
Glycoconjugates - DiVA portal144063/FULLTEXT01.pdf · 2005-11-16 · Glycoconjugates Gal galactose...
78
Glycoconjugates Solid-phase synthesis and biological applications Fredrik Wallner Akademisk avhandling Som med vederbörligt tillstånd av rektorsämbetet vid Umeå uni- versitet för avläggande av filosofie doktorsexamen framläggs till of- fentligt försvar i Stora hörsalen, KB3B1, KBC, lördagen den 10 de- cember 2005, kl. 10.00. Avhandlingen kommer att försvaras på sven- ska. Fakultetsopponent: Docent Ulf Ellervik, Organisk kemi, Lunds uni- versitet.
Transcript of Glycoconjugates - DiVA portal144063/FULLTEXT01.pdf · 2005-11-16 · Glycoconjugates Gal galactose...

Glycoconjugates
Solid-phase synthesis and biologicalapplications
Fredrik Wallner
Akademisk avhandling
Som med vederbörligt tillstånd av rektorsämbetet vid Umeå uni-versitet för avläggande av filosofie doktorsexamen framläggs till of-fentligt försvar i Stora hörsalen, KB3B1, KBC, lördagen den 10 de-cember 2005, kl. 10.00. Avhandlingen kommer att försvaras på sven-ska.
Fakultetsopponent: Docent Ulf Ellervik, Organisk kemi, Lunds uni-versitet.

Department of Chemistry, Organic ChemistryUmeå UniversitySE-901 87 Umeå,Sweden
c© 2005 Fredrik WallnerISBN: 91-7305-984-6
Printed in Sweden by Solfjädern Offset AB, Umeå 2005

TitleGlycoconjugates – Solid-phase synthesis and biological applications
AuthorFredrik Wallner, Department of Chemistry, Organic Chemistry, Umeå University, SE-901 87 Umeå,Sweden.
AbstractGlycoconjugates are biologically important molecules with diverse functions. They consist of carbohydra-tes of varying size and complexity, attached to a non-sugar moiety as a lipid or a protein. Glycoconjugatestructures are often very complex and their intricate biosynthetic pathways makes overexpression difficult.This renders the isolation of pure, structurally defined compounds from natural sources cumbersome.Therefore, to better address questions in glycobiology, synthetic glycoconjugates are an appealing alter-native. In addition, synthetic methods allow for the preparation of non-natural glycoconjugates that canenhance the understanding of the influence of structural features on the biological responses.
In this thesis, synthetic methods for the preparation of glycoconjugates, especially glycolipid ana-logues, have been developed. These methods make use of solid-phase chemistry and are amenable tolibrary synthesis of series of similar compounds. Solid-phase synthesis is a technique where the startingmaterial of the reaction is attached to small plastic beads through a linker. This allows large excess ofreagents to speed up the reactions and the sometimes difficult purifications of intermediate products arereduced to simple washings of the beads.
One problem with solid-phase synthesis is the difficulties to monitor the reactions and characterize theintermediate products. Gel-phase 19F-NMR spectroscopy, using fluorinated linkers and protecting groups,is an excellent tool to overcome this problem and to monitor solid-phase synthesis of e.g. glycoconjugates.Two novel fluorinated linkers for the attachment of carboxylic acids have been developed and are presentedin the thesis. These linkers can be cleaved with both acids of varying strengths and nucleophiles likehydroxide ions, and they are stable to glycosylation conditions. In addition, a novel filter reactor forsolid-phase synthesis was designed. The reactor fits into an ordinary NMR spectrometer to facilitate thereaction monitoring with gel-phase 19F-NMR spectroscopy.
The biological applications of the synthesized glycolipids were demonstrated in two different settings.The CD1d restricted binding of glycolipids carrying the monosaccharide α-GalNAc as carbohydrate couldbe detected on viable cells of mouse origin. CD1d is one of several antigen presenting molecules (theCD1 proteins) that presents lipids and glycolipids to circulating T-cells that in turn can initiate an immuneresponse. The CD1 molecules are relatively sparsely investigated, and the method to measure glycolipidbinding on viable cells, as described in the thesis, has the possibility to greatly enhance the knowledge ofthe structural requirements for CD1-binding.
Serine-based neoglycolipids with terminal carboxylic acids were used to prepare glycoconjugate arrayswith covalent bonds to secondary amines on microtiter plates. Carbohydrate arrays have great possibilitiesto simplify the study of interactions between carbohydrates and e.g. proteins and microbes. The usefulnessof the glycolipid arrays constructed in the thesis was illustrated with two lectins, RCA120 from Ricinuscommunis and BS-1 from Bandeiraea simplicifolia. Both lectins bound to the array of neoglycolipids inagreement with their respective specificity for galactosides.
Glycobiology is a large area of great interest and the methods described in this thesis can be used toanswer a variety of glycoconjugate-related biological questions.
KeywordsGlycoconjugate, Glycolipid, Solid-phase synthesis, Glycosylation, Gel-phase 19F-NMR spectroscopy,Fluorinated linker, Carbohydrate array, CD1
ISBN: 91-7305-984-6

To Anna and Thea...

Solid-phase synthesis and biological applications
List of Papers
This thesis is based on the following papers, which are referred to inthe text by their Roman numerals.
I Wallner, F. K., Chen, L., Moliner, A., Jondal, M., and Elof-sson, M. (2004) Loading of the Antigen-Presenting ProteinCD1d with Synthetic Glycolipids. ChemBioChem, 5:437-444.
II Wallner, F. K., Norberg, H. A., Johansson, A. I., Moge-mark, M., and Elofsson, M. (2005) Solid-phase synthesis ofserine-based glycosphingolipid analogues for preparationof glycoconjugate arrays. Organic & Biomolecular Chemistry,3:309-315.
III Wallner, F. K., Spjut, S., Boström, D., and Elofsson M.(2005) A Novel, Acid-Labile, Fluorinated Linker for Solid-Phase Organic Chemistry. Manuscript.
IV Wallner, F. K. and Elofsson M. (2005) An NMR Tube FilterReactor for Solid-Phase Synthesis and Gel-Phase 19F-NMRSpectroscopy. Submitted.
Reprints were made with kind permission from the publishers.
- v -

Solid-phase synthesis and biological applications
List of abbreviations
aq. aqueous
atm atmosphere
BH3·DMS borane dimethyl sulfide complex
BS-1 Bandeiraea simplicifolia lectin
BSA bovine serum albumin
CD1 cluster of differentiation 1
cDNA complementary deoxyribonucleic acid
DBU 1,8-diazabicyclo[5.4.0]undec-7-ene
DC dendritic cell
DIC N,N’-diisopropyl carbodiimide
DMAP N,N-dimethylaminopyridine
DMF N,N-dimethylformamide
DMSO dimethylsulfoxide
EDC 1-(3-dimethylaminopropyl)-3-ethylcarbodiimidehydrochloride
FACS fluorescent-activated cell sorting
FITC fluorescein isothiocyanate
Fmoc 9-fluorenylmethoxycarbonyl
Fmoc-Gly-OH N-Fmoc-glycine
Fmoc-p-F-Phe-OH N-Fmoc-para-fluoro-phenylalanine
- vii -

Glycoconjugates
Gal galactose
GalNAc N-acetyl-galactosamine
HIV human immunodeficiency virus
HOAt 1-hydroxy-7-azabenzotriazole
HOBt 1-hydroxybenzotriazole
HRP horseradish peroxidase
LC/MS liquid chromatography, mass spectrometry
MeIm N-methyl imidazole
MFI mean fluorescence intensity
mFPh meta-fluorophenyl
MHC Major Histocompatibility Complex
MSNT 1-(2-mesitylenesulfonyl)-3-nitro-1H-1,2,4-triazole
NHS N-hydroxysuccinimide
NIS N-iodosuccinimide
NMR nuclear magnetic resonance
oFBn orto-fluorobenzyl
PBS phosphate buffered saline
pFBz para-fluorobenzoyl
RCA120 Ricinus communis agglutinin
SDS sodium dodecyl sulfate
TFA trifluoroacetic acid
TfOH trifluoromethane sulphonic acid
THF tetrahydrofuran
TLC thin-layer chromatography
- viii -

Solid-phase synthesis and biological applications
Contents
1 Glycoconjugates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11.1 Glycolipids . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
1.1.1 CD1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41.2 Glycoconjugate synthesis . . . . . . . . . . . . . . . . . . . . . . . . . 6
2 Solid-phase synthesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92.1 Gel-phase 19F-NMR spectroscopy . . . . . . . . . . . . . . . . . . . 92.2 Linkers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
3 Carbohydrate arrays . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 134 Aims of the thesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 175 Glycolipid synthesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
5.1 Synthesis of α-N-acetyl-galactosamine glycolipids (pa-per I) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
5.2 Solid-phase glycosylations in glycolipid synthesis (pa-per II) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
6 Glycolipid loading of CD1d (paper I) . . . . . . . . . . . . . . . . . . . 277 Preparation of glycolipid arrays (paper II) . . . . . . . . . . . . . . 338 Design, synthesis, and evaluation of fluorinated linkers . . . 37
8.1 Synthesis of a new fluorinated monoalkoxy linker (pa-per II) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
8.2 Design, synthesis, and evaluation of a fluorinated di-alkoxy linker (paper III) . . . . . . . . . . . . . . . . . . . . . . . . . . 38
9 Design of an NMR tube filter reactor for solid-phase synthesisand gel-phase 19F-NMR spectroscopy (paper IV) . . . . . . . . . . 47
10 Concluding remarks and future perspectives . . . . . . . . . . . . 5111 Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
- ix -


Solid-phase synthesis and biological applications
1 Glycoconjugates
Glycoconjugates are carbohydrates covalently linked to a non-sugarmoiety such as a lipid or protein. They are often found on the outsideof cell membranes and are for example involved in specific recogni-tion events between cells.
Oligosaccharides, together with nucleic acids and proteins consti-tute the three major biopolymers. While nucleic acids and proteinsare linear polymers, oligosaccharides are often branched in theirstructure.1 Each monosaccharide has several hydroxyl groups thatother monosaccharides can be coupled to and the glycosidic link-age can have two different stereoisomers, the α and β configura-tions. In addition, each monosaccharide can exist in two differentring forms, the furanose and the pyranose, and have further sub-stituents on any of the hydroxyls. Together, this means that thepossible ways for two monosaccharides to connect are numerous (seefigure 1.1) and, consequently, large amounts of biological informa-tion can be encoded in a rather short saccharide.2 For example, over38,000 different trisaccharides can be formed out of three D-hexoses.3Despite this capacity for information storage, oligosaccharides werelong thought to act only as a physical defense, energy storage, orto change the physical properties of proteins and lipids. Today, itis known that carbohydrate-protein and carbohydrate-carbohydrateinteractions are important in many biological processes like devel-opment,4 immune responses,5 and microbial infections6 as well asprotein folding.1
While the biosynthesis of proteins and nucleic acids follows a tem-plate, the assembly of the oligosaccharides is more complex and de-pends on which glycosylating enzymes that are active in the cell andif they are capable of glycosylating the substrate.7 These enzyme re-actions do not always reach completion, resulting in a set of oligosac-charides instead of a single isomer. Two copies of the same protein,synthesized in the same cell, may have somewhat different glycosy-lation patterns. These proteins are referred to as two glycoforms8
- 1 -

Glycoconjugates
OHOH
OHO
α-D-galactopyranose β-D-galactopyranose α-D-galactofuranose
β-Galabioseα-D-galactopyranosyl-(1-4)-β-D-galactopyranose
a)
b)
OHOH
O
OH
HOHO
OHHO
OHO
HO
OHHO
OH
OHO
HO
OH
OH
O
O
HOHO
OHHO
Figure 1.1: The combinatorial possibilities of saccharides. a) Three isomers ofD-galactose, with the possible attachment points marked on the α-pyranose. b)β -Galabiose, one of the 73 possible disaccharides that can be formed from twoD-galactose units.
and even though there are several glycoforms of a protein in a givencell, the set of glycoforms, or the glycan profile, is often very charac-teristic for a cell type and/or condition.7 It is well established thatthe glycosylation pattern of a cell changes with the development ofcancer and inflammation, with the first examples as early as 1968.9These changes can be used to diagnose or treat the disease. Severalexamples exist where immunizations with glycoconjugates are usedin tumor immunotherapy and different vaccines are in phase I to IIIclinical trials.7 Here, one of the large obstacles is that the individ-ual glycans are endogenous and often exist also in embryonal stagesand/or in low amounts in normal tissue. However, the specific glyco-sylation patterns with elevated or reduced levels of certain glycansare different between healthy cells and cancer cells.7 On the otherhand, microbial pathogens often have completely different glycansthan humans and carbohydrate based vaccines are used against sev-
- 2 -

Solid-phase synthesis and biological applications
eral pathogens and much efforts are made on many more.10 As aresult of the difference in carbohydrate structures of humans andpathogens, an immune response raised by a vaccine against a micro-bial pathogen is more likely to be specific than a cancer vaccine.
The information in oligosaccharides is often decoded by lectins,carbohydrate recognizing proteins other than antibodies andenzymes,11 but also antibodies4 and T-cells12 can recognize specificcarbohydrate structures. The interaction between proteins andcarbohydrates is mostly very week and multiple simultaneousbinding events are required for a biological response.13 Thisphenomenon, called multivalency, has a number of physiologicaladvantages such as a graded response,14 context-dependentinteractions,4 fast on/off rates,15 and resistance to shear stress.13 Atthe same time, it complicates the use of small molecules to interferewith, and study, the interactions and therefore multivalent ligandsare increasingly used.13
1.1 Glycolipids
Glycoconjugates where the carbohydrate is attached to a lipid arecalled glycolipids. The combination of a polar carbohydrate and anon-polar lipid makes glycolipids very amphiphilic and they are al-most always found in cell membranes, preferably on the outside.16
In animals, the majority of glycolipids are glycosphingolipids, wherethe carbohydrate is attached to a sphingoid base that is acylatedwith a fatty acid16 (see figure 1.2a). The carbohydrate parts of theglycosphingolipids are however very diverse, and more than 200 nat-ural glycosphingolipids have been identified.17 Lower organisms, e.g.bacteria and protozoa, have a more heterogeneous collection of gly-colipids, unique both in the carbohydrate and the lipid parts (figure1.2b),16, 18 although the total amounts are not as high.
The glycolipids have similar biological responses as the other gly-coconjugates. The placement of their carbohydrate in close proximityto the cell membrane has led to a common use as attachment pointsfor microbes such as uropathogenic E. coli19 and HIV.20
- 3 -

Glycoconjugates
O
OHO
HN
OH
O
a)
b)
OO
HOHO
OH
O
OH
HOHO
O
OH
OH
Figure 1.2: Examples of glycolipids. a) β -Glucosyl-ceramide, a glycosphin-golipid. b) A stereoisomer of glucose monomycolate IIa, a glycolipid frommycobacteria.
1.1.1 CD1While the presentation of protein and peptide antigens to T-cellsthrough Major Histocompatibility Complex (MHC) molecules is wellstudied,21 the T-cell recognition of lipids and glycolipids was recentlydiscovered.22, 23 This recognition is moderated by the CD1 molecules,a class of antigen-presenting molecules with structural similaritiesto MHC class I molecules.24 The CD1 proteins have a hydropho-bic groove with deep pockets well suited to bind lipids. The polarheads of the lipids protrude from the protein to be recognized by aT-cell (figure 1.3).25–30 The human CD1 proteins can be divided intotwo groups depending on the sequence similarities. Group 1 con-sists of CD1a, b, and c, while CD1d is the only member of group 231
and CD1e seem to be intermediate or possibly a third group.32 Bothgroup 1 and 2 CD molecules participate in both the innate,33, 34 ornatural, and the adaptive,33, 35 or acquired, immune systems. Group2 is however reported to be involved in an earlier stage of the im-mune response.36
- 4 -

Solid-phase synthesis and biological applications
Figure 1.3: Schematic drawing of CD1 presentation of a glycolipid to a T-cell.
The different CD1 molecules have somewhat different binding mo-tifs, regarding the lengths and number of the lipid chains. Althoughall four studied CD1 proteins (a-d) can bind the self glycolipid sul-fatide with two lipid chains of about 15-30 carbons each,37, 38 CD1calso presents single chained glycolipids39 and CD1b is capable ofbinding very long-chained lipids like mycolic acid with up to 80 car-bons in one chain27 and possibly also lipids with more than twochains.25 In addition to their somewhat different binding motifs,the CD1 molecules have diverse trafficking pathways in the cell tosurvey a larger variety of lipids.40
In recent years, much knowledge has been gained about the roleof CD1 molecules in the immune system. Still, our understandingof CD1-restricted immunity is far from complete and considerableefforts are made and needed in this area.
- 5 -

Glycoconjugates
1.2 Glycoconjugate synthesisThe presence of several glycoforms and glycolipids of similar, butnot identical, composition in biological samples makes isolation ofpure, structurally defined glycoconjugates cumbersome.10 This im-poses a need for synthetic glycoconjugates and neoglycoconjugates.The large variability of glycans also makes their synthesis complexand steps need to be taken to ensure the regio- and stereoselectiv-ity of the reactions.41 In chemical synthesis of saccharides the se-lectivity is mostly controlled via protecting groups. Masking of thehydroxyls that should not react results in regioselectivity and thecorrect protecting group can also give stereoselectivity in the glyco-side formation.41, 42 As an example, the two galactose units 1 and 2will form the β -disaccharide 3 while galactoside 4, where the esterat O-2 in 1 is changed to an ether, will mainly react with 2 to formα-disaccharide 5 (see scheme 1.1). The protecting groups will also
OSPhMeBnO
BnO
OBnBnO
1 2
NISTfOHO
SPhMeBnOBzO
OBnBnO
OOBnBnO
BnO
OBnHO
OOBnBnO
BnO
OBnOOBnO
BzO
OBnBnO
OOBnBnO
BnO
OBn
O
O
BnOBnO
OBnBnO
+
3
4 2
NISTfOHO
OBnBnOBnO
OBnHO
+
5
Scheme 1.1: Protecting groups in glycosylations
have an effect on the reactivity of the saccharides,43 where electronwithdrawing groups deactivate both donors and acceptors, making 4more reactive than 1. In addition, the stereoselectivity and the reac-tivity can be influenced by changes in temperature and solvents.10
Carbohydrate chemistry has received interest for over 100 yearsbut, in spite of recent advances, glycoconjugate synthesis is still a dif-
- 6 -

Solid-phase synthesis and biological applications
ficult and time-consuming process. The preparation of starting ma-terials often require numerous steps and considerable efforts have tobe made on optimizations of glycosylation conditions.
- 7 -


Solid-phase synthesis and biological applications
2 Solid-phase synthesis
Since the pioneering work of Merrifield,44–47 solid-phase organic syn-thesis has become a subject of great interest. By attaching the start-ing material onto small plastic beads, two big advantages are ob-tained. The reactions can be sped up and brought to completion withthe use of relatively large excess of reactants and the purificationof intermediates is reduced to a simple filtering and washing proce-dure, where unreacted reactants and reagents can be removed fromthe resin-bound product.5 However, the attachment to the beads alsomakes it difficult to analyze the intermediates and monitor the re-actions.5 For peptides, simple methods have been devised to mon-itor the progress of the reaction,48 and their relatively pure reac-tion chemistry, with few byproducts, diminish the need to analyzeresin-bound intermediates. This has made solid-phase synthesis astandard method for peptides as well as oligonucleotides and auto-mated synthesizers have been developed.48, 49 Commercially avail-able building blocks can thus be combined in a routine fashion, andsynthetic peptides and oligonucleotides have been important in an-swering a wide range of biological questions. For more complicatedmolecules, like oligosaccharides, the lack of reaction monitoring andanalysis techniques has hampered the development.5 Lately, sometechniques for on-resin analysis have been developed and efficientmethods for glycosylations have even made automated solid-phasesynthesis of carbohydrates possible.50 However, since different gly-cosylations behave differently,43 detailed optimizations of reactionconditions, often in solution,51 are still required, and solid-phaseoligosaccharide synthesis is far from routine (see section 1.2).
2.1 Gel-phase 19F-NMR spectroscopyOne good method to monitor solid-phase reactions is gel-phase 19F-NMR spectroscopy. 19F has several desirable properties as a nucleusfor gel-phase NMR spectroscopy.52 It has a high sensitivity and 100%
- 9 -

Glycoconjugates
natural abundancy,53 leading to a good signal to noise ratio in a shortacquisition time. The large polarizability of the fluorine atom leadsto a large distribution of the chemical shifts reducing peak overlap-ping.53 Gel-phase NMR spectra suffer from rather broad signalsdue to highly hindered motion and dipolar couplings and the largechemical shift dispersion is essential to receive detailed informationfrom the spectra.54 In addition, fluorine is not present in solid-phaseresins for supported synthesis and hence no interfering signals willarrive from the resin.
These properties have led to the use of 19F-NMR spectroscopy tomonitor the progress of solid-phase reactions with the beginningin 1980.55 Later it was found that TentaGel, with its flexiblepoly(ethylene glycol) spacers, gives rise to spectra of highresolution.56 This meant that also small chemical shift changescan be monitored, and byproducts such as diastereomers can bequantified.57 The influence of flexible spacers on the spectra qualitywas recently confirmed by a thorough study of the influence ofresins and solvents in gel-phase 19F-NMR spectroscopy.58
To be able to use 19F-NMR spectroscopy, fluorine atoms must bepresent in the reactions. This can be accomplished with a fluori-nated linker, protecting groups, reactants or a combination of thethree. The strategy based on fluorinated protecting groups is espe-cially well suited for carbohydrate synthesis where protecting groupsare essential (see section 1.2). Fluorinated versions of several com-mon protecting groups are commercially available and their use insolid-phase carbohydrate synthesis in combination with fluorinatedlinkers have been demonstrated.57–61
In addition, fluorinated molecules have received an increased in-terest in medicinal chemistry during the last two decades.62 Theinsertion of a fluorine atom into a molecule influences its reactiv-ity,63 metabolic stability, physiochemical and conformational prop-erties, and protein binding.62 The presence of a fluorine atom alsopermits 19F-NMR spectroscopy to be used to study binding interac-tions of small molecules to proteins.53, 64–67 The combined use of 19F-NMR spectroscopy in solid-phase synthesis of libraries of fluorinatedmolecules and screening has however not been well explored.
- 10 -

Solid-phase synthesis and biological applications
2.2 LinkersSolid-phase synthesis typically requires the use of a linker throughwhich the starting material can be attached to the resin andfrom which the product later can be cleaved. Such a linker needsto be stable during the reaction conditions and cleavable underconditions that does not affect the product. A large variety of linkershave been devised throughout the years68, 69 since Merrifield’schloromethylated nitro-benzyl linker44 (for examples, see figure2.1). Essentially all protecting groups can be adopted to be a linker,and linkers exist that are cleaved by for example acid, base, light,reduction, and oxidation, some during very mild conditions whileothers require a more harsh treatment. Among the acid sensitivelinkers, it is common to make use of the resonance stabilization ofbenzylic cations. The original papers44, 45 by Merrifield discussedthis point and also described how to increase the acid stabilitythrough electron withdrawing groups. Wang later took advantage ofthe opposite when he created a more acid labile linker by includinga phenol ether in the Merrifield linker70 (see figure 2.1, compound7). This linker, later named the Wang linker, has become thestandard linker for the synthesis of peptide acids and is also used ina large variety of other solid supported organic reactions.68 Additionof more electron donating or resonance stabilizing groups can ina similar way further increase the acid lability, as in the SASRINlinker 8 (figure 2.1).
HOO
HO
O
HO
OMe
6Merrifield resincleaved with HF
7Wang linker
cleaved with 50% TFA
8SASRIN linker
cleaved with 1% TFA
Figure 2.1: Three linkers used to immobilize carboxylic acids and examples oftheir respective cleavage methods.68
Incorporation of a fluorine atom into the linker makes it usefulfor 19F-NMR spectroscopy.71 It can be used as an internal standardand is often sensitive enough to give direct information about subse-
- 11 -

Glycoconjugates
quent reactions, such as linker loading and product cleavage. Fluori-nated linkers described in the literature include fluorinated versionsof several commonly used linkers that can be cleaved with acid,71–74
nucleophiles,57–60, 71–76 oxidation,61 and metal assisted chemistry,77, 78
as well as a linker that relies on more product specific cleavage condi-tions79 (for examples, see figure 2.2). Since fluorine is electron with-
O
HO
TFA/CH2Cl2, 3:7room temperatureF
O
HN
O
HO TFA/water/thioanisole/ethanedithiol87.5:5:5:2.5, 60°C
orLiOH 1 M in THF:MeOH:water, 3:1:1
0°C - room temperatureF
O
HN
O
LiOH, 40 mM in THF/water, 3:1room temperatureF
HO4
9
10
11
Figure 2.2: Three fluorinated linkers used to immobilize carboxylic acids andexamples of their respective cleavage methods.57, 72, 73
drawing, the fluorinated versions of the Wang linker require moreharsh conditions for acidic cleavage than the original.72
Gel-phase 19F-NMR spectroscopy has been demonstrated as a use-ful technique to monitor solid-phase synthesis on fluorinated linkers.Several such linkers, with a broad panel of cleavage conditions, havebeen devised, derivatized, and used, e.g. in the synthesis of smallmolecule libraries and oligosaccharides.
- 12 -

Solid-phase synthesis and biological applications
3 Carbohydrate arrays
Nucleotide microarrays are important tools in the post-genomic pro-cess to analyze gene transcription. On one glass plate, the tran-scripts from the whole human genome can be analyzed from a smallsample.80 In addition, protein microarrays are increasingly usedto study protein interactions with other proteins, nucleic acids andsmall molecules.81 The large structural variations in oligosaccha-rides and glycoconjugates (see chapter 1), together with the relativedifficulty to access large amounts of pure glycans make carbohydratemicroarrays a promising technique in glycobiology.82
Increasing efforts are made to construct carbohydrate arrays andboth commercial83–85 and non-commercial86 arrays are now avail-able. In general, a carbohydrate array is constructed by attachingoligosaccharides or glycoconjugates in an ordered fashion to a solidsurface (see figure 3.1). To this carbohydrate array another sub-stance, such as a lectin or an antibody, can be added and its bindingcan be measured, usually by a fluorescent marker or a colorimetricmethod. By indirect measurement, the effect of enzymes or smallmolecule inhibition of a binding can also be studied. In addition,viable cells or bacteria can bind to the array for analysis.87
To demonstrate the usefulness of carbohydrate arrays severalgroups have prepared arrays with a multitude of attachmenttechniques. In 1992, Shao biotinylated glycans derived fromovalbumin and added them to microtiter plates, precoated withstreptavidin.88 The formed glycan array was used to study bindingspecificities of lectins. Streptavidin coated microtiter plates andbiotin conjugated carbohydrates have later for example been usedto investigate the influence of the linker on carbohydrate-proteininteractions89 and the specificity of cell receptors related to HIVinfections.86 In addition, other non-covalent linkages have beendemonstrated. Neoglycolipids, glycolipids, and polysaccharideshave been adsorbed on microtiter plates,90, 91 nitrocellulose,92–94 andmodified plastic95 and glass slides.96 A variation with synthetic
- 13 -

Glycoconjugates
O
O O
O OO O
O
O O
O
O O
O
O
Plate
Carbohydratearray
O O
Lectin bindingstudy
Inhibitorscreening
Enzyme specificitystudy
Bacterialanalysis
O
Labeled lectin
Glycosylatingenzyme
Glycosylatingsubstrate
Microbe
Inhibitor oflectin binding
Modifiedcarbohydrate array
Figure 3.1: Carbohydrate array construction and applications.
- 14 -

Solid-phase synthesis and biological applications
fluorous carbohydrates on modified glass slides was recentlyreported.97 As an interesting extension of the non-covalent methods,several reports have been made on the use of simple chemistry tocreate neoglycolipids directly in microtiter plates from modifiedsaccharides and reactive lipids coated in the wells.98–101
The exclusivity of the glycans makes reusable arrays desirable. Toaccomplish that, and to increase the general stability of the array,covalent attachment of the carbohydrate is an interesting option. Inaddition, almost all covalent methods result in a site-specific bind-ing of the glycan to the solid surface.87 This gives a higher con-trol over the actual saccharide epitope that is presented by the ar-ray and a higher signal to noise ratio. An increasing proportion ofthe prepared carbohydrate arrays uses a covalent attachment to mi-crotiter plates,102–106 modified glass slides,83, 84, 107–121 or fiber opticmicrospheres.122 Among the bond forming reactions are amide for-mations, reductive aminations, Diels-Alder reactions, 1,3-dipolar cy-cloadditions and more. Most of the reactions rely on specific groupspresent on the glycan, imposing the need of synthetic compoundsor derivatization of natural carbohydrates, often with destruction ofthe monosaccharide at the reducing end. An interesting exceptionis the use of hydrazide or aminoxy derivatized glass slides to formglycosylamines directly from reducing saccharides.121
In the construction of carbohydrate arrays, the glycan density isof importance. It should be high enough to allow multivalent pro-tein carbohydrate interactions. It is however also reported that atoo high density can diminish binding, probably due to steric inter-actions.108 Low non-specific binding of proteins to the array is also ofgreat significance to decrease the noise in the measurements.123
Even though carbohydrate arrays have the possibility to displayup to tens of thousands of glycans110 most reports so far use con-siderably fewer different glycans to demonstrate the utility of thearray format and the immobilization method. Ligand profiling ofseveral proteins capable of binding the HIV protein gp120,86, 106, 114
as well as other proteins involved in the immune system,94 has beenreported by several groups. One interesting report found that theligand specificity of a protein changed with the ligand density.107 Atlow surface density the lectin from Bauhinia purpurea bound prefer-ably to a thiol conjugate of the disaccharide β -galactosyl-(1-3)-β -N-acetyl-2-amino-2-deoxy-galactose (12 figure 3.2) while at high densi-
- 15 -

Glycoconjugates
12 13
OHO
HO
OHHO
OSRO
NH
OHHO
O
OHO
HO
OHHO
OSR
NH
OH
HO
O
O
Figure 3.2: Ligands to the lectin from Bauhinia purpurea.
ties, it preferred α-galactosyl-(1-3)-α-N-isovaleroyl-2-amino-2-deoxy-glucose (13). Angeloni et al. made a glycoprofiling of model cell ex-tracts of Caco-2 intestinal cells that were arrayed and profiled witha panel of lectins and could show changes in the glycan expressiondepending on differentiation and treatment with 3’-sialyllactose.119
Both prokaryotic116 and eukaryotic83 cells have been shown to ad-here to carbohydrate arrays. Detection and profiling of bacterialcarbohydrate binding could be useful as a mean to diagnose dis-eases.116 The interaction between carbohydrate arrays and enzymeshas been used to, among others, study the substrate specificity of β -1,4-galactosyltransferase,109 synthesize a tetrasaccharide on an ar-ray117 and screen for inhibitors to a α-1,3-fucosyltransferase.99 Alsoinhibitors to lectin carbohydrate interactions have been studied, e.g.by Houseman and Mrksich.109 A few studies are reported with morethan 100 carbohydrates and in one as much as 200 different glycanswere arrayed and used for ligand profiling of nine proteins as well ashuman serum and intact virus.115
Carbohydrate arrays have great potential answering questions inglycobiology. In spite of the public availability of such arrays, theyhave however not yet reached their full potential and the applica-tions are relatively few. Streamlined synthesis of both natural andnon-natural glycoconjugates and array preparation will improve theutility with arrays custom made to answer a specific question.
- 16 -

Solid-phase synthesis and biological applications
4 Aims of the thesis
Initially, we set out to study the CD1 binding of glycolipids, andtheir use in immunizations. The first goal was to establish syntheticmethods to use for preparation of glycolipid libraries and a methodto measure glycolipid binding to CD1d molecules on viable mousecells. With these methods at hand, the next steps would be to pre-pare the libraries, and develop cell free methods to measure CD1dbinding. Immunizations using glycolipids that bind well to CD1 andcarry different immunologically relevant carbohydrates would be aninteresting continuation.
However, after the first paper was finished, we changed focus to-wards preparation of glycoconjugate arrays to obtain a streamlinedprocess including synthesis, reaction monitoring, purification, andarray manufacturing. The goal was to develop synthetic methodsand equipment that allow synthesis of libraries of glycolipid ana-logues. The preparation and use of glycoconjugate arrays formedfrom these glycolipid analogues were also to be demonstrated.
- 17 -


Solid-phase synthesis and biological applications
5 Glycolipid synthesis
5.1 Synthesis of α-N-acetyl-galactosamine glycolipids(paper I)
In order to study glycolipid binding to CD1d molecules on viablemurine cells (see chapter 6), glycolipids with specific carbohydrateswere needed. The glycan should not be normally expressed on thecells in question, to allow detection with antibodies, and shouldpreferably be simple and easy to work with. In this light, we choseto work with α-N-acetyl-galactosamine (α-GalNAc, see figure 5.1).α-GalNAc is a common monosaccharide, present e.g. in mucins,where it is linked to serine or threonine. However, apart fromthe blood group determinant A, terminal α-GalNAcs are rare inhealthy humans. Some cancers are associated with an increase inthe levels of terminal α-GalNAc, and terminal α-GalNAc connectedto a serine or threonine is referred to as the Tn antigen. To confer
NH
O
m
14a, m=115a, m=5
O
O
HOAcHN
OHHO
NH
O
14b
O
O
HOAcHN
OHHO
Figure 5.1: α-GalNAc containing glycolipids.
- 19 -

Glycoconjugates
CD1d-binding, the commercial lipids 2-(n-hexadecyl)-stearic acidand palmitic acid were chosen to be attached to the α-GalNActhrough a spacer (see figure 5.1). 2-(n-hexadecyl)-stearic acid is ananalogue to the mycolic acids (see figure 1.2), that are known tobind to CD1 proteins. Two different lengths of the spacer were usedto ensure that the carbohydrate protruded enough from the CD1dmolecules to be recognized by monoclonal antibodies.
In 1893, Emil Fischer reported a way to synthesize glycosides of al-cohols by boiling the sugar, together with an acid, in the alcohol thatworks as both acceptor and solvent.124 This process leads to the ther-modynamically most stable glycoside, most often the α-pyranose.The protocol works very well for simple, liquid, low-boiling, and inex-pensive alcohols. In the case of α-GalNAc, more complex alcohols arefrequently glycosylated with 2-azido-2-deoxy-galactose donors, giv-ing the appropriate α-glycoside.125 Later, the azide is reduced to anamine, which in turn is acetylated. An example of a synthetic routeis shown in scheme 5.1.
16
2 steps
17
Glycosylation
18Reduction andacetylation
19
Deprotection
20
OAcO
OAcAcO
O
Br
AcON3
OAcAcO
O
OR
AcON3
OAcAcO
O
OR
AcOAcHN
OAcAcO
O
OR
HOAcHN
OHHO
Scheme 5.1: Example of a synthesis of α-GalNAc glycosides.
To simplify this synthesis, we aimed to use a modified Fischer syn-thesis, using an aprotic solvent and Fmoc-protected amino-alcoholsin modest excess. It was found that refluxing GalNAc with five equiv-alents of N-Fmoc-2-amino-ethanol and four equivalents of the Lewisacid boron trifluoride etherate in THF for 19 hours gave the wantedα-glycoside 22 in 34% yield (see scheme 5.2). The reaction couldalso be performed with four equivalents N-Fmoc-6-amino-hexanol
- 20 -

Solid-phase synthesis and biological applications
and 10% p-toluene sulfonic acid in THF/toluene, resulting in some-what lower yields of 23. Experiments were also made using mi-crowave assisted heating of the reactions and a small experimentaldesign resulted in conditions that looked promising according to an-alytical LC/MS on crude products. However, the isolated yields wereonly ∼15% and a hard, glassy solid was formed around the wallsof the reactors. Most probably the starting material decomposed orpolymerized under the high temperatures and acidic conditions. Thismethod was therefore abandoned. However, a recent report demon-strates successful microwave assisted Fischer glycosylations usingsimple alcohols.126
O
OHHO
AcHN
OHHO O
O
HOAcHN
OHHO
NHFmocm
O
O
HOAcHN
OHO
NHFmocm
O
O
HOAcHN
OHO
NHm R
O
O
O
HOAcHN
OHHO
NHm
R
O
a) b)
d)
c)
21 22, m=123, m=5
24, m=125, m=5
26, m=127, m=5
14, m=115, m=5
a, R=C(C16H33)2b, R=C15H31
Scheme 5.2: Synthesis of GalNAc-bearing lipids. a) Fmoc-ω-aminoalcohol,Lewis acid, aprotic solvent. b) 2-chlorotrityl chloride polystyrene resin, DMAP,CH2Cl2. c) i. Piperidine, 20% in DMF. ii. Palmitic acid or 2-(n-hexadecyl)-stearic acid, DIC, HOAt. d) 5% TFA in CH2Cl2.
The formed α-GalNAc glycosides were then to be attached to the
- 21 -

Glycoconjugates
fatty acids 2-(n-hexadecyl)-stearic acid and palmitic acid. Acyla-tion of the deprotected amines in solution turned out to be very dif-ficult, likely due to the different solubilities of the sugar and theacid. Instead the GalNAc derivatives 22 and 23 were attached toa polystyrene resin, derivatized with 2-chlorotrityl chloride. Theresin, glycoside and N,N-dimethylaminopyridine (DMAP) were dis-solved/suspended in CH2Cl2 and the mixture was stirred slowly atreflux for 24 hours. This yielded resins 24 and 25 with approximately0.2 mmol glycoside per gram according to Fmoc analysis. The gly-coside was most likely attached to the resin through the primary6-hydroxyl group. Deprotection of the amine using piperidine (20%in DMF) and acylation with palmitic acid or 2-(n-hexadecyl)-stearicacid yielded the resin-bound glycolipids 26 and 27 in a straightfor-ward manner. The formed glycolipids were cleaved from the solidsupport using 5% TFA in CH2Cl2 and, after chromatography, thepure lipids 14a, 14b, and 15a were obtained in 66%, 75%, and 30%yield respectively, based on Fmoc determination on resins 24 and 25.
Three glycolipids containing the monosaccharide α-N-acetyl-galactosamine were prepared in a short synthesis. The syntheticroute, based on the immobilized spacer glycosides 24 and 25, is wellsuited for library synthesis where the spacer and lipid parts can bevaried more extensively.
5.2 Solid-phase glycosylations in glycolipid synthesis(paper II)
Serine-based neoglycolipids (see 29-31, figure 5.2) are analoguesof glycosphingolipids like 28 and have been used in severalstudies.127–150 Glycosphingolipids are normally present in cellmembranes (see section 1.1) where the lipid tails are inserted intothe membrane and the polar head is exposed at the surface forrecognition events. The serine and its adjacent amide bonds mimicthe polar head of the ceramide and the serine-based neoglycolipidshave been shown to mimic the sphingolipid e.g. in inhibition ofHIV invasion,140, 141, 143 ceramide glycanase transglycosylations,132
and activation of β -glucocerebrosidase.146 There are also indicationsthat a serine-based glycolipid have similar bioactivities, althoughto a lesser degree, as the CD1d-binding α-galactosylceramideKR7000.149
- 22 -

Solid-phase synthesis and biological applications
HN
OH
O
OO
HO
HO
OH
OH
28
HN
O
O
HN
OH
OHO
HO
OH
OH
29, β, m=1, n=130, β, m=7, n=1131, α, m=1, n=1
On
mO
Figure 5.2: A β -galactosyl ceramide (28) and serine-based analogues 29-31.
To explore the possibilities for solid-phase glycosylations ofserine based ceramide analogues and preparation of glycoconjugatearrays of the formed neoglycolipids three different glycolipidswere synthesized (29-31, scheme 5.3). Linker 32 was attachedon ArgoGel R©-NH2 with N,N’-diisopropyl carbodiimide (DIC) and1-hydroxy-7-azabenzotriazole (HOAt) in DMF. The free alcoholof the linker resin 33 was esterified with an Fmoc-ω-amino acidwith the help of 1-(2-mesitylenesulfonyl)-3-nitro-1H-1,2,4-triazole(MSNT), and N-methyl imidazole (MeIm). Continued amideformations with Fmoc-serine and a single stranded fatty acidusing DIC and 1-hydroxybenzotriazole (HOBt) gave the solidsupported lipids 36 and 37. The amide formations were monitoredwith bromophenol blue to ensure near quantitative yields andthe esterfication was confirmed to be quantitative by gel-phase19F-NMR spectroscopy.
Investigations on the solid-phase glycosylation of the lipids 36and 37 were made with both α- and β -selective glycosylations usingthe previously described thioglycoside donors 4157 and 42.60 Thedonor 41 has a benzoate ester at the 2-hydroxyl group, enablingneighboring group participation (see section 1.2) which in turn leadsto good β -selectivity. The resins 36 and 37 were thus subjected tofour equivalents of 41 together with N-iodosuccinimide (NIS) and
- 23 -

Glycoconjugates
c)
b)
34, m=135, m=7
36, m=1, n=137, m=7, n=11
38, β, R=R'=pFBz, m=1, n=139, β, R=R'=pFBz, m=7, n=1140, α, R=oFBn, R'=mF-benzylidene, m=1, n=1
29, β, m=1, n=130, β, m=7, n=1131, α, m=1, n=1
e)
d)
OpFBzO
pFBzO
OpFBzpFBzO
SPhMe
OoFBnO
oFBnOSPhMe
OO
mFPh
41
42
a)HO
FO
OH
O32
HO
FO
NH-Argogel
O
=HO
F
33
O
FOFmocHN
mO
FOHN
mO
HN
O
nHO
O
FOHN
mO
HN
O
nO
RORO
OR'R'O
O
OH
OHN
mO
HN
O
nO
HOHO
OHHO
O
Scheme 5.3: Synthesis of serine-based glycolipids. a) ArgoGel R©-NH2 resin,DIC, HOAt. b) Fmoc-ω-amino acid, MSNT, MeIm. c) i. Piperidine 20% inDMF. ii. Fmoc-Ser-OH, DIC, HOBt. iii. Piperidine 20% in DMF. iv. Fattyacid, DIC, HOBt. d) 2 x 4 eq. 41 or 5 eq. 42, NIS, TfOH. e) i. TFA/water 9:1,60 ◦C. ii. NaOMe or i. LiOH. ii. H2 (g), Pd/C.
- 24 -

Solid-phase synthesis and biological applications
Entry Solvent Temperature (◦C) Yield (%) α/β -ratio1 CH2Cl2 -40 53 2:12 CH2Cl2 room temperature quant. 2:13 CH2Cl2/THF -40 45 2:14 CH2Cl2/THF room temperature quant. 4:1
Table 5.1: Investigation on α-selective glycosylations. Reactions were per-formed with 5 eq. of donor 42.
trifluoromethane sulfonic acid (TfOH). The mixture was shaken atroom temperature, protected from light, for 3.5 hours to give theglycolipids 38 and 39 in ∼50% yield. Repeating the reaction onceincreased the yield to ∼75%. The yields were determined from19F-NMR spectra that also showed β /α-selectivities greater than 5:1.The donor 42 lacks the participating group and should react to formmostly α-glycosides (see section 1.2). To improve the selectivity,α-glycosylations are often performed at low temperatures. Initially,this strategy was used for the formation of 40 using five equivalentsof donor 42, NIS, and TfOH. The reaction was performed in CH2Cl2at -40 ◦C with a yield of ∼50% that increased to ∼85% uponrepetition. The selectivity was rather modest (2:1) with a third,unknown product at equimolar ratios to the β -anomer. Solid-phasereactions are greatly simplified if performed at room temperatureand therefore the possibilities for α-selective glycosylations atroom temperature were investigated (see table 5.1). Repeating thereaction at room temperature made the yield quantitative after onesingle glycosylation but the selectivity remained the same. Ethersolvents are known to have an α-directing effect42 and running thereaction at room temperature in CH2Cl2/THF (1:1) increased theselectivity to 4:1. The third, unknown product was still formed inequimolar ratios to the β -anomer. At low temperature the effectof the ether solvent could not be detected. This investigation ofdifferent reaction conditions could be performed quickly and easily,thanks to the fluorinated linker and protecting groups. The higheryields of the α-glycolipid 40, compared to 38 and 39, are most likelydue to the higher reactivity of the more electron-rich donor 42compared to 41.43
The β -glycolipids were cleaved from the resin using trifluoroacetic
- 25 -

Glycoconjugates
acid (TFA) and water (9:1) at 60 ◦C. Debenzoylation produced theglycolipids 29 and 30 in 11% and 26% total yields respectively, basedon the initial loading capacity of the ArgoGel R© resin. One differencebetween the on-resin and the cleaved yields could be the presence oflow reactivity amino groups on the resin, lowering the effective load-ing. The yields are comparable to those obtained in other solid-phaseglycosylations of serine.58 Subjecting the resin bound glycolipid 40 tothe same cleavage conditions resulted in a complex product mixture,where the benzylidene group was cleaved and most likely the ben-zyls had migrated. Instead, the glycolipid 31 was formed from basicester hydrolysis with LiOH, followed by deprotection with catalytichydrogenation, in a total yield of 22%. Also the β -anomer of theprotected glycolipid was purified and its structure was confirmed byNMR spectroscopy. Unfortunately, the unknown product could notbe isolated from the cleavage mixture and remain elusive.
Three different serine-based glycolipids, analogues to glycosphin-golipids, were synthesized on solid-phase. Fluorinated protectinggroups and a fluorinated linker made reaction monitoring and on-resin analysis of diastereomeric ratios with 19F-NMR spectroscopypossible. Thanks to the on-resin analysis, the α-selectivity of one re-action could easily be improved, without the need for solution-phaseoptimizations or cleavage from the resin.
- 26 -

Solid-phase synthesis and biological applications
6 Glycolipid loading of CD1d (paper I)
The investigations of lipid binding to CD1 molecules (see section1.1.1) has so far mostly been based on T-cell recognition of glycolipid-CD1 complexes. In those methods, the obtained signal depends onboth the CD1 binding of the lipid and the T-cell response, leadingto a complex interpretation of the data. In addition they are badlysuited to measure non-specific binding of glycolipids in the cell mem-brane. Some studies have been made on lipid binding to solubleor surface-bound CD1 molecules using surface plasmon resonance,however predominantly with heavily modified lipids.151–153
As an alternative method to measure glycolipid binding to CD1molecules on viable cells the use of carbohydrate specific antibodies,selective for the glycolipid, was imagined. Five different glycolipids,14a, 14b, and 15a, prepared as described in section 5.1, and the pre-viously synthesized 43154 and 44,155 carrying three different carbohy-drates were used to develop this method (see figure 6.1). The studywas performed with CD1d-bearing mouse cells of different origins.The surfaces of these cells normally lack all the carbohydrate struc-tures used in the glycolipids, enabling the specific recognition withmonoclonal antibodies (data not shown). Viable cells were incubatedwith the glycolipids in PBS buffer containing 5% DMSO and, afterwashing, with monoclonal antibodies specific for the carbohydrate orCD1d. Secondary antibodies labeled with fluorescein isothiocyanate(FITC) and fluorescent-activated cell sorting (FACS) were used toquantify the monoclonal antibodies. A dose-response relationshipfor the glycolipid loading of mouse EL-4 cells, carrying CD1d pro-teins, was established using the galabiose lipid 43 (see figure 6.2).The CD1d selectivity of the binding was investigated using all fiveglycolipids and wild-type as well as β2-microglobulin-knockout EL-4cells. The knockout cells lack all β2-microglobulin-associated pro-teins, including CD1d. Significantly higher responses were obtainedfor the wild-type cells with all glycolipids (figure 6.3) indicating aCD1d-restricted loading. A monoclonal CD1d-specific antibody con-
- 27 -

Glycoconjugates
NH
O
NH
O
m
14b
14a, m=115a, m=5
O
O
HOAcHN
OHHO
O
O
HOAcHN
OHHO
OHO
HO
OHO
O
O
N
OHHO
O
O
AcHNHO
OH
HO
OH
OHN
O
C7H15
C6H13
OH
OHO
HO
OHHO
OOHO
HO
OHO
SO
O
SO
O
44
43
Figure 6.1: Glycolipids used in CD1d loading experiments.
- 28 -
Solid-phase synthesis and biological applications
010203040506070
0 5 50 500
MFI
[43] (µg/ml)
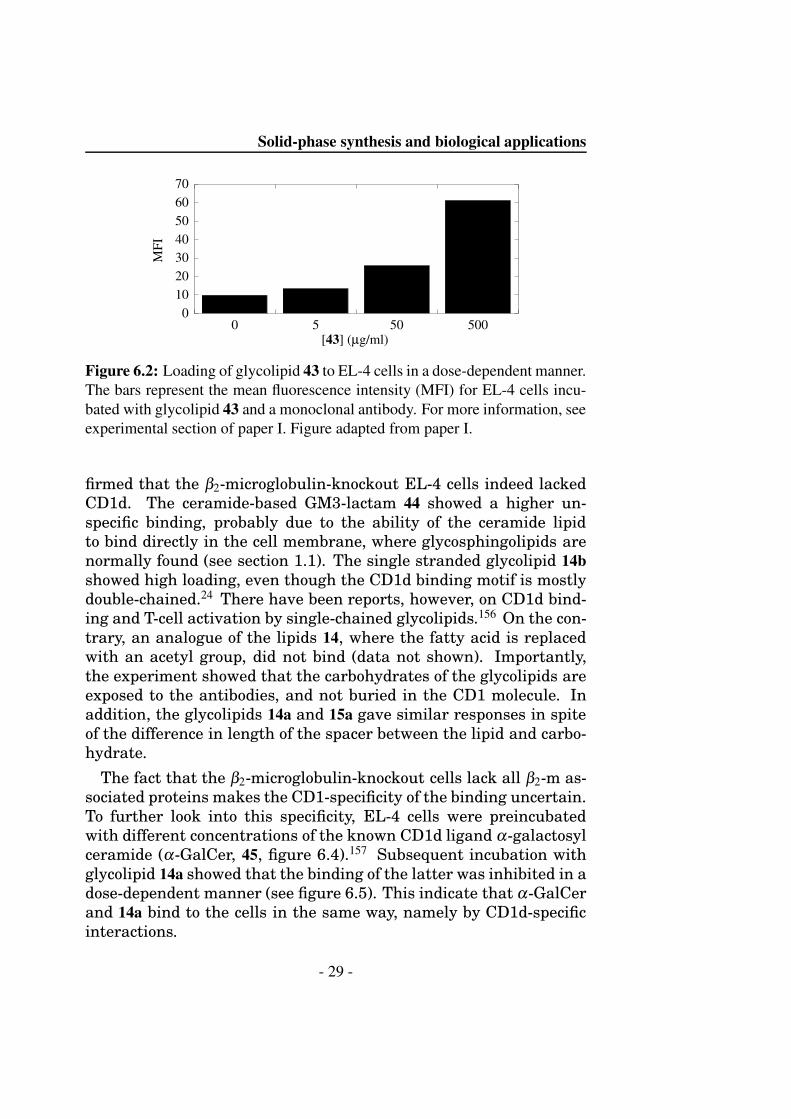
Figure 6.2: Loading of glycolipid 43 to EL-4 cells in a dose-dependent manner.The bars represent the mean fluorescence intensity (MFI) for EL-4 cells incu-bated with glycolipid 43 and a monoclonal antibody. For more information, seeexperimental section of paper I. Figure adapted from paper I.
firmed that the β2-microglobulin-knockout EL-4 cells indeed lackedCD1d. The ceramide-based GM3-lactam 44 showed a higher un-specific binding, probably due to the ability of the ceramide lipidto bind directly in the cell membrane, where glycosphingolipids arenormally found (see section 1.1). The single stranded glycolipid 14bshowed high loading, even though the CD1d binding motif is mostlydouble-chained.24 There have been reports, however, on CD1d bind-ing and T-cell activation by single-chained glycolipids.156 On the con-trary, an analogue of the lipids 14, where the fatty acid is replacedwith an acetyl group, did not bind (data not shown). Importantly,the experiment showed that the carbohydrates of the glycolipids areexposed to the antibodies, and not buried in the CD1 molecule. Inaddition, the glycolipids 14a and 15a gave similar responses in spiteof the difference in length of the spacer between the lipid and carbo-hydrate.
The fact that the β2-microglobulin-knockout cells lack all β2-m as-sociated proteins makes the CD1-specificity of the binding uncertain.To further look into this specificity, EL-4 cells were preincubatedwith different concentrations of the known CD1d ligand α-galactosylceramide (α-GalCer, 45, figure 6.4).157 Subsequent incubation withglycolipid 14a showed that the binding of the latter was inhibited in adose-dependent manner (see figure 6.5). This indicate that α-GalCerand 14a bind to the cells in the same way, namely by CD1d-specificinteractions.
- 29 -

Glycoconjugates
0
10
20
30
40
50
CD1d 14a 14b 15a
MFI
010203040506070
43
MFI
0
50
100
150
200
250
44M
FI
Figure 6.3: Selective binding of glycolipids to CD1d positive EL-4 cells. Thebars represent the mean fluorescence intensity (MFI) for EL-4 (black bars) andβ2m –/– EL-4 cells (grey bars) incubated with a glycolipid and a monoclonalantibody. For more information, see experimental section of paper I. Figureadapted from paper I.
HN
OH
O C23H47O
HO
HO
OH
HO
45
OOH
Figure 6.4: The known CD1d-binding glycolipid KRN7000, an α-galactosylceramide (α-GalCer)
Dendritic cells (DCs) are specialized antigen-presentingmolecules, and preloaded with a glycolipid antigen they arecapable of inducing a strong immune response upon injection intomice.158 DCs from B6 and CD1d-negative B6 mice were loaded withglycolipid 14a as before. Again, a dose-dependent loading, specific for
- 30 -

Solid-phase synthesis and biological applications
0
5
10
15
Control 0.002 0.2 20
MFI
[44445555] (µg/ml)
Figure 6.5: α-GalCer (45) inhibits binding of glycolipid 14a to CD1d positiveEL-4 cells. The bars represent the mean fluorescence intensity (MFI) for EL-4cells incubated with different concentrations of α-GalCer 45 followed by gly-colipid 14a and a monoclonal antibody. For more information, see experimentalsection of paper I. Figure adapted from paper I.
the wild-type DCs, was noted (figure 6.6). Since CD1d-negative B6mice are true CD1d-knockouts, the possible interaction with otherβ2-microglobulin-associated proteins was ruled out. Also thymocytesfrom B6 mice could be loaded with the α-GalNAc containing lipids14a, b, and 15a (data not shown).
0
5
10
15
20
25
0 0.05 0.5 5 50
MFI
[14a] (µg/ml)
Figure 6.6: Glycolipid 14a binds selectively to CD1d positive DCs in a dose-dependent manner. The bars represent the mean fluorescence intensity (MFI)for DCs from B6 (black bars) and CD1d –/– B6 (grey bars) mice incubatedwith glycolipid 14a and a monoclonal antibody. For more information, seeexperimental section of paper I. Figure adapted from paper I.
Using synthetic glycolipids and monoclonal antibodies, CD1d spe-cific and dose-dependent glycolipid loading of viable cells was con-firmed. This FACS based method is rapid and estimates CD1 binding
- 31 -

Glycoconjugates
independent of T-cell activation. The method is therefore suitable inscreening for lipids capable of binding CD1. The glycolipids used inthe study all have immunologically relevant saccharides and can beused to investigate the capacity of these conjugates to evoke a CD1drestricted immune response. In addition, three new CD1d-bindinglipid motives were discovered.
- 32 -

Solid-phase synthesis and biological applications
7 Preparation of glycolipid arrays (paper II)
To start exploring the possibilities of glycolipid microarrays,glycolipids prepared through solid-phase synthesis were covalentlybound to amine groups in microtiter plates. The water-solubleneoglycolipids 29 and 31 (figure 7.1, see also section 5.2),with terminal carboxylic acid functionalities, and commercialmicrotiter plates with secondary amines tethered to the plasticsurface (CovaLinkTM) were used in the study. The glycolipids
29
31
HN
O
HN
OH
O
O
OO
HOHO
OHHO
HN
O
HN
OH
O
O
O
HOHO
OHHO
O
Figure 7.1: Water soluble glycosphingolipid analogues used to construct gly-colipid arrays.
were serially diluted in the wells and coupled to the amineswith an aqueous solution of N-hydroxysuccinimide (NHS) and1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride(EDC). After extensive washing and blocking of the wells withbovine serum albumin (BSA) the carbohydrates were detected withlabeled lectins. Biotin-labeled agglutinin from Ricinus communis(RCA120) has affinity for α- and β -Gal and horseradish peroxidase(HRP)-conjugated lectin from Bandeiraea simplicifolia (BS-1) for
- 33 -

Glycoconjugates
α-Gal and α-GalNAc.159 To the RCA120 lectin, HRP-conjugatedavidin was added and both lectins were then detected throughHRP-catalysed oxidation of o-phenylenediamine and absorbancemeasurements of the resulting 2,3-diaminophenazine. The RCA120recognized both glycolipids 29 and 31 with similar response curves(see figure 7.2), even though its affinity for β -Gal is four timeshigher than for α-Gal.159 This indicates that the measurements of
0.00.20.40.60.81.01.2
0.01 0.1 1 10
Abs
450
[lipid] (mM)
Figure 7.2: Binding of lectin RCA120 to a carbohydrate array with differentconcentrations of 29 (black circles) and 31 (grey squares). The graph shows themean and standard deviations from duplicate columns, with the lipid concentra-tion in the coupling solutions on the x-axis. The mean from eight blank wellswere subtracted from all points. For more information, see the experimentalsection of paper II. Figure adapted from paper II.
lectin binding to the carbohydrate array are qualitative rather thanquantitative. The α-selective BS-1 only recognized the α-galactosyllipid 31 as expected (see figure 7.3). The overall signal for BS-1is lower, probably due to the signal-amplifying properties of thebiotin-avidin construct for RCA120. In a previous study, using thelactose derivative of 29 and 31 and a variant of the microtiter plateswith primary amines, the lactose-binding lectin from Erythrinacorallodendron also bound in a similar fashion to the lectins in thepresent study.127 Importantly, the plates could be regeneratedthrough washing with a 10% SDS solution, followed by reblockingwith BSA, as shown in figure 7.4. Since the synthetic glycolipids areprecious, and the preparation of the plates rather time-consuming,the possibility to regenerate plates is valuable.
The neoglycolipids 29 and 31, prepared by solid-phase synthesis,were covalently attached to microtiter plates to form arrays of neo-glycolipids of varying density. The protein-binding properties of the
- 34 -

Solid-phase synthesis and biological applications
arrays were tested with two different lectins, RCA120, specific for α-and β -galactose, and BS-1, specific for α-galactose and α-GalNAc.Both lectins bound to the array of neoglycolipids in agreement withtheir respective specificity.
-0.10.00.10.20.30.40.50.6
0.01 0.1 1 10
Abs
450
[lipid] (mM)
Figure 7.3: Binding of lectin BS-1 to a carbohydrate array with different con-centrations of 29 (black circles) and 31 (grey squares). The graph shows themean and standard deviations from duplicate columns, with the lipid concen-tration in the coupling solutions on the x-axis. The mean from eight blank wellswere subtracted from all points. For more information, see the experimentalsection of paper II. Figure adapted from paper II.
0.0
0.5
1.0
1.5
2.0
a b c
Abs
450
Figure 7.4: Regeneration of CovaLinkTM wells containing 29. a) Detectionwith RCA120 before regeneration. b) Detection without lectin after strippingwith 10% SDS. c) Renewed detection with RCA120. The bars show the meanand standard deviations from duplicate wells, with the mean from two blankwells subtracted from all points. For more information, see the experimentalsection of paper II. Figure adapted from paper II.
- 35 -


Solid-phase synthesis and biological applications
8 Design, synthesis, and evaluation offluorinated linkers
For solid-phase reaction monitoring with gel-phase 19F-NMR spec-troscopy, fluorinated linkers are crucial. So far, relatively few flu-orinated linkers have been described in the literature (see section2.2) and no such linkers are commercially available. This chapterdescribes the synthesis and application of two novel linkers for im-mobilization of carboxylic acids. Both linkers can be cleaved witheither acids of varying strength or nucleophiles, e.g. hydroxide.
8.1 Synthesis of a new fluorinated monoalkoxy linker(paper II)
The linker 46 (see figure 8.1) is a fluorinated analogue to the classicWang linker and has been applied in solid-phase synthesis moni-tored with gel-phase 19F-NMR spectroscopy.71, 72 When the startingmaterial for the synthesis of that linker no longer was commerciallyavailable, a new synthetic route towards such a linker was needed.The availability of 3-fluoro-4-hydroxy-benzoic acid (47) prompted
OOH
O
46
HO
F
Figure 8.1: Fluorinated monoalkoxy linker developed by Svensson et al.71, 72
synthesis of (2-fluoro-4-(hydroxymethyl)-phenoxy)-acetic acid (32)in three steps (see scheme 8.1). Reduction of the acid 47 usingtrimethyl borate and borane dimethyl sulfide complex (BH3·DMS)proceeded well, in nearly quantitative yields according to thin-layer
- 37 -

Glycoconjugates
chromatography (TLC). The crude alcohol 48 was alkylated withethyl bromoacetate and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU).After chromatographic purification the ester 49 was obtained in 57%yield over two steps. Finally, the ester was hydrolyzed with LiOH togive the linker 32 in 90% yield.
HO
O
OHF
a) HO
OHF
b) HO
OF
OEt
O
c) HO
OF
OH
O
47 48 49
32
Scheme 8.1: Synthesis of a fluorinated monoalkoxy benzyl alcohol linker. a)B(OMe)3, BH3·DMS, THF. b) ethyl bromoacetate, DBU, MeCN, reflux. c)LiOH, THF/MeOH/water 3:1:1.
The linker 32 was prepared in 51% total yield in a three step syn-thesis, amenable to gram scale preparation. The formed linker is sta-ble to glycosylating conditions and can be cleaved under both acidic(TFA/water 9:1, 60 ◦C) and basic (LiOH, 20–33 mM, THF/water 1:2)conditions as described in section 5.2.
8.2 Design, synthesis, and evaluation of a fluorinateddialkoxy linker (paper III)
The acidic conditions used to cleave a carboxylic acid from thelinker 32 are rather harsh, as seen in section 5.2, and can poseproblems when the product is not sufficiently stable. On the otherhand, also the basic conditions can be troublesome, e.g. serineglycosides can undergo β -elimination.160 Therefore, a fluorinatedlinker from which carboxylic acids can be cleaved using milder
- 38 -

Solid-phase synthesis and biological applications
acid is desired. Addition of a second alkoxy-group to the linker32 should make it more acid labile due to stabilization of thebenzylic cation (see section 2.2). A retrosynthetic analysis indicatedthat (6-fluoro-4-(hydroxymethyl)-3-methoxy-phenoxy)-acetic acid(58) could be synthesized from a trifluorobenzoic acid derivativewith nucleophilic aromatic substitutions as key steps. Bothtert-butyl 2,4,5-trifluorobenzoate161, 162 (50, scheme 8.2) and2,4,5-trifluorobenzonitrile161, 163–167 (51, scheme 8.3) have beenreported as electrophiles in such reactions. Based on this, astrategy using the benzoate 50 and methyl glycolate was initiallyexplored (see scheme 8.2). Unfortunately, the nucleophilic
t-BuO
O
FF
a) b)
HO
OF
OH
O
50
58
F
t-BuO
O
OF
F
OMe
O
t-BuO
O
OF
OMe
OMe
O
OMe
Scheme 8.2: Attempted synthesis of linker 58 from benzoate 50 and methylglycolate. a) Methyl glycolate, t-BuOK, THF, 0 ◦C. b) MeOH, t-BuOK, THF,0 ◦C - room temperature.
substitutions, especially with methoxide, were low yielding dueto trans esterifications and decomposition. Instead, the use ofbenzyl alcohol as nucleophile for introduction of the oxygen at thepara-position was investigated. Again, trans esterifications causedproblems with the benzoate 50. Benzonitrile 51 on the other hand,gave good yields in the nucleophilic aromatic substitutions, usingpotassium tert-butoxide (t-BuOK) as base, and was therefore usedin the continued synthesis (scheme 8.3). The dialkoxy benzonitrile53 was thus obtained in 67% yield over two steps. Hydrolysis ofthe nitrile in refluxing NaOH in ethanol/water 4:3 and catalytichydrogenolysis of the benzylether furnished the benzoic acid 55 in
- 39 -

Glycoconjugates
NC
FF
a) b)
HO
OF
OH
O
51
58
FNC
OBnF
FNC
OBnF
OMe
OMe
52 53
OBnF
OMe
HO
O
OHF
OMe
HO
OHF
OMe
HO
O
HO
OF
OEt
O
OMe
c)
e)d)
54 55 56
57
g)f)
Scheme 8.3: Synthesis of linker 58 from benzonitrile 51. a) BnOH, t-BuOK, THF, -78 ◦C – room temperature. b) MeOH, t-BuOK, THF, -50 ◦C– 0 ◦C. c) NaOH (aq.), EtOH, reflux. d) H2 (g, 1 atm.), Pd/C, AcOH. e)B(OMe)3, BH3·DMS, THF. f) ethyl bromoacetate, DBU, MeCN, reflux. g)LiOH, THF/MeOH/water 3:1:1.
53% total yield from benzonitrile 51. The acid 55 is an analogueto 47, the starting material in the synthesis of the monoalkoxylinker 32 (see section 8.1). Unfortunately, the extra methoxy groupdisturbed the continued synthesis. The reduction to the benzylalcohol could be performed in good yields (∼90%) but the productwas very sensitive, probably towards oxidation, and was only brieflypurified and characterized before continuation of the synthesis.Alkylation of the phenolic oxygen with ethyl bromoacetate andDBU in the same conditions as for 48 surprisingly yielded thedimer 59 as the main product (see figure 8.2). Since dimer 59 andits monomer 57 have very similar spectroscopic properties, andboth gave the benzylic cations as main peaks in LC/MS analysis,
- 40 -

Solid-phase synthesis and biological applications
OF
OEt
O
OMe
59
O
OF
EtO
O
OMe
Figure 8.2: The dimer formed in alkylation of 56.
x-ray crystallography was used to verify the structures (data notshown). Reduction of the reaction time diminished the formation ofthe dimer and the monomer 57 could be isolated in 66% yield overtwo steps. Finally, the linker 58 was received in 81% yield by basicester hydrolysis. In total, 58 was synthesized in seven steps fromtrifluorobenzonitrile 51 with an overall yield of 29%.
To test the cleavage conditions for linker 58, a benzoate-terminated dipeptide was synthesized and cleaved (scheme 8.4). Thelinker 58 and its analogue 32 were attached to TentaGel HL-NH2through amide bonds formed with DIC and HOBt. Capping of anyremaining amines with acetic anhydride and pyridine followedby deesterification with NaOMe in MeOH gave the linker-resins60 and 61. To these, Fmoc-glycine (Fmoc-Gly-OH) was esterifiedusing 1-(2-mesitylenesulfonyl)-3-nitro-1H-1,2,4-triazole (MSNT)and N-methyl imidazole (MeIm) in quantitative yields according to19F-NMR spectroscopy. Continued amide formations using DIC andHOBt attached Fmoc-para-fluoro-phenylalanine (Fmoc-p-F-Phe-OH)and, after deprotection, 2,4-difluorobenzoic acid. All couplingsproceeded quantitatively according to 19F-NMR spectroscopy.However, both linkers were cleaved to a small extent during thereactions and when the solid-supported peptides 64 and 65 wereready, 11% and 8% free linker was present on the respectiveresins. The non-fluorinated versions of 58 (such as 8, figure 2.1) areconsidered very acid labile and carboxylic acids are cleaved fromthem using as little as 1% TFA in CH2Cl2. The electron withdrawingproperties of the fluorine, and the fact that fluorinated versionsof the Wang-resin require more harsh cleaving conditions thanthe original,72 prompted the examination of 5% TFA in CH2Cl2 ascleaving solution for 58. The resins were submitted to the cleavingsolution for 30 minutes at room temperature. After extensive
- 41 -

Glycoconjugates
OF
OH
O
R
32, R=H58, R=OMe
HO
OF
HN
O
R
60, R=H61, R=OMe
HO
OF
HN
O
R
62, R=H63, R=OMe
O
OFmocHN
OF
HN
O
R
64, R=H65, R=OMe
O
OHN
ONH
OF
F
a)
c)b)
=TentaGel HL-NH2
F
Scheme 8.4: Synthesis of a small peptide for investigation of cleavage condi-tions. a) TentaGel HL-NH2, DIC, HOBt. b) Fmoc-Gly-OH, MSNT, MeIm. c)i. Piperidine, 20% in DMF. ii. Fmoc-p-F-Phe-OH, DIC, HOBt. iii. Piperidine,20% in DMF. iv. 2,4-difluorobenzoic acid, DIC, HOBt.
- 42 -

Solid-phase synthesis and biological applications
washings, 19F-NMR spectroscopy showed ∼25% cleavage from thedialkoxy linker (i.e resin 65) and none from 64. Repeated cleavagefor 1.5, 2, and 3 hours in sequence showed continued cleavage fromresin 65 while resin 64 remained unaffected (see figure 8.3). Afterthe full 7 hours, ∼85% of the peptide had been cleaved from resin65. Preliminary studies indicated that the acid lability of the linker46 (see sections 8.1 and 2.2) is intermediate to 32 and 58.
-20
0
20
40
60
80
1000 1 2 3 4 5 6 7
Cle
aved
pep
tide
(%)
Time (h)
Figure 8.3: Investigation of peptide acid cleavage from resins 64 (black circles)and 65 (grey squares). The resins were subjected to 5% TFA in CH2Cl2 for 0.5,1.5, 2, and 3 hours in sequence. After each period, the resins were washedextensively and 19F-NMR spectra were recorded. The ratio between the linkerand the peptide was so measured after a total of 0.5, 2, 4, and 7 hours.
To further examine the usefulness of the linker 58, its stability tosolid-phase glycosylation with thioglycosides was studied. The pep-tide 66 was synthesized in a manner analogous to 65. The serineresidue in the peptide was then glycosylated using the previouslydescribed glycosyl donor 67,58 activated with NIS and TfOH (seescheme 8.5). Repeated glycosylations (2 x 2 equivalents) furnishedthe resin-bound glycopeptide 68 in 73% yield according to 19F-NMRspectroscopy. This is a rather high yield considering the relativelyfew equivalents of donor used. Many solid-phase glycosylations areroutinely carried out with 2 x 5 equivalents donor.10 No cleavage ofthe peptide could be detected under these conditions. The glycopep-tide was cleaved from the solid-phase with concurrent deprotection ofthe fluoro-benzylidene group by using TFA/water 9:1 at room temper-ature for 2 hours. After preparative LC/MS the partially protectedglycopeptide 69 was received in 13% based on the initial loading ca-pacity of TentaGel HL-NH2. Debenzoylation was performed using
- 43 -

Glycoconjugates
LiOH in MeOH/water 4:1 to give the deprotected glycopeptide in 55%yield. Unfortunately the 1H NMR spectra of 25 were slightly impureand the anomeric proton could not be assigned. However, a 1H-13Ccorrelation spectrum showed one major product with the anomeric13C shift at 103.8 ppm, indicating the β -anomer.
In conclusion the linker 58 was prepared in 29% yield from tri-fluorobenzonitrile 51 using nucleophilic aromatic substitutions. Thelinker can be cleaved with mild acid (5% TFA in CH2Cl2) and is suf-ficiently stable for glycosylations. The linker 58 therefore is an im-portant complement to the less acid sensitive monoalkoxy linker 32.
- 44 -

Solid-phase synthesis and biological applications
OF
HN
O
OMe
66
O
OHN
ONH
O
=TentaGel HL-NH2
OHHN
ONH
O
OSPhMepFBzO
pFBzO
OO
mFPh
a)
67
OF
HN
O
OMe
68
O
OHN
ONH
OHN
ONH
O
O
O
pFBzO
pFBzO
OO mFPh
69, R=pFBz70, R=H
OH
OHN
ONH
OHN
ONH
O
O
O
RO
RO
OHOH
b)
c)
F
F
F
Scheme 8.5: Solid-phase glycosylation of a peptide bound to linker 58 and itscleavage and deprotection. a) NIS, TfOH. b) TFA/water 9:1, 2 h. c) LiOH (20mM in MeOH/water 4:1).
- 45 -


Solid-phase synthesis and biological applications
9 Design of an NMR tube filter reactor forsolid-phase synthesis and gel-phase19F-NMR spectroscopy (paper IV)
When monitoring solid-phase reactions with 19F-NMR spectroscopythe transfer of resin between the reaction vessel and an NMR tubewith the following preparation of the gel-phase is a both impracticaland time-consuming step. The modification of an NMR tube to asolid-phase reactor was anticipated to address these problems.
The reactor was constructed out of an ordinary medium-thick (0.8mm) walled borosilicate glass tube with 5 mm outer diameter (i, fig-ure 9.1). Since gel-phase NMR spectra have rather large line widths,the high precision of specialized NMR-tubes was not needed and thecost was thereby reduced. A hollow teflon plug (ii) with a teflon filter(iii) was inserted into the glass tube, making the reactor. For reac-tions, this reactor was attached to a circulating pump through twoteflon adapters (figure 9.1a and f). At the bottom of the reactor, ateflon Luer adapter (iv) covered the plug ii and the base of the tube i.This Luer adapter was connected to the pump via an ordinary Luersyringe needle carrying a teflon hose. At the top of the tube, anotherteflon adapter (v) was attached, with an inserted steel tube carryinga teflon hose. By using this setup, reaction solutions could be circu-lated through resin placed in the reactor (figure 9.1f). For washings,the top adapter (v) was instead connected to a container for wash-ing solvents and the pump outlet was diverted to a waste (see figure9.1g). At the time for NMR spectroscopy, CDCl3 was added to forma gel-phase, the two adapters iv and v were removed and the holein plug ii was closed by the teflon stopper vi (see figure 9.1e). Thereactor was now ready to be inserted into an NMR spectrometer andgel-phase spectra showed similar line-width to those recorded in nor-mal NMR tubes.58
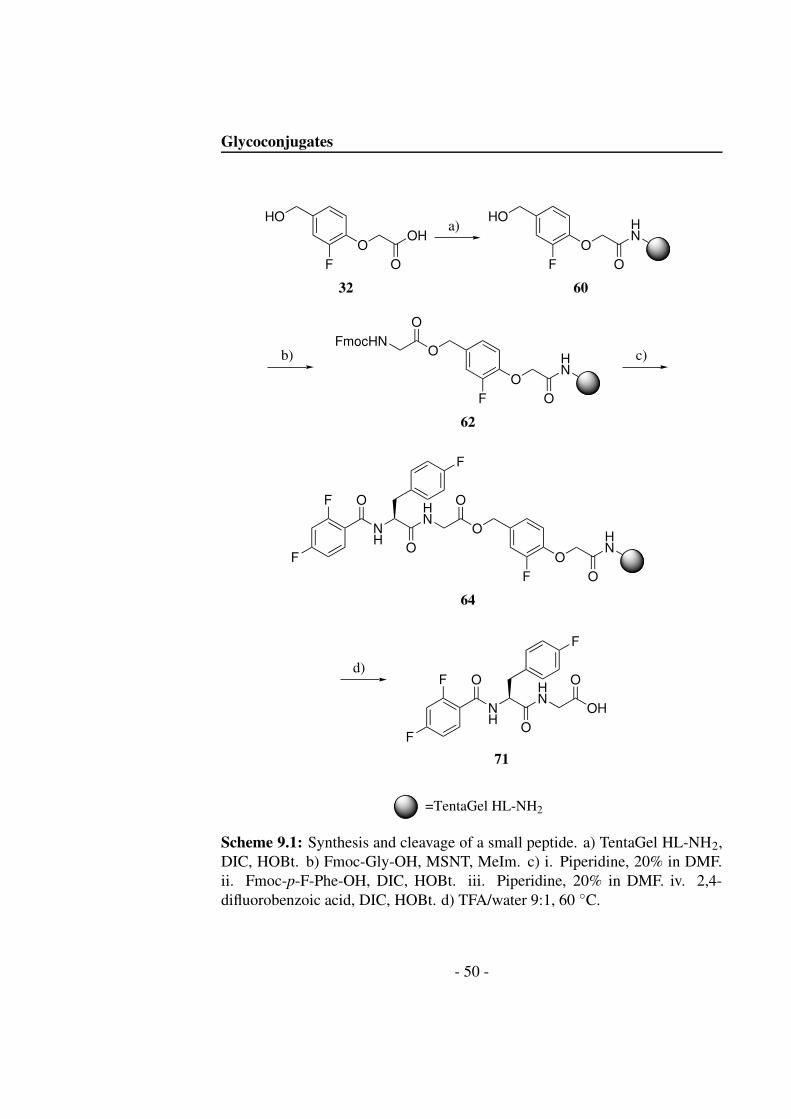
To test the applicability of the reactor, the N-benzoylated dipep-tide 71 was synthesized. The synthesis was performed on TentaGel
- 47 -

Glycoconjugates
Figure 9.1: The NMR tube filter reactor. a) The reactor during a reaction. b)The reactor ready for NMR. c) The teflon adapters. d) The teflon filter, plug,and stopper. e) Schematic drawing of the reactor ready for NMR. f) Schematicdrawing of the reactor during synthesis. g) Schematic drawing of the reactorduring washing. Figure adapted from manuscript IV
- 48 -

Solid-phase synthesis and biological applications
HL-NH2 with the linker 32 (scheme 9.1), essentially as for the test ofcleavage reactions of linker 58 (see section 8.2). The same buildingblocks and reagents led up to resin-bound peptide 64. However, dur-ing this full synthesis, including gel-phase NMR spectroscopy, theresin was never removed from the reactor. All reactions were quan-titative according to 19F-NMR spectroscopy, but as for the synthesisin section 8.2 a small amount of the peptide was cleaved from thelinker during the synthesis. When the peptide was ready, the resinwas removed from the reactor to allow for heating and the peptidewas cleaved with TFA/water 9:1 at 60 ◦C. Purification with prepara-tive LC/MS gave the peptide in 37% yield based on the initial loadingcapacity of TentaGel HL-NH2.
A solid-phase filter reactor that can be inserted into an NMR spec-trometer was constructed and its usefulness was demonstrated withthe synthesis of a small peptide. The resin-bound peptide was con-structed without removal of the resin from the reactor during thefull synthesis, including NMR spectroscopy. The gel-phase 19F-NMRspectra were of similar quality to those obtained in standard NMRtubes. Gel-phase 19F-NMR spectroscopy is a good method to mon-itor solid-phase synthesis and with the described reactor the resinhandling is simplified and the synthesis and analysis process is thusstreamlined.
- 49 -
Glycoconjugates
OF
OH
O
32
HO
OF
HN
O
60
HO
OF
HN
O
62
O
OFmocHN
OF
HN
O
64
O
OHN
ONH
OF
F
a)
c)b)
=TentaGel HL-NH2
71
OH
OHN
ONH
OF
F
d)
F
F
Scheme 9.1: Synthesis and cleavage of a small peptide. a) TentaGel HL-NH2,DIC, HOBt. b) Fmoc-Gly-OH, MSNT, MeIm. c) i. Piperidine, 20% in DMF.ii. Fmoc-p-F-Phe-OH, DIC, HOBt. iii. Piperidine, 20% in DMF. iv. 2,4-difluorobenzoic acid, DIC, HOBt. d) TFA/water 9:1, 60 ◦C.
- 50 -

Solid-phase synthesis and biological applications
10 Concluding remarks and futureperspectives
Glycoconjugates are important molecules with diverse biologicalfunctions. The complex structure and biosynthesis of glycoconju-gates make the isolation of pure, structurally defined compoundsfrom natural sources cumbersome. Therefore, to better addressquestions in glycobiology, the use of synthetic glycoconjugates is anappealing alternative.
In this thesis, synthetic methods for the preparation of glycolipidanalogues have been developed. These methods are amenable tolibrary synthesis and make use of solid-phase chemistry. The bio-logical applications of the glycolipids were demonstrated in two dif-ferent settings. The CD1 restricted binding of glycolipids carryingα-GalNAc as carbohydrate was detected on viable cells of mouse ori-gin and glycoconjugate arrays was prepared using serine-based neo-glycolipids. The usefulness of the arrays was illustrated with twolectins, RCA120, specific for α- and β -galactose, and BS-1, specific forα-galactose and α-GalNAc. Both lectins bound to the array of neo-glycolipids in agreement with their respective specificity.
Gel-phase 19F-NMR spectroscopy, using fluorinated linkers andprotecting groups, is an excellent tool for monitoring solid-phase syn-thesis of e.g. glycoconjugates. Two novel fluorinated linkers for theattachment of carboxylic acids have been developed and are pre-sented in the thesis. These linkers can be cleaved with both acidsof varying strengths and nucleophiles like hydroxide ions, and theyare stable to glycosylation conditions. In addition, a novel filter re-actor for solid-phase synthesis was designed. The reactor fits intoan ordinary NMR spectrometer to facilitate the reaction monitoringwith gel-phase 19F-NMR spectroscopy.
The projects in the thesis have several interesting openings forcontinued research. In the CD1-related project (section 5.1 and chap-ter 6) the first thing to do would be to synthesize a library of α-GalNAc containing glycolipids with different lipid composition. This
- 51 -

Glycoconjugates
library can be evaluated for CD1 binding using the method describedin chapter 6 with the possible extension of competitive experimentsto obtain more quantitative responses. Also the serine-based gly-colipids from section 5.2 would be exciting to test for CD1-binding.To follow up, immunization studies on mouse and preferably alsoguinea pigs with some immunologically relevant glycolipids is of in-terest. Guinea pigs are of special interest in such experiments be-cause of their full CD1 repertoire in contrast to the mouse that onlyhave CD1d molecules.
The synthetic methods developed in section 5.2 and chapters 8 and9 can be used to prepare serine-based glycolipid libraries suitable forarray preparation. In addition to the microtiter approach describedin chapter 7, also glass slides with an amine surface can be used forattachment and hence additional miniaturizations can be achievedthrough the use of equipment and techniques developed for oligonu-cleotide and cDNA microarrays. To synthesize more complex gly-colipids, enzymatic modifications of array-bound glycolipids are anappealing alternative.
The fluorinated dialkoxy linker 58 have, apart from the librarysynthesis mentioned above, a number of possible applications. Onewould be to investigate direct glycosylations on the linker and fur-ther oligosaccharide synthesis. Acidic cleavage of the linker-boundglycoside should give the hemiacetal that could be further deriva-tized, e.g. to give a glycosyl donor for glycoconjugate synthesis. Theattachment and cleavage of alcohols would also be interesting to in-vestigate. Non-fluorinated benzaldehyde linkers can be used to im-mobilize amines through reductive amination and the formed aminecan be acylated and cleaved. The oxidation of the alcohol in resin-bound 58 will result in a resin that may be used in a similar way.This would enable e.g. synthesis of serine-based glycolipids withoutthe terminal carboxylic acid.
Regarding the NMR tube filter reactor, more reactions need to betested. The first thing to investigate would be glycosylation reac-tions. Also other, more harsh reaction conditions, e.g. the acidiccleavage from linker 58, are of interest. Finally, more automatedmethods for reactions and washings would be appealing.
In summary, the methods and equipment described in this thesiscan be used to answer a variety of questions in glycobiology such asstructural requirements for CD1 binding and specificity of carbohy-
- 52 -

Solid-phase synthesis and biological applications
drate recognizing proteins from bacteria and virus.
- 53 -


Solid-phase synthesis and biological applications
11 Acknowledgments
Det finns väldigt många att tacka här i världen. Här tänkte jagbegränsa mig till de som har någonting med avhandlingsarbetet attgöra men det är fortfarande alltför många för att jag ska kommaihåg alla. Så ett alldeles särskilt tack till de som blir bortglömda!
Till att börja med vill jag tacka Mikael! Det har varit både roligaoch spännande år och jag har särskilt uppskattat att du har haft tidnär jag har kommit med frågor. Du har också haft en förmåga attentusiasmera och pigga upp när data inte tyckt som jag. . .
Jag vill också tacka Jan och Björn för introduktionen tillkolhydratkemi. Alla sockerbagare, Brodde, Petter, Lotta, Moge, Sussi,Tomas?, Majeic, Shaji, Baquer, Anna L, Sara, socker är gott!
Alla exjobbare och projektarbetare i och kring projektet förtjänarockså ett stort tack för allt slit, framförallt för alla saker jag intebehövt misslyckas med :-) Nåväl, tack PJ, Kicki, Henke, Rupa, Am-paro, Johan, Matilda, Anders, Sandra, Annika, P-A, Lennart.
Mikael, Liying och Annalena på KI, tack för ett gott samarbete och föratt jag fått komma och prova på själv!
Tack Sara, som tar över, du fick slita i början. . .
Mikael, Moge, Sussi, Maciej och Anna, tack för korrekturläsningen. Nihittade mycket, men jag är säker på att en del återstår att se och jaghar nog stoppat in fler fel sen ni fick läsa.
Pelle, Bert, Carina, och Ann-Helén för all hjälp med alla praktiskabestyr.
Gamla och nuvarande rums- och labkamrater, Björn, Lotta, Shaji,Hasse, Neas, Henke, Anna, Veronica, Sussi, Maciej och alla exjobbare
- 55 -

Glycoconjugates
för prat och tjat både nu och då.
Alla andra på avdelningen, för att det är lätt att fika lite för länge. . .
Göstasgängen, med ingredienser som Trulsson, Koffe, Björn, Lotta,Tomas, Mattias, PJ, och många fler. En gång i tiden var det nästangott!
Alla i kp96 med omnejd för att studietiden kändes så rätt.
Övriga vänner som förgyller min vardag!
Mamma, pappa, Jon, Ida, Noah och Gustav för att ni trott på mig ävenom ni inte har förstått vad jag säger.
Familjen Bergstrand för att ni tagit emot mig och blivit familj ni med.
Tack Gud, för inspiration, peppning och för att jag hållit mig friskunder skrivandet! Stort tack till alla som bett och ber för mig, Annaoch Thea.
Anna och Thea, mina tjejer! Utan er vore det inget värt! Tack för attni stått ut de senaste veckorna.
- 56 -

Solid-phase synthesis and biological applications
References
[1] C.-H. Wong, Protein glycosylation: New challenges and opportunities. Jour-nal of Organic Chemistry, 2005, 70, 4219–4225.
[2] A. Imberty and S. Pérez, Structure, conformation, and dynamics of bioac-tive oligosaccharides: Theoretical approaches and experimental validations.Chemical Reviews, 2000, 100, 4567–4588.
[3] R. A. Laine, A calculation of all possible oligosaccharide isomers bothbranched and linear yields 1.05 X 1012 structures for a reducing hexasac-charide: the Isomer Barrier to development of single-method saccharide se-quencing or synthesis systems. Glycobiology, 1994, 4, 759–767.
[4] N. O. L. Seto and S. V. Evans, Specificity in protein-carbohydrate recogni-tion. Current Organic Chemistry, 2000, 4, 411–427.
[5] P. H. Seeberger and W.-C. Haase, Solid-phase oligosaccharide synthesis andcombinatorial carbohydrate libraries. Chemical Reviews, 2000, 100, 4349–4393.
[6] K. A. Karlsson, Microbial recognition of target-cell glycoconjugates. CurrentOpinion in Structural Biology, 1995, 5, 622–635.
[7] D. H. Dube and C. R. Bertozzi, Glycans in cancer and inflammation – Poten-tial for therapeutics and diagnostics. Nature Reviews Drug Discovery, 2005,4, 477–488.
[8] R. A. Dwek, Glycobiology: Toward understanding the function of sugars.Chemical Reviews, 1996, 96, 683–720.
[9] S.-i. Hakomori and W. T. Murakami, Glycolipids of hamster fibroblastsand derived malignant-transformed cell lines. Proceedings of the NationalAcademy of Sciences of the United States of America, 1968, 59, 254–261.
[10] P. H. Seeberger and D. B. Werz, Automated synthesis of oligosaccharides asa basis for drug discovery. Nature Reviews Drug Discovery, 2005, 4, 751–763.
[11] M. Ambrosi, N. R. Cameron, and B. G. Davis, Lectins: tools for the molecularunderstanding of the glycocode. Organic & Biomolecular Chemistry, 2005,3, 1593–1608.
[12] B. Holm, J. Bäcklund, M. A. F. Recio, R. Holmdahl, and J. Kihlberg, Gly-copeptide specificity of helper T cells obtained in mouse models for rheuma-toid arthritis. ChemBioChem, 2002, 3, 1209–1222.
[13] C. R. Bertozzi and L. L. Kiessling, Chemical glycobiology. Science, 2001, 291,2357–2364.
[14] M. Mammen, S.-K. Choi, and G. M. Whitesides, Polyvalent interactions inbiological systems: Implications for design and use of multivalent ligandsand inhibitors. Angewandte Chemie-International Edition, 1998, 37, 2754–
- 57 -

Glycoconjugates
2794.[15] J. Holgersson, A. Gustafsson, and M. E. Breimer, Characteristics of protein-
carbohydrate interactions as a basis for developing novel carbohydrate-based antirejection therapies. Immunology and Cell Biology, 2005, 83, 694–708.
[16] W. H. Evans and J. M. Graham, Membrane structure and function. In focus,IRL Press at Oxford University Press, Oxford, 1989.
[17] Y. Lavie, H.-t. Cao, S. L. Bursten, A. E. Giuliano, and M. C. Cabot, Accumu-lation of glucosylceramides in multidrug-resistant cancer cells. The Journalof Biological Chemistry, 1996, 271, 19530–19536.
[18] J. Chan, T. Fujiwara, P. Brennan, M. McNeil, S. J. Turco, J.-C. Sibille,M. Snapper, P. Aisen, and B. R. Bloom, Microbial glycolipids: Possible vir-ulence factors that scavenge oxygen radicals. Proceedings of the NationalAcademy of Sciences of the United States of America, 1989, 86, 2453–2457.
[19] B. B. Finlay and S. Falkow, Common themes in microbial pathogenicity re-visited. Microbiology and Molecular Biology Reviews, 1997, 61, 136–169.
[20] N. Yahi, S. Baghdiguian, H. Moreau, and J. Fantini, Galactosyl ceramide(or a closely related molecule) is the receptor for human immunodeficiencyvirus type 1 on human colon epithelial HT29 cells. Journal of Virology, 1992,66, 4848–4854.
[21] E. R. Unanue, Perspective on antigen processing and presentation. Im-munological Reviews, 2002, 185, 86–102.
[22] Y. Dutronc and S. A. Porcelli, The CD1 family and T cell recognition of lipidantigens. Tissue Antigens, 2002, 60, 337–353.
[23] S. Joyce and L. Van Kaer, CD1-restricted antigen presentation: an oily mat-ter. Current Opinion in Immunology, 2003, 15, 95–104.
[24] M. Brigl and M. B. Brenner, CD1: Antigen presentation and T cell function.Annual Review of Immunology, 2004, 22, 817–890.
[25] S. D. Gadola, N. R. Zaccai, K. Harlos, D. Shepherd, J. C. Castro-Palomino,G. Ritter, R. R. Schmidt, E. Y. Jones, and V. Cerundolo, Structure of hu-man CD1b with bound ligands at 2.3 Å, a maze for alkyl chains. NatureImmunology, 2002, 3, 721–726.
[26] D. M. Zajonc, M. A. Elsliger, L. Teyton, and I. A. Wilson, Crystal structureof CD1a in complex with a sulfatide self antigen at a resolution of 2.15 Å.Nature Immunology, 2003, 4, 808–815.
[27] T. Batuwangala, D. Shepherd, S. D. Gadola, K. J. C. Gibson, N. R. Zaccai,A. R. Fersht, G. S. Besra, V. Cerundolo, and E. Y. Jones, The crystal struc-ture of human CD1b with a bound bacterial glycolipid. The Journal of Im-munology, 2004, 172, 2382–2388.
[28] D. M. Zajonc, C. Cantu III, J. Mattner, D. Zhou, P. B. Savage, A. Bendelac,I. A. Wilson, and L. Teyton, Structure and function of a potent agonist forthe semi-invariant natural killer T cell receptor. Nature Immunology, 2005,6, 810–818.
[29] B. Giabbai, S. Sidobre, M. D. M. Crispin, Y. Sanchez-Ruìz, A. Bachi, M. Kro-nenberg, I. A. Wilson, and M. Degano, Crystal structure of mouse CD1dbound to the self ligand phosphatidylcholine: A molecular basis for NKTcell activation. The Journal of Immunology, 2005, 175, 977–984.
[30] M. Koch, V. S. Stronge, D. Shepherd, S. D. Gadola, B. Mathew, G. Ritter,
- 58 -

Solid-phase synthesis and biological applications
A. R. Fersht, G. S. Besra, R. R. Schmidt, E. Y. Jones, and V. Cerundolo, Thecrystal structure of human CD1d with and without α-galactosylceramide.Nature Immunology, 2005, 6, 819–826.
[31] F. Calabi, J. M. Jarvis, L. Martin, and C. Milstein, Two Classes of CD1Genes. European Journal of Immunology, 1989, 19, 285–292.
[32] C. Angénieux, J. Salamero, D. Fricker, J.-P. Cazenave, B. Goud, D. Hanau,and H. de la Salle, Characterization of CD1e, a third type of CD1 moleculeexpressed in dendritic cells. The Journal of Biological Chemistry, 2000, 275,37757–37764.
[33] M. Sugita and M. B. Brenner, T lymphocyte recognition of human group 1CD1 molecules: Implications for innate and acquired immunity. Seminarsin Immunology, 2000, 12, 511–516.
[34] K. Benlagha and A. Bendelac, CD1d-restricted mouse Vα14 and humanVα24 T cells: lymphocytes of innate immunity. Seminars in Immunology,2000, 12, 537–542.
[35] S. M. Behar and S. Cardell, Diverse CD1d-restricted T cells: diverse pheno-types, and diverse functions. Seminars in Immunology, 2000, 12, 551–560.
[36] J. E. Gumperz and M. B. Brenner, CD1-specific T cells in microbial immu-nity. Current Opinion in Immunology, 2001, 13, 471–478.
[37] A. Shamshiev, H.-J. Gober, A. Donda, Z. Mazorra, L. Mori, and G. De Libero,Presentation of the same glycolipid by different CD1 molecules. The Journalof Experimental Medicine, 2002, 195, 1013–1021.
[38] A. Jahng, I. Maricic, C. Aguilera, S. Cardell, R. C. Halder, and V. Kumar,Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. The Journal of ExperimentalMedicine, 2004, 199, 947–957.
[39] D. B. Moody, T. Ulrichs, W. Mühlecker, D. C. Young, S. S. Gurcha, E. Grant,J.-P. Rosat, M. B. Brenner, C. E. Costello, G. S. Besra, and S. A. Porcelli,CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacteriumtuberculosis infection. Nature, 2000, 404, 884–888.
[40] V. Briken, D. B. Moody, and S. A. Porcelli, Diversification of CD1 proteins:sampling the lipid content of different cellular compartments. Seminars inImmunology, 2000, 12, 517–525.
[41] T. Ziegler, Protecting group strategies for carbohydrates, In Carbohydratechemistry (G.-J. Boons, ed.). Blackie Academic and Professional, London,1998, 21–45.
[42] G. H. Veeneman, Chemical synthesis of O-glycosides, In Carbohydrate chem-istry (G.-J. Boons, ed.). Blackie Academic and Professional, London, 1998,98–174.
[43] Z. Zhang, I. R. Ollmann, X.-S. Ye, R. Wischnat, T. Baasov, and C.-H. Wong,Programmable one-pot oligosaccharide synthesis. Journal of the AmericanChemical Society, 1999, 121, 734–753.
[44] R. B. Merrifield, Solid phase peptide synthesis. I. The synthesis of atetrapeptide. Journal of the American Chemical Society, 1963, 85, 2149–2154.
[45] R. B. Merrifield, Solid phase peptide synthesis. II. The synthesis ofbradykinin. Journal of the American Chemical Society, 1964, 86, 304–305.
[46] R. B. Merrifield, Solid-phase peptide synthesis. III. An improved synthesis
- 59 -

Glycoconjugates
of bradykinin. Biochemistry, 1964, 3, 1385–1390.[47] R. B. Merrifield, Solid phase peptide synthesis. IV. The synthesis of
methionyl-lysyl-bradykinin. Journal of Organic Chemistry, 1964, 29, 3100–3102.
[48] G. B. Fields, Z. Tian, and G. Barany, Principles and practice of solid-phasepeptide synthesis, In Synthetic peptides: A user’s guide (G. A. Grant, ed.).W. H. Freeman and Company, New York, 1992, 77–183.
[49] M. H. Caruthers, Gene synthesis machines: DNA chemistry and its uses.Science, 1985, 230, 281–285.
[50] O. J. Plante, E. R. Palmacci, and P. H. Seeberger, Automated solid-phasesynthesis of oligosaccharides. Science, 2001, 291, 1523–1527.
[51] K. R. Love and P. H. Seeberger, Solution syntheses of protected type II Lewisblood group oligosaccharides: Study for automated synthesis. Journal ofOrganic Chemistry, 2005, 70, 3168–3177.
[52] M. Mogemark, Solid-phase glycoconjugate synthesis: On-resin analysis withgel-phase 19F NMR spectroscopy. PhD thesis, Umeå University, 2005.
[53] T. Tengel, T. Fex, H. Emtenäs, F. Almqvist, I. Sethson, and J. Kihlberg, Useof 19F NMR spectroscopy to screen chemical libraries for ligands that bindto proteins. Organic & Biomolecular Chemistry, 2004, 2, 725–731.
[54] M. Dal Cin, S. Davalli, C. Marchioro, M. Passarini, O. Perini, S. Provera, andA. Zaramella, Analytical methods for the monitoring of solid phase organicsynthesis. Il Farmaco, 2002, 57, 497–510.
[55] S. L. Manatt, C. F. Amsden, C. A. Bettison, W. T. Frazer, J. T. Gudman, B. E.Lenk, J. F. Lubetich, E. A. McNelly, S. C. Smith, D. J. Templeton, and R. P.Pinnell, A fluorine-19 NMR approach for studying Merrifield solid-phasepeptide syntheses. Tetrahedron Letters, 1980, 21, 1397–1400.
[56] A. Svensson, T. Fex, and J. Kihlberg, Use of 19F NMR spectroscopy to eval-uate reactions in solid phase organic synthesis. Tetrahedron Letters, 1996,37, 7649–7652.
[57] M. Mogemark, M. Elofsson, and J. Kihlberg, Monitoring solid-phase glyco-side synthesis with 19F NMR spectroscopy. Organic Letters, 2001, 3, 1463–1466.
[58] M. Mogemark, F. Gårdmo, T. Tengel, J. Kihlberg, and M. Elofsson, Gel-phase 19F NMR spectral quality for resins commonly used in solid-phaseorganic synthesis; a study of peptide solid-phase glycosylations. Organic &Biomolecular Chemistry, 2004, 2, 1770–1776.
[59] M. Mogemark, M. Elofsson, and J. Kihlberg, Fluorinated protective groupsfor on-resin quantification of solid-phase oligosaccharide synthesis with 19FNMR spectroscopy. ChemBioChem, 2002, 3, 1266–1269.
[60] M. Mogemark, M. Elofsson, and J. Kihlberg, Solid-phase synthesis of α-Galepitopes: On-resin analysis of solid-phase oligosaccharide synthesis with19F NMR spectroscopy. Journal of Organic Chemistry, 2003, 68, 7281–7288.
[61] M. Mogemark, L. Gustafsson, C. Bengtsson, M. Elofsson, and J. Kihlberg,A fluorinated selenide linker for solid-phase synthesis of n-pentenyl glyco-sides. Organic Letters, 2004, 6, 4885–4888.
[62] H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander, and M. Stahl, Fluorine in medicinal chemistry. ChemBioChem,2004, 5, 637–643.
- 60 -

Solid-phase synthesis and biological applications
[63] P. Sjölin and J. Kihlberg, Use of fluorobenzoyl protective groups in synthe-sis of glycopeptides: β -elimination of O-linked carbohydrates is suppressed.Journal of Organic Chemistry, 2001, 66, 2957–2965.
[64] C. Dalvit, M. Flocco, M. Veronesi, and B. J. Stockman, Fluorine-NMR compe-tition binding experiments for high-throughput screening of large compoundmixtures. Combinatorial Chemistry & High Throughput Screening, 2002, 5,605–611.
[65] C. Dalvit, P. E. Fagerness, D. T. A. Hadden, R. W. Sarver, and B. J. Stock-man, Fluorine-NMR experiments for high-throughput screening: Theoreti-cal aspects, practical considerations, and range of applicability. Journal ofthe American Chemical Society, 2003, 125, 7696–7703.
[66] C. Dalvit, E. Ardini, M. Flocco, G. P. Fogliatto, N. Mongelli, and M. Veronesi,A general NMR method for rapid, efficient, and reliable biochemical screen-ing. Journal of the American Chemical Society, 2003, 125, 14620–14625.
[67] C. Dalvit, G. Papeo, N. Mongelli, P. Giordano, B. Saccardo, A. Costa,M. Veronesi, and S. Y. Ko, Rapid NMR-based functional screening and IC50measurements performed at unprecedentedly low enzyme concentration.Drug Development Research, 2005, 64, 105–113.
[68] F. Guillier, D. Orain, and M. Bradley, Linkers and cleavage strategies insolid-phase organic synthesis and combinatorial chemistry. Chemical Re-views, 2000, 100, 2091–2157.
[69] A. J. Wills and S. Balasubramanian, Recent developments in linker designand application. Current Opinion in Chemical Biology, 2003, 7, 346–352.
[70] S.-S. Wang, p-Alkoxybenzyl alcohol resin and p-alkoxybenzyloxycarbonyl-hydrazide resin for solid phase synthesis of protected peptide fragments.Journal of the American Chemical Society, 1973, 95, 1328–1333.
[71] A. Svensson, K.-E. Bergquist, T. Fex, and J. Kihlberg, Fluorinated linkersfor monitoring solid-phase synthesis using gel-phase 19F NMR spectroscopy.Tetrahedron Letters, 1998, 39, 7193–7196.
[72] A. Svensson, T. Fex, and J. Kihlberg, Preparation of fluorinated linkers: Useof 19F NMR spectroscopy to establish conditions for solid-phase synthesis ofpilicide libraries. Journal of Combinatorial Chemistry, 2000, 2, 736–748.
[73] J. M. Salvino, S. Patel, M. Drew, P. Krowlikowski, E. Orton, N. V. Kumar,T. Caulfield, and R. Labaudiniere, Synthesis of a new fluoro-Wang resinfor solid-phase reaction monitoring by 19F NMR spectroscopy. Journal ofCombinatorial Chemistry, 2001, 3, 177–180.
[74] P. Lakshmipathi, C. Crévisy, and R. Grée, Reaction monitoring in LPOSby 19F NMR. Study of soluble polymer supports with fluorine in spacer orlinker components of supports. Journal of Combinatorial Chemistry, 2002,4, 612–621.
[75] J. M. Salvino, N. V. Kumar, E. Orton, J. Airey, T. Kiesow, K. Crawford,R. Mathew, P. Krolikowski, M. Drew, D. Engers, D. Krolikowski, T. Her-pin, M. Gardyan, G. McGeehan, and R. Labaudiniere, Polymer-supportedtetrafluorophenol: A new activated resin for chemical library synthesis.Journal of Combinatorial Chemistry, 2000, 2, 691–697.
[76] H. Emtenäs, K. Åhlin, J. S. Pinkner, S. J. Hultgren, and F. Almqvist, Designand parallel solid-phase synthesis of ring-fused 2-pyridinones that targetpilus biogenesis in pathogenic bacteria. Journal of Combinatorial Chem-
- 61 -

Glycoconjugates
istry, 2002, 4, 630–639.[77] Y. Pan and C. P. Holmes, A traceless perfluoroalkylsulfonyl (PFS) linker for
the deoxygenation of phenols. Organic Letters, 2001, 3, 2769–2771.[78] Y. Pan, B. Ruhland, and C. P. Holmes, Use of a perfluoroalkylsulfonyl (PFS)
linker in a ”traceless” synthesis of biaryls through Suzuki cleavage. Ange-wandte Chemie-International Edition, 2001, 40, 4488–4491.
[79] M. Hourdin, G. Gouhier, A. Gautier, E. Condamine, and S. R. Piettre, De-velopment of cross-linked polystyrene-supported chiral amines featuring afluorinated linker for gel-phase 19F NMR spectrometry monitoring of reac-tions. Journal of Combinatorial Chemistry, 2005, 7, 285–297.
[80] M. N. Kronick, Creation of the whole human genome microarray. ExpertReview of Proteomics, 2004, 1, 19–28.
[81] H. Zhu and M. Snyder, Protein chip technology. Current Opinion in Chemi-cal Biology, 2003, 7, 55–63.
[82] M. D. Disney and P. H. Seeberger, Carbohydrate arrays as tools for the gly-comics revolution. Drug Discovery Today: TARGETS, 2004, 3, 151–158.
[83] L. Nimrichter, A. Gargir, M. Gortler, R. T. Altstock, A. Shtevi, O. Weisshaus,E. Fire, N. Dotan, and R. L. Schnaar, Intact cell adhesion to glycan microar-rays. Glycobiology, 2004, 14, 197–203.
[84] M. Schwarz, L. Spector, A. Gargir, A. Shtevi, M. Gortler, R. T. Altstock, A. A.Dukler, and N. Dotan, A new kind of carbohydrate array, its use for profilingantiglycan antibodies, and the discovery of a novel human cellulose-bindingantibody. Glycobiology, 2003, 13, 749–754.
[85] Lundonia Biotech AB, http://www.lundonia.com/.[86] Y. Guo, H. Feinberg, E. Conroy, D. A. Mitchell, R. Alvarez, O. Blixt, M. E.
Taylor, W. I. Weis, and K. Drickamer, Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR.Nature Structural & Molecular Biology, 2004, 11, 591–598.
[87] I. Shin, S. Park, and M.-r. Lee, Carbohydrate microarrays: An advancedtechnology for functional studies of glycans. Chemistry - A European Jour-nal, 2005, 11, 2894–2901.
[88] M.-C. Shao, The use of streptavidin-biotinylglycans as a tool for character-ization of oligosaccharide-binding specificity of lectin. Analytical Biochem-istry, 1992, 205, 77–82.
[89] C. Leteux, M. S. Stoll, R. A. Childs, W. Chai, M. Vorozhaikina, and T. Feizi,Influence of oligosaccharide presentation on the interactions of carbohy-drate sequence-specific antibodies and the selectins – Observations with bi-otinylated oligosaccharides. Journal of Immunological Methods, 1999, 227,109–119.
[90] C. Galustian, A. M. Lawson, S. Komba, H. Ishida, M. Kiso, and T. Feizi,Sialyl-Lewisx sequence 6-O-sulfated at N-acetylglucosamine rather than atgalactose is the preferred ligand for L-selectin and de-N-acetylation of thesialic acid enhances the binding strength. Biochemical and Biophysical Re-search Communications, 1997, 240, 748–751.
[91] M. C. Bryan, O. Plettenburg, P. Sears, D. Rabuka, S. Wacowich-Sgarbi, andC.-H. Wong, Saccharide display on microtiter plates. Chemistry & Biology,2002, 9, 713–720.
[92] S. Fukui, T. Feizi, C. Galustian, A. M. Lawson, and W. Chai, Oligosaccharide
- 62 -

Solid-phase synthesis and biological applications
microarrays for high-throughput detection and specificity assignments ofcarbohydrate-protein interactions. Nature Biotechnology, 2002, 20, 1011–1017.
[93] D. Wang, S. Liu, B. J. Trummer, C. Deng, and A. Wang, Carbohydratemicroarrays for the recognition of cross-reactive molecular markers of mi-crobes and host cells. Nature Biotechnology, 2002, 20, 275–281.
[94] C. Galustian, C. G. Park, W. Chai, M. Kiso, S. A. Bruening, Y.-S. Kang,R. M. Steinman, and T. Feizi, High and low affinity carbohydrate ligandsrevealed for murine SIGN-R1 by carbohydrate array and cell binding ap-proaches, and differing specificities for SIGN-R3 and langerin. InternationalImmunology, 2004, 16, 853–866.
[95] W. G. T. Willats, S. E. Rasmussen, T. Kristensen, J. D. Mikkelsen, andJ. P. Knox, Sugar-coated microarrays: A novel slide surface for the high-throughput analysis of glycans. Proteomics, 2002, 2, 1666–1671.
[96] Y. Miura, H. Sato, T. Ikeda, H. Sugimura, O. Takai, and K. Kobayashi,Micropatterned carbohydrate displays by self-assembly of glycoconjugatepolymers on hydrophobic templates on silicon. Biomacromolecules, 2004, 5,1708–1713.
[97] K.-S. Ko, F. A. Jaipuri, and N. L. Pohl, Fluorous-based carbohydrate mi-croarrays. Journal of the American Chemical Society, 2005, 127, 13162–13163.
[98] F. Fazio, M. C. Bryan, O. Blixt, J. C. Paulson, and C.-H. Wong, Synthesis ofsugar arrays in microtiter plate. Journal of the American Chemical Society,2002, 124, 14397–14402.
[99] M. C. Bryan, L. V. Lee, and C.-H. Wong, High-throughput identification offucosyltransferase inhibitors using carbohydrate microarrays. Bioorganic &Medicinal Chemistry Letters, 2004, 14, 3185–3188.
[100] F. Fazio, M. C. Bryan, H.-K. Lee, A. Chang, and C.-H. Wong, Assembly ofsugars on polystyrene plates: a new facile microarray fabrication technique.Tetrahedron Letters, 2004, 45, 2689–2692.
[101] G.-L. Huang, T.-C. Liu, M.-X. Liu, and X.-Y. Mei, The application of quantumdots as fluorescent label to glycoarray. Analytical Biochemistry, 2005, 340,52–56.
[102] S. Zielen, M. Bröker, N. Strnad, L. Schwenen, P. Schön, G. Gottwald, andD. Hofmann, Simple determination of polysaccharide specific antibodiesby means of chemically modified ELISA plates. Journal of ImmunologicalMethods, 1996, 193, 1–7.
[103] A. Satoh, K. Kojima, T. Koyama, H. Ogawa, and I. Matsumoto, Immobiliza-tion of saccharides and peptides on 96-well microtiter plates coated withmethyl vinyl ether-maleic anhydride copolymer. Analytical Biochemistry,1998, 260, 96–102.
[104] M. C. Bryan, F. Fazio, H.-K. Lee, C.-Y. Huang, A. Chang, M. D. Best, D. A.Calarese, O. Blixt, J. C. Paulson, D. Burton, I. A. Wilson, and C.-H. Wong,Covalent display of oligosaccharide arrays in microtiter plates. Journal ofthe American Chemical Society, 2004, 126, 8640–8641.
[105] M. Mizuno, M. Noguchi, M. Imai, T. Motoyoshi, and T. Inazu, Interactionassay of oligosaccharide with lectins using glycosylasparagine. Bioorganic& Medicinal Chemistry Letters, 2004, 14, 485–490.
- 63 -

Glycoconjugates
[106] D. A. Calarese, H.-K. Lee, C.-Y. Huang, M. D. Best, R. D. Astronomo, R. L.Stanfield, H. Katinger, D. R. Burton, C.-H. Wong, and I. A. Wilson, Dis-section of the carbohydrate specificity of the broadly neutralizing anti-HIV-1 antibody 2G12. Proceedings of the National Academy of Sciences of theUnited States of America, 2005, 102, 13372–13377.
[107] N. Horan, L. Yan, H. Isobe, G. M. Whitesides, and D. Kahne, Nonstatisticalbinding of a protein to clustered carbohydrates. Proceedings of the NationalAcademy of Sciences of the United States of America, 1999, 96, 11782–11786.
[108] B. T. Houseman and M. Mrksich, The role of ligand density in the enzymaticglycosylation of carbohydrates presented on self-assembled monolayers ofalkanethiolates on gold. Angewandte Chemie-International Edition, 1999,38, 782–785.
[109] B. T. Houseman and M. Mrksich, Carbohydrate arrays for the evaluation ofprotein binding and enzymatic modification. Chemistry & Biology, 2002, 9,443–454.
[110] S. Park and I. Shin, Fabrication of carbohydrate chips for studyingprotein-carbohydrate interactions. Angewandte Chemie-International Edi-tion, 2002, 41, 3180–3182.
[111] B. T. Houseman, E. S. Gawalt, and M. Mrksich, Maleimide-functionalizedself-assembled monolayers for the preparation of peptide and carbohydratebiochips. Langmuir, 2003, 19, 1522–1531.
[112] M. Köhn, R. Wacker, C. Peters, H. Schröder, L. Soulère, R. Breinbauer, C. M.Niemeyer, and H. Waldmann, Staudinger ligation: A new immobilizationstrategy for the preparation of small-molecule arrays. Angewandte Chemie-International Edition, 2003, 42, 5830–5834.
[113] E. A. Smith, W. D. Thomas, L. L. Kiessling, and R. M. Corn, Surface plasmonresonance imaging studies of protein-carbohydrate interactions. Journal ofthe American Chemical Society, 2003, 125, 6140–6148.
[114] E. W. Adams, D. M. Ratner, H. R. Bokesch, J. B. McMahon, B. R. O’Keefe,and P. H. Seeberger, Oligosaccharide and glycoprotein microarrays as toolsin HIV glycobiology: Glycan-dependent gp120/protein interactions. Chem-istry & Biology, 2004, 11, 875–881.
[115] O. Blixt, S. Head, T. Mondala, C. Scanlan, M. E. Huflejt, R. Alvarez, M. C.Bryan, F. Fazio, D. Calarese, J. Stevens, N. Razi, D. J. Stevens, J. J. Ske-hel, I. van Die, D. R. Burton, I. A. Wilson, R. Cummings, N. Bovin, C.-H.Wong, and J. C. Paulson, Printed covalent glycan array for ligand profilingof diverse glycan binding proteins. Proceedings of the National Academy ofSciences of the United States of America, 2004, 101, 17033–17038.
[116] M. D. Disney and P. H. Seeberger, The use of carbohydrate microarrays tostudy carbohydrate-cell interactions and to detect pathogens. Chemistry &Biology, 2004, 11, 1701–1707.
[117] S. Park, M.-r. Lee, S.-J. Pyo, and I. Shin, Carbohydrate chips for studyinghigh-throughput carbohydrate-protein interactions. Journal of the Ameri-can Chemical Society, 2004, 126, 4812–4819.
[118] D. M. Ratner, E. W. Adams, J. Su, B. R. O’Keefe, M. Mrksich, and P. H.Seeberger, Probing protein-carbohydrate interactions with microarrays ofsynthetic oligosaccharides. ChemBioChem, 2004, 5, 379–382.
[119] S. Angeloni, J. L. Ridet, N. Kusy, H. Gao, F. Crevoisier, S. Guinchard,
- 64 -

Solid-phase synthesis and biological applications
S. Kochhar, H. Sigrist, and N. Sprenger, Glycoprofiling with micro-arraysof glycoconjugates and lectins. Glycobiology, 2005, 15, 31–41.
[120] M. B. Biskup, J. U. Müller, R. Weingart, and R. R. Schmidt, New methods forthe generation of carbohydrate arrays on glass slides and their evaluation.ChemBioChem, 2005, 6, 1007–1015.
[121] M.-r. Lee and I. Shin, Facile preparation of carbohydrate microarraysby site-specific, covalent immobilization of unmodified carbohydrates onhydrazide-coated glass slides. Organic Letters, 2005, 7, 4269–4272.
[122] E. W. Adams, J. Ueberfeld, D. M. Ratner, B. R. O’Keefe, D. R. Walt,and P. H. Seeberger, Encoded fiber-optic microsphere arrays for probingprotein-carbohydrate interactions. Angewandte Chemie-International Edi-tion, 2003, 42, 5317–5320.
[123] M. Mrksich, A surface chemistry approach to studying cell adhesion. Chem-ical Society Reviews, 2000, 29, 267–273.
[124] E. Fischer, Ueber die glucoside der alkohole. Berichte der Deutschen chemis-chen Gesellschaft, 1893, 26, 2400–2412.
[125] J. Banoub, P. Boullanger, and D. Lafont, Synthesis of oligosaccharides of2-amino-2-deoxy sugars. Chemical Reviews, 1992, 92, 1167–1195.
[126] L. F. Bornaghi and S.-A. Poulsen, Microwave-accelerated Fischer glycosyla-tion. Tetrahedron Letters, 2005, 46, 3485–3488.
[127] M. Elofsson, J. Broddefalk, T. Ekberg, and J. Kihlberg, Synthesis of a water-soluble serine-based neoglycolipid which can be covalently linked to solidphases. Carbohydrate Research, 1994, 258, 123–133.
[128] L. Clary, J. Greiner, C. Santaella, and P. Vierling, Synthesis of single- anddouble-chain fluorocarbon and hydrocarbon β -linked galactose amphiphilesderived from serine. Tetrahedron Letters, 1995, 36, 539–542.
[129] Y. Nagao, T. Nekado, K. Ikeda, and K. Achiwa, Synthesis of ganglioside GM3and GM4 analogs having mimics of ceramide moieties and their binding ac-tivities with influenza virus A. Chemical & Pharmaceutical Bulletin, 1995,43, 1536–1542.
[130] H. Yoshida, K. Ikeda, K. Achiwa, and H. Hoshino, Synthesis of sulfatedcerebroside analogs having mimicks of ceramide and their anti-human im-munodeficiency virus type 1 activities. Chemical & Pharmaceutical Bulletin,1995, 43, 594–602.
[131] K. Ikeda, T. Asahara, K. Achiwa, and H. Hoshino, Synthesis of N-acetylglucosaminyl- and N-acetylgalactosaminylceramides as cerebrosideanalogs and their anti-human immunodeficiency virus type 1 activities.Chemical & Pharmaceutical Bulletin, 1997, 45, 402–405.
[132] S.-I. Nishimura and K. Yamada, Transfer of ganglioside GM3 oligosaccha-ride from a water soluble polymer to ceramide by ceramide glycanase. Anovel approach for the chemical-enzymic synthesis of glycosphingolipids.Journal of the American Chemical Society, 1997, 119, 10555–10556.
[133] M. S. Mulligan, R. L. Warner, J. B. Lowe, P. L. Smith, Y. Suzuki,M. Miyasaka, S. Yamaguchi, Y. Ohta, Y. Tsukada, M. Kiso, A. Hasegawa,and P. A. Ward, In vitro and in vivo selectin-blocking activities of sulfatedlipids and sulfated sialyl compounds. International Immunology, 1998, 10,569–575.
[134] K. Yamada, E. Fujita, and S.-I. Nishimura, High performance polymer sup-
- 65 -

Glycoconjugates
ports for enzyme-assisted synthesis of glycoconjugates. Carbohydrate Re-search, 1998, 305, 443–461.
[135] L. Clary, C. Gadras, J. Greiner, J.-P. Rolland, C. Santaella, P. Vierling, andA. Gulik, Phase behavior of fluorocarbon and hydrocarbon double-chainhydroxylated and galactosylated amphiphiles and bolaamphiphiles. Long-term shelf-stability of their liposomes. Chemistry and Physics of Lipids,1999, 99, 125–137.
[136] K. D. McReynolds, M. J. Hadd, and J. Gervay-Hague, Synthesis of biotiny-lated glycoconjugates and their use in a novel ELISA for direct comparisonof HIV-1 gp120 recognition of GalCer and related carbohydrate analogues.Bioconjugate Chemistry, 1999, 10, 1021–1031.
[137] T. Nishikawa, J. Nishida, R. Ookura, S.-I. Nishimura, S. Wada, T. Karino,and M. Shimomura, Mesoscopic patterning of cell adhesive substratesas novel biofunctional interfaces. Materials Science & Engineering, C:Biomimetic and Supramolecular Systems, 1999, 10, 141–146.
[138] T. Nishikawa, J. Nishida, R. Ookura, S.-I. Nishimura, S. Wada, T. Karino,and M. Shimomura, Honeycomb-patterned thin films of amphiphilic poly-mers as cell culture substrates. Materials Science & Engineering, C:Biomimetic and Supramolecular Systems, 1999, 8-9, 495–500.
[139] K. Yamada, S. Matsumoto, and S.-I. Nishimura, Efficient synthesis ofnon-natural ganglioside (pseudo-GM3) and fluorescent labeled lysoGM3 onthe basis of polymer-assisted enzymic strategy. Chemical Communications,1999, 507–508.
[140] B. Faroux-Corlay, L. Clary, C. Gadras, D. Hammache, J. Greiner, C. San-taella, A.-M. Aubertin, P. Vierling, and J. Fantini, Synthesis of single-and double-chain fluorocarbon and hydrocarbon galactosyl amphiphiles andtheir anti-HIV-1 activity. Carbohydrate Research, 2000, 327, 223–260.
[141] E. Fujita and S.-I. Nishimura, Density dependent interaction of polymericanalogs of β -galactosyl ceramide with gp120 of human immunodeficiencyvirus 1. Carbohydrate Letters, 2000, 4, 53–60.
[142] T. Nishikawa, J. Nishida, R. Ookura, S.-I. Nishimura, V. Scheumann, M. Zi-zlsperger, R. Lawall, W. Knoll, and M. Shimomura, Web-structured films ofan amphiphilic polymer from water in oil emulsion: Fabrication and char-acterization. Langmuir, 2000, 16, 1337–1342.
[143] B. Faroux-Corlay, J. Greiner, R. Terreux, D. Cabrol-Bass, A.-M. Aubertin,P. Vierling, and J. Fantini, Amphiphilic anionic analogues of galactosyl-ceramide: Synthesis, anti-HIV-1 activity, and gp120 binding. Journal ofMedicinal Chemistry, 2001, 44, 2188–2203.
[144] J. Nishida, K. Nishikawa, S.-I. Nishimura, S. Wada, T. Karino, T. Nishikawa,K. Ijiro, and M. Shimomura, Preparation of self-organized micro-patternedpolymer films having cell adhesive ligands. Polymer Journal, 2002, 34, 166–174.
[145] K. Yoshiizumi, F. Nakajima, R. Dobashi, N. Nishimura, and S. Ikeda, Stud-ies on scavenger receptor inhibitors. Part 1: Synthesis and structure-activityrelationships of novel derivatives of sulfatides. Bioorganic & MedicinalChemistry, 2002, 10, 2445–2460.
[146] K. Fukunaga, M. Yoshida, F. Nakajima, R. Uematsu, M. Hara, S. Inoue,H. Kondo, and S.-I. Nishimura, Design, synthesis, and evaluation of β -
- 66 -

Solid-phase synthesis and biological applications
galactosylceramide mimics promoting β -glucocerebrosidase activity in ker-atinocytes. Bioorganic & Medicinal Chemistry Letters, 2003, 13, 813–815.
[147] N. Nagahori, K. Niikura, R. Sadamoto, K. Monde, and S.-I. Nishimura, Syn-thesis of photopolymerizable glycolipids and their application as scaffoldsto immobilize proteins with a micron-sized pattern. Australian Journal ofChemistry, 2003, 56, 567–576.
[148] K. Niikura, N. Osuga, N. Nagahori, R. Sadamoto, M. Shiono, N. Iwasaki,K. Monde, A. Minami, and S.-I. Nishimura, Fluorescent glyconanoparticlesas a sensitive device to monitor sugar-involving molecular events. PolymerJournal, 2004, 36, 209–218.
[149] G.-T. Fan, Y.-s. Pan, K.-C. Lu, Y.-P. Cheng, W.-C. Lin, S. Lin, C.-H. Lin, C.-H.Wong, J.-M. Fang, and C.-C. Lin, Synthesis of α-galactosyl ceramide and therelated glycolipids for evaluation of their activities on mouse splenocytes.Tetrahedron, 2005, 61, 1855–1862.
[150] A. Toda, K. Yamada, and S.-I. Nishimura, An engineered biocatalystfor the synthesis of glycoconjugates: Utilization of β1,3-N-acetyl-D-glucosaminyltransferase from Streptococcus agalactiae type Ia expressed inEscherichia coli as a fusion with maltose-binding protein. Advanced Synthe-sis & Catalysis, 2002, 344, 61–69.
[151] W. A. Ernst, J. Maher, S. Cho, K. R. Niazi, D. Chatterjee, D. B. Moody, G. S.Besra, Y. Watanabe, P. E. Jensen, S. A. Porcelli, M. Kronenberg, and R. L.Modlin, Molecular interaction of CD1b with lipoglycan antigens. Immunity,1998, 8, 331–340.
[152] O. V. Naidenko, J. K. Maher, W. A. Ernst, T. Sakai, R. L. Modlin, and M. Kro-nenberg, Binding and antigen presentation of ceramide-containing glycol-ipids by soluble mouse and human CD1d molecules. The Journal of Experi-mental Medicine, 1999, 190, 1069–1079.
[153] T. Sakai, O. V. Naidenko, H. Iijima, M. Kronenberg, and Y. Koezuka, Syn-theses of biotinylated α-galactosylceramides and their effects on the im-mune system and CD1 molecules. Journal of Medicinal Chemistry, 1999,42, 1836–1841.
[154] G. Magnusson, S. Ahlfors, J. Dahmén, K. Jansson, U. Nilsson, G. Noori,K. Stenvall, and A. Tjörnebo, Prespacer glycosides in glycoconjugate chem-istry. Dibromoisobutyl glycosides for the synthesis of neoglycolipids, neo-glycoproteins, neoglycoparticles, and soluble glycosides. Journal of OrganicChemistry, 1990, 55, 3932–3946.
[155] M. Wilstermann, L. O. Kononov, U. Nilsson, A. K. Ray, and G. Magnusson,Synthesis of ganglioside lactams corresponding to GM1-, GM2-, GM3-, andGM4-ganglioside lactones. Journal of the American Chemical Society, 1995,117, 4742–4754.
[156] L. Brossay, O. Naidenko, N. Burdin, J. Matsuda, T. Sakai, and M. Kronen-berg, Cutting edge: Structural requirements for galactosylceramide recog-nition by CD1-restricted NK T cells. The Journal of Immunology, 1998, 161,5124–5128.
[157] T. Kawano, J. Cui, Y. Koezuka, I. Toura, Y. Kaneko, K. Motoki, H. Ueno,R. Nakagawa, H. Sato, E. Kondo, H. Koseki, and M. Taniguchi, CD1d-restricted and TCR-mediated activation of Vα14 NKT cells by glycosylce-ramides. Science, 1997, 278, 1626–1629.
- 67 -

Glycoconjugates
[158] S.-i. Fujii, K. Shimizu, M. Kronenberg, and R. M. Steinman, Prolonged IFN-γ-producing NKT response induced with α-galactosylceramide-loaded DCs.Nature Immunology, 2002, 3, 867–874.
[159] I. J. Goldstein and R. D. Poretz, In The lectins: Properties, functions, andapplications in biology and medicine (I. E. Liener, N. Sharon, and I. J. Gold-stein, eds.). Academic Press, Inc., Orlando, 1986, 119–196.
[160] P. Sjölin, M. Elofsson, and J. Kihlberg, Removal of acyl protective groupsfrom glycopeptides: Base does not epimerize peptide stereocenters, and β -elimination is slow. Journal of Organic Chemistry, 1996, 61, 560–565.
[161] K. M. Wells, Y.-J. Shi, J. E. Lynch, G. R. Humphrey, R. P. Volante, and P. J.Reider, Regioselective nucleophilic substitutions of fluorobenzene deriva-tives. Tetrahedron Letters, 1996, 37, 6439–6442.
[162] P. R. Guzzo, R. N. Buckle, M. Chou, S. R. Dinn, M. E. Flaugh, A. D. Kiefer Jr.,K. T. Ryter, A. J. Sampognaro, S. W. Tregay, and Y.-C. Xu, Preparation of 8-amido-2-dimethylamino-1,2,3,4-tetrahydro-2-dibenzofurans and several flu-orinated derivatives via [3,3]-sigmatropic rearrangement of O-aryloximes.Journal of Organic Chemistry, 2003, 68, 770–778.
[163] A. Tomazic and J. B. Hynes, Synthesis of some novel halogenated 2,4-diaminoquinazolines. Journal of Heterocyclic Chemistry, 1992, 29, 915–20.
[164] R. B. Silverman and W. P. Hawe, SAR studies of fluorine-substituted ben-zylamines and substituted 2-phenylethylamines as substrates and inactiva-tors of monoamine oxidase B. Journal of Enzyme Inhibition, 1995, 9, 203–15.
[165] A. J. Bridges and H. Zhou, Synthesis of [1]benzothieno[3,2-d]pyrimidinessubstituted with electron donating substituents on the benzene ring. Jour-nal of Heterocyclic Chemistry, 1997, 34, 1163–1172.
[166] I. M. Bell, J. M. Erb, R. M. Freidinger, S. N. Gallicchio, J. P. Guare,M. T. Guidotti, R. A. Halpin, D. W. Hobbs, C. F. Homnick, M. S. Kuo,E. V. Lis, D. J. Mathre, S. R. Michelson, J. M. Pawluczyk, D. J. Pettibone,D. R. Reiss, S. Vickers, P. D. Williams, and C. J. Woyden, Developmentof orally active oxytocin antagonists: Studies on 1-(1-{4-[1-(2-methyl-1-oxidopyridin-3-ylmethyl)piperidin-4-yloxy]-2-methoxybenzoyl}piperidin-4-yl)-1,4-dihydrobenz[d][1,3]oxazin-2-one (L-372,662) and related pyridines.Journal of Medicinal Chemistry, 1998, 41, 2146–2163.
[167] C.-A. Chen, R.-H. Yeh, and D. S. Lawrence, Design and synthesis of a fluo-rescent reporter of protein kinase activity. Journal of the American Chemi-cal Society, 2002, 124, 3840–3841.
- 68 -


















