process and so be
used to bring function to this natural scaffold.63 Naturally
occurring fibrous collagens form right-handed triple helices
due to underlying Pro-Hyp-Gly sequence repeats (where Hyp
is hydroxyproline), Fig. 1d. The CMPs are short, synthetic
peptides made up of such repeats. Yu and colleagues reason
that CMPs should associate with partly denatured collagen,
and demonstrate this as a series of fluorophore-labelled CMPs
with varying melting temperatures (TM): long, for instance
(ProHypGly)10-based CMPs with high TMs interact with
collagen films equilibrated at 80 1C, but not at 25 1C;
shortening the core sequence, to (ProHypGly)6, lowers the
TM to 25 1C, and this modified CMP interacts with collagen
films at 30 1C. Importantly in the second case, the temperature
is lower than the TM of collagen itself (37 1C), indicating that
denaturation of CMP is more critical for the interaction than
denaturation of collagen. To study the binding site of CMPs,
(ProHypGly)7 has been conjugated to gold nanoparticles via a
cysteine residue.64 This shows preferential decoration of type I
collagen fibres in the gap regions, which arise from staggered
packing of the triple-helices along the long fibre axis, and
appear as dark bands every 67 nm in negative-stain TEM. The
group suggest that the gaps correspond to regions low in
hydroxyproline, and that the CMPs supplement this acting
as triple-helix stabilisers and, hence, have increased affinities
for these regions. CMPs may be exploited in tissue engineer-
ing. For example, it is possible to make hybrid PEG-CMP
hydrogels, which retain cell-secreted collagen, thus allowing
the cells to grow in an environment more-closely mimicking
the natural extracellular matrix.65 Further modifications to
these gels using CMPs have been tested, including the addition
of multiple anionic charges to the N-terminus to recruit
VEGF.66 The VEGF-CMP charged complex is released from
the gel at a sustained rate, allowing the development of Hu-
man Umbilical Vein Endothelial Cells (HUVEC) with a
tubular morphology indicative of three-dimensional growth.
Non-covalent decoration using complementary pairing.
Related to Stupp’s earlier PA work, Li and Stupp have
engineered new PAs after Yamada and co-workers.67 In these
Fig. 5 Non-covalent decoration of self-assembling fibres with amyloid-like structure. (a) Bi-Da and Bi-ADa dodecyl anchors bind non-covalently to
F1-peptide fibres via hydrophobic interactions. (b) Replacing the dodecyl chain with Ile-Ile units allows the anchors to bind with a regular
periodicity. Reproduced in part from ref. 17 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright r 2010.
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

3472 Chem. Soc. Rev., 2010, 39, 3464–3479 This journal is c The Royal Society of Chemistry 2010
constructs, fatty acid-like dialkyl-chains and monoalkyl chains
are linked through a tripeptide unit. These wedge-shaped
molecules assemble to nanofibres in organic solvents burying
the monoalkyl moieties and leaving the dialkyl-chains
exposed. In this case, Li and Stupp append thymine to the
terminus of one of the latter chains. This can then be
non-covalently decorated with gold nanoparticles functiona-
lised with diaminopyridine (DAP), which forms comple-
mentary hydrogen bonds with the base, to render long, linear
gold nanowires.
Returning to water-soluble PAs, Guler et al. and Stupp,
have decorated yet another variant of the PA nanofibres
post-assembly using a biotin-avidin label.68 These designs
differ in a number of respects from the original PAs: first,
the alkyl chain, which drives assembly of the rod-like micelles,
is appended to a C-terminal side-chain of a central poly-
peptide; second, at the N-terminus of the polypeptide two
branches are spurred off from lysine side chains. These
branches are RGDS peptides with N-terminal biotins. The
resulting PAs are again wedge-shaped and form nanofibres
that gel. The surface biotin moieties can then be decorated
with BODIPY-NeutrAvidin.
2.3 Route 3: templating hard inorganic materials on soft
biomolecular assemblies
The final approach to decorating self-assembled peptide- and
protein-based biomaterials that we discuss is templating. This
bridges the gap between soft, self-assembled scaffolds, which
afford control over assembly and nanostructure at the 2-D and
3-D levels, and systems that cannot, or do not readily self-
assemble, but bring new functions to the resulting composite
materials. In general terms, this is another broad field,69 which
we cannot do justice to here, so we present a small number of
illustrative examples.
In templating, usually inorganic materials—though there
are examples for organic and polymeric materials—are deposited
on the surfaces of the scaffolding materials. This is either done
passively, which usually relies on recruitment of charges akin
to layer-by-layer deposition,70,71 or more-actively, which often
involves the inclusion of thiols (usually cysteine residues) into
the peptide and protein scaffolds. Materials that have been
combined with biomolecular assemblies in this way include,
gold and silver nanoparticles and metals, clays and silica,
and polyelectrolytes. The challenge is to template these in a
controlled fashion, to form functional conducting wires or
biomimetic substitutes for example.
Route 3a: towards metallic nanowires and related bio-
inorganic structures. Building on amyloid-like assemblies,
Reches and Gazit have exploited the self-assembling properties
of aromatic dipeptides to create silver nanowires with uniform
dimensions, Fig. 6.72 At high concentrations, diphenylalanine
precipitates out of hexafluoro-2-propanol as self-assembled
hollow nanotubes with outer diameters of B100 nm. The
hollow of these tubes can be filled with silver ions, which are
subsequently reduced to silver metal using citric acid as a mild
reducing agent. Moreover, the outer peptidic shell can
be removed with proteinase K to leave free-standing silver
nanowires of B20 nm diameter and almost microns in length.
Thus, the authors have succeeded in passively casting metal
wires using a peptide scaffold.
Similarly, Scheibel et al. use a variant of the N-terminal-to-
middle, ‘‘NM’’, region of yeast prion protein Sup35p, which
assembles into fibres B10 nm wide and up to microns in
length, to template gold nanoparticles.76 In this case, the initial
recruitment is actively encouraged by using a cysteine-
modified variant of NM. Again, metal wires are produced
by reduction of either silver or gold ions from solution. In this
case, the resulting silver and gold nanowires coat the surface of
the fibres, and are B100 nm across. With both metals, the
wires show low-resistance and ohmic metallic properties.
Further on this theme, other early pioneering studies in this
area come from Belcher and co-workers. Along with others,
such as Naik,69 this group show how metal recruitment can be
specified and controlled more tightly using peptide aptamers
selected using phage display against the target metal or
inorganic material.77 In these studies, the bacteriophage M13
is used both as a vehicle for peptide selection, and as a scaffold
for display and decoration, Fig. 7. M13 is a single-stranded
DNA virus widely used in molecular biology. The virus
particles are filaments with dimensions B7 nm in diameter
and B1 mm in length, which display a number of different
proteins on their surfaces in copy numbers ranging from
B3–3000. The genes for these proteins can be modified to
allow peptides and proteins to be displayed on the viral
surfaces; hence the term phage display. Belcher, Naik and
others have used this system to display libraries of peptides
and to select from these those that specifically and tightly bind
inorganic surfaces. The cunning tricks that allow this are:
(1) that binding phage can be enriched and amplified through
rounds of selection and infection into Escherichia coli; and
(2) that the sequences of the selected peptides can be deter-
mined via DNA sequencing as they are barcoded in the viral
DNA. In this way, Belcher’s team, for example, has developed
phage-templated silver,78 and gold and cadmium nanowires.79
Recently, the group has focussed on developing more-
ambitious phage-templated systems as a basis for highly
conductive lithium-ion batteries,80 and porphyrin assemblies
for light-driven water oxidation.81
Other recent examples of note in this general area include:
the production of metal-insulator-metal, trilayered, coaxial
nanocables from Gazit’s group.85 In this case, the afore-
mentioned diphenylalanine tubes that sequester silver are
supplemented by cysteine-containing peptides that bind the
surface of the structures to provide a second, outer template
for metalation. Recently, Ostrov and Gazit have turned to the
prokaryotic tubulin homologue, and filamentous protein Z
(FtsZ), which, in the presence of GTP or calcium, assembles
into nanometre-scale fibres. Through genetic engineering,
various metal-binding peptides with different metal-binding
affinities have been fused to the N-terminus of the protein,
allowing a series of nanowires to be generated.86 Mitraki et al.
used the self-assembling octapeptide peptide Asn-Ser-Gly-Ala-
Ile-Thr-Ile-Gly (NSGAITIG) from the fibre protein as a
template. This forms amyloid-like structures, and N-terminal
cysteine variants can be used to sequester gold, silver and
platinum nanoparticles to impressively high coverage.74 Yet
another structure has been explored as a template by Raines
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

This journal is c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 3464–3479 3473
and co-workers. They use collagen-like self-assembling peptides
and electroless silver plating (silver enhancement) to generate
nanowires. The basic collagen repeat of Pro-Hyp-Gly is
modified by replacing one of the hydroxyprolines by lysine,
which, in turn, is used to recruit amine-reactive gold nano-
particles, followed by silver enhancement.87
Route 3b: templating minerals on soft materials. As already
covered, the original PAs from Hartgerink and Stupp template
the mineralisation of hydroxyapatite via the phosphoserine
incorporated in the peptide component.3,88 PAs also assemble
into gels when incubated with calcium, which decorates the
outside of the fibres to form nucleation points for hydroxy-
apatite mineralization. This system has been expanded further
to template hydroxyapatite crystals in 3-D using the enzyme
alkaline phosphatase.89 Additional rigidity and stability can be
added to the system by filling these amphiphile gels into
titanium foam scaffolds.47
Similarly, Hartgerink et al. have used PAs bearing an RGD
motif and a enzyme-cleavable sequence to encapsulate two
types of dental stem cells.90 When grown in the presence of
osteogenic supplements, these PAs support growth of both
soft cells and osteoblastic cells, which mineralize the matrix,
making this a useful scaffold for dental tissue regeneration.
On a related theme, Aggeli and co-workers have used their
amyloid-like b-tapes as a scaffold to promote hydroxyapatite
crystallization and tooth mineralization.91
4a. Anecdotes from studies towards multi-component and
decorated SAFs
Over the last decade, we have explored the design, assembly
and application of a Self-Assembling peptide-based Fibre
(SAF) system.7,92–94 Unlike the majority of the systems
described above, which tend to employ a single self-assembling
molecule, the basic SAFs comprise two peptides that only
assembly when mixed. The peptides are of completely de novo
design, based on well-established sequence-to-structure
relationships for a-helical coiled coils.4 A key difference
between the SAFs and natural, or other designed coiled coils
is that they are designed to form offset, or staggered hetero-
dimers.7 These so-called sticky ended dimers are the building
blocks for fibrillogenesis. We have performed a detailed
analysis of the folding and assembly of the SAFs (at 100 mMin each peptide, 20 1C, pH 7):95 essentially, the two peptides
combine instantly to form the sticky ended dimer; this is
followed by a lag phase on the order of tens of minutes during
Fig. 6 Self-assembly and functionalisation of peptide nanotubes. (a) Diphenylalanine assembles into peptide nanotubes. (b) Schematic of metal
nanowires and coaxial cables templated from these nanotubes. (c) SEM image of vertically aligned peptide nanotubes useful for photoptic
applications. Reproduced in part from ref. 73, copyright r the American Chemical Society. (d) TEM image platinum-coated NSGAITIG
nanotubes (adapted from ref. 74). (e) Expression and self-assembly of filamentous protein Z (FtsZ) into nanofibres. (f) TEM image of FtsZ
filaments coated with silver particles. Reproduced in part from ref. 75 with permission of Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim,
copyright r 2010.
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

3474 Chem. Soc. Rev., 2010, 39, 3464–3479 This journal is c The Royal Society of Chemistry 2010
which aggregates comprisingB8 sticky ended dimers nucleate;
over the next few hours fibrillogenesis occurs both longi-
tudinally and laterally from these nuclei; subsequent fibrillo-
genesis is essentially one-dimensional leading to matured fibres
B50 nm thick and tens of microns in length. In addition, we
have a good working model for the molecular structure and
organisation in the equilibrium, matured fibres.93 This design
concept, together with our understanding of the mechanism of
assembly and the equilibrium structures of fibres, place us in a
strong position to build on and exploit the SAF systems;
specifically, they allow considerable control over assembly,
and present possibilities for introducing additional compo-
nents either at the start of folding and assembly, or during
fibrillogenesis, Fig. 8.
Rather than seeking decoration of the SAFs per se, we
began by investigating the addition of non-standard, or special
peptides, to the standard SAF mixtures in order to alter the
morphology of the fibres. This included introducing T-shaped
peptides and half-peptides connected through flexible linkers
to bring branches and kinks, respectively, to otherwise straight
and stiff fibres.96–98 One surprise from this early research
was that the special peptides incorporated into the fibres only
Fig. 7 Using M13 bacteriophage virus to create ordered nanostructures. (a) Nonocrystalline nickel, rhodium and cerium templated on M13 for
production of hydrogen (adapted from ref. 82). (b) Schematic and TEM images of M13-templated gold and silver nanowires. Reproduced in part
from ref. 83 with permission of the American Chemical Society, copyrightr 2010. (c) SEM image of Kevlar fibres coated with M13 phage bearing
a peptide that interacts with gold nanoparticles (reproduced in part from ref. 84 with permission of WILEY-VCH Verlag GmbH & Co. KGaA,
Weinheim, copyright r 2007.
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

This journal is c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 3464–3479 3475
sparingly; in some cases although stoichiometric mixtures of the
two parent peptides and the specials used to initiate fibrillo-
genesis (that is, 1 : 1 : 1 mixtures), the resulting fibres incorporate
special peptides to massively sub-stoichiometric levels. We
understand this now from our structural studies:93 the indivi-
dual SAFs are crystalline, and as such are not permissive of
incorporating specials that are significantly different from the
parent peptides; in other words, the introduction of special
peptides is like introducing defects into a crystal lattice. None-
theless, these new peptides do insert into, and do cause the
desired, albeit limited, morphological changes.
Furthermore, using this strategy, we were also able to
incorporate peptides harbouring biotin and FLAG-peptide
tags into the fabric of the SAFs.99 These allowed the sub-
sequent recruitment of streptavidin and anti-FLAG antibodies,
respectively, which could be visualised using nanogold probes
and TEM. Finally in this approach, we have demonstrated
that fluorophore-labelled peptides assembled into the fibres.100
This facilitates direct visualisation of the fibres by light
microscopy (LM), which, in turn, has led us to explore the
epitaxial (polar) growth of the fibres,100 and the late stages of
the kinetics of assembly.95 In these cases, fluorophores can be
introduced after peptide synthesis via bioconjugation, or
during peptide synthesis using modified amino acids. One note
of caution, however, because the SAFs carry an overall
positive charge they recruit negatively charged fluorescein
non-specifically; therefore, exclusively we use positively
charged rhodamine to label peptides.
As with the aforementioned systems, the SAFs can be
used as templates for the deposition of inorganic materials.
Fig. 8 Functionalisation of Self-Assembling peptide Fibres (SAFs). (a) SAF-p1 and SAF-p2a, which when mixed together in equimolar
concentrations at pH 7.4 assemble to form long straight fibres tens of microns long and tens of nanometres thick. (b) Non-covalent decoration
using short 11-residue peptides (SAF-tags),19 which bind via electrostatic interactions. Here decoration is visualised using rhodamine attached to
the N-terminus of the tag. (c) A click-chemistry approach for post-assembly decoration of fibres incorporating azide side chains. Here decoration is
followed by reacting the surface azide moieties with biotin-alkyne, followed by visualisation with 5 nm streptavidin nanogold.
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

3476 Chem. Soc. Rev., 2010, 39, 3464–3479 This journal is c The Royal Society of Chemistry 2010
We have demonstrated this by depositing silica on both
standard linear SAFs and some of the morphological
variants.101 Here, and in contrast to amyloid-like and
PA-based systems in particular—where the templating
material has to be removed by calcination102,103—the SAF
templates can be removed under mild, ambient conditions by
proteolysis to leave silica replicas.
Returning to decoration with specific chemical and biological
moieties: most recently we have explored two other routes to
decorating the SAFs (ref. 19 and Mahmoud et al. &Woolfson,
unpublished work). This has been done for several reason:
(1) to tackle the aforementioned problem of low levels of
inclusion of special peptides into SAFs, in particular, to
increase these to stoichiometric incorporation of labels and
the subsequent high degrees of decoration; (2) to improve the
biocompatibility of the labelling; and (3) to begin addressing
issues in temporal and spatial control of functionalisation.
Both methods are post-assembly routes (Route 2 in the above
rationale), and therefore demand mild conditions.
The first approach is entirely non-covalent (Route 2b), with
the aim of engineering small peptides that bind the outer
surface of the matured SAFs.19 The rationale is that, as the
fibres exhibit a high degree of structural order and carry an
overall positive charge, one might be able to select peptides
that bind the surfaces. Our initial attempts used peptide
libraries displayed on M13 phage. This failed for a variety of
reasons related to the generally acknowledged stickiness of
M13 phage, which led to high background binding. However,
this, along with the aforementioned observations with
fluorophores, suggested an approach employing short,
negatively charged, synthetic peptides. Briefly, we find that
peptides of the type DEDEDE bind strongly to the fibres;
neutral peptides, AQAQAQ, bind less weakly; and positively
charged peptides, KRKRKR, do not bind at all. Moreover,
the DE- and AQ-based peptides can be tagged, and used to
recruit cargo to the SAFs, as demonstrated with nanogold
particles visualised by TEM, and with fluorophores visualised
directly in solution by LM, Fig. 8b.19 The advantage of these
so-called SAF-tags is that they could be readily added to target
proteins for recruitment to the SAFs. A disadvantage is the
potential for highly dynamic and non-specific binding with
such simple sequences; though in the studies we have
conducted we see long-lived binding and very little evidence
for non-specific interactions.
Our second approach involves small chemically reactive
groups suitable for biocompatible ‘‘click’’ reactions being
directly introduced into the SAF peptides during synthesis
(Route 2a). The modified peptides are then co-assembled with
standard SAF peptides to produce matured fibres. These can
then be decorated with a variety of organic and peptidic
reagents harbouring a complementary click group. To demon-
strate this, we have synthesised SAF peptides incorporating
azide and alkene side chains. Importantly, and unlike our
earlier approaches,99 the modified peptides incorporate
stoichiometrically with the standard SAF peptides. The
resulting modified SAFs can be functionalised through
conjugation with copper-catalysed azide-alkyne and thiol-ene
click reactions, respectively. This allows high levels of
incorporation of the modified peptides (up to 100%), subsequent
high coverage of the SAFs with cargo to be achieved, and the
proportion of the label to be varied. Moreover, as the afore-
mentioned click reactions are orthogonal and both modified
peptides can be co-incorporated into a single fibre preparation,
dual functionalisation of the SAFs is possible. We have
demonstrated all of these possibilities by incorporating either
or both nanogold and fluorophore labels, Fig. 8c. Of course,
this approach requires the synthesis of non-proteinogenic
peptides, which are not amenable to standard recombinant
DNA expression. However, as the incorporation rates are very
high, relatively small amounts of the modified peptides are
required to get respectable levels of decoration, and, so, these
could be used sub-stoichiometrically with standard peptides;
also, side chains that incorporate groups for the click
reactions, and diazo transfer reagents that work in water are
becoming more available.104 Therefore, in our view, this is the
most promising approach to making functionalised SAFs that
we have investigated so far.
4b. Decoration of other coiled-coil-based fibrous systems
A number of other groups have presented designs for fibrous
materials based on a-helical, and in particular coiled-coil
building blocks,6,105,8–10 and some of these have decorated
or functionalised their systems. Notably, Kajava and collea-
gues offer a system similar in some respects to the SAFs;
however, it is based on pentameric coiled coils, and the
individual helices ‘‘slip’’ to give sticky ended assemblies, which
then associate end-to-end and side-by-side to form thickened
fibres depending on the conditions. In a modified design they
incorporate the integrin-binding RGD sequence, to render
fibres that support cell growth in culture.106 In another
SAF-related design approach, Dublin and Conticello describe
a sticky ended trimeric coiled coil with buried histidine
residues. In this case, these imidazole side chains coordinate,
and facilitate the preparation of silver nanowires shrouded in
peptide fibre.107
Conclusion
Through this review, we hope to have outlined the current
activities and trends in decorating, or functionalising, soft
biomaterials, in particular fibrous assemblies. We confess that,
whilst we appreciated that there was a strong and growing
base of literature in the area, we hadn’t expected to find the
depth and breadth of new, exciting and varied research that we
did. Indeed, this review, and the literature cited in it, could
have been expanded considerably had we had the time and
space to do so. As a result of the growth of this field and its
associated literature, rather than simply list and review the
papers that we came across—say, chronologically, by materials
type, or research group—we have tried as far as possible to see
past individual papers and onto generalisations that are
emerging. Specifically, we have categorised the work in terms
of routes to functionalising fibrous biomaterials. Clearly there
are limits to this: some examples are not so easily pigeon-
holed, others span more than one route, and, of course, there
will be future ground-breaking pieces of work that challenge
dogma, and force us to reconsider the categorisation.
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

This journal is c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 3464–3479 3477
Nonetheless, we find this analysis useful, and we hope that
others will find it helpful too.
During the course of our reading and writing, it became
clear that common themes, approaches and methods that
represent best practice are emerging in the field. For example:
(1) the exploitation of natural self-assembling systems, or
peptides derived from these often present a good starting
point. However, this can present restrictions in terms of
control over self-assembly, and also asks the question, just
how does one functionalise materials pre- or post-assembly
without interfering with the self-assembly process itself? This
raises the next two key points from our perspective. (2) That
the design or engineering of the self-assembling and functional
components of the system should be orthogonal. And, related
to this, (3) that this orthogonality should be designed or
engineered into the system, or at least considered, from the
outset of materials development. Perhaps the best examples
that illustrate these two points to date are the peptide amphi-
philes from Stupp and colleagues. (4) Rational peptide design
also represents a good approach in these respects, but it does
rely on having good rules that relate sequence, structure and
assembly. Our understanding for certain protein-folding
motifs, such as the coiled coil, is headed in the right direction,
but this is by no means the case universally for protein folding.
Finally (5) there are now many examples of designed and
engineered self-assembled systems, and it is likely that some
will be better suited to appending certain functions and in
specific applications than others. It would seem prudent, to
choose the right tool for the job in this respect, and not to be
wedded to any one material type. Ideally, as the field develops
we would have an open-source or synthetic-biology ethos, and
a toolkit of basic materials with which to build will emerge.
Somewhat related to this, it is quite understandable that
different groups have adopted similar strategies to demon-
strate that they have achieved decoration. The common
methods are ‘‘functionalisation’’ with a fluorophore of some
description followed by visualisation using light microscopy;
or the addition of gold nanoparticles (GNPs) followed by
visualisation using electron microscopy. Clearly, these are the
best options for demonstrating that decoration has been
achieved, but usually they do not represent functionalisations
in themselves; though examples such as the formation of
metallic nanowires following the recruitment of gold nano-
particles are cases where functional materials are being
developed in this way. Nonetheless, true functionalisation
would be to impart some biological activity such as cell-
binding properties onto the scaffolds. Of course, there is strong
research in this endeavour. However, this is and must remain
one of the tenets of both materials science and synthetic
biology: that is, whilst as a community we must develop the
very best understanding of natural and design self-assembling
systems, as materials scientist and/or synthetic biologists, we
must consider applications that put our materials to use.
In the case of soft fibrous biomaterials, the key applications
areas that are being explored here are scaffolds for 3D cell
culture and tissue engineering, and as sacrificial templates for
the organisation of functional, often hard, materials that are
less readily self-assembled. In many respects, the studies that
we have highlighted have laid the groundwork for this
development, and it is now time to translate further this basic
research and deliver functionalised materials for specific real-
life applications. There are many challenges ahead, including
issues of large-scale materials production, biocompatibility of
methods used in functionalisation, and, for tissue engineering,
immunogenicity of the final materials. However, we feel that
these and other issues are being tackled and will be surmounted.
In summary, the current state of basic research in functional
fibrous biomaterials is buoyant, and provides a strong basis
for translation into real-life applications.
References
1 O. S. Makin and L. C. Serpell, FEBS J., 2005, 272, 5950–5961.2 J. L. Jimenez, E. J. Nettleton, M. Bouchard, C. V. Robinson,
C. M. Dobson and H. R. Saibil, Proc. Natl. Acad. Sci. U. S. A.,2002, 99, 9196–9201.
3 J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294,1684–1688.
4 D. N. Woolfson, Adv. Protein Chem., 2005, 70, 79–112.5 E. H. C. Bromley, K. Channon, E. Moutevelis and
D. N. Woolfson, ACS Chem. Biol., 2008, 3, 38–50.6 S. Kojima, Y. Kuriki, T. Yoshida, K. Yazaki and K. Miura, Proc.
Jpn. Acad., Ser. B, Phys. Biol. Sci., 1997, 73, 7–11.7 M. J. Pandya, G. M. Spooner, M. Sunde, J. R. Thorpe,
A. Rodger and D. N. Woolfson, Biochemistry, 2000, 39,8728–8734.
8 S. A. Potekhin, T. N. Melnik, V. Popov, N. F. Lanina,A. A. Vazina, P. Rigler, A. S. Verdini, G. Corradin andA. V. Kajava, Chem. Biol., 2001, 8, 1025–1032.
9 Y. Zimenkov, V. P. Conticello, L. Guo and P. Thiyagarajan,Tetrahedron, 2004, 60, 7237–7246.
10 K. L. Lazar, H. Miller-Auer, G. S. Getz, J. Orgel andS. C. Meredith, Biochemistry, 2005, 44, 12681–12689.
11 H. Dong, S. E. Paramonov and J. D. Hartgerink, J. Am. Chem.Soc., 2008, 130, 13691–13695.
12 D. N. Woolfson and M. G. Ryadnov, Curr. Opin. Chem. Biol.,2006, 10, 559–567.
13 D. N. Woolfson, Biopolymers, 2010, 94, 118–127.14 L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2008, 41,
1674–1684.15 H. G. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94,
1–18.16 Z. H. Nie and E. Kumacheva, Nat. Mater., 2008, 7, 277–290.17 A. Miyachi, T. Takahashi, S. Matsumura and H. Mihara,
Chem.–Eur. J., 2010, 16, 6644–6650.18 A. J. Baldwin, R. Bader, J. Christodoulou, C. E. MacPhee,
C. M. Dobson and P. D. Barker, J. Am. Chem. Soc., 2006, 128,2162–2163.
19 Z. M. Mahmoud and D. N. Woolfson, Biomaterials, 2010, DOI:10.1016/j.biomaterials.2010.06.041.
20 B. L. Nilsson, M. B. Soellner and R. T. Raines, Annu. Rev.Biophys. Biomol. Struct., 2005, 34, 91–118.
21 K. Channon and C. E. MacPhee, Soft Matter, 2008, 4, 647–652.22 O. S. Makin, P. Sikorski and L. C. Serpell, Curr. Opin. Chem.
Biol., 2006, 10, 417–422.23 K. J. Channon, G. L. Devlin and C. E. MacPhee, J. Am. Chem.
Soc., 2009, 131, 12520–12521.24 E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269.25 R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675.26 T. P. J. Knowles, T. W. Oppenheim, A. K. Buell, D. Y. Chirgadze
and M. E. Welland, Nat. Nanotechnol., 2010, 5, 204–207.27 S. L. Gras, A. K. Tickler, A. M. Squires, G. L. Devlin,
M. A. Horton, C. M. Dobson and C. E. MacPhee, Biomaterials,2008, 29, 1553–1562.
28 A. Horii, X. M. Wang, F. Gelain and S. G. Zhang, PLoS One,2007, 2, e190, DOI: 10.1371/journal.pone.0000190.
29 Y. Kumada and S. G. Zhang, PLoS One, 2010, 5, e10305, DOI:10.1371/journal.pone.0010305.
30 L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick,R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

3478 Chem. Soc. Rev., 2010, 39, 3464–3479 This journal is c The Royal Society of Chemistry 2010
J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104,7791–7796.
31 D. A. Salick, J. K. Kretsinger, D. J. Pochan and J. P. Schneider,J. Am. Chem. Soc., 2007, 129, 14793–14799.
32 A. Firth, A. Aggeli, J. L. Burke, X. B. Yang and J. Kirkham,Nanomedicine, 2006, 1, 189–199.
33 S. Kyle, A. Aggeli, E. Ingham and M. J. McPherson, TrendsBiotechnol., 2009, 27, 423–433.
34 S. Kasai, Y. Ohga, M. Mochizuki, N. Nishi, Y. Kadoya andM. Nomizu, Biopolymers, 2004, 76, 27–33.
35 Y. Ohga, F. Katagiri, K. Takeyama, K. Hozumi, Y. Kikkawa,N. Nishi and M. Nomizu, Biomaterials, 2009, 30, 6731–6738.
36 K. Channon, E. H. C. Bromley and D. N. Woolfson, Curr. Opin.Struct. Biol., 2008, 18, 491–498.
37 U. Baxa, V. Speransky, A. C. Steven and R. B. Wickner, Proc.Natl. Acad. Sci. U. S. A., 2002, 99, 5253–5260.
38 S. C. Kundu, B. C. Dash, R. Dash and D. L. Kaplan, Prog.Polym. Sci., 2008, 33, 998–1012.
39 A. Leal-Egana and T. Scheibel, Biotechnol. Appl. Biochem., 2010,55, 155–167.
40 E. Bini, C. W. P. Foo, J. Huang, V. Karageorgiou, B. Kitchel andD. L. Kaplan, Biomacromolecules, 2006, 7, 3139–3145.
41 A. W. Morgan, K. E. Roskov, S. Lin-Gibson, D. L. Kaplan,M. L. Becker and C. G. Simon, Biomaterials, 2008, 29,2556–2563.
42 C. M. Elvin, A. G. Carr, M. G. Huson, J. M. Maxwell,R. D. Pearson, T. Vuocolo, N. E. Liyou, D. C. C. Wong,D. J. Merritt and N. E. Dixon, Nature, 2005, 437, 999–1002.
43 P. S. Nadaud, M. Sarkar, B. Wu, C. E. MacPhee, T. J. Maglieryand C. P. Jaroniec, Protein Expression Purif., 2010, 70, 101–108.
44 D. A. Harrington, E. Y. Cheng, M. O. Guler, L. K. Lee,J. L. Donovan, R. C. Claussen and S. I. Stupp, J. Biomed. Mater.Res., Part A, 2006, 78a, 157–167.
45 M. A. Greenfield, J. R. Hoffman, M. O. de la Cruz andS. I. Stupp, Langmuir, 2010, 26, 3641–3647.
46 R. N. Shah, N. A. Shah, M. M. D. Lim, C. Hsieh, G. Nuber andS. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3293–3298.
47 T. D. Sargeant, S. M. Oppenheimer, D. C. Dunand andS. I. Stupp, J. Tissue Eng. Regener. Med., 2008, 2, 455–462.
48 A. Mata, Y. Geng, K. J. Henrikson, C. Aparicio, S. R. Stock,R. L. Satcher and S. I. Stupp, Biomaterials, 2010, 31, 6004–6012.
49 S. Ghanaati, M. J. Webber, R. E. Unger, C. Orth, J. F. Hulvat,S. E. Kiehna, M. Barbeck, A. Rasic, S. I. Stupp andC. J. Kirkpatrick, Biomaterials, 2009, 30, 6202–6212.
50 L. Aulisa, H. Dong and J. D. Hartgerink, Biomacromolecules,2009, 10, 2694–2698.
51 K. M. Galler, L. Aulisa, K. R. Regan, R. N. D’Souza andJ. D. Hartgerink, J. Am. Chem. Soc., 2010, 132, 3217–3223.
52 I. C. Reynhout, J. Cornelissen and R. J. M. Nolte, J. Am. Chem.Soc., 2007, 129, 2327–2332.
53 T. B. Yu, J. Z. Bai and Z. B. Guan, Angew. Chem., Int. Ed., 2009,48, 1097–1101.
54 A. J. Dirks, S. S. van Berkel, H. I. V. Amatdjais-Groenen,F. Rutjes, J. Cornelissen and R. J. M. Nolte, Soft Matter, 2009,5, 1692–1704.
55 E. K. Schillinger, E. Mena-Osteritz, J. Hentschel, H. G. Bornerand P. Bauerle, Adv. Mater., 2009, 21, 1562–1567.
56 M. A. Gauthier and H. A. Klok, Chem. Commun., 2008,2591–2611.
57 H. G. Borner, Prog. Polym. Sci., 2009, 34, 811–851.58 H. R. Marsden and A. Kros,Macromol. Biosci., 2009, 9, 939–951.59 J. M. Baskin and C. R. Bertozzi, Aldrichimica Acta, 2010, 43,
15–23.60 C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater.,
2009, 8, 659–664.61 H. Kodama, S. Matsumura, T. Yamashita and H. Mihara, Chem.
Commun., 2004, 2876–2877.62 S. Matsumura, S. Uemura and H. Mihara, Chem.–Eur. J., 2004,
10, 2789–2794.63 A. Y. Wang, X. Mo, C. S. Chen and S. M. Yu, J. Am. Chem. Soc.,
2005, 127, 4130–4131.64 X. Mo, Y. J. An, C. S. Yun and S. M. Yu, Angew. Chem., Int. Ed.,
2006, 45, 2267–2270.65 H. J. Lee, J. S. Lee, T. Chansakul, C. Yu, J. H. Elisseeff and
S. M. Yu, Biomaterials, 2006, 27, 5268–5276.
66 A. Y. Wang, S. Leong, Y. C. Liang, R. C. C. Huang, C. S. Chenand S. M. Yu, Biomacromolecules, 2008, 9, 2929–2936.
67 L. S. Li and S. I. Stupp, Angew. Chem., Int. Ed., 2005, 44,1833–1836.
68 M. O. Guler, S. Soukasene, J. F. Hulvat and S. I. Stupp, NanoLett., 2005, 5, 249–252.
69 M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev.,2008, 108, 4935–4978.
70 L. L. del Mercato, P. Rivera-Gil, A. Z. Abbasi, M. Ochs,C. Ganas, I. Zins, C. Sonnichsen and W. J. Parak, Nanoscale,2010, 2, 458–467.
71 D. J. Evans, Inorg. Chim. Acta, 2010, 363, 1070–1076.72 M. Reches and E. Gazit, Science, 2003, 300, 625–627.73 N. Amdursky, M. Molotskii, D. Aronov, L. Adler-Abramovich,
E. Gazit and G. Rosenman, Nano Lett., 2009, 9, 3111–3115.74 E. Kasotakis, E. Mossou, L. Adler-Abramovich, E. P. Mitchell,
V. T. Forsyth, E. Gazit and A. Mitraki, Biopolymers, 2009, 92,164–172.
75 N. Ostrov and E. Gazit, Angew. Chem., Int. Ed., 2010, 49,3018–3021.
76 T. Scheibel, R. Parthasarathy, G. Sawicki, X. M. Lin, H. Jaegerand S. L. Lindquist, Proc. Natl. Acad. Sci. U. S. A., 2003, 100,4527–4532.
77 S. W. Lee, C. B. Mao, C. E. Flynn and A. M. Belcher, Science,2002, 296, 892–895.
78 K. T. Nam, Y. J. Lee, E. M. Krauland, S. T. Kottmann andA. M. Belcher, ACS Nano, 2008, 2, 1480–1486.
79 Y. Huang, C. Y. Chiang, S. K. Lee, Y. Gao, E. L. Hu, J. DeYoreo and A. M. Belcher, Nano Lett., 2005, 5, 1429–1434.
80 Y. J. Lee, H. Yi, W. J. Kim, K. Kang, D. S. Yun, M. S. Strano,G. Ceder and A. M. Belcher, Science, 2009, 324, 1051–1055.
81 Y. S. Nam, T. Shin, H. Park, A. P. Magyar, K. Choi, G. Fantner,K. A. Nelson and A. M. Belcher, J. Am. Chem. Soc., 2010, 132,1462–1463.
82 B. Neltner, B. Peddie, A. Xu, W. Doenlen, K. Durand, D. S. Yun,S. Speakman, A. Peterson and A. Belcher, ACS Nano, 2010, 4,3227–3235.
83 Y. J. Lee, Y. Lee, D. Oh, T. Chen, G. Ceder and A. M. Belcher,Nano Lett., 2010, 10, 2433–2440.
84 C. Y. Chiang, C. M. Mello, J. J. Gu, E. Silva, K. J. Van Vliet andA. M. Belcher, Adv. Mater., 2007, 19, 826–832.
85 O. Carny, D. E. Shalev and E. Gazit, Nano Lett., 2006, 6,1594–1597.
86 N. Ostrov and E. Gazit, Angew. Chem., Int. Ed., 2010, 49,3018–3021.
87 D. Gottlieb, S. A. Morin, S. Jin and R. T. Raines, J. Mater.Chem., 2008, 18, 3865–3870.
88 L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke andS. I. Stupp, Chem. Rev., 2008, 108, 4754–4783.
89 E. D. Spoerke, S. G. Anthony and S. I. Stupp, Adv. Mater., 2009,21, 425–430.
90 H. W. Jun, V. Yuwono, S. E. Paramonov and J. D. Hartgerink,Adv. Mater., 2005, 17, 2612–2617.
91 J. Kirkham, A. Firth, D. Vernals, N. Boden, C. Robinson,R. C. Shore, S. J. Brookes and A. Aggeli, J. Dent. Res., 2007,86, 426–430.
92 A. M. Smith, E. F. Banwell, W. R. Edwards, M. J. Pandya andD. N. Woolfson, Adv. Funct. Mater., 2006, 16, 1022–1030.
93 D. Papapostolou, A. M. Smith, E. D. T. Atkins, S. J. Oliver,M. G. Ryadnov, L. C. Serpell and D. N. Woolfson, Proc. Natl.Acad. Sci. U. S. A., 2007, 104, 10853–10858.
94 E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall,A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell,M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600.
95 E. H. C. Bromley, K. J. Channon, P. J. S. King, Z. N. Mahmoud,E. F. Banwell, M. F. Butler, M. P. Crump, T. R. Dafforn,M. R. Hicks, J. D. Hirst, A. Rodger and D. N. Woolfson,Biophys. J., 2010, 98, 1668–1676.
96 M. G. Ryadnov and D. N. Woolfson, Angew. Chem., Int. Ed.,2003, 42, 3021–3023.
97 M. G. Ryadnov and D. N.Woolfson,Nat.Mater., 2003, 2, 329–332.98 M. G. Ryadnov and D. N. Woolfson, J. Am. Chem. Soc., 2005,
127, 12407–12415.99 M. G. Ryadnov and D. N. Woolfson, J. Am. Chem. Soc., 2004,
126, 7454–7455.
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online

This journal is c The Royal Society of Chemistry 2010 Chem. Soc. Rev., 2010, 39, 3464–3479 3479
100 A. M. Smith, S. F. A. Acquah, N. Bone, H. W. Kroto,M. G. Ryadnov, M. S. P. Stevens, D. R. M. Walton andD. N. Woolfson, Angew. Chem., Int. Ed., 2005, 44, 325–328.
101 S. C. Holmstrom, P. J. S. King, M. G. Ryadnov, M. F.Butler, S. Mann and D. N. Woolfson, Langmuir, 2008, 24,11778–11783.
102 J. E. Meegan, A. Aggeli, N. Boden, R. Brydson, A. P. Brown,L. Carrick, A. R. Brough, A. Hussain and R. J. Ansell, Adv.Funct. Mater., 2004, 14, 31–37.
103 V. M. Yuwono and J. D. Hartgerink, Langmuir, 2007, 23,5033–5038.
104 S. F. M. van Dongen, R. L. M. Teeuwen, M. Nallani, S. S. vanBerkel, J. Cornelissen, R. J. M. Nolte and J. C. M. van Hest,Bioconjugate Chem., 2009, 20, 20–23.
105 D. E. Wagner, C. L. Phillips, W. M. Ali, G. E. Nybakken,E. D. Crawford, A. D. Schwab, W. F. Smith and R. Fairman,Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12656–12661.
106 V. Villard, O. Kalyuzhniy, O. Riccio, S. Potekhin, T. N. Melnik,A. V. Kajava, C. Ruegg and G. Corradin, J. Pept. Sci., 2006, 12,206–212.
107 S. N. Dublin and V. P. Conticello, J. Am. Chem. Soc., 2008, 130,49–51.
Publ
ishe
d on
02
Aug
ust 2
010.
Dow
nloa
ded
by U
nive
rsity
of
Bri
stol
on
12/1
0/20
15 1
4:30
:42.
View Article Online


















