
GLOMERULOPATHY clinical categories ➲ Acute nephritic syndrome: haematuria, red blood cell casts,...
109
GLOMERULOPATHY clinical categories ➲ Acute nephritic syndrome: haematuria, red blood cell casts, proteinuria, oliguria, hypertension, edema, circulatory congestion ➲ Rapidly progressive nephritic syndrome: relentlessly progressive glomerulonephritis resulting in ESRF within weeks ➲ Recurrent gross haematuria
-
Upload
preston-hutchinson -
Category
Documents
-
view
217 -
download
0
Transcript of GLOMERULOPATHY clinical categories ➲ Acute nephritic syndrome: haematuria, red blood cell casts,...

GLOMERULOPATHYclinical categories
➲ Acute nephritic syndrome: haematuria, red blood cell casts, proteinuria, oliguria, hypertension, edema, circulatory congestion
➲ Rapidly progressive nephritic syndrome: relentlessly progressive glomerulonephritis resulting in ESRF within weeks
➲ Recurrent gross haematuria

GLOMERULOPATHYclinical categories
➲ Asymptomatic glomerulopathy: proteinuria, haematuria, or both – without clinical symptoms
➲ Chronic nephritic (glomerular) syndrome: glomerular disease that progress in chronic renal failure
➲ Nephrotic syndrome

GLOMERULOPATHYstructural characteristics
➲Acute damage (diffuse or segmental): - proliferation of epithelial, endothelial and mesangial cells; - exudation of polymorphonuclear leukocytes in the glomerulus; - necrosis of glomerular capillaries

GLOMERULOPATHYstructural characteristics
➲ Chronic damage: - proliferation of cellular elements (epithelium, endothelium, mesangium); - membranous involvement with thickening of glomerular basement membrane (GBM); - sclerosis of the glomerulus; - tubular atrophy, nephrosclerosis, interstitial scarring (in ESRF)

GLOMERULOPATHYpathogenesis
➲ Immunologic reactions➲ Vascular diseases ➲ Abnormalities in coagulation➲ Metabolic defects➲ Hereditary factors➲ Unknown factors

GLOMERULOPATHYimmunologic mechanism
➲ Circulating immune complex – mediated disease
➲ Antitissue antibody – mediated disease➲ Cell- mediated disease➲ Disease associated with activation of
alternative complement pathway

Physiology of protein excretion
➲ Protein filtration through the glomerulus is dependent on the protein size, shape and electrical charge

Physiology of protein excretion
➲ Protein charge
●At physiological pH, most proteins are negatively charged
●Since the basement membranes are also negatively charged, most proteins are retained

Physiology of protein excretion
➲ Protein size
●Proteins greater than 40kDa are almost completely retained
●Thus, only small proteins, e.g. retinol-binding protein, ß2 microglobulin, passes into the ultrafiltrate

Physiology of protein excretion
➲ However, most of the filtered proteins are reabsorbed by the proximal tubules.
➲ Consequently, very little plasma protein appears in the urine
➲ Normally < 150mg/24hours

“Physiological” proteinuria
➲ In some non-pathological situations, a higher than normal urine protein level is found:
● A concentrated spot urine● Exercise● Orthostatic proteinuria● Contamination e.g. from vagina

Classification
➲ Tubular proteinuria●Tubular dysfunction●Overflow proteinuria
➲ Glomerular proteinuria●Selective proteinuria●Non-selective proteinuria●microalbuminuria

Tubular proteinuria
➲ This occurs when glomerular function is intact, but protein is lost to the urine either because of:
●Tubular dysfunction●Overflow

Tubular proteinuria
➲ Tubular dysfunction●The tubules are damaged and cannot function
properly●Therefore, the small MW proteins that are
normally filtered are not reabsorbed by the tubules
●The small MW proteins include: retinol-binding protein, ß2 microglobulin, lysozyme, light chains, haemoglobin, myoglobin

Tubular proteinuria
➲ Tubular dysfunction●Pyelonephritis●Acute tubular necrosis●Papillary necrosis e.g. analgesic
nephropathy●Heavy metal poisoning●SLE●Fanconi’s syndrome

Tubular proteinuria
➲ Overflow proteinuria
●Occurs when the concentration of one of the small MW proteins is so high that the filtered load exceeds the tubular reabsorptive capacity
●Thus, the excess filtered load appears in the urine

Tubular proteinuria
➲ Overflow proteinuria
●Bence Jones proteinuria●Myoglobinuria●Haemoglobinuria

Glomerular proteinuria
➲ When there is glomerular dysfunction, proteins > 40kDa can escape into the urine
➲ The most common form of proteinuria

Glomerular proteinuria
➲ Causes●Glomerulonephritis●Diabetes mellitus●Multiple myeloma●Amyloidosis●SLE●Pre-eclampsia●Penicillamine, gold

Definitions
➲ Proteinuria●Urine protein excretion > 150mg/day
➲ Microalbuminuria●Urine [albumin] > 30mg/day but not detectable
by urine dipstick➲ Nephrotic syndrome
●Urine protein excretion > 3.5g/day (with hypoalbuminaemia, oedema and hyperlipidaemia)

Nephrotic Syndrome (NS)
Is not a disease but a group of signs and symptoms seen in patients with heavy proteinuria
presents with oedema proteinuria usually > 3.5g / 24hrs (>0.05g / kg /
24hrs in children) serum albumin < 30g/l other features: hyperlipidaemia, and
hypercoaguable state

NS pathophysiology proteinuria: due to an increase in glomerular
permeability hypoalbuminuria: occurs when liver synthesis cannot
keep up with urine losses oedema mechanism is complex and still in dispute:
primary salt and water retention associated with reduced renal function as well as reduced plasma oncotic pressure are primary factors (overfill and underfill)
hyperlipidaemia: increased liver synthesis hypercoagulation: increased fibrinogen and loss of
antithrombin III

Clinical Features in NS - Thrombosis
➲ Serious risk of thrombosis➲ Increased fibrinogen concentration➲ Antithrombin III concentration reduced➲ NS patients resistant to heparin➲ Platelets hyperaggregable➲ Increased blood viscosity

NS - laboratory Features
➲Hct may be elevated➲Hyponatremia is common➲Plasma creatinine is elevated in
33% of patients

NS laboratory- Plasma Protein
➲ Albumin● Hypoalbuminemia due to loss via the kidney
● Urinary excretion● Proximal tubular cells catabolism
➲ Immunoglobulins● IgG levels reduced● IgM levels elevated● IgM-IgG-Switching

NS laboratory- Hyperlipidemia
➲ Increased synthesis of cholesterol, triglycerides and lipoproteins
➲ Decreased catabolism of lipoproteins● Decreased activity of lipoprotein lipase
➲ Decreased LDL receptor activity➲ Increased urinary loss of HDL➲ Lp(a) levels are elevated

Primary glomerular diseases commonly causing the nephrotic syndrome
minimal change disease focal and segmental glomerulosclerosis membranous glomerulonephritis proliferative glomerulonephritis (various
histology and less common cause)membranoproliferative (mesangiocapillary)focal proliferativediffuse proliferativemesangial proliferative

Other causes of the nephrotic syndrome 1
Systemic diseasesdiabetes mellitusamyloidosisSLE and other connective tissue diseasesHIV/AIDS
nephrotoxinsnsaidsmercury poisoningpenicillaminegold salts

Other causes of the nephrotic syndrome 2
Allergiesbee stingpollenspoison ivy
Circulatory effectscongestive cardiac failureconstrictive pericarditisrenal vein thrombosis (cause or result?)
Neoplasticleukaemiasolid tumours

NS epidemiology

NS treatment- Diet
➲ Low protein●Decreases albuminuria●Malnutrition
➲ Salt restriction (Na+< 60 mmol/24 hrs)●During edema
➲ Calorie control●Steroids

NS treatment
water restriction diuretics (if not volume depleted) reduced protein diet (controversial) treat infections prophylaxis for thrombosis specific therapy
corticosteroidsimmunosuppression

NS treatment- Albumin
➲ Controversial➲ Indication- Hypovolemia
●Abdominal pain●Hypotension●Oliguria●Renal insufficiency

NS complications
➲ Mortality●1940’s- 40% 1 year mortality●Now 1-2%●Main cause of death
●Infection●Thrombosis

Corticosteroids Initiation in NS
➲ High dose steroids●2 mg/kg/day (max 80 mg)●60 mg/m2 (max 80 mg)
➲ 3 accepted protocols➲ 80% respond within 2 weeks

Steroid Toxicity
➲ Cushingoid habitus➲ Obesity➲ Striae➲ Hirsutism➲ Acne➲ Growth failure➲ Avascular necrosis➲ Osteoporosis

Steroid Toxicity
➲ Peptic ulceration➲ Pancreatitis➲ Posterior lens opacities➲ Myopathy➲ Increased ICP➲ Susceptibility to infection

Options for Alternative Therapy in NS
➲ Alkylating Agents●Nitrogen mustard●Cyclophosphamide●Chlorambucil
➲ Levamisole➲ Cyclosporine

Indications for Alternative Therapy in NS
➲ Relapse on Prednisone Dosage >0.5 mg/kg/alt day plus:●Severe steroid side effects●High risk of toxicity- diabetes●Unusually severe relapses
➲ Relapses on Prednisone Dosage >1.0 mg/kg/alt day

Acute Nephritic Syndrome
Syndrome characterised in typical cases by:
haematuria oliguria oedema hypertension reduced GFR proteinuria fluid overload

Clinical Features of the Acute Nephritic Syndrome
haematuria is usually macroscopic with pink or brown urine (like coca cola)
oliguria may be overlooked or absent in milder cases
oedema is usually mild and is often just peri-orbital- weight gain may be detected
hypertension common and associated with raised urea and creatinine
proteinuria is variable but usually less than in the nephrotic syndrome

Etiology of the Nephritic Syndrome
➲ Most common cause is acute post infectious glomerulonephritis
➲ group A beta haemolytic streptococci of certain serotypes important in NZ
➲ IgA disease and Henoch-Schonlein purpura, crescentic glomerulonephritis and SLE can also present in this way

Complications of the Nephritic Syndrome
Hypertensive encephalopathy (seizures, coma)
Heart Failure (pulmonary oedema)
Uraemia requiring dialysis

Acute poststreptococcal GN
➲ Archetype of acute nephritic syndrome➲ Proliferative character➲ Only certain varietes of beta-hemolytic
streptococci (nephritogenic strains) induce abnormalities in kidneys
➲ Production of nonspecific evidence of streptococcal exposure (elevated antistreptolisin titers)
➲ Antibody production and immune complexes

Acute poststreptococcal GN
➲ Kidneys are enlarged, edematosus, pale➲ Electron-dense deposits on the epithelial
side of GBM➲ Reduced GFR ➲ Elevation of urea and creatinine is
characteristic➲ Urine: reduced in volume, concentrated,
reddish brown; contains as much as 2 to 4 mg/day of protein

Acute poststreptococcal GN
➲ More common in males than females and most frequent between the ages of 3 and 7 years
➲ Classically, 10 days after sore throat➲ Acute nephritic syndrome➲ Gross hematuria and fever➲ Worse prognosis in adults

Definition of glomerulonephritis
Glomerulonephritides are supposedly immunologically mediated glomerular diseases,
often, but not always,
inflammatory in nature

Glomerular inflammation
1. Exsudation of neutrophils and/or macrophages
2. Proliferation of mesangial and/or endothelial cells

Ultrastructural changes in non-proliferative vs. proliferative glomerulonephritides

Mechanisms of glomerular damage

Simplified classification of primary glomerulonephritides
1. Nonproliferative- minimal change disease
- focal segmental glomerulosclerosis - membranous nephropathy
2. Proliferative - IgA nephropathy
- membranoproliferative GN

Ultrastructural changes in glomerular capillaries in different glomerular diseases

Podocytes and slit diaphragms

Major causes of podocyte effacement
1. Slit diaphragm and its lipid raft nephrin, podocin1. Podocyte cytoskeleton -actinin1. Adhesion of podocyte to GBM
-dystroglycan, 1-integrins4. Loss of podocyte electronegative charge podocalyxin

Non-proliferative glomerulopathies
Damage to the glomerular capillary wall resulting in:
1. nephrotic selective proteinuria - minimal change disease2. nephrotic non-selective proteinuria with
microscopic hematuria - focal segmental glomerulosclerosis
- idiopathic membranous nephropathy

Primary glomerulonephritides as a cause of nephrotic syndrome Korbet et al., Am. J. Kidney Dis., 1996, 27: 647 - 651

Indications for Biopsy
➲ Pretreatment● Recommended
●Onset age < 6 months●Macroscopic hematuria●Microscopic hematuria and HTN●Low C3●Renal failure
● Discretionary●Onset between 6-12 months or > 12 years●Persistent HTN of hematuria

Indications for Biopsy
➲ Post treatment●Steroid resistance●Frequent relapsers

Minimal change disease

Minimal change disease

Pathogenesis of minimal change disease
1. circulating permeability factor(hemopexin?)
1. decreased synthesis of glomerular polyanions (heparan sulfate) by podocytes
2. impaired adhesion of podocytes to GBM( -dystroglycan, 1-integrins?)
4. expression of TGF1 detectable almost only in steroid resistant MCD and FSGS

Minimal change disease
1. full-blown nephrotic syndrome with selective proteinuria
2. hematuria, hypertension and reduced renal function uncommon
3. absence of glomerular abnormalities on LM and IF
4. fusion of epithelial cells foot processes on electron microscopy

Minimal change disease-prevalence among nephrotic patients
Children - 85 – 95%
Young adults - 50%
Adults > 40 years - 20 – 25%

Classification of patients with minimal change disease based on response to corticosteroids
1. Steroid responsive (sensitive)develop complete remission of proteinuria
within 8 – 12 weeks of treatment (in adults remission should develop within
16 weeks)2. Steroid dependent
develop relapse during tapering of steroids or within 2 weeks after cessation of therapy
3. Steroid resistant fail to respond to steroid treatment at all

Definitions
➲ Steroid Dependence- Two consecutive relapses occurring during corticosteroid treatment or within 14 days of its cessation
➲ Steroid Resistance- Failure to achieve response in spite of 4 weeks of prednisone 60 mg/m2*day

Clinical course of MCD in children
1. Remission - 90%a. no relapses - 20%
b. infrequent relapses - 40%c. frequent relapses and
steroid dependent - 30%2. Resistance to steroids - 10%
a. response to alternative treatment - 8%
b. refractory to any kind of treatment - 2%
In adults, initial response rate is lower, relapses and steroid dependence are less frequent

Therapy of MCD in children – current recommendations
1. Initially course of prednisone 60 mg/m2 for 4-6 weeks with 40 mg/m2 every alternate day for another 4-6 weeks
2. Relapses treated in a similar way, but tapering of prednisone starts when urine becomes protein free
3. Frequent relapsers and steroid dependent patients treated either by cyclophosphamide 2 mg/kg/day for 8 weeks or by cyclosporine 5 mg/kg/day for 6-12 months
4. Treatment of steroid resistant patients is usually unsatisfactory

Therapy of MCD – modifications in adults
1. Initially course of prednisone 1mg/kg for 8-16 weeks or for one week after remission is achieved, then several weeks (one month) 1 mg/kg on alternate days, thereafter corticosteroids are slowly tapered during several months
2. Relapses treated in a similar way3. Frequent relapsers and steroid dependent
patients treated either by CPH 2 mg/kg/day for 8 weeks or by CyA 5 mg/kg/day for 6-12 months
4. Treatment of steroid resistant patients is usually unsatisfactory

Mild FSGS

Moderate FSGS

Tip lesion in early FSGS

Collapsing FSGS

Etiology of FSGS
1. Primary FSGSa. glomerular tip lesionb. collapsing glomerulopathy
2. Secondary FSGSa. healing focal lesions (FSGN)b. hyperfiltration in residual nephrons
- agenesis of one kidney- vesicoureteral reflux- morbid obesity
c. damage to epithelial cells- HIV nephropathy- heroin nephropathy

Classification of FSGS
1. Genetic FSGSa. podocinb. -actinin
2. Immunologicmechanisms not yet identified
3. Viral FSGSa. HIVb. hepatitis C
4. Toxic FSGSa. heroinb. pamidronate

Pathogenesis of primary FSGS
1. Late onset congenital FSGSdeficiency of podocyte proteins (podocin, -actinin, CD2AP, et al.)
2. Circulating permeability factorsa. imunoglobulin, or Ig-like moleculeb. protein of MW about 30-50 kDa
c. factor inhibiting inducible NO synthase in mesangial cells (hemopexin)
3. Deficient inhibitors of permeability factors lost in urineapolipoproteins of HDL complex (e.g. apo J, apo E2 and apo E4)

Permeability factors in MCD and FSGSGlassock, J Am Soc Nephrol, 2003, 14: 541 - 543
1. Permeability factors in MCD and FSGS may be different2. Among PF described in MCD (e.g. heparanase, VEGF)
hemopexin is best characterized (Cheung et al., Kidney Int, 2000, 57: 1512 – 1520)
3. In FSGS 30-50 kD weakly anionic, heat labile, protease-sensitive factor inhibiting NO production in mesangial cells was identified with the Palb assay (Sharma et al., Kidney Int, 2000, 58: 1073 - 1079).
4. This PF is increased also in pts with genetic mutation of podocin (Carraro et al., JASN, 2002, 13: 1946 - 1952).

Serial estimates of permeability factors in FSGSCattran et al., J Am Soc Nephrol, 2003, 14: 448 - 453
1. Serum permeability activity assessed in 27 pts with FSGS treated either by cyclosporine or placebo before and after 26 weeks of treatment (Cattran et al., Kidney Int., 1999, 56: 2220 – 2226)
2. Proteinuria decreased in cyclosporine treated patients from 7.2 to 3.1 g/day and did not change in pts on placebo (from 9.5 to 7.4 g/day)
3. Serum permeability activity changed neither in cyclosporine (from 0.31 to 0.46), nor in placebo (from 0.41 to 0.36) treated pts
4. Antiproteinuric effect of cyclosporine seemed to be independent on changes of Palb

Focal segmental glomerulosclerosis
1. Asymptomatic proteinuria or full blown nephrotic syndrome
2. Hypertension, microscopic hematuria and decreased renal function common
3. Slowly progressive disease – 50% 10-year renal survival
4. Sclerosis of segments of glomerular tuft
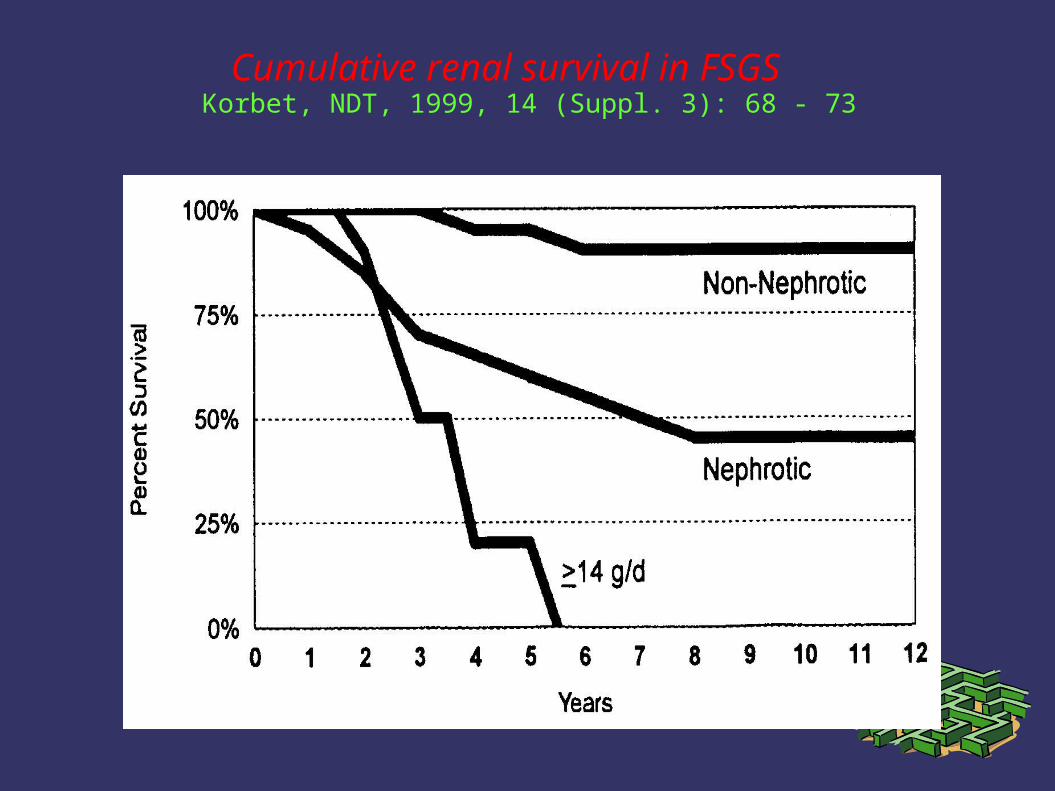
Cumulative renal survival in FSGS Korbet, NDT, 1999, 14 (Suppl. 3): 68 - 73

Cumulative renal survival in FSGS Korbet, NDT, 1999, 14 (Suppl. 3): 68 - 73

Treatment of primary FSGS – current recommendations
1. Response to corticosteroids may increase from only 10-30% up to 60% with longer treatment with higher dose (60 mg/m2 at least 3 months, patients should be considered steroid resistant after 6 months)
2. Cyclosporine may reduce proteinuria and lower the risk of progression to ESRD even in steroid resistant patients, treatment should be long (at least 6 months), relapses after cyclosporine withdrawal common
3. Cytotoxics remain only second-line therapy, the evidence for their effect in steroid resistant patients is not conclusive

Membranous nephropathy

Membranous nephropathy

Membranous nephropathy

Membranous nephropathy
1. Secondary- infections
(hepatitis B, syphilis, malaria)- drugs
(organic gold, penicillamine, NSAID)
- neoplasms(carcinomas, e.g. Colon, lung, or
stomach, and lymphomas)- systemic lupus erythematosus
2. Idiopathic

Idiopathic membranous nephropathy
1. Membranous nephropathy represents 15-25% of adult nephrotic syndrome
2. Nephrotic proteinuria is present in about 80% of patients, remaining patients have asymptomatic proteinuria
3. Microscopic hematuria is common4. Hypertension and chronic renal failure are
uncommon at presentation, but may develop during follow-up
5. Histology – subepithelial deposits along often thickened GBM

Natural course of idiopathic membranous nephropathy
1. Spontaneous remission may develop in about one third of patients
2. Nephrotic syndrome persists in another third of patients
3. Only 20-30% of patients progress to ESRD during 20-30 years of follow up

Cyclosporine in steroid-resistant MNCattran et al., Kidney Int., 2001, 59: 1484 - 1490
complete or partial remission developed after 26 weeks in 75% of pts treated by CyA vs. in 22% of pts treated by placebo
during 52 weeks relapse developed in 43% of pts treated by CyA and 40% of pts treated by placebo
at the end of follow-up in remission was 39% of pts treated by CyA and 13% of pts treated by placebo

Treatment of idiopathic membranous nephropathy – current
recommendations
1. Corticosteroids should not be used a sole therapy
2. Azathioprine is not effective in reversing or stabilizing progressive renal insufficiency
3. Cytotoxics induce prolonged remission of nephrotic syndrome and improve renal survival, their use should be reserved for patients with progressive disease
4. Cyclosporine seems to be effective in progressive renal insufficiency

Guidelines for the treatment of IMN Cattran, Kidney Int., 2001, 59: 1983 - 1994

Proliferative glomerulonephritides
1. Microscopic hematuria and/or bouts of macroscopic hematuria - mesangial proliferation
(IgA nephropathy)
2. Microscopic hematuria with proteinuria - mesangial proliferation with peripheral
expansion of mesangium (membranoproliferative GN)

Ultrastructural changes in glomerular capillaries in mesangio- and membranoproliferative GN

IgA nephropathy –mesangioproliferative glomerulonephritis

IgA nephropathy

Pathogenesis of IgA nephropathyGómez-Guerrero et al., Kidney Int, 2002, 62: 715 - 717
1. Aberrantly O-glycosylated IgA1 with exposed GalNAc may be recognized as antigens by IgG
2. Circulating immune complexes of IgA1 and IgG and/or IgA1 and soluble FcRI (CD89) were identified in pts with IgA nephropathy
3. Except from ASGP-R, CD89, Fc/R and TfR mesangial cells may express further, not yet described IgA receptors
4. Patients with IgA nephropathy have increased expression of megsin (serine protease inhibitor – serpin). Overexpression of megsin leads to progressive mesangial matrix expansion

IgA nephropathy
1. Commonest glomerulonephritis in Europe (20-40% of primary glomerulonephritides)
2. Typical clinical presentation – asymptomatic microscopic hematuria or episodes of parainfectious macroscopic hematuria
3. Natural history is not benign – at least 20% of patients develop ESRD during 20 years

IgA nephropathy –negative prognostic factors
a. clinicalhypertension
proteinuria (> 1 g/24 hrs)decreased renal function at presentation
b. histologic glomerulosclerosisinterstitial fibrosis vascular sclerosis

IgA nephropathy - treatment
1. Strict control of hypertension with ACE inhibitors
2. Fish oil in patients with slowly progressive course of renal insufficiency
3. Corticosteroids in proteinuric patients with preserved renal function
4. Cytotoxics in patients with progressive renal insufficiency

Corticosteroids in IgA nephropathy: long-term results
Pozzi et al., J Am Soc Nephrol, 2004, 15: 157 - 163
1. Secondary analysis of a multicenter, randomized, controlled trial of 86 adult IgAN treated for 6 months either with intravenous methylprednisolone followed by oral steroids of supportive therapy
2. Ten-year renal survival was significantly better in the steroid than in the control group (97% vs. 53%, p=0.0003)
3. Proteinuria decreased in patients who did not double baseline serum creatinine and increased in progressive patients

Evidence-based recommendations for IST in IgAN: handle with caution
Floege, Nephrol Dial Transplant, 2003, 18: 241 - 245
1. In low risk pts with Pu < 1.5 g/day and normal GFR, steroid therapy may reduce proteinuria, but its effect on long-term outcome is uncertain
2. In pts with Pu 1 – 3.5 g/day and preserved renal function 6 month steroid course is indicated
3. In pts with progressive renal failure as long as serum creatinine does not exceed 250 mol/l steroids plus cytotoxics are recommended

Membranoproliferative glomerulonephritis

Membranoproliferative glomerulonephritis
Type I – a. Secondary
– infection (visceral abscesses, endocardisis,
infected ventriculoatrial shunts, malaria)
- systemic diseases (type III-IV of lupus nephritis)
- paraproteinemias ( LCDD, cryoglobulinemia)
- thrombotic microangiopathies ( HUS/TTP, APS)
b. IdiopathicType II – dense deposit disease

Membranoproliferative glomerulonephritis –type I

Membranoproliferative glomerulonephritis –type II (dense deposit disease)

Idiopathic MPGN type I
1. relatively rare in developed countries2. occurs in younger adults3. presents with nephrotic syndrome and
microscopic hematuria4. slowly progressive disease – 50% 10-
year renal survival5. treatment of nephrotic adults is
controversial (corticosteroids, or antiplatelet agents?)

MPGN type II (dense deposit disease)
1. very rare disease2. nephritic factor with
hypocomplementemia3. more expressed nephritic features
and more aggressive course4. no effective therapy

Therapy of glomerular diseases
1. Drugs and procedures with relatively well defined indications
corticosteroidscytotoxics (CPH, chlorambucil)cyclosporine symptomatic treatment
(ACEI, AIIA, and other antihypertensives,NSAIDSlipid lowering drugs)

Therapy of glomerular diseases
2. Drugs and procedures with limited experience and not well defined indications
mycophenolate mofetiltacrolimusrapamycin
intravenous immunoglobulinsmonoclonal antibodies (e.g. infliximab,
rituximab)soluble cytokine receptors (e.g. etanercept)
plasma exchangeimmunoadsorption

Conclusions
1. Patients suffering from primary GN are endangered by:a. complications of nephrotic syndromeb. progression to ESRF
2. Urinary findings are important, but renal biopsy remains essential for diagnosis, treatment and assesment of outcome
3. primary GN are treatable diseases, patients should be treated according to available evidence
4. further progress in treatment depends on better understanding of their pathogenesis


















