
GENETICS OF ANTIBIOTIC RESISTANCE IN STAPHYLOCOCCUS SPECIES · genetics of antibiotic resistance in...
83
GENETICS OF ANTIBIOTIC RESISTANCE IN STAPHYLOCOCCUS SPECIES A SCHOLARLY PAPER PRESENTED TO THE DEPARTMENT OF NATURAL SCIENCES IN CANDIDACY FOR THE DEGREE OF MASTER OF SCIENCE By ERICA L. CONNER NORTHWEST MISSOURI STATE UNIVERSITY MARYVILLE, MISSOURI APRIL, 2016
Transcript of GENETICS OF ANTIBIOTIC RESISTANCE IN STAPHYLOCOCCUS SPECIES · genetics of antibiotic resistance in...

GENETICS OF ANTIBIOTIC RESISTANCE IN STAPHYLOCOCCUS SPECIES
A SCHOLARLY PAPER PRESENTED TO
THE DEPARTMENT OF NATURAL SCIENCES IN CANDIDACY FOR THE DEGREE OF
MASTER OF SCIENCE
By ERICA L. CONNER
NORTHWEST MISSOURI STATE UNIVERSITY MARYVILLE, MISSOURI
APRIL, 2016

Genetics of Antibiotic Resistance 2
Running Head: GENETICS OF ANTIBIOTIC RESISTANCE
Genetics of Antibiotic Resistance
In Staphylococcus Species
Erica L. Conner
Northwest Missouri State University
SCHOLARLY PAPER APPROVED
Scholarly Advisor Date
Dean of Graduate School Date

Genetics of Antibiotic Resistance 3
I. Introduction
A. General Problem of Antibiotic Resistance to Medicine
Antibiotics are chemical substances used to prevent and treat infections caused by
microorganisms, such as bacteria, parasites and fungi. Antibiotic medications play a role
by suppressing or inhibiting the growth of microorganisms (Sanders et al. 2011). There
are more than 150 antibiotics currently available, but only 125 are effectively used for
treatment (Zuchora-Walske 2014). Antibiotics have benefitted society (Fabbretti et al.
2011) by providing treatments for tuberculosis, gonorrhea, methicillin-resistant
Staphylococcus aureus (MRSA), bronchitis, urinary infections, and many others (Tindall
et al. 2013). Yet, antibiotic resistance has become a worldwide issue that has expanded
and persisted among all nations on earth. New strains of antibiotic-resistant microbes
migrate easily, and they often create a “worldwide threat” to society (Levy 2002).
Developing countries have been impacted greatly due to malnutrition, poor living
conditions, and lack of medical sources (Alanis 2005). Over-the-counter antibiotics can
be obtained more easily in developing countries than in developed nations where their
access is more restricted (Alanis 2005). Resistant strains evolved during times of
environment instability, such as war, and migrations introduced new strains in
communities. Some antibiotics are no longer effective or efficient to treat infections
(Baquero and Campos 2003). The biggest challenge is when the most clinically
significant pathogens become resistant to most widely effective drugs (Lee et al. 2013).
One of the settings in which antibiotic resistance is most concerning is a hospital. In
the United States (US), numerical data for antibiotic resistance have been collected from

Genetics of Antibiotic Resistance 4
hospitalized patients since 2002 (Collins 2008). Yearly, more than 35 million US patients
require hospital care due to infection or disease (Wenzel and Edmond 2001).
Additionally, exposure to microorganisms in intensive care units presents a 60% risk of
acquiring nosocomial infections. In other words, these infections that are caused by
pathogens that acquired resistance to antibiotics (Haddadin et al. 2002). Nearly 2 million
patients develop hospital-acquired infections during treatment, 55% of which involve
antibiotic-resistant bacteria (Stone 2009).
Some bacteria, such as methicillin-resistant Staphylococcus aureus (MRSA), have
become resistant to antibiotics and spread throughout hospitals, clinical settings, and
other areas. Nowadays, there are only a few effective drugs that can control MRSA
(David and Daum 2010). In the U.S., National Nosocomial Infections Surveillance (NNIS)
stated that approximately 60% of S. aureus infections derived from intensive care units
were methicillin resistant, and the number of infections rose from 29% in 2005 to 49% in
2009 (Sydnor and Perl 2011). Vancomycin resistance in Enterococcus faecium
infections increased from 9,829 infections in 2000 to roughly 22,000 infections in 2006
(Sydnor and Perl 2011). With restricted options to cure infections, healthcare providers
propose to increase medical fees and introduce patients to medications with potentially
harmful? side effects (David and Daum 2010). Without effective antimicrobial agents,
any individual with an infection is approximately 70% more likely to die (Frieden et al.
2013).
Researchers have seen a consistent and rapid increase in antibiotic resistance,
especially in recent years. Sydnor (2011) stated that hospital epidemics can be
minimized if accurate surveillance of nosocomial infections are installed, best practices

Genetics of Antibiotic Resistance 5
are implemented to prevent and treat them, and health care personnel are trained to
avoid transmission of infectious microbes from patient to patient (Sydnor et al. 2011). As
of 2011, the Netherlands was reported to have the lowest antibiotic consumption rate in
Europe, and this country believes antibiotic resistance can be prevented by reducing the
dosage of antibiotics prescribed to a patient (Vandenbroucke - Grauls 2014).
Community environments have also shown signs of bacterial infections that are
resistant to antibiotics. Vancomycin resistance in Enterococcus faecium infections have
shown an increase percentage of 124% between 2000 to 2006 (Sydnor and Perl 2011).
For instance, Mycobacterium tuberculosis is a community-acquired pathogen
responsible for tuberculosis (TB), which infects 30% of the human population and
accounts for nearly 2 million deaths annually (Jensen et al. 2005). Until 2002,
streptomycin was routinely used to treat patients with tuberculosis (Gillespie 2002). Drug
resistance developed because M. tuberculosis was continuously exposed to a single
drug. Gillespie studies (2002) showed that a combination of isoniazid, pyrazinamide,
rifampin, and ethambutol drugs were able to control and prevent tuberculosis from
expanding (Gillespie 2002). As of 2014, a combination of multiple drugs diminished the
number of spontaneous mutations and increased the treatment rate by 48% (Fonseca et
al. 2015). Hence, antibiotic-resistant pathogens have become a major concern not only
in the healthcare facility, but also in community settings (Tomasz 1994).
B. Mortality Rate
Mortality and morbidity rates are escalating due to the prevalence of antibiotic
resistance strains in intensive care units (Hanberger et al. 2014). Non-resistant strains
are nonpathogenic, while antibiotic resistant strains are pathogenic, leading to an

Genetics of Antibiotic Resistance 6
increase death rate (Hanberger et al. 2014). In the US, about 2 million people
developed bacterial infections from 2005 through 2008 that were resistant to more than
one antibiotic, resulting in an estimated 99,000 deaths (Kallen et al. 2010). In 2011, the
EU had nearly 2 million people with nosocomial infections that lead to 200,000 deaths. In
the EU, housing of vulnerable patients within the same area increases the probability of
resistant strains to pass from one person to another (Guggenbichler et al. 2011).
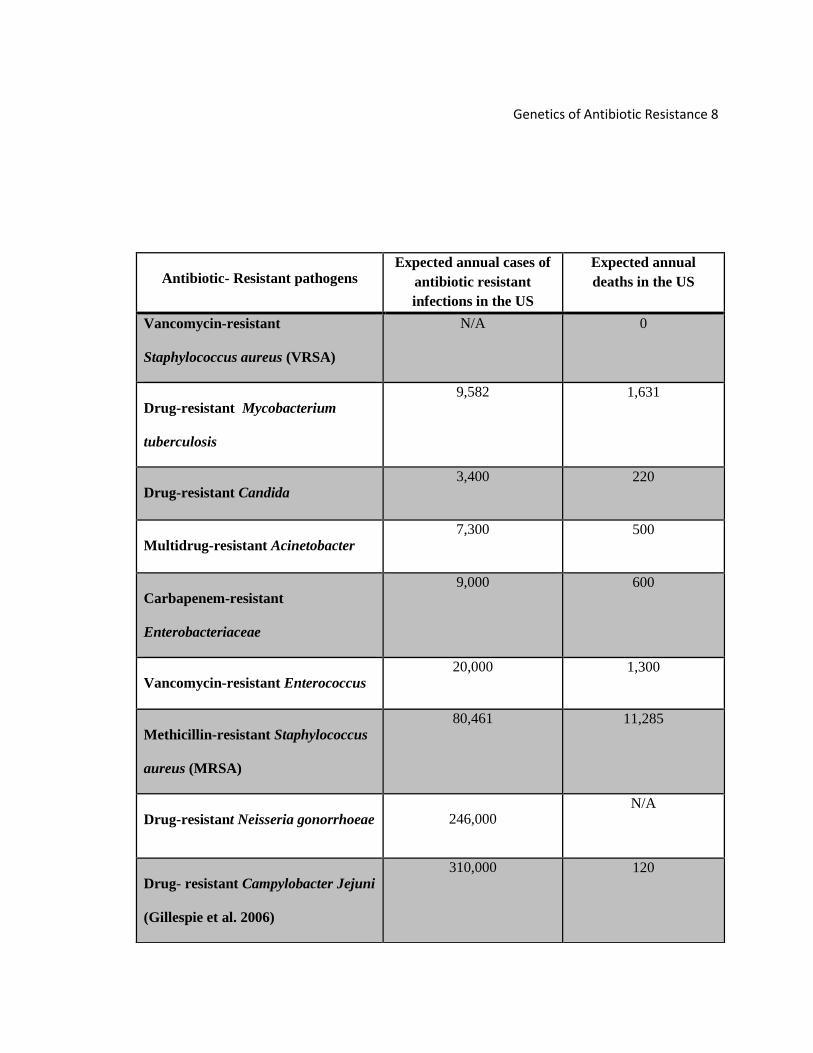
Based on Table 1, multi-drug resistance (MDR) M. tuberculosis (TB) is one of the
three organisms that causes the highest mortality in humans. Highly diverse populations
and poor sanitation leads to development of multidrug resistance, which contributes to
95% of mortality rates in low- and middle-income states (Drobniewski et al. 2015). As of
2007 in India, MDR TB was twice as common in TB patients living with HIV versus in TB
patients without HIV because patients were showing spontaneously resistant mutants in
reserve drugs, such as ofloxacin (Isaakidis et al. 2011).
Despite the fact that MDR TB is curable, its treatment depends upon extensive
chemotherapy that is exceptionally costly for low-income nations. Efforts to prevent the
spread of the infection highly depend on the socioeconomic status of the country (Olson
et al. 2012). Implementation of new programs, effective screening tests, and efficient
therapy options assist reducing the mortality rates in US patients. Maintaining the low
mortality of TB in the U.S. will demand continued prevention and control efforts,
specifically fast diagnosis, guaranteed accomplishment of treatment, and efficient and
complete reporting (Geiter 2000).
Vancomycin-resistant Enterococcus (VRE) infections are also emerging as
serious health risks. VRE is the fourth most common antibiotic-resistant pathogen with

Genetics of Antibiotic Resistance 7
the highest expected annual causes in the US and the third most common with the
highest annual deaths. Several drugs have been used effectively against VRE, but its
mortality rate (Table 1) still spiked (Cho et al. 2013). A patient who contracts VRE is
considered immunosuppressed because he/she has been previously exposed to the
organism during a major surgery or other medical procedures, which have been treated
with multiple antibiotics (Collins 2008). These infections pose a 10% risk of death in
patients with graft transplant, but almost 70% in those with endocarditis, tumors, and
liver transplants (Kapur et al. 2000). Nonetheless, some authorities are more skeptical
and tend to admit that patients might have other medical conditions that could lead to the
increase of mortality and not necessarily attribute all mortalities to VRE infections (Cho
et al. 2013). A potential reason for high mortality rates among VRE infections is because
VRE can potentially transfer genetic traits to S. aureus, another organism with high
fatality rates (Cetinkaya et al. 2000).
According to Table 1, drug-resistant Campylobacter jejuni and Neisseria
gonorrhoeae infections are the most common cases of antibiotic resistant infections,
followed by MRSA which continues to be the number one antibiotic resistant pathogens
with highest mortality rate in the US. Carbapenem-resistant Enterobacteriaceae (CRE)
infections are relatively more predominant than MDR Acinobacter infections by showing
100 more annual deaths in the US (Gupta et al. 2011). Vancomycin-resistant
Staphylococcus aureus (VRSA) infections have shown no mortality rate in the US in
comparison with drug-resistant Candida. Drug-resistant Candida infections contribute to
the leading cause of invasive candidiasis due to the delays on finding appropriate and
efficient antifungal therapeutic agents (Pfaller and Diekema 2007).
Genetics of Antibiotic Resistance 8
Antibiotic- Resistant pathogens Expected annual cases of
antibiotic resistant
infections in the US
Expected annual
deaths in the US
Vancomycin-resistant
Staphylococcus aureus (VRSA)
N/A 0
Drug-resistant Mycobacterium
tuberculosis
9,582 1,631
Drug-resistant Candida 3,400 220
Multidrug-resistant Acinetobacter 7,300 500
Carbapenem-resistant
Enterobacteriaceae
9,000 600
Vancomycin-resistant Enterococcus 20,000 1,300
Methicillin-resistant Staphylococcus
aureus (MRSA)
80,461 11,285
Drug-resistant Neisseria gonorrhoeae 246,000 N/A
Drug- resistant Campylobacter Jejuni
(Gillespie et al. 2006)
310,000 120

Genetics of Antibiotic Resistance 9
Table 1 - Comparison of annual antibiotic resistant infections with annual deaths in U.S patients due to several types of bacteria (most data condensed from text in (Frieden 2013)).
C. Health Care Costs
Antibiotic resistance is a financial burden on the healthcare system. Physicians
are obligated to treat individual patients with the most effective drugs for an infection.
Health care institutions believe that hospital expenses are a form of reimbursing
physicians for their service to society (Roberts et al. 2009). Patients, on the other hand,
demand better healthcare service and better quality of life for them and for those around
them (Roberts et al. 2009).
Antibiotics for bacterial infections are costly, and sometimes hospitalization is
required until the infection is cleared, further raising costs (Roberts et al. 2009). In the
US, Ventola’s (2015) statistics showed that antibiotic-resistant infections incur roughly
$20 billion in annual healthcare costs and estimated that patients with antibiotic-resistant
infections cost from $19,000 to $29,000. According to a study by Dellit (2007), VRE
infections required up to 17 days of hospital care with an average cost of $27,000.
Antibiotic-resistant Pseudomonas aeruginosa infections incurred hospital fees of
$54,081 per patient, compared to $22,116 for those infected with antibiotic-susceptible
strains (Slama 2008).
S. aureus infections are expensive to treat, and MRSA infections are more costly
than those that are methicillin-sensitive (Singh et al. 2006). Inpatient treatment, which
includes overall/basic hospitalization costs, antibacterial drugs, preliminary exams, and
imaging will total close to $35,000 for patients with resistant strains (Filice 2010).

Genetics of Antibiotic Resistance 10
Assuming that 15% of the US patients become infected annually, costs for only MRSA
infections would total approximately $45 million (Filice 2010).
Currently, several elements have played a vital role in the cost of controlling
infection. The cost to create a new antibiotic is recently calculated at $1 billion (Slama
2008). Not only are novel antibiotics needed, but surveillance within each hospital must
be augmented (Slama 2008). Hospital administration plays a vital role on controlling
resistant pathogens, as well as enforcing rules to limit resistant strains from spreading.
Hospitals or medical centers can only initiate surveillance programs or policy changes to
improve infection control if there is financial support. Without financial support, resistant
pathogens will continue to develop and spread, and the quality of health care worldwide
will be drastically affected (Slama 2008).

Genetics of Antibiotic Resistance 11
II. Methods Used by Bacteria to Acquire Antibiotic Resistance
A. Basis for Antibiotic Resistance
Numerous mechanisms (illustrated in Figure 1) are used by bacteria to avoid
antibiotic treatment, such as reducing drug uptake, actively pumping antibiotics outside
of the cell, enzymatic modification of antibiotics, altering antibiotics’ target sites,
overproduction of antibiotic’s target, and metabolic bypass of the antibiotic’s target
(Wilcox 2004).
Figure 1 - Different antibiotic-resistance strategies (Wilcox 2004).

Genetics of Antibiotic Resistance 12
Decreased drug intake is an antibiotic-resistance mechanism used by bacteria
(Lee et al. 2013). Aminoglycosides are one of the main antimicrobial classes that are
addressed by reduced drug uptake (Lee et al. 2013). Resistance to aminoglycosides,
which block protein synthesis, can be acquired by modifying the number and structure of
the porin channels that allow passage of the antibiotic into the cell (Bonomo and Szabo
2006). Bacteria tend to be resistant to the number of aminoglycosides molecules present
in the cytoplasm (Ramirez and Tolmasky 2010). As a result, aminoglycoside uptake will
decrease, and they cannot enter the cytoplasm, bind to ribosomes and inhibit protein
synthesis (Ramirez and Tolmasky 2010).
Actively pumping drugs outside of the cell is accomplished by efflux pumps, as
demonstrated in Figure 1 (Webber and Piddock 2003). Efflux pumps are energy-
dependent (active transport) mechanisms that move antibiotics and other elements
outside of the cell (Ramirez and Tolmasky 2010). Pumps can be specific to a single
antibiotic or function with multiple targets (Nikaido 2009). Pumps that are functional
against multiple antibiotics are called multidrug efflux pumps and can lead to multidrug
resistance (Nikaido 2009). Genetic elements encoding efflux pumps may be expressed
from chromosomes and plasmids (Weinstein 2005). As an example, quinolones are a
class of antibiotics responsible for inhibiting DNA gyrase and type IV topoisomerase -
two enzymes important for bacterial DNA replication (Collin et al. 2011). Efflux pumps
can remove quinolones from the cell before their concentration is sufficient to inhibit
DNA metabolism (Drlica et al. 2009). Often times, this mechanism is employed with both
Gram-positive (e.g. Staphylococcus sp.) and Gram-negative (e.g. Pseudomonas
aeruginosa) bacteria (Drlica et al. 2008).

Genetics of Antibiotic Resistance 13
Enzymatic modification of antibiotics utilizes enzymes that are capable of altering
or destroying the antibiotic’s structure before it reaches its target (Figure 1). Enzymes
are selective due to their different specificities for their target (Mahon et al. 2014). A
common example of enzymatic modification of an antibiotic is found with aminoglycoside
resistance. Enzymes in aminoglycoside-resistant bacteria catalyze the modification at
different hydroxyl and ammonium groups of the 2-deoxystreptamine nucleus (nucleus
linked to various sugars through glycosidic linkages) by the addition of an acetyl, a
nucleotidyl, or phosphate group by acetyltransferases, nucleotidetransferases, or
phosphotransferases, respectively (Ramirez and Tolmasky 2010). Addition of these
functional groups (illustrated on Figure 2) to an antibiotic blocks their interaction with
ribosomes, leaving the bacterium drug resistant (Ramirez and Tolmasky 2010).
Figure 2 –Aminoglycoside Resistance: Mechanism of Action (Jia et al. 2013)

Genetics of Antibiotic Resistance 14
Bacteria may also alter target sites of antibiotics. Isolates of Mycobacterium
tuberculosis have become resistant to ciprofloxacin due to mutations of gyrA and gyrB
that combine to form DNA gyrase (Aubry et al. 2006). DNA gyrase has a crucial function
in DNA replication, where it nicks DNA, introduces negative supercoils, then re-joins
DNA (Collin et al. 2011). Quinolones, such as ciprofloxacin bind to GyrA and interfere
with its ability to cut DNA strands and reconnect them (Collin et al. 2011). However,
some strains of Mycobacterium tuberculosis produce mutant proteins that have changed
their physical structure, blocking ciprofloxacin from binding and inhibiting its function
(Collin et al. 2011).
Another example of altering antibiotics’ target sites to gain resistance can be
found with enzymes responsible for peptidoglycan synthesis. Peptidoglycan synthesis
during cell growth requires the activity of penicillin binding proteins (PBPs) that break
and reform peptide interbridges between peptidoglycan polymers (Kohanski et al. 2010).
PBPs can be targeted by β-lactam antibiotics (e.g. penicillin and methicillin) as an
effective means for halting cell growth (Kohanski et al. 2010). Upon structural
modification of the PBPs through mutations, PBPs have low affinity for these antibiotics,
making them useless (Kohanski et al. 2010).
Resistance to carbapenems can occur when bacteria produce carbapenem-
hydrolyzing enzymes (carbapenemases) that fall into two categories (serine
carbapenemases and metallo-β-lactamases) based on the reactive site of the enzymes
(Bush 2010). Serine carbapenemases also known as class A carbapenemases, undergo
a substitution that changes the active site (Queenan and Bush 2007). On the other hand,

Genetics of Antibiotic Resistance 15
metallo--β-lactamases (class B carbapenemases) cleave the beta-lactam ring of the
antibiotic and disrupt its activity (Queenan and Bush 2007).
Overproduction of an enzyme that is targeted by an antibiotic is a fourth
resistance mechanism employed by antibiotic-resistant bacteria (Nikaido 2009). Enzyme
overproduction can be used in response to sulfonamides, which mimic p-aminobenzoic
acid (PABA), a substrate of dihydropterate synthetase (Behera 2010). If PABA
conversion to folic acid by dihydropterate synthetase (DHPS) is blocked by
sulfonamides, biosynthesis of purines and several amino acids is blocked (Behera
2010). A cell can create an overabundance of PABA, diluting the antibiotic’s
concentration against its target molecule (Behera 2010). Therefore, a smaller fraction of
dihydropterate synthetase molecules will bind PABA, instead of the sulfonamide
antibiotic, preserving some enzyme function (Behera 2010).
A gene’s promoter site (sequence that initiates a gene’s transcription into
mRNA) can be altered to increase production of an antibiotic’s substrate (Nikaido 2009).
For example, trimethoprim functions by obstructing a different step in folic acid
production (Vilcheze and William 2012). Trimethoprim binds to an enzyme known as,
dihydrofolate reductase, which suppresses the reduction of dihydrofolic acid to
tetrahydrofolic acid (Vilcheze and William 2012). Trimethoprim resistance results from
overproduction of the enzyme dihydrofolate reductase (DHFR). If the promoter is altered,
more DHFR gene will be transcribed and translated into above-normal levels of DHFR
(Vilcheze and William 2012). This excess of DHFR enzyme compensates for those
enzyme molecules inhibited by trimethoprim and retains net activity in the cell (Vilcheze
and William 2012).

Genetics of Antibiotic Resistance 16
B. Transfer of Antibiotic Resistance
Genetic diversity in bacterial populations is a result of mutations, vertical gene
transfer, and horizontal gene transfer (Bennett 2008). Specific mechanisms that allow
the horizontal transfer of DNA material between bacteria are transformation,
transduction and conjugation (Bennett 2008).
1. Vertical gene transfer
The evolution of pathogenic bacteria is associated with innumerous influencing
factors dictated by their population genetics. Vertical gene transfer (VGT) or vertical
evolution is a mechanism in which genetic material is transmitted from a mother cell to
daughter cells during cell division (Vogan and Higgs 2011). VGT and mutation are the
mechanisms responsible for Darwin’s principles of evolution and natural selection and
the basic principles of Mendel’s work: spontaneous mutations on the bacterial
chromosome confer resistance to a member of the bacterial population (Gogarten et al.
2002). Mutations are considered rare, but bacterial growth rates are so rapid that
resistance can become dominant in a population very quickly (Davies and Davies 2010).
2. Horizontal gene transfer
Horizontal gene transfer (HGT) is defined as the transfer of genetic material
between individual bacteria of the same or different species (Aminov 2011). HGT relies

Genetics of Antibiotic Resistance 17
on three fundamental mechanisms in terms of genetic exchange: transduction,
transformation or conjugation (Aminov 2011).
3. Transduction
Transduction is the passage of genes from one bacterium to another via bacteria-
specific viruses called bacteriophages (Huddleston 2014). Bacteriophages replicate by
infecting bacterial cells and using them as a host to produce more virus particles. Upon
multiplication, virus particles accumulate, burst the cell and are released into the
surrounding environment to begin another round of infection (Huddleston 2014).
Bacteriophages are generally very host-specific, often infecting only small numbers of
strains within a species and rarely infecting more than one species. Therefore,
transduction is an HGT method that is viable for only closely related species.
Transduction occurs within the lytic cycle, which could be delayed due to a
lysogenic cycle. During a lysogenic cycle, phage DNA is combined into the
bacterial chromosome (known as a prophage) and remains dormant for some
length of time (Blackstock 2014). Usually, due to environmental cues (UV
damage to the cell, starvation, etc.), the phage genome will undergo excision
from the bacterial chromosome and begin the lytic cycle (Russell et al. 2013).
Defective excision steps play a crucial role because assembling phage heads
can accidentally and randomly package host DNA, instead of its own DNA
(Koneman 2006). Subsequent infection of host cells by these phage particles
would transfer DNA from the previous host to the new host. This also means that

Genetics of Antibiotic Resistance 18
this phage particle is defective for replication, and the next host cell will not die as
a result of infection. Therefore, transduction can be associated with antibiotic
resistance because antibiotic resistance genes are just as likely to be
accidentally packaged and transduced as any other gene (Koneman 2006).
4. Conjugation
Plasmid-mediated conjugation takes place in Bacillus subtilis, Streptococcus
lactis, and Enterococcus faecalis but is not present as frequently in Gram-positive as
Gram-negative bacteria (Mayer 2010). Plasmids can carry genes needed for formation
of a sex pilus, and, therefore, conjugation. Overall, conjugation is the most common
mechanism of horizontal gene transfer in bacteria (Huddleston 2014). Conjugation, as
the term indicates, is a physical linkage between two bacteria by direct cell-to-cell
contact. A sex pilus (tube-like appendage) is required for bacterial conjugation in Gram-
negative bacteria (Bennett 2008). Conjugation takes place when DNA is transferred
directly from a donor to a recipient bacterium through the pilus (Parija 2009). Transferred
genetic material can contain not only the genes required to make pili, but other
segments of the donor chromosome, as well. If antibiotic-resistance genes are among
those transferred, conjugation will lead to the spread of antibiotic resistance (Griffiths et
al. 2000). Not all bacteria are capable of producing sex pili, but conjugation can exist
between bacteria of different species (Griffiths et al. 2000).
Gram-positive cells are not known to produce sex pili, but conjugation is still
possible. In Gram-positive bacteria sticky surface molecules are formed in order to bring
the two bacteria together in what is known as a mating bridge (Grohmann et al. 2003).
This bridge allows cells to aggregate, and DNA can then be transferred from the donor
to the recipient cells.

Genetics of Antibiotic Resistance 19
5. Transformation
Transformation is a natural process of HGT, during which genetic material in the
environment is transferred across the cytoplasmic membrane, leading to integration of
foreign DNA within the host chromosome (Huddleston 2014). The ability to acquire
naked, foreign DNA is referred to as natural competence (Chen and Dubnau 2004).
During the natural transformation process, foreign DNA links to a DNA translocase
protein made by the host and enters the host cell (Domingues et al. 2012). Only one
single-stranded DNA molecule is retained by the host cell, while the other is degraded by
nucleases. The translocated, single-stranded DNA may then be integrated into the
bacterial chromosome by a RecA-mediated transformation, if the foreign DNA was linear
(Domingues et al. 2012). However, plasmid DNA (small, closed-circular DNA; discussed
later) may be maintained as a plasmid in the host’s cytoplasm. Acquired plasmid or
chromosomal DNA will continue to replicate and be expressed along with the host’s
original DNA (Bennett 2008). If the foreign DNA contained genes that encode antibiotic
resistance, the “transformed” cell is now resistant and can transfer this gene to its
daughter cells (Giedraitiene et al. 2011).
6. Transposons
Transposons are also important sources of antibiotic resistance acquired via
horizontal gene transfer (Huddleston 2014). Transposons are mobile DNA elements that
integrate into the chromosomal or plasmid DNA of prokaryotes (Lodish 2000). In other
words, transposons have the ability to move a short piece of DNA from one location to
another within a cell. Inverted repeats (IR) are short, palindromic sequences of

Genetics of Antibiotic Resistance 20
nucleotides that read in reverse direction and flank the gene(s) within the transposon to
facilitate recombination into the host DNA (Bousios et al. 2010). Between the inverted
repeats is the gene for transposase, whose protein product detects the ends of the
insertion sequences (Bousios et al. 2010). Transposases do not usually move
transposons to specific sequences in the chromosome; therefore it moves (hops) the
transposon into random positions (Alberts et al. 2002). In some transposons, the original
transposon leaves a copy of itself at the progenitor site while replicating itself in other
locations in a process called, replicative transposition (Skipper et al. 2013). Gene
sequences disrupted by transposition are no longer capable of forming their intended
products and are “knocked out” (Hickman et al. 2011)
The simplest transposable elements are known as insertion sequences, which
carry only the transposase genes necessary to insert into and excise itself from a
molecule of DNA (Hickman et al. 2011). However, standard transposons typically
contain inverted repeats, a transposase gene and unrelated genes encoding metal-
antibiotic resistance markers, proteins needed for causing disease, or simply metabolic
genes (Juhas et al. 2009). Insertion sequences can also lead to formation of composite
transposons (Wagner 2006). Composite transposons are normally a pair of insertion
sequence elements that flank a sequence of DNA composed of one or more genes,
often coding for antibiotic and/or metal resistance (Wagner 2006). The terminal insertion
sequence is able to transpose by itself, which can be seen in transposon Tn5, Tn9 and
Tn10 families (Wagner 2006). Also, the Tn3 family of transposases contains genes for a
transposase, β-lactamase and resolvase (enzyme used for excising the transposon from
the host DNA) in which all are located in between the inverted terminal repeats (Mayers

Genetics of Antibiotic Resistance 21
2009). β-lactamase is a resistance gene used for the β-lactam antibiotics, including
penicillin and its derivate (Mayers 2009).
7. Bacterial plasmids
Plasmids are independently-replicating, double-stranded DNA molecules that are
separate from chromosomal DNA in prokaryotic cells (Murray et al. 2012). Plasmids are
generally up to approximately 10 kilobases in size and are often considered mini-
chromosomes (Bennett 2008). Plasmids can be described as having narrow- or broad-
host range (Gelder et al. 2008). Narrow-host range (NHR) plasmids are the most
common plasmids in nature. A limited number of NHR plasmids have been developed in
specific hosts to be second chromosomes due to their stability in a particular strain and
an inability to replicate in other strains or species. This stability allows NHR plasmid size
to successfully increase by acquiring genes from the host’s chromosome due to
transposition and recombination events (Gelder et al. 2008). Broad-host-range (BHR)
plasmids can be transferred to and replicate in relatively diverse and distant bacterial
species than can NHR plasmids (Gelder et al. 2008). Hence, NHR plasmids seemed to
play a role as genetic storage for the same group of species, while BHR act as an active
gene transporter between diverse species (Gelder et al. 2008). Occasionally, plasmids
possess characteristics that provide some selective advantage to the host bacterium,
such as virulence elements, adherence and antibiotic resistance genes (Mahon et al.
2014). The expansion of plasmids is definitely something that the public health sector
wants to avoid (Gelder et al. 2008). With better technology, researchers will be able to
analyze the epidemiology of the plasmids.

Genetics of Antibiotic Resistance 22
C. Development of Antibiotic Resistance
Antibiotics function either by altering the bacterial structures or by interfering with
fundamental physiological mechanisms of the bacteria (e.g. growth, DNA replication,
gene expression, etc.) (Kohanski et al. 2010). Bacteria may acquire or be innately
resistant to an antibiotic because they have no sites where molecules can enter the cell,
because efflux pumps expel the antibiotic, or the cell is capable of producing enzymes
that interfere with and destroy the antibiotics (Fernandez and Handock 2012). There are
numerous reasons that can lead to the survival of the bacteria, regardless which drug is
used to prevent infections from developing (Davies and Davies 2010).
The use and misuse of antibiotics is another factor that allows the expansion of
resistant organisms. To make matters worse, clinicians prescribe antibiotics that perhaps
are unnecessary, and patients increase the problem by requesting drugs for every
infection (Weckx 2012). Diseases for which antibiotics are prescribed, like sinus
infections, are expanding, especially at day-care settings (Oxford et al. 2013). With an
increase in day-care environments, infections will surge; therefore, more antibiotics will
be used, leading to an increased spread of drug resistance. Viral infections (e.g.
influenza or cold) are often treated with unnecessary drugs at patients’ demands,
increasing chances of creating resistant bacterial strains that can evolve. Even when
drugs are prescribed correctly, patients fail to use them appropriately. Therefore, the
weak organisms are killed, but the strongest will persist and propagate within the
population (Weckx 2012).
Even when antibiotics are used correctly, problems can arise. Travel increases
the probability of transferring drug resistance from one country to another at jet speed.
Hospitalized patients often times are being medicated with several drugs, which create a

Genetics of Antibiotic Resistance 23
favorable situation for multi-resistant strains to develop and thrive. Hospitalized patients
often have compromised immune systems, which increases their vulnerability to many
infections, in particular those who are antibiotic-resistant (Oxford et al. 2013).
To conclude, a variety of factors such as population growth, usage and misuse of
antibiotics have all contributed to development of antibiotic resistance. Hopefully, the
evolution of resistance can be stopped eventually, if society begins to change its
behavior and attitude towards infectious diseases. Precautions should be taken serious
for not only the patients’ benefit, but for the good of all humankind (Davies and Davies
2010).

Genetics of Antibiotic Resistance 24
III. The Genus Staphylococcus
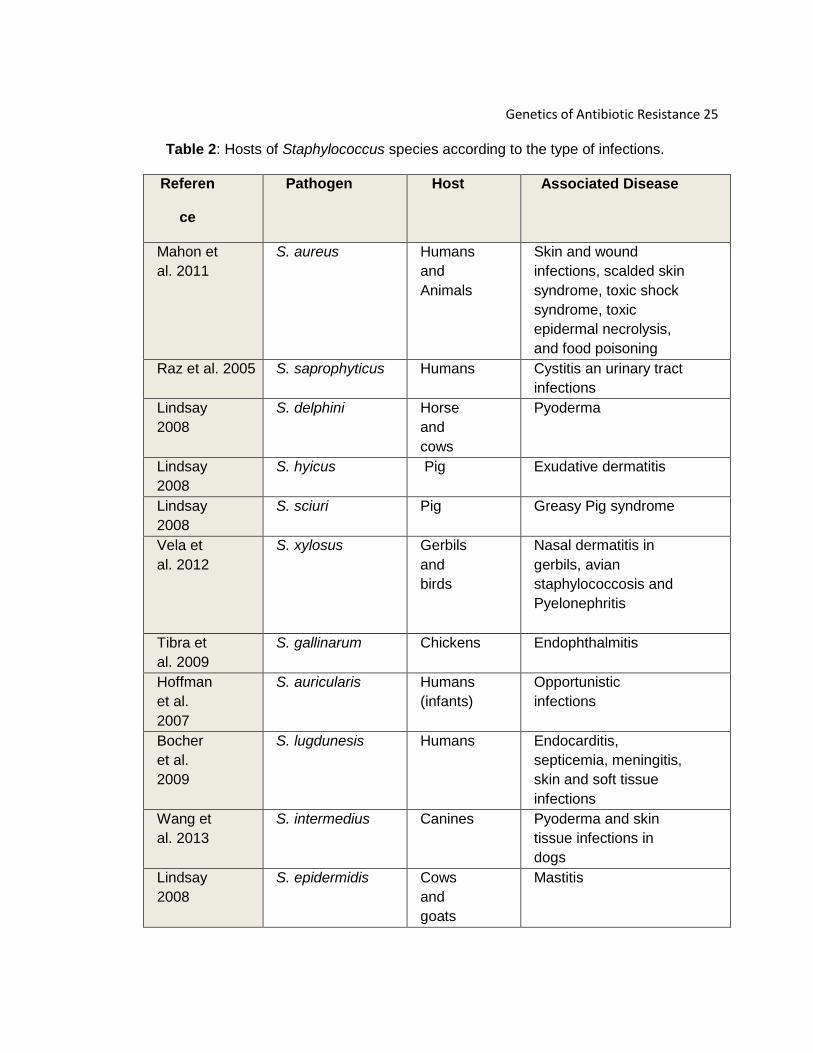
A. Natural Habitat and Host Range
Worldwide, Staphylococcus species are notorious human pathogens and
are the focus of comprehensive studies among researchers. More than 40
Staphylococcus species cause diseases known to be clinically and economically
significant (Lindsay 2008). Public health has become threatened by antibiotic-
resistance strains that pass among humans, and it is thought that species or their
habitats might correlate with type of infection (Lindsay 2008). There is a wide host
range among species that belong to the genus Staphylococcus. For instance, S.
aureus can be isolated from horses, rabbits, humans, and ruminants and cause
skin infections, septic arthritis, and others (Lindsay 2008).Overall, the pathogens
among the staphylococcus species can be colonize among man and other
mammals. Depending upon the host, infections can range from a mild skin
infections to a more severe type of syndrome. Table 2 shows the host range and
infections caused by several species of staphylococci.
Genetics of Antibiotic Resistance 25
Table 2: Hosts of Staphylococcus species according to the type of infections.
Referen
ce
Pathogen Host Associated Disease
Mahon et
al. 2011
S. aureus Humans
and
Animals
Skin and wound
infections, scalded skin
syndrome, toxic shock
syndrome, toxic
epidermal necrolysis,
and food poisoning
Raz et al. 2005 S. saprophyticus Humans Cystitis an urinary tract
infections
Lindsay
2008
S. delphini Horse
and
cows
Pyoderma
Lindsay
2008
S. hyicus Pig Exudative dermatitis
Lindsay
2008
S. sciuri Pig Greasy Pig syndrome
Vela et
al. 2012
S. xylosus Gerbils
and
birds
Nasal dermatitis in
gerbils, avian
staphylococcosis and
Pyelonephritis
Tibra et
al. 2009
S. gallinarum
Chickens Endophthalmitis
Hoffman
et al.
2007
S. auricularis Humans
(infants)
Opportunistic
infections
Bocher
et al.
2009
S. lugdunesis Humans Endocarditis,
septicemia, meningitis,
skin and soft tissue
infections
Wang et
al. 2013
S. intermedius Canines Pyoderma and skin
tissue infections in
dogs
Lindsay
2008
S. epidermidis Cows
and
goats
Mastitis

Genetics of Antibiotic Resistance 26
B. How are Staphylococcus Infections Acquired?
An individual is exposed to diverse strains of microorganisms daily
without causing any infections. Human microbiota are usually nonpathogenic, as
long as they remain within their normal habitat (Reid et al. 2011). When normal
microbiota are displaced, infections can occur. Human skin is one of the
favorable sites for pathogens to establish a direct interaction and potentially
cause infections (Ki and Rotstein, 2008). Infections associated with staphylococci
can range from minor skin problems to life-threatening systemic infections (Ki and
Rotstein, 2008). A variety of conditions may be associated with staphylococcal
infections. Therefore treatment depends on the site and severity of the illness (Ki
and Rotstein, 2008).
1. Bacteremia
Bacteremia, the presence of bacteria in the blood, is one of the main routes
taken to destabilize the immune system of an individual (Delost 2014). Bacteremia
cases begin showing signs of fever, chills and hyperventilation but at late stages
can travel through the internal organs (heart, brain and lungs), catheters and
implanted devices, such as pacemakers (Delost 2014). Bacteria enter the
bloodstream through small cuts and wounds, following by the adhesion to inner
membranes of the tissues, which subsequently triggers a cascade of infections
(Cabell et al. 2003).
2. Food Poisoning Food poisoning is derived from ingesting contaminated products.
Symptoms such as nausea, vomiting, diarrhea, dehydration, and low blood

Genetics of Antibiotic Resistance 27
pressure can present (Argudin et al. 2010). Often times, negligent food workers
contract staphylococcal infections and continue to process food without further
precautions. Once S. aureus is present inside of the human body, it can trigger
infections (Argudin et al. 2010). S. aureus is capable of forming a broad variety of
toxins, such as staphylococcal enterotoxins (SEs) responsible for creating potent
biological effects, such as damage of gastrointestinal tract (Dinges et al. 2000).
Usually SEs are active in low amounts and are capable of resisting heat and
cannot be eliminated by cooking. Acid-resistant SEs can withstand the pH of the
digestive tract (Loir et al. 2003).
3. Skin
Staphylococcal infections are normally skin infections, such as boils,
impetigo, and staphylococcal scalded skin syndrome (SSSS) (Proft 2013). Usually,
bacteria invade superficial layers of the skin via open wounds or cuts. Boils, for
instance, are skin infections created by pockets of pus that developed in a hair
follicle or oil gland (Murray et al. 2008). The skin over the infected site normally
shows some redness and swelling (Murray et al. 2008). In this case, the boil
breaks open and pus drains and causes other infections. Boils generally develop
under the arms or around the groin (Ibler and Kromann 2014). SSSS forms blisters
due to the production of toxins that disrupts the epidermis, leaving the affected
area exposed to harmful pathogens. Clinically, SSSS is seen to spread throughout
large portions of the body (Bukowski et al. 2010). A defined area of infection
created by SSSS is commonly diagnosed as impetigo (Bukowski et al. 2010).

Genetics of Antibiotic Resistance 28
C. Risk Factors for Infections
1. Recent Hospitalization
Regardless of intense efforts implemented by infection control programs,
bacteria remain persistent in hospital settings where they can infect susceptible
people, primarily those with weakened immune systems (Sydnor and Perl 2011).
Infections can occur before, during, or after surgery if certain precautions are not
followed (Sydnor and Perl 2011). In a hospital setting, staphylococci infect critically
ill patients more than any other organism because of the way they spread.
Generally staphylococci, in particular S. aureus, diffuse by skin-to-skin contact
between doctors, nurses, or visitors (Barnes 2010). Normally, someone who
contracts a staphylococcal infection, such as MRSA, can stay hospitalized for up to
10 days (Barnes 2010). Therefore, precautions such as maintaining proper
personal protective equipment to protect patients and physicians must be followed
(Barnes 2010). However, what makes staphylococci significant is that several
species have become resistant to antibiotics. Methicillin-resistant S. aureus
(MRSA) is a prime example of antibiotic resistance to methicillin and penicillin
(Nikaido 2009). With limited treatments, it becomes extremely difficult to control the
spread of infection. The best way to minimize the problem is to maintain good
hygiene between health care workers and extending these protocols to visitors and
patients (Barnes 2010).
2. Residing in a Long-Term-Care Facility
MRSA is 20% more common in long-term-care facilities (LTCFs) than it is
in hospitals (Mazur and Gudiol 2009). Urinary tract infections are the number one

Genetics of Antibiotic Resistance 29
risk found among elderly that reside in the LTCFs, followed by respiratory and
gastrointestinal infections (Bonono 2000). As a whole, these diseases comprise
94% of the infections in a LTCF due to the lack of training among staff and proper
care of residents (Engelhart et al. 2005). Some individuals admitted to a care
facility are likely to carry MRSA and have the ability to spread it to other patients
(Bonono 2000). As the population ages and technology advances to provide better
care for patients, both the number and proportion of people residing in LTCFs will
increase (Bonono 2000). Hospital-acquired infections differ from those contracted
in community settings because it is an older population of patients with a broad
range of disabilities (Mathei et al. 2007). Some characteristics of a nursing-home
setting promote the growth of infectious diseases. Residents tend to be clustered
in confined areas and daily activities usually involve groups (Mathei et al 2007).
Also, healthcare workers might have limited training in how to control infectious
diseases. Understaffing is also a common problem in LTCFs, and caregivers are
often too occupied to worry about infectious control (Mathei et al. 2007).
3. Indwelling devices
As opportunistic pathogens, staphylococci can migrate from medical
equipment to an individual’s internal organs (Otto 2008). Dialysis, catheters,
pacemakers and prosthetic devices are examples of invasive, indwelling devises
that are used routinely in hospital facility (Guggenbichler et al. 2011). As an
example, S. epidermidis can directly and indirectly cause infections, despite its role
as a constituent of human skin’s normal microbiota. Patients with weakened and
debilitated immune systems are more vulnerable to S. epidermidis infections when
introduced to the organism via the implantation of medical devices (Otto 2008).

Genetics of Antibiotic Resistance 30
According to Baddour, in 2003 roughly 53% and 20% of the patients became
infected with S. aureus and coagulase-negative staphylococci (CoNS),
respectively, as a result of prosthetic vascular graft. Indwelling devices side-step
the normal defense mechanism of the epidermal surface and provide a habitat in
which pathogens can proliferate protected from the patient’s immune defenses
(Mehta et al. 2014). Therefore, proper maintenance and cleaning of these devices
could reduce the risk of staphylococcus infections (Mehta et al. 2014).
D. Physiology of How Staphylococci Cause Disease
1. Human Pathogens
Staphylococci are known to be opportunistic pathogens, meaning that they
take advantage of a wound or weakened immune system within a host. Infections
are predominantly found in hair follicles, sutures, and even along the respiratory
tract (Mahon et al. 2011). Upon entering skin or mucous membranes,
staphylococci are able to produce and secrete several components that allow them
to survive in the host and cause damage to host tissues (Coates et al. 2014).
Pathogenicity of staphylococci can be attributed to various virulence factors,
including surface proteins (Protein A), exfoliatin toxins, and biofilm formation
(Crossley et al. 2009).
Staphylococcus aureus has on its cell wall a surface protein that is referred
to as Protein A, which is capable of attaching to a fragment of the antibody
Immunoglobulin G (Mahon et al. 2011). IgG is a protein synthesized by B cells that
protects the internal tissues and binds to foreign substances upon entering the

Genetics of Antibiotic Resistance 31
body (Janeway et al. 2001). Once bound by IgG, foreign bodies are typically
engulfed and destroyed by phagocytic white blood cells. However, Protein A of S.
aureus binds in the wrong orientation to the IgG surface (Tattlybaeva et al. 2013).
As a result, white blood cells show decreased ability to ingest and kill them, and
potential for diseases increases (Tattlybaeva et al. 2013).
Exfoliatin Toxins (ETs), also referred to as epidermolytic toxins, are known
to cause SSSS (affecting large parts of the skin) and can have implications in
bullous impetigo (affecting limited areas of the skin) (Bukowski et al. 2010). Serine
proteases are enzymes present in ETs responsible for cleaving desmosomal
cadherins on the extracellular layer of the skin (Bukowski et al. 2010).
Desmosomes are cell junctions composed of cadherins linked to catenins that
interact to form strong mechanical attachments between basement membrane and
epithelial cells (Garrod and Chidgey 2007). Disruption of desmosomal cadherins
causes cell attachments to weaken and, as a result, clinical manifestation of SSSS
ensues (Garrod and Chidgey 2007). During early stages of SSSS, fever, fatigue
and malnutrition are often followed by erythematous rash (large fluid blisters).
Bullous impetigo refers to large liquid filled blisters, specifically present at the site
of infection (Bukowski et al. 2010). Diagnosis involves PCR or randomly amplified
polymorphic DNA analysis (Bukowski et al. 2010). Strains producing ETs are
strictly treated intravenously with antibiotics. ET-producing strains contain the eta
gene, which encodes for exfoliative toxin A (Magni 2010), is only positive in 10% of
MRSA cases (Bukowski et al. 2010). Hence, a vast range of antibiotics can still be
effective, according to the patient’s needs (Bukowski et al. 2010).

Genetics of Antibiotic Resistance 32
Biofilms formed by staphylococci are responsible for the microorganisms’
adherence to each other in an aqueous matrix (Schommer et al. 2011). Adherence
takes place due to a secreted matrix, referred to as extracellular polymeric
substance (EPS). EPS can be made of extracellular components such as DNA,
proteins, and polysaccharides, although polysaccharides and proteins are most
common and abundant (Flemming et al. 2007). Biofilms on living or nonliving
surfaces show increased resistance to antibiotics and disinfectants. Infections in
cystic fibrosis patients and indwelling medical devices become problematic when
biofilms are involved (Joo and Otto 2012).
Development of bacterial pathogenicity depends greatly upon the formation
of the biofilm and the interaction between bacteria and the host. Biofilms are
capable of hiding bacteria from antimicrobial agents and affect the host immune
system during and after the infection (Joo and Otto 2012). Diagnoses of biofilms
can be accomplished by performing microscopy and culture studies (Hassan et al.
2011). Current treatment involving a combination of a StaphVAX (polysaccharide
conjugate vaccine) and vancomycin usually shows effectively reduced signs of
infection. However, further treatments must be studied to provide a better and
efficient approach (Brady et al. 2011).
2. Non-Human Pathogens
As referred to in Table 2, several species of Staphylococcus are animal
pathogens or promote infections that impact humans and animals. Pathogenicity of
these species is also due to a variety of opportunistic infections including skin
lesions, pyoderma, and mastitis.

Genetics of Antibiotic Resistance 33
S. hyicus is found predominantly associated with pigs, particularly along
nasal and vaginal mucosa (Dworkin et al. 2006). S. hyicus causes exudative
epidermatitis, more commonly referred to as Greasy Pig Disease (Casanova et al.
2011). Greasy Pig Disease, as the name suggests, causes greasy, peeling skin
due to the production of an ET that forms blisters that disrupts spinosum
epithelium cells. The spinosum epithelium becomes fragile and ruptures, allowing
entry of pathogens (Fudaba et al. 2005). To control the spread of infections, this
disease can be treated using a combination of procaine penicillin G and novobiocin
for a period of 5 days (Park et al. 2013).
S. intermedius is naturally present among skin microbiota of all canines,
and no pathogenicity is known for humans (Tanner et al. 2000). S. intermedius
directly affects canine skin flora but indirectly impacts their central nervous system
(Koneman 2006). Infections that occur at the central nervous system level are
poorly understood. The brain barrier is an extremely complex membrane capable
of preventing the entry of invasive pathogens. Toxins and other organisms may
enter through the skull and the selective membranes, but that rarely occurs
because of the brain membrane isolation (Durdik et al. 2009). Development of
pyoderma occurs when S. intermedius invades the superficial layer of the skin and
hair follicles without formation of a scab (Futagawa-Saito et al. 2006). Without a
scab or coating, the epidermis becomes too fragile, superficial scale is produced,
and debris will accumulate in and around hair follicles, leading to hair loss
(Zachary and McGavin 2013). Infections normally respond well to cefotaxime
therapy (Durdik et al. 2009).

Genetics of Antibiotic Resistance 34
S. lentus is primarily found in animals that are used as a source of aliment,
such as cattle, poulty, and goats (Schwendener and Perreten 2012). Rarely, S.
lentus can transmit infections to humans, but most commonly this organism can
inflame the breast tissues of sheep and goats, leading to pain, redness and
soreness (Schwendener and Perreten 2012). Once the breast is infected, milk
becomes very cloudy due to the presence of clots. Normal milk contains
predominately epithelial and white blood cells (Rupp et al. 2008). If mammary
gland infection occurs, the number of neutrophils will increase in the bloodstream
(Rupp et al. 2008). Hence, by performing a somatic cell count it is possible to
determine that cows and/or goats with high cell counts will have greater risk to
contract clinical mastitis in comparison to those with lower somatic cell counts
(Rupp et al. 2008). Treatment should be focused on culture results obtained from
milk samples (Pieterse and Todorov 2010). Dry-off (non-lactating) period is an
efficient method to cure mastitis (Pieterse and Todorov 2010). Antibacterial agents
may be utilized during the non-lactating period in the infected animal. Once the
dry-off period is completed, prophylactic antibiotics can be administered to prevent
subsequent infections (Pieterse and Todorov 2010).

Genetics of Antibiotic Resistance 35
E. Diagnosis
1. Selective Testing for Identification of Staphylococcus species Staphylococci testing is accomplish by doing several and selective
laboratory examinations. The schema for identification of Staphylococcus species
relies primarily on microscopic confirmation of a Gram-positive coccus, followed by
a catalase-positive test (Mahon et al. 2011). Catalase-positive cultures then
undergo coagulase testing. Coagulase-positive results are seen mainly in S.
aureus, whereas coagulase-negative cultures (CoNS) are seen mostly in other
staphylococcus species, such in S. epidermidis (Foster 1996). CoNS can then be
examined for oxidase/bacitracin susceptibility. Cultures resistant to
oxidase/bacitracin will proceed with novobiocin susceptibility test (Mahon et al.
2011). Most CoNS are novobiocin-sensitive, whereas S. saprophyticus is known to
be novobiocin-resistant (Mahon et al. 2011).
2. Salt Tolerance and Mannitol Fermentation
Many media are utilized for identification of staphylococci, but certain
media are more useful than others (Tille 2013), such as Mannitol Salt Agar (MSA),
Blood Agar (BA), and Muller-Hinton Agar (MHA). MSA is a selective and
differential media with 7.5% of NaCl concentration that favors the growth of Gram-
positive bacteria. Any isolates forming colonies on MSA are considered salt-
tolerant. MSA also differentiates mannitol fermentation to acids by changing color
from red to yellow. Non-fermenters of mannitol leave the medium red (Mahon et al.
2011). Staphylococcus aureus ferments mannitol and the medium turns into

Genetics of Antibiotic Resistance 36
yellow color while other non-pathogenic staphylococci will not ferment mannitol
and the media will remain red in color (Mahon et al. 2011).
3. Catalase Catalase is an enzyme that catalyzes the conversion of hydrogen peroxide
into water and oxygen. Detection of catalase is a key feature for differentiation of
staphylococci from streptococci. Bacteria are characterized as catalase positive
when bubbles are formed when cells contact hydrogen peroxide. Staphylococcus
species are catalase-positive, whereas species of Streptococcus are catalase-
negative (Mahon et al. 2011).
4. Coagulase Reaction
Staphylococcus and Yersinia are two of the few genera that can produce
coagulase (Lackie 2010). Coagulase is an enzyme that converts fibrinogen to
fibrin, which forms clots that encapsulate cells. Coagulase occurs when clumping
factor binds to the cell walls of bacteria that react with fibrinogen. Then fibrinogen
dissolves, allowing cells to clump together (Kateete et al. 2010). Coagulase
differentiates staphylococcal isolates, and it can be classified as CoNS or
coagulase-positive staphylococcus (CPS) (Foster 1996). CoNS isolates are the
most frequent in medical microbiology laboratory because they survive in dead or
decomposing matter, but CPS isolates are considered to be the most pathogenic
(Foster 1996).
5. Clumping Factor
Clumping factors are heat-stable proteins that reside in the cell wall of S.
aureus (Tille 2013). These proteins cause clumping of fibrinogen or fibrin.

Genetics of Antibiotic Resistance 37
Disruption of the bacterial cell wall allows the release of clumping factor, which can
then act upon the fibrinogen in the plasma, allowing blood cells to aggregate (Tille
2013). Despite the fact that clumping factor and coagulase might have similarities,
they prove to be distinct from each other. Coagulase is an enzyme created by
many bacteria that distinguishes S. aureus from CoNS species (Becker et al.
2014). The confusion occurs because clumping factor is a portion of coagulase
that is bound to the bacterial cell wall and links directly to fibrinogen. Clumping
factor and coagulase act independently from each other (Josefsson et al. 2001).
6. Hemolysins Hemolysins are one of many features utilized to differentiate staphylococcal
species from other organisms. Hemolysins are microbial proteins that can lyse red
blood cells (RBCs) by disrupting their cell membrane. Many hemolysins are pore-
forming toxins which induce lysis of RBCs by creating pores in their cytoplasmic
membrane (Thompson et al. 2011). This can be visually seen by performing a
blood agar test (Nayak et al. 2013).
Blood Agar (BA) is a nutritive medium composed of tryptic soy agar
amended with 5-10% of sheep, rabbit, or horse blood and is used to grow a variety
of bacteria. The identification of each species would depend on the hemolytic
activity (Mahon et al. 2011). If RBCs are completely destroyed by a hemolysin and
a clear zone forms around the colonies. This is referred to as β-hemolysis. In the
agar, partial destruction shows a greenish color underneath the colony because
bacteria contain hydrogen peroxide that converts hemoglobin (red) into
methemoglobin (green). Non-hemolytic or γ-hemolysis is the result of no

Genetics of Antibiotic Resistance 38
destruction of RBCs and therefore, the agar does not undergo any changes
(Engelkirk et al. 2011).
S. haemolyticus, S. lugdunensis and S. aureus can produce hemolytic
zones around the colonies (Mahon et al. 2011). Others such as, S. epidermidis are
referred to be non-hemolytic (Mahon et al. 2011). However, S. epidermidis shows
partial destruction of hemoglobin in the RBCs, referred to as α-hemolysis
(Engelkirk et al. 2011).
7. Production of Deoxyribonucleases Testing for enzymes which degrade DNA, deoxyribonucleases (DNAses), is
essential to the identification of potentially pathogenic bacteria (Kateete et al. 2010).
Testing involves streaking the organisms on an agar medium that also contains DNA
and a dye which changes color in the presence of the degraded DNA (Tille 2013).
Several staphylococcal species are able to produce this enzyme, such as S. aureus, S.
epidermidis, S. intermedius (Pammi et al. 2013), and S. hyicus (Bhatia and Zahoor
2007).
8. Thermostable Nuclease (TNase)
TNase is an enzyme that is produced only by a few species of
Staphylococcus (Enoch et al. 2008), including S. aureus, S. hyicus and S.
intermedius. TNase testing is performed by placing blood broth from the blood
culture in heated environment (60 °C) and then transfering the suspension to an
agar medium for up to 4 hours at room temperature. If the DNA has been
degraded a clearing zone will form, demonstrating the TNase activity (Enoch et al.
2008).

Genetics of Antibiotic Resistance 39
9. Sensitivity to Novobiocin and Methicillin
Mueller-Hinton Agar (MH) with 2% NaCl makes the agar selective to
staphylococcus species (Mahon et al. 2011). MH contains acid digest of Casein,
beef extract, starch and agar, specifically to select pathogenic Neisseria species
(Mahon et al. 2011). The translucent appearance of the agar helps determine the
susceptibility to antimicrobial agents. The Kirby-Bauer method continues to be a
very common method to measure bacterial resistance. If bacteria growth is
inhibited in the presence of a certain antibiotic, a zone of inhibition (no visible
growth) is formed (Mahon et al. 2011).
Novobiocin is an antimicrobial agent discovered in Streptomyces niveus
that disrupts the normal ATP formation and its activity (Leboffe and Pierce 2012).
Most CoNS can be distinguished by using the novobiocin test. Novobiocin
sensitivity is tested by placing a 5-μg disk containing the antibiotic onto Muelher-
Hinton agar (MH) for 24 hours at room temperature (Leboffe and Pierce 2012). A
novobiocin-susceptible isolate forms a zone of inhibition greater than 16 mm in
diameter around the disk. If the organism is resistant to novobiocin, no zone would
be formed. S. saprophyticus is often used as a positive control because it is
resistant to novobiocin (Leboffe and Pierce 2012).
The Kirby-Bauer test differentiates from novobiocin testing by using a
choice of antibiotics that should be directly applied into the MH and then classified
as being Resistant (R), Intermediate (I) or Susceptible (S) according to the
organism in the study.

Genetics of Antibiotic Resistance 40
S. aureus is said to be resistant to methicillin upon determining that no
visible growth occurred in the agar (Mahon et al. 2011). Based on Clinical
Laboratory and Standards Institute guidelines, S. aureus is resistance to methicillin
when the minimal inhibitory concentration (MIC) is > 4 mg/L and susceptible when
the MIC is ≤ 4 mg/L (Brown et al. 2005).
F. Treatment
Staphylococci are considered to be the most pathogenic organisms in
healthcare facilities, as well as in society (Mahon et al. 2011). Without prevention,
they can impact the health of an individual at any age. The choice of an
antimicrobial agent should follow certain criteria, such as drug sensitivity, site of
infection and disease condition. The two main types of treatment used are surgery
or antibiotic chemotherapy (Leekha et al. 2011). In the majority of cases, patients
that require surgical treatment also need antibiotics. However, each individual
requires personalized treatment based upon their medical condition (Leekha et al.
2011).
Some CoNS, such as S. epidermidis, are known to cause UTIs and serious
inflammation but have become resistant to drugs such as methicillin, gentamicin,
ampicillin, and erythromycin (Villari et al. 2000). Currently, S. epidermidis is treated
by using vancomycin or rifampin (Villari et al. 2000). Nearly 20% of UTIs are
derived from S. saprophyticus, which are novobiocin resistance, and are treatable
with multiple antibiotics (Raz et al. 2005). S. haemolyticus is considered to be the
most antibiotic-resistant organism among the CoNS family (Barros et al. 2012).
Strains have become resistant to multiple drugs such as penicillins, ampicillin,

Genetics of Antibiotic Resistance 41
gentamicin, and ciprofloxacin (Barros et al. 2012). Vancomycin was reported to
treat patients that were infected with S. haemolyticus (Biavasco et al. 2000).
CoNS tend to create problems when it comes to animal infections, as well
(Mahon et al. 2011). Pigs typically contract S. hyicus, which causes exudative
epidermidis (Casanova et al. 2011). This disease is highly contagious to other pigs
and potentially to humans through skin exfoliation (Casanova et al. 2011). No
satisfactory treatment has yet been determined (Park et al. 2013). However, the
use of topical antibiotics (procaine penicillin G and novobiocin) appeared to be the
most efficient way to prevent infection from spreading (Park et al. 2013).

Genetics of Antibiotic Resistance 42
IV. Pathogenesis of Staphylococcus aureus
A. Mode of Pathogenicity
Staphylococcus aureus is a well-known bacterium for its commensal role in animal
health. It is commensal because it can harmlessly colonize a host’s nasal cavities,
armpits, vagina, or pharynx (Jenkins et al. 2015). Commensal bacteria protect the
superficial layer of our body from colonization of potentially pathogenic bacteria. For
instance, S. aureus produces bacteriocins (toxins created by bacteria that target other
bacteria) that impede pathogenic Staphylococcus epidermidis from increasing in number
(Chiller et al. 2001).
A versatile, pathogenic bacterium is formed when an organism successfully
overcomes the normal host defense protection and causes disease (Chiller et al. 2001).
S. aureus causes superficial lesions, such as boils or furuncles, systemic infections
(endocarditis and osteomyelitis), food poisoning, and toxic shock syndrome (Jenkins et
al. 2015). Infections may result from a combination of actions between multiple genetic
factors or single virulence factors (Jenkins et al. 2015). Infections commonly result from
wounds that allow staphylococci to access neighboring tissues and the bloodstream.
Infections can remain localized or become systemic, depending upon the interaction
between virulence factors and host-defense mechanisms (Anderson et al. 2012).
1. Adherence to Host Tissue
S. aureus is found primarily in the nares, and nearly 20% of the human
population is considered to be carriers for this pathogen, which increases risk of
contracting an infection (Naber 2009). Cells are capable of binding to host cells via
extracellular-matrix elements with the support of microbial-surface-components

Genetics of Antibiotic Resistance 43
recognizing adhesive-matrix molecules (MSCRAMM), which are a collection of adhesive
molecules (Clarke and Foster 2006). MSCRAMM adhere to fibrinogen (protein that
assists with the formation of blood clots) via different binding sites (Cognasse et al.
2015). Consequently, MSCRAMM function during early stages of infections. Due to the
broad range of MSCRAMM types, a variety of S. aureus strains may initiate an array of
diseases (Bien et al. 2011). For instance, coagulase is a type of adhesive molecules in
the MSCRAMM which links to prothrombin to produce staphylothrombin (Cognasse et al.
2015). Staphylothrombin converts fibrinogen into fibrin, leading to platelet aggregation
and allowing adherence of S. aureus to platelets (Vanassche et al. 2012). Fibrin clots
protect cells from phagocytosis, and, as a result, they will persist and cause staph
infections (Cheng et al. 2010). All clinical isolates of S. aureus are known to be
coagulase-positive, making it a classical phenotypic marker for identifying this bacterium
(Cheng et al. 2010).
2. Invasion of Host Tissues
The invasion of S. aureus into host tissues activates production of a wide variety
of extracellular proteins, each with a distinct role (Bien et al. 2011). Alpha-toxin, also
referred to as alpha hemolysin, is one of several pore forming toxins and a critical
virulence factor for S. aureus against platelets, erythrocytes, and monocytes (Berube
and Wardenburg 2013). S. aureus produces alpha-hemolysin as a single molecule to
facilitate the linkage with eukaryotic cell membranes (Frank et al. 2012). On target
membranes, its subunits oligomerize to create heptameric rings with a central pore
(Berube and Wardenburg 2013). The central pore formed on the target membrane can
now permit leakage of simple ions (e.g. Ca2+) that would result in apoptosis (Berube and
Wardenburg 2013).

Genetics of Antibiotic Resistance 44
Beta-toxin is composed of sphingomyelinase, a fatty-acid enzyme responsible for
catalyzing sphingomyelin into ceramide and phosphorylcholine (Kurek et al. 2013).
Sphingomyelinase is found in animal cell membranes capable of targeting membranes
rich in this lipid (Kurek et al. 2013). Beta-toxin lyses erythrocytes by crossing the lipid
membrane and promoting the release of their contents (Fournier and Philpott 2005).
Normally, beta-toxin is expressed by S. aureus strains in non-human infections to a
greater extent than in human isolates.
Delta-toxin, despite its small size, is produced by 97% of strains of S. aureus
(Dinges et al. 2000). It is also speculated that nearly 60% of coagulase-negative
staphylococci (CoNS) form delta-toxin, especially S. epidermidis (Noble 2004). Delta-
toxin is said to be lytic to a variety of cells by the disintegration of their membranes.
However, its pathogenicity remains unknown (Turnidge et al. 2002).
Gamma-toxin and Panton-Valentine-Leukocidin (PVL) are additional toxins
produced by S. aureus. Each individual toxin creates secreted proteins, called S and F
subunits.
S subunit stands for slow-eluting proteins during purification on an ion exchange
column responsible for identifying the cell specificity of the cytolysin (Sabour and
Griffiths 2010). S class is created as a single molecule that ended up linking tightly to
other S molecules, forming a multi-complex structure. This complex structure can then
attach and produce tiny pores on the membranes of the neutrophils (Sabour and Griffiths
2010). F subunit refers to fast-eluting proteins during purification on an ion-exchange
column. F subunit attaches primarily to red blood cells and then to S subunits to
perforate their membranes (Fluit and Schmitz 2003). The difference between the

Genetics of Antibiotic Resistance 45
Gamma toxin and PVL toxin is that gamma toxin binds strongly to red blood cells, while
PVL binds to white blood cells, specifically neutrophils (Nilsson et al. 1999). Overall, S
and F subunits act together in the destruction of membranes on both toxins (Fluit and
Schmitz 2003). Gamma-toxin is present in 99% of S. aureus strains whereas only 2%-
3% of strains produce PVL toxins (Lydyard et al. 2009).
3. Avoidance of Host Defenses
S. aureus can interfere with innate and adaptive immune responses in numerous
ways (Liu 2009). Capsular polysaccharide (CP) is composed of a variety of
polysaccharides present on the surface of S. aureus, and the majority of strains express
CPs in vivo or in vitro (Turnidge et al. 2002). In the presence of CPs, phagocytosis is
inhibited by preventing cell-surface structure identification by receptors on the surface of
a phagocyte. Therefore, CPs prevent complement activation and prevent S. aureus from
being recognized by the host’s immune system (Mahon et al. 2014). As a result, the
host defense mechanism no longer fights against the infection (Moroni et al. 2011).
Superantigens belong to a class of antigens that initiate a non-specific T-cell
response to release cytokines, causing severe inflammation (Proft and Fraser 2003).
Enterotoxins belong to the family of superantigens that stimulate T-cell production due to
the interaction of the major histocompatibility complex class II molecules on antigen
presenting cells and T cell receptors (Proft and Fraser 2003). Among numerous
enterotoxins, staphylococcal superantigens A, B, and C contain superantigenic features.
Enterotoxins have been associated with autoimmune diseases, such as systemic lupus
erythematosus, and rheumatic arthritis, and are involved in abnormal immunologic
stages, such as psoriasis and atopic dermatitis (Turnidge et al. 2002). S. aureus avoids
the immune system because it expresses different types of superantigens that corrupt

Genetics of Antibiotic Resistance 46
the normal humoral immune response, leading and immunosuppression (Foster 2005).
Superantigens help S. aureus avoid the host immune system by stimulating a large
amount of T cells, which secrete large amount of interleukin-2. Interleukin- 2 is a protein
that, in large amounts, can induce vomiting. Overstimulation by superantigens is not
always beneficial to the host and can lead to shock and death (Freeman-Cook et al.
2006).
Toxic shock syndrome toxin is caused by a superantigen known to be produced by
Staphylococcus aureus (Mahon et al. 2014). These toxins are not processed by APCs,
but instead TSS toxins attach directly to the outside portion of an MHC II antigen on
APCs and to the outside portion of the T cell receptor of the T helper cells.
Superantigens are non-specific, so they can bind without antigen specificity and
stimulate many helper T cells. Excessive amounts of interleukin- 2 produced by T cells
will enter the bloodstream, instead of only acting locally as they normally do.
Overproduction of interleukin-2 circulating in the bloodstream initially induces vomiting
and nausea, but left untreated can become fatal (Freeman-Cook et al. 2006).

Genetics of Antibiotic Resistance 47
A. Modes of Antibiotic Resistance
The spread of antibiotic resistance and the occurrence of multiple drug resistant
pathogenic bacteria have negatively impacted the medical field due to the lack of
effective therapeutic treatment for bacterial infections, such as S. aureus (Lowry 2003).
S. aureus can develop resistance to antibiotics by altering the antibiotic target, trapping
the antibiotic, or through efflux pumps (Pantosti et al. 2007).
1. Alteration of the Antibiotic Target
S. aureus tolerates the cytotoxic activity of β-lactam antibiotics by modifying the
normal penicillin-binding proteins, such as PBP2a (Stapleton and Taylor 2002). PBP2a
modification is best seen in methicillin-resistant S. aureus (MRSA) due to the mecA gene
that is responsible for encoding PBP2a. MRSA forms PBP2a as a fifth PBP, in addition
to four PBPs found in all S. aureus strains (Zapun et al. 2008). β-lactams have a
reduced affinity for PBP2a, and enzyme function will not be disturbed even if it is in the
presence of methicillin or any other β-lactam (Drawz and Bonomo 2010).
Vancomycin-resistant strains can also modify antibiotic targets D-Alanyl-D-
Lactate of peptidoglycan precursors (Kwun et al. 2013). According to Figure 3, D-Ala-D-
Lac production requires VanS, a membrane detector of vancomycin that regulates the
levels of phosphorylation of VanR. VanR is a transcriptional inducer of the van operon
composed of membrane bound histidine kinase and cytoplasmic response regulator
vanHAX (Depardieu et al. 2007). VanH is a dehydrogenase that converts pyruvate to D-
Lac, while VanA acts as a ligase which assemblies the formation of an ester bond
between D-Ala and D-Lac precursors (Kwun et al. 2013). The resistance takes place
because vancomycin does not interact with D-Ala-D-Lac in peptidoglycan (Courvalin

Genetics of Antibiotic Resistance 48
2006). VanX is an enzyme composed of two peptides that cleaves the normal D-Ala-D-
Ala peptidoglycan. Lastly, VanY is a D,D-carboxypeptidase that cleaves the terminal D-
Ala precursors that are formed if D-Ala-D-Ala is not broken by VanX (Cetinkaya et al.
2000). Once D-Ala-D-Ala is substituted for the D-Ala-D-Lac in peptidoglycan production,
S. aureus strains are resistant to vancomycin (Cetinkaya et al. 2000).
Figure 3- Mechanism of action of vancomycin resistance in S. aureus (Hughes 2003).
2. Exclusion of the Antibiotic
Daptomycin is an antibiotic created by Streptomyces roseosporus, a soil
actinomycete, that is used to treat systemic infections with Gram-positive bacteria
(Steenbergen et al. 2005). Daptomycin is composed of a 13-amino-acid-lipopeptide
chain that is trapped by cytoplasmic membranes, which leads to membrane perforation,
K+ efflux (Steenbergen et al. 2005), and apoptosis of the cell (Straus and Hancock

Genetics of Antibiotic Resistance 49
2006). It is believed that resistance to daptomycin arises due to a modification of
bacterial cell walls and might be correlated to vancomycin resistance in vancomycin-
intermediate S. aureus (VISA) strains (Camargo et al. 2008). VISA strains show
peptidoglycan layers that are joined together to prevent vancomycin and daptomycin
access to the cell membrane (Yu et al. 2012). Daptomycin is a large molecule and is
unable to cross through the porin channels of modified walls (Kohanski et al. 2010).
3. Efflux Pumps
Efflux pumps are mechanisms that transport elements, like antibiotics, from the
interior to the exterior of the cell (Webber and Piddock 2003). Some efflux pumps are
specific to one antibiotic while others can accommodate a variety of antibiotics
(multidrug) and be responsible for a multitude of bacterial resistance (Nikaido 2009). S.
aureus is known to have 30 efflux pumps for quinolones (Costa et al. 2013). NorA is an
efflux pump for fluoroquinolones in S. aureus (Costa et al. 2013). norA is a chromosomal
gene that encodes for three alleles that differ by up to 10% in their nucleotide sequences
(Costa et al. 2013). According to the literature, flouroquinolone resistance has increased
with the overexpression of norA gene. The resistance to fluoroquinolones can be due to
mutations in the promoter region of the norA gene through the action of regulatory
proteins (Costa et al. 2003). Thus, if mutations take place at the norA promoter region,
transcription level is affected and consequently, overexpression of the gene associated
with a mutation will occur. As a result, the efflux pump suffered a misfolding due to the
inability of the regulator protein to bind to the promoter and act against S. aureus (Couto
et al. 2008).

Genetics of Antibiotic Resistance 50
4. Mode of mecA gene
SCCmec stands for staphylococcal cassette chromosome mec, which is part of a
family of mobile genetic elements that encode genomic islands with a methicillin-
resistance gene known as mecA (Katayama et al. 2000). SCCmec elements that do not
have mecA gene have cassette chromosome recombinase (ccr) gene instead (Ito 2009).
ccr genes code for recombinases that allow deletions or insertions to take place in the
SCCmec chromosome in order to obtain the correct orientation for transcription (Zong
2013). mec complex carries copies of plasmids with resistant genes while ccr complex is
responsible for the mobility of SCCmec. Five types of ccr and four classes of mec
complex (mecA being the most important) work together to form SCCmec cassette
(Katayama et al. 2003). All methicillin-resistant S. aureus (MRSA) strains harbor at least
one SCCmec element in their chromosome, some of which include a mecA gene that
encodes PBP2a (Lowry 2003). PBP2a is a protein which binds to peptidoglycan and
disrupts the peptide bond in cross-linkages of peptidoglycan chains. Penicillin-binding
proteins have a natural affinity for penicillin, methicillin, and other β-lactam/β-lactam-like
antibiotics (Drawz and Bonomo 2010). When PBPs are bound to one of these
antibiotics, they no longer catalyze cross-linkage of peptidoglycan in the cell wall. This
weakens the cell walls, and cells are more likely to lyse (Drawz and Bonomo 2010).
However, PBP2a shows a decreased affinity to these drugs, conferring antibiotic
resistance (Stapleton and Taylor 2002).

Genetics of Antibiotic Resistance 51
MRSA strains have the ability to activate mecA expression in the presence of β-
lactam antibiotics and repress mecA expression in their absence (Stapleton and Taylor
2002). In reference to Figure 4, if methicillin, penicillin or any other β-lactam is not
present, mecA expression is repressed by MecI, which binds to the promoter of mecA
and inhibits transcription (Stapleton and Taylor 2002). In the presence of β-lactams,
expression of mecA is activated by MecR1, a cell surface protein whose role is to detect
β-lactams. Upon detection, the MecR1 inhibitor is disrupted by deoxyribozymes which
reduce mecRI and PBP2a mRNAs expression, and thus facilitate expression of mecA
(Meng et al. 2009).

Genetics of Antibiotic Resistance 52
Figure 4- Mechanism of action of mecA gene in the presence and absence of beta-
lactam antibiotics (Arede et al. 2012).

Genetics of Antibiotic Resistance 53
5. Specific Treatment for Staphylococcus aureus
S. aureus infections are the most alarming of all staphylococci. In the US, nearly
60% of infections with S. aureus are caused by methicillin-resistant strains, and this
bacterium is thought to be the most difficult to cure (Sakoulas and Moellering 2008). As it
was previously stated, strains can cause food poisoning, toxic shock syndrome, MRSA,
MSSA, and skin infections (Turnidge et al. 2002). Despite the fact that these diseases
are serious, they are worsened because they are also resistant to a number of
antibiotics. Treatment options are limited, complex, and expensive (Sakoulas and
Moellering 2008). Without effective antibiotics, it becomes nearly impossible to treat
such complicated infections (Davies and Davies 2010). Forrest and Tamura (2010)
believe that a combination of antibiotics can improve the efficacy of the treatment
against MRSA versus the use of a single monotherapy.
1. Methicillin-Resistant Staphylococcus aureus (MRSA)
To effectively reduce and stop MRSA development, it is imperative to test clinical
isolates against antibiotics using disk-diffusion (Kirby-Bauer) tests on agar plates (Skov
et al. 2006). Kirby-Bauer tests determine an isolate’s sensitivity to a certain antibiotic. In
the presence of an efficient antibiotic, a zone of inhibition will appear on agar plates
(Skov et al. 2006). Vancomycin, for example, is considered an efficient antibiotic to treat
MRSA infections (Appelbaum 2007). On the other hand, methicillin disks show no zone
of inhibition, indicating that bacteria are uninhibited (Skov et al. 2006). Antibiotic therapy
should be continually administered for 72 hours to MRSA patients until multiple sets of
blood cultures are obtained, even if the symptoms have improved. Early stoppage of
therapy can allow MRSA to survive and develop further antibiotic resistance (Hughes

Genetics of Antibiotic Resistance 54
2008). Overall, each patient may require a personalized approach based on the type and
severity of infection and status of the individual (Hughes 2008).
For patients with severe MRSA infections (more prone to death), a combination
of two or more drugs, like vancomycin, linezolid, and sulfamethoxazole-trimethoprim is
considered to be the most efficient treatment (Deresinski 2009). Minor skin infections
may respond well to mupirocin. Mupirocin is utilized mainly as a topical agent that
specifically and irreversibly binds to isoleucyl tRNA synthetase, thus preventing protein
formation (Gisby and Bryant 2000). The effectiveness of mupirocin has been observed in
surgical and bronchopulmonary infections (Watanabe et al. 2001).
For patients with endocarditis, teicoplanin is known to be the most efficient
treatment. The recommended target is more than 20 mg/L of plasma reached by
teicoplanin prior to the next dose administered, which is refered to as trough
concentration (Ueda et al. 2014). Long-term use of teicoplanin does not lead to
resistance, but isolates can become resistance to vancomycin (Cetinkaya et al. 2000).
Alternative drugs are available for patients with less severe infections. Normally, a
combination of oral therapy with two active agents, such as rifampin or fluoroquinolones,
can be prescribed (Leekha et al. 2011). Oral therapeutic antimicrobial agents are
discouraged for long-term use because of risk for occurrence of resistance and adverse
effects of the antibiotics (Turnidge et al. 2002).
2. Methicillim-Susceptible Staphylococcus aureus (MSSA)
In the 1990s, methicillin-susceptible S. aureus (MSSA) strains were studied and
known from infections in previously healthy patients. (David et al. 2011). Currently,
MSSA infections are treated with penicillinase-resistant penicillins, such as dicloxacillin

Genetics of Antibiotic Resistance 55
and oxacillin in oral or injectable form (Rayner and Munckhof 2005). A wide range of
researchers have adopted these drugs because penicillinase-resistant penicillins destroy
bacteria and show a reduced incidence of adverse reactions (Fair and Tor 2014).
Studies have shown that outpatients with serious staphylococcal infections have
presented satisfactory outcomes (Turnidge et al. 2002).
Cephalosporins have been a useful alternative to penicillinase-resistant
penicillins due to their stability against staphylococcal β-lactamase. Patients with a
history of mild allergies or intolerance to penicillins have been able to use
cephalosporins as a treatment (Turnidge et al. 2002). Nonetheless, cephalosporins are
not considered a safe choice for patients who suffer from severe allergies, such as
angioedema or anaphylaxis. This can be due to partial cross-allergenicity between
penicillins and cephalosporins which can create severe hypersensitivity (Rayner and
Munckhoff 2005). In these cases, clindamycin or vancomycin are known to be most
effective treatments (Rayner and Munckhof 2005).
Currently, β-lactams combined with β-lactam inhibitors, such as amoxicillin/
clavulanic acid, or ampicillin/sulbactam are effective against Gram-negative anaerobes.
This combination of drugs gives a broader spectrum, a considerable advantage when
staphylococci are present in mixed infections with Gram-negative bacteria, including
anaerobes (Turnidge et al. 2002).

Genetics of Antibiotic Resistance 56
V. Conclusion
Antibiotics developed to answer the demands of the global society. Antibiotics
became one of the resources used to treat and prevent diseases. During World War II,
penicillin saved millions of soldiers and ultimately changed the course of history (Ventola
2015). Until 1950s, antibiotics were responding well to the demands of society and
controlled nearly all major bacterial infections during WWII (Ventola 2015). Bacteria
began to survive in the presence of the most common antibiotics used in the medicine
era. Today, several studies have shown and prove that inappropriate and unnecessary
use of antibiotics plays a very crucial role in the expansion of antibiotic resistance
(McEachran et al. 2015).
Bacteria are capable of passing genetic material from one bacterium to another,
leading to the appearance of mutations. Mutations when developed can confer
resistance to antibiotics. Despite warnings from Ventola and other researchers,
antimicrobial agents continue to be misused and overprescribed all over the world
(Ventola 2015). In the U.S., data collected in 2010 states that up to 50% of antibiotics
where inappropriately prescribed to the patients (Ventola 2015). Zdziarski study shows
that unnecessary drug prescription has led to foodborne and waterborne infections,
gastrointestinal complications, antibiotic oversensitivity, ecosystem modification, and
disturbances in human and animal evolution (Zdziarski et al. 2003).
Further studies should investigate how to minimize antibiotic resistance by
controlling the over-prescription of drugs. Not every disease requires antibiotics. In the
UK and Thailand, self-medication is easily available over the counter, and without

Genetics of Antibiotic Resistance 57
evaluation people continue to abuse antibiotics (Pechere 2001). However, the
Netherlands is one of the northern European countries with the lowest incidence of
antibiotic resistance because laws have been implemented prohibiting over the counter
drugs. For instances, physicians are the only ones allowed to prescribe drugs that are
used to treat the infections. If a patient has common flu or cold, antibiotics cannot be
prescribed (Rossignoli and Calvenna 2007). In livestock settings, the European Union
committee implemented drug prescription forms in order to control the usage of
antibiotics in food animals and farms. A threshold limit determined by the European
committee is controlled to ensure that the usage of antibiotics are been met in farm
animals (Bartlett et al. 2013). Also, educating the general public of the risks associated
with misuse of drugs can facilitate and control the bacterial spread (Grigoryan et al.
2010). By following hygiene guidelines and effective disinfection procedures in the
community and in the hospital, the Netherlands believes that it can play a vital role in
minimizing the risks of antibiotic resistance (Deurenberg et al. 2006).
In the US, staphylococcal infections have imposed severe health conditions and
economic burden in over 2 million people every year (Stone et al. 2005). In 2005, after
hand hygiene compliance was implemented, successful results arose in the US hospitals
for MRSA but have not yet been seen in community settings (Carlet et al. 2012). On the
other hand, Netherlands communities found that MRSA skin abscesses less than 5 cm
in size are effectively treated with an incision and drainage instead of using antibiotics.
Scandinavia has followed similar measures to control antibiotic resistance and has the
lowest MRSA prevalence of all European countries, at only 0.6% (Deurenberg et al.
2006). Even though in the US laws have been implemented, guidelines have been
established and educational programs have been campaigned, society still has

Genetics of Antibiotic Resistance 58
preconceived habits concerning antimicrobial agents and their side effects. The scientific
community would highly benefit if further research could be done on how to effectively
educate society, pharmacists, physicians, nurses, and parents of every nation (Carlet et
al. 2012). National and international committees should together propose effective
strategies to address antibiotic resistance among humans and animals, in particular the
interaction between livestock feed and the environment (McEachran et al. 2015).

Genetics of Antibiotic Resistance 59
VI. Acknowledgement
Through the years there have been many blockades and detours in the journey.
Without the help of my family, friends and colleagues, I would have never been able to
achieve my goal of obtaining a master’s degree.
First and foremost I give thanks to my loving and supportive family. My parents
and my brother for always believing in me, for their unconditional support and
understanding. My parents thought me to be persistent, dedicated and extremely hard
worker and never give up when detours appear in the way. After living such an
adventure life at young age, I learnt how to survive and how to succeed, and my
commitment leads me to where I am today. I am truly blessed and grateful for what my
parents have thought me, in order to reach my goals.
Second, I want to share my deepest gratitude to my husband. I truly appreciate
his understanding during many late evenings spent studying, reading, and writing that
consumed my time over this past few years.
For this scholarly paper, I would like to thank my advisor: Dr. James Campbell,
for his time and interest toward my project. I am very appreciative of the support and
guidance given to me by Dr. James Campbell. His guidance and support were critical to
the success of this paper. I am forever grateful for all his patience and time committed
to reviewing and commenting on the numerous drafts that I presented.

Genetics of Antibiotic Resistance 60
VII. References
Alanis A. 2005. Resistance to Antibiotics: Are we in the post-antibiotic Era?
Archives of Medical Research 36(6): 697-705.
Alberts B, Johnson A, Lewis J. 2002. Molecular Biology of the Cell. 4th Edition.
New York: Garland Science.
Aminov R. 2011. Horizontal gene exchange in environmental microbiota. Front
Microbiol 2: 158.
Anderson A, Miller A, Donald R, Scully I, Nanra J, Cooper D, Jansen K. 2012.
Development of a multicomponent Staphylococcus aureus vaccine designed to counter
multiple bacterial virulence factors. Hum Vaccin Immunother 8(11): 1585-1594.
Appelbaum P. 2007. Microbiology of Antibiotic Resistance in Staphylococcus
aureus. Clin Infect Dis 45(supplement 3): S165-S170.
Arede P, Milheiriço C, Lencastre H, Oliveira D. 2012. The Anti-Repressor MecR2
Promotes the Proteolysis of the mecA Repressor and Enables Optimal Expression of β-
lactam Resistance in MRSA. PLoS Pathog 8(7): e1002816.
Argudin M, Mendoza M, Rodicio M. 2010. Food Poisoning and Staphylococcus
aureus enterotoxins. Toxins (Basel) 2(7): 1751–1773
Aubry A, Veziri N, Cambau E, Truffot-Pernot C, Jarlier V, Fisher M. 2006. Novel
Gyrase Muttions in Quinolone-Resistant and Hypersusceptible Clinical Isolates of
Mycobacterium tuberculosis: Functional Analysis of Mutant Enzymes. Antimicrob Agents
Chemother 50(1): 104-112.
Baddour L, Bettmann M, Bolger A, Epstein A, Ferrieri P, Gerber M, Gewitz M,
Jacobs A, Levison M, Newburger J, Pallasch T, Wilson W. Baltimore R, Falace D,

Genetics of Antibiotic Resistance 61
Shulman S, Tani R, Taubert K. 2003. Nonvalvular Cardiovascular Device-Related
Infections. Circulation 108: 2015-2031.
Baquero F, Campos J. 2003. The tragedy of the commons in antimicrobial
chemotherapy. Revista Espanola de Quimioterapia. 16(1):11-3.
Barnes B. 2010. Negligence, medical malpractice, vicarious liability, or patient
responsibility: Who should pay when a patient contracts MRSA from a Healthcare
facilty? Indiana Health Law Review 7: 335.
Barros E, Ceotto H, Bastos M, Santos K, Giambiagi-deMarval M. 2012.
Staphylococcus haemolyticus as an Important Hospital Pathogen and Carrier of
Methicillin Resistance Genes. J Clin Microbiol 50(1): 166-168.
Bartlett J, Gilbert D, Spellberg B. 2013. Seven ways to preserve the miracle of
anitbiotics. Clin Infect Dis 56(10):1445-1450.
Becker K, Heilmann C, Peters G. 2014. Coagulase-Negative Staphylococci. Clin
Microbiol Rev 27(4): 870-926.
Behera D. 2010. Textbook of Pulmonary Medicine. Jaypee Brothers Publishers
1: 269.
Bennett P. 2008. Plasmid encoded antibiotic resistance: acquisition and transfer
of antibiotic resistance genes in bacteria. Br J Pharmacol 153(1): S347-S357.
Berube B, Wardenburg J. 2013. Staphylococcus aureus α-Toxin: Nearly a
Century of Intrigue. Toxins 5: 1140-1166.
Bhatia A, Zahoor S. 2007. Staphylococcus aureus Enterotoxins: A Review.
Journal of Clinical and Diagnostic Research 3:188-197.

Genetics of Antibiotic Resistance 62
Biavasco F, Vignaroli C, Lazzarini R, Varaldo P. 2000. Glycopeptide
Susceptibility profiles of Staphylococcus haemolyticus Bloodstream isolates. Antimicrob
Agents Chemother 44(11): 3122.
Bien J, Sokolova O, Bozko P. 2011. Characterization of virulence factors of
Staphylococcus aureus: Novel function of known virulence factors that are implicated in
activation of airway epithelial proinflammatory response. Journal of Pathogens doi:
10.4061/2011/601905.
Blackstock J. 2014. Guide to Biochemistry. Butterworth-Heinemann. Pp 234.
Blondal K, Rahu K, Altraja A, Viiklepp P, Rhau M. 2013. Overall and cause-
specific mortality among patients with tuberculosis and multidrug resistant tuberculosis.
Int J Tuber Lung Dis 17(7): 961-8.
Bocher S, Tonning B, Skov R, Prag J. 2009. Staphylococcus lugdunensis, a
common cause of skin and soft tissue infections in the community. J Clin Microbiol 47(4):
946-950
Bonomo R, Szabo D. 2006. Mechanism of multidrug resistance in Acinetobacter
species and Pseudomonas aeruginosa. Clin Infect Dis 43:S49-S56.
Bonono R. 2000. Multiple Antibiotic-resistant Bacteria in Long-term-care Facilities:
An emerging problem in the practice of Infectious disease. Clin Infect Dis 31(6): 1414-
1422.
Bousios A, Darzentas N, Tsaftaris A, Pearce S. 2010. Highly conversed motifs in
non –coding regions of Sirevirus retrotransposons: the Key for their pattern of
distribuition within and across plants? BMC Genomics 11:89.

Genetics of Antibiotic Resistance 63
Brady R, Graeme A, O’May, Leid J, Prior M, Costerton J, Shirtliff. 2011.
Resolution of Staphylococcus aureus Biofilm Infection Using Vaccination and Antibiotic
Treatment. Infect Immun 79(4):1797-1803.
Brown D, Edwards D, Hawkey P, Morrison D, Ridgway G, Towner K, Wren M.
2005. Guidelines for the laboratory diagnosis and susceptibility testing of methicillin-
resistant Staphylococcus aureus (MRSA). Journal of Antimicrobial Chemotherapy 56:
1000-1018.
Bukowski M, Wladyka B, Dubin G. 2010. Exfoliative Toxins of Staphylococcus
aureus. Toxins (Basel) 2(5): 1148–1165.
Bush K. 2010. Bench-to-bedside review: The role of β-lactamases in antibiotic-
resistant Gram-negative infections. Crit Care 14(3): 224.
Cabell C, Abrutyn E, Karchmer A. 2003. Bacterial endocarditis: The Disease,
treatment and prevention. Circulation 107:e185-187.
Camargo I, Neoh H, Cui L, Hiramatsu K. 2008. Serial Daptomycin Selection
Generates Daptomycin-nonsusceptible Staphylococcus aureus Strains with a
Heterogeneous Vancomycin-Intermediate Phenotype. Antimicrob Agents Chemother
52(12): 4289-4299.
Carlet J, Jarlier V, Harbarth S, Voss A, Goossens H, Pittet D. 2012. Ready for a
world without antibiotics? The Pensières Antibiotic Resistance Call to Action.
Antimicrobial Resistance and Infection Control 1:11.
Casanova C, Iselin L, Sendi P. 2011. Staphylococcus hyicus bacteremia in a
Farmer. J Clin Microbiol 49(12): 4377-4378.
Centinkaya Y, Falk P, Mayhall C. 2000. Vancomycin-Resistant Enterococci. Clin
Microbiol Rev 13(4):6866-707.

Genetics of Antibiotic Resistance 64
Chen I, Dubnau D. 2004. DNA uptake during bacterial transformation. Nat. Rev.
Microbiol 2 (3): 241–9.
Cheng A, McAdow M, Kim H, Bae T, Missiakas D, Schneewind O. 2010.
Contribution of Coagulase towards Staphylococcus aureus Disease and Protective
Immunity. PLOS Pathog 6(8): e1001036.
Chiller K, Selkin B, Murakawa G. 2001. Skin Microflora and Bacterial Infections of
the Skin. Journal of Investigative Dermatology Symposium Proceeding 6:170-174.
Cho S, Lee D, Choi S, Kwon J, Kim S, Choi J, Park Y, Choi J, Yoo J. 2013.
Impact of vancomycin resistance on mortality in neutropenia patients with enterococcal
bloodstream infection: a retrospective study. BMC Infect Dis 13:504.
Clarke S, Foster S. 2006. Surface adhesins of Staphylococcus aureus. Adv
Microb Physiol 51:187-224
Coates R, Moran J, Horsburgh M. 2014. Staphylococci: colonizers and pathogen
of human skin. Future Microbiol 9(1): 75-91.
Cognasse H, Damien P, Chabert A, Pozzetto B, Cognasse F, Garraud O. 2015.
Platelets and Infections- complex interactionsw with bacteria. Front Immunol 10:3389.
Cole S. 2014. Who will develop new antibacterial agents? Phil Trans R Soc
369(1645): 1471-2970.
Collin F, Karkare S, Maxwell A. 2011. Exploiting bacterial DNA gyrase as a drug
target: current state and perspectives. Appl Microb Biotechnol 92(3): 479-497.
Collins S. 2008. Preventing Health Care-Associated Infections. In: Hughes G.
Patient Safety and Quality: An Evidence-Based Handbook for Nurses. Agency for
Healthcare Research and Quality (US): 41.

Genetics of Antibiotic Resistance 65
Costa S, Viveiros M, Amaral L, Couto I. 2013. Multidrug Efflux Pumps in
Staphylococcus aureus: an Update. Open Microbiol J 7: 59-71.
Courvalin P. 2006. Vancomycin Resistance in Gram-positive cocci. Clin Infect
Dis 42(supplement 1): S25- S34.
Couto I, Costa S, Viveiros M. 2008. Efflux-mediated response of Staphylococcus
aureus exposed to ethidium bromide. J Antimicrob Chemother 62:504–13.
Crossley K, Jefferson K, Archer G, Fowler V. 2009. Staphylococci in Human
Disease. Second edition. Oxford (UK). Wiley-Blackwell.
David M, Boyle-Vavra S, Zychowski D, Daum R. 2011. Methicillin-Susceptible
Staphylococcus aureus as a Predominantly Healthcare-Associated Pathogen: A
Possible Reversal of Roles? PLoS One 6(4): e18217.
David M, Daum R. 2010. Community-Associated Methicillin-Resistant
Staphylococcus aureus: Epidemiology and Clinical Consequences of an Emerging
Epidemic. Clin Microbiol Rev 23(3): 616-687.
Davies J, Davies D. 2010. Origins and evolution of antibiotic resistance. Microbiol
Mol Biol Rev 74(3): 417-433.
Dellit T, Owens R, McGowan J, Gerding D, Weinstein R, Burke J,Huskins W,
Paterson D, Fishman N, Carpenter C, Brennan P, Billeter M, Hooton T. 2007. Infectious
Diseases Society of America and the Society for Healthcare Epidemiology of America
Guidelines for Developing an Institutional Program to Enhance Antimicrobial
Stewardship. Clin Infect Dis 44(2): 159-177.
Delost M. 2014. Introduction to Diagnostic Microbiology for the Laboratory
Sciences. Burlington: MA. Jones & Bartlett Publishers.

Genetics of Antibiotic Resistance 66
Depardieu F, Podglajen I, Leclercq R, Collatz E, Courvalin P. 2007. Modes and
Modulations of Antibiotic Resistance Gene Expression. Clin Microbiol Rev 20(1): 79-114.
Deresinski S. 2009. Vancomycin in Combination with Other Antibiotics for the
Treatment of Serious Methicillin-Resistant Staphylococcus aureus Infections. Clin Infect
Dis 49(7): 1072-1079.
Deurenberg R, Vink C, Kalenic S, Friedrich A, Bruggeman C, Stobberingh E.
2006. The molecular evolution of methicillin-resistant Staphylococcus aureus. Clinical
Microbiology and Infection 13(3): 222-235.
Dinges M, Orwin P, Schlievert P. 2000. Exotoxins of Staphylococcus aureus. Clin
Microbiol Rev 13(1): 16-34.
Domingues S, Harms K, Fricke W, Johnsen P, Silva G. 2012. Natural
Transformation Facilitates Transfer of Transposons, Integrons and Gene Cassettes
between Bacterial Species. PLoS Pathog 8(8): e1002837.
Drawz S, Bonomo R. 2010. Three Decades of β-Lactamase Inhibitors. Clin
Microbiol 23(1): 160-201.
Drlica K, Hiasa H, Kerns R, Malik M, Mustaev A, Zhao X. 2009. Quinolones:
Action and Resistance Updated. Curr Top Med Chem 9(11): 981-998.
Drlica K, Malik M, Kerns RJ, Zhao X. 2008. Quinolone-mediated bacterial death.
Antimicrob Agents Chemother 52:385–92.
Drobniewski F, Cooke M, Jordan J. 2015. Systematic review, meta-analysis and
economic modeling of molecular tests for antibiotic resistance in tuberculosis.
Southampton (UK): NIHR Journals library 19.34.

Genetics of Antibiotic Resistance 67
Durdik P, Fedor M, Jesenak M, Hamzikova J, Knotkova H, Banovcin P. 2009.
Staphylococcus intermedius – rare pathogen of acute meningitis. International Journal of
Infectious Diseases 14(3): e236-238.
Dworkin M, Falkow S, Rosenberg E, Schleifer K, Stackebrandt E. 2006. The
Prokaryotes: Volume 4 Bacteria: Firmicutes, Cyanobacteria. 3rd edition. Minneapolis
(MN). Springer.
Engelhart S, Hanses-Derendorf L, Exner M, Kramer M. 2005. Prospective
surveillance for healthcare-associated infections in German nursing home residents. J
Hosp Infect. 60(1):46
Engelkirk P. Duben-Engelkirk J, Gwendolyn R, Burton W. 2011. Burton’s
Microbiology for the Health Sciences. 9th Edition. Lippincott Williams & Wilkins:
Baltimore, MD.
Enoch D, Cooke F, Guha S, Brown N. 2008. Thermostable nuclease: a study of
clinical impact. J Antimicrob Chemother 61(3): 754-755.
Fabbretti A, Gualerzi C, Brandi L. 2011. How to cope with the quest for new
antibiotics. FEBS Lett 585(11):1673-81.
Fair R, Tor Y. 2014. Antibiotics and Bacterial Resistance in the 21st Century.
Perspect Medicin Chem 6:25-64.
Fernandez L, Handcock R. 2012. Adaptive and Mutational Resistance: Role of
Porins and Efflux Pumps in Drug Resistance. Clin Microbiol 25(4): 661-681.
Filice G, Nyman J, Lexau C, Lees C, Bockstedt L, Como-Sabetti K, Lesher L,
Lynfield R. 2010. Excess Costs and Utilization Associated with Methicillin Resistance for
Patients with Staphylococcus aureus Infection. Infection Control and Hospital
epidemiology 31:4.

Genetics of Antibiotic Resistance 68
Flemming H, Neu T, Wozniak D. 2007. The EPS matrix: The “House of Biofilm
Cells” J Bacteriol 189(22): 7945-7947.
Fluit C, Schmitz F. 2003. MRSA: current perspectives. Horizon Scientific Press.
Norfolk: England.
Fonseca J, Knight G, McHugh T. 2015. The complex evolution of antibiotic
resistance in Mycobacterium tuberculosis. International Journal of Infectious Diseases
32: 94-100.
Forrest G, Tamura K. 2010. Rifampin Combination Therapy for
Nonmycobacterial Infections. Clin Microbiol Rev 23(1): 14-34.
Foster T. 1996. Staphylococcus. In: Baron S. Medical Microbiology. 4th edition.
Galveston (TX). Available from: http://www.ncbi.nlm.nih.gov/books/NBK8448.
Foster T. 2005. Immune evasion by staphylococci. Nat Rev Microbiol 3: 948-958.
Fournier B, Philpott D. 2005. Recognition of Staphylococcus aureus by the Innate
Immune System. Clin Microbiol Rev 18(3): 521-540.
Frank K, Zhou T, Moreno-Vinasco L, Hollett B, Garcia J, Wardenburg J. 2012.
Host Response Signature to Staphylococcus aureus Alpha-Hemolysin Implicates
Pulmonary Th17 Response. Infect Immun 80(9): 3161-3169.
Freeman-Cook L, Freeman-Cook K, Alcamo E, Heymann D. 2006.
Staphylococcus aureus infections. Infobase Publishing: USA.
Frieden T. Antibiotic Resistance Threats in the United States. U.S Department of
Health and Human Services. CDC. 2013. 114 p.
Fudaba Y, Nishifuji K, Andresen L, Yamaguchi T, Komatsuzawa H, Amagai M,
Sugai M. 2005. Staphylococcus hyicus exfoliate toxins selectively digest porcine
desmoglein 1. Microbial Pathogenesis 39: 171-176.

Genetics of Antibiotic Resistance 69
Futagawa-Saito K, Ba-Thein W, Sakurai N, Fukuyasu T. 2006. Prevalence of
virulence factors in Staphylococcus intermedius isolates from dogs and pigeons. BMC
Veterinary Research 2:4.
Garrod D, Chidgey M. 2007. Desmosomal structure, composition and function.
Apical Junctional Complexes Part I 1778(3): 572-587.
Geiter L. 2000. Ending Neglect: The Elimination of Tuberculosis in the United
States. Washington (DC): National Academies Press.
Gelder L, Williams J, Ponciano JM, Sota M, Top EM. 2008. Adaptive plasmid
evolution results in host-range expansion of a broad-host-range plasmid. Genetics. 178:
2179-2190.
Giedraitiene A, Vitauskiene A, Naginiene R, Pavilonis A. 2011. Antibiotic
resistance mechanisms of clinically important bacteria. Medicina (Kaunas) 47(3): 137-
46.
Gillespie I, O’Brien S, Frost J, Tam C, Tompkins D, Neal K, Syed Q, Farthing M.
2006. Investigating vomiting and/or bloody diarrhea in Campylabacter Jejuni infection. J
Med Microbiol 55(6):741-6.
Gillespie S. 2002. Evolution of Drug Resistance in Mycobacterium tuberculosis:
Clinical and Molecular Perspective. Antimicrob Agents Chemother 46(2): 267-274.
Gisby J, Bryant J. 2000. Efficacy of a New Cream Formulation of Mupirocin:
Comparison with Oral and Topical agents in Experiemental skin infections. Antimicrob
Agents Chemother 44(2): 255-260.
Gogarten P, Doolittle W, Lawrence J. 2002. Prokaryotic Evolution in Light of
Gene transfer. Mol Biol Evol 19(12): 2226-2238.

Genetics of Antibiotic Resistance 70
Griffiths A, Miller J, Suzuki D. 2000. An Introduction to Genetic Analysis 7th
edition. New York: W. H. Freeman.
Grigoryan L, Monnet D, Haaijer-Ruskamp F, Bonten M, Lundborg S, Verheij T.
2010. Self-Medication with Antibiotics in Europe: A case for action Curr Drug Safety 5(4):
329-332.
Grohmann E, Muth G, Espinosa M. 2003. Conjugative Plasmid Transfer in Gram-
Positive Bacteria. Microbiol Mol Rev 67(2): 277-301.
Guggenbichler J, Assadin O, Boeswald M, Kramer A. 2011. Incidence and clinical
implication of nosocomial infections associated with implantable biomaterials- catheters,
ventilator-associated pneumonia, urinary tract infections. GMS Krankenhhyg Interdiszip
6(1): Doc18.
Gupta N, Limbago B, Patel J, Kallen A. 2011. Carbapenem-Resistant
Enterobacteriaceae: Epidemiology and Prevention. Clin Infect Dis 53(1): 60-67.
Haddadin A, Fappiano S, Lipsett P. 2002. Methicillin resistant Staphylococcus
aureus (MRSA) in the intensive care unit 78: 385-392.
Hanberger H, Antonelli M, Holmborn M, Lipman J, Pickkers P, Leone M, Rello J,
Sakr Y, Walter S, Vabhems P, Vincent J. 2014. Infections, antibiotic treatment and
mortality in patients admitted to ICUs in countries considered to have high levels of
antibiotic resistance compared to those with low levels. BMC Infect Dis 22(14): 513.
Hassan A, Usman J, Kaleen F, Omair M, Khalid A, Igbal M. 2011. Evaluation of
different detection methods of biofilm formation in the clinical isolates. Braz J Infect Dis
15(4): 305-11.
Hickman A, Chandler M, Dyda F. 2011. Integrating prokaryotes and eukaryotes:
DNA transposases in light of structure. Crit Rev Biochem Mol Biol 45(1): 50-69.

Genetics of Antibiotic Resistance 71
Hoffman D, Brown G, Lombardo F. 2007. Early-onset sepsis with Staphylococcus
auricularis in an extremely low-birth weight infant - an uncommon pathogen. Journal of
perinatology: official journal of the California Perinatal Association 27(8): 519–20.
Huddleston J. 2014. Horizontal gene transfer in the human gastrointestinal tract:
potential spread of antibiotic resistance genes. Infect Drug Resist 7:167-176.
Hughes D. 2003. Exploiting genomics, genetics and chemistry to combat
antibiotic resistance. Nature Reviews Genetics 4: 432-441.
Ibler K, Kromann C.2014. Recurrent furunculosis – challenges and management:
a review. Clin Cosmet Invest Dermatol 7:59-64.
Isaakidis P, Cox H, Varghese B, Montaldo C, Da Silva E, Mansoor H,
Ladomirska J, Sotgiu G, Migliori G, Pontali E, Saranchuk P, Rodrigues C, Reid T. 2011.
Ambulatory multi-drug resistant tuberculosis treatment outcomes in a cohort of HIV-
infected patients in a slum setting in Mumbai, India. PLoS One 6(12):e28066.
Ito T. 2009. Classifiction of Staphylococcal Cassette Chromosome mec
(SCCmec): Guidelines for reporting Novel SCCmec elements. Antimicrob. Agents
Chemother 53(12): 4961-4967.
Janeway C, Travers P, Walport M. 2001. Immunology: The Immune System in
Health and Disease. 5th edition. New York (NY): Garland Science.
Jenkins A, Diep B, Mai T, Nhung V, Warrener P, Suzich J, Stover C, Sellman B.
2015. Differential Expression and Roles of Staphylococcus aureus Virulence
Determinants during Colonization and Disease. mBio 6(1): e022 2-14.
Jensen P,Lambert L, Iademarco M, Ridzon R. 2005. Guidelines for preventing
the transmission of Mycobacterium tuberculosis in health-care settings, 2005. MMWR
Recommend. Rep. 54:1-141.

Genetics of Antibiotic Resistance 72
Jia X, Zhang J, Sun W, He W, Jiang H, Chen D, Murchie A. 2013. Riboswitch
Control of Aminoglycoside Anitbiotic Resistance. Cell 152(1-2): 68-81.
Joo H, Otto M. 2012. Molecular basis of in vivo biofilm formation by bacterial
pathogens. Chem Biol 19(12): 1503-13.
Josefsson E, Hartford O, O’Brien L, Patti J, Foster T. 2001. Protection against
Experimental Staphylococcus aureus Arthritis by Vaccination with Clumping factor A, a
novel virulence determinant. J Infect Dis 184(12): 1572-1580.
Juhas M, Meer J, Gaillard M, Harding R, Hood D, Crook D. 2009. Genomic
Islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev
33(2): 376-393.
Kallen A, Mu Y, Bulens S, Reingikd A, Petit S, Gershman K, Ray S, Harrison L,
Lynfield R, Dumyati G, Townes J, Schaffner W, Patel P, Fridkin S. 2010. Health care
associated invasive MRSA infections, 2005-2008. The Journal of the American Medical
Association, 304(6), 641-647.
Kapur D, Dorsky D, Feingold J, Bona R, Edwards R, Aslanzadeh J, Tutschaka P,
Bilgrami S. 2000. Incidence and outcome of vancomycin-resistant enterococcal
bacteremia following autologous peripheral blood stem cell transplantation. Bone
Marrow Transplantation 25(2): 147-152.
Katayama Y, Ito T, Hiramatsu K. 2000. A New Class of Genetic Element,
Staphylococcus Cassette Chromosome mec, Encodes Methicillin Resistance in
Staphylococcus aureus. Antimicrob Agents Chemother 44(6): 1549.
Katayama Y, Takeuchi F, Ito T, Ma X, Ui-Mizutani Y, Kobayashi I, Hiramatsu K.
2003. Identification in Methicillin-Susceptible Staphylococcus hominis of an active

Genetics of Antibiotic Resistance 73
primordial mobile genetic element for the staphylococcal cassette chromosome mec of
Methicillin-Resistant Staphylococcus aureus. J Bacteriol 185(9): 2711-2722.
Kateete D, Kimani C, Katabazi F, Okeng A, Okee M, Nanteza A, Joloba M,
Najjuka F. 2010. Identification of Staphylococcus aureus: DNase and Mannitol salt agar
improve the efficiency of the tube coagulase test. Ann Clin Microbiol Antimicrob 9: 23.
Keshavjee S, Gelmanova I, Pasechnikov A, Mishustin S.Andreew Y, Yedilbayev
A, Murlherjee J, Rich M, Nardell E, Farmer P, Kim J, Shin S. 2008. Treating Multidrug-
Resistant Tuberculosis in Tomsk, Russia. Ann N Y Acad Sci 1136:1-11.
Ki V, Rotstein C. 2008. Bacterial skin and soft tissue infections in adults: A review
of their epidemiology, pathogenesis, diagnosis, treatment and site of care. Can J Infect
Dis Med Microbiol 19(2):173-184.
Kohanski M, Dwyer D, Collins J. 2010. How antibiotics kill bacteria: from targets
to networks. Nat Rev Microbiol 8(6): 423–435.
Koneman E. 2006. Koneman’s Color Atlas and Textbook of Diagnostic
Microbiology. 6th edition. Baltimore (MD). Lippincott Williams & Wilkins.
Kurek K, Lukaszuk B, Piotrowska D, Wiesiolek P, Chabowska A, Zendzian-
Piotrowska M. 2013. Metabolism, Physiological Role, and Clinical Implications of
Sphingolipids in Gastrointestinal Tract. Biomed Res Int 908907.
Kwun M, Novotna G, Hesketh A, Hill L, Hong H. 2013. In Vivo Studies Suggest
that Induction of VanS-Dependent Vancomycin Resistance Requires Binding of the Drug
to d-Ala-d-Ala Termini in the Peptidoglycan Cell Wall. Antimicrob Agents Chemother
57(9): 4470-4480.
Lackie J. 2010. Oxford: Dictionary of biomedicine. New York: NY. Oxford
University Press.

Genetics of Antibiotic Resistance 74
Leboffe M, Pierce B. 2012. Microbiology: Laboratory Theory & Application. Third
Edition. US. Morton Publishing Company.
Lee C, Cho H, Jeong B, Lee S. 2013. Strategies to Minimize Antibiotic
Resistance. Int J Environ Res Public Health 10(9): 4274-4305.
Leekha S, Terrell C, Edson R. 2011. General Principles of Antimicrobial Therapy.
Mayo Clin Proc 86(2): 156-167.
Levy S. 2002. Factors impacting on the problem of antibiotic resistance. J
Antimicrob Chemother 49(1):25-30.
Lindsay J. Staphylococcus: Moleculear Genetics. Norfolk, UK: Caister Academic
Press; 2008. 255 p.
Liu G. 2009. Molecular Pathogenesis of Staphylococcus aureus infection. Pediatr
Res 65 (5 Pt 2): 71R-77R.
Lodish H, Berk A, Zipursky S. 2000. Mobile DNA. Molecular Cell Biology. 4th
edition. New York: W. H. Freeman.
Loir Y, Baron F, Gautier M. 2003. Staphylococcus aureus and food poisoning.
Genet Mol Res 2(1): 63-76.
Lowry F. 2003. Antimicrobial resistance: the example of Staphylococcus aureus.
J Clin Invest 111(9): 1265-1273.
Lydyard P, Cole M, Holton J, Irving W, Porakishvili N, Venkatesan P, Ward K.
2009. Case Studies in Infectious Disease: Staphylococcus aureus. Garland Science:
608.
Magni M. 2010. Detection of Bacteria, Viruses, Parasites and Fungi: Bioterrotism
Prevention. Springer Science & Business Media, ISBN: 978904818543-6.

Genetics of Antibiotic Resistance 75
Mahon C, Donald C, Manuselis G. 2014. Textbook of Diagnostic Microbiology.
Elsevier 5(12): 270.
Mahon C, Lehman D, Manuselis G. 2011. Textbook of Diagnostic Microbiology
fifth edition. Elseier, ISBN: 9780323089890.
Mathei C, Niclaes L, Suetens C, Jans B, Buntinx F. 2007. Infections in residents of
nursing homes. Infectious disease clinics of North America 21(3): 761-772.
Mayer G. 2010. Exchange of genetic information. Microbiology and Immunology
Online 8(6).
Mayers D. 2009. Antimicrobial drug resistance: mechanism of drug resistance.
Human Press 1(8): 81.
Mazur A, Gudiol F. 2009. Methicillin- Resistant Staphylococcus aureus in long-
term-care facilities. Clin Microbiol Infect 7:26-30.
McEachran A, Blackwell B, Hanson J, Wooten K, Mayer G, Cox S, Smith P.
2005. Antibiotics, bacteria, and antibiotic resistance genes: aerial transport from cattle
feed yards via particulate matter. Environ Health Perspect 123:337–343
Mehta Y, Gupta A, Todi S, Myatra S, Samaddar D, Patil V, Bhattacharya P,
Ramasubban S. 2014. Guidelines for prevention of hospital acquired infections. Indian J
Crit Care Med 18(3): 149-163.
Meng J, Wang H, Hou Z, Fu J, Ma X, He G, Xue X, Jia M, Luo X. 2009. Novel
Anion Liposome-Encapsulated Antisense Oligonucleotide Restores Susceptibility of
Methicillin-Resistant Staphylococcus aureus and Rescues Mice from Lethal Sepsis by
targeting mecA. Antimicrob Agents Chemother 53(7): 2871-2878.
Moroni P, Pisoni G, Cremonesi P, Castiglioni B. 2011. Molecular Detection of
Human bacterial Pathogens: Staphylococcus. CPR Press 28: 309- 311.

Genetics of Antibiotic Resistance 76
Murray P, Rosenthal K, Pfaller M. 2008. Medical Microbiology. Philadelphia: PA.
Mosby Elsevier.
Murray P, Rosenthal K, Pfaller M. 2012. Medical Microbiology. Elsevier Health
Sciences pp 960.
Naber C. 2009. Staphylococcus aureus Bacteremia: Epidemiology,
Pathophysiology, and Management Strategies. Clin Infect Dis 48(4): S231-S237.
Nayak A, Green B, Beezhold D. 2013. Fungal Hemolysins. Med Mycol 51(1): 1-
16.
Nicasio A, Kuti J, Nicolau D. 2008. The current state of multidrug-resistant Gram-
negative bacilli in North America. Pharmacotherapy 28:235-249.
Nikaido H. 2009. Multidrug Resistance in Bacteria. Annu Rev Biochem 78:119-
146.
Nilsson I, Hartford O, Foster T, Tarkowski A. 1999. Alpha-toxin and Gamma-toxin
jointly promote Staphylococcus aureus virulence in Murine septic arthritis. Infect Immun
67(3): 1045-1049.
Noble W. 2004. The Skin Microflora and Microbial Skin Disease. Cambridge
University Press. Cambrigde, UK.
Olson N, Davidow A, Winston C, Chen M, Gazmararian J, Katz D. 2012. A
national study of socioeconomic status and tuberculosis rates by country of birth, United
States, 1996–2005. BMC Public Health 12: 365.
Otto M. 2008. Staphylococcus epidermidis – the “accidental” pathogen. Nat Rev
Microbiol 7(8): 555-567.

Genetics of Antibiotic Resistance 77
Oxford J, Goossens H, Schedler M, Sefton A, Sessa, Van der Velden A. 2013.
Factors influencing inappropriate antibiotic prescription in Europe. Educ Prim Care 24:
291-3.
Pammi M, Liang R, Hicks J, Mistretta T, Versalovi J. 2013. Biofilm extracellular
DNA enhances mixed species biofilms of Staphylococcus epidermidis and Candida
albicans. BMC Microbiology 13: 257.
Pantosti A, Sanchini A, Monaco M. 2007. Mechanism of antibiotic resistance in
Staphylococcus aureus. Future Microbiol 2(3): 323-34.
Papp-Wallace K, Endimiani A, Taracila M, Bonomo R. 2011. Carbapenems:
Past, Present, and Future. Antimicrob Agents Chemother 55(11): 4943.
Parija. 2009. Textbook of Microbiology & Immunology. Elsevier India. pp 580.
Park J, Friendship R, Poljak Z, Weese J, Dewey C. 2013. An investigation of
exudative epidermitis (greasy pig disease) and antimicrobial resistance patterns of
Staphylococcus hyicus and Staphylococcus aureus isolated from clinical cases. Can Vet
J 54(2): 139-144.
Pechere J. 2001. Patients Interviews and Misuse of antibiotics. Clin Infect Dis
33(3): S170-S173.
Pfalller M, Diekema D. 2007. Epidemiology of Invasive Candidiasis: a Persistent
Public Health Problem. Clin Microbiol Rev 20(1): 133-163.
Pieterse R, Todorov S. 2010. Bacteriocins- exploring alternatives to antibiotics in
mastitis treatment. Braz J Microbiol 41(3): 542-562.
Proft T, Fraser J. 2003. Bacterial Superantigens. Clin Exp Immunol 133(3): 299-
306.

Genetics of Antibiotic Resistance 78
Proft T. 2013. Bacterial Toxins: Genetics, Cellular Biology and Practical. Norfold:
UK. Horizon Scientific Press.
Queenan A, Bush K. 2007. Carbapenemases: the versatile β-Lactamases. Clin
Microbiol Rev 20(3): 440-458.
Ramirez M, Tolmasky M. 2010. Aminoglycoside Modifying Enzymes. Drug Resist
Update. 13(6): 151-171.
Rayner C, Munckhof W. 2005. Antibiotics currently used in the treatment of
infections caused by Staphylococcus aureus. Intern Med J 35(2): S3-16.
Raz R, Colodner R, Kunun C. 2005. Who Are You? – Staphylococcus
saprophyticus? Clin Infect Dis 40(6): 896-898.
Reid G, Younes J, Van der Mei H, Gloor G, Knight R, Busscher H. 2011.
Microbiota restoration: natural and supplemented recovery of human microbial
communities. Nature Reviews Microbiology 9: 27-38.
Roberts R, Hota B, Ahmad I, Scott R, Foster S, Abbasi F, Schabowski S, Kampe
L, Ciavarella G, Supino M, Naples J, Cordell R, Levy S, Weisntein R. 2009. Hospital and
Societal Costs of Antimicrobial-Resistant Infections in a Chicago Teaching Hospital:
Implications for Antibiotic Stewardship. Clin Infect Dis 49(8): 1175-1184.
Rossignoli A, Clavenna A. 2007. Antibiotic prescription and prevalence rate in the
outpatient paediatric population: analysis of surveys published during 2000-2005. Eur J
Clin Pharmacol 63:1099-1106.
Rupp R, Bergonier D, Dion S, Hygoneng M, Aurel R, Robert-Graunie C, Foucras
G. 2008. Response to somatic cell count-based selection for mastitis resistance in a
divergent selection experiment in sheep. J Dairy Sci 92(3): 1203-19.

Genetics of Antibiotic Resistance 79
Sabour P, Griffiths M. 2010. Bacteriophages in the Control of Food- and
waterborne pathogens. American Society for Microbiology Press. Washington, DC.
Sakoulas G, Moellering R. 2008. Increasing Antibiotic Resistance among
Methicillin-Resistant Staphylococcus aureus Strains. Clin Infect Dis 46: S360-S367.
Sanders M, McKenna K, Lewis L, Quick G. 2011. Mosby’s Paramedic Textbook.
4th ed. Massachusetts: Jones & Barlett Learning.
Schommer N, Christner M, Hentschke M, Ruckdeschel K, Aepfelbacher M, Rohde
H. 2011. Staphylococcus epidermidis Uses Distinct Mechanisms of Biofilm Formation to
Interfere with Phagocytosis and Activation of Mouse Macrophage-Like Cells 774A.1.
Infect Immnun 79(6): 2267-2276.
Schwendener S, Perreten V. 2012. New MLSb resistance gene erm (43) in
Staphylococcus lentus. Antimicrob Agents Chemother 56(9): 4746-4752.
Singh A, Goering R, Simjee S, Foley S, Zervos M. 2006. The application of
molecular techniques to the study of hospital infection. Clin Microbiol Rev 19(3): 512-30.
Skipper K, Andersen P, Sharma N, Mikkelsen J. 2013. DNA transposon-based
gene vehicles – scenes from an evolutionary drive. Journal of Biomedical Science 20:92.
Skov R, Smyth R, Larsen A, Bolmstrom A, Karlsson A, Mills K, Frimodt-Moller N,
Kahlmeter G. 2006. Phenotypic Detection of Methicillin Resistance in Staphylococcus
aureus by Disk Diffusion Testing and Etest on Mueller-Hinton Agar. J Clin Microbiol
44(12): 4395-4399.
Slama T. 2008. Gram-negative antibiotic resistance: there is a price to pay.
Critical Care 12(4): S2.
Stapleton P, Taylor P. 2002. Methicillin resistance in Staphylococcus aureus. Sci
Prog 85(Pt 1): 57-72.

Genetics of Antibiotic Resistance 80
Steenbergen J, Alder J, Throne G, Tally F. 2005. Daptomycin: a lipopetide
antibiotic for the treatment of serious Gram-positive infections. J Antimicrob Chemother
55(3): 283-288.
Stone P, Braccia D, Larson E. 2005. Systematic review of economic analyses of
health care-associated infections. Am J Infect Control 33:501-509
Stone P. 2009. Economic burden of healthcare-associated infections: an
American perspective. Expert Rev Pharmacoecon Outcomes Res 9(5): 417-422. ,
Braccia D, Larson E. 2005. Systematic review of economic analyses of health care-
associated infections. Am J Infect Control 33:501-509.
Straus S, Hancock R. 2006. Mode of action of the new antibiotic of Gram-positive
pathogens daptomycin: Comparison with cationic antimicrobial peptides and
lipopeptides. Biochimica et Biophysica Acta 1758(9): 1215-1223.
Sydnor M, Emily R, Perl T. 2011. Hospital Epidemiology and Infection Control in
Acute-Care Settings. Clin Microbiol Rev. 24(1): 141–173.
Tanner M, Evertt C, Youvan D. 2000. Molecular Phylogenetic Evidence for
Noninvasive Zoonotic Transmission of Staphylococcus intermedius from a Canine Pet to
a Human. J Clin Microbiol 38(4): 1628-1631.
Tattlybaeva E, Nikiyan H, Vasilchenko A, Deryabin D. 2013. Atomic force
microscopy recognition of protein A on Staphylococcus aureus cell surfaces by labelling
with IgG–Au conjugates. Beilstein J Nanotechnol 4: 743-749.
Thompson J, Cronin B, Bayley H, Wallace M. 2011. Rapid assembly of a
multimeric membrane protein pore. Biophys J 101(11): 2679–83.

Genetics of Antibiotic Resistance 81
Tibra N, Jalali S, Reddy A, Narayanan R, Agarwal R. 2009. Traumatic
endophthalmitis caused by Staphylococcus gallinarum. Journal of Medical Microbiology
59(3): 365–366.
Tille P. 2013. Bailey & Scott’s Diagnostic Microbiology. Thirteenth edition.St.
Louis: MO. Elsevier.
Tindall W, Sedrak M, Boltri J. 2013. Patient-Centered Pharmacology.
Philadelphia: F.A Davis Company.
Tomasz A. 1994. Multiple-antibiotic resistant pathogenic Bacteria. The New
England Journal of Medicine 330(17): 1247-1251.
Turnidge J, Rao N, Chang F, Fowler V, Kellie S, Arnold S, Lee B, Tristan A.
2002. Staphylococcus aureus. Antimicrobial Therapy and vaccines 2: 631-58.
Ueda T, Takesue Y, Nakajima K, Wada Y, Tsuchida T, Takahshi Y, Ishihara M,
Kimura T, Uchino M, Ikeuchi H. 2014. High-dose regimen to achieve novel target trough
concentration in teicoplanin. J infect Chemother 20(1): 43-7.
Vanassche T, Kaushot A, Verhaehen J, Peetermans W, Van Ryn J, Schneewind
O, Hoylaerts M, Verhamme P. 2012. Fibrin formation by staphylothrombin facilitates
Staphylococcus aureus-induced platelet aggregation. Thromb Haemost 107(6): 1107-21.
Vandenbroucke-Grauls. 2014. Antimicrobial resistance in the Netherlands: a
natural experiment? Front Public Health 2(5).
Vela J, Hildebrandt K, Metcalfe A, Rempel H, Bittman S, Topp E, Diara M. 2012.
Characterization of Staphylococcus xylosus isolated from broiler chicken barn
bioaerosol. Poultry Science 91(12): 3003–3012.
Ventola C. 2015. The Antibiotic Resistance Crisis. P T 40(4): 277-283.

Genetics of Antibiotic Resistance 82
Vilcheze C, Williams J. 2012. The Combination of Sulfamethoxazole,
Trimethoprim, and Isoniazid or Rifampin Is Bactericidal and Prevents the Emergence of
Drug Resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 56(10):
5142-5148.
Villari T, Sarnataro, Iacuzio. 2000. Molecular Epidemiology of Staphylococcus
epidermidis in a Neonatal Intensive Care Unit over a Three-Year Period.” Journal of
Clinical Microbiology. 38(5): 1740-1746
Vogan A, Higgs P. 2011. The advantages and disadvantages of Horizontal gene
transfer and the emergence of the first species. Biol Direct 6(1): 10.1186/1745-6150-6-1.
Wagner A. 2006. Cooperation is Fleeting in the World of transposable elements.
PloS Comput Biol 2(12): e162.
Wang N, Neilan A, Klompas M. 2013. Staphylococcus Intermedius Infections:
Case Report and Literature Review. Infect Dis Rep 5(1): e3.
Watanabe H, Masaki H, Asoh N, Watanabe K, Oishi K, Kobayashi S, Sato A,
Sugita R, Nagatake T. 2001. Low Concentrations of Mupirocin in the Pharynx following
Intranasal Application May Contribute to Mupirocin Resistance in Methicillin-Resistant
Staphylococcus aureus. J Clin Microbiol 39(10): 3775-3777.
Webber M, Piddock L. 2003. The importance of efflux pumps in bacterial
antibiotic resistance. J Antimicrob Chemother 51(1): 9-11.
Weckx L. 2012. Antibiotics: from use to abuse. Braz J Ortohinolaryngol 78(2): 2-
2.
Weinstein R. 2005. Efflux Pump and Nosocomial Antibiotic Resistance: A primer
for hospital Epidemiologists. Clin Infect Dis 40(12): 1811-1817.

Genetics of Antibiotic Resistance 83
Wenzel R, Edmond M. 2001. The impact of hospital-acquired bloodstream
infections. Emerg Infect Dis 7(2): 174-177.
Wilcox S. 2004. Cationic Peptides: A new hope. The Science Creative Quarterly
< http://www.scq.ubc.ca/cationic-peptides-a-new-hope/>. Accessed 2014 July 20.
Wisplinghoff H. 2004. Nosocomial bloodstream infections in US hospitals:
analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect
Dis 39:309-317.
Yu R, Dale S, Yamamura D, Stankus V, Lee C. 2012. Daptomycin-
nonsusceptible, vancomycin-intermediate, methicillin-resistant Staphylococcus aureus
endocarditis, Can J Infect Dis Med Microbiol 23(2): e48.
Zachary J, McGavin M. 2013. Pathologic Basis of Veterinary Disease. Fifty
Edition. St. Louis: MO. Elsevier
Zapun A, Contrearas-Martel C, Vernet T. 2008. Penicillin-binding proteins and β-
lactam resistance. Oxford University Press 32(2): 361-385.
Zdziarski P, Simon K, Majda J. 2003. Overuse of high stability antibiotics and its
consequences in public and environmental health. Acta Microbiologica Polonica 52(1):5-
13.
Zong Z. 2013. Characterization of a complex context containing mecA but lacking
genes encoding cassette chromosome recombinases in Staphylococcus haemolyticus.
BMC Microbio 13: 64.
Zuchora-Walske C. 2014. Antibiotics. North Mankato (MN): ABDO Publishing
Company.


















