Genetic parameters and response to selection in blue mussel (Mytilus galloprovincialis) using a...
7
Genetic parameters and response to selection in blue mussel (Mytilus galloprovincialis) using a SNP-based pedigree Thuy T.T. Nguyen a,b, ⁎, Ben J. Hayes b , Brett A. Ingram c a School of Life and Environmental Sciences, Deakin University, Geelong Campus Waurn Ponds, Geelong, Victoria 3216, Australia b BioSciences Research Division, Department of Environment and Primary Industries, Bundoora, Victoria 3080, Australia c Fisheries Victoria, Department of Environment and Primary Industries, Alexandra, Victoria 3714, Australia abstract article info Article history: Received 2 July 2013 Received in revised form 18 November 2013 Accepted 18 November 2013 Available online 28 November 2013 Keywords: Blue mussel Genetic parameters Heritability Selection response Breeding programme In this study, we estimated genetic parameters and realised response to selection in the second generation of a breeding programme of the blue mussel (Mytilus galloprovincialis). A total of 77 full-sib families were produced and reared communally after the fertilisation stage. To assist the reconstruction of a pedigree from these families, a panel of single nucleotide polymorphisms (SNPs) was developed de novo from genomic sequences. A total of 227 out of 432 SNPs were validated. We used only SNPs with polymorphic information content greater than or equal to 0.10 (i.e. 179 SNPs) for family identification. The Bayesian approach using Cervus-type model could as- sign 92.5% of offspring to the intended parent pairs, which is a significant improvement compared to previously used microsatellites. Likely as a result of both improved parentage assignment and greater depth of pedigree, es- timates of heritability of economic traits increased compared to that reported in the first generation. In the sec- ond generation, estimates for heritability of total weight (TW), shape (SH), meat yield as ratio between meat weight and total weight (MY1), and meat yield as ratio between meat weight and the sum of meat weight and shell weight (MY2) were 0.35 ± 0.09, 0.64 ± 0.10, 0.23 ± 0.08 and 0.46 ± 0.10, respectively. Realised selection response (compared to wild–wild matings) from the selection decisions in the first generation of the breeding programme were positive and up to 10%, indicating that further genetic gains can be achieved through this family-based breeding programme. © 2013 Elsevier B.V. All rights reserved. 1. Introduction The blue mussel, Mytilus galloprovincialis, is recognised as an impor- tant aquaculture species in many countries, including Australia. In Victoria (Australia), the traditional farming method relying on collec- tion of natural spat has shifted towards the use of hatchery-produced seed since the success of the artificial propagation of the species in 2008 (Ingram et al., 2013; Jahangard et al., 2010). As a result, annual production of mussel culture in Victoria increased from 449 t in 2008/ 09 to 951 t in 2010/11 (Ingram et al., 2013). The success of hatchery production of the blue mussel, M. galloprovincialis, in 2008 enabled the establishment of a family-based selective breeding programme for the species, based on a founder population of 74 full-sib families (Nguyen et al., 2011). These mussels were reared communally and microsatellite markers were used to iden- tify families in order to improve the accuracy in estimation of genetic parameters. A total of 48 individuals (G 1 ) from each sex were selected from the first generation based on their breeding values for total weight, shape and meat yield as broodstock for the second generation (G 2 ). Although microsatellites were proven to be useful in family identifi- cation in blue mussel, the resolution was low (62.6% of mussels could be assigned to single families) (Nguyen et al., 2011). With recent advances in genome sequencing, coupled with significant reduction in associated costs, we aimed to develop a panel of single nucleotide polymorphisms (SNPs) for blue mussel. This panel of SNPs was tested for efficiency in family identification for the G 2 mussels in this study. The aim of the present study were three-fold: 1) to develop a SNP panel for parentage assignment in blue mussel using the Illumina se- quencing technology; 2) to estimate genetic parameters for total weight, shape and meat yield in the G 2 population, the results from which will be used to select mussels to generate the third generation (G 3 ), and 3) to estimate selection response, by comparing the perfor- mance of these traits in the offspring generated by the selected line with those from non-selected parents. 2. Methods 2.1. Mussel spawning and culture The design of the current experiment is schematically illustrated in Fig. 1. A total of 77 families were generated in June 2010, including 43 families of G 1 ♂ ×G 1 ♀ (SS), 5 families of G 1 ♂ × wild ♀ (SW), 5 Aquaculture 420–421 (2014) 295–301 ⁎ Corresponding author at: BioSciences Research Division, Department of Environment and Primary Industries, Bundoora, Victoria 3080, Australia. Tel.: +61 3 9479 2251; fax: +61 3 9032 7119. E-mail address: [email protected] (T.T.T. Nguyen). 0044-8486/$ – see front matter © 2013 Elsevier B.V. All rights reserved. http://dx.doi.org/10.1016/j.aquaculture.2013.11.021 Contents lists available at ScienceDirect Aquaculture journal homepage: www.elsevier.com/locate/aqua-online
Transcript of Genetic parameters and response to selection in blue mussel (Mytilus galloprovincialis) using a...

Aquaculture 420–421 (2014) 295–301
Contents lists available at ScienceDirect
Aquaculture
j ourna l homepage: www.e lsev ie r .com/ locate /aqua-on l ine
Genetic parameters and response to selection in blue mussel(Mytilus galloprovincialis) using a SNP-based pedigree
Thuy T.T. Nguyen a,b,⁎, Ben J. Hayes b, Brett A. Ingram c
a School of Life and Environmental Sciences, Deakin University, Geelong Campus Waurn Ponds, Geelong, Victoria 3216, Australiab BioSciences Research Division, Department of Environment and Primary Industries, Bundoora, Victoria 3080, Australiac Fisheries Victoria, Department of Environment and Primary Industries, Alexandra, Victoria 3714, Australia
⁎ Corresponding author at: BioSciences Research Divisioand Primary Industries, Bundoora, Victoria 3080, Austfax: +61 3 9032 7119.
E-mail address: [email protected] (T.T.T. N
0044-8486/$ – see front matter © 2013 Elsevier B.V. All rihttp://dx.doi.org/10.1016/j.aquaculture.2013.11.021
a b s t r a c t
a r t i c l e i n f oArticle history:Received 2 July 2013Received in revised form 18 November 2013Accepted 18 November 2013Available online 28 November 2013
Keywords:Blue musselGenetic parametersHeritabilitySelection responseBreeding programme
In this study, we estimated genetic parameters and realised response to selection in the second generation of abreeding programme of the blue mussel (Mytilus galloprovincialis). A total of 77 full-sib families were producedand reared communally after the fertilisation stage. To assist the reconstruction of a pedigree from these families,a panel of single nucleotide polymorphisms (SNPs) was developed de novo from genomic sequences. A total of227 out of 432 SNPs were validated. We used only SNPs with polymorphic information content greater than orequal to 0.10 (i.e. 179 SNPs) for family identification. The Bayesian approach using Cervus-type model could as-sign 92.5% of offspring to the intended parent pairs, which is a significant improvement compared to previouslyusedmicrosatellites. Likely as a result of both improved parentage assignment and greater depth of pedigree, es-timates of heritability of economic traits increased compared to that reported in the first generation. In the sec-ond generation, estimates for heritability of total weight (TW), shape (SH), meat yield as ratio between meatweight and total weight (MY1), and meat yield as ratio between meat weight and the sum of meat weight andshell weight (MY2)were 0.35 ± 0.09, 0.64 ± 0.10, 0.23 ± 0.08 and 0.46 ± 0.10, respectively. Realised selectionresponse (compared to wild–wild matings) from the selection decisions in the first generation of the breedingprogramme were positive and up to 10%, indicating that further genetic gains can be achieved through thisfamily-based breeding programme.
© 2013 Elsevier B.V. All rights reserved.
1. Introduction
The blue mussel,Mytilus galloprovincialis, is recognised as an impor-tant aquaculture species in many countries, including Australia. InVictoria (Australia), the traditional farming method relying on collec-tion of natural spat has shifted towards the use of hatchery-producedseed since the success of the artificial propagation of the species in2008 (Ingram et al., 2013; Jahangard et al., 2010). As a result, annualproduction of mussel culture in Victoria increased from 449 t in 2008/09 to 951 t in 2010/11 (Ingram et al., 2013).
The success of hatchery production of the blue mussel,M. galloprovincialis, in 2008 enabled the establishment of a family-basedselective breeding programme for the species, based on a founderpopulation of 74 full-sib families (Nguyen et al., 2011). These musselswere reared communally andmicrosatellitemarkerswere used to iden-tify families in order to improve the accuracy in estimation of geneticparameters. A total of 48 individuals (G1) from each sex were selectedfrom thefirst generationbased on their breeding values for totalweight,shape and meat yield as broodstock for the second generation (G2).
n, Department of Environmentralia. Tel.: +61 3 9479 2251;
guyen).
ghts reserved.
Althoughmicrosatellites were proven to be useful in family identifi-cation in bluemussel, the resolutionwas low (62.6% ofmussels could beassigned to single families) (Nguyen et al., 2011).With recent advancesin genome sequencing, coupled with significant reduction in associatedcosts, we aimed to develop a panel of single nucleotide polymorphisms(SNPs) for blue mussel. This panel of SNPs was tested for efficiency infamily identification for the G2 mussels in this study.
The aim of the present study were three-fold: 1) to develop a SNPpanel for parentage assignment in blue mussel using the Illumina se-quencing technology; 2) to estimate genetic parameters for totalweight, shape and meat yield in the G2 population, the results fromwhich will be used to select mussels to generate the third generation(G3), and 3) to estimate selection response, by comparing the perfor-mance of these traits in the offspring generated by the selected linewith those from non-selected parents.
2. Methods
2.1. Mussel spawning and culture
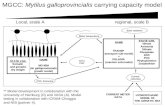
The design of the current experiment is schematically illustrated inFig. 1. A total of 77 families were generated in June 2010, including 43families of G1 ♂ × G1 ♀ (SS), 5 families of G1 ♂ × wild ♀ (SW), 5

43 familiesSelected F1 x Selected
(SS)
24 familiesWild x Wild
(WW)
5 familiesSelected F1 x Wild
(SW)
5 familiesWild x Selected F1
(WS)
3 million fertilised eggs per familyEvenly settled on rope, nursed and grow-out until one year old
Randomly collected mussels(2,496 individuals)
Larger size and rounder shape from each rope
~20%~80%
Potential broodstock candidates
(1,112 individuals)
YES
Culled (not used)
NO
• Tagged (both sides)• Measured length, width
and total weight• Sedated prior to collection
of tissue for genotyping
• Measured length, width and total weight
• Shucked to measure shell weight and meat weight
• Collection of tissue for genotyping
Fig. 1. Steps involved in selection of potential broodstock and phenotypic measurements undertaken in the breeding programme for blue mussel in 2011.
296 T.T.T. Nguyen et al. / Aquaculture 420–421 (2014) 295–301
families of wild ♂ × G1 ♀ (WS), and 24 families of wild ♂ × wild ♀(WW). Wild broodstock were included in the experiment to introducenew genetic material into the selected line, and for comparison withfamilies generated from the selected G1 individuals. Due to thefact that blue mussel in Australia may contain species other thanM. galloprovincialis (Westfall and Gardner, 2010), we used the approachdescribed by Inoue et al. (1995) and concluded that the wild musselsused in the current experiment are M. galloprovincialis. The G1 musselswere confirmed to be M. galloprovincialis previously (Nguyen et al.,2011).
Steps involved in spawning and rearing of mussels were describedpreviously (Nguyen et al., 2011). In brief, mussels were induced tospawn and single pair matingwas undertaken to create full-sib families.Approximately 3 million fertilised eggs from each family were pooledfor communal incubation. Larvae were reared in nine 1000 l tanks(15 million/tank). After 23 days, pediveligers were transferred to nine5000 l tanks (7.5 million/tank) for settlement onto ropes. Once spatreached approximately 1.0 mm in length, 480 ropes with a density of
7954–11,333 spat/m of rope were distributed to each of four differentfarms in Port Phillip Bay, Victoria (144°21′23″–145°07′32″E; 37°50′22″–38°22′15″S) for growing-out at four locations: Grassy Point (GP),Pinnace (PI), Mornington (MO) and Clifton Springs (CS). Musselgrown in the first three locations were graded into large, medium andsmall sizes and re-settled onto ropes once during the grow-out cycle,whereas those at CS were not.
2.2. Measurement of phenotypic characters and tissue sampling
When offspring of the above families reached 12 months of age,sampling was conducted to collect phenotypic data and tissue forgenotyping. We followed the procedures described in Nguyen et al.(2011). A total of 10 mussel ropes were collected to sample for pheno-typic data, including one rope of each grade from GP, PI and MO farms,and one rope from the CS farm. Sampling was undertaken between16/06/2011 to 05/07/2011 and spread over 11 days.

297T.T.T. Nguyen et al. / Aquaculture 420–421 (2014) 295–301
Fig. 1 shows the process involved in mussel sampling. From eachmussel rope, we firstly sampled approximately 20% of the length ofthe rope to select mussels which were larger and rounder as potentialbroodstock. These mussels were tagged with a glue-on shellfish tag(type FPN; Hallprint, Hindmarsh Valley, SA, Australia) on each side ofthe shell, the shell length and width and total weight were measured,then the mussels were sedated for tissue sampling. With the remaining80% of the rope, we randomly sampled mussels and measured wholeweight, length and width then shucked, measured meat and shellweight separately and collected mantel tissue for DNA analysis. Tissuesamples were frozen at−20 °C until required.
2.3. Genome sequencing, assembly and SNP discovery
A single female bluemussel from the G2 populationwas used for ge-nome sequencing. Approximately 5 μg of genomic DNA extracted frommantel tissue of thismussel was fragmented to ~250 bpwith Covaris S2(Covaris Inc., USA) following the manufacturer's instructions. The re-maining steps of library preparation followed the protocol providedby Illumina (Illumina Inc., USA). All sequencing runs were performedon the Hi-Seq2000 platform (Illumina Inc., USA). A total of 150.6 Gb of101-bp paired end sequences were obtained.
Quality control of sequences involved several steps. Firstly, we usedkmers_remove_clonal program in the libngs package (https://github.com/sylvainforet/libngs) to remove PCR duplicates in the sequences inorder to reduce the effects of fault coverage estimate during assembly.Secondly, the package AdapterRemoval (Lindgreen, 2012) was used toremove adapter residues in the sequences, join overlapping pairedreads, trim Ns and low quality bases. Finally, we identified phiX reads(which were added for sequencing control purposes and were notflagged/filtered under our system)by aligning them to the phiX genomeusing a combination of the Burrows–Wheeler Aligner (BWA) tool (Liand Durbin, 2009) and mrFast (Alkan et al., 2009) and removed themusing an in-house script. After removing PCR duplicates, joining shortpaired reads and removing adapter residues, filtering and trimming, atotal of ~96.90 Gb high quality paired-end data were used for assembly.
Velvet version 1.1.06 (Zerbino and Birney, 2008)was used to assem-ble the paired-end data de novo. We tested a range of k-mer = 57–71 inVelvet and obtained the largest and N50 contig sizes at k-mer = 65.This assembly generated ~2.3 million contigs with a N50 contig size of846 bp, and the longest contig length of 39,846 bp. The estimated cov-erage and cut-off coverage values were 17.04.
BLAST search (Altschul et al., 1990) against possible contaminationsources such as human, bacteria, fungi, virus and bovine genomedatabases were conducted. Contigs that significantly matched thesedatabases (at least 95% of contig length aligned at E-value b 0.001)were removed from the assembly. We identified 0, 181, 28 210,468 and 0 contigs that significantly matched each of the databases,respectively; and that at least 95% of the length of the contig se-quence was aligned. When contaminants were excluded, the ge-nome was left with ~1.59 Gb. The current assembly was depositedat DDBJ/EMBL/GenBank under the accession APJB00000000 and theBioProject ID PRJNA178783. The version described in this paper isthe first version, APJB01000000.
We used Novoalign v2.08.02 (Novocraft Technologies, http://www.novocraft.com/) to align all paired-end reads to the remaining contigs.Alignment in SAM format of the reads was processed using SAMtoolsv0.1.18 (Li et al., 2009) to filter and report high quality SNP positions.SNP detection was performed using SAMtools ‘mpileup’ command.Since one of our purposes was to design a SNP panel for genotyping,we applied the first filter to select only SNPs having no adjacent SNPsor indels within the 110 bp flanking region. Subsequent filtering stepswere carried out using SAMtools ‘varFilter’ command specifying mini-mum strand bias P-value of 0.05, proximity to indels (100), coveragedepth min/max cut-offs (5/20), mapping quality (50), and minimumoverall quality (20).
In order to avoid flanking regions of a SNP being one of repetitive el-ements, we masked the assembly with repetitive elements using thehomology-based approach. For this purpose, RepeatMasker (http://www.repeatmasker.org) and the known Repbase library (version17.07, 23 July 2012) were used. Secondly, RepeatScout (Price et al.,2005) was used to identify de novo repetitive elements. The resulting li-brary was then used to mask the assembly with RepeatMasker. SNPswith 110 bp flanking regions being masked were filtered. Only oneSNP per contig was chosen. After all filtering steps, a total of 1248SNPs were sent to GeneSeek (http://www.neogen.com/, NE, USA) forprimer and multiplexing design as well as genotyping.
2.4. SNP multiplexing and genotyping
MassARRAY®Designer software was used to automatically designPCR and iPLEX single base extension primers for multiplexed assays.Nine multiplexes obtained from 432 SNP sequences (i.e. 48 SNPs perplex), were genotyped using Sequenom iPLEX® Platinum platform(Sequenom Inc., San Diego, CA, USA). Briefly, genomic DNA samples(5–10 ng/μl), extracted using DNeasy 96 Blood & Tissue kit (QIAGENPty Ltd, Australia), were used to amplify regions containing SNPs. Unin-corporated dNTPs were neutralised using shrimp alkaline phosphatase.Extended reaction (iPLEX Platinum) was then performed using three-tier primer mixing method. The products of the extended reactionwere desalted and transferred on to a SpectroCHIP® array. The sequencedifference at the single nucleotide level was detected as an allele-specificdifference in mass between extension products, using MassARRAY®MALDI-TOF mass spectrometer. The results were automatically loadedinto a database and exported into tab-delimitated text format.
DNA from the sequenced mussel was included in one well of each ofthe 40 96-well sample plates before genotyping for control purposes.Mean error rate per allelewas estimated using this 40 replicates followingPompanon et al. (2005). Loci with allele error rate N0.0125 (at least onemismatched allele in 40 replicates) were excluded from further analyses.
2.5. Family identification
We used the R package MasterBayes (Hadfield et al., 2006) for ped-igree reconstruction. Only genotypic information was used to assignparentage. The Cervus-type ofmodel (Kalinowski et al., 2007)wasfittedto the data, but confidence in parent–offspring relationships is assessedat the individual instead of at the population level. Genotyping errorrates were set to 0.005 per allele. Twomismatched alleles were allowedfor each offspring and a parent. Following this Bayesian approach, a cat-egorical pedigree was generated in which only assignments with atleast 95% confidence were accepted.
2.6. Estimation of phenotypic and genetic parameters
The traits of interest to the Victorian mussel industry are: 1) totalweight (TW) — the weight in grams at harvest of whole closed musselincluding shell, flesh and enclosed water; 2) shell shape (SH) — theratio of width to length; and 3) meat yield (MY1) — the weight of themeat divided by total weight. In addition, we also included anothermeat yield trait (MY2) — the ratio of meat weight to the sum of shellweight and meat weight (i.e. enclosed water removed).
Amulti-trait animalmodel was fitted to the data as follows (Westellet al., 1988):
y ¼ Xbþ ZQgþ Zuþ e
where:
y are the vectors of phenotypes of only randomly sampled indi-viduals. Only families with 20 offspring or more were used;y = (TW, SH, MY1, MY2);

298 T.T.T. Nguyen et al. / Aquaculture 420–421 (2014) 295–301
b are the vector of fixed effects, including farm locations(4 levels = GP, PI, MO and CS), linear and quadratic effectof covariable rope density (mussels/m), sampling date(11 levels = 11 sampling days), grading practice (2 levels =grading or not grading), and sex (2 levels = male andfemale).
g are the vectors of effects of three genetic groups (both sireand dam from wild, only sire from wild, and only dam fromwild);
u are additive genetic effect of the animal, assumed to be dis-tributed as u ~ N(0, A ⊗ G), where A is the relationship ma-trix derived from pedigree for all animals, and G is a 4 × 4matrix of additive variance and covariance.
e is the residual term, assumed to be distributed as e ~ N(0,I ⊗ R), where I is an identity matrix and R is a matrix of theresidual variance and covariance.
X and Z design matrices for fixed and random animal effects and re-spectively. ZQ relates animals to genetic groups. The variance covari-ance structure as given by:
varuþ Qg
ge
24
35 ¼
Αpp⊗G Apg⊗G 0Agp⊗G Agg⊗G 0
0 0 I⊗R
24
35
where:
App is the submatrix of the inverse of the relationship matrix cor-responding to relationship among animals in the pedigree;
Apg is the submatrix of relating animals in the pedigree with theircorresponding genetic groups;
Agp is the submatrix of the inverse of the relationship matrix cor-responding to relationships between corresponding geneticgroups and animals in the pedigree; and
App is the submatrix of the inverse of the relationship matrix cor-responding to relationships among genetic groups.
Variance components, including residual and genetic covariances,and heritabilities were estimated with ASReml (Gilmour et al., 2002).With the inclusion of the A matrix in the model, ASReml takes into ac-count unbalanced family sizes.
Realised selection response for each trait were obtained by compar-ing mean breeding values of randomly sampled animals produced bythe SS with effects of the group that had both sire and dam from thewild. This was also compared to the theoretical gain that could beachieved by using the formula (Falconer, 1983):
ΔG ¼ i � h � σg
where i is the intensity of selection, h2 is the heritability and g is thestandard deviation of genetic variance of the trait.
050
100
150
200
Fa
Num
ber
of o
ffspr
ing
assi
gned
Fig. 2. Number of offspring assigned to 77 families
3. Results
3.1. SNP validation and genotyping
GeneSeek designed a number of multiplexes from these 1248 se-quences and the first nine multiplexes (48 SNPs each) were used toassay 432 SNPs. Out of these, 38 SNPs failed to PCR. The remaining384 SNPs were scored for 3711 samples (including 40 replicates of thesequenced mussel) with an average call rate of 0.915.
We found 125 homozygous lociwhen examining the 40 replicates ofthe sequenced mussel. We also found 23 loci which showed missingdata in eight replicates or more. In the remaining 236 loci, the errorrate per allele per locus was greater than 0.0125 in eight loci. When allof these problematic loci were excluded, 228 loci remained for furtherexamination in the larger dataset.
When examining the dataset containing 3761 mussel samples, weidentified one locus with all heterozygous genotypes. This is likely be-cause the SNP is located at one of the repetitive regions of the genome.We excluded individuals with a SNP call rate less than 0.800, yieldingthe final dataset of 3635 individuals with genotypes from 227 SNPloci, with an average call rate of 0.924. When loci with PIC b0.10 wereremoved, we obtained the final dataset of 179 loci for 3485 offspringand 154 parents, with average individual call rate of 0.927. This set of179 SNPs was deposited in the dbSNP database, with SNP ID numbers(ss#) ranging from 836314862 to 836315040.
3.2. Parentage assignment
MasterBayes using the Cervus type model could assign parentage toall offspring with probability exceeding 0.95. Out of 3485 offspring,92.5% (3224 individuals) could be assigned to the parent pairs as inour mating plan. The remaining offspring were assigned to sires anddams which were not paired according to our mating plans and there-fore excluded before estimation of genetic parameters. Offspring wereassigned to all 77 families, and the number of mussels assigned toeach family is shown in Fig. 2. On average 47 mussels were assignedto each family, with a range of 1–177 individuals per family. Twelvefamilieswhich had less than 20 offspring assigned to themwere exclud-ed from ASReml analysis.
3.3. Descriptive statistics of traits and estimates of fixed effects
High levels of phenotypic variation were observed in all traits. Totalweight (TW) showed a large range of values (2.3–54.2 g, mean ±SD = 21.99 g ± 7.85). Shape (SH) was also highly variable (range:0.339–0.632, mean ± SD = 0.545 ± 0.028). The two meat yield traits(MY1 and MY2) had a mean ± SD of 0.265 ± 0.056 and 0.448 ±0.044, respectively.
Among the fixed effects, farm and sex were significant for all traits(P b 0.001 in all cases, Table 1). Female mussels showed better TW,
mily
that were in accordance with the mating plan.

Table 1Significant fixed effects (P b 0.001).
Effect Trait F value
Farm (GP, PI, MO, CS) All traits 67.7Sampling dates TW, MY1, MY2 19.3Rope density (linear) TW, MY1, MY2 49.4Sex (male and female) All traits 31.5
Table 3Phenotypic correlation (above the diagonal) and genetic correlation (below the diagonal)between traits. Numbers in parentheses are standard errors.
TW SH MY1 MY2
TW – −0.07 (0.06) 0.01 (0.00) −0.06 (0.06)SH −0.06 (0.17) – −0.02 (0.06)MY1 −0.13 (0.22) 0.35 (0.18) – 0.27 (0.07)MY2 0.23 (0.19) 0.00 (0.00) 0.85 0.09 –
299T.T.T. Nguyen et al. / Aquaculture 420–421 (2014) 295–301
but lower SH, MY1 andMY2 thanmales. In addition, sampling date andrope density also significantly (P b 0.001) affected TW, MY1 and MY2.
3.4. Phenotypic and genetic parameters and response to selection
Analysis of variance components showed the presence of significantadditive genetic variance and phenotypic variance in the SS crosses.These estimates and heritabilities of the four traits are presented inTable 2. SH showed the greatest heritability (0.64 ± 0.10), followedby MY2 (0.46 ± 0.10), TW (0.35 ± 0.09) and MY1 (0.23 ± 0.08).
Genetic and phenotypic correlations between traits are given inTable 3. MY1 had a moderate genetic correlation (0.35 ± 0.18) withSH and a strong correlation with MY2 (0.85 ± 0.09). As for phenotypiccorrelations, a moderate positive phenotypic correlation (0.27 ± 0.07)was observed between MY1 and MY2.
Realised selection responses are shown in Table 4. In percentage, thegenetic gains obtained from the first generation of our mussel breedingprogramme were 10%, 3%, 2.3% and 1.1% for TW, SH, MY1 and MY2, re-spectively. These responses were similar to the theoretical expectationsfor TW, SH and MY1; but less than expected in MY2.
4. Discussion
This study reports the estimates of genetic and phenotypic parame-ters as well as selection response in the second generation of a bluemussel breeding programme in Victoria, Australia. We developed apanel of SNPs using genomic sequences generated by the massive-parallel sequencing technology, which was more effective in familyidentification than previously used microsatellites in this population.Estimates of heritability for all traits were greater compared to that inthe first generation. We also obtained positive selection response inthe traits of interest.
4.1. De novo SNP discovery
This is one in a rare number of studies in bivalves or aquaculture spe-cies in general that use the next generation sequencing technologies inSNP development for genotyping. In the current blue mussel genomeassembly, we estimated a sequence polymorphism rate of 0.39%which is lower compared to the 4th generation inbred (0.73%) or awild (1.30%) Pacific oyster (Zhang et al., 2012). However, identifyinghigh quality SNPs with adequate conserved flanking region for primerdesign was still a challenge.
We found 38 out of 422 SNPs (9%) failed to PCR. A number of factorswere identified to influence the success rate in SNP development suchas sequence quality, sequencing depth, minor allele frequency cut-off
Table 2Variance components and heritabilities of the four traits under study.
Trait Additive geneticvariance(σA
2)
Phenotypicvariance(σP
2)
Heritability ± standarderror
TW 12.07 34.38 0.35 ± 0.09SH 0.00045 0.00070 0.64 ± 0.10MY1 0.00054 0.00232 0.23 ± 0.08MY2 0.00056 0.00123 0.46 ± 0.10
and SNP flanking sequence composition (Andreassen et al., 2010;Wang et al., 2008). In our study,we have ensured stringent sequencefil-tering and reasonable genome coverage (~40 folds). As such, the SNPsthat failed to amplify could be due to the inability of primers to alignwith the sequences due to sequence mismatch, or presence of non-annotated SNPs in the vicinity of the targeted SNPs. This is very likelyin our study aswe sequenced only a singlemussel, and as such polymor-phisms between individuals could not be taken into account in our fil-tering process. It highlights the need to sequence more than oneindividual in de novo SNP discovery.
An additional 125 SNPs (29.6%) were foundmonomorphic in the se-quenced mussel. We investigated these SNPs and found low alignmentdepth (5–6 read depth). This, coupled with the fact that the sequencingtechnologies such as Illumina, though producing sequences with highfidelity, are prone to errors (Kelley et al., 2010; Salmela, 2010). We didnot perform an error correction step prior to assembly and this mayserve as an additional cause of the false SNPs observed herein.Performing error correction to sequence reads along with a slight in-crease in read depth may help to reduce the number of false SNPs. Thevalidated rate achieved in the present study (52.8%) is similar to that ob-served in silver-lipped pearl oyster, Pinctada maxima (55.7%) (Joneset al., 2013), given the differences in approaches and methods used.
4.2. Family identification
In this study, we used 179 SNPs which had the PIC N0.10 to recon-struct the pedigree for mussels which were communally reared. All off-spring could be assigned to their parents with aminimumprobability of0.95. However, 92.5% of individuals could be assigned to expected par-ent pairs which was in concordance with the mating plan. This assign-ment rate is significantly increased compared to that achieved in thefirst generation of mussel breeding (62.5%) (Nguyen et al., 2011), andsimilar to that observed in other studies (Dupont-Nivet et al., 2008;Fishback et al., 2002; Gheyas et al., 2009; Herlin et al., 2007; Vandeputteet al., 2004).
A small proportion of offspring (7.5%) was assigned to unintendedparent pairs. This could be due to two reasons. Firstly, in our breedingprotocol, although efforts weremade to remove possible contaminationof gametes fromothermussel in the broodstock treatment tank, by rins-ingwith distilledwater prior tomating, it is still likely that gametes shedby other broodstock were inadvertently introduced into the matingcontainer. Secondly, it is likely that 30 min of fertilisation was inade-quate to eliminate active but unfertilised eggs and sperms from eachpair, which could have the opportunity to do so at the incubationstage where a pool of unfertilised gametes were present, leading to
Table 4Realised and theoretical selection responses in four traits.
Trait Realised selection response Theoretical selection response
TW (g) 2.12 2.19SH 0.016 0.015MY1 0.006 0.005MY2 0.005 0.008

300 T.T.T. Nguyen et al. / Aquaculture 420–421 (2014) 295–301
the presence of half-sib animals to the intended full-sib families. Theabove suggest the need for precautionary steps that should be adoptedin comparable studies of this nature in the future.
4.3. Heritabilities
Estimates of heritability observed in the present study were im-proved for all traits in the present study compared to those reportedin Nguyen et al. (2011). This could be attributed to the increase in par-entage assignment rate and in turn number of records used to estimategenetic parameters, as well as using two generations of pedigree ratherthan one. Heritability of the new meat yield trait (MY2, 0.46 ± 0.10)was higher than that ofMY1.MY2 is probably a better trait to be includ-ed in the selection index given that it is closer to the trait consumerswillexperience.
Estimate of heritability of TW in this study is higher than that report-ed previously in Mytilus. Heritability of TW in Mytilus chilensis rangedfrom 0.001 to 0.040 for a comparable culture period, and from 0.08 to0.15 if cultured for 22 months (Alcapán et al., 2007). However, the her-itability estimated in our study is similar to that in the Pacific oyster,Crassostrea gigas (0.35 ± 0.17) (Kong et al., 2013). It is noted that stan-dard error of heritabilities in our study ismuch lower compared to thosecited herein, reflecting the larger numbers of records used in our study.
4.4. Response to selection
We predicted the genetic gains for the four traits from the first gen-eration of selection, and then compared these with the selection re-sponses (Table 4). The data revealed that the realised genetic gainswere positive, indicating that selection is following the intended direc-tion. Low progress in improvement of MY2 (1.1%), MY1 (2.3%) and SH(3.0%) was observed compared to that in TW (10%), and it is probablybecause SH, MY1 andMY2 are ratio traits. The observed level of geneticgain in TW falls within the range reported in other aquaculture species,including bivalves such as Sydney rock oyster, Saccostrea commercialis(Nell et al., 1999) and European oyster, Ostrea edulis (Newkirk andHaley, 1983), or temperate (Gjedrem, 2000) and tropical (Ponzoniet al., 2005) fish species.
5. Conclusions
We report genetic parameters and response to selection in the sec-ond generation of the Australian blue mussel breeding programmewhich was established in 2008. We successfully developed a SNPpanel de novo from genomic sequence data that can be effectivelyused for reconstruction of pedigree. Success rate in parentage assign-ment using this SNP panel wasmuch higher compared to that resultingfrom previously developedmicrosatellites. As a result, estimates of her-itability for economic traits in blue mussel were greatly improved andmore reliable. Selection responses in all traits were positive and up to10%, confirming that further improvement of these traits can beachieved through this family-based breeding programme. Integrationof this information into a selective breeding programme for musselshas the potential to enhance production and marketabality. Musselswill reach market size in a shorter time. Harvested mussels will berounder and have a higher meat yield, which are desired by consumers.
Acknowledgements
This study was funded by Fisheries Victoria, Department of Environ-ment and Primary Industries, Victoria (DEPI) with support from theVictorian mussel farming industry through the Victorian ShellfishHatchery Pty Ltd. (VSH) (Queenscliff, Victoria). We thank NathanO'Mahony (FV), Samad Jahangard (FV) andMikeWilliams (VSH) for ac-tivities associated with mussel husbandry and assisting with samplecollection throughout the project. Dr. Steve Petrovski is acknowledged
for his kind assistance in library preparation and sequencing. CharlotteAnderson and Elizabeth Ross provided support in bioinformatics. Wethank Dr. Jennie Pryce, Dr. Mekonnen Haile-Mariam and Dr. OscarGonzalez-Recio for various discussions on the use of animal model.DNA extraction and bioinformatics related activities were undertakenusing the facilities and resources at the BioSciences Research Division(DEPI). Comments from an anonymous reviewer helped us improvethe manuscript.
ReferencesAlcapán, A.C., Nespolo, R.F., Toro, J.E., 2007. Heritability of body size in the Chilean blue
mussel (Mytilus chilensis Hupé 1854): effects of environment and ageing. Aquac.Res. 38, 313–320.
Alkan, C., Kidd, J.M., Marques-Bonet, T., Aksay, G., Antonacci, F., Hormozdiari, F., Kitzman,J.O., Baker, C., Malig, M., Mutlu, O., Sahinalp, S.C., Gibbs, R.A., Eichler, E.E., 2009. Per-sonalized copy number and segmental duplication maps using next-generation se-quencing. Nat. Genet. 41, 1061–1067.
Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J., 1990. Basic local alignmentsearch tool. J. Mol. Biol. 215, 403–410.
Andreassen, R., Lunner, S., Hoyheim, B., 2010. Targeted SNP discovery in Atlantic salmon(Salmo salar) genes using a 3′UTR-primed SNP detection approach. BMC Genomics11, 706.
Dupont-Nivet, M., Vandeputte, M., Vergnet, A., Merdy, O., Haffray, P., Chavanned, H.,Chatain, B., 2008. Heritabilities and GxE interactions for growth in the European seabass (Dicentrarchus labrax L.) using a marker-based pedigree. Aquaculture 275,81–87.
Falconer, D.S., 1983. Introduction to Quantitative Genetics. Longman Group Limited, NewYork.
Fishback, A.G., Danzmann, R.G., Ferguson, M.M., Gibson, J.P., 2002. Estimates of genetic pa-rameters and genotype by environment interactions for growth traits of rainbowtrout (Oncorhynchus mykiss) as inferred using molecular pedigrees. Aquaculture206, 137–150.
Gheyas, A.A., Woolliams, J.A., Taggart, J.B., Satta, M.A., Das, T.K., McAndrew, B.J., Penman,D.J., 2009. Heritability estimation of silver carp (Hypophthalmichthys molitrix) harvesttraits using microsatellite based parentage assignment. Aquaculture 294, 187–193.
Gilmour, A.R., Gogel, B.J., Cullis, B.R., Welham, S.J., Thompson, R., 2002. ASReml User GuideRelease 1.0. VSN International Ltd, Hemel Hempstead, UK.
Gjedrem, T., 2000. Genetic improvement of cold-water fish species. Aquac. Res. 31, 25–33.Hadfield, J.D., Richardson, D.S., Burke, T., 2006. Towards unbiased parentage assignment:
combining genetic, behavioural and spatial data in a Bayesian framework. 15, 3730.Herlin, M., Taggart, J.B., McAndrew, B.J., Penman, D.J., 2007. Parentage allocation in a com-
plex situation: a large commercial Atlantic cod (Gadus morhua) mass spawning tank.Aquaculture 272, S195–S203.
Ingram, B.A., Williams, M., Jahangard, S., Thomas, J., Shipley, M., Mercer, J., O'Mahony, N.,2013. Hatchery manual for the production of the blue mussel, Mytilusgalloprovincialis, spat, Fisheries Victoria Internal Report. Department of PrimaryIndustries, Queenscliff.
Inoue, K., Waite, J., Matsouka, M., Odo, S., Harayama, S., 1995. Interspecific variation inadhesive protein sequences of Mytilus edulis, M. galloprovincialis, and M. trossulus.Biol. Bull. 189, 370–375.
Jahangard, S., Williams, M., Mercer, J.A., Ab Rahim, E., Ingram, B.A., 2010. A technical re-port on hatchery production of blue mussel Mytilus galloprovincialis at the VictoriaShellfish Hatchery (VSH). Department of Primary Industries, Queenscliff.
Jones, D.B., Jerry, D.R., Forêt, S., Konovalov, D.A., Zenger, K.R., 2013. Genome-wide SNP val-idation and mantle tissue transcriptome analysis in the silver-lipped pearl oyster,Pinctada maxima. Mar. Biotechnol. http://dx.doi.org/10.1007/s10126-013-9514-3(Online early).
Kalinowski, S.T., Taper, M.L., Marshall, T.C., 2007. Revising how the computer programCERVUS accommodates genotyping error increases success in paternity assignment.Mol. Ecol. 16, 1099–1106.
Kelley, D., Schatz, M., Salzberg, S., 2010. Quake: quality-aware detection and correction ofsequencing errors. Genome Biol. 11, R116.
Kong, N., Li, Q., Yu, H., Kong, L.-F., 2013. Heritability estimates for growth-related traits inthe Pacific oyster (Crassostrea gigas) using a molecular pedigree. Aquac. Res. http://dx.doi.org/10.1111/are.12205, 1-10 (Online early).
Li, H., Durbin, R., 2009. Fast and accurate short read alignment with Burrows–Wheelertransform. Bioinformatics 25, 1754–1760.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G.,Durbin, R., 1000 Genome Project Data Processing Subgroup, 2009. The sequencealignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079.
Lindgreen, S., 2012. AdapterRemoval: easy cleaning of next generation sequencing reads.BMC Res. Notes 5, 1–7.
Nell, J.A., Smith, I.R., Sheridan, A.K., 1999. Third generation evaluation of Sydney rock oys-ter Saccostrea commercialis (Iredale and Roughley) breeding lines. Aquaculture 170,195–203.
Newkirk, G.F., Haley, L.E., 1983. Selection for growth rate in the European oyster, Ostreaedulis: response of second generation groups. Aquaculture 33, 149–155.
Nguyen, T.T.T., Hayes, B.J., Guthridge, K., Ab Rahim, E., Ingram, B.A., 2011. Use of amicrosatellite-based pedigree in estimation of heritabilities for economic traitsin Australian blue mussel, Mytilus galloprovincialis. J. Anim. Breed. Genet. 128,482–490.
Pompanon, F., Bonin, A., Bellemain, E., Taberlet, P., 2005. Genotyping errors: causes, con-sequences and solutions. Nat. Rev. Genet. 6, 847–859.

301T.T.T. Nguyen et al. / Aquaculture 420–421 (2014) 295–301
Ponzoni, R.W., Hamzah, A., Tan, S., Kamaruzzaman, N., 2005. Genetic parameters and re-sponse to selection for live weight in the GIFT strain of Nile tilapia (Oreochromisniloticus). Aquaculture 247, 203–210.
Price, A.L., Jones, N.C., Pevzner, P.A., 2005. De novo identification of repeat families in largegenomes. Bioinformatics 21, i351–i358.
Salmela, L., 2010. Correction of sequencing errors in a mixed set of reads. Bioinformatics26, 1284–1290.
Vandeputte, M., Kocour, M., Mauger, S., Dupont-Nivet, M., de Guerry, D., Rodina, M., Gela,D., Vallod, D., Chevassus, B., Linhart, O., 2004. Heritability estimates for growth-related traits using microsatellite parentage assignment in juvenile common carp(Cyprinus carpio L.). Aquaculture 235, 223–236.
Wang, S., Sha, Z., Sonstegard, T., Liu, H., Xu, P., Somridhivej, B., Peatman, E., Kucuktas, H.,Liu, Z., 2008. Quality assessment parameters for EST-derived SNPs from catfish.BMC Genomics 9, 450.
Westell, R.A., Quaas, R.L., Van Vleck, L.D., 1988. Genetic groups in an animalmodel. J. DairySci. 71, 1310–1318.
Westfall, K.M., Gardner, J.P.A., 2010. Genetic diversity of Southern hemisphere blue mus-sels (Bivalvia: Mytilidae) and the identification of non-indigenous taxa. Biol. J. Linn.Soc. 101, 898–909.
Zerbino, D.R., Birney, E., 2008. Velvet: algorithms for de novo short read assembly using deBruijn graphs. Genome Res. 18, 821–829.
Zhang, G., Fang, X., Guo, X., Li, L., Luo, R., Xu, F., Yang, P., Zhang, L., Wang, X., Qi, H., Xiong,Z., Que, H., Xie, Y., Holland, P.W.H., Paps, J., Zhu, Y., Wu, F., Chen, Y., Wang, J., Peng, C.,Meng, J., Yang, L., Liu, J., Wen, B., Zhang, N., Huang, Z., Zhu, Q., Feng, Y., Mount, A.,Hedgecock, D., Xu, Z., Liu, Y., Domazet-Loso, T., Du, Y., Sun, X., Zhang, S., Liu, B.,Cheng, P., Jiang, X., Li, J., Fan, D., Wang, W., Fu, W., Wang, T., Wang, B., Zhang, J.,Peng, Z., Li, Y., Li, N., Wang, J., Chen, M., He, Y., Tan, F., Song, X., Zheng, Q., Huang, R.,Yang, H., Du, X., Chen, L., Yang, M., Gaffney, P.M., Wang, S., Luo, L., She, Z., Ming, Y.,Huang, W., Zhang, S., Huang, B., Zhang, Y., Qu, T., Ni, P., Miao, G., Wang, J., Wang, Q.,Steinberg, C.E.W., Wang, H., Li, N., Qian, L., Zhang, G., Li, Y., Yang, H., Liu, X., Wang,J., Yin, Y., Wang, J., 2012. The oyster genome reveals stress adaptation and complexityof shell formation. Nature 490, 49–54.