
Glycerol Dehydration to Acrolein in the Context of New Uses of Glycerol
20
CRITICAL REVIEW www.rsc.org/greenchem | Green Chemistry Glycerol dehydration to acrolein in the context of new uses of glycerol Benjamin Katryniok, a,b S´ ebastien Paul, a,b,c Virginie Belli` ere-Baca, d Patrick Rey e and Franck Dumeignil* a,b Received 9th July 2010, Accepted 5th October 2010 DOI: 10.1039/c0gc00307g Catalytic dehydration of glycerol to acrolein has the potential to valorise the glut of crude glycerol issuing from biodiesel production. This reaction requires catalysts with appropriate acidity, and intensive research activities have been focused on the application of families of catalysts including zeolites, heteropolyacids, mixed metal oxides and (oxo)-pyrophosphates, as their acidic properties are well-known. Nevertheless, their deactivation by coking remains the main obstacle in the way of large-scale industrial applications. Considering this important issue, various technologies have been proposed for regenerating the catalysts. This review shows that a well-balanced combination of an appropriate catalytic system together with an adapted regeneration process could put large-scale industrial applications within reach. 1 Introduction The finite reserves of fossil-fuel feedstocks have encouraged intensive research activities for developing substitutes/additives for fuels such as biodiesel, bioethanol or biokerosene. Due to their origin from biomass, they have the strong advantage of a lower carbon footprint than fuels derived from fossil-fuel resources. However, from an economic point of view, it is not yet possible to produce any of these biofuels competitively. Nevertheless, political decisions have pushed the production of fuels from bioresources in order to be able to fulfill the CO 2 reduction objectives fixed by the Kyoto climate protocol. 1 For example, the European Union has planned a progressive increase in the mandatory proportion of bioethanol blended in gasoline, and of biodiesel blended in diesel fuel. The blended amounts must reach 10% and 7% by 2015, respectively. 2 The alternative gasoline production route using vegetable oils and fats has attracted the attention of many academic and industrial researchers. 3 The raw materials for biodiesel production are vegetable oils and fats – from canola, soy, corn, etc. – and a mono-alcohol (usually methanol), which is used to cleave the fatty acids from their glycerol backbone to yield the desired fatty acid esters (Scheme 1). These esters can be directly used as biodiesels, but they are commonly blended with diesels derived from fossil-fuels to meet the regulations. Scheme 1 Transesterification reaction of vegetable oils to yield biodiesel. a Univ. Lille Nord de France, F-59000, Lille, France. E-mail: [email protected]; Fax: +33 (0)3.20.43.65.61; Tel: +33 (0)3.20.43.45.38 b CNRS UMR8181, Unit´ e de Catalyse et Chimie du Solide, UCCS, F-59655, Villeneuve d’Ascq, France c ECLille, F-59655, Villeneuve d’Ascq, France d RHODIA France, 52 Rue de la Haie Coq, 93308, Aubervilliers, France e ADISSEO France SAS, Antony Parc 2-10, 92160, Antony, France The capacity of biodiesel production is expanding all over the world. For example, in 2008, the USA produced 2.3 million tonnes of biodiesel, while the EU produced 7.8 million tonnes (Fig. 1), and these values are expected these to double by 2012. 4,5 This growth is accompanied by a significant increase in glycerol production, as this a significant by-product (~10 wt%) of the biodiesel process (Scheme 1). Simple projections enable one to forecast that 1.54 million tonnes of glycerol will be generated worldwide in 2015, 6 all of which will have to be efficiently processed in order to achieve a sustainable business. Fig. 1 Comparative evolution of the quantities of biodiesel produced in the EU and the US. 5,7 Depending on the process used for the cleavage of the fatty acids, the purity of the crude glycerol can vary greatly. The crude glycerol obtained from most of the conventional biodiesel processes contains ca. 80 wt% of glycerol, but it also contains water, methanol, traces of fatty acids as well as various inorganic and organic compounds (called ‘MONG’, denoting ‘Matter Organic Non-Glycerol’) (Table 1). As a consequence, in most of the cases, crude glycerol must be purified by an expensive distillation step prior to further use, to meet the requirements of the downstream processes. The proportion of glycerol that is refined is actually steadily decreasing, due to the high cost of the distillation step, together with a rapid growth of the quantity of crude glycerol produced, and also (primarily) because of the absence of any market able to absorb the massive overproduction (Fig. 2). In fact, an increase in the price of glycerol – together with a sustainable and significant demand – would automatically result in a decrease in This journal is © The Royal Society of Chemistry 2010 Green Chem., 2010, 12, 2079–2098 | 2079 Published on 12 November 2010 on http://pubs.rsc.org | doi:10.1039/C0GC00307G View Article Online / Journal Homepage / Table of Contents for this issue
-
Upload
bernyke-contracy -
Category
Documents
-
view
112 -
download
9
Transcript of Glycerol Dehydration to Acrolein in the Context of New Uses of Glycerol
CRITICAL REVIEW www.rsc.org/greenchem | Green Chemistry
Glycerol dehydration to acrolein in the context of new uses of glycerol
Benjamin Katryniok,a,b Sebastien Paul,a,b,c Virginie Belliere-Baca,d Patrick Reye and Franck Dumeignil*a,b
Received 9th July 2010, Accepted 5th October 2010DOI: 10.1039/c0gc00307g
Catalytic dehydration of glycerol to acrolein has the potential to valorise the glut of crude glycerolissuing from biodiesel production. This reaction requires catalysts with appropriate acidity, andintensive research activities have been focused on the application of families of catalysts includingzeolites, heteropolyacids, mixed metal oxides and (oxo)-pyrophosphates, as their acidic propertiesare well-known. Nevertheless, their deactivation by coking remains the main obstacle in the way oflarge-scale industrial applications. Considering this important issue, various technologies have beenproposed for regenerating the catalysts. This review shows that a well-balanced combination of anappropriate catalytic system together with an adapted regeneration process could put large-scaleindustrial applications within reach.
1 Introduction
The finite reserves of fossil-fuel feedstocks have encouragedintensive research activities for developing substitutes/additivesfor fuels such as biodiesel, bioethanol or biokerosene. Due totheir origin from biomass, they have the strong advantage ofa lower carbon footprint than fuels derived from fossil-fuelresources. However, from an economic point of view, it is notyet possible to produce any of these biofuels competitively.Nevertheless, political decisions have pushed the production offuels from bioresources in order to be able to fulfill the CO2
reduction objectives fixed by the Kyoto climate protocol.1 Forexample, the European Union has planned a progressive increasein the mandatory proportion of bioethanol blended in gasoline,and of biodiesel blended in diesel fuel. The blended amountsmust reach 10% and 7% by 2015, respectively.2
The alternative gasoline production route using vegetableoils and fats has attracted the attention of many academicand industrial researchers.3 The raw materials for biodieselproduction are vegetable oils and fats – from canola, soy, corn,etc. – and a mono-alcohol (usually methanol), which is used tocleave the fatty acids from their glycerol backbone to yield thedesired fatty acid esters (Scheme 1). These esters can be directlyused as biodiesels, but they are commonly blended with dieselsderived from fossil-fuels to meet the regulations.
Scheme 1 Transesterification reaction of vegetable oils to yieldbiodiesel.
aUniv. Lille Nord de France, F-59000, Lille, France.E-mail: [email protected]; Fax: +33 (0)3.20.43.65.61;Tel: +33 (0)3.20.43.45.38bCNRS UMR8181, Unite de Catalyse et Chimie du Solide, UCCS,F-59655, Villeneuve d’Ascq, FrancecECLille, F-59655, Villeneuve d’Ascq, FrancedRHODIA France, 52 Rue de la Haie Coq, 93308, Aubervilliers, FranceeADISSEO France SAS, Antony Parc 2-10, 92160, Antony, France
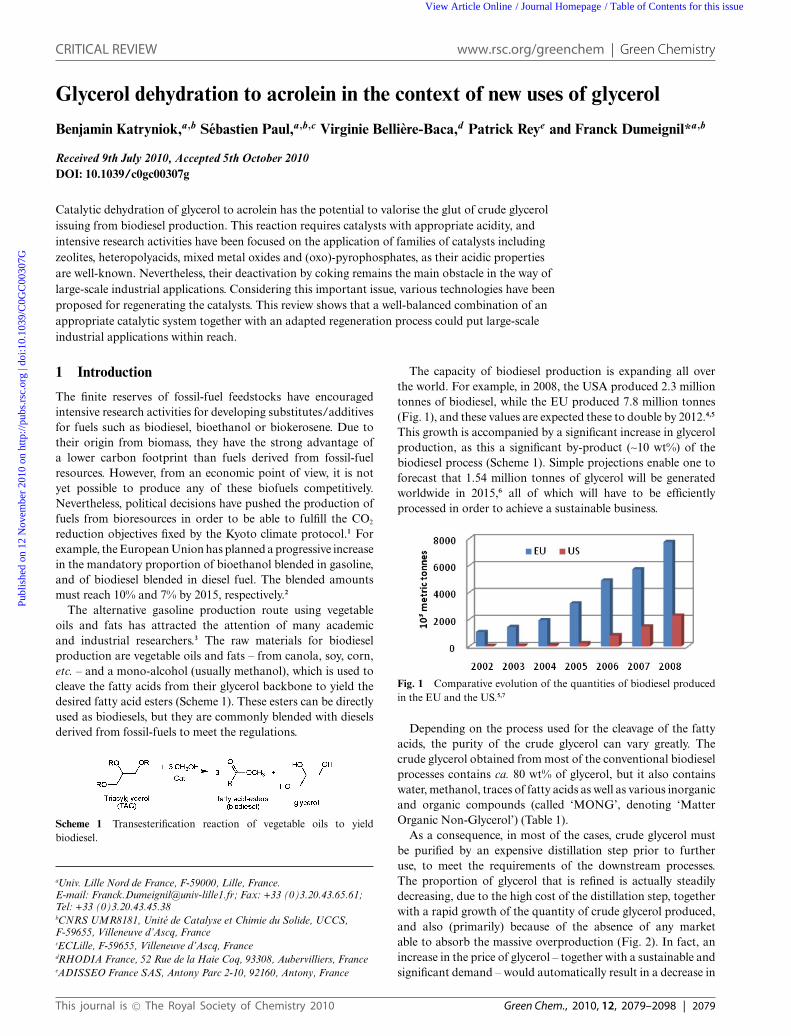
The capacity of biodiesel production is expanding all overthe world. For example, in 2008, the USA produced 2.3 milliontonnes of biodiesel, while the EU produced 7.8 million tonnes(Fig. 1), and these values are expected these to double by 2012.4,5
This growth is accompanied by a significant increase in glycerolproduction, as this a significant by-product (~10 wt%) of thebiodiesel process (Scheme 1). Simple projections enable one toforecast that 1.54 million tonnes of glycerol will be generatedworldwide in 2015,6 all of which will have to be efficientlyprocessed in order to achieve a sustainable business.
Fig. 1 Comparative evolution of the quantities of biodiesel producedin the EU and the US.5,7
Depending on the process used for the cleavage of the fattyacids, the purity of the crude glycerol can vary greatly. Thecrude glycerol obtained from most of the conventional biodieselprocesses contains ca. 80 wt% of glycerol, but it also containswater, methanol, traces of fatty acids as well as various inorganicand organic compounds (called ‘MONG’, denoting ‘MatterOrganic Non-Glycerol’) (Table 1).
As a consequence, in most of the cases, crude glycerol mustbe purified by an expensive distillation step prior to furtheruse, to meet the requirements of the downstream processes.The proportion of glycerol that is refined is actually steadilydecreasing, due to the high cost of the distillation step, togetherwith a rapid growth of the quantity of crude glycerol produced,and also (primarily) because of the absence of any marketable to absorb the massive overproduction (Fig. 2). In fact, anincrease in the price of glycerol – together with a sustainable andsignificant demand – would automatically result in a decrease in
This journal is © The Royal Society of Chemistry 2010 Green Chem., 2010, 12, 2079–2098 | 2079
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online / Journal Homepage / Table of Contents for this issue
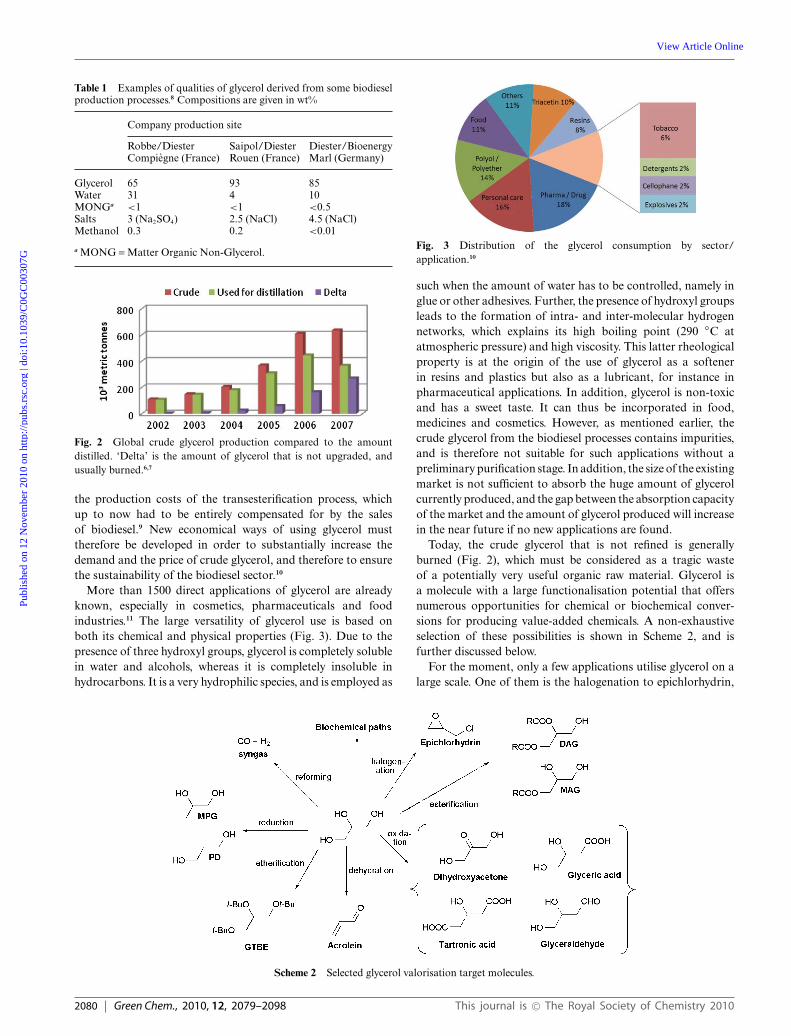
Table 1 Examples of qualities of glycerol derived from some biodieselproduction processes.8 Compositions are given in wt%
Company production site
Robbe/DiesterCompiegne (France)
Saipol/DiesterRouen (France)
Diester/BioenergyMarl (Germany)
Glycerol 65 93 85Water 31 4 10MONGa <1 <1 <0.5Salts 3 (Na2SO4) 2.5 (NaCl) 4.5 (NaCl)Methanol 0.3 0.2 <0.01
a MONG = Matter Organic Non-Glycerol.
Fig. 2 Global crude glycerol production compared to the amountdistilled. ‘Delta’ is the amount of glycerol that is not upgraded, andusually burned.6,7
the production costs of the transesterification process, whichup to now had to be entirely compensated for by the salesof biodiesel.9 New economical ways of using glycerol musttherefore be developed in order to substantially increase thedemand and the price of crude glycerol, and therefore to ensurethe sustainability of the biodiesel sector.10
More than 1500 direct applications of glycerol are alreadyknown, especially in cosmetics, pharmaceuticals and foodindustries.11 The large versatility of glycerol use is based onboth its chemical and physical properties (Fig. 3). Due to thepresence of three hydroxyl groups, glycerol is completely solublein water and alcohols, whereas it is completely insoluble inhydrocarbons. It is a very hydrophilic species, and is employed as
Fig. 3 Distribution of the glycerol consumption by sector/application.10
such when the amount of water has to be controlled, namely inglue or other adhesives. Further, the presence of hydroxyl groupsleads to the formation of intra- and inter-molecular hydrogennetworks, which explains its high boiling point (290 ◦C atatmospheric pressure) and high viscosity. This latter rheologicalproperty is at the origin of the use of glycerol as a softenerin resins and plastics but also as a lubricant, for instance inpharmaceutical applications. In addition, glycerol is non-toxicand has a sweet taste. It can thus be incorporated in food,medicines and cosmetics. However, as mentioned earlier, thecrude glycerol from the biodiesel processes contains impurities,and is therefore not suitable for such applications without apreliminary purification stage. In addition, the size of the existingmarket is not sufficient to absorb the huge amount of glycerolcurrently produced, and the gap between the absorption capacityof the market and the amount of glycerol produced will increasein the near future if no new applications are found.
Today, the crude glycerol that is not refined is generallyburned (Fig. 2), which must be considered as a tragic wasteof a potentially very useful organic raw material. Glycerol isa molecule with a large functionalisation potential that offersnumerous opportunities for chemical or biochemical conver-sions for producing value-added chemicals. A non-exhaustiveselection of these possibilities is shown in Scheme 2, and isfurther discussed below.
For the moment, only a few applications utilise glycerol on alarge scale. One of them is the halogenation to epichlorhydrin,
Scheme 2 Selected glycerol valorisation target molecules.
2080 | Green Chem., 2010, 12, 2079–2098 This journal is © The Royal Society of Chemistry 2010
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

which is an important intermediate for epoxy resins. The processuses hydrochloric acid in the presence of organic acids (caprylicacid – Solvay/acetic acid – Dow)12 as catalysts working in thegaseous phase at 180–220 ◦C under a pressure of 1–5 bar.13
This technology was commercialised in 2007 by Solvay, whichoperates an existing production facility in France that wasformerly used to produce glycerol from epichlorhydrin. Anothercommercialised process for large-scale consumption of glycerolis the reforming over Pt–Rh catalysts to yield syngas. Thislatter can either be used in the Fischer–Tropsch process forthe synthesis of alkanes (Biomass to Liquid, BtL) or for thesynthesis of methanol as performed by BioMCN in Delfzijl(Netherlands) with a capacity of 200 kt/year.14–16 From thistechnology is derived the sustainable production of hydrogen– an energy vector with an expected high potential in the nearfuture – through the water-gas shift process. As another option,the etherification to glycerol tert-butyl ether (GTBE) targetsthe classical petrol-based economy. Indeed, this compoundis an environmentally friendly additive in gasoline, and canbeneficially substitute for the problematic methyl tert-butyl ether(MTBE). Nevertheless, the associated process is still far fromreaching industrial applications.17
The selective reduction and esterification of glycerol aretwo processes with potential impacts on glycerol consumptionat a medium scale. The former can be used to yield eitherpropylene glycol (MPG) or 1,3-propanediol (PD), which arevaluable intermediates in the polymer industry. The latter targetsthe esters of glycerol – namely, monoacylglycerol (MAG) anddiacylglycerol (DAG) – that find applications as emulsifiers,e.g., in food (margarines and sauces) or in cosmetics. Thisprocess can either be catalysed by a conventional catalyst or byenzymes (lipase-type).18 Recently, the production of MPG hasbeen commercialised by Synergy Chemicals with a productioncapacity of 30 kt/year.10,19
A small-scale application, but aiming at the synthesis ofhigh-value fine chemicals, consists on the partial oxidation ofglycerol to carboxylic acids (i.e., glyceric acid, tartronic acid andketomalonic acid), aldehydes (i.e., glyceraldehyde) or ketones(i.e., dihydroxyacetone). The main challenge is to find oxidationcatalysts that are highly selective to the target molecule, amonga large quantity of possible reaction products. One example isthe selective oxidation to dihydroxyacetone over Bi/Pt catalysts,giving 37% yield at 70% conversion of glycerol.20 Oxidationcan also be performed using genetically modified bacteria orelectrochemical reactions.21,22
However, one of the most promising routes to glycerolvalorisation lies in its catalytic dehydration to acrolein, whichis an important industrial intermediate for the chemical and theagro-industries. The synthesis of acrolein is currently based onthe selective oxidation of propene over complex multicomponentBiMoOx-based catalysts (Scheme 3). The selectivity reached inthis process is close to 85% at 95% conversion.23 New approacheswith propane as a starting material are currently being exploredat the laboratory scale, but they suffer from insufficient yields,which are incompatible with further commercialisation.24 Con-
Scheme 3 Catalytically-assisted selective oxidation of propene.
sidering the petrochemical feedstock depletion issues, sustain-able resources will become more and more competitive, not tomention their positive effect in terms of impact on the climate.Within this context, a massive bioresourced and sustainableacrolein production is an important challenge, which has beenaccepted by academic and industrial researchers. An economicsstudy has shown that competitive production of acrolein fromglycerol could be possible if the price of glycerol became lessthan ca. 300 US$/t.25 In January 2010, refined glycerol still cost500–550 US$/t, but crude glycerol was only around 100 US$/t.This makes this latter a potentially very competitive raw materialfor acrolein production, even though some technical difficultiesin the application of crude glycerol have still to be solved, inparticular avoiding catalyst poisoning by the impurities.26
Due to its toxicity, acrolein is usually directly converted tothe desired high value-added derivatives. Most acrolein is usedfor the synthesis of acrylic acid, which is used, for example, as astarting material for synthesising sodium polyacrylate (Scheme4). According to its physical properties, this polymer, classified asa superabsorbent, finds uses in hygiene products, such as diapers.The annual worldwide market for this superabsorbent polymer(SAP) is estimated at 1.9 million tonnes for 2010.27
Scheme 4 Industrial synthesis of sodium polyacrylate, a superab-sorbent polymer.
The second largest consumer of acrolein is represented by thesynthesis of DL-methionine via 3-methylthio-propionaldehydeas an intermediate (Scheme 5).28 DL-Methionine is an essentialamino-acid, which cannot be synthesised by living organisms,and is widely used in meat production to accelerate animalgrowth. The annual worldwide production capacity of DL-methionine is about 500 000 tonnes.29 As natural methioninesources like plants and microorganisms provide concentrationsthat are too low, it has to be synthesised on the industrial scale.It is estimated that the global demand will increase by 3–7% inthe near future.30
Scheme 5 Chemical pathways for the industrial manufacture of DL-methionine.
In the following sections, we will present the state-of-the-art on the sustainable production of acrolein by gas-phasedehydration of glycerol. In fact, since our last review on thedehydration of glycerol,31 the number of publications in this
This journal is © The Royal Society of Chemistry 2010 Green Chem., 2010, 12, 2079–2098 | 2081
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online
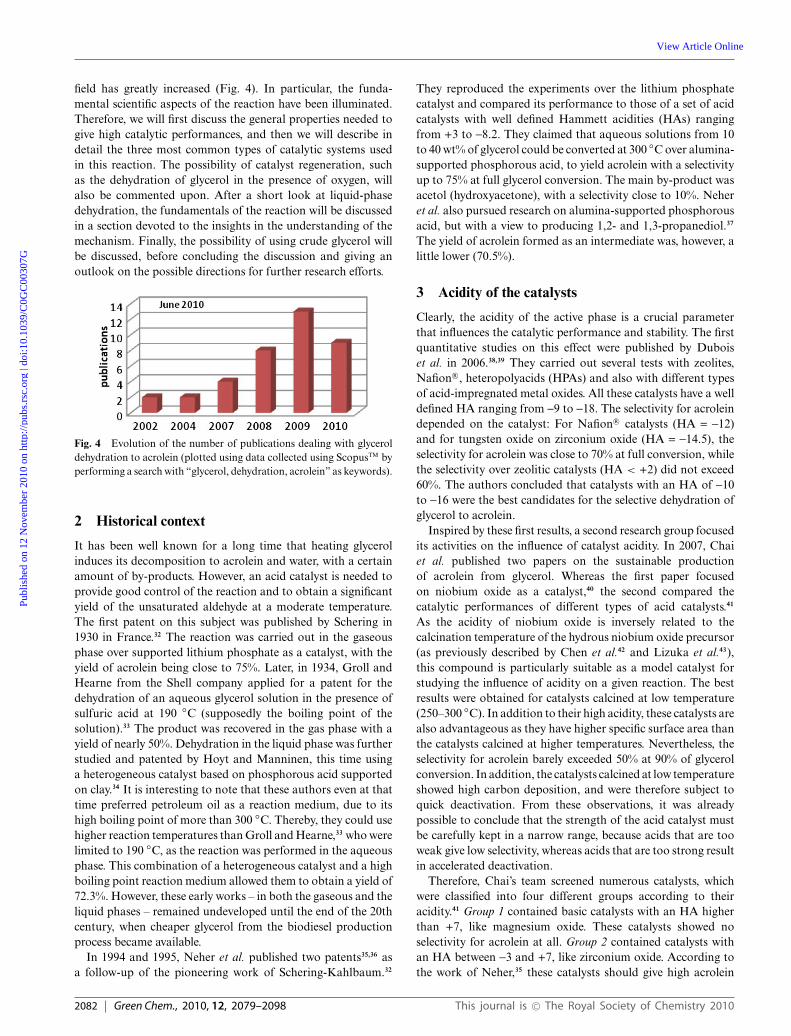
field has greatly increased (Fig. 4). In particular, the funda-mental scientific aspects of the reaction have been illuminated.Therefore, we will first discuss the general properties needed togive high catalytic performances, and then we will describe indetail the three most common types of catalytic systems usedin this reaction. The possibility of catalyst regeneration, suchas the dehydration of glycerol in the presence of oxygen, willalso be commented upon. After a short look at liquid-phasedehydration, the fundamentals of the reaction will be discussedin a section devoted to the insights in the understanding of themechanism. Finally, the possibility of using crude glycerol willbe discussed, before concluding the discussion and giving anoutlook on the possible directions for further research efforts.
Fig. 4 Evolution of the number of publications dealing with glyceroldehydration to acrolein (plotted using data collected using ScopusTM byperforming a search with “glycerol, dehydration, acrolein” as keywords).
2 Historical context
It has been well known for a long time that heating glycerolinduces its decomposition to acrolein and water, with a certainamount of by-products. However, an acid catalyst is needed toprovide good control of the reaction and to obtain a significantyield of the unsaturated aldehyde at a moderate temperature.The first patent on this subject was published by Schering in1930 in France.32 The reaction was carried out in the gaseousphase over supported lithium phosphate as a catalyst, with theyield of acrolein being close to 75%. Later, in 1934, Groll andHearne from the Shell company applied for a patent for thedehydration of an aqueous glycerol solution in the presence ofsulfuric acid at 190 ◦C (supposedly the boiling point of thesolution).33 The product was recovered in the gas phase with ayield of nearly 50%. Dehydration in the liquid phase was furtherstudied and patented by Hoyt and Manninen, this time usinga heterogeneous catalyst based on phosphorous acid supportedon clay.34 It is interesting to note that these authors even at thattime preferred petroleum oil as a reaction medium, due to itshigh boiling point of more than 300 ◦C. Thereby, they could usehigher reaction temperatures than Groll and Hearne,33 who werelimited to 190 ◦C, as the reaction was performed in the aqueousphase. This combination of a heterogeneous catalyst and a highboiling point reaction medium allowed them to obtain a yield of72.3%. However, these early works – in both the gaseous and theliquid phases – remained undeveloped until the end of the 20thcentury, when cheaper glycerol from the biodiesel productionprocess became available.
In 1994 and 1995, Neher et al. published two patents35,36 asa follow-up of the pioneering work of Schering-Kahlbaum.32
They reproduced the experiments over the lithium phosphatecatalyst and compared its performance to those of a set of acidcatalysts with well defined Hammett acidities (HAs) rangingfrom +3 to -8.2. They claimed that aqueous solutions from 10to 40 wt% of glycerol could be converted at 300 ◦C over alumina-supported phosphorous acid, to yield acrolein with a selectivityup to 75% at full glycerol conversion. The main by-product wasacetol (hydroxyacetone), with a selectivity close to 10%. Neheret al. also pursued research on alumina-supported phosphorousacid, but with a view to producing 1,2- and 1,3-propanediol.37
The yield of acrolein formed as an intermediate was, however, alittle lower (70.5%).
3 Acidity of the catalysts
Clearly, the acidity of the active phase is a crucial parameterthat influences the catalytic performance and stability. The firstquantitative studies on this effect were published by Duboiset al. in 2006.38,39 They carried out several tests with zeolites,Nafion R©, heteropolyacids (HPAs) and also with different typesof acid-impregnated metal oxides. All these catalysts have a welldefined HA ranging from -9 to -18. The selectivity for acroleindepended on the catalyst: For Nafion R© catalysts (HA = -12)and for tungsten oxide on zirconium oxide (HA = -14.5), theselectivity for acrolein was close to 70% at full conversion, whilethe selectivity over zeolitic catalysts (HA < +2) did not exceed60%. The authors concluded that catalysts with an HA of -10to -16 were the best candidates for the selective dehydration ofglycerol to acrolein.
Inspired by these first results, a second research group focusedits activities on the influence of catalyst acidity. In 2007, Chaiet al. published two papers on the sustainable productionof acrolein from glycerol. Whereas the first paper focusedon niobium oxide as a catalyst,40 the second compared thecatalytic performances of different types of acid catalysts.41
As the acidity of niobium oxide is inversely related to thecalcination temperature of the hydrous niobium oxide precursor(as previously described by Chen et al.42 and Lizuka et al.43),this compound is particularly suitable as a model catalyst forstudying the influence of acidity on a given reaction. The bestresults were obtained for catalysts calcined at low temperature(250–300 ◦C). In addition to their high acidity, these catalysts arealso advantageous as they have higher specific surface area thanthe catalysts calcined at higher temperatures. Nevertheless, theselectivity for acrolein barely exceeded 50% at 90% of glycerolconversion. In addition, the catalysts calcined at low temperatureshowed high carbon deposition, and were therefore subject toquick deactivation. From these observations, it was alreadypossible to conclude that the strength of the acid catalyst mustbe carefully kept in a narrow range, because acids that are tooweak give low selectivity, whereas acids that are too strong resultin accelerated deactivation.
Therefore, Chai’s team screened numerous catalysts, whichwere classified into four different groups according to theiracidity.41 Group 1 contained basic catalysts with an HA higherthan +7, like magnesium oxide. These catalysts showed noselectivity for acrolein at all. Group 2 contained catalysts withan HA between -3 and +7, like zirconium oxide. According tothe work of Neher,35 these catalysts should give high acrolein
2082 | Green Chem., 2010, 12, 2079–2098 This journal is © The Royal Society of Chemistry 2010
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

selectivities, but, in practice, the selectivity did not exceed 30%,while the performances remained quite stable for 10 h on stream.Group 3, which was more promising, contained catalysts with anHA between -8 and -3. In this group, we find alumina-supportedphosphorous acid next to alumina-supported HPAs, niobiumoxide (calcined at 400–500 ◦C), as well as HZSM zeolite andpure alumina. In good agreement with previous results,38,39 theselectivity was generally higher than that observed for Group 2catalysts, with the exception of pure alumina and niobium oxidecalcined at 500 ◦C. Interesting results were also observed overthe alumina-supported phosphotungstic HPA and over a mixedphase of tungsten oxide/zirconium oxide, with approximately70% selectivity at 70% conversion in both cases after 2 h on-stream. Unfortunately, these catalysts showed poor stability,and their performance significantly decreased during time on-stream (for both catalysts, the glycerol conversion dropped from68–69% after 1 h under reactant flow to 23–25% at 10 h, theselectivity being more-or-less unaffected at roughly 66–70%).The other catalysts were on the whole less efficient, and gaveselectivities of 35–55% at conversions of 55–100% after 10 h on-stream. In Group 4 were included catalysts with a HA less than-8, like Hb-zeolite, niobium oxide calcined at 350 ◦C, and alsoalumina-silicate as well as sulfonated zirconium oxide. Thesecatalysts were less selective to acrolein than those of Group 3,but their performances were less altered with time on-stream.As an example, the glycerol conversion over an aluminosilicatedropped only from 94% after 1 h under reactant flow to 75%after 10 h, the acrolein selectivity being more-or-less unchangedat roughly 43–46%. Nevertheless, like niobium oxide, Group 4catalysts were also subject to detrimental coke formation, witha carbon deposit of 100–400 mg per gram of catalyst after tenhours on-stream.
In addition to the acidic strength of the catalyst, the type of theacid sites present at their surfaces has an important influence ontheir catalytic performance. When Chai et al. classified the cata-lysts according to their strength,41 they mixed up Brønsted andLewis acid catalysts without differentiation. Whereas Brønstedacids donate a proton (as for the aforementioned silicotungsticacid, protonated zeolites or phosphorous acid), Lewis acidsare acceptors for electron pairs (as for niobium oxide or purealumina). From the catalytic results obtained by Chai et al.over these two different types of acids, one can conclude thatthey do not obey the same reaction pathway, as the authorssystematically obtained rather low selectivities for acrolein overLewis acids.41 A more detailed study on the different catalyticbehaviors of Brønsted and Lewis acids in this reaction wasproposed by Alhanash et al.,44 who compared a pure Brønstedacid catalyst (acidic caesium salts of phosphotungstic acid) toa pure Lewis acid catalyst (tin-chromium mixed oxide). Theyshowed that:
∑ Lewis acid catalysts need a higher reaction temperature tobe activated, due to a higher activation energy compared toBrønsted acid catalysts;
∑ Lewis acid catalysts give a larger selectivity for ace-tol, which is the major by-product observed during glyceroldehydration.
These results were confirmed by Kim et al., who comparedHZSM-5 aluminosilicates with different ratios of alumina andsilica.45
According to Alhanash et al.44 the reaction over Brønsted-type acid catalysts starts with the protonation of the secondaryhydroxyl group of glycerol by a proton from a Brønsted site, aspostulated by Buhler for homogenous reaction media (Scheme6a).87 The resulting intermediate is transformed by release of ahydronium ion (H3O+) followed by a keto–enol rearrangement togive 3-hydroxypropionaldehyde and – after a second dehydrationstep – acrolein. The reacted Brønsted acid site can be regenerated(re-protonated) by the aforementioned released hydronium ion.The reaction over Lewis acid sites proceeds in a completelydifferent way (Scheme 6b), as the glycerol interacts via concertedtransfer of a terminal hydroxyl group on one of the metal centersand migration of the secondary proton to the other metal center.Consequently, 2,3-dihydroxypropene and pseudo-Brønsted sitesare formed. The enol will tautomerise to acetol, whereasthe pseudo-Brønsted site can either catalyse the dehydrationreaction, as in the case of the aforementioned Brønsted sites,or can regenerate the initial Lewis site by thermal dehydration.Thereby, one can explain why Lewis acid sites generally showhigher selectivity for acetol than Brønsted acid sites.
Scheme 6 Reaction mechanism over basic catalysts, as proposed byKinage et al.44
As catalysts usually contain both acidic and basic sites,the influence of the basicity must also be considered. Kinageet al. investigated the selective dehydration of glycerol to acetoland proposed a reaction mechanism over basic catalysts.46 Incontrast to the mechanisms proposed over acid catalysts, thereaction over basic catalysts does not begin with a dehydrationstep but with a dehydrogenation step (Scheme 7). The resulting2,3-dihydroxypropanal can either further react via consecutivedehydration and hydrogenation to yield acetol, or can form – viaa retro-aldol reaction – formaldehyde and hydroxyacetaldehyde,which can be subsequently hydrogenated to ethylene glycol. Withthis reaction mechanism, one can explain the high selectivity foracetol observed by Chai et al. over basic catalysts.41
In this section, we have seen that the strength and the type ofthe catalyst acidity are of utmost importance for obtaining highcatalytic performances. In the following sections, we will furtherdiscuss in detail the three most widely used catalytic systems for
This journal is © The Royal Society of Chemistry 2010 Green Chem., 2010, 12, 2079–2098 | 2083
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

Scheme 7 Reaction mechanism over protonated zeolite (H-MFI) catalyst as proposed by Yoda et al.46
the dehydration of glycerol: supported HPAs, zeolites, and thefamily of metal oxides, phosphates and pyrophosphates.
4 Supported heteropolyacids
The use of supported inorganic acids such as phosphorousacid or HPAs is one possibility to reach high performancesin the dehydration of glycerol to acrolein, as shown by Neherand Chai.35,36,41 However, when using supported catalysts, newparameters in addition to acidity must be taken into account,namely the influence of the support on the active-phase prop-erties, the dispersion extent of the active phase on the support,and the distribution of the pore size of the support.
In 2008 and 2009, Chai et al. published two papers dealingwith zirconia- and silica-supported HPA catalysts.47,48 Thesestudies showed that the nature of the catalyst support has asignificant effect on the thermal stability and on the dispersionof the Keggin-type active phase. The use of zirconia as a supportled to better results, as far as the deactivation of the catalystis concerned. The Keggin-anion density at the surface of thesupport was identified as being key for tuning the activity andthe selectivity of the HPA for acrolein production, which isin good agreement with the conclusions of Ning et al., whoused activated carbon-supported silicotungstic acid catalysts.49
The latter claimed that a 10 wt% silicotungstic acid-supportedcatalyst gives the best acrolein space-time yield ever reportedin the literature (namely 68.5 mmol acrolein h-1 g-1), but thisvalue is misleading, as the calculation is based on the mass ofactive phase and not on the mass of catalyst. This promisingperformance is attributed to the good dispersion of the HPAon the support surface and also to the relative quantities ofstrong acid sites. Our team also recently used silica-supportedsilicotungstic acid as a catalyst, but on supports that were
grafted with zirconia before impregnation of the active phase.50
We showed that the strong electronic interaction between thezirconia and the heteropolyanion results in a decreased strengthof the Brønsted acid sites, whereby the long-term catalyticperformances were increased in comparison to bare silica (69%acrolein vs. 24% respectively, after 24 h on-stream) due to lesscoking. Nevertheless, it is worth mentioning that the quantity ofgrafted zirconia and active phase necessitates careful balancingin order to avoid the introduction of undesired Lewis-acidcharacter into the catalyst, which results in decreased selectivityfor acrolein as shown by Alhanash et al.44
The influence of the pore size distribution of the support wasdiscussed for the first time in 2007 by Tsukuda and coworkers,51
who worked with silica-supported acids. They focused theirstudy on the influence of the textural properties of the poroussupport on the selectivity, and used three commercial silicaswith pore sizes of 3, 6 and 10 nm, namely CARiACT Q3, Q6and Q10. The active phase was either a HPA or a conventionalinorganic acid like phosphorous or boric acid. Good resultswere obtained using Keggin-type HPAs such as phosphotungsticacid (H3PW12O40) or silicotungstic acid (H4SiW12O40) depositedon the 6 nm silica support. At full conversion, the selectivityfor acrolein was 65% and 74%, respectively, whereas themolybdenum-based homologous HPA (i.e., H3PMo12O40) didnot yield high selectivity (34%) (Table 2). When a catalyst basedon silicotungstic acid over silica with 10 nm pore diameter wasused, the selectivity could be further increased to 86%, whereasfor a support with smaller pores (namely 3 nm) the selectivityslightly decreased to 67%. Furthermore, it was observed thatthe small pore size of 3 nm resulted in a decreased catalyticactivity (55% conversion), which most probably originates fromenhanced coking resulting from steric limitations within thecatalyst framework. The influence of temperature was also
Table 2 Catalytic results obtained over various supported heteropolyacidsa
Catalyst T react (◦C) mcat (g) TOS (h) C (%) Sacrolein (%) Sacetol (%) Y acrolein (%) STY (mmol g-1 h-1) Ref.
15 wt% PW/SiO2 315 0.3 10 18 57 7 10 0.6 4715 wt% PW/ZrO2 315 0.63 10 76 76 12 58 1.6 4710 wt% SiW/AC 330 0.38 5 93 67 8 62 11.1 4920 wt% PW/Al2O3 275 0.3 5 99 52 8 51 5.1 5220 wt% SiW/Al2O3 275 0.3 5 98 64 8 63 6.3 5230 wt% PMo/Q6 325 0.3 5 98 65 7 64 3.8 5130 wt% PW/Q6 325 0.3 5 100 33 5 33 2.0 5130 wt% SiW/Q6 325 0.3 5 100 74 7 74 4.4 5130 wt% SiW/Q10 275 0.3 5 98 86 7 84 5.1 51CsPW 275 0.3 1 100 98 nc 98 49.0 44
a T react = reaction temperature; mcat = mass of catalyst; TOS = time on stream; C = conversion of glycerol; Sacrolein = selectivity for acrolein; Sacetol =selectivity for acetol; Y acrolein = yield of acrolein; STY = space-time yield; nc = not communicated; PW = H3PW12O40; SiW = H4SiW12O40; CsPW =Cs2.5H0.5PW12O40; AC = active carbon; Q6 = CARiACT-Q6; Q10 = CARiACT-Q10.
2084 | Green Chem., 2010, 12, 2079–2098 This journal is © The Royal Society of Chemistry 2010
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

studied, 275 ◦C being optimal in terms of acrolein yield, whichis consistent with earlier studies on Brønsted acid catalysts.
Similar results were obtained by Atia et al., who investigatedthe catalytic performance of the silicotungstic acid supportedon alumina with 5 nm and 12 nm pore sizes.52 The reactionconditions for the catalytic tests were chosen such as to matchthose used by Tsukuda et al.,51 for the sake of easier comparison.The selectivity for acrolein increased from 65% for the aluminasupport with a 5 nm pore size to over 85% for the aluminasupport with a 12 nm pore size, both at full conversion andfor catalysts loaded with 20 wt% of active phase (Table 2).Furthermore, Atia et al. found the same optimum reactiontemperature as Tsukuda, since the alumina-based catalysts alsoshowed complete conversion for a temperature higher than275 ◦C.
These results show that the size of the pores has a directinfluence on the selectivity of the catalysts. If steric limitations –like pores that are too narrow – hinder the rapid desorptionand diffusion of the reactants and of the products in theporous network of the catalyst, then condensation is more likelyto occur, which results in lower selectivity for acrolein anddeactivation of the catalyst due to carbon deposition (coke).
From the aforementioned studies, one can identify twoimportant parameters that govern the catalytic performancesof supported inorganic acids: (i) the dispersion of the activephase and (ii) the textural properties of the support. These twoparameters were further investigated by Chai et al. and Satoet al., who focused on supported HPAs and on their catalyticperformances with regard to the textural properties of the poroussupport.53–55 They carried out a huge number of performancetests, by varying both the loading of active phases and thenature of the supports. Here also, phosphomolybdic acid wasunselective compared to phosphotungstic acid and silicotungsticacid.51 With 20–30 wt% silicotungstic acid deposited on twoporous silicas with pore sizes of 6 and 10 nm, respectively, theyobtained a selectivity for acrolein up to ca. 87% at 275 ◦C inboth cases. Smaller pore diameters (3 nm) or a higher reactiontemperature (300 ◦C) always led to a decrease in the catalyticperformance. It is worth mentioning that the influence of theBET specific surface area of the support was also studied, butthis proved to have only a small effect.
Whereas Chai and Tsukuda51,53 showed that silicotungsticacid and phosphotungstic acid gave the best performances,due to their higher acidities, the group of Bruckner et al.56
recently decided to focus their investigations on supported phos-phomolybdic (H3PMo12O40) and the vanado-phosphomolybdic(H4PMo11VO40) acids. Using electron paramagnetic resonancespectroscopy (EPR), they showed that the Keggin structureof the molybdenum compounds was partially destroyed uponimpregnation on silica-alumina supports and that furthercalcination at 350 ◦C led to total decomposition to simplemetal oxides. These catalysts were then tested in the glyceroldehydration reaction, and the oxidation state of the metal atomswas followed by Operando EPR. The catalytic performance wasclose to that already observed for these compounds by Chaiet al.,53 that is to say quite poor (the selectivity for acrolein didnot exceed 25%). On the other hand, the Operando analysisof the catalyst revealed that the oxidation state of the metalswas 3+ for vanadium and 4+ for molybdenum, which indicates
that they were reduced during the reaction. When oxygen wasco-fed, the reduction degree of vanadium was limited to 4+and that of molybdenum to 5+. Furthermore, the coking wasslowed down, and the authors claimed that the carbon waspreferentially deposited on the vanadium centers. The carbonspecies constituting the coke layer was also analysed. Accordingto the EPR signal, the carbon was mainly in the sp3 hybridisationstate, which corresponds to non-double-bonded C–C chains, likefrom the radical polymerisation of the C C bond in acrolein.Furthermore, if oxygen was present in the feed, the formationof C O carbonyl groups was observed.
Another idea for using heteropolycompounds was inves-tigated by Dubois et al., who used salts of HPAs ob-tained by complete or partial substitution of the protonsof HPAs by metal cations.57 They used silicotungstic orphosphotungstic acid as bases, and prepared their respec-tive salts using elements such as caesium, rubidium, calciumor bismuth, or transition metals like zirconium, lanthanum,iron or hafnium. The partially neutralised caesium-containingHPAs (Cs2.5H0.5PW12O40 and Cs2.5H1.5SiW12O40) and rubidium-containing HPAs (Rb2.5H0.5PW12O40) required significantly lowerreaction temperatures (260–280 ◦C vs. 300–350 ◦C) than thetotally proton-free compounds, and further showed higherselectivity for acrolein in oxydehydration (90% vs. 50–70%). Thebest result was reported for Cs2.5H1.5SiW12O40, with a 93.1% yieldof acrolein. Nevertheless, when this catalyst was used withoutco-feeding of oxygen the catalytic performance was much lower,with an acrolein yield of no more than 40%. Comparable resultswith caesium salts of phosphotungstic acid were reported byAlhanash et al., who also obtained acrolein yields of up to98% during the first hour of reaction, which is the highestperformance ever reported, with a productivity up to 49 mmolg-1 h-1 (Table 2).44 Nevertheless, these authors admit that thecatalyst deactivates rapidly, and that excellent performancesclaimed were observed only for the first few hours of reaction.
In conclusion, all these results suggest that the use ofKeggin-type heteropolycompounds is a highly promising meansof obtaining high catalytic performances. They offer manypossibilities for tuning their acidity due to the possible variationsin their composition, which can be adjusted by modifying thecentral atom, the addenda atoms and the counter-ions. Whenthese compounds are deposited on a support, it is also possibleto adjust an additional parameter, as one can even control thepore size of the catalyst, and thus reduce diffusion limitations.
5 Zeolites
The use of supported heteropolycompounds is not the only wayof obtaining good catalytic performances. Zeolitic structures arepromising, and have been well studied since the work of Duboiset al.39
In addition to ZSM-5 and b-zeolite (already patented byDubois et al.39), Li and Zhuang et al. studied other zeolitestructures such as MCM-49, MCM-22, MCM-56 and ZSM-11.58,59 The obtained acrolein yields were in the range of 70–85%for all the catalysts, with nearly no decrease in the catalyticperformance during 400 h of reaction.
In 2007, Okuno and coworkers patented metallosilicatecatalysts with an MFI structure, a typical structure of zeolites
This journal is © The Royal Society of Chemistry 2010 Green Chem., 2010, 12, 2079–2098 | 2085
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online
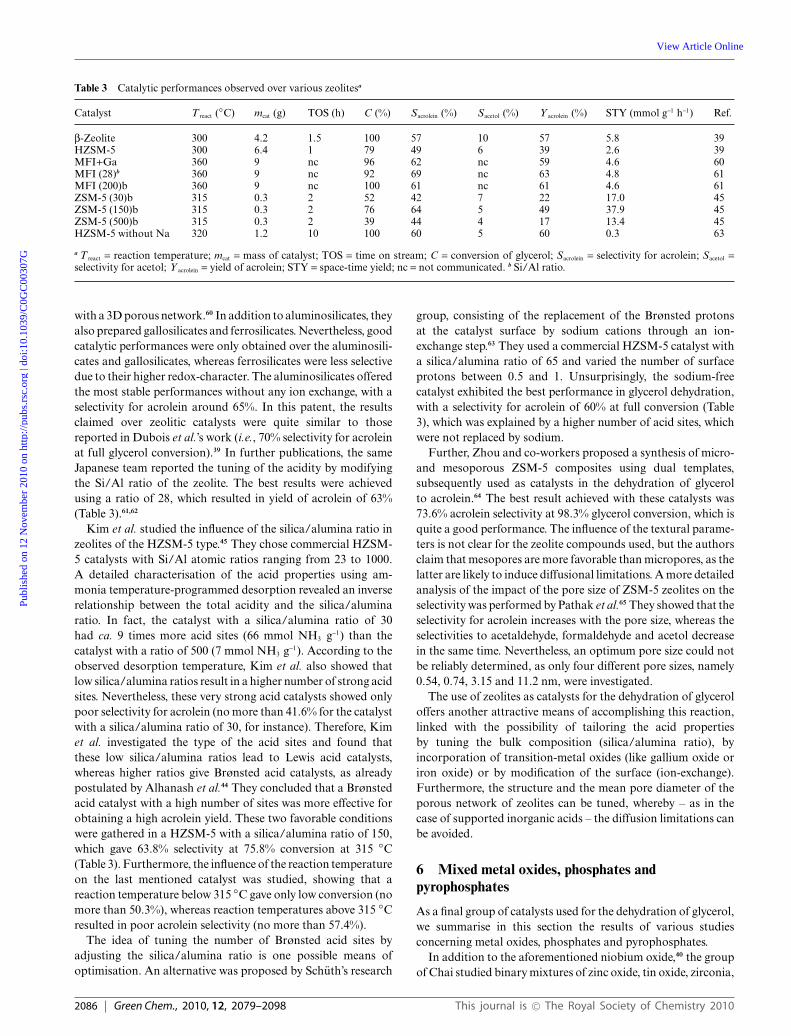
Table 3 Catalytic performances observed over various zeolitesa
Catalyst T react (◦C) mcat (g) TOS (h) C (%) Sacrolein (%) Sacetol (%) Y acrolein (%) STY (mmol g-1 h-1) Ref.
b-Zeolite 300 4.2 1.5 100 57 10 57 5.8 39HZSM-5 300 6.4 1 79 49 6 39 2.6 39MFI+Ga 360 9 nc 96 62 nc 59 4.6 60MFI (28)b 360 9 nc 92 69 nc 63 4.8 61MFI (200)b 360 9 nc 100 61 nc 61 4.6 61ZSM-5 (30)b 315 0.3 2 52 42 7 22 17.0 45ZSM-5 (150)b 315 0.3 2 76 64 5 49 37.9 45ZSM-5 (500)b 315 0.3 2 39 44 4 17 13.4 45HZSM-5 without Na 320 1.2 10 100 60 5 60 0.3 63
a T react = reaction temperature; mcat = mass of catalyst; TOS = time on stream; C = conversion of glycerol; Sacrolein = selectivity for acrolein; Sacetol =selectivity for acetol; Y acrolein = yield of acrolein; STY = space-time yield; nc = not communicated. b Si/Al ratio.
with a 3D porous network.60 In addition to aluminosilicates, theyalso prepared gallosilicates and ferrosilicates. Nevertheless, goodcatalytic performances were only obtained over the aluminosili-cates and gallosilicates, whereas ferrosilicates were less selectivedue to their higher redox-character. The aluminosilicates offeredthe most stable performances without any ion exchange, with aselectivity for acrolein around 65%. In this patent, the resultsclaimed over zeolitic catalysts were quite similar to thosereported in Dubois et al.’s work (i.e., 70% selectivity for acroleinat full glycerol conversion).39 In further publications, the sameJapanese team reported the tuning of the acidity by modifyingthe Si/Al ratio of the zeolite. The best results were achievedusing a ratio of 28, which resulted in yield of acrolein of 63%(Table 3).61,62
Kim et al. studied the influence of the silica/alumina ratio inzeolites of the HZSM-5 type.45 They chose commercial HZSM-5 catalysts with Si/Al atomic ratios ranging from 23 to 1000.A detailed characterisation of the acid properties using am-monia temperature-programmed desorption revealed an inverserelationship between the total acidity and the silica/aluminaratio. In fact, the catalyst with a silica/alumina ratio of 30had ca. 9 times more acid sites (66 mmol NH3 g-1) than thecatalyst with a ratio of 500 (7 mmol NH3 g-1). According to theobserved desorption temperature, Kim et al. also showed thatlow silica/alumina ratios result in a higher number of strong acidsites. Nevertheless, these very strong acid catalysts showed onlypoor selectivity for acrolein (no more than 41.6% for the catalystwith a silica/alumina ratio of 30, for instance). Therefore, Kimet al. investigated the type of the acid sites and found thatthese low silica/alumina ratios lead to Lewis acid catalysts,whereas higher ratios give Brønsted acid catalysts, as alreadypostulated by Alhanash et al.44 They concluded that a Brønstedacid catalyst with a high number of sites was more effective forobtaining a high acrolein yield. These two favorable conditionswere gathered in a HZSM-5 with a silica/alumina ratio of 150,which gave 63.8% selectivity at 75.8% conversion at 315 ◦C(Table 3). Furthermore, the influence of the reaction temperatureon the last mentioned catalyst was studied, showing that areaction temperature below 315 ◦C gave only low conversion (nomore than 50.3%), whereas reaction temperatures above 315 ◦Cresulted in poor acrolein selectivity (no more than 57.4%).
The idea of tuning the number of Brønsted acid sites byadjusting the silica/alumina ratio is one possible means ofoptimisation. An alternative was proposed by Schuth’s research
group, consisting of the replacement of the Brønsted protonsat the catalyst surface by sodium cations through an ion-exchange step.63 They used a commercial HZSM-5 catalyst witha silica/alumina ratio of 65 and varied the number of surfaceprotons between 0.5 and 1. Unsurprisingly, the sodium-freecatalyst exhibited the best performance in glycerol dehydration,with a selectivity for acrolein of 60% at full conversion (Table3), which was explained by a higher number of acid sites, whichwere not replaced by sodium.
Further, Zhou and co-workers proposed a synthesis of micro-and mesoporous ZSM-5 composites using dual templates,subsequently used as catalysts in the dehydration of glycerolto acrolein.64 The best result achieved with these catalysts was73.6% acrolein selectivity at 98.3% glycerol conversion, which isquite a good performance. The influence of the textural parame-ters is not clear for the zeolite compounds used, but the authorsclaim that mesopores are more favorable than micropores, as thelatter are likely to induce diffusional limitations. A more detailedanalysis of the impact of the pore size of ZSM-5 zeolites on theselectivity was performed by Pathak et al.65 They showed that theselectivity for acrolein increases with the pore size, whereas theselectivities to acetaldehyde, formaldehyde and acetol decreasein the same time. Nevertheless, an optimum pore size could notbe reliably determined, as only four different pore sizes, namely0.54, 0.74, 3.15 and 11.2 nm, were investigated.
The use of zeolites as catalysts for the dehydration of glyceroloffers another attractive means of accomplishing this reaction,linked with the possibility of tailoring the acid propertiesby tuning the bulk composition (silica/alumina ratio), byincorporation of transition-metal oxides (like gallium oxide oriron oxide) or by modification of the surface (ion-exchange).Furthermore, the structure and the mean pore diameter of theporous network of zeolites can be tuned, whereby – as in thecase of supported inorganic acids – the diffusion limitations canbe avoided.
6 Mixed metal oxides, phosphates andpyrophosphates
As a final group of catalysts used for the dehydration of glycerol,we summarise in this section the results of various studiesconcerning metal oxides, phosphates and pyrophosphates.
In addition to the aforementioned niobium oxide,40 the groupof Chai studied binary mixtures of zinc oxide, tin oxide, zirconia,
2086 | Green Chem., 2010, 12, 2079–2098 This journal is © The Royal Society of Chemistry 2010
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

Table 4 Catalytic performances observed over metal oxides, phosphates and oxophosphates, with and without co-feedinga
Catalyst T react (◦C) mcat (g) Co-feed TOS (h) C (%) Sacrolein (%) Sacetol (%) Y acrolein (%) STY (mmol g-1 h-1) Ref.
Nb2O5-350 315 0.56 — 10 75 47 10 35 1.3 40Nb2O5-400 315 0.57 — 10 88 51 12 45 1.6 40Nb2O5-500 315 0.61 — 10 91 35 14 32 1.2 40TiAl-600 315 0.6 — 2 86 43 17 37 1.1 66SAPO-11 280 0.2 — 1 66 62 10 41 0.9 67SAPO-34 280 0.2 — 1 59 72 7 42 0.9 67VPO-700 300 0.2 O2 10 81 95 9 77 6.9 68VPO-800 300 0.2 O2 10 100 80 6 80 7.1 68VPO-900 300 0.2 O2 10 97 58 15 56 5.0 68Fex(PO4)y 280 0.8 — 5 100 92 0 92 4.1 729 wt% WO3/ZrO2 300 17 — 5 100 74 11 74 1.9 3819 wt% WO3/ZrO2 280 5 — nc 83 69 10 57 9.3 79
a T react = reaction temperature; mcat = mass of catalyst; co-feed = nature of the co-feeding gas; TOS = time on stream; C = conversion of glycerol;Sacrolein = selectivity for acrolein; Sacetol = selectivity for acetol; Y acrolein = yield of acrolein; STY = space-time yield; nc = not communicated; MxOy-***indicates the calcination temperature (in ◦C); TiAl = TiO2/Al2O3 binary mixture; SAPO = silica-alumina phosphate; VPO = vanadium oxophosphate.
titania, alumina and silica.66 Whereas the catalytic performancesof these mixed oxides remained rather low, with acrolein yieldsnot exceeding 36%, the study of the acidity and of the texturalparameters clearly showed that the less acid catalysts haveincreased selectivity for acetol, and that small pore diametersinduce a decrease in the selectivity for acrolein.
Suprun et al. published a paper on phosphate-modifiedtitania, alumina and silica/alumina (SAPO),67 and found astrong influence of the acidic and textural parameters on thecatalytic performances. As in the case of supported inorganicacids and of zeolites, a relationship between the total acidity andthe selectivity for acrolein was observed. In fact, the most acidiccatalyst, a silica-alumina phosphate, showed a selectivity foracrolein up to 72% (Table 4). Furthermore, Suprun et al. pointedout that microporous materials (5–6 A) are less active dueto internal diffusion limitations, and tend to induce increasedcarbon deposition, as observed on SAPO-34.
A similar catalytic system, namely vanadium oxophosphatesand oxo-pyrophosphates, was studied by Wang et al.68,69 Asin the case of niobium oxide, the acidity of these compoundscan be tuned by modifying their calcination temperature, butin an inverse relationship, as high calcination temperaturesresult in a larger number of acid sites. The catalysts weretested in the glycerol dehydration reaction, with co-feedingof oxygen. The best result was obtained using a vanadiumpyrophosphate calcined at 800 ◦C, which gave a selectivityfor acrolein of 66% at full conversion (Table 4). Nevertheless,this result was not obtained over the catalyst with the largestnumber of acid sites. Actually, as the specific surface decreasedfor high calcination temperatures due to sintering of the solid,the selectivity consequently dropped drastically in this case.Dubois extended the study of pyrophosphates using boron andaluminium as cations, and observed that the boron-containingcompounds give the highest acrolein yields, with a maximum of77.8%.70
Other metal pyrophosphates were studied by Liu and cowork-ers, who used rare earth metals such as lanthanum, neodymiumand cerium.71 All the considered pyrophosphates exhibitedquite similar catalytic performances, with the best results beingachieved over Nd4(P2O7)3. As in the case of the aforementionedvanadium pyrophosphates, the acidity of the neodymium com-
pounds increases with the calcination temperature, whereby theoptimum in the catalytic performance (80% yield of acrolein)was found for a calcination temperature of 500 ◦C.
The use of phosphates instead of pyrophosphates was studiedby Deleplanque et al.72 They prepared iron phosphates usingdifferent preparation techniques, such as hydrothermal reactionor precipitation. The highest acrolein yield without oxygen co-feeding was 92.1% (Table 4). In the so-called ‘oxy-dehydration’reaction, i.e., with co-feeding of molecular oxygen, the selectivityfor acrolein decreased to 62.5% at full conversion, due to the totaloxidation to carbon oxide species (COx) as an important sidereaction. Therefore, Dubois investigated the addition of group 1and group 2 metal cations to these catalytic formulations, suchas caesium, strontium and potassium.73 Doping with caesium in-creased the acrolein yield to 72.2%, which was thus significantlyhigher than that observed over the undoped iron phosphate(62.5%), but still lower than the yield obtained without oxygenco-feeding (92.1%). The idea of doping the phosphate specieswas also investigated by Matsunami et al., who used phosphate-modified silica with alkali salts. Nevertheless, in this latter case,the yield of acrolein did not exceed 67%.74,75
In 2006, Dubois et al. proposed the use of tungsten oxide onzirconia as a catalyst for glycerol dehydration. Total conversionof glycerol was achieved at 300 ◦C with a catalyst containing9 wt% of tungsten oxide, with a yield of acrolein of 74%.38
Redlingshofer et al. further studied this binary metal oxidesystem.76,77 The reaction was carried out at a lower temperature(260 ◦C). Good performances were observed with an acroleinyield between 77 and 79%. Nevertheless, the catalysts tendedto deactivate on-stream, with the acrolein yield decreasing at arate of 5% every 10 h. Additionally, these authors describedthe possibility of regenerating the catalysts by oxygen post-treatment at 350 ◦C for 5 h. Bythis treatment, they claimedthat the initial catalytic performances could be recovered.78
This well-known combination of tungsten oxide on zirconiawas further studied by Ulgen et al.79 They showed that the acidityof the solid increases with the amount of tungsten oxide. For thecatalyst containing 19 wt% of tungsten oxide, an acrolein yieldof 57% was observed (Table 4).
Furthermore, they studied the influence of the calcinationtemperature on the physical properties of the catalysts. Whereas
This journal is © The Royal Society of Chemistry 2010 Green Chem., 2010, 12, 2079–2098 | 2087
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

the temperature treatment had only a low impact on the acidicproperties, the number of basic sites significantly decreased whenthe calcination temperature increased. Moreover, the texturalparameters were quite dramatically modified, as the sinteringled to larger pores but reduced specific surface areas. For thesetwo reasons, the thermal treatment had in any case a positiveimpact on the catalytic performance, as the decrease in basicityled to the suppression of acetol formation, and the larger poresize reduced the deactivation extent due to coking, as internaldiffusion limitations were lower.
The above-described family of mixed metal oxides, phosphatesand pyrophosphates is rather heterogeneous. Some compounds,like niobium oxide, tungsten oxide and pyrophosphates, offer thepossibility of controlling their acid strength via the calcinationstep. This thermal treatment generally also has an influence onthe specific surface area and the pore size due to sintering.In comparison to the aforementioned zeolites and supportedHPAs, the control of the physical properties of the mixed metaloxides and phosphates seems nevertheless less straightforward.
7 Regeneration of spent catalyst
From the studies reported above, it is clear that very efficientcatalysts for the dehydration of glycerol to acrolein can beprepared. Unfortunately, these catalysts quickly deactivate, mostprobably because of the formation of coke, which makes itdifficult to straightforwardly use them in industrial plants atthe current time. Up to now, only a few studies have beendevoted to the characterisation of carbon deposits and to thesites responsible in these catalytic systems. Erfle et al. used FTIRspectroscopy to characterise the spent catalyst and observedC O and C C signals.56 They attributed these signals to cyclicanhydride species and claimed that the Brønsted acid sites werethe centers of coke formation. Suprun and coworkers confirmedthat the acid sites were responsible for the formation of carbondeposits on phosphate catalysts.67 They further found that thequantity of coke increased with higher reaction temperaturesand also with decreasing the pore diameter of the catalyst (Table5). For example, the coke loading on SAPO-11 increased from4.6 wt% at 280 ◦C to 9.5 wt% at 320 ◦C. From the ammoniadesorption experiments, one can also see that the acid strengthincreases in the order Al2O3-PO4 ª TiO2-PO4 < SAPO-11 �SAPO-34, which correlates well with the amount of carbondeposit found over these catalysts. Indeed, the quantity ofdeposited carbon increased for all the reaction temperaturesin the same order.
Furthermore, the hydrogen-to-carbon (H/C) ratio for thecarbon species was determined by elementary analysis. All thecatalysts showed a decrease in the H/C ratio at higher reactiontemperatures, meaning that the carbon deposit became moredeficient in hydrogen (Table 5). For example, the H/C ratio overSAPO-34 decreased from 0.62 at 280 ◦C to 0.47 at 320 ◦C.This can be ascribed to the formation of highly condensed,unsaturated carbonaceous aromatic species. Solutions still haveto be found to avoid or to limit the coke deposit on the catalystsurface or at least to optimise the regeneration of the catalysts.
Although a fundamental approach is still needed for studyingefficient means for limiting the coke deposition during thereaction, at least three kinds of solutions have been proposed sofar for continuously regenerating the catalysts: (i) co-injection(co-feeding) of oxygen or hydrogen with the gas feed to yieldin situ regeneration;39 (ii) cyclic regeneration of the used catalystby injection of a flow or of pulses of air or oxygen;83 and(iii) circulation of the catalyst in a moving-bed reactor withregeneration in a separate parallel vessel (as for the FCCprocess).25 Whereas the first option is associated with the risk ofgenerating explosive conditions and/or to oxidise the variousreaction products, the second alternative is accompanied bythe disadvantage of a loss in productivity. The third optiondoes not suffer from these drawbacks, but the constructionand the operation of a circulating bed reactor implies serioustechnological difficulties that can only be overtaken by skilledpersons through fine process control.
The idea of working with air in the reactant mixture was firstproposed by Dubois for catalysts of the zeolite type.39 To avoidthe explosivity range, the oxygen fraction must never overpass7 vol%. Therefore, the composition of the reaction feed wasadjusted to 6% of oxygen for 4.5% of glycerol, the remaining89.5% being steam. Thereby, the authors claim to reduce catalystdeactivation and even to inhibit the formation of by-productssuch as acetol in these conditions.
The same process was further used for vanadiumpyrophosphates,69 boron phosphates,70 iron phosphates,72 tung-sten oxide on zirconia79 and caesium salts of phosphotungsticacid.57 As a general trend, the selectivity for by-products suchas acetol, acetaldehyde and formaldehyde decreased, whereasthe formation of organic acids, like acrylic acid, acetic acidand formic acid, increased, due to subsequent oxidation of thealdehydes formed. For some catalysts, like zeolites, vanadiumoxophosphates, and tungsten oxide on zirconia, the selectivityfor acrolein was not or only slightly affected. In the case ofthe caesium salt of phosphotungstic acid, an increase in theselectivity for acrolein by a factor of two was even observed (93%
Table 5 Correlation between acidity and the carbon deposit for phosphate catalystsa
T react = 280 ◦C T react = 300 ◦C T react = 320 ◦C
Catalyst Ac (mmol NH3 g-1) Dp (A) SBET (m2 g-1)Coke loading(wt%)
H/Cratio
Coke loading(wt%)
H/Cratio
Coke loading(wt%)
H/Cratio
Al2O3-PO4 295 111 118 2.4 0.54 3.9 0.52 5.8 0.49TiO2-PO4 258 101 38 3.1 0.55 6.8 0.50 8.9 0.48SAPO-11 1330 6 172 4.6 0.68 7.4 0.55 9.5 0.40SAPO-34 498 5 49 9.6 0.62 12.7 0.54 16.2 0.47
a Ac = total acidity; Dp = pore diameter; SBET = specific surface area; T react = reaction temperature; SAPO = silica-alumina-phosphate.
2088 | Green Chem., 2010, 12, 2079–2098 This journal is © The Royal Society of Chemistry 2010
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

Table 6 Comparison of catalytic performance observed in glycerol dehydration, with and without co-feedinga
Catalyst T react (◦C) mcat (g) Co-feed TOS (h) C (%) Sacrolein (%) Sacetol (%) Y acrolein (%) STY (mmol g-1 h-1) Ref.
b-Zeolite 300 4.2 O2 3 100 57 0 57 5.8 39b-Zeolite 300 4.2 — 1.5 100 57 10 57 5.8 39HZSM-5 300 6.4 O2 1 88 46 2 40 2.7 39HZSM-5 300 6.4 — 1 79 49 6 39 2.6 39VPO-800 300 0.2 O2 nc 100 65 6 65 5.8 68VPO-800 300 0.2 — nc 95 60 12 57 5.1 68BPO 280 0.3 O2 nc 100 65 8 65 0.8 70BPO 280 0.3 — nc 96 81 6 78 0.9 70Fex(PO4)y 280 1.3 O2 5 100 63 0 63 1.7 72Fex(PO4)y 280 0.8 — 5 100 92 0 92 4.1 7219 wt% WO3/ZrO3 280 5 O2 nc 73 74 8 54 8.9 7919 wt% WO3/ZrO2 280 5 — nc 83 69 10 57 9.3 79CsPW 260 23 O2 nc 100 93 nc 93 3.0 57CsPW nc 23 — 1 83 47 3 39 0.7 570.5% Pd-CsPW 275 0.3 H2 5 79 96 1 76 37.9 44CsPW 275 0.3 — 1 100 98 nc 98 49.0 44WO3/ZrO2 275 9.2 SO2 24 87 69 7 60 1.2 81WO3/ZrO2 275 9.2 — 24 69 71 9 49 1.0 81
a T react = reaction temperature; mcat = mass of catalyst; Co-feed = nature of the eventual co-feeding gas; TOS = time on stream; C = conversion ofglycerol; Sacrolein = selectivity for acrolein; Sacetol = selectivity for acetol; Y acrolein = yield of acrolein; STY = space-time-yield; nc = not communicated;VPO = vanadium oxophosphate calcined at 800 ◦C; BPO = boron phosphate; CsPW = Cs2.5H0.5PW12O40.
vs. 47%; Table 6). On the other hand, for boron phosphates andiron phosphates, the co-feeding of oxygen led to a significantdecrease in the selectivity for acrolein, which was ascribed tothe over-oxidation to carbon oxides (CO/CO2). To facilitate theactivation of molecular oxygen, doping with metal cations likecaesium, potassium, strontium, silver and platinum was probedby Dubois, which led to an acrolein selectivity of around 70%.73
A comparable approach was followed by Kasuga et al., whoused protonated MFIs as catalysts.80 When they injected air inthe gas reactant feed, the selectivity for acrolein on unmodifiedcatalysts was rather low and did not exceed 45% after 24 hunder reactant flow. To optimise the regeneration, Kasuga et al.thus also proposed to modify the MFI protonated-zeolite witha small amount of metal (Pt, Pd, Ru, Cu, Ir or Au). By thismean of doping, they claimed that it is possible to accelerate thesplitting of the dioxygen used during regeneration.80 The bestresults were obtained with 0.1 wt% Pt and 1 wt% Au (80.7 and79.7% of acrolein yield, respectively, at full glycerol conversionafter 150 min under reaction conditions).
The same concept to reduce the carbon deposit, but this timeby co-feeding hydrogen, was investigated by Alhanash et al.,who used caesium salts of HPAs.44 Following a similar principleas that proposed by Kasuga et al.,80 they probed the doping withsmall quantities of noble metals such as ruthenium, palladiumand platinum. Thereby, they could increase the catalyst activityby a factor of two after 5 h of reaction (conversion of 79% vs.41%; Table 6) without any significant impact on the selectivityfor acrolein (96% vs. 98%). The best results were obtained withpalladium on the caesium salt of phosphotungstic acid, overwhich a productivity of 37.9 mmol g-1 h-1 was achieved.
Dubois proposed a third kind of feed additive for enhancingthe long-lasting performances of tungsten oxide on zirconia.Instead of oxygen or hydrogen, he injected 250 ppm of sulfurdioxide into the feed in order to slow down the deactivation.81 Infact, the addition of SO2 even induced a slight positive impacton the selectivity for acrolein (73% vs. 72% without SO2) and
significantly inhibited the formation of acetol (selectivity of0.2% vs. 2.4% without SO2). After 24 h on-stream, the catalyticperformance observed when co-feeding SO2 were significantlyimproved, still with an excellent conversion of 87% (initially100%), whereas it was 69% in the absence of SO2 (Table 6).
The results obtained using co-feeding of oxygen are quitedifferent and strongly depend on the studied catalytic system. Asmentioned earlier, the oxygen concentration must be kept below7 mol% in order to stay out of the explosion limits. Wang et al.studied the influence of the oxygen concentration on the catalyticperformance of vanadium phosphate catalysts. They observedthat too little (as well as too much) oxygen has a negative impacton the selectivity for acrolein.69 In fact, low quantities of oxygenled to larger amounts of acetol, whereas high concentrationsinduced an increase in the selectivity for acetaldehyde, aceticacid, acrylic acid and – of course – to carbon oxides, whichis explained by the successive over-oxidation of the products.The exceptions where the co-feeding process is unfavorable tothe selectivity for acrolein – as over iron phosphate and boronphosphate – can be easily explained by the redox character ofthe catalysts, which promotes oxidation processes. In these cases,co-feeding with hydrogen might be a good way to overcome thisproblem. The process using co-feeding of sulfur dioxide mightalso be an interesting approach, even if one should keep in mindits toxicity, which is certainly the most important drawback ofthis method. Furthermore, this reagent may be incompatiblewith some catalytic systems, due to some possible poisoningissues.
The second process for regenerating the spent catalyst –namely, periodic regeneration – was studied by Arita et al., whoregenerated a used H-ZSM5 catalyst under an air flow. Theyevidenced the possibility of recovering the initial performances.82
However, the exothermicity of the regeneration led to theformation of hot-spots. Consequently, the temperature duringregeneration exceeded the reaction temperature by more than100 ◦C, at which the catalyst risks thermal decomposition.
This journal is © The Royal Society of Chemistry 2010 Green Chem., 2010, 12, 2079–2098 | 2089
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

Therefore, this technique is not applicable for temperature-sensitive catalytic systems.
A more academic approach was followed by Atia et al.Their preliminary work involved long-term runs (up to 300 h),and they found that over alumina-supported heteropolyacidcatalysts the selectivity for acrolein remained quite stable,whereas glycerol conversion more-or-less linearly decreased.This effect is typically observed for deactivation by depositionof coke on the catalyst surface, and is linked with the progressivedecrease in the number of accessible active sites. To verify thishypothesis, spent catalysts were regenerated under a flow of 1%of oxygen in nitrogen at 325 ◦C for 24 h. After this treatment, theregenerated catalyst showed performances identical to those ofthe fresh catalyst. To quantify the coking effect, the spent catalystwas also analysed by thermogravimetry, which straightforwardlyconfirmed the hypothesis of carbon deposition.52
Meanwhile, Corma et al. adopted the idea of a circulatingbed reactor,25,83 which was previously disclosed in a Duboispatent.38,39 These authors studied the opportunity of injectingcrude glycerol directly into FCC plants. An advantage wouldbe the use of existing facilities and therefore avoiding havingto invest in building specific infrastructures. Note that this ideawas already proposed, but under another form, by Dubois, whopointed out the possibility of injecting glycerol into propyleneoxidation plants to concomitantly yield acrolein from bothsources.84 In the FCC plants, the heat recovered by the burningof coke could be used to provide the energy necessary forthe evaporation of glycerol. Corma et al. concluded that anautothermal process is possible in that case. Nevertheless, theauthors did not limit their view to the dehydration reaction, butalso investigated the possibilities for reforming glycerol at 500–600 ◦C to produce ethylene and propylene. In this approach,the glycerol feedstock may be an alternative or a complement tonaphtha cracking.
An original approach was followed by Kasuga et al., whofocused their investigations on various possibilities for pre-treating catalysts (protonated MFI zeolite or Alox alumina).85
Three types of pretreatments were tested: (i) a flow of acetol,water and nitrogen; (ii) a flow of acrolein, water and nitrogen;and (iii) a flow of acetol, water and air. Irrespective of thepretreatment method, the Alox catalyst always led to verypoor acrolein yields, while over the MFI catalyst all these
pretreatments induced higher acrolein selectivity in the firstfew minutes after switching to the reactant flow. However,after 150 min on-stream, the catalytic performances returnedto values similar to those obtained without any preliminarypretreatment, which means that this effect is only effective in theearly stages of the reaction. Despite this, the authors pointedout the possibility of recovering the initial performance byregenerating the catalysts at 500 ◦C under a flow of air.
8 Dehydration of glycerol in the liquid phase
As mentioned in the introduction, the first experimental resultson dehydration of glycerol in the liquid phase were reportedby Groll in 1936,33 and by Hoyt and Manninen in 1951,34 in thepresence of either homogenous or heterogeneous catalysts. Sincethese studies, less than a dozen articles and patents have beenpublished on liquid-phase dehydration, while there has been aboom in the number of publications dealing with the gas-phasereaction. In the following section, we give a brief overview ofthe dehydration reaction in the liquid phase, and point out themajor drawbacks of this process.
Ramayya et al. studied the dehydration of glycerol in batchexperiments using an aqueous solution of sulfuric acid, andconditions close to the critical point of water (namely P = 22.1MPa, and T = 374 ◦C).86 (Near) supercritical water offers theadvantage that its physical properties, like the dielectric constantor the ion product, can be adjusted by varying temperature andpressure. The mixture was heated in a batch reactor at 350 ◦C,pressurised to 34.5 MPa and acidified with 5 mM sulfuric acid.In these conditions, and at a residence time of 25 s, the conversionof glycerol and the selectivity for acrolein were 55% and 86%,respectively (Table 7). Considering the fact that sulfuric acid issoluble in supercritical water, this process can be classified ashomogeneous catalysis with, the consequent catalyst–reactionmixture separation issues. Furthermore, a batch process is notapplicable for large-scale production of acrolein.
Nevertheless, the idea of using supercritical water as a reactionmedium was a new aspect, and Buhler et al. decided to study itin more detail in 2001.87 Instead of working in a batch reactor,they built a flow-type reactor and used supercritical water bothas a solvent and a catalytic system. They observed variousdecomposition products such as acetaldehyde, formaldehyde,
Table 7 Catalytic performances observed for glycerol dehydration in the liquid phasea
Catalyst T react (◦C) P (MPa) Solvent Reactor type C (%) S (%) Y (%) Ref.
H2SO4 190 0.1 Water Batch nc nc 49 33H3PO3/clay 300 0.1 Paraffin Batch nc nc 72 34H2SO4 350 34.5 Water Batch 55 86 47 86No catalyst 356 45 Water Continuous 71 38 27 87ZnSO4 360 25 Water Continuous 50 75 38 88ZnSO4 360 34 Water Continuous 62 59 37 89H2SO4 400 34.5 Water Continuous 92 81 74 90KHSO4 280 0.1 Paraffin Continuous 97 82 80 91MgSO4 280 0.1 Paraffin Continuous 92 56 52 915 wt% H3PO4/Al2O3 280 0.1 Paraffin Continuous 89 57 51 91FePO4 320 0.1 Xylene Continuous 87 78 68 93CuPO4 280 0.1 Sulfolane Continuous 98 85 84 93
a T react = reaction temperature; P = pressure; C = conversion of glycerol; S = selectivity for acrolein; Y = yield of acrolein; nc = not communicated.
2090 | Green Chem., 2010, 12, 2079–2098 This journal is © The Royal Society of Chemistry 2010
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

allylic alcohol and propionaldehyde, as well as acrolein. Theacrolein yield varied between 10 and 27% depending on thereaction conditions, with the best result (27%) obtained at areaction temperature of 350 ◦C under 45 MPa pressure (Table 7).As mentioned earlier, these tests were carried out in the absenceof any catalyst or acid component, which explains the poor yieldsin acrolein.
Recently, a combination of the ideas of Hoyt and Manninen34
and of Ramaya et al.,86 namely using a supported catalyst insupercritical water as a reaction medium, was proposed by Ottand coworkers.88 They investigated the dehydration of glycerol ina batch reactor under supercritical conditions using zinc sulfateas a catalyst. The choice of zinc sulfate as a catalyst is quitesurprising, as this compound is not known as a strong acidcatalyst, which is typically needed for this reaction. However,they justified their choice considering material stress issues:water in its supercritical form is very corrosive, and thereforerequires the use of special and expensive steel grades for reactors;this corrosive power would become even stronger if acidiccompounds were subsequently added to the medium, leadingto unacceptable reactor material stress. In these conditions, theauthors showed that supercritical conditions are not necessarilythe optimal ones. Actually, the best catalytic performance,namely 75% selectivity at 50% conversion, was obtained in thenear-critical region of water (310 ◦C at 25 MPa) (Table 7). Thismight be explained by the degradation of acrolein when it issubjected to temperatures that are too high. In 2007, Lehr et al.also published results on the dehydration of biomass-derivedpolyols in sub- and super-critical water.89 As far as glycerol isconcerned, they reported 59% glycerol conversion with almost60% selectivity for acrolein over zinc sulfate as a catalyst undersubcritical water conditions, like Ott et al. (Table 7).88
To determine if the dehydration of glycerol in the liquid phasemight offer a sustainable source of acrolein at an industrial scale,Watanabe et al. attempted to optimise the acrolein yield.90 Theycombined the supercritical conditions in a flow-type reactor withsulfuric acid as a catalyst. Several blank tests performed in theabsence of a catalyst gave low glycerol conversions, but highacrolein selectivities with a yield up to 16%. The best resultswere obtained at 400 ◦C under 34.5 MPa with a sulfuric acidconcentration of 5 mM. In these conditions, the yield of acroleinwas 74% at a glycerol conversion around 92% (Table 7). One ofthe main issues in this work was to increase the formation rateof acrolein without modifying its decomposition rate, which wasachieved by adding acid in supercritical conditions.
In 2007, Suzuki et al. and Yoshimi et al. revamped the ideaof Hoyt and Manninen,34 using a liquid reaction medium withhigh boiling point at atmospheric pressure.91,92 Potassium sulfateand bisulfate were used as catalysts in a batch reactor, wherethe glycerol solution was added dropwise in a paraffin solutionmaintained at 280 ◦C. The produced acrolein was continuouslyevaporated from the reaction mixture and was recovered in anaqueous solution, and yields close to 80% were achieved. Thistechnique was further adapted by Takanori et al., who used ironor copper phosphate as catalysts in high-boiling organic solvents(e.g. n-octane, m-xylene or sulfolane) as reaction media.93,94 Theyobtained (to our knowledge) the highest ever reported acroleinyield in the liquid phase, over copper sulfate in sulfolane (84%;Table 7).
Thus, we have seen that the dehydration of glycerol in theliquid phase (at atmospheric pressure or in near-critical orsupercritical conditions) leads to the formation of acrolein. Theapplication of supercritical conditions is an especially interestingconcept – the change in the physical properties of water inducingdehydration of glycerol even in the absence of a catalyst –but, unfortunately, the yield of acrolein remains rather low inthis case. On the other hand, the combination of acids andnear-critical or supercritical conditions results in much betterperformances, but also in the generation of an extremely corro-sive medium. Therefore, the reaction vessel, which must resisthigh temperature and pressure, must be specifically designed,implying important investment/maintenance costs, which is amajor drawback for industrial applications.
Recent results show that the initial idea of using high-boilingliquids as reaction media at atmospheric pressure seems morepreferable. Furthermore, the proposed process in this latter case,where the reactant is constantly fed into the reaction mixtureand the acrolein is separated by evaporation upon formation,can be classified as a continuous process. Nevertheless, theaccumulation of heavier by-products in the reaction mixtureseems quite probable in this case. Moreover, the adaptation ofthis kind of process for industrial application is a real challenge,as the results from laboratory scale (300 mL) cannot easily betransferred to plant size. In conclusion, one can state that a morepractical process seems to still be a topical issue of researchbefore commercial applications of liquid-phase dehydration ofglycerol to acrolein can be reliably envisaged.
9 Reaction mechanism
Whereas the research for an efficient catalyst can follow (at leastin an initial screening step) a more-or-less applied approach,a more fundamental aspect lies in the understanding of thereaction mechanism, which further enables back-optimisationof the catalytic formulations. This requires the identification ofthe intermediate steps and the explanation of the formation ofthe by-products. A first proposal was made by Buhler et al. forthe glycerol activation in the liquid phase.87 Two pathways wereclaimed, either via an ionic or a radical mechanism (Scheme8). The ionic reaction (Scheme 8a) begins with the protonationof glycerol either on one primary hydroxyl group or on thesecondary hydroxyl group. Consequently, the elimination of awater molecule leads to the formation of a carbocation. In thecase of the secondary carbocation, the only possible productafter release of H3O+ is acrolein, whereas either acrolein oracetaldehyde + formaldehyde can be formed starting from theprimary carbocation. The radical pathway (Scheme 8b) startswith the abstraction of a hydrogen atom from a primary carbonusing an OH∑ radical. The resulting radical species further losesan hydroxyl radical (OH∑), which leads to the formation ofacrolein.
Tsukuda et al. – who performed the dehydration in the gaseousphase – developed a more formal reaction framework (Scheme9).51 The first dehydration step leads to the formation of twoenols, which are in tautomeric equilibrium with the correspond-ing ketone (acetol) and aldehyde (3-hydroxypropionaldehyde).The latter may subsequently react in a second dehydrationreaction to yield acrolein, or undergoes a retroaldol reaction to
This journal is © The Royal Society of Chemistry 2010 Green Chem., 2010, 12, 2079–2098 | 2091
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

Scheme 8 Reaction pathways to the formation of acrolein considering(a) an ionic pathway and (b) a radical pathway.87
liberate formaldehyde + acetaldehyde, which – in the presence ofoxygen – can easily be oxidised to acetic acid. In agreement withthe previous proposal of Buhler et al.,87 Tsukuda et al. confirmedthat the key to obtain high acrolein selectivity lies in the controlof the first dehydration step, whereby the formation of 3-hydroxypropionaldehyde must be favored over the formation ofacetol, which is identified as the main by-product of the process.
The same mechanism has been further amended by Chaiet al. (Scheme 9),41 who added two possible hydrogenationsteps, one starting from acrolein and the other from acetolto yield allylic alcohol and 1,2-propanediol, respectively. Inthe catalytic system studied, these by-products can indeed beobserved. Furthermore, the hydrogenation of formaldehyde tomethanol or its decomposition to CO + H2 are mentioned.
Scheme 9 Mechanism proposed by Tsukuda et al. and further com-pleted by Chai et al.41,51
These two first proposals were the first approaches forexplaining the observed by-products from glycerol dehydration.Successively, Corma et al.25 postulated a more complex interwo-ven reaction framework (Scheme 10), where the main reactionsteps are the same as those previously described by Tsukuda andChai. In order to achieve a deeper understanding and specificallyinvestigate the secondary reactions, the authors used acetol as
Scheme 10 Reaction network proposed by Corma et al.25
a reactant at 350 ◦C. The products were classified into threegroups: acids (9.4%), other aldehydes (52.0%) and coke (27.7%).Furthermore, the formation of low amounts of acetone (4.0%),acetaldehyde (1.4%) and carbon monoxide (1.2%) was observed.Nevertheless, the conversion of acetol did not exceed 25%, whichmeans that this compound is quite stable and does not easilyundergo consecutive reactions. This explains why it is usuallyidentified as the major by-product in the reaction of dehydrationof glycerol to acrolein.
Even though only small amounts of acetone were observed,Corma et al. investigated the consecutive reactions starting fromthis compound by repeating their method. The resulting productdistribution was dominated by unsaturated hydrocarbons likebutene (27.9%), propylene (1.7%), C5 + C6–8 aromatics (6.6%)and – as a product of these compounds – coke (31.9%). Otheridentified products were various acids (20.2%) and aldehydes(7.3%), but, as in the case of hydroxylacetone, the conversionof acetone remained low and did not exceed 14%. Nevertheless,with their findings, they enlarged the reaction scheme proposedby Chai and Tsukuda by adding further side-reactions ema-nating from acetol. Thereby, they explained the formation ofoligomers and coke by consecutive reactions starting from acetolvia acetone and acetaldehyde.
The idea of investigating the consecutive reactions startingfrom a by-product was picked up by Suprun et al. for 3-hydroxypropionaldehyde and acetol.67 They could thus confirmthe retroaldol reaction previously postulated by Tsukuda,51
which leads to the formation of formaldehyde and acetaldehydeissuing from 3-hydroxypropionaldehyde. Additionally, signifi-cant amounts of coke were found, which was ascribed to theformation of cyclic C6 compounds identified by GC–MS analysis(Scheme 11a). The authors further identified numerous furanderivatives as products from acetol (Scheme 11b).
2092 | Green Chem., 2010, 12, 2079–2098 This journal is © The Royal Society of Chemistry 2010
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online
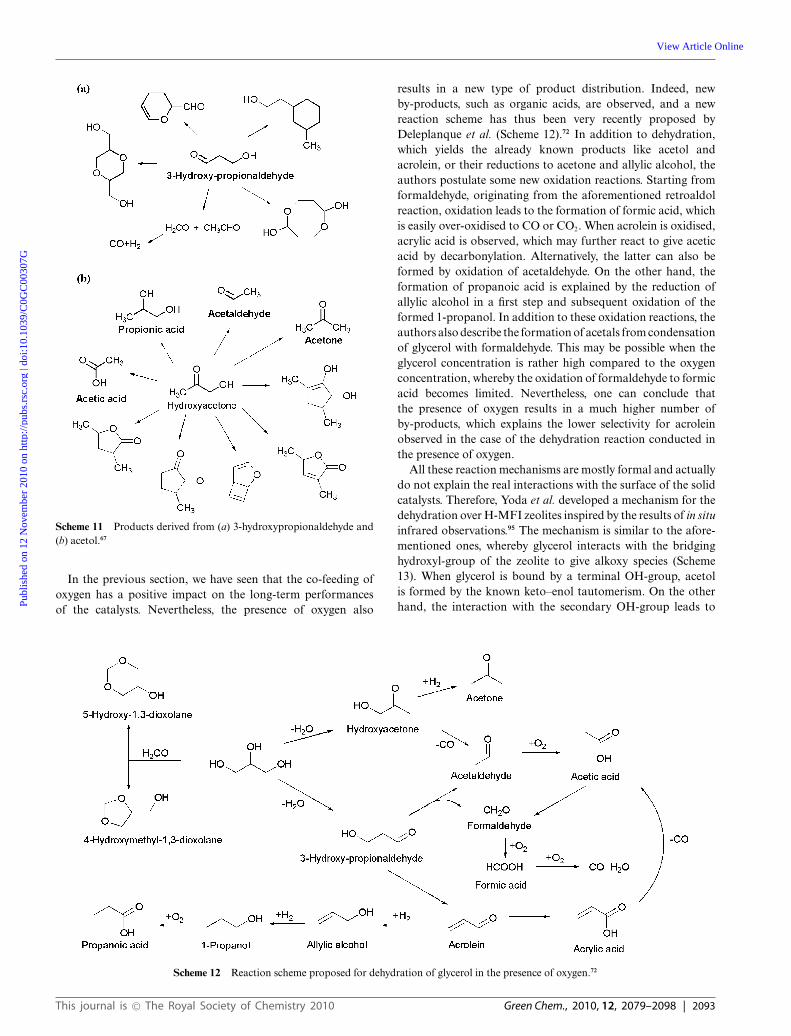
Scheme 11 Products derived from (a) 3-hydroxypropionaldehyde and(b) acetol.67
In the previous section, we have seen that the co-feeding ofoxygen has a positive impact on the long-term performancesof the catalysts. Nevertheless, the presence of oxygen also
results in a new type of product distribution. Indeed, newby-products, such as organic acids, are observed, and a newreaction scheme has thus been very recently proposed byDeleplanque et al. (Scheme 12).72 In addition to dehydration,which yields the already known products like acetol andacrolein, or their reductions to acetone and allylic alcohol, theauthors postulate some new oxidation reactions. Starting fromformaldehyde, originating from the aforementioned retroaldolreaction, oxidation leads to the formation of formic acid, whichis easily over-oxidised to CO or CO2. When acrolein is oxidised,acrylic acid is observed, which may further react to give aceticacid by decarbonylation. Alternatively, the latter can also beformed by oxidation of acetaldehyde. On the other hand, theformation of propanoic acid is explained by the reduction ofallylic alcohol in a first step and subsequent oxidation of theformed 1-propanol. In addition to these oxidation reactions, theauthors also describe the formation of acetals from condensationof glycerol with formaldehyde. This may be possible when theglycerol concentration is rather high compared to the oxygenconcentration, whereby the oxidation of formaldehyde to formicacid becomes limited. Nevertheless, one can conclude thatthe presence of oxygen results in a much higher number ofby-products, which explains the lower selectivity for acroleinobserved in the case of the dehydration reaction conducted inthe presence of oxygen.
All these reaction mechanisms are mostly formal and actuallydo not explain the real interactions with the surface of the solidcatalysts. Therefore, Yoda et al. developed a mechanism for thedehydration over H-MFI zeolites inspired by the results of in situinfrared observations.95 The mechanism is similar to the afore-mentioned ones, whereby glycerol interacts with the bridginghydroxyl-group of the zeolite to give alkoxy species (Scheme13). When glycerol is bound by a terminal OH-group, acetolis formed by the known keto–enol tautomerism. On the otherhand, the interaction with the secondary OH-group leads to
Scheme 12 Reaction scheme proposed for dehydration of glycerol in the presence of oxygen.72
This journal is © The Royal Society of Chemistry 2010 Green Chem., 2010, 12, 2079–2098 | 2093
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online
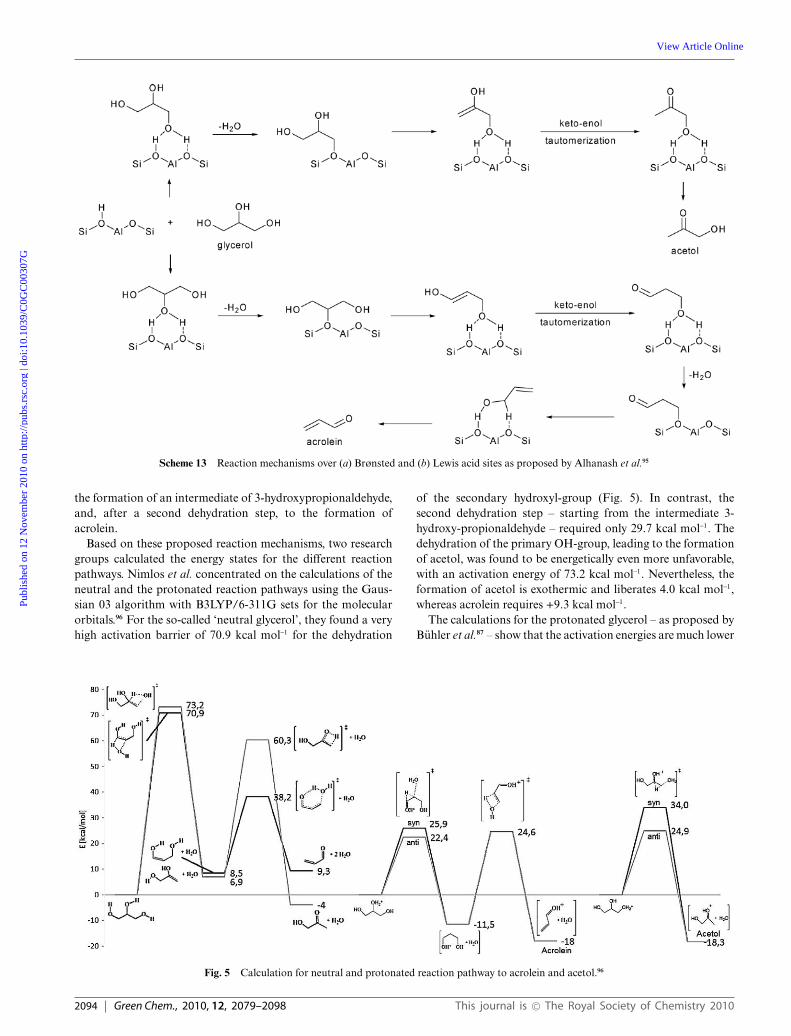
Scheme 13 Reaction mechanisms over (a) Brønsted and (b) Lewis acid sites as proposed by Alhanash et al.95
the formation of an intermediate of 3-hydroxypropionaldehyde,and, after a second dehydration step, to the formation ofacrolein.
Based on these proposed reaction mechanisms, two researchgroups calculated the energy states for the different reactionpathways. Nimlos et al. concentrated on the calculations of theneutral and the protonated reaction pathways using the Gaus-sian 03 algorithm with B3LYP/6-311G sets for the molecularorbitals.96 For the so-called ‘neutral glycerol’, they found a veryhigh activation barrier of 70.9 kcal mol-1 for the dehydration
of the secondary hydroxyl-group (Fig. 5). In contrast, thesecond dehydration step – starting from the intermediate 3-hydroxy-propionaldehyde – required only 29.7 kcal mol-1. Thedehydration of the primary OH-group, leading to the formationof acetol, was found to be energetically even more unfavorable,with an activation energy of 73.2 kcal mol-1. Nevertheless, theformation of acetol is exothermic and liberates 4.0 kcal mol-1,whereas acrolein requires +9.3 kcal mol-1.
The calculations for the protonated glycerol – as proposed byBuhler et al.87 – show that the activation energies are much lower
Fig. 5 Calculation for neutral and protonated reaction pathway to acrolein and acetol.96
2094 | Green Chem., 2010, 12, 2079–2098 This journal is © The Royal Society of Chemistry 2010
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

Fig. 6 Calculated energies for the glycerol dehydration over MFI-zeolite.97
than for the non-protonated one. Starting from the protonatedsecondary hydroxyl group of glycerol, the activation energy ofthe transition state of the first dehydration is only 22.4 kcal mol-1
vs. 70.9 kcal mol-1 without proton. The second dehydrationstep, which is calculated to 24.6 kcal mol-1 in the presenceof a proton, is also 5.1 kcal mol-1 lower than without (29.7kcal mol-1). Furthermore, the reaction pathway for protonatedglycerol leads to the exothermic formation of acrolein with anenergy release of 18.0 kcal mol-1. As in the case of the non-protonated reaction, the formation of acetol requires a higheractivation energy of 24.9 kcal mol-1 compared to that requiredfor the formation of acrolein (22.4 kcal mol-1). Moreover, thedifferences in the final energy states of the two products are quitelow (acetol: -18.3 kcal mol-1; acrolein: -18.0 kcal mol-1). Fromthese results, one can conclude that the formation of acetol isthermodynamically less favored than the formation of acrolein,due to the associated higher barrier of activation energy.Nevertheless, the difference is rather small, which suggeststhat it might be difficult to entirely suppress the formation ofacetol.
The calculations based on the results obtained for the reactionover ZSM-5 zeolite catalysts– as proposed by Yoda et al.95 – wereperformed by Kongpatpanich et al., who used Gaussion 03 withM06-2X/6-31G set for the zeolite 12T cluster model.97 For thefirst dehydration step, they found an activation energy slightlyhigher than that obtained by Nimlos et al. for the protonatedglycerol (41.4 kcal mol-1 vs. 22.4 kcal mol-1 (Fig. 6).96 Theactivation energy of the second dehydration was calculated at38.5 kcal mol-1 and the final energy state was +19.5 kcal mol-1.It is difficult to explain these differing results and, unfortunately,Kongpatpanich has not yet calculated the reaction pathwayleading to the formation of acetol.
The mechanisms described give quite good explanations forthe observed products in the glycerol dehydration reaction, suchas acrolein, acetol, formaldehyde, acetaldehyde, 1,2-propanedioland coke. The recently published mechanisms over basic cata-lysts and over the different types of acid catalysts are the firstattempts to explain the different product selectivities observedover these types of catalysts.
10 Direct use of crude glycerol as a reactant: animportant economic issue
As mentioned in the introduction, the price of glycerol drasti-cally depends on the required technical grade. Whereas refinedglycerol cost between 500 and 600 US$ t-1 in January 2010, crudeglycerol could be obtained at the same time for only 200 US$t-1.26 Nevertheless, the glycerol market has previously proven tobe highly volatile, with costs up to 1800 US$ t-1 for refined glyc-erol and up to 500 US$ t-1 for crude glycerol in 2007–2008.26 Itshould also be mentioned that glycerol availability and price alsodepend on political decisions concerning biodiesel productionand agricultural subsidies. Therefore, making possible the useof crude glycerol is a crucial issue for achieving a sustainableand economically viable process for the production of acrolein.However, as previously mentioned, crude glycerol is generallycontaminated with by-products issued from biodiesel process(inorganic traces like salts and organic traces like water, esters,fatty acids and alcohols). The use of crude glycerol as a feedstockmay therefore induce serious difficulties, either by poisoning ofthe catalysts or by causing plugs due to deposition of high-boiling organic materials or inorganic salts. To our knowledge,at this time no catalytic test using this type of feedstock hasbeen reported in the literature – all the disclosed studies werecarried out using diluted aqueous solutions prepared fromrefined glycerol. Furthermore, it has been found that the yieldof acrolein strongly depends on the glycerol concentration inthe reactor feed, with a decrease in the yield with increasingconcentration.35,36 However, the use of crude glycerol is of primeeconomical importance, and different ideas have already beenproposed to avoid the costly distillation of crude glycerol and toovercome the obstacles raised by its use as a feedstock. Kijenskiet al. proposed a modified evaporation system, where the crudeglycerol is brought into contact with an inert liquid at hightemperature.98 The glycerol is evaporated and fed to the reactordiluted in an inert carrier gas, whereas the impurities remain inthe inert liquid. The authors describe an example of a processin which a solution of 75 wt% of glycerol with up to 3 wt%impurities is added to silicon-oil heated to 330 ◦C.
This journal is © The Royal Society of Chemistry 2010 Green Chem., 2010, 12, 2079–2098 | 2095
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

To facilitate the use of crude glycerol, Dubois and Sereshikiet al. instead suggest a fluidised inert solid heated to hightemperature.99,100 This leads to the evaporation of glycerol,whereas the impurities remain inside the fluidised bed, whichcan be continuously regenerated. The regeneration may consistin a flushing step with water to dissolve inorganic salts followedby a thermal treatment to burn off the organic deposits. Asan example, the authors describe a process whereby a solutionof 18 wt% glycerol with 2 wt% sodium chloride is put intocontact with a fluidised bed of silica particles. The fluidisationis performed using nitrogen at 310 ◦C. The author claimsa separation/recovery of 99.9% of the introduced sodiumchloride.
Another process enabling the use of crude glycerol was alsoproposed by Dubois.101 As the evaporation of crude glycerolat high temperature induces reactions between glycerol andMONGs, this author proposed the possibility of forming acetalsby reacting glycerol with the acrolein in a first step (Scheme14). These acetals have lower boiling points than glycerol (185–240 ◦C vs. 290 ◦C), and thus the aforementioned undesired side-reactions are said to be reduced. The formed acetals are thenevaporated in the second step and subsequently dehydrated overan acid catalyst in the third step, to finally obtain acrolein. Theacrolein formed is partially recycled for the acetalisation in thefirst step.
Scheme 14 Process based on an intermediate acetalisation step topurify crude glycerol.
In conclusion, one can see that the use of crude glycerolnecessitates a supplementary upstream step at the level ofthe reactant evaporation. All the proposed concepts have incommon the fact that inorganic impurities like salts are retained,whereby the poisoning of the catalyst is inhibited. Nevertheless,we have to mention that important progress has been madeconcerning the improvement of the quality of the glycerol issuingfrom biodiesel production processes.102 The early processes werebased on homogenous catalytic cleavage of the vegetable oilsand fats in the presence of a base – for example, one of thenew processes developed by the French company Axens usesa heterogeneous catalyst.103 This ‘Esterfip-H’ process providesexceptionally high glycerol purities close to 98%, with only afew traces of inorganic salts.104 Furthermore, the costs generated
by the use of heterogeneous catalytic systems are claimed to belower than those from the conventional homogeneous process,as the leaching of the catalyst is negligible in this case. Thisprocess is planned or already in application in six units in Europe,and North & South America, with a total capacity of nearly1 million tonnes per year. Nevertheless, for the near future,both homogenous and heterogeneous processes will still haveto deal with the availability of different grades of crude glycerolon the global market. Therefore, the question concerning theeconomic sense of investing (or not) in a supplementary unit forthe evaporation of crude glycerol is difficult to answer at thisstage.
11 Conclusion
The depletion of the petrol feedstock and the global warmingcaused by CO2 emission have led to a change in the energypolicies. Biofuels have been proposed as an alternative to fossilfuels for reducing the impact of greenhouse gases. Due topolitical directives, the production of bioethanol for blendingwith gasoline and the production of biodiesel have drasticallyincreased. As a consequence, the market has been flooded withcrude glycerol co-produced in the biodiesel production process.As glycerol is a highly functionalisable molecule, many processesfor its valorisation have been recently published. In particular,the catalytic dehydration to acrolein – a precursor of sodiumpolyacrylate used as a superabsorbent polymer and of DL-methionine used for cattle feeding – is of high economic interest.This reaction is catalysed over Brønsted and Lewis acid sitesaccording to two different mechanisms, which are still a matterof debate and will undoubtedly be refined in the near future.Furthermore, the picture of the influence of the acidity on thecatalytic performances has become clearer, showing that optimalacidic strength is needed for high and constant performance.
As far as gas-phase dehydration is concerned, a large variety ofhighly efficient catalysts has been proposed, which can be dividedinto three different categories: (i) supported HPAs; (ii) zeolites;and (iii) mixed-metal oxide type catalysts, including phosphates.The best catalytic performance has, up to now, been achievedwith caesium salts of HPAs, yielding up to 98% acrolein in thevery early stages of the reaction. On zeolitic and mixed oxidecatalysts, the acrolein yield is generally slightly lower (around 70to 80%) and the temperature needed to reach high conversionsis higher (around 300 ◦C vs. 275 ◦C for HPAs). Nevertheless, thefamily of zeolites and supported HPAs offers many possibilitiesfor tuning acidity and textural parameters, which makes possiblefine-tuning to yield tailor-made catalysts.
However, the main problem remains rapid catalyst deactiva-tion due to carbon deposition. Up to now, no catalyst has had ahalf-life of more than a few days, which is of course not sufficientfor commercial applications. Therefore, various approaches havebeen published to regenerate the coked catalysts. These are: theuse of a fluidised circulating catalyst, which is continuouslyregenerated in a parallel reactor; the introduction of oxygen,hydrogen or sulfur dioxide into the reaction feed for continuousregeneration of the catalyst inside the reactor; and discontinuousregeneration by alternating the glycerol feed and the oxygen feed.It was even proposed to dope the catalysts with noble metals forfacilitating the splitting of the dioxygen used for regeneration. At
2096 | Green Chem., 2010, 12, 2079–2098 This journal is © The Royal Society of Chemistry 2010
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

this time, the co-feeding of oxygen seems to be the most practicalway for extending catalyst life-time, as this technique requiresrelatively low investment and is easy compared to pulse-wiseregeneration or to a fluidised process. Nevertheless, this methodis not applicable for all kinds of catalysts, as redox propertiesmay lead to significant decrease in the selectivity for acrolein.
Liquid-phase dehydration was well studied with the rise ofresearch into supercritical fluids. Nevertheless, a dead-end hasbeen reached, as dehydration just in supercritical water withoutacid catalyst results in low acrolein yields, while the combinationof supercritical conditions with acids is problematic in termsof material stress on the reaction vessel, which would lead tosignificant investment costs. The second – and more promising– route is the use of high-boiling solvents containing solidcatalysts. With this technology, a quasi-continuous process canbe achieved, even if scale-up to the industrial scale remains risky.As a consequence, the dehydration of glycerol in the liquid phaseappears to be a rather uneconomic process.
Finally, we have shown the need for the use of crude glycerolas a feedstock in regard to economic aspects. We have notcome across any attempt to directly use crude glycerol overcatalytic formulations, but two promising upstream technologieshave been presented, which aim to eliminate the impuritiesof the crude glycerol during evaporation and thus avoid thecostly crude glycerol distillation. As the number of publicationsconcerning this kind of technology is still very limited, it isdifficult at the moment to foresee which of them is the mostpromising. Furthermore, the quality of crude glycerol frombiodiesel production is constantly increasing, reaching a purityup to 98% without any inorganic salts, which makes industrialapplications of the proposed purification techniques ratherimprobable, at least in the long-term.
References1 United Nations Framework Convention on Climate Change,
http://unfccc.int.2 Directive 2003/30/EC of the European Parliament and of the
council of 05/08/2003 on the promotion of the use of biofuelsor other renewable fuels for transport.
3 L. C. Meher, D. V. Sagar and S. N. Naik, Renew. Sustain. EnergyRev., 2006, 10, 248–268.
4 Eurostat European Commission, http://epp.eurostat.ec.europa.eu.5 U.S. National Biodiesel Board, http://www.biodiesel.org.6 Global Industry Analysts, http://www.strategyr.com/Glycerin_
Market_Report.asp.7 European Biodiesel Board, http://www.ebb-eu.org.8 J.-L. Dubois and G. S. Patience (Arkema), WO2009044051, 2009.9 M. J. Haas, A. J. Mc Aloon, W. C. Yee and T. A. Foglia, Biores.
Tech., 2006, 97, 671–678.10 M. Pagliaro and M. Rossi, in The Future of Glycerol, 2nd edn (RSC
Green Chemistry Book Series), 2010.11 C. S. Callam, S. J. Singer, T. L. Lowary and C. M. Hadad, J. Am.
Chem. Soc., 2001, 123, 11743.12 D. Siano, E. Santacesaria, V. Fiandra, R. Tesser, G. Di
Nuzzi, M. Di Serio and M. Nastasi (ASER), WO 2006111810,2006.
13 D. Schreck, W. Kruper, F. Varjian, M. Jones, R. Campbell, K.Kearns, B. Hook, J. Briggs and J. Hippler (DOW Chemicals), WO2006020234, 2006.
14 D. A. Simonetti, J. Ross-Hansen, E. L. Kunkes, R. R. Soares and J.A. Dumesic, Green Chem., 2007, 9, 1073.
15 R. R. Soares, D. A. Simonetti and J. A. Dumesic, Angew. Chem.Int. Ed., 2006, 45, 3982.
16 BioMNC, http://www.biomnc.eu.
17 M. Di Serio, L. Casale, R. Tesser and E. Santacesaria, Energy Fuels,2010, DOI: 10.1021/ef901230r, in press.
18 N. O. V. Sonntag, J. Am. Oil. Chem. Soc., 1992, 75, 795.19 Synergy Chemicals, http://www.senergychem.com.20 H. Kimura, Appl. Catal. A: Gen., 1993, 105, 147–158; H. Kimura,
K. Tsuto, T. Wakisaka, Y. Kazumi and Y. Inaya, Appl. Catal. A:Gen., 1993, 96, 217–228.
21 K. Nabe, N. Izuo, S. Yamada and I. Chibata, Appl. Environ.Microbiol., 1979, 38, 1056.
22 R. Ciriminna, G. Palmisano, C. Della Pina, M. Rossi and M.Pagliaro, Tetrahedron Lett., 2006, 47, 6993.
23 G. W. Keulks, L. D. Krenzke and T. M. Notermann, Adv. Catal.,1978, 27, 183.
24 M. M. Lin, Appl. Catal. A: Gen., 2001, 207, 1.25 A. Corma, G. W. Huber, L. Sauvanaud and P. O’Connor, J. Catal.,
2008, 257, 163–171.26 ICIS Market-reporter, 6 January 2010, http://www.icis.com.27 Global Industry Analysts, http://www.strategyr.com/Super_
Absorbent_Polymers_Market_Report.asp.28 A. Yamamoto, Encyclopaedia of Chemical Technology, 3rd edn,
1978, vol. 2, p. 403.29 M. P. Malveda, H. Janshekar, K. Yokose, CEH Marketing Research
Report – SRI Consulting, Major Amino Acids, June 2006.30 M. P. Malveda, H. Janshekar, K. Yokose, Chemical Economics
Handbook-SRI Consulting, June 2006, 502.5000 B.31 B. Katryniok, S. Paul, M. Capron and F. Dumeignil, Chem-
SusChem., 2009, 2, 719–730.32 Schering-Kahlbaum FR695931, 1930.33 H. Groll and G. Hearne (Shell), US 2042224, 1936.34 H. Hoyt and T. Manninen (US Ind. Chemicals. Inc.), US 2558520,
1951.35 A. Neher, T. Haas, A. Dietrich, H. Klenk and W. Girke (Degussa),
DE 4238493, 1994.36 A. Neher, T. Haas, A. Dietrich, H. Klenk and W. Girke (Degussa),
US 5387720, 1995.37 A. Neher and T. Haas (Degussa), US 5426249, 1995.38 J.-L. Dubois, C. Duquenne, W. Hoelderich and J. Kervennal
(Arkema), WO 2006087084, 2006.39 J.-L. Dubois, C. Duquenne and W. Hoelderich (Arkema),WO
2006087083, 2006.40 S.-H. Chai, H.-P. Wang, Y. Liang and B.-Q. Xu, J. Catal., 2007, 250,
342–349.41 S.-H. Chai, H.-P. Wang, Y. Liang and B.-Q. Xu, Green Chem., 2007,
9, 1130–1136.42 Z. H. Chen, T. Lizuka and K. Tanabe, Chem. Lett., 1984, 13, 1085.43 T. Lizuka, K. Ogasawara and K. Tanabe, Bull. Chem. Soc. Jpn.,
1983, 56, 2927.44 A. Alhanash, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl.
Catal. A: Gen., 2010, 378, 11–18.45 Y. T. Kim, K.-D. Jung and E. D. Park, Microporous Mesoporus
Mater., 2010, 131, 28–36.46 A. K. Kinage, P. P. Upare, P. Kasinathan, Y. K. Hwang and J.-S.
Chang, , Catal. Commun., 2010, 11, 620–623.47 S.-H. Chai, H.-P. Wang, Y. Liang and B.-Q. Xu, Green Chem., 2008,
10, 1087–1093.48 S.-H. Chai, H.-P. Wang, Y. Liang and B.-Q. Xu, Appl. Catal. A:
Gen., 2009, 353, 213–222.49 L. Ning, Y. Ding, W. Chen, L. Gong, R. Lin, Y. Lu and Q. Xin,
Chin. J. Catal., 2008, 29(3), 212–214.50 B. Katryniok, S. Paul, M. Capron, C. Lancelot, V. Belliere-Baca, P.
Rey and F. Dumeignil, Green Chem., 2010, 12, 1922–1925.51 E. Tsukuda, S. Sato, R. Takahashi and T. Sodesawa, Catal.
Commun., 2007, 8, 1349–1353.52 H. Atia, U. Armbruster and A. Martin, J. Catal., 2008, 258, 71–
82.53 B.-Q. Xu, S.-H. Chai, T. Takahashi, M. Shima, S. Sato and R.
Takahashi (Nippon Catalytic Chem. Ind.), WO 2007058221, 2007.54 T. Sato and R. Takahashi (Nippon Catalytic Chem. Ind.), JP
2008088149, 2008.55 H. Jo, S.-H. Chai, T. Takahashi and M. Shima (Nippon Catalytic
Chem. Ind.), JP 2007137785, 2007.56 S. Erfle, U. Armbruster, U. Bentrup, A. Martin and A. Bruckner,
Appl. Catal. A, 2010, DOI: 10.1016/j.apcata.2010.04.042, in press.57 J.-L. Dubois, Y. Magatani and K. Okumura (Arkema), WO
2009127889 and WO 2009128555, 2009.
This journal is © The Royal Society of Chemistry 2010 Green Chem., 2010, 12, 2079–2098 | 2097
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online

58 X.-Z. Li (Shanghai Huayi Acrylic Acid Co), CN 101070276, 2007.59 A. Zhuang, C. Zhang , S. Wen, X. Zhao and T. Wu (Shanghai Huayi
Acrylic Acid Co), CN 101225039, 2008.60 M. Okuno, E. Matsunami, T. Takahashi, H. Kasuga, M. Okada
and M. Kirishik (Nippon Catalytic Chem. Ind.), WO 2007132926,2007.
61 M. Okuno, E. Matsunami, T. Takahashi and H. Kasuga (NipponCatalytic Chem. Ind.), JP 2007301505, 2007.
62 M. Okuno, E. Matsunami, T. Takahashi and H. Kasuga (NipponCatalytic Chem. Ind.), JP 2007301506, 2007.
63 C.-J. Jia, Y. Liu, W. Schmidt, A.-H. Lu and F. Schuth, J. Catal.,2010, 269, 71–79.
64 C.-J. Zhou, C.-J. Huang, W.-G. Zhang, H.-S. Zhai, H.-L. Wu andZ. S. Chao, Stud. Surf. Sci. Catal., 2007, 165, 527–530.
65 K. Pathak, K. M. Reddy, N. N. Bakhshi and A. K. Dalai, Appl.Catal. A, 2010, 372, 224–238.
66 L.-Z. Tao, S.-H. Chai, Y. Zuo, W.-T. Zheng, Y. Liang and B.-Q. Xu,Catal. Today, 2010, DOI: 10.1016/j.cattod.2010.03.073, in press.
67 W. Suprun, M. Lutecki, T. Haber and H. Papp, J. Mol. Catal. A:Chem., 2009, 309, 71–78.
68 F. Wang, J.-L. Dubois and W. Ueda, J. Catal., 2009, 268, 260–267.69 F. Wang, J.-L Dubois and W. Ueda, Appl. Catal. A, 2010, 276,
25–32.70 J.-L. Dubois (Arkema), WO2010046227, 2010.71 Q. Liu, Z. Zhang, Y. Du, J. Li and X. Yang, Catal. Lett., 2008,
127(3-4), 419–428.72 J. Deleplanque, J.-L. Dubois, J.-F Devaux and W. Ueda, Catal.
Today, 2010, DOI: 10.1016/jcattod.2010.04.012, in press.73 J.-L. Dubois (Arkema), WO2009044081, 2009.74 E. Matsunami, T. Takahashi and H. Kasuga (Nippon Catalytic
Chem. Ind.), JP 2007268363, 2007.75 E. Matsunami, T. Takahashi and H. Kasuga (Nippon Catalytic
Chem. Ind.), JP 2007268364, 2007.76 H. Redlingshoefer, C. Weckbecker, K. Huthmacher and A. Doer-
flein (Evonik Degussa), WO 2008092533, 2008.77 H. Redlingshoefer, C. Weckbecker, K. Huthmacher and A. Doer-
flein (Evonik Degussa), WO 2008092533, 2008.78 H. Redlingshoefer, C. Weckbecker, K. Huthmacher and A. Doer-
flein (Evonik Degussa), WO 2008092534, 2008.79 A. Ulgen and W. Hoelderich, Catal. Lett., 2009, 131, 122–128.
80 H. Kasuga and M. Okada (Nippon Catalytic Chem. Ind.), JP2008137950, 2008.
81 J.-L. Dubois (Arkema), WO 2009156664, 2009.82 Y. Arita, H. Kasuga and M. Kirishiki (Nippon Catalytic Chem.
Ind.), JP 2008110298, 2008.83 P. O’Connor, C. Corma, G. Huber and L. Savanaud (Bioecon.
Internat. Holding), WO 2008052993, 2008.84 J.-L. Dubois (Arkema), FR 2897058, 2007.85 H. Kasuga (Nippon Catalytic Chem. Ind.), JP 2008137952, 2008.86 S. Ramayya, A. Brittain, C. De Almeida, W. Mok and M. J. Antal,
Fuel, 1987, 66, 1364.87 W. Buhler, E. Dinjus, H. J. Ederer, A. Kruse and C. Mas, J.
Supercritical Fluids, 2001, 22, 37–53.88 L. Ott, M. Bicker and H. Vogel, Green Chem., 2006, 8(2), 214–220.89 V. Lehr, M. Sarlea, L. Ott and H. Vogel, Catal. Today, 2007, 121(1-
2), 121–129.90 M. Watanabe, T. Iida, Y. Aizawa, T. M. Aida and H. Inomata,
Biores. Technol., 2007, 98, 1285.91 N. Suzuki and M. Takahashi (KAO Corp.), JP 2006290815, 2006.92 Y. Yoshimi, Y. Masayuki, A. Torakichi and A. Takanori (Showa
Denko), JP2009179569, 2009.93 A. Takanori and Y. Masayuki (Showa Denko), JP2009292773, 2009.94 A. Takanori and Y. Masayuki (Showa Denko), JP2009292774, 2009.95 E. Yoda and Y. Ootawa, Appl. Catal. A, 2009, 360, 66–70.96 M. R. Nimlos, S. J. Blanksby, X. Qian, M. E. Himmel and D. K.
Johnson, J. Phys. Chem. A., 2006, 110, 6145–6156.97 K. Kongpatpanich, T. Nanok, B. Boekfa and J. Limtrekul, Prepr.
Pap. Am. Chem. Soc. Div. Pet. Chem., 2010, 55(1), 115–118.98 J. Kijenski, A. Migdal, O. Osawaru and E. Smigiera (Inst. Chemii
Przemyslowe), EP 1860090, 2007.99 J.-L. Dubois (Arkema), WO 2008129208, 2008.
100 B. R. Sereshiki, S.-J. Balan, G. S. Patience and J.-L. Dubois, Ind.Eng. Chem. Res., 2010, 49, 1050–1056.
101 J.-L. Dubois (Arkema), WO2009081021, 2009.102 C. Perego and D. Bianchi, Chem. Eng. J., 2010,
DOI: 10.1016/j.cej.2010.01.036, in press.103 R. Stern, G. Hillion, J-J. Rouxel and L. Serge (Institut Francais du
Petrol), US 5908946, 1999.104 L. Bournay, D. Casanave, B. Delfort, G. Hillion and J. A. Chodorge,
Catal. Today, 2005, 106, 190–192.
2098 | Green Chem., 2010, 12, 2079–2098 This journal is © The Royal Society of Chemistry 2010
Publ
ishe
d on
12
Nov
embe
r 20
10 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C0G
C00
307G
View Article Online


















