
Genetic changes of chromosome region 15q11-q13 in Prader...
108
GENETIC CHANGES OF CHROMOSOME REGION 15q11-q13 IN PRADER-WILLI AND ANGELMAN SYNDROMES IN FINLAND HANNALEENA KOKKONEN Department of Clinical Genetics, University of Oulu University Hospital of Oulu OULU 2003
Transcript of Genetic changes of chromosome region 15q11-q13 in Prader...

GENETIC CHANGES OF CHROMOSOME REGION 15q11-q13 IN PRADER-WILLI AND ANGELMAN SYNDROMES IN FINLAND
HANNALEENAKOKKONEN
Department of Clinical Genetics,University of Oulu
University Hospital of Oulu
OULU 2003


HANNALEENA KOKKONEN
GENETIC CHANGES OF CHROMOSOME REGION 15q11-q13 IN PRADER-WILLI AND ANGELMAN SYNDROMES IN FINLAND
Academic Dissertation to be presented with the assent ofthe Faculty of Medicine, University of Oulu, for publicdiscussion in the Auditorium 3 of the University Hospitalof Oulu, on May 23rd, 2003, at 12 noon.
OULUN YLIOPISTO, OULU 2003

Copyright © 2003University of Oulu, 2003
Supervised byProfessor Jaakko Leisti
Reviewed byProfessor Emeritus Pertti AulaDocent Harriet von Koskull
ISBN 951-42-7027-4 (URL: http://herkules.oulu.fi/isbn9514270274/)
ALSO AVAILABLE IN PRINTED FORMATActa Univ. Oul. D 726, 2003ISBN 951-42-7026-6ISSN 0355-3221 (URL: http://herkules.oulu.fi/issn03553221/)
OULU UNIVERSITY PRESSOULU 2003

Kokkonen, Hannaleena, Genetic changes of chromosome region 15q11-q13 in Prader-Willi and Angelman syndromes in Finland Department of Clinical Genetics, University of Oulu, P.O.Box 5000, FIN-90014 University of Oulu,Finland; University Hospital of Oulu, P.O.Box 10, FIN-90029 OYS, Finland 2003
Abstract
The Prader-Willi (PWS) and Angelman (AS) syndromes are clinically distinct developmentaldisorders which are caused by genetic defects in the imprinted domain at chromosome 15q11-q13,resulting in the loss of paternal (PWS) or maternal (AS) gene function. In this study, the geneticchanges of 15q11-q13 and their possible inheritance in Finnish PWS (n=76) and AS (n=47) patientsare described. The diagnosis could be confirmed by laboratory methods in all PWS and in 43 (91%)AS patients.
A deletion of 15q11-q13 accounted for 76% of the PWS and 67% of the AS patients in whom aspecific genetic defect had been established. The origin of deletion was always paternal in PWS andmaternal in AS. In PWS, deletions of four different sizes were detected, while in AS only type I or IIdeletions were found. The smallest overlap of deletions/critical region detected was from locusD15S13 to locus D15S10 in PWS and from locus D15S128 to locus D15S12 in AS. A rare recurrenceof del(15)(q11q13) due to maternal germ line mosaicism is described.
Maternal uniparental disomy of chromosome 15 accounted for 21% of PWS patients and paternalUPD for 2% of AS patients. In PWS, most UPD cases were due to errors in maternal meiosis (87%),most commonly leading to maternal heterodisomy (MI error). In AS, a rare error in the secondsegregation of paternal meiosis was found. UPD was associated with advanced maternal age, themean being 34.6 years.
Imprinting defects were found in 3% of PWS (two non-IC-deletions) and 11% of AS (IC deletionin one sib pair and three non-IC-deletions) patients. In the case with IC deletion, the mutation wasseen in several generations. The non-deletion cases were thought to be due to a de novo prezygotic orpostzygotic error. In the non-deletion PWS cases, the maternally imprinted paternal chromosomeregion was shown to have been inherited from the paternal grandmother, while in AS the paternallyimprinted maternal chromosome region had been inherited from either the maternal grandfather orthe maternal grandmother. The region of IC involved in AS was defined to be 1.15 kb.
Five (11%) AS patients with normal DNA methylation test results had a UBE3A mutation. One ofthe two novel missense mutations (902A→C) had been inherited from the mosaic mother, while themutation 975T→C was a new one. De novo deletions 1930delAG and 3093delAAGA have also beendescribed previously, suggesting that these sites may be mutation hotspots in UBE3A.
Identification of different genetic aetiologies with different recurrence risks is essential for geneticcounselling, and close co-operation between clinicians and the laboratory is required both fordiagnosis and for the detection of possible inheritance.
Keywords: Angelman syndrome, genetics, genomic imprinting, Prader-Willi syndrome


Acknowledgements
This study was carried out at the Department of Clinical Genetics, Oulu University Hospital and University of Oulu, during the years 1989-2000. I wish to express my sincere gratitude to all those who have participated in this project.
I owe my deepest gratitude to my supervisor, Emeritus Professor Jaakko Leisti, M.D., Ph.D., former head of the Department of Clinical Genetics, for introducing me to the fascinating field of medical genetics and for providing the good reseach facilities for my work. His expertise, combined with his friendly attitude and patience have helped and encouraged me many times during these years.
I wish to express my warm gratitude to Professor Jaakko Ignatius, M.D.,Ph.D., current head of the Clinical Genetics, for his supportive attitude towards my research work.
I wish to warmly thank Emeritus Professor Pertti Aula, M.D., Ph.D., and Docent Harriet von Koskull, Ph.D., for their review and valuable comments on the manuscript of this thesis. I also wish to thank Sirkka-Liisa Leinonen, Lic.Phil., for her careful revision of the language of this thesis and the original papers number IV and V, Malcolm Hicks, M.A., for revising the language of original paper number I and Ms. Katri Takalahti for her help in typing the references of this thesis.
I wish to warmly acknowledge my collaborators and co-authors Doctor Karin Buiting, M.D., Ph.D., Professor Bernhard Horsthemke, M.D., Ph.D., Professor Robert Nicholls, M.D., Ph.D., Docent Marketta Kähkönen, Ph.D., and Ms. Katrin Rapakko, B.Sc., for their valuable contributions to this work.
I owe my warm thanks to all my research colleagues, past and present, for creating a friendly and inspiring atmosphere over these years. My special thanks are due to my closest colleague and friend, Ms. Marja-Leena Väisänen, Ph.D., with whom I have been able to share the good and bad moments. Her empathy and encouragement have been invaluable for me during this project. I warmly thank Docent Robert Winqvist, Ph.D., whose profound knowledge and experience in the field of molecular genetics and his supportive attitude towards my work have been valuable to me. I also wish to give my warm thanks to my fellow student and colleague Ms. Ritva Haataja, Ph.D, whose help I have been able to rely on many times.
I wish to warmly thank the whole staff of the Department of Clinical Genetics for their kind help in the course of this study. Especially I thank Ms. Riitta Mattlar for the

collection of most blood samples from the catchment area of Oulu University Hospital, Ms. Nina Pitkänen, M.Sc., for her assistance in high-resolution studies, Ms. Aila Haataja, Ms. Riitta Keskitalo, Ms. Irma Kurvinen, Ms. Sirkka Laine, Ms. Soili Pohjola, Ms. Hilkka Sahavirta, M.Sc., Ms. Liisa Sova and Ms. Tarja Stenius, for their help and support, and for creating a friendly atmosphere in which to work.
I also wish to thank all the doctors who have referred patients to this study. Without them this work would not have been possible.
I wish to express my sincere gratitude to all the patients and their families who have participated in this study.
My loving thanks are due to all members of my family, especially my mother Ulpu Kokkonen and her companion Raimo Manninen, and my family-in-law, for their encouragement and support in so many ways. I also wish to thank all my friends for the pleasant moments of leisure in the world outside the laboratory.
Finally, I wish to present my dearest thanks to Markku Lohvansuu for his care and unfailing support and to our children Helmi and Urho, whose unselfish love has given me the strength to complete this thesis.
This work was financially supported by The Rinnekoti Reseach Foundation, The Alma and K.A. Snellman Foundation, Oulu, Finland, and Oulu University Hospital.
Oulu, April 2003
Hannaleena Kokkonen

Abbreviations
AS Angelman syndrome ASCR Angelman syndrome chromosome region AS-IC Angelman syndrome imprinting centre region ATP10C human aminophopholipid-transporting ATPase gene bp base pair BrdU bromodeoxyuridine cM centiMorgan CSGE Conformation-sensitive gel electrophoresis del deletion DNA deoxyribonucleic acid FISH fluorescence in situ hybridization q long arm of the chromosome GBG G bands with bromodeoxyuridine using Giemsa hect homologous to the E6-AP carboxyl termines HERC2 HERC2-encoding gene IC imprinting centre ID imprinting defect kb kilobase LTP long term potentiation MAGEL2 a necdin-like gene (NDNL1) MI meiosis first segregation MII meiosis second segregation NDN necdin-encoding gene PCR polymerase chain reaction PWCR Prader-Willi chromosome region PWS Prader-Willi syndrome PWS-IC Prader-Willi imprinting centre region RFLP restriction fragment length polymorphism RNA ribonucleic acid mRNA messenger ribonucleic acid RT-PCR reverse transcriptase-PCR SNURF SNRPN upstream open reading frame

SNRPN small nuclear ribonucleoprotein polypeptide N gene SSCP single strand conformation polymorphism SRO smallest region of deletion overlap STR short tandem repeat UPD uniparental disomy UBE3A E6AP ubiquitin protein ligase 3A gene UTR untranslated region UV ultraviolet ZNF127 a zinc-finger gene

List of original publications
This thesis is based on the following original articles, which are referred to in the text by their Roman numerals:
I Kokkonen H, Kähkönen M, Leisti J (1995) A molecular and cytogenetic study in Finnish Prader-Willi patients. Hum Genet 95:568-571.
II Buiting K, Dittrich B, Gross S, Lich C, Färber C, Buchholz T, Smith E, Reis A, Burger J, Nöthen M, Barth-Witte U, Janssen B, Abeliovich D, Lerer I, Ouweland van den A, Halley D, Schrander-Stumpel C, Smeets H, Meinecke P, Malcolm S, Gardner A, Lalande M, Nicholls R, Friend K, Schulze A, Matthijs G, Kokkonen H, Hilbert P, Maldergem L, Glover G, Carbonell P, Willems P, Gillessen-Kaesbach G, Horsthemke B (1998) Sporadic Imprinting Defects in Prader-Willi syndrome and Angelman syndrome: Implications for Imprint-Switch Models, Genetic Counseling and Prenatal Diagnosis. Am J Hum Genet 63:170-180.
III Ohta T, Buiting K, Kokkonen H, McCandless S, Heeger S, Driscoll DJ, Cassidy SB, Horsthemke B, Nicholls RD (1999) Molecular Mechanism of Angelman Syndrome in Two Large Families Involves an Imprinting Mutation. Am J Hum Genet 64:385-396.
IV Kokkonen H, Leisti J (2000) An unexpected recurrence of Angelman syndrome suggestive of maternal germ-line mosaicism of del(15)(q11q13) in a Finnish family. Hum Genet 107:83-85.
V Rapakko K*, Kokkonen H*, Leisti J (2003) UBE3A gene mutations in Finnish Angelman Syndrome Patients Detected by Conformation-sensitive Gel Electrophoresis. Submitted.
The original articles are referred to in the text by their Roman numerals. In addition, some unpublished data are presented.


Contents
Abstract Acknowledgements Abbreviations List of original publications Contents 1 Introduction ...................................................................................................................15 2 Review of the literature .................................................................................................17
2.1 Prader-Willi and Angelman syndromes ..................................................................17 2.1.1 History .............................................................................................................17 2.1.2 Clinical features...............................................................................................18
2.2 Genetic basis of Prader-Willi and Angelman syndromes........................................21 2.2.1 Structure of the proximal long arm of chromosome 15 (15q) .........................22 2.2.2 Genetic imprinting of chromosome 15 ............................................................22
2.2.2.1 General ................................................................................................22 2.2.2.2 Imprinted domain of the chromosome region 15q11-q13 ...................24
2.2.3 Candidate genes for Prader-Willi syndrome....................................................28 2.2.4 Candidate genes for Angelman syndrome .......................................................30
2.3 Known genetic defects causing Prader-Willi or Angelman syndromes ..................33 2.3.1 Deletions of chromosome region 15q11-q13...................................................34 2.3.2 Uniparental disomy of chromosome 15...........................................................37 2.3.3 Imprinting defects of chromosome region 15q11-q13.....................................39 2.3.4 UBE3A gene mutations ...................................................................................40 2.3.5 Other structural chromosomal rearrangements involving chromosome 15 .....41
2.4 Genotype-phenotype correlations ...........................................................................42 2.5 Inheritance and risks of recurrence of Prader-Willi syndrome and Angelman syndrome ...............................................................................................43

2.6 Laboratory diagnostics of Prader-Willi and Angelman syndromes ........................46 2.6.1 Cytogenetic detection of Prader-Willi and Angelman syndromes ...................46
2.6.1.1 Detection of deletions at 15q11-q13....................................................46 2.6.1.2 Detection of uniparental disomy of chromosome 15...........................47 2.6.2 Molecular methods.................................................................................48 2.6.2.1 Detection of deletions at 15q11-q13....................................................48 2.6.2.2 Detection of uniparental disomy of chromosome 15...........................48 2.6.2.3 DNA methylation test as a screening method for the detection of an abnormal imprinting pattern.......................................................49 2.6.2.4 Detection of imprinting centre (IC) mutations in imprinting defects .................................................................................................50 2.6.2.5 Mutation analyses of the UBE3A gene ...............................................51 2.6.2.6 Other aspects of laboratory diagnosis..................................................51
2.6.3 Diagnostic approaches.....................................................................................51 3 Purposes of the present study ........................................................................................53 4 Materials and methods...................................................................................................54
4.1 Subjects (I-V) .........................................................................................................54 4.1.1 Prader-Willi and Angelman patients ................................................................54 4.1.2 Family and control materials ...........................................................................55
4.2 Laboratory analyses................................................................................................55 4.2.1 Cytogenetic and fluorescence in situ hybridization analyses (I, III, IV) .........56 4.2.2 DNA extraction (I, II, III, IV, V)......................................................................56 4.2.3 RFLP and quantitative analyses (I, II, III, IV) .................................................56 4.2.4 Microsatellite analyses (I, II, III, IV)...............................................................57 4.2.5 DNA methylation analyses (I, II, III, IV) ........................................................57 4.2.6 Mutation analyses of the imprinting centre (IC) at 15q11-q13 (II, III)............58 4.2.7 Mutation analyses of the UBE3A gene (V) .....................................................58
4.2.7.1 Conformation-sensitive gel electrophoresis (CSGE) analysis.............58 4.2.7.2 DNA sequencing .................................................................................59
5 Results ...........................................................................................................................60 5.1 General aspects (I, IV, V)........................................................................................60 5.2 Characteristics of the deletions at 15q11-q13 (I, IV)..............................................61
5.2.1 Size variation of the deletion ...........................................................................61 5.2.2 Detection of deletions......................................................................................62 5.2.3 Origin of deletion ............................................................................................63
5.3 Uniparental disomy of chromosome 15 (I, IV).......................................................63 5.4 Imprinting defect in the Prader-Willi and Angelman syndromes (II, III, IV) .........64
5.4.1 DNA methylation and haplotype analysis .......................................................64 5.4.2 Imprinting mutation analysis ...........................................................................65
5.5 Identification of UBE3A gene mutations (V)..........................................................66 6 Discussion .....................................................................................................................67
6.1 General ...................................................................................................................67 6.2 Deletions.................................................................................................................68
6.2.1 Detection of deletions......................................................................................68 6.2.2 Size variation of the deletion ...........................................................................69

6.3 Uniparental disomy.................................................................................................70 6.4 Imprinting defects...................................................................................................72 6.5 UBE3A mutations ...................................................................................................73 6.6 Inheritance of defects and recurrence of Prader-Willi and Angelman syndromes .............................................................................................78 6.7 Laboratory evaluation of the Prader-Willi and Angelman syndromes....................80
7 Concluding remarks.......................................................................................................83 References


1 Introduction
Prader-Willi syndrome (PWS) (MIM 176270) and Angelman syndrome (AS) (MIM 105830) are clinically distinct developmental disorders, both with an incidence of 1/10 000 – 1/15 000 livebirths (Cassidy et al 2000). Despite this, they are genetically related: both disorders are caused by a loss of function of gene(s) in the chromosome region 15q11-q13, which are subject to genomic imprinting and expressed from the paternal (PWS) or maternal (AS) allele(s) only. The imprinting of the 15q11-q13 domain is regulated by an imprinting centre (IC), which has a bipartite structure (Buiting et al. 1995, Dittrich et al. 1996a). The PWS-IC includes a promoter of the SNURF-SNRPN gene (Sutcliffe et al. 1994, Ohta et al. 1999) and controls the imprint switch in the paternal germ line, while the more proximal AS-IC controls the imprint switch in the maternal germ line (Dittrich et al. 1996a).
The imprinted 15q11-q13 domain includes several paternally and two known maternally expressed genes. In contrast to Prader-Willi syndrome, which is likely to be a contiguous gene syndrome, Angelman syndrome is basically thought to arise from the deficiency of a single gene, UBE3A (Kishino et al. 1997, Matsuura et al. 1997). In both disorders, the loss of gene function of 15q11-q13 may be the consequence of either an interstitial deletion (75%), uniparental disomy (UPD) (PWS 24%, AS 2%) or a deficiency in the imprinting process (= an imprinting defect, ID) (1% in PWS, 3% in AS), whereas in AS, mutations of the UBE3A gene may also cause the syndrome (10%). In about 10% of Angelman patients, the genetic defect has remained unknown.
Recurrence of the Prader-Willi and Angelman syndromes in the affected families is rare. Neither the common interstitial deletion nor uniparental disomy, which cover almost all PWS and about 77% of AS defects, are associated with an increased recurrence risk if the parental chromosomes have been structurally normal. Almost all of the families with recurrence of PWS studied to date have had a deletion in the imprinting centre (Ohta et al. 1999). In AS, in addition to the inherited IC deletions, part of the UBE3A mutations has been hereditary (Fang et al. 1999). In these cases, the recurrence risk may be as high as 50%
Due to the discrepancies in the recurrence risks of different defects causing PWS and AS, identification of the specific change is essential. The initial laboratory studies were done by high-resolution chromosome analysis, by which the interstitial deletion of 15q11-q13 was detected in most but not all cases. For the detection of deletions,

16
quantitative analysis with 15q11-q13 specific DNA probes (Tantravahi et al. 1989, Nicholls et al. 1989a, Robinson et al. 1991) and fluorescence in situ hybridization (Kuwano et al. 1992, Mutirangura et al. 1993a, Delach et al. 1994, White and Knoll 1995) have been shown to be much more accurate, and they are now routinely used in many laboratories. For the detection of uniparental disomies, the chromosome 15 specific restriction fragment length polymorphism (RFLP) (Nicholls et al.1989b) was used first, but this method was later replaced by more informative microsatellite marker analyses (Mutirangura et al. 1993b, Robinson et al. 1993a). These FISH and DNA methods greatly improved the diagnosis of PWS and AS, but were still laborious and failed to recognize patients with an imprinting defect. During the years 1992 – 1996, three different tests based on the methylation status of imprinted genes (= DNA methylation analysis) were developed (Driscoll et al. 1992, Dittrich et al. 1992, Sutcliffe et al. 1994, Glenn et al. 1996). DNA methylation analyses were particular useful in confirming the diagnosis of PWS or AS, because they detected all patients with a deletion, uniparental disomy or imprinting defect (i.e. almost all PWS and 80% of AS cases), although they were unable to distinguish between these changes. The specific defect can be identified by fluorescence in situ hybridization or quantitative DNA analysis with 15q11-q13 specific probes (deletions) and/or chromosome 15 specific microsatellite analysis (uniparental disomies and imprinting defects). Angelman patients with UBE3A mutations cannot be identified by the methods described above. To identify this group of patients, sequencing of the abnormal product detected by a mutation screening method (e.g. SSCP) (Kishino et al. 1997) or direct sequencing of amplified genomic DNA (Matsuura et al. 1997) has been used.
In the present study, the primary aim was to find out the genetic changes of the chromosome region 15q11-q13 and to study the origin and nature of the different defects in Finnish Prader-Willi and Angelman patients as well as to study the inheritance of these syndromes. A further purpose was to improve the diagnostic approach to these syndromes.

2 Review of the literature
2.1 Prader-Willi and Angelman syndromes
2.1.1 History
The Prader-Willi (PWS) and Angelman (AS) syndromes are two clinically distinct developmental disorders, each with a characteristic cognitive, behavioural and neurological phenotype. The prevalence of both syndromes is approximately 1/10 000 – 1/15 000 individuals (Burd et al 1990, Clayton-Smith & Pembrey 1992, Petersen et al. 1995, Steffenburg et al. 1996, Buckley et al. 1998, Vercesi et al. 1999), and they occur in both sexes and in all ethnic groups.
The Prader-Willi syndrome was first described in 1956 by three Zürich paediatricians, by whom the disorder was also named (Prader, Labhardt & Willi, 1956). The first case report of a PWS patient, however, had probably already been published 70 years earlier by J L Down (Down 1887). He described a mentally subnormal woman with short stature, small feet and hands, extreme obesity and primary amenorrhea and called this condition “polysarcia” (Down 1887). Angelman syndrome (AS) was first described in 1965 by an English paediatrician Harry Angelman (Angelman 1965), who reported three children with a similar pattern of mental retardation, seizures, ataxia, easily provoked laughter, absent speech and dysmorphic facial features, and he called them ‘puppet children’. In subsequent reports, this name was elaborated as the ‘happy puppet syndrome’ (Bower & Jeavons 1967), but because the term ‘happy puppet’ was considered derogatory by the majority of parents, the name Angelman syndrome is now preferred (Williams & Frias 1982).
The first report of the involvement of a D group chromosome translocation in Prader-Willi syndrome dates back to 1963 (Buchler et al. 1963). Additional translocations were found subsequently, and after the introduction of chromosome banding, it became obvious that chromosome 15 was involved in all instances (Hawkley & Smithies 1976, Fraccaro et al. 1977, Zuffardi et al. 1978, Kucerova et al. 1979, Guanti 1980). This led to

18
high-resolution studies of chromosome 15 in patients with PWS, and de novo deletions of 15q11-q13 were first described in 1981 (Ledbetter et al. 1981) (Fig 1). By studying the inherited heteromorphisms of chromosome 15, the deleted 15q was shown to be paternal in origin in all informative cases (Butler & Palmer 1983, Mattei et al. 1983, Niikawa & Ishikiriyama 1985, Butler et al. 1986), and the exclusively paternal origin of the deletions was later confirmed by molecular marker analysis (Nicholls et al. 1989a, Robinson et al. 1991, Trent et al. 1991). In 1987, studies of individuals with Angelman syndrome showed that they also had a deletion in the same 15q11-q13 region (Magenis et al. 1987, Kaplan 1987). The deletions in both syndromes were shown to be indistinguishable by molecular analysis (Donlon 1988). The observation that two such clearly diverse phenotypes could be caused by a similar chromosomal deletion could not be explained by the classical concepts of genetics. This apparent paradox was, however, resolved when it turned out that, in Angelman syndrome, the partially deleted chromosome 15 was always derived from the mother (Knoll et al. 1989a,b, Imaizumi et al. 1990, Hamabe et al. 1991b, Clayton-Smith et al. 1992). This parent-of-origin effect on the phenotype is now known to be attributable to the phenomenon of genomic imprinting (Hall 1990). Since then, other molecular mechanisms causing either PWS or AS have been discovered, the basic reason in all cases being the absence or lack of expression of the paternal (PWS) or maternal (AS) contribution of the imprinted region 15q11-q13.
Fig. 1. Chromosome pair 15 with microdeletion in 15q11-q13 in one homologue (arrow) (550 bands; G-banding).
2.1.2 Clinical features
Prader-Willi syndrome (MIM 176270) is a developmental disorder characterized by severe hypotonia and feeding difficulties in early childhood. By later infancy, most

19
children develop an insatiable appetite (hyperphagia) and become morbidly obese unless strict external control is imposed. Gross motor milestones and language are delayed. All individuals with PWS have some degree of intellectual impairment, ranging from borderline to moderate mental retardation. Hypogonadism manifests as genital hypoplasia, incomplete pubertal development and infertility in both sexes. Characteristic facial features, including a narrow bifrontal diameter, almond-shaped palpebral fissures and a down-turned mouth, often evolve over time. Behavioural problems, particularly temper tantrums, are common. Frequently observed features in PWS further include decreased foetal movement and infantile lethargy, sleep disturbance, short stature, hypopigmentation, small hands and feet, narrowed hands with straight ulnar border, estropia/myopia, thick, viscous saliva and skin picking (Prader et al. 1956, Butler 1990, Holm 1993, Cassidy 1997a). Most of the manifestations seen in PWS are related to functional hypothalamic deficiency (Cassidy & Schwartz 1998).
Angelman syndrome (MIM 105830) is characterized by severe developmental delay or mental retardation, severe speech impairment with minimal to no use of words, gait ataxia and/or tremulousness of the limbs and a unique behaviour with an inappropriate happy demeanour that includes frequent laughing, smiling and excitability. In addition, the presence of microcephaly, a flat occiput, seizures and abnormal EEG with a characteristic pattern of large-amplitude slow-spike waves are common. The dysmorphic facial features (large mouth and widely spaced teeth, pointed and prognathic chin, thin upper lip and midfacial hypoplasia) are not apparent at birth, but evolve during infancy and childhood. Hypopigmentation of the skin, hair and eyes compared to other family members is seen in a majority but not all AS patients. Developmental delay is evident by 6 – 12 months, but forward progression occurs (Angelman 1965, Williams et al. 1995a, b). In the pathogenesis of AS, deficiencies of Purkinje cells and hippocampal neurons are suggested to account for the ataxia and tremor and the learning deficits and epilepsy, respectively (Jiang et al. 1998, 1999).
Appropriate management may have a significant positive impact on the health and quality of life of Prader-Willi patients. However, many features are subtle or non-specific and some change with age, making a clinical diagnosis of PWS difficult. In Angelman syndrome, too, some manifestations may be absent or late to emerge, and the syndrome may hence not be considered or diagnosed until later in childhood (Cassidy et al. 2000). To facilitate the diagnosis of these syndromes, consensus diagnostic criteria for the PWS (Table 1) (Holm et al. 1993) and AS (Table 2) (Williams et al. 1995a) have been published.

20
Table 1. Diagnostic Criteria for Prader-Willi syndrome. Scoring: major criteria are weighted at one point each. Minor criteria are weighed at one half point. Children ≤ 3 years of age: five points are required for diagnosis, four of which should come from the major group. Children 3 years of age to adulthood: total score of eight is necessary for the diagnosis. Major criteria must comprise ≥ five points of the total score (Holm et al. 1993).
Major criteria 1. Neonatal and infantile central hypotonia with poor suck, gradually improving with age 2. Feeding problems in infancy with need for special feeding techniques and poor weight gain/failure to thrive 3. Excessive or rapid weight on weight-for-length chart (excessive is defined as crossing two centle channels) after 12 months but before 6 years of age; central obesity in the absence of intervention 4. Characteristic facial features with dolichocephaly in infancy, narrow face or bifrontal diameter, almond-shaped eyes, smail-appearing mouth with thin upper lip, down turned corners of the mouth (3 or more required) 5. Hypogonadism-with any of the following, depending on age: a. Genital hypoplasia (male: scrotal hypoplasia, cryptorchidism, small penis and/or testes for age (<5th percentile); female: absence or severe hypoplasia or labia minora and/or clitoris b. Delayed or incomplete gonadal maturation with delayed pubertal signs in the absence of intervention after 16 years of age (male: small gonads, decreased facial and body hair, lack of voice change; female: amenorrhea/oligoamenorrhea after age 16 6. Global developmental delay in a child younger than 6 years of age; mild to moderate mental retardation or learning problems in older children 7. Hyperphagia/food foraging/obsession with food 8. Deletion 15q11-q13 on high resolution (> 650 band) or other cytogenetic/molecular abnormality of the Prader-Willi chromosome region, including maternal disomy Minor criteria 1. Decreased fetal movement of infantile lethargy or weak cry in infancy, improving with age 2. Characteristic behavior problems: temper tantrums, violent outbursts and obsessive/compulsive behaviour; tendency to be argumentative, oppositional, rigid, manipulative, possesive, and subborn; perseverating, stealing, and lying (5 or more of these symptoms required) 3. Sleep disturbance or sleep apnea 4. Short stature for genetic background by age 15 (in the absence of growth hormone intervention) 5. Hypopigmentation (fair skin and hair compaired to family) 6. Small hands (<25th percentile) and/or feet (<10th percentile) for height age 7. Narrow hands with straight ulnar border 8. Eye abnormalities (esotropia, myopia) 9. Thick viscous saliva with crusting at corners of the mouth 10. Speech articulation defects 11. Skin picking Supportive findings (increase the certainity of diagnosis but are not scored) High pain threshold, decreased vomiting, temperature instability in infancy or altered temperature sensitivity in older children and adults,.scoliosis and/or kyphosis, early adrenarche, osteoporosis, unusual skill with jigsaw puzzles, and normal neuromuscular studies

21
Table 2. Clinical features of Angelman syndrome, grouped by relative frequency of occurrence (Williams et al. 1995a)
Consistent (100%) 1. Developmental delay, functionally severe speech impairment, minimal or no use of words; receptive and nonverbal communication skills higher than verbal ones 2. Movement or balance disorder, usually ataxia of gait and/or tremulous movement of limbs 3. Behavioral uniqueness: any combination of frequent laughter/smiling; apparent happy demeanor; easily excitable personality, often with hand flapping movements; hypermotoric behavior; short attention span Frequent (more than 80%) 1. Delayed, disproportionate growth in head circumference, usually resulting in microcephaly (absolute or relative) by 2 years of age 2. Seizures, onset usually < 3 years of age 3. Abnormal EEG, characteristic pattern with large amplitude slow-spike waves (usually 2 to 3 Hz) facilitated by eye closure Associated (20 % to 80%) 1. Flat occiput 2. Occipital groove 3. Protruding tongue 4. Tongue thrusting; suck/swallowing disorders 5. Feeding problems during infancy 6. Prognathia 7. Wide mouth, wide-spaced teeth 8. Frequent drooling 9. Excessive chewing/mouthing behaviors 10. Strabismus 11. Hypopigmented skin, light hair and eye color (compaired with family), seen only in deletion cases 12. Hyperactive lower extremity deep tendon reflexes 13. Uplifted, flexed arm position especially during ambulation 14. Increased sensitivity to heat 15. Sleep disturbance 16. Attraction to/fascination with water
2.2 Genetic basis of Prader-Willi and Angelman syndromes
The structure of the proximal long arm of chromosome 15 and the imprinting of the chromosome region 15q11-q13 creates the background for the formation of the deletions common to the Prader-Willi and Angelman syndromes and explains the different mechanisms by which these syndromes may arise, respectively.

22
2.2.1 Structure of the proximal long arm of chromosome 15 (15q)
Chromosome 15 is an acrocentric chromosome with satellite-rich heterochromatic centromere and stalk regions. The pericentromeric region, located adjacent to the alpha-satellite arrays of the centromeres, contains paralogous copies of chromosomal regions duplicated and translocated from other locations within the human genome, including partial copies of the immunoglobulin heavy chain (IgH) V and D segments (Tomlinson et al. 1994, Nagaoka et al. 1994), the neurofibromatosis type 1 (NF 1) pseudogenes (Legius et al. 1992, Purandare et al. 1995, Regnier et al. 1997, Kehrer-Sawatzki et al. 1997, Barber 1998) and the GABRA5 pseudogenes (Ritchie et al. 1998). The presence of these large repeat units between the centromere and the region most commonly deleted in the Prader-Willi and Angelman syndromes and the differences in the conformation of the euchromatic region (Browne et al. 1997, Cook et al. 1997) make it difficult to distinguish between normal and duplicated chromosome regions 15q11-q13 with normal light microscopy.
The 15q11-q13 region appears to be particularly labile, as it is frequently associated with a variety of cytogenetic rearrangements, including deletions in the Prader-Willi and Angelman syndromes, chromosomal translocations (Butler 1990, Sun et al. 1996), inverted duplications (inv dup (15)) (Robinson et al. 1993b, Huang et al. 1997, Wandstrat et al. 1998), interstitial duplications and triplications (Clayton-Smith et al. 1993a, Schinzel et al. 1994, Cassidy et al. 1996, Browne et al. 1997, Repetto et al. 1998) and inversions (Clayton-Smith et al. 1993b). This multiplicity of chromosomal rearrangements involving 15q11-q13 was originally suggested to be due to tandem or inverted DNA repeats predisposing this segment to structural instability (Donlon et al. 1986). Previously, the proximal 15q was shown to contain duplicated sequences, i.e. END repeats, localized at or near the breakpoints of the 15q11-q13 deletion (Ji et al. 1999, Amos Landgraft et al. 1999). The END repeats include the D15F37 (= MN7) sequence, also localized at 15q11-q13 (Buiting et al. 1998a), and represent large genomic duplications of a novel HERC2 gene located at the distal PWS/AS breakpoint (Ji et al. 1999, 2000). The END-repeat units are postulated to mediate misalignment and unequal crossing-over between these repeats, resulting in deletions and duplications, and they may also predispose the region to inversions (Amos-Landgraft et al. 1999). The products of these homologous recombination events will be dependent on the orientation of the DNA elements, the type of DNA strand exchange and whether the rearrangements are inter- or intrachromosomal (Christian et al. 1999).
2.2.2 Genetic imprinting of chromosome 15
2.2.2.1 General
Genetic (genomic) imprinting refers to differential expression of genes, depending upon the parent of origin of the genetic information (Hall 1990). Imprinting is a normal process

23
and does not affect the structure of DNA. Rather, it is a epigenetic phenomenon, in which a specific DNA region is marked (i.e. imprinted) in a sex-specific manner that determines whether or not it is expressed during gene transcription (Driscoll et al 1994, Lalande et al. 1996, Cassidy & Schwartz 1998, Everman & Cassidy 2000, Reik & Walter 2001), and its role is probably that of regulating growth and development during both gestation and the early postnatal phase (Khan & Wood 1999). Imprinting can be regulated in a tissue-specific manner during development (DeChiara et al. 1991, Vu & Hoffman 1994, Szabo & Mann 1995, Ekstrom et al. 1994, Lee et al. 1997, Lerchner et al. 1997) or genes can be imprinted at any time or any stage of development (Szabo & Mann 1995, Tremblay et al. 1995).
The following minimal set of criteria for a gametic imprint has been proposed: 1) it must occur either during gametogenesis or in the zygote, prior to fusion of the two gametes, while the maternal and paternal chromosomes are still physically separate in the gametes and the alleles can be differentially modified, 2) the imprint must be stably maintained as cells divide and differentiate. The imprint may remain identical to the original imprint on the gametic chromosomes or may be a secondary derivative of that imprint, and 3) it must be recognized by the transcriptional machinery, so as to result in monoallelic expression, and 4) the imprint must be capable of being erased and reset during the production of germ cells in such a way that the appropriate sex-specific imprint is transmitted to the progeny (Bartolomei & Tilghman 1997, Pfeiffer 2000).
Multiple elements have been hypothesized to convey a parent of the origin imprint, the differential patterns of allele-specific DNA methylation in somatic tissues being the most clearly established alternative (McGrath & Solter 1984, Reik et al. 1987). Almost all imprinted genes have differentially methylated regions (DMRs), and either the active or the inactive allele can be methylated (Mann & Lovell-Badge 1984). However, recent studies have implicated a wide range of gene-specific and chromatin domain features in the regulation of imprinted gene expression in somatic cells (Tilghman 1999, Lee et al. 1999, Smilinich et al. 1999, Greally et al. 1999, Nicholls 2000), including differential histone H4 and H3 acetylation (Grunstein et al. 1997, Saitoh et al. 2000, Fulmer-Smentek & France 2001, Jenuwein & Allis 2001) and methylation (Nielsen et al. 2001, Xin et al. 2001, Fournier et al. 2002), nuclease sensitivity (Schweizer et al. 1999) and nuclear matrix association as well as the presence of G-rich direct-repeat sequences in or near CpG islands, oppositely imprinted antisense RNA transcripts (Rouguelle et al. 1998a) and asynchronous DNA replication (Kitsberg et al. 1993, Knoll et al. 1994, Gunaratne et al. 1995, LaSalle & Lalande 1995, Kawame et al. 1995) as well as homologous chromosome association (LaSalle & Lalande 1996). Although it is likely that a combination of these elements operates to create an imprint, as observed in mice (Reik & Walter 2001), their respective roles remain unknown (Nicholls 2000).
After the first suggestion that the mammalian genome might include imprinted genes in mouse (McGrath & Solter 1983, Surani & Barton 1983, McGrath & Surani 1984, Surani et al. 1984, Surani et al. 1986) and the identification of the first endogenous imprinted gene (the insulin-like growth factor 2 gene (Igf2)) (DeChiara et al. 1991), more than 50 imprinted genes have been identified in the combined human and mouse genomes, probably representing 5-20% of those predicted to be imprinted (Morison & Reeve 1998, Falls et al. 1999, Tilghman 1999, Beaudet et al. 2002). These genes are not distributed as single genes throughout the genome, but have a tendency to cluster

24
together. Most of these clusters contain both maternally and paternally imprinted genes (Bartolomei & Tilghman 1997). Although it is apparent that the imprinting of adjacent genes is jointly regulated, multiple mechanisms among and within clusters may operate (Brannan & Bartolomei 1999). In man, imprinted genes have been described in chromosomes 1, 6, 7, 11, 13, 14, 15, 19, 20 and in X-chromosome (Morison et al. 2001, Beaudet et al. 2002). Thus far, two major clusters of imprinted genes are known in the human genome: a 1 Mb region at 11p15, encompassing the Beckwith-Wiedeman (BW) region (Lee et al. 1999), and a 2 Mb cluster at the 15q11-q13 region, encompassing the Prader-Willi syndrome and Angelman syndrome loci (Schweizer et al. 1999, Falls et al. 1999). In addition to the Beckwith-Wiedeman, Prader-Willi and Angelman syndromes, Russel-Silver syndrome (chromosome 7) (Kotzot et al. 1995) and Albright hereditary osteodystrophy (chromosome 20) (Hayward et al. 1998) are known to associate with imprinted genes. Imprinted genes have also been shown to contribute to language development and social skills (Skuse et al. 1997) and probably to other complex behavioural phenotypes in humans, including schizophrenia, alcohol preference, and bipolar affective disorder (Nicholls 2000). Furthermore, imprinting may have an important role in the pathogenesis of cancer, because distruption in the monoallelic expression of imprinted genes is suggested to be the most common mutation associated with cancer (Feinberg 2000, Pfeiffer 2000).
2.2.2.2 Imprinted domain of the chromosome region 15q11-q13
Proximal 15q contains a 2 Mb domain that is subject to genomic imprinting and affected in patients with either Prader-Willi or Angelman syndrome. This domain contains a cluster of paternally expressed genes and transcripts, whose absence cause PWS, and at least two maternally expressed genes, of which the mutations in UBE3A have already been shown to cause AS. Based on a small group of Prader-Willi and Angelman patients with small deletions resulting in a regional imprinting defect, it has been suggested that imprinting of this domain is regulated by an imprinting centre (IC) (Buiting et al. 1995) (see Fig. 6 p. 29). The IC has been localized to the SNRUF-SNRPN locus and appears to have a bipartite structure (Reis et al. 1994, Buiting et al. 1995, Dittrich et al. 1996a, Saitoh et al.1996, Horsthemke et al. 1997). The PWS-IC is defined by the smallest region of deletion overlap (PWS-SRO) in the PWS families with an imprinting defect, and it is less than 4.3-kb in size, spanning the first exon 1 and the promoter of the SNURF-SNRPN locus (Sutcliffe et al. 1994, Ohta et al. 1999). The 880 bp AS-IC maps ~35 kb upstream of the SNRPN exon 1, immediately distal to an alternative 5’ exon of SNRPN called u4 (or BD3 or IC3) (Buiting et al. 1995, Saitoh et al. 1996, Buiting et al. 1999a), and it is defined by the smallest region of deletion overlap (AS-SRO) in the AS families with an imprinting defect. The AS-IC contains one (= u5) of several alternative upstream exons of SNURF-SNRPN, probably playing a role in maternal imprinting (Fäber et al. 1999). AS-IC is hypermethylated on both parental alleles (Schumacher et al 1998), whereas PWS-IC exhibits maternally specific methylation in humans (Sutcliffe et al. 1994, Glenn et al. 1996) and mice (Shemer et al. 1997, Gabriel et al. 1998).

25
Deletions of PWS-IC appear to block the maternal to paternal imprint switch in the paternal germ line, and deletions of AS-IC are associated with a block of the paternal to maternal imprint switch in the maternal germ line (Dittrich et al. 1996a), suggesting that this bipartite imprinting centre regulates the initiation of the parental imprint switch in both the male and the female germ lines (Nicholls et al. 1998). Several models have been presented to explain the mechanisms of gamete-specific imprinting events at 15q11-q13 (Buiting et al. 1995, Dittrich et al. 1996a, Ferguson-Smith 1996, Bürger et al. 1997, Tilghman et al. 1998, Nicholls et al. 1998, Ohta et al. 1999, Brannan and Bartolomei 1999). Based on these models and previous expression data of the UBE3A antisense transcript (Rouguelle et al. 1998a) as well as mouse studies, Shemer et al. (2000) have presented a model for regional control of PWS/AS imprinting, in which they propose that, during oogenesis, a trans-acting factor binds to the AS-IC sequence and confers methylation and silencing on the SNRPN promoter, which, in turn, leads to methylation and inactivation of genes on the maternal allele. In the paternal germ line, the trans-acting factor is probably absent and PWS-IC remains unmethylated. The spreading of undermethylation and the generation of a stable active state on the paternal chromosome take place by an as-yet-unknown mechanism (Fig. 2) (Shemer et al. 2000). The UBE3A gene is tissue-specifically imprinted with maternal expression in human brain. The ability to inactivate paternal UBE3A in brain may be explained by the recent discovery of a paternally expressed antisense transcript overlapping UBE3A but proximal to it relative to PWS-IC (Rouguelle et al. 1998a). The UBE3A imprinting might be an indirect effect of the paternal-specific expression of the antisense transcript UBE3A-AS in a manner similar to what has been suggested for Ig2r (Wutz et al.1997). In keeping with this hypothesis, UBE3A is biallelic in all tissues in which the antisense transcript is not expressed, but results in expression of UBE3A specifically on the maternal allele in human brain, where the antisense transcript is expressed (Rouguelle et al 1998a). In this model, the role of the AS-IC and PWS-IC elements is to regulate the expression of paternal-specific genes, and maternal expression of UBE3A occurs only by default, thus representing a secondary effect of imprinting. Other possible mechanisms to silence paternal UBE3A include the roles for chromatin structure, insulators or transcriptional enhancer competition (Tilghman, 1999).

26
Fig. 2. Model of gene regulation at the imprinted 15q1-q13 domain. In the female germ line (A), gamete-specific imprinting factors activates AS-IC leading to de novo methylation of the adjacent PWS-IC. In the male germline (B), the AS-IC is not functional and the PWS-IC remains unmethylated, activating the genes at the 15q11-q13 domain. Mat = maternal, pat = paternal, = active gene, = inactive gene. Modified from Shemer et al. (2000).
Recent experiments demonstrate that PWS-IC is not only required for the establishment of the paternal imprint in the paternal germ line, but also for the postzygotic maintenance of this imprint in both humans and mice (Bielinska et al. 2000). The control of imprint switching has been assumed to function via DNA methylation. However, it was shown by El-Maarri et al. (2001) that the incorrect maternal methylation imprint in PWS-IC deletion patients was established de novo after fertilization, indicating that the SNURF-SNRPN exon 1 region is not a control element for a germ line methylation switch, but essential for maintaining the paternal methylation imprint during embryonic development. Similar data were obtained from a mouse model harbouring a microdeletion of the Snrpn exon 1 region (Yang et al. 1998). Moreover, it was found that CpG-rich regions in SNURF-SNRPN are hypomethylated in unfertilized human oocytes, indicating that the normal maternal methylation imprints in 15q11-q13 are also established very late during ovulation or after fertilization, when the two genomes are still separate pronuclei (El-Maarri et al. 2001). This is in contrast to mouse eggs, where the corresponding Snrpn exon 1 region is heavily methylated (Shemer et al. 2000, El-Maarri et al. 2001), indicating that an important aspect of imprinting may have evolved divergently between mouse and human (El-Maarri et al. 2001).
A mechanism additional to DNA methylation that could distinguish between the two parental chromosomes in the zygote could be alterations in chromatin structure, which mark the male and female alleles. The imprinting centre could regulate the initiation of the parental imprint switch in the male and female germ lines, leading to the establishment of heterochromatic-like DNA at the IC in oocytes and euchromatic-like DNA at the IC during spermatogenesis (Greally 1999, Schweitzer et al. 1999, Nicholls 2000). These differences could then be recognized after fertilization and translated into stable DNA methylation imprints. Another possibility might be that methylation differences at other, as yet undetected loci carry the parental chromosomal mark, which
matAS-IC PWS-IC
ZNF127 MAGEL2 NDNSNURF-
SNRPN PAR5 IPW PAR1 A3EBU UBE3A
AS-ICpat
PWS-IC
A)
B)

27
leads to de novo methylation at neighbouring loci in the early embryo (El-Maarri et al. 2001). However, the PWS-IC region has shown to be especially CpG-rich and to be conserved between human and mouse (Chaillet et al. 1991). It has also shown to carry the minimal elements for imprinting determined by transgenic studies (Shemer et al. 2000) and epigenetic activity in Drosophila Melanogaster (Lyko et al. 1998). Furthermore, the close proximity and/or orientation of the AS-IC to the PWS-IC has shown to be essential for the establishment of a maternal imprint (Buiting et al. 2001). These findings support an important role of the IC region in setting up and maintaining imprinting in both paternal and maternal alleles. Furthermore, the important influence of the late oocyte and zygote cytoplasm on the setting and maintenance of imprints also seems essential (Reik & Walter 2001).
Recently, it has been demonstrated that PWS-IC shows parent-specific complementary methylation patterns of histone H3 lysine 9 (Lys9) and H3 lysine 4 (Lys4) with either a maternal or a paternal copy methylated, respectively, and it has been suggested that H3 Lys9 methylation would be a candidate maternal gametic imprint for this region (Xin et al. 2001). Furthermore, during spermatogenesis, histones are removed from chromatin and replaced by protamines (Braun et al. 2001), providing a mechanism of erasing the chemically stable histone-methylation imprint from the maternally inherited chromosome 15 in the male germ line (Xin et al. 2001) (Fig.3). The maintenance of a histone modification imprint after fertilization throughout the life span has been suggested to be somatically propagated by inheritance of a silenced chromatin marked with methyl Lys9 H3 during chromosome replication, by association of methyl Lys9 H3 and the heterochromatin protein HP1 and by association of HP1 and the mammalian H3 Lys9 methyltransferase SUVAR39H1. This protein complex might allow H3 methyltransferase associated with an unreplicated chromosome region to methylate histones associated with newly synthesized DNA at a nearby replication fork (Bannister et al. 2001, Xin et al. 2001).
Of the several different theories proposed to explain the evolution of genetic imprinting, the genetic conflict model has been considered the most likely. According to this theory imprinting has evolved because of the conflicting interests of maternal and paternal genes in relation to the transfer of nutrients from the mother to her offspring (Moore & Haig 1991). This parent-offspring ‘conflict’ causes the enhancers of prenatal and postnatal growth to be of paternal origin, while growth suppressors are of maternal origin (Moore & Haig 1991). Based on this theory Nicholls et al. (1999) have proposed that the selection for imprinting in 15q11-q13 has probably resulted in a postnatal growth advantage in individuals with a paternally derived gene, given the failure-to-thrive phenotype of neonates with Prader-Willi syndrome (Cassidy 1997a) and mouse model pups (Cattanach et al. 1992, Yang et al. 1998, Gabriel et al. 1999). They suggested that the selection pressure might have been operating on the SNURF exons only, and that most genes within the imprinted 15q11-q13 domain might display imprinting simply as a consequence of recent evolutionary acquisition by the domain or as a consequence of the spreading effect of the imprinting mechanism (Nicholls 2000).
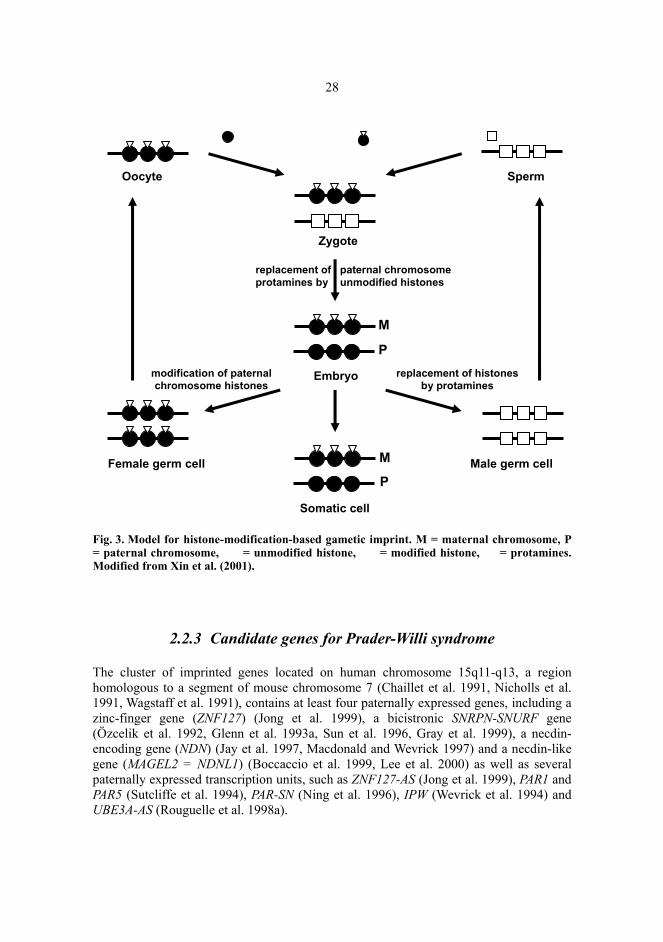
28
Fig. 3. Model for histone-modification-based gametic imprint. M = maternal chromosome, P = paternal chromosome, = unmodified histone, = modified histone, = protamines. Modified from Xin et al. (2001).
2.2.3 Candidate genes for Prader-Willi syndrome
The cluster of imprinted genes located on human chromosome 15q11-q13, a region homologous to a segment of mouse chromosome 7 (Chaillet et al. 1991, Nicholls et al. 1991, Wagstaff et al. 1991), contains at least four paternally expressed genes, including a zinc-finger gene (ZNF127) (Jong et al. 1999), a bicistronic SNRPN-SNURF gene (Özcelik et al. 1992, Glenn et al. 1993a, Sun et al. 1996, Gray et al. 1999), a necdin-encoding gene (NDN) (Jay et al. 1997, Macdonald and Wevrick 1997) and a necdin-like gene (MAGEL2 = NDNL1) (Boccaccio et al. 1999, Lee et al. 2000) as well as several paternally expressed transcription units, such as ZNF127-AS (Jong et al. 1999), PAR1 and PAR5 (Sutcliffe et al. 1994), PAR-SN (Ning et al. 1996), IPW (Wevrick et al. 1994) and UBE3A-AS (Rouguelle et al. 1998a).
Oocyte
Zygote
Embryo
Somatic cell
Female germ cell
replacement of paternal chromosome protamines by unmodified histones
modification of paternal chromosome histones
replacement of histones by protamines
M
M
P
P
Sperm
Male germ cell

29
Although numerous paternally expressed genes and transcripts that reside in the candidate region have been identified, their individual contributions to the development of PWS have not been established. The only identified protein products are those for the SNURF-SNRPN, NDN, MAGEL2 and ZNF127 genes, of which SNURF-SNRPN is the best candidate and likely to cause some of the features of Prader-Willi syndrome.
SNURF-SNRPN is a complex locus containing at least 148 exons on human chromosome 15 (Runte et al. 2001). The first ten exons are transcribed into a 1.4-kb bicistronic mRNA (Gray et al. 1999, Runte et al. 2001). The exons 1-3 contain SNURF (SNRPN upstream open reading frame) (Gray et al. 1999), which encodes a highly basic 71-amino acid protein that is nuclear-localized. The SNRPN gene spanning the exons 4-10 encodes the small nuclear ribonucleoprotein polypeptide N (SmN), which is located in the spliceosome involved in mRNA splicing (Gray et al. 1999). Furthermore, there are at least two alternative 5’ upstream start sites (u1B and u1A) and several upstream exons, which are spliced onto the exon 2 of the SNURF-SNRPN gene (Dittrich et al. 1996a, Färber et al. 1999), and at least 138 additional downstream exons (Buiting et al. 1996, Lee and Wevrick 2000, Wirth et al. 2001, Runte et al. 2001) have been detected so far. Both SNURF and SNRPN are translated in normal human tissues, but not in patients with PWS (Glenn et al. 1993a, Reed and Leff 1994, Gray et al. 1999). The unique location of the SNURF-SNRPN gene in the imprinting centre, the SNURF exons being completely contained within the 4.3 kb PWS-IC, implies that this genetic locus is ultimately associated with the cis regulation of all imprinted genes in a 2-Mb domain within the region 15q11-q13. Based on this and the identification of two typical PWS patients with balanced translocation interrupting SNURF-SNRPN not affecting the expression of other maternally imprinted genes in 15q11-q13, it has been suggested that the loss of SNURF and/or SNRPN function might lead to the observed neonatal failure-to-thrive phenotype (Nicholls et al. 1999). However, the SNRPN-SNURF translation unit has also been shown to serve as the start site for the UBE3A antisense transcript and to be a host for multiple snoRNA genes (Runte et al. 2001), and it has been recently suggested that a loss of expression of the snoRNAs (PWCR1/HBII-85 cluster and the HBII-43A) in the proposed ~121 kb minimal region for PWS located within the SNRPN-SNURF locus might be responsible for much or all of the phenotype of PWS (Gallagher et al. 2002).
The other genes assumed to be involved in the PWS phenotype are the paternally expressed ZNF127, NDN and MAGEL2 genes, which map to PWCR proximal to SNURF-SNRPN (Jay et al. 1997, MacDonald & Wervick 1997, Nakada et al. 1998, Jong et al. 1999, Boccaccio et al. 1999, Lee et al. 2000). The ZNF127 gene encodes a protein with a RING (C3HC4) zinc-finger and multiple C3H zinc-finger motifs, and it is predicted function as a ribonucleoprotein (Jong et al. 1999). As the DNA methylation imprint of ZNF127 is complete only in brain and germ cells, it is suggested that these are critical tissues for ZNF127 function, and the ZNF127 gene may be responsible for the behavioural abnormalities and obesity or the hypogonadism and infertility in PWS.
The NDN and the MAGEL2 genes belong to the MAGE-NDN gene family, the NDN gene encoding a protein homologous to the mouse necdin protein (Jay et al. 1997, Macdonald & Wevrick 1997) and MAGEL2 gene encoding a putative protein of 500 amino acids homologous to the MAGE proteins and NDN (Boccaccio et al. 1999, Lee et al. 2000). The NDN gene is solely expressed in post-mitotic murine neurones, implicating involvement in the control of cell growth or neuronal differentiation, and in humans,

30
normal mRNA expression is highest in the hypothalamus (Macdonalds & Wevrick 1997, Sutcliffe et al. 1997a, Watrin et al. 1997). Based on these studies, it has been postulated that the loss of expression of the NDN gene in human brain may contribute to the hypothalamic defect and developmental delay that are characteristic of PWS (Jay et al. 1997). MAGEL2 is also expressed by the paternal allele in human brain, suggesting a potential role in the aetiology of PWS (Boccaccio et al. 1999).
Although almost all of the paternally expressed genes or transcripts have been assumed to be candidates for Prader-Willi syndrome, mice deficient in Zfp127 (see Jong et al. 1999), Ndn (Gérard et al. 1999, Tsai et al.1999a), Snurf (Tsai et al. 1999b), Snrpn (Yang et al. 1998) and Ipw (see Nicholls 1999) are phenotypically normal and do not show excess perinatal lethality associated with feeding difficulties equivalent to infants with PWS. An exception is the ambiguous data on the Ndn gene, probably due to the different mouse strains used in these studies (Nicholls et al. 1999). One of these mouse models of PWS suggests that paternal deficiency of the Snrpn-Ipw interval may be responsible for the severe phenotypic aspects of PWS (Tsai et al. 1999b). This interval (DR region) encodes multiple snoRNA genes (Cavaille et al. 2000, Meguro et al. 2001) and is part of the large SNURF-SNRPN locus (Runte et al. 2001).
Ambiguous data obtained with Ndn-deficient mice (Gérard et al. 1999, Tsai et al. 1999a) as well as recently recognized opposite methylation of the imprinting centre in human and mouse oozytes (Shemer et al. 2000, ElMaarri et al. 2001) demonstrate the difficulty of producing and interpreting any animal model of human disease and underscore the need for many more mice to be bred to generate appropriate animal models (Nicholls 1999). Furthermore, no behavioural studies have been performed on these mice. However, Prader-Willi syndrome mice with uniparental disomy (Cattanach et al. 1992) or a large 4 Mb deletion affecting the region of the mouse genome corresponding to human 15q11-q13 and deletion of Snurf-Snrpn (IC deletion mice) (Yang et al. 1998) all died at a few days of age due to a failure to thrive. Currently, PWS can, at best, be characterized as a contiguous gene syndrome involving multiple paternally expressed genes.
2.2.4 Candidate genes for Angelman syndrome
In contrast to Prader-Willi syndrome, only two maternally expressed genes, UBE3A (Rouguelle et al. 1997, Vu and Hoffman 1997) and ATP10C (Meguro et al. 2001b, Herzing et al. 2001), are located in the imprinted 15q11-q13 domain. Of these, mutations of UBE3A have been shown to cause Angelman syndrome (Kishino et al. 1997, Matsuura et al. 1997).
The E6AP ubiquitin protein ligase 3A gene (UBE3A) was identified in 1993 based on its ability to associate with the E6 oncoprotein of the human papillomavirus and to selectively degrade p53 (Scheffner et al. 1990, Huibregtse et al. 1991). Although the UBE3A gene was mapped to the AS chromosome region as early as 1994, preliminary studies in human lymphoblast and skin fibroblast cell lines (Nakao et al. 1994) as well as whole mouse brain and testis failed to yield evidence of imprinted expression (Sutcliffe et

31
al. 1997b). Soon after the discovery of UBE3A mutations (Kishino et al. 1997, Matsuura et al. 1997), however, the UBE3A gene was found to exhibit tissue-specific imprinting with maternal allele expression in hippocampal neurons, Purkinje cells and olfactory mitral cells in mice (Albrecht et al. 1997, Jiang et al. 1998a,b) and in human brain (Rouguelle et al. 1997, Vu and Hoffman 1997), providing conclusive evidence of the role of UBE3A in the pathogenesis of AS.
The UBE3A gene spans approximately 120 kb on genomic DNA, with transcription oriented from telomere to centromere (Fig. 4) (Yanamoto et al. 1997, Kishino & Wagstaff 1998). The gene consists of ten protein-coding exons (exons 7 to 16), six to nine non-coding exons in the 5’ untranslated region (UTR) (Yamamoto et al. 1997, Kishino et al. 1997, Rouguelle et al. 1997, Vu & Hoffman 1997, Kishino & Wagstaff 1998) and an additional 2.0 kb region of 3’UTR (Kishino & Wagstaff, 1998). The 5’-end of the gene displays alternative splicing and accounts for the production of nine adult and two fetal transcripts (Yamamoto et al. 1997, Kishino et al. 1997, Vu & Hoffman 1997, Kishino & Wagstaff 1998). These mRNA subtypes encode three protein isoforms, differing in the use of the initiation codon and, therefore, over their amino-terminal regions. It has been suggested that the different amino-termini of the UBE3A isoforms could function to generate different specificities within the same protein (Rouguelle et al. 1998a).
The product of the UBE3A gene is an E6-AP ubiquitin protein ligase (Scheffner et al. 1993), which contains two independent, separable functions: coactivation of the nuclear hormone receptor superfamily (Nawaz et al. 1999) and ubiquitin-ligase activity (Huitbregtse et al. 1993a,b). In the pathogenesis of Angelman syndrome, only a defect in the ubiquitin-proteosome protein degradation pathway seems to result in the AS phenotype (Nawaz et al. 1999).
Fig. 4. Schematic structure of the UBE3A gene. Exons 1 – 6 are untranslated exons, ATG = initiate codon.
The ubiquitin-proteasome system is an important regulatory mechanism, whereby proteins are marked for degradation by the attachment of multiubiquitin chains, which targets the selected proteins to the 26S proteosome for destruction (Glotzer et al. 1991, Schwob et al. 1994, Scheffner et al. 1995, Cohen-Fix et al. 1996, Rouguelle et al. 1998a, Hochstrasser 1996, Ciechanover & Schwartz 1998, Hershko & Ciehanover 1998, Vu & Sakamoto 2000). The cytoplasmic ATP-dependent ubiquitin-proteosome system has a broad range of substrates, such as cell cycle and division regulators, mitotic cyclins, cyclin-dependent kinase (CDK) inhibitors, ion channels, tumour suppressors, transcription factors and a myriad of other proteins (Hershko & Ciechanover 1998), which indicates its important role in cell cycle progression, apoptosis, immune response,
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
ATG

32
development, transcriptional regulation, signal transduction and receptor down-regulation (Vu & Sakamoto, 2000).
The E6-AP ubiquitin protein ligase is a member of the group of E3 enzymes, which are important in substrate recognition and ubiquitin transfer. This protein of approximately 120 kD has at least six functional domains, of which three are involved in ubiquitin-ligase activity: the E6-binding domain (amino acids 391 - 408), the p53-binding domain (aa 280-781) and the hect (homologous to the E6-AP carboxyl terminus) domain (carboxyl-terminal 350 amino acids). The highly conserved hect domain is encoded by the exons 9-16 (Huibregtse et al. 1995), and it is of functional importance as it contains the minimal region necessary for the ubiquitination and degradation of the target protein (the extreme carboxyl-terminal 88 amino acid segment) (Huitbregtse et al 1993b).
In addition to ubiquitination, E6-AP alone and/or in conjunction with a ubiquitin-conjugating enzyme defines the substrate specificity of ubiquitin transfer (Scheffner et al. 1995). Subsequently, in addition to p53, E6-AP has been shown to independently mediate the degradation of HHR23A (a protein homologous to the yeast DNA repair factor RAD23), MCM-7 (a protein implicated in chromosomal replication), Bak, BIK and E6-AP itself (Kumar et al. 1997, Vu & Sakamoto 2000), but the substrates might number up to hundreds.
The substrate essential to the pathogenesis of Angelman syndrome is not yet known. However, the lack of ubiquitination of target proteins in tissues where the paternal allele for UBE3A is silenced could lead to a failure to degrade these proteins or to other functional alterations of target proteins (Hochstrasser 1996), leading to the phenotype. Recent studies of a mouse model for maternal UBE3A deficiency (Ube3a knockout mice) demonstrated motor dysfunction, inducible seizures and a defect in contextual learning and hippocampal long-term potentiation (LTP). LTP is generally considered the most likely candidate cellular mechanism for learning and memory, especially in the limbic system (Nayak & Browning 1999, Stevens 1998). Furthermore, the elevated cytoplasmic levels of p53 in Purkinje cells and some hippocampal neurons in Ube3a knockout mice (Jiang et al. 1998b) as well as in a deceased AS patient (Jay et al. 1991, Jiang et al. 1998b) suggest that the expression of UBE3A is imprinted in this cell type also in humans. Based on these studies, the maternal deficiency of UBE3A in Purkinje cells is suggested to account for the ataxia and tremor seen in patients with AS, and the deficiency in hippocampal neurons is thought to explain the learning deficits and epilepsy (Jiang et al. 1998b, 1999). The candidate proteins in Purkinje cells and proteins implicated in LTP in hippocampus can be evaluated as potential targets for ubiquitination by E6-AP.
Recently, an other maternally expressed gene, ATP10C, has been mapped telomeric to the UBE3A locus within the chromosome region 15q11-q13 (Meguro et al. 2001b, Herzing et al. 2001). This gene encodes a novel member of a subfamily of P-type ATPase with a well-defined phosphorylation domain (DKTGT(L/1)T. The expression of ATP10C has been shown to be absent in AS patients with a maternal deletion of 15q11-q13 or an imprinting defect, and it has been proposed that this gene might be involved in the varied manifestations of Angelman syndrome (Meguro et al. 2001b, Herzing et al. 2001). However, to determine whether mutations or a loss of function of this gene could explain a substantial number of AS patients with no identifiable molecular defect will remain to be resolved.

33
A 3.5 kb sense transcript whose promoter is embedded in the 3’-UTR of the UBE3A gene has also previously been detected in the AS critical region, and it has been suggested that mutations in this candidate transcript could account, at least in part, for the patients without mutations in UBE3A (Roguelle et al. 1998a). In addition, an antisense transcript that spans at least the 3’-half of UBE3A and the region downstream of the gene has also been identified (Roguelle et al. 1998a). The antisense transcript is expressed exclusively by the paternal allele in brain, but is transcriptionally silent in other tissues where UBE3A and the sense transcript display biallelic expression, and this transcript has been proposed to control tissue-specific imprinting of the UBE3A gene by excluding the paternal allele-specific UBE3A expression in brain, as discussed earlier (Roguelle et al. 1998a,b, Brannan & Bartolomei 1999, Shemer et al. 2000).
2.3 Known genetic defects causing Prader-Willi or Angelman syndromes
Prader-Willi syndrome and Angelman syndrome are, according to our current knowledge, due to a loss of function of imprinted genes in the chromosome region 15q11-q13. In the case of PWS, the absent contribution to this region is always paternal, leading to a loss of expression of paternally transcribed genes. In AS, the maternal contribution of 15q11-q13 is missing, causing the syndrome via a lack of expression of maternally transcribed gene(s). In both syndromes, the loss of gene function may be due to three shared genetic defects: microdeletion, uniparental disomy (UPD) and an imprinting defect, whereas Angelman syndrome has also been described to arise from a mutation in a single gene, UBE3A (Fig. 5).
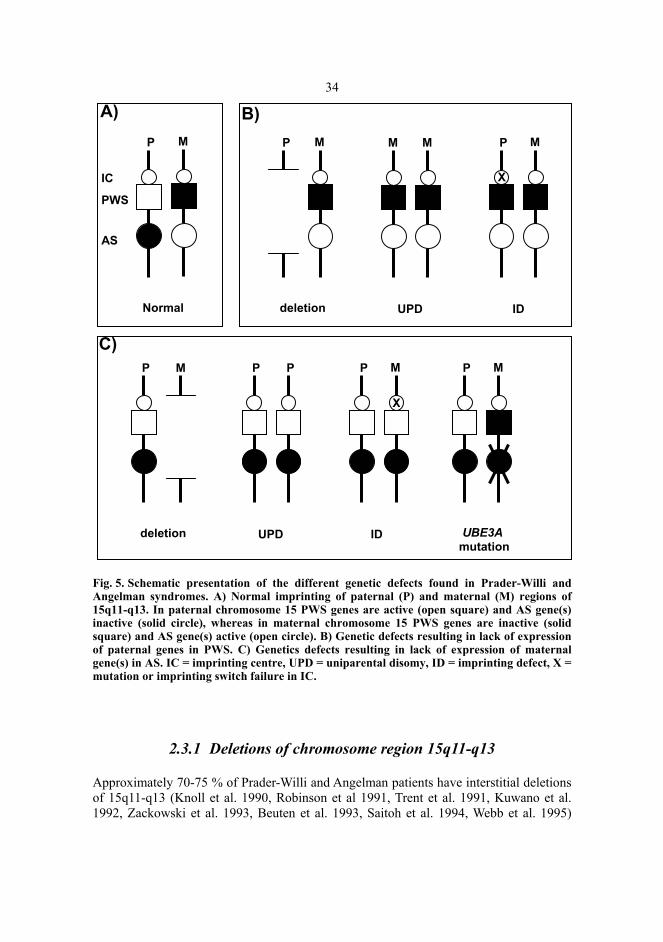
34
Fig. 5. Schematic presentation of the different genetic defects found in Prader-Willi and Angelman syndromes. A) Normal imprinting of paternal (P) and maternal (M) regions of 15q11-q13. In paternal chromosome 15 PWS genes are active (open square) and AS gene(s) inactive (solid circle), whereas in maternal chromosome 15 PWS genes are inactive (solid square) and AS gene(s) active (open circle). B) Genetic defects resulting in lack of expression of paternal genes in PWS. C) Genetics defects resulting in lack of expression of maternal gene(s) in AS. IC = imprinting centre, UPD = uniparental disomy, ID = imprinting defect, X = mutation or imprinting switch failure in IC.
2.3.1 Deletions of chromosome region 15q11-q13
Approximately 70-75 % of Prader-Willi and Angelman patients have interstitial deletions of 15q11-q13 (Knoll et al. 1990, Robinson et al 1991, Trent et al. 1991, Kuwano et al. 1992, Zackowski et al. 1993, Beuten et al. 1993, Saitoh et al. 1994, Webb et al. 1995)
P M M M M P
X
Normal deletion UPD ID
AS
PWS
IC
A) B)
P P P P M MM P
UBE3A mutation
IDUPDdeletion
C)
X
P M

35
(Fig. 1, p. 12). Although there are some exceptions, the vast majority (≥ 95%) of both paternal (PWS) and maternal (AS) deletions are remarkably homogeneous in size (∼ 4 Mb), with two alternative proximal and one distal breakpoint region (Christian et al. 1995, Amos-Landgraf et al. 1999). The proximal deletion breakpoint commonly lies either between D15S18 and the centromere or between the D15S18 and D15S9 loci. The distal breakpoint has been mapped between the D15D12 and D15S24 loci (Knoll et al. 1990, Robinson et al. 1991, Kuwano et al. 1992, Chan et al. 1993, Robinson et al. 1993b, Christian et al. 1995, Robinson et al.1998, Amos-Landgraft et al. 1999) (Fig. 6).
Fig. 6. Genetic map of human chromosome region 15q11-q13. Common deletion breakpoints (BP1-BP3) are shown by zigzag lines. IC = imprinting centre.
The clustered breakpoints detected in the Prader-Willi and Angelman syndromes suggest that the deletions originate from unequal crossover between repeated DNA units (Amos-Landgraf et al. 1999, Christiansen et al. 1999). Low-copy repeats have been identified in the genomic regions implicated in the DiGeorge/velo-cardiofacial syndromes (del(22)(q11)) (Halford et al. 1993), Williams syndrome (del(7)(q11.2)) (Perez et al. 1996) and Smith-Magenis syndrome (del(17)(p11.2)) (Chen et al. 1997), and homologous recombination of flanking repeat gene clusters have been expected to be the mechanism of common contiguous gene deletion syndromes (Chen et al. 1997, Shaffer et al. 2000).
D15S18
D15S542
IC
BP1
BP2
ZNF127
D15S11
D15S13
PW71 (D15S63) SNURF-SNRPN D15S128
D15S122UBE3A
D15S10
D15S113
D15S97GABRB3
GABRA5
GABRG3
D15S156P (D15S12)
HERC2
D15S24
t l
BP3
PWS
AS
non-
imprinted
q11.2 q13
Chr 15
ATP10C

36
Previously, duplicated sequences (i.e. END repeats) at the chromosome 15q11 and 15q13 were detected (Buiting et al. 1999b, Ji et al. 1999, Amos-Landgraft et al. 1999, Christian et al. 1999), and the common deletion sizes observed in PWS and AS were also thought to be generated by unequal crossing-over between misaligned proximal and distal END repeat homologues. Such interchromosomal misalignment and crossing-over within repeats during meiosis is expected to result in the formation of reciprocal duplication/deletion products, as is the case in, for example, Charcot-Marie-Tooth syndrome type 1A (CMT1A, dup(17)(p12)) and hereditary neuropathy with liability to pressure palsies (HNPP, del(17)(p12)) (Pentao et al. 1992, Lupski 1998). The rarity of the reported cases of interstitial duplications 15 (Pettigrew et al. 1987, Clayton-Smith et al. 1993a, Repetto et al. 1998, Browne et al. 1997, Robinson et al. 1998a, Thomas et al. 1999) suggests that an intrachromosomal mechanism may also be a common cause of deletions of 15q11-q13, although the rarity of duplication cases might also indicate that a milder phenotype causes them to be ascertained much less often. The involvement of an intrachromosomal mechanism as a cause of deletion was confirmed by two studies, where both intra- and interchromosomal 15q11-q13 exchanges were shown to occur between the sister chromatids during paternal meiosis in PWS (Carrozzo et al. 1997, Robinson et al. 1998a). In intrachromosomal events, the deletion probably arises due to the formation of an intrachromosomal foldback or a stem-loop intermediate structure between inverted duplicons, with exclusion or deletion of the intervening loop (Sachs et al. 1995, Christian et al. 1999, Amos-Landgraft et al. 1999). In AS, only interchromosomal deletion events have been observed so far (Robinson et al. 1998a), although the absence of intrachromosomal changes is probably due to the small sample size analyzed (n=3) up till now.
Despite the common deletion sizes in Prader-Willi syndrome and Angelman syndrome, the critical regions for these disorders are separate, being more telomeric in AS. The first evidence of distinct regions in these disorders was demonstrated in a Japanese family, where transmission of a small deletion to a woman from the father resulted in a normal phenotype, while transmission of this deletion to her children resulted in AS (Hamabe et al. 1991b, Saitoh et al. 1992). Since PWS was not the outcome of a paternal deletion, the PWS and AS gene(s) were concluded to be located at separate loci. In addition, this family provided strong support for genomic imprinting, since the deletion exerted a phenotypic effect when inherited maternally, but was harmless when inherited through the male germ line (Nicholls 1993). Detailed mapping using rare familial translocations and other rearrangements (Saitoh et al. 1992, Buiting et al. 1993, Reis et al. 1993, Greger et al. 1994, Nelen et al. 1994, Michaelis et al. 1995) has revealed the smallest region of deletion overlap (SRO) for PWS to be approximately 100-200 kb in size and to include the SNRPN, PAR-5 and PAR-7 genes (Butler et al. 1996), while the SRO for AS deletions lies between the D15S122 and D15S113 loci, being roughly 300 kb in size (Greger et al. 1994). This interval includes the UBE3A and ATP10C genes (Fig. 6).
De novo deletions of 15q11-q13 are quite common. Given the incidence of Prader-Willi and Angelman syndromes (1/10 000 – 1/ 15 000) and the frequency of deletions in both syndromes (75%), it can be suggested that a 15q11-q13 deletion occurs in about 1/8000 liveborns and is thus a relatively frequent cause of genetic rearrangements in birth defects. The HERC2 sequence just telomeric to the distal breakpoint of the PWS and AS

37
deletions and some related sequences that comprise END repeats have been shown to include a CpG island promoter (Ji et al. 1999, Amos-Landgraft et al. 1999) and to be transcribed at high levels in male and female germ cells, being probably one factor that enhances the likehood of 15q11-q13 recombination and facilitates misalignment in germ line tissues (Amos-Landgraft et al. 1999). Environmental factors have also been suggested to have caused deletions in the paternally derived chromosome. Strakowski and Butler (1987) found that an increased incidence (21 %) of fathers of children with PWS were employed in occupations where hydrocarbons had been used at the time of conception compared with the fathers of children with the Down or fragile X syndromes (12%), and Cassidy et al. (1989) found that approximately half of the fathers had been exposed to hydrocarbons around conception. In AS, no environmental factors have been reported.
2.3.2 Uniparental disomy of chromosome 15
Uniparental disomy (UPD) arises when a child receives both copies of a chromosome (or only a small segment of chromosome) from one of the parents and none from the other parent (Engel 1996, 1997). The frequency of UPD has been estimated to be 2.8/10 000 conceptions (Engel 1980), and it has been reported for all chromosomes except for chromosomes 12, 18 and 19 (Hurst & McVean 1997, Kotzot 1999). Uniparental disomy may occur both as heterodisomy, where sequences from both homologues from the transmitting parent are present, and as isodisomy, in which two genetically identical segments from one parental homologue are present. The origin of UPD depends primarily of non-disjunction (ND) events that may occur in either meiosis I (MI) or meiosis II (MII) segregation. Uniparental disomy may arise through multiple mechanisms, including: 1) trisomic zygote rescue (fertilization of a disomic egg with normal sperm, resulting in a trisomic foetus with a subsequent loss of the paternal chromosome (or vice versa)), 2) gamete complementation (fertilization of a disomic egg with sperm nullisomic for the same chromosome (or vice versa)), 3) compensatory UPD (a somatic event leading to the replacement of an abnormal or absent chromosome with the normal homologue by post-zygotic duplication) and 4) mitotic errors (mitotic non-disjunction and duplication leading to isodisomy or somatic recombination) (Spence et al. 1988, Cassidy et al. 1992, Engel et al 1993, Engel 1996) (Fig. 7). Uniparental disomy may lead to either normal or abnormal development. In man, it may result in clinical conditions due to either homozygosity for recessive mutations (isodisomy) or the presence of imprinted genes in the chromosome involved (iso- or heterodisomy) (Woodage et al. 1994a, Ledbetter & Engel 1995, Robinson et al. 2000), as is the case in Prader-Willi syndrome and Angelman syndrome.
The first report of maternal uniparental disomy in Prader-Willi syndrome was published in 1989 (Nicholls et al. 1989b). Maternal uniparental disomy (i.e. both chromosomes 15 inherited only from the mother) has now been shown to occur in approximately 25% of Prader-Willi patients (Robinson et al. 1991, Mascardi et al. 1992, Woodage et al. 1994b). The inheritance of both chromosomes 15 from the mother only

38
leads to an absence of expression of paternal genes at 15q11-q13, which leads to the phenotype. Most PWS cases with UPD show maternal heterodisomy for chromosome 15 and are mainly thought to result from postzygotic correction of a trisomic embryo (Robinson et al. 1991, Mascardi et al. 1992, Mutirangura et al. 1993b, Robinson et al. 1993a, Robinson et al. 1998a, Robinson et al. 2000, Fridman et al. 2000a). The loss of one of the three chromosomes from a trisomic cell would be expected to result in maternal UPD in 1/3 of cases. In the trisomy rescue scenario, there is also a possibility that, instead of a complete loss of the extra chromosome, a remnant of the chromosome will persist in the derivative cell. This has been described in some Prader-Willi or Angelman patients with UPD15 and a small additional chromosome derived from chromosome 15 (inv dup (15)) (Robinson et al. 1993c).
Fig. 7. Different mechanisms leading to uniparental disomy (UPD). Adapted from Spence et al. (1988).
The first report of maternal uniparental disomy in Prader-Willi syndrome was published in 1989 (Nicholls et al. 1989b). Maternal uniparental disomy (i.e. both chromosomes 15 inherited only from the mother) has now been shown to occur in approximately 25% of Prader-Willi patients (Robinson et al. 1991, Mascardi et al. 1992, Woodage et al. 1994b). The inheritance of both chromosomes 15 from the mother only leads to an absence of expression of paternal genes at 15q11-q13, which leads to the phenotype. Most PWS cases with UPD show maternal heterodisomy for chromosome 15 and are mainly thought to result from postzygotic correction of a trisomic embryo (Robinson et al. 1991, Mascardi et al. 1992, Mutirangura et al. 1993b, Robinson et al. 1993a, Robinson et al. 1998a, Robinson et al. 2000, Fridman et al. 2000a). The loss of one of the three chromosomes from a trisomic cell would be expected to result in maternal UPD in 1/3 of
NORMAL TRISOMIC ZYGOTE RESCUE
GAMETE COMPLEMENTATION
COMPENSATORY UPD
MITOTIC ERROR
disomy
non-disjunction
non-disjunction duplication
duplication
UPD UPD UPD UPD partial UPD
SOMATIC
TISSUE
ZYGOTE
GAMETES
trisomy UPD
disomy
monosomy
mitotic recombination
or gene conversion

39
cases. In the trisomy rescue scenario, there is also a possibility that, instead of a complete loss of the extra chromosome, a remnant of the chromosome will persist in the derivative cell. This has been described in some Prader-Willi or Angelman patients with UPD15 and a small additional chromosome derived from chromosome 15 (inv dup (15)) (Robinson et al. 1993c).
In paternal uniparental disomy, which accounts for 2-5% of the Angelman patients (Malcolm et al. 1991, Knoll et al. 1991), both chromosomes 15 are derived only from the father, leading to the absence of maternal gene expression of 15q11-q13. In Angelman syndrome, the primary mechanism producing paternal UPD is post-zygotic mitotic duplication of paternal chromosome 15 (monosomy rescue) (Robinson et al. 1993a, Bottani et al. 1994, Chan et al. 1993, Prasad & Wagstaff 1997, Harpey et al. 1998, Robinson et al. 2000, Fridman et al. 2000a,b).
The maternal heterodisomy detected in Prader-Willi syndrome is due to a maternal non-disjunction event in meiosis I, and, as in trisomy 21 (Antonarakis et al. 1992), the probability of maternal UPD has been shown to increase exponentially with maternal age (Robinson et al. 1991, Mascardi et al. 1992, Robinson et al. 1993d, Mitchell et al. 1996, Robinson et al. 1996, Ginsburg et al. 2000, Robinson et al. 1998b, Fridman et al. 2000a). In Angelman syndrome, too, postzygotic errors have been associated with increased maternal age, since the primary event is the maternal meiotic non-disjunction leading to a nullisomic egg fertilized by normal sperm and, later, duplication of the paternal chromosome in the zygote (Robinson et al. 2000, Fridman et al. 2000a). The age-specific risk for UPD15 in women aged 40 or older has been calculated to be approximately 1/3400 livebirths (Robinson et al. 1996).
Other syndromes where disruption of the normal imprinting pattern due to uniparental disomy has been described are, transient neonatal diabetes mellitus (paternal UPD 6) (James et al. 1995), Silver-Russel syndrome (maternal UPD 7) (Kotzot et al. 1995), Beckwith-Wiedemann syndrome (paternal UPD 11p15.5) (Henry et al. 1993) and specific conditions due to maternal UPD 2, both maternal and paternal UPD 14, maternal UPD 16 (see Kotzot 1999), maternal UPD 20 (Salafsky et al. 2001) and UPD X (Yorifuji et al. 1998). On the other hand, maternal UPD 10, maternal and paternal UPD 21 and maternal and paternal UPD 22 are associated with a normal phenotype (see Kotzot 1999). Uniparental disomy may also occur within a portion of cells in all individuals simply as a consequence of somatic recombination occurring during mitotic cell divisions. This may be an important step in cancer development (see Tyco et al. 1999) and a factor contributing to other late-onset diseases (Aviv &Aviv 1998).
2.3.3 Imprinting defects of chromosome region 15q11-q13
In approximately 2-4% of Prader-Willi and Angelman patients, the loss of function of imprinted genes in proximal 15q is caused by an imprinting defect (ID) (Buiting et al. 2003). These patients have mutations in the 15q11-q13 region, which affect genomic imprinting in such a way that the patients with PWS have a maternal imprint on the paternal chromosome and fail to express paternally derived genes. Correspondingly, the

40
AS gene(s) appear(s) to be silenced by a paternal imprint on the maternal chromosome (Nicholls et al 1998, Mann & Bartolomei 1999, Falls et al. 1999, Khan & Wood 1999, Muir 2000). Imprinting defects can be divided into two molecular classes: those arising from deletions in the imprinting control centre (IC) (Sutcliffe et al. 1994, Buiting et al. 1995, Saitoh et al. 1996, Schuffenhauer et al. 1996, Teshima et al. 1996, Ohta et al. 1999, Buiting et al.1999a, 2000, Bielinska et al. 2000, McEntagart et al. 2000, Ming et al. 2000) and those without an apparent deletion in the imprinting centre (Bürger et al. 1997, Schulze et al. 1997, Ohta et al. 1999).
Microdeletions in the imprinting centre (IC), varying in size from 5 to 200 kb, have been found in 15-20% of the patients with imprinting defects (Ohta et al. 1999, Buiting et al. 1999a, 2000). In Prader-Willi syndrome, deletions encompassing a 4.3 kb PWS-IC of the bipartite IC on the paternal chromosome 15 prevent the establishment or maintenance of the paternal imprint, resulting in a paternal chromosome carrying a maternal imprint (Sutcliffe et al. 1994, Buiting et al. 1995, Schuffenhauer et al. 1996, Teshima et al. 1996, Ohta et al. 1999a, Bielinska et al. 2000, Buiting et al. 2000, McEntagart et al. 2000, Ming et al. 2000). In Angelman syndrome, deletions affecting the 880 bp AS-IC approximately 35 kb upstream of the PWS-IC on the maternal chromosome 15 prevent the establishment of a maternal imprint, thus resulting in the expression of paternal genes on the maternally derived chromosome 15 (Buiting et al. 1995, Dittrich et al.1996, Saitoh et al. 1996, Buiting et al. 1999a). Most of the imprinting deletions have been familial, with more than one affected child (Sutcliffe et al. 1994, Buiting et al. 1995, Saitoh et al. 1996, Teshima et al. 1996, Ohta et al. 1999, Buiting et al. 1999a, 2000, McEntagart et al. 2000, Ming et al. 2000).
Other cases that have been identified as imprinting defects have not been associated with detectable deletions in the imprinting centre (Bürger et al. 1997, Schulze et al. 1997, Ohta et al. 1999). Familial recurrence has not been reported for this group of patients. It has been suggested that, in these cases, a de novo or random failure to switch the opposite sex imprint to a sex-specific imprint during parental gametogenesis and/or fertilization could lead to the syndrome (Bürger et al. 1997).
In Angelman syndrome, an exceptional imprinting defect due to a maternal paracentric inversion has also been described (Buiting et al. 2001). This inversion was shown to disrupt the imprinting centre by removing AS-IC from PWS-IC in the centre of the imprinted 15q11-q13 domain close to its proximal border, preventing the establishment of a maternal imprint (Buiting et al. 2001).
2.3.4 UBE3A gene mutations
Approximately 5% - 10% of Angelman patients have been found to have a mutation in the UBE3A gene (Malzac et al. 1998, Fang et al. 1999, Baumer et al. 1999, Russo et al. 2000, Lossie et al. 2000). So far, over 40 different disease-causing mutations in the coding region of the UBE3A gene have been reported (Kishino et al. 1997, Matsuura et al. 1997, Malzac et al. 1998, Tsai et al. 1998, Fung et al. 1998, Fang et al. 1999, Baumer et al. 1999, Ouweland van der et al. 1999, Russo et al. 2000, Lossie et al. 2001). The vast majority of UBE3A mutations are protein-truncating mutations (84%, 30 frameshift and 7

41
nonsense). Missense mutations comprise 9% of the mutations (n=4), and 7% produce a protein product with a changed number of amino acids (n=3). The protein-truncating mutations probably contribute to the phenotype both by reducing UBE3A mRNA concentrations and by causing any translation products to be catalytically inactive because of the absence of an important extreme C terminus of the E6-AP protein necessary for catalytic function (Huibregtse et al. 1995, Malzac et al. 1998). Most of the AS-associated missense and single amino acid insertion or deletion mutations also affect the functionally important hect domain, and a majority of these mutations have been shown to map to the broad catalytic cleft located at the junction of 2 hect domain lobes, interfering with the ubiquitin-thioester bond formation (Huang et al. 1999).
Most of the reported UBE3A mutations cluster within exon 9, which covers more than 50% of the coding region (1247 bp) (Kishino & Wagstaff 1998), or within the much smaller exon 16 (121 bp) of the UBE3A gene (Kishino et al.1997, Matsuura et al. 1997, Malzac et al. 1998, Tsai et al. 1998, Fung et al. 1998, Fang et al. 1999, Baumer et al. 1999, Ouweland van der et al. 1999, Russo et al. 2000, Lossie et al. 2001). In contrast, no mutations have been detected so far in the non-coding exons or the exons 13 and 14. Furthermore, mutations have been identified much more often in familial (75%-80%) than in sporadic (14%-23%) cases, probably due to the more accurate diagnosis of Angelman syndrome in familial cases (Malzac et al. 1998, Fang et al. 1999).
2.3.5 Other structural chromosomal rearrangements involving chromosome 15
Structural abnormalities of the chromosome region 15q11-q13, such as inversions and translocations, account for less than 4% of Prader-Willi syndrome cases and less than 1% of Angelman syndrome cases (Khan & Wood 1999). Most of these rearrangements have been associated with a deletion of 15q11-q13 (Duckett et al. 1984, Elder et al. 1985, Fernandez et al. 1987, Ledbetter et al. 1987, Butler 1990, Cuoco et a.1990, Hulten et al. 1992, Smeets et al. 1992, Qumsiyeh et al. 1992, Webb et al. 1992, Chan et al. 1993, Reeve et al. 1993, Vickers et al. 1994, Jauch et al. 1995, Spinner et al. 1995, Suzuki et al. 1996, Burke et al. 1996, Wenger et al. 1997, Eliez et al. 1997, Bettio et al. 1997, Krajewska et al. 1998), uniparental disomy (Nicholls et al 1989b, Smeets et al. 1992, Robinson et al. 1993a, 1994, Freeman et al. 1993, Smith et al. 1993, Smith et al. 1994, Toth-Fejel et al. 1996, Smith et al. 1997a, Bettio et al. 1997, Park et al. 1998) or an imprinting defect (Buiting et al. 2001), and only five translocations in PWS (Sun et al. 1996, Schulze et al. 1996, Conroy et al. 1997, Kuslich et al. 1999, Wirth et al. 2001) and one inversion in AS (Greger et al. 1997) without these changes have been described. In PWS four of these five balanced translocations were inherited from the father (Sun et al. 1996, Kuslich et al. 1999, Schulze et al. 1996, Conroy et al. 1997), while one was a de novo change (Wirth et al. 2001). In all cases, the translocation was located within the large polycistronic SNURF-SNRPN locus. Two of these patients had a typical PWS phenotype, and in both the translocation disturbed the SNURF portion of the SNURF-SNRPN transcript (t(15;19) and t(4;15)) (Sun et al. 1996, Kuslich et al. 1999). These patients transcribed multiple paternally expressed genes and transcripts within the 15q11-

42
q13 region, suggesting that these translocations might uniquely affect the SNURF-SNRPN gene, leading to PWS (Sun et al. 1996, Kuslich et al. 1999). Three patients with atypical PWS phenotypes had balanced translocations (t(9;15), t(2;15) and t(X;15)) with a breakpoint in a previously identified translocation breakpoint cluster between SNURF-SNRPN and IPW (Schulze et al. 1996, Conroy et al. 1997, Wirth et al. 2001). In contrast to typical translocation patients, these patients did not express IPW or PAR-1. The SNURF-IPW interval includes the DR region (Cavaille et al. 2000, Meguro et al. 2001a), which has been suggested to function as a structural regulatory element maintaining the paternal chromosome in a largely unmethylated state in somatic cells (Meguro et al. 2001a). While the translocation breakpoint cluster is located within this region (Wirth et al. 2001), these translocations might disturb the function of the DR region corresponding to some aspects of PWS.
In the case of Angelman syndrome, the maternally inherited paracentric inversion had one breakpoint within the UBE3A gene, inactivating the maternally active allele leading to the AS phenotype in this patient (Greger et al. 1997, Kishino et al. 1997).
2.4 Genotype-phenotype correlations
In Prader-Willi syndrome, patients with deletion and uniparental disomy have displayed subtle phenotypic differences in many cases (Lai et al. 1993, Gillessen-Kaesbach et al. 1995a, Mitchell et al. 1996, Saitoh et al. 1997, Cassidy et al. 1997c, Gunay et al. 1997, Rogan et al. 1998, Dykens and Cassidy 1999). Patients with UPD usually fulfil the criteria for classical PWS, but the manifestations have generally been reported to be somewhat milder than those of patients with deletions. In addition to minor physical differences, slightly, although significantly, higher IQ values have been observed among patients with maternal UPD compared to those with paternal deletion (Dykens &Cassidy 1999). Furthermore, two atypical patients with UPD were shown to express some of the chromosome 15 alleles that are ordinarily silent in typical PWS patients, suggesting that the milder phenotype in these patients might be due to the expression of some paternal genes as a consequence of relaxed imprinting (Rogan et al. 1998). The phenotype of the PWS cases with an imprinting defect could be predicted to be equivalent to the UPD cases. So far, clinical analyses of seven PWS patients with ID from three families have been published (Lubinsky et al. 1987, Östravick et al. 1992, Saitoh et al. 1997). These patients fulfilled the clinical criteria for PWS, but lacked hypopigmentation, which has been shown to be due to a deficiency of the non-imprinted P gene in both PWS and AS patients with deletion of 15q11-q13 (Gardner et al. 1992, Lee et al. 1994). The P gene is a tyrosine transporter gene important for the development of pigment of skin, hair and irides (Spritz et al. 1997). P protein deficiency also causes strabismus, which is often seen in patients with deletion (Wiesner et al. 1987, King et al. 1993).
Certain phenotypic differences have also been reported in Angelman patients with different genetic causes, although most of the patients with different defects have been considered typical (Bottani et al. 1994, Gillessen-Kaesbach et al. 1995, Bürger et al. 1996, Smith et al. 1996, 1997a, Moncla et al. 1999a,b). Several reports suggest more severe and frequent seizures, ataxia and lower cognitive function in AS patients with

43
maternal deletions (Williams et al. 1995b, Smith et al. 1996, Minassian et al. 1998, Lossie et al. 2001). It has been suggested that the more severe epilepsy and seizure phenotype seen in these cases might be due to a deletion of a cluster of GABAA receptor genes within the 15q11-q13 domain (DeLorey et al. 1998, Minassian et al. 1998). The 15q11-q13 deletion has also been suggested to lead a reduced number of GABRB3 receptors affecting brain development (Holopainen et al. 2001). The same chromosomal region is deleted on the paternal chromosome in Prader-Willi syndrome, and these patients do not have seizures or other AS features. This discrepancy could be easily explained by paternal imprinting. However, the status of the genomic imprinting of a cluster of GABAA receptor genes is uncertain, showing paternal imprinting with maternal gene expression in placenta (Kubota et al. 1994), maternal imprinting in rodent-human cell hybrids (Meguro et al. 1997) and biallelic expression in brain, the latter finding arguing against a role for these genes in the molecular pathogenesis of AS (Rougeulle & Lalande 1998b). Instead of imprinting, the possible role of anomalous GABAA neurotransmission in AS might involve a threshold effect with a concomitant reduction in GABRB3 gene expression accompanying a loss of maternally inherited UBE3A function (Nicholls 1998).
Angelman patients with paternal uniparental disomy or an imprinting defect have been less prone to show microcephaly and ataxia, and they also appear to have less severe or frequent seizures. Furthermore, they have greater mental facility with rudimentary communication, such as signing or gesturing (Bottani et al. 1994, Gillessen-Kaesbach et al. 1995a, Bürger et al. 1996, Smith et al. 1997a, 1998, Saitoh et al. 1997, Minassian et al. 1998, Giellessen-Kaesbach et al. 1999, Dupont et al. 1999, Fridman et al. 2000b, Lossie et al. 2001). The milder symptoms in UPD and ID patients have been suggested to be a consequence of the UBE3A gene showing incomplete imprinting in brain (Albrecht et al. 1997, Rougeulle et al. 1997, Vu & Hoffman 1997), thus resulting in a double dose of paternal allelic expression of this gene.
Milder Angelman syndrome phenotypes have also been associated with UBE3A mutations and with less severe ataxia, epilepsy and microcephaly in some cases compared to the phenotypes involving deletion (Minassian et al. 1998, Moncla et al. 1999a, 1999b). When the symptoms and different types of UBE3A mutation (i.e. truncating mutations and missense mutations affecting the hect domain of E6-AP ligase) were compared, no differences were observed in the phenotype, and it is still unclear whether missense mutations, which do not directly involve the hect domain, may lead to milder forms of the disorder, as was suggested in a patient with missense mutation (C21Y) in exon 8 (Matsuura et al. 1997).
2.5 Inheritance and risks of recurrence of Prader-Willi syndrome and Angelman syndrome
Most Prader-Willi and Angelman patients have been sporadic, and familial recurrence has been described only in families with structural chromosomal rearrangements, imprinting defects with a deletion in the imprinting centre or, in Angelman syndrome, UBE3A

44
mutations. In the case of IC and UBE3A mutations, the risk for an affected child may be increased even in distant relatives, since the mutation can theoretically be passed silently in the family by the mechanism of inheritance of imprinted diseases.
Based on an estimation derived from cumulative data of many large surveys of Prader-Willi and Angelman cases, the risk of recurrence in families where a proband has a common de novo deletion without chromosomal rearrangement is less than 1% (Horsthemke et al. 1996, Connerton-Moyer et al. 1997, Cassidy et al. 1997b). In contrast, in rare families with small deletions, the recurrence risk might be as high as 50% if the deletion encompassing only the Prader-Willi syndrome chromosome region (PWCR) is inherited from the father or if the mother is a carrier of the deletion encompassing only the Angelman syndrome chromosome region (ASCR) (Hamabe et al. 1991b, Saitoh et al 1992). Other patrilineal (PWCR deletions) and matrilineal (ASCR deletions) relatives may also be at risk to have affected children or grandchildren due to the imprinting inheritance mechanism.
If the deletion identified in a proband is due to unbalanced rearrangement, the risk to the family depends on whether the rearrangement is inherited or de novo (Horsthemke et al. 1996, Stalker & Williams 1998a), being less than 1% in the latter case. Instead, parental transmission of unbalanced rearrangement may lead to 15q11-q13 deletions due to abnormal meiotic segregation and will result in case-specific recurrence risks (Hulten et al. 1991, Smeets et al. 1992). Furthermore, parental translocation may predispose to the formation of de novo deletions with normal chromosomes due to unequal crossing-over between derivative and normal chromosomes 15, as was described in two Prader-Willi patients (Horsthemke et al. 1996).
Based on the lack of recurrence among all known cases of uniparental disomy for chromosome 15, the experience of UPD in other disorders and theoretical consideration regarding the mechanism of UPD, the risk of recurrence in families where the syndromes are due to uniparental disomy without translocation is less than 1% (Stalker & Williams 1998a). In families where the parent of the affected child carries a balanced translocation raising slightly the probability of occurrence of non-disjunction at meiosis, the recurrence risk is case-specific.
Families with an imprinting control centre (IC) deletion have a 50% recurrence risk if one of the parents (father in PWS, mother in AS) carries the deletion (Buiting et al. 1995, Ishikawa et al. 1996, Reis et al. 1994, Teshima et al. 1996, Saitoh et al. 1997, Ming et al. 2000). Other male (in PWS) or female (in AS) relatives who carry a known deletion in IC are also at 50% risk of having an affected child, whereas female (PWS) or male (AS) relatives who carry the mutation are at risk to have affected grandchildren through sons (PWS) and daughters (AS) who inherit the mutation (Fig 8). In contrast, all imprinting defects without IC deletion have been sporadic and thus probably represent a de novo defect in the imprinting process in 15q11-q13 (Bürger et al. 1997, Buiting et al. 2003). Therefore, the risk in such families is less than 1%.
UBE3A mutations, which are detected in approximately 10% of Angelman patients, may be inherited or represent new mutations (Matsuura et al. 1997, Kishino et al. 1997, Fung et al. 1998, Malzac et al. 1998, Tsai et al. 1998, Fang et al. 1999, Ouweland van der et al. 1999, Russo et al. 2000, Lossie et al. 2001). When the proband’s mother has a UBE3A mutation, the risk of recurrence is 50%. Female relatives of the mother who carry the same mutation also have a 50% risk of having a child with AS. Male relatives of the

45
mother who have the defect are not at risk of having affected children, but are at risk of having affected grandchildren through their daughters who have inherited the defect from them (Fig. 8).
Both somatic and gonadal mosaicism in parents may increase the risk of recurrence in families with patients as a result of IC deletion (PWS and AS) (Saitoh et al. 1996, Gilbert et al. 1997, Bielinska et al. 1999) or UBE3A gene mutations (AS) (Malzac et al. 1998). In the case of parental mosaicism, the recurrence risk depends on the ratio of mutant to normal germ cells (Stalker et al. 1998b). The theoretical risk of gonadal mosaicism for the deletion of 15q11-q13 has been estimated to be less than 0.5% (Gardner & Sutherland 1996), but should be taken into consideration in genetic counselling.
PWS AS
= healthy carries = affected
Fig. 8. Hypothetical pedigrees describing the inheritance of imprinting mutation in PWS, and imprinting or UBE3A mutation in AS. In PWS, the mutation is passed through female silently but causes PWS when inherited from the father. In AS, the mutation is passed silently through male carriers and causes AS only when inherited from the mother.
The majority of Angelman cases without identifiable molecular aberration have been sporadic, but there are some reports of families with more than one affected sibling, and the theoretical possibility of a 50% recurrence risk cannot be excluded (Stalker & Williams 1998a).
The recurrence risks in families with different genetic mechanisms are summarized in Table 3.

46
Table 3. Percentages of different genetic defects and the estimated recurrence risks in Prader-Willi (PWS) and Angelman (AS) syndromes.
Genetic defect Percentage Recurrence risk deletion of 15q11-q13 70-75% <1% unbalanced rearrangement of 15q11-q13 <1% increased (case-specific) uniparental disomy of chromosome 15 ~25% (PWS)
3-7% (AS) < 1%
UPD due to unbalanced rearrangement <1% increased (case-specific) mutation in imprinting centre 1-3% 50% if present in father (PWS) or
mother (AS) other imprinting defect 1-3% <1% UBE3A mutation ~10% (AS) 50% if present in mother no identifiable molecular abnormality ~10% (AS) theoretically up to 50%
2.6 Laboratory diagnostics of Prader-Willi and Angelman syndromes
Although the involvement of chromosome 15 in Prader-Willi patients with translocation became obvious during the late 1970’s (Hawkley & Smithies 1976, Zuffardi et al. 1978, Kucerova et al. 1979, Guanti 1980) after the introduction of chromosome banding (Dutrillaux & Lejeune 1971), the laboratory diagnosis of PWS first become possible when de novo deletions of 15q11-q13 were detected by high-resolution chromosome studies in patients with PWS (Ledbetter et al. 1981) and, few years later, also in Angelman patients (Magenis et al. 1987). In a significant number of patients, however, the genetic defect remained unresolved until the development of molecular techniques.
2.6.1 Cytogenetic detection of Prader-Willi and Angelman syndromes
2.6.1.1 Detection of deletions at 15q11-q13
Chromosome 15 is prone to deletions due to repeat sequences flanking the q11-q13 region. These microdeletions could usually be detected by high-resolution analysis of chromosome 15. This analysis is based on a chemical block at an earlier stage of the cell cycle, so that when cultures are subsequently released from the block, the cells proceed in synchrony into complete division. With knowledge of the cell cycle, careful timing allows harvesting of cultures with a high proportion of prophase, prometaphase or early metaphase cells (Yunis 1976).
The most commonly used chemical blocking agent has been methotrexate (MTX), which inhibits the synthesis of folic acid required for the production of thymidine, which, in turn, is required for DNA synthesis at the G1/S interface. The MTX block can be

47
released by washing and adding thymidine (Yunis 1976) or bromodeoxyuridine (BrdU) (Pai & Thomas 1980), which has the added advantage of mildly inhibiting chromosome condensation. Other chemical agents, such as Fluorodeoxyuridine (FUdR), have also been used to block the mitotic cycle and to synchronize the cells (Webber & Carson 1983).
Although the interstitial deletion of 15q11-q13 was usually detectable by high-resolution chromosome analysis, both false-negative and false-positive results have been obtained in more specific molecular and fluorescence in situ hybridization analyses (Robinson et al. 1991, Trent et al. 1991, Beuten et al. 1993, Chan et al. 1993, Delach et al. 1994, Saitoh et al. 1994a,b, Butler 1995, Petersen et al. 1995, Webb et al. 1995, White & Knoll 1995, Teshima et al. 1996, Erdel et al. 1996), and high-resolution chromosome analysis has been shown to be insufficient for deletion detection. However, cytogenetic methods are still useful in the identification of translocations predisposing to 15q11-q13 deletions or uniparental disomy.
Fluorescence in situ hybridization (FISH) has been shown to be a much more accurate method for the detection of 15q11-q13 deletions than conventional cytogenetic methods (Kuwano et al. 1992, Delach et al. 1994, Butler 1995, Smith et al. 1995, Teshima et al. 1996, Dewald et al. 1996). FISH is based on in situ hybridization of a nucleic acid probe into nucleic acids within cytological preparations. For the detection of microdeletions, biotin- and digoxigenin-labelled locus-specific probes within the common deletion boundaries (e.g., D15S11, SNRPN, D15S10, GABRB3) were mostly used in conjunction with secondary reagents (e.g., avidin or antidigoxigenin antibodies conjugated to fluorescein or rhodamine), displayed now by commercial directly fluorophorated probes. These probes have the advantage that they can be visualized immediately after the posthybridization washes. For the detection of deletion and the identification of cases where the deletion is due to translocation, two-colour FISH using a probe within the typical deletion boundaries and a centromeric probe has been shown to be optimal (see ASHG/ACMG report 1996).
2.6.1.2 Detection of uniparental disomy of chromosome 15
The detection of uniparental disomy from cytological preparations has been based on the observation that an imprinted domain in 15q11-q13 displays asynchronous replication timing between homologous loci in a higher percentage of cells than probes from non-imprinted regions, the paternally derived locus replicating earlier than the maternal one at D15S9, D15S63, SNRPN, D15S10 and GABRB3, and the maternal locus replicating earlier than the paternal one at GABRA5 (Kitsberg et al. 1993, Knoll et al. 1994, LaSalle & Lalande 1995). By using a technique for detecting the replication of specific genomic sites in interphase nuclei by FISH, single-sequence hybridization results in two discrete signals in G1 cells (unreplicated chromosomes) and two pairs of discrete signals in G2 cells (both homologues replicated), while asynchronous replication is visualized as a single hybridization signal (singlet) on one chromosome and a doublet on the other chromosome (G1-G2 cells). In this analysis, UPD patients are recognized based on their significantly lower population of asynchronously replicating cells (3% - 11%) compared

48
to patients with biparental inheritance (21% - 36%) (White et al. 1996). This test can be used on interphase cells from the same hybridized cytological preparation as that used for deletion detection. The test is, however, not commonly used for diagnostic purposes.
2.6.2 Molecular methods
2.6.2.1 Detection of deletions at 15q11-q13
The isolation of molecular probes specific for chromosome 15q11-q13 (Donlon et al. 1986) has enabled more accurate detection or exclusion of deletions in this region. For this purpose, quantitative Southern blot hybridization of HindIII-digested DNA with 15q11-q13 specific probes showing no polymorphism has been used (Latt et al. 1986, Tantravahi et al. 1989). The evaluation of the copy number in this area is done by simultaneous hybridization of a control probe detecting DNA sequences at some other chromosome than 15. The gene doses at 15q11-q13 are determined by comparing the ratios of the hybridization densities of the chromosome 15 probe to the control probe in densitometric analysis from at least two exposures (Tantravahi et al. 1989, Imaizumi et al. 1990, Hamabe et al. 1991a,b, Zackowski et al. 1993). In quantitative Southern blot analysis with the cDNA probe hN4HS containing sequences complementary to the SNRPN gene located at 15q12 and to the pseudogene SNRPN1 located at chromosome 6, the latter can be used as an internal control in the determination of the dosage of the chromosome 15 specific fragment (Özçelik et al. 1992).
Restriction fragment length polymorphisms (RFLP) have also been used in the detection of deletions (Nicholls et al. 1989a). In this analysis, the dosages of each allele at 15q11-q13 in a test sample are compared to the dosages of a normal control heterozygote. RFLP analysis has been shown to simplify the determination of copy number in the hemizygote state compared to an analysis in the absence of polymorphism. For the detection of deletions, a combination of RFLPs and a non-chromosome 15 control probe has also been used (Robinson et al. 1991).
In some cases, the detection of deletions has been based on the transmission of polymorphic alleles from parents to the proband, the absence of an obligate parental allele in the proband being indicative of a deletion (Gregory et al. 1990). In such analysis, the use of polymorphic markers only at 15q11-q13 may lead to misinterpretation, as isodisomy and deletions yield similar results. Therefore, markers outside the 15q11-q13 region should also be used.
2.6.2.2 Detection of uniparental disomy of chromosome 15
Identification of uniparental disomy has been based on the observation of the inheritance of parental alleles to the patient. The presence of only either maternal (PWS) or paternal (AS) alleles of chromosome 15 is indicative of uniparental disomy (Mutirangura et al.

49
1993b, Ledbetter & Engel 1995). In the detection of UPD, the use of RFLPs has been replaced by more informative microsatellite or short tandem repeat (STR) markers with a large number of alleles easily detectable by the polymerase chain reaction (PCR). Both markers within and outside the PWS/AS critical region are used, and the analysis requires DNA samples from the patient and both parents.
2.6.2.3 DNA methylation test as a screening method for the detection of an abnormal imprinting pattern
Several different regions of imprinted 15q11-q13 have been shown to exhibit different maternal and paternal methylation, which can be used in the diagnosis of Prader-Willi syndrome and Angelman syndrome (Driscoll et al. 1992, Dittrich et al. 1992, Sutcliffe et al. 1994, Glenn et al. 1996). The parent-specific DNA methylation imprint of the AS/PWS region has been typically determined by using Southern hybridization and methylation-sensitive restriction enzymes with DN34/ZNF127 (Driscoll et al. 1992), PW71B (Dittrich et al. 1992) and/or KB17 (Glenn et al. 1996) as a probe (Fig 9). This test is based on the observation that, at these loci, the maternally inherited segment is extensively methylated, while the paternally inherited contribution is unmethylated. Double-digested DNA with two different endonucleases, of which one is methylation-sensitive, not cleaving the DNA sequence when the cytosine residues are methylated, makes it possible to distinguish the maternally and paternally derived alleles (Dittrich et al. 1996b, Gillessen-Kaesbach et al. 1995b, Kubota et al. 1996a). If the DNA methylation test demonstrates only the methylated maternal allele, PWS is confirmed, while in the case of AS, only the unmethylated paternal allele is observed. Parental samples are not required for this test. In addition to Southern blot analysis, the development of methylation-specific PCR (MSP) assays now allows more rapid analysis of these syndromes (Kosaki et al. 1997, Kubota et al. 1997, Zeschnighk et al. 1997, Chotai & Payne 1998). The DNA methylation test identifies patients with deletion, uniparental disomy or imprinting defect, but it does not distinguished between these changes, and further testing of patients with an abnormal parent-specific DNA methylation imprint has been required to establish the molecular class of these syndromes.

50
Fig. 9. DNA methylation test in the diagnosis of Prader-Willi and Angelman syndromes. DNA was digested with XbaI and NotI and probed with KB17. PWS patient is missing the paternally derived 0.9 kb band, whereas AS patient is missing the maternally derived 4.2 kb band. N1-N3 = normal result.
2.6.2.4 Detection of imprinting centre (IC) mutations in imprinting defects
In patients with imprinting defects, i.e. those with abnormal methylation imprinting and exclusion of deletions and UPD, possible mutations in the imprinting centre (IC) have been searched for by Southern blot analysis using several IC-specific probes spanning the imprinting centre (deletions), by sequencing of IC exons or by heteroduplex analysis (HA) of IC3/AS-IC and PWS-IC, followed by sequencing of abnormal products (point mutations, nucleotide insertions/deletions) (Buiting et al 1995). The screening for mutation by heteroduplex analysis is based on the different mobilities of homoduplexes (containing two wild-type alleles) and heteroduplexes (containing a mutant and a wild-type allele) on non-denaturing polyacrylamide gel (see Glavac & Dean 1995).

51
2.6.2.5 Mutation analyses of the UBE3A gene
To identify UBE3A mutations in Angelman patients, both direct sequencing of amplified genomic DNA (Matsuura et al. 1997, Fung et al. 1998, Tsai et al. 1998, Fang et al. 1999, Lossie et al. 2001) and sequencing of the abnormal products detected by single-strand conformation polymorphism (SSCP) have been used (Kishino et al. 1997, Malzac et al. 1998, Baumer et al. 1999). Sequencing of the gene is a reliable, but expensive and laborious method. SSCP is a commonly used mutation screening method, which is based on the defined secondary structure of a single-stranded DNA under specific conditions, and where an altered sequence leads to mobility shifts in electrophoresis (Orita et al. 1989a,b). The sensitivity of SSCP is best with fragments of less than 200 bp, being 60% – 95% (see Cotton 1993, Eng & Vijg 1997). In UBE3A mutation screening, 27 different primer pairs have been used (Malzac et al. 1998).
2.6.2.6 Other aspects of laboratory diagnosis
Gene expression detected by PCR of reverse-transcribed RNA (RT-PCR) has been used as a tool for diagnosing Prader-Willi syndrome (Wevrick & France 1996). This test is based on the fact that the SNRPN gene is expressed only from the paternally derived chromosome 15 and thus not in patients with PWS. PCR is done on the first strand cDNA product of reverse transcription with primers corresponding to the SNRPN gene and control primers amplifying the DNA sequences outside chromosome 15. The SNRPN expression test has been shown to be rapid and sensitive for the detection of PWS, but it cannot be used in molecular diagnosis of Angelman syndrome.
The laboratory diagnosis of PWS or AS has been mostly done from blood samples, although, in some rare cases, diagnostic testing has also been done from chorionic villus samples (CVS) or amniotic fluid cells. In prenatal diagnosis, the DNA methylation test using the KB17 probe has been shown to give reliable results even in early pregnancies. Instead, the D15S63 locus detected by the PW71 probe is hypomethylated in both maternal and paternal chromosome 15 in CVS and some amniotic fluid samples and is thus unreliable (Kubota et al. 1996b, Slater et al. 1997, Glenn et al. 2000). In addition to families with known increased recurrence risks of PWS or AS, prenatal diagnosis should be offered if trisomy 15 mosaicism is detected on CVS or amniocentesis, if cytogenetic deletion is suspected when analyzing these samples or if a de novo translocation involving chromosome 15 or a dicentric chromosome 15 marker is detected (Christian et al. 96).
2.6.3 Diagnostic approaches
The diagnostic approaches different laboratories use differ from each other. Some laboratories begin their examinations with chromosome and FISH analyses to detect deletions, followed by the DNA methylation test if the deletion is not found. However,

52
beginning with the DNA methylation test, deletions as well as uniparental disomy and imprinting defects can be recognized. In the case of an abnormal methylation imprint, quantitative DNA or FISH analysis and microsatellite analysis are recommended to distinquish between these changes. If deletion is found, also chromosomes 15 of the father (PWS) or the mother (AS) should be examined by using FISH (see the statement of the American Society of Human Genetics and American College of Medical Genetics 1996).
In the case of imprinting defect, the mutation analysis of the imprinting centre should be done to detect families with high recurrence risk. Furthermore, in Angelman syndrome, if no deletion, uniparental disomy, or imprinting defect is found in the patient with typical symptoms, the mutation analysis of UBE3A gene should be performed (Stalker et al. 1998).

3 Purposes of the present study
At the time when this study was initiated, many Prader-Willi patients and some Angelman patients were shown to have a deletion of the chromosome region 15q11-q13, and the deletions in PWS were strongly suspected to be of paternal origin. During the years 1989 – 1991, it was confirmed that the origin of the deletion was exclusively paternal in PWS and maternal in AS, respectively. However, in approximately 30 % of PWS and AS cases, the genetic defect remained unknown, and towards the end of the year 1989, the first report of uniparental disomy as a cause of Prader-Willi syndrome was published. Later, it became obvious that both syndromes may result from several different mechanisms, such as deletion, uniparental disomy, imprinting defect and, in Angelman syndrome, a mutation in a single gene, and that in both syndromes the basic reason is the lack of expression of paternal (PWS) or maternal (AS) gene(s) of the imprinted chromosome 15 region q11-q13. Although the Prader-Willi and Angelman syndromes are predominantly sporadic diseases, some families with more than one affected child have been described. The specific aims of the present study were: 1. To find out and describe the genetic changes in the chromosomal region 15q11-q13
in Finnish Prader-Willi and Angelman patients and to study the possible inheritance of these syndromes.
2. To study the size and parental origin of the deletions observed at 15q11-q13 in the Prader-Willi and Angelman syndromes.
3. To study the nature and origin of uniparental disomy of chromosome 15 in the Prader-Willi and Angelman syndromes.
4. To study the role and origin of imprinting defects in the chromosome region 15q11-q13 in PWS and AS.
5. To study the nature and origin of mutations of the UBE3A gene in Finnish Angelman syndrome patients.
6. To assess the role of different methods in the diagnosis of Prader-Willi and Angelman syndromes.

4 Materials and methods
Detailed descriptions of the subjects and methods have been presented in the original articles (I – V).
4.1 Subjects (I-V)
4.1.1 Prader-Willi and Angelman patients
The patient series studied was collected during the years 1989 - 2000 from amongst the patients referred to the Genetics Laboratory at Oulu University Hospital for a genetic diagnosis of either Prader-Willi or Angelman syndrome.
At the initial stages of the study, most of the diagnostic samples came from Northern Finland, but later on, especially samples for Angelman syndrome were increasingly received from all parts of the country. The referring clinical units included departments of clinical genetics, paediatrics, neuropaediatrics and institutions for mentally handicapped subjects. The patients had been seen by clinicians experienced in the diagnosis of developmental disorders. The patients from Northern Finland had been seen at the Department of Clinical Genetics at Oulu University Hospital or at the outreach clinics in its catchment area.
As to Prader-Willi syndrome, at the first stage of the study in the years 1989 to 1994, a group of 41 patients suspected of having the syndrome were available for detailed laboratory tests. By using the diagnostic criteria proposed by Holm et al (1993) (Table 1), modified by excluding genetic abnormalities in 15q11-q13 from the list and using a six-point score as the diagnostic limit for PWS in children aged over three years, 27 patients were included as typical PWS patients, and 14 atypical patients were excluded. The subsequent PWS cases were collected from a more heterogeneous set of laboratory referrals; all patients with an observed genetic defect were either seen at the outpatient department or evaluated based on information obtained from the referring physicians

55
and/or hospital records. The ultimate PWS series consisted of altogether 76 patients. The ages of the patients ranged between one week and 38 years.
The Angelman syndrome series consisted of 47 patients. The clinical features of the patients were assessed during their visit to the Department of Clinical Genetics, or if seen elsewhere, from hospital records and/or laboratory remittance, except in the case of UBE3A mutation analysis, where the clinical assessment was done either at the Department of Clinical Genetics or in collaboratorion with the referring physicians outside Northern Finland, and all were considered to fulfil the diagnostic criteria proposed by Williams et al. (1995a) (Table 2). The ages of the AS patients ranged from 4 months to 37 years.
4.1.2 Family and control materials
To assess the origin and inheritance of the observed genetic changes, the parents and also other selected relatives were included in the study. Altogether 113 parents of PWS and 56 parents of AS patients and 14 other relatives of AS patients were studied (183 individuals).
In UBE3A mutation analysis, DNA of 100 unrelated control individuals (= 200 alleles) was investigated for the presence of the UBE3A mutations detected in the AS patients.
4.2 Laboratory analyses
Several different methods were used in this study. The high-resolution chromosome analysis of 15q11-q13 was used to detect a deletion or other structural changes, and it was later replaced by fluorescence in situ hybridization (FISH) analysis in selected cases. Restriction fragment length polymorphism (RFLP) together with quantitative analysis was used to identify deletions and uniparental disomy and to determine the size of the deletion, and RFLP analysis was used to detect the parental origin of the deletion or UPD. Microsatellite analysis was used to determine the size of the deletion detected by quantitative analysis, to identify uniparental disomy and to investigate its origin, to detect the parental origin of a deletion or UPD, to identify imprinting defects from patients with abnormal methylation imprint and for haplotype analysis of these patients and their families. DNA methylation analysis was made on the patients with no deletion or uniparental disomy, to identify the patients with imprinting defects. Furthermore, this analysis was also made on all the other patients to find out how different probes could be used to recognize patients with different types of deletion, UPD or imprinting defect and to estimate their use in the diagnosis of PWS and AS. Later, this method was used as a primary test for the recognition of patients with either PWS or AS. Detection of the possible specific mutation at the imprinting centre (IC) was done in collaboration with German colleagues. For the detection and identification of specific UBE3A mutations, conformation-sensitive gel electrophoresis (CSGE) and sequencing were used, respectively.

56
4.2.1 Cytogenetic and fluorescence in situ hybridization analyses (I, III, IV)
A high-resolution chromosome analysis was performed using peripheral blood and the GBG method (G bands with bromodeoxyuridine using Giemsa, Dutrillaux & Viegas-Pequignot 1981). To find prometaphases with chromosomes 15 at greater than 650-band accuracy, the chromosome preparations were screened under a normal light microscope. At least five chromosome pairs 15 were analyzed and photographed with a chromosome analyzer (CytoScan, Applied Imaging).
Fluorescence in situ hybridization (FISH) analysis was done on either prometaphase or ordinary chromosome preparations. One-colour FISH was performed with digoxigenin-labelled D15S10/PML and SNRPN/PML probes obtained from Oncor, observing the protocols recommended by the manufacturer. For dual-colour FISH studies, a biotin-labelled D15S11/PML and digoxigenin-labelled D15S10/PML probes (Oncor) and a D15Z1/SNRPN/PML directly labelled dual-colour probe set (Vysis) were used in accordance with the protocols provided by the manufacturers. Analyses through a single band pass filter set for FITC, TRITC and DAPI and figures were done using the CytoVisionTM system (Applied Imaging) connected to an epifluorescence microscope (Leica).
4.2.2 DNA extraction (I, II, III, IV, V)
Genomic DNA was isolated from venous blood lymphocytes by a conventional phenol/chloroform extraction method (Sambrook et al. 1989).
4.2.3 RFLP and quantitative analyses (I, II, III, IV)
Both in RFLP and quantitative studies, the following nine chromosome 15q11-q13 specific probes were used: IR39 (D15S18), ML34 (D15S9), IR4-3R (D15S11), 189-1 (D15S13), 3-21 (D15S10), IR10-1 (D15S12) (Donlon et al. 1986), hN4HS (SNRPN) (Özcelik et al. 1992), 28β-3H (GABRB3) (Wagstaff et al. 1991b) and CMW-1 (D15S24) (Rich et al. 1988). For quantitative studies, the chromosome 13 and 19 specific probe H2-26 (Lalande et al. 1984) and pCKMM3’ (Coerwinkel-Driessen et al. 1988) were used as control probes, with the exception of the probe hN4Hs, which detects DNA sequences at the SNRPN (15q12) and SNRPNP1 (6pter-p21) loci, the latter serving as an internal control. RFLP and quantitative analyses were done using standard methods (Southern 1975). The probes were radiolabelled with [α -32P]-dCTP by nick-translation or random priming, using a commercial kit (Boehringer Mannheim), and hybridized to the DNA fragments bound either onto nitrocellulose (Schlecher and Schull) or nylon (Magnagraph, MSI) membranes. After post-hybridization washes, autoradiography was performed using

57
1 – 7 days exposures. Deletions were determined by visual inspection and/or by densitometric scanning of autoradiograms (Laser Scanner, Molecular Dynamics, Ins.). The average density ratio of the chromosome 15 and the control probe bands within a single electrophoretic lane was calculated in order to determine the copy number at each locus (corrected for background levels). Dosages of each locus in non-PWS/AS individuals were normalized to a control value of 1.0 ± 0.2. Dosages of patients were determined relative to this control value. Deletion was considered when value was ≤ 0.6.
The origin of the defect was detected by RFLP analysis by observing the lack of the paternal (PWS) or maternal (AS) allele.
4.2.4 Microsatellite analyses (I, II, III, IV)
In the microsatellite studies, nine chromosome 15q11-q13 specific markers (D15S542, D15S11, D15S128, D15S122, D15S113, GABRB3, D15S97, GABRA5, D15S156) and six markers distal to the 15q11-q13 region (D15S165, D15S118, CYP-19, FES, D15S125, D15S87) were used. The oligonucleotide primer sequences and the polymerase chain reaction (PCR) conditions were obtained from the Genome Data Base (http://gdbwww.gdb.org/) or the report of the second International Chromosome 15 Workshop (Malcom et al. 1994).
Polymerase chain reactions were carried out using alternatively non-radioactive (I) or radioactive methods (II, III, IV). In the non-radioactive protocol, the amplified PCR products were resolved by electrophoresis on 8 % or 10 % polyacrylamide sequencing gels, and the gels were silver-stained according to the manufacturer’s instructions (Silver Stain Kit, Bio-Rad). In the radioactive protocol, a radiolabelled nucleotide [α -32P]-dCTP was included during the amplification of target DNA. After electrophoresis of the PCR products on 7 % polyacrylamide sequencing gel, the gel was transferred to blotting paper and autography was performed.
4.2.5 DNA methylation analyses (I, II, III, IV)
Southern blot analyses of the parent-of-origin DNA methylation imprints were done using the probes DN34, PW71B and KB17 (Table 4). In Prader-Willi syndrome the paternal fragment and in Angelman syndrome the maternal fragment is either diminished (DN34) or lacking (PW71B, KB17).
The physical map order of the different RFLP, quantitative, DNA methylation and microsatellite loci used in this study is: centromere – D15S18 - D15S542 - ZNF127/D15S9 - D15S11 - D15S13 - D15S63 - SNRPN - D15S128 - D15S122 - D15S10 - D15S113 - GABRB3/D15S97 - GABRA5 - D15S156 - D15S12 - CMW-1 - D15S165 - D15S118 - CYP-19 - FES - D15S125 - D15S87 - telomere.

58
Table 4. Probes used in the parent-of-origin DNA methylation analysis. Mat = maternal imprint, pat = paternal imprint.
Probe Locus Enzymes Fragments Reference DN34 ZNF127 EcoRI/HpaII 4.3-kb (mat)
4.0-kb (mat) 3.5-kb (pat)
Driscoll et al.1992
PW71B D15S63 HindIII/HpaII 6.6-kb (mat) 4.7-kb (pat)
Dittrich et al. 1992
KB17 SNRPN exon 1
XbaI/NotI
4.2-kb (mat) 0.9-kb (pat)
Glenn et al. 1996
4.2.6 Mutation analyses of the imprinting centre (IC) at 15q11-q13 (II, III)
Detection of the possible specific mutation at the imprinting centre (IC) was done collaboratively at the Institute of Human Genetics, University of Essen, Germany. Deletion analysis of IC was performed by means of quantitative Southern blot analysis with a battery of probes from the IC (λ71.13, λ48.18, λ48.45, λ48.19, 48.I, λ48.2, 48.43, λ48.II, λ48.4I, λ48.6, λ48.8,λ49.39, λ48.24, λ48.3, λ48.29, λ48.22, λ48.33, λ48.25, λ48.17, λ48.40, λ48.28, λ48.26 and λ12 (Buiting et al. 1995).
For mutation screening, IC exons were amplified by PCR, sequenced on both strands as described by Bürger et al. (1997) and analyzed on an ABI 377A DNA sequencer (Applied Biosystems).
IC3/AS-IC and PWS-IC (PWS-IC-C, PWS-IC-T) were analyzed by heteroduplex analysis. After PCR amplification, the products were digested to produce smaller fragments. These fragments were subjected to heteroduplex analysis on high-resolution polyacrylamide gels, as described by Lohmann et al. (1996). Abnormal bands were sequenced.
BglII/MspI-digested DNA and the KB17 probe were used for the analysis of the SNRPN locus to study the grandpaternal origin of the affected chromosome 15 in the cases with imprinting defects.
4.2.7 Mutation analyses of the UBE3A gene (V)
4.2.7.1 Conformation-sensitive gel electrophoresis (CSGE) analysis
Conformation-sensitive gel electrophoresis (CSGE) was used to define the alterations in all protein-coding exons of UBE3A (exons 8 to 16), with the exception of exon 7, and the

59
corresponding slice site junctions. CSGE is based on the different mobilities of homo- and heteroduplexes formed by heat denaturation and subsequent reannealing of the two forms of DNA and electrophoreses in mildly denaturing agents enhancing the conformational changes in the heteroduplex (Ganguly & Williams 1997, Körkkö et al. 1998). The primers were designed to be at least 60 bp of both the 5’- and the 3’-flanking sequences of the target sequence, to detect correctly all the mismatches. Exon 8 was amplified in two and exon 9 in five parts, the others could be amplified in one part. The primer sequences, the annealing temperatures and the sizes of the PCR products are presented in Table 2 in article V. After amplification of the target DNA by PCR, the products were denatured at 98°C for 5 min and annealed at 68°C for 30 min, to allow formation of DNA heteroduplexes, and stored at 4°C. The PCR products were electrophoresed in mildly denaturing 10% polyacrylamide gel, stained with ethidium bromide, visualized with a UV transilluminator and photographed for analysis. All the band shifts were verified by repetition of PCR and CSGE.
In the case of suspected mosaicism, at least three independent CSGE analyses of the mother and the child were done. The dosage of the abnormal allele was compared to the dosage of the normal allele in the samples of both the mother and the child.
4.2.7.2 DNA sequencing
Samples showing variant bands after mutation analysis by CSGE were sequenced using the original primers. Exon 7 was also analyzed by sequencing, using the primers described by Kishino & Wagstaff (1998) for SSCP analysis. PCR products were analyzed either by manual sequencing (Cyclist Exo¯Pfu DNA Sequencing Kit Stratagene) or by an automated DNA sequencer (the Li-Cor IR2 4200-S DNA Analysis system, (Li-Cor Inc.)). In manual sequencing, [α -35S]-dATP was used as a radioactive label. The products were electrophoresed on 6 % polyacrylamide sequencing gel and analyzed after autoradiography from Biomax X-ray films. For automated DNA sequencing, IRD700 (pentamethine carbocyanide dye) and IRD800 (heptamethine cyanine dye) labelled nucleotides were used in PCR reactions.
In the cases where both primers were inside the exon, the sequence of the mutant allele was compared with the UBE3A sequence and with that of the processed pseudogene UBE3AP2 (Kishino & Wagstaff 1998), to verify that the mutation affected UBE3A.
In the cases of suspected mosaicism, at least two independent instances of sequencing of the abnormal fragments of the mother and the child were done.

5 Results
5.1 General aspects (I, IV, V)
A genetic defect affecting the chromosome region 15q11-q13 could be shown in 76 of the 90 patients with clinically suspected Prader-Willi syndrome. The remaining fourteen suspected PWS patients had normal results in DNA studies. None of them fulfilled the criteria of the syndrome when scored according to Holm et al. (1993). In Angelman syndrome, 43 of the 47 patients showed a genetic abnormality, while in four clinically typical AS patients the genetic cause of the syndrome remained unsolved (Table 5).
Table 5. Distribution of the observed genetic changes at 15q11-q13 in 76 Finnish Prader-Willi and 47 Angelman syndrome patients (UPD = uniparental disomy, n = number of patients)
Type of change Prader-Willi syndrome n %
Angelman syndrome n %
Deletion 53 69.7 31 66.1 UPD 15 19.7 1 2.1 Imprinting defect 2 2.6 5 10.6 UBE3A mutation 5 10.6 Not known 4 8.5 Incompletely characterized1 6 8.0 1 2.1 1 = parental DNA samples were not available
Among the Prader-Willi patients, a specific change causing the syndrome could be determined in 70 out of 76 cases. Most of them (76 %) had a deletion at 15q1-q13, and 21% showed uniparental disomy for chromosome 15. Two patients with imprinting defects were found (3 %). Among the molecularly incompletely characterized PWS patients, five had a methylation pattern typical of PWS, but no deletion at the SNRPN locus, indicating the presence of either uniparental disomy or an imprinting defect. One patient had an abnormal methylation pattern. The DNA samples of these six families were not available for further studies.

61
Among the 46 Angelman patients whose molecular tests could be completed, a specific change was identified in 42 cases, while no changes were found in four cases. Deletions accounted for 67.4 % of the etiologic changes, while uniparental disomy was present in only one patient (2%). In 10.9 %, the underlying defect was an imprinting defect, and another 10.9 % had a mutation in the UBE3A gene. No molecular abnormalities were seen in 8.7 % of the cases. Furthermore, one incomplete characterized patient showed a paternal methylation imprint at 15q11-q13, but no deletion at the SNRPN locus, suggesting either uniparental disomy or a defect in the imprinting process. The DNA samples of her family were not available for further studies.
All of the Prader-Willi patients appeared sporadic. Of the Angelman patients, recurrence was identified in two families (4.4%). In one family (ASFIV), two affected children had an imprinting mutation inherited from the maternal grandfather. In the other family (ASFV), an interstitial deletion at 15q11-q13 was detected in both affected sibs, most probably due to germ line mosaicism of the mother. Furthermore, two inherited changes in the region 15q11-q13 were detected: a mutation in an UBE3A gene in one AS patient (AS4) and a deletion proximal to the PWS and AS region in one atypical patient (PAS-1), both inherited from their mothers.
5.2 Characteristics of the deletions at 15q11-q13 (I, IV)
5.2.1 Size variation of the deletion
During the first years of this study, the size of the deletion was determined in detail by quantitative, RFLP and microsatellite analyses, in order to understand the size variation and to restrict the chromosomal region for the purposes of defining the PWS and AS regions more accurately.
The size of the deletion was determined in 24 Prader-Willi patients. Four types of deletion were found: a large deletion including the loci from D15S18 to D15S12 (type I, n = 5), a deletion including the loci from D15S9 to D15S12 (type II, n = 16), a deletion involving the loci from D15S11 to GABRB3 (type III, n = 1), and a small deletion including the loci from D15S11 to D15S10 (type IV, n = 2). The most common deletions were type II (67 %) and type I (21 %) deletions. In the subsequently studied 29 PWS patients, the presence of a deletion at 15q11-q13 was determined with the hN4HS (SNRPN) probe, which maps inside the smallest region of deletion overlap in PWS.
In Angelman syndrome, the size of the deletion was studied in nineteen patients. Eight patients were studied with all of the 15q11-q13 specific markers used, and four of them had type I and four type II deletions, respectively. The proximal breakpoints of the type I deletion were proximal to the locus D15S18 and those in the type II deletion between the loci D15S18 and D15S9. The distal breakpoints in both deletion types were located between the loci D15S12 and D15S24. The breakpoints of these two deletion types were similar to those seen in PWS patients. In the subsequent 11 patients studied with the markers from D15S9 to D15S24, at least the type II deletion, encompassing the region

62
from D15S9 to D15S12, was found. In the remaining fourteen AS patients, the deletion was detected by using the hN4HS probe.
A small deletion outside the Prader-Willi and Angelman chromosome region was detected in one patient (PAS-1) originally studied for features of Prader-Willi syndrome. He was first found to have a paternal imprint only at the D15S63 locus in the DNA methylation test. Subsequent quantitative, RFLP and microsatellite analyses with 20 different markers and DNA methylation analysis with additional probes confirmed a deletion including the loci from D15S13 to D15S63 both in this patient and in his mother in their maternally derived chromosome 15. A known deletion polymorphism reported at D15S63 was excluded by PCR analysis as described by Buiting et al. (1999c).
5.2.2 Detection of deletions
High-resolution chromosome analysis and DNA methylation studies were applied to compare their ability to detect or exclude a deletion diagnostically. Furthermore, in three families, fluorescence in situ hybridization (FISH) analysis was used to confirm the presence/absence of a deletion detected by molecular analysis and to exclude possible complex chromosomal rearrangements.
In Prader-Willi patients subjected to high-resolution chromosome analysis, the type I deletion was detected in all of the five cases, whereas the type II deletion (n=15) was observed in only 10/13 cases studied (77 %). The type III and IV deletions as well as the small maternal deletion encompassing the loci D15S13 – D15S63 (patient PAS1) were too small to be detected by high-resolution methods. When high-resolution chromosome analysis was applied to six Angelman patients, a deletion could be observed in all of the three type I cases but in only one of the three type II cases.
Of the fourteen Prader-Willi patients who did not fulfil the criteria of typical PWS, thirteen had normal chromosomes in both molecular and high-resolution studies. In one of the clinically atypical PWS patient with a suspected cytogenetic deletion, too, the observed abnormality could be shown to be a polymorphism by molecular and FISH analyses.
In DNA methylation analysis, all of the different deletion types could be recognized by the probes PW71 (D15S63) and KB17 (SNRPN), with the exception of the small deletion between the loci D15S13 and D15S63 in an additional patient PAS-1, which was observed only by PW71. Furthermore, this deletion and the deletion types III or IV showed a normal biparental imprint with the probe DN34, which detects DNA sequences proximal to these deletions.
In the family with two Angelman children due to a large deletion at 15q11-q13 (family ASFV), a molecularly detected deletion was also revealed by FISH analysis with SNRPN and D15S10 as probes, while the chromosomes 15 of the parents were normal. In one Angelman family with a suspected imprinting defect (family ASFIV), FISH analysis with D15S10 as a probe was used to exclude a small deletion in the smallest region of deletion overlap in AS in both the affected sibs and their mother. No deletion was found, which was consistent with the molecular analyses. The FISH analysis of the family with a small

63
deletion proximal to the Prader-Willi and Angelman chromosome region did not show deletions at the loci D15S11, SNRPN and D15S10 either in the patient (PAS-1) or in his parents, confirming the boundaries detected by molecular analyses.
5.2.3 Origin of deletion
The parental origin of the affected chromosome 15 was determined in all of the 53 Prader-Willi and 31 Angelman deletion cases. In the early phases of the study, RFLP analysis was able to reveal the parental origin of the deletion in 21/23 PWS (91 %) and 13/13 AS (100 %) patients, while later microsatellite and DNA methylation analyses were always informative. The affected chromosome 15 was always paternal in PWS and maternal in AS patients.
In one Angelman family (ASFV), an identical interstitial type I deletion at the maternal chromosome 15 was unexpectedly detected in both affected sibs. In haplotype analysis, these patients showed an identical, maternally derived haplotype distal to the deletion region, which was also present in an unaffected brother. Both parents’ chromosomes 15 were cytogenetically normal, and no mosaicism of the 15q11-q13 deletion (one-colour FISH) or structural changes involving chromosome 15 (dual-colour FISH analysis) could be detected in fluorescence in situ hybridization studies of the mother. These findings suggest that the recurrent deletion in this family originated from germ line mosaicism in the gonads of the phenotypically healthy mother.
5.3 Uniparental disomy of chromosome 15 (I, IV)
The inheritance of the RFLP and later polymorphic microsatellite markers of chromosome 15 were used to detect uniparental disomy among the patients in whom no deletion was found. The origin of the patient’s uniparental disomy was inferred from the state of the centromere markers, which were heterozygous in the parent of origin. The heterodisomic state of these markers (non-reduction) indicates an MI error, while their isodisomy (reduction) indicates non-disjunction at the MII stage or a postzygotic event if all markers encompassing the entire chromosome 15 showed reduction to homozygosity (Robinson et al. 1993a).
The analysis detected fifteen Prader-Willi patients (patients 24 – 27 in article I and eleven additional patients) and one Angelman patient with uniparental disomy of chromosome 15 (article IV). The parental origin of UPD was always maternal in PWS and paternal in AS.
In Prader-Willi syndrome, the origin of uniparental disomy was meiotic in 13 cases, mitotic in one case and non-informative in one case. The majority of UPD patients (n = 9) showed non-reduction (i.e. heterodisomy) of the most proximal informative marker (D15S9 – D15S11), which indicates maternal non-disjunction in the first meiotic division (MI error). Five of these cases showed heterodisomy throughout the entire chromosome

64
with no evidence of recombination, while two cases showing some loci in hetero- and other isodisomy were detected. Two cases were shown to have heterodisomy throughout the Prader-Willi chromosome region, but the study of uniparental disomy in more distal regions was not informative. One patient showed reduction (i.e. isodisomy) of the proximal marker D15S11 and heterodisomy of the distal part of chromosome 15, indicating an MII error. In three patients, the most proximal informative locus was located at 5 cM (D15S10, one case) or 5.6 cM (D15S113, two cases) from the locus D15S18, all showing heterozygosity for the respective locus, indicating an error in either meiosis I or II segregation. One patient showed reduction to homozygosity of all informative markers, which implies rather a mitotic origin of the uniparental isodisomy due to mitotic duplication of the maternal chromosome 15 than an MII error in maternal meiosis. In one patient, two proximal markers showed uniparental disomy, but were not informative of hetero-/isodisomy. Other informative markers showed isodisomy, implying either an MI error or, more probably, a mitotic origin of UPD. The only Angelman patient with paternal disomy showed isodisomy of the proximal markers and heterodisomy at the distal part of chromosome 15, suggesting an error of paternal meiosis II. In both syndromes, the entire chromosome 15 was involved in the uniparental disomy, as shown by an analysis of the most telomeric locus D15S87 and other markers throughout chromosome 15.
The parents in the uniparental disomy cases (n=16) were older compared to those with reportedly normal births in Finland. The mean maternal and paternal ages were 34.5 and 36.8 years, respectively, in the maternal UPD15 cases and 38 years for both parents in the paternal UPD15 case.
5.4 Imprinting defect in the Prader-Willi and Angelman syndromes (II, III, IV)
5.4.1 DNA methylation and haplotype analysis
To identify imprinting defects, DNA methylation analysis (probes DN34, PW71 and/or KB17) and microsatellite markers (D15S11, D15S128, D15S122, D15S113, D15S97, GABRB3, GABRA5, D15S118, CYP-19) were used. Analysis of the RFLP markers of 15q11-q13 was also done in one AS family (ASFIV) in the early stages of this study.
In two unrelated Prader-Willi patients, DNA methylation analysis of the loci D15S63 and SNRPN revealed a loss of the unmethylated (paternal) band, and no deletion could be found. Microsatellite analysis showed biparental inheritance of chromosome 15q11-q13 for the markers D15S128 and D15S113 for a patient from the family PWSFI and D15S11, D15S122 and D15S97 for a patient from the family PWSFII, respectively, indicating the presence of an imprinting defect. Although the paternal grandparents of these patients were not available for tests, a combined test for DNA methylation (probe KB17) and an HpaII/MspI polymorphism showed that the affected chromosome could be

65
determined to have been inherited from the paternal grandmother in both cases (unpublished results).
In Angelman syndrome, five patients (patient ASID10 from the AS family I (article II) and the patients ASF3 and ASF4 from the AS family V (article III) and two additional patients) showed a loss of the methylated (maternal) band in a DNA methylation test. Four of them were analyzed with all of the three methylation-sensitive probes, and one patient (ASFIII) only with the KB17 probe because there was not enough DNA for further Southern analysis. None of these patients had a deletion of 15q11-q13. Microsatellite analysis revealed biparental inheritance of chromosome 15q11-q13 for the markers D15S11 and D15S97 in a patient from the family ASFI, the markers D15S122 and D15S118 in a patient from the family ASFII, the markers D15S122 and D15S128 in a patient from the family ASFIII and the markers D15S11 (RFLP), D15S128, D15S97, GABRB3, GABRA5 and D15S12 (RFLP) in both of the affected sibs from the family ASFIV, indicating the presence of an imprinting defect.
In the analyses of the grandparental origin of the affected chromosome 15, both the haplotype analysis and the combined test for DNA methylation and an HpaII/MspI polymorphism showed that the affected chromosome came from the maternal grandfather in the AS family I (ASFI). In the AS family II (ASFII), only haplotype analysis was informative, showing the affected chromosome 15 to have been inherited from the maternal grandmother. In both of these families, the healthy sib had a different maternal 15q11-q13 haplotype compared to the affected sib. In the AS family III (ASFIII), where the maternal grandparents were deceased, the affected child and his healthy brother shared a similar haplotype inherited from the mother, reducing the risk for recurrence.
In the family ASFIV with two affected children, the haplotype associated with Angelman syndrome was shared by the affected sibs, their healthy mother, two maternal uncles and the maternal grandfather. A combined test for DNA methylation and an HpaII/MspI polymorphism also showed the same grandpaternal chromosome 15 in these individuals. Methylation studies of the parents were normal.
5.4.2 Imprinting mutation analysis
Imprinting mutation analysis was done collaboratively in the Institute of Human Genetics in Germany. Quantitative Southern blot analysis of the exon 1 region of the SNRPN gene in the Prader-Willi patients and Angelman patients from the families ASFI and ASFII and quantitative Southern blot analysis of the critical region in the AS IC-deletion patients did not show deletion in the imprinting centre. Combined haplotype and mutation analysis data suggest that these patients belong to a group of patients with a spontaneous imprinting defect with a low risk of recurrence. The patient from the AS family III was not available for IC mutation analysis.
In the IC mutation analyses of the Angelman family IV, all individuals associated with the AS haplotype showed a ~19-kb deletion in IC AS-SRO with the proximal breakpoint in the L48.6I EcoRI fragment and the distal breakpoint in the L48.33II EcoRI fragment. Combined haplotype and IC mutation analysis data showed that the microdeletion had

66
been inherited by the two affected children from the maternal grandfather through the healthy carrier mother.
5.5 Identification of UBE3A gene mutations (V)
UBE3A mutation analysis was done on nine clinically diagnosed Angelman patients with no deletion, uniparental disomy or imprinting defect. Conformation-sensitive gel electrophoresis detected DNA alterations in six of the nine patients: four in exon 9 and two in exon 16. After sequencing, three of these turned out to be deletions that cause frameshifting and premature chain termination: a 2-bp deletion in exon 9 (1930delAG) in patient AS1 and a 4-bp deletion in exon 16 (3093delAAGA) in patients AS2 and AS3. The parents of the patients had normal CSGE results.
Two disease-causing point mutations were detected, both within the first 344 bases of exon 9. Patient AS4 had an A to C transversion (902A→C) causing a threonine to proline change at position 106. Her mother showed a similar but fainter abnormal band in the three independent CSGE analyses, and by sequencing she was shown to have the same T106P missense mutation in a mosaic form. The patient AS5 had a T to C transition (975T→C), which causes an isoleucine to threonine change at position 130. Neither of his parents had the mutation. Neither of these mutations was found in the northern Finnish control population.
Patient AS6 had a G to A transition (1118G→A), which causes an alanine to threonine change at position 178. The same change was detected in her father and in one of the 100 control individuals, suggesting that the change is rather a polymorphism than a causative mutation.

6 Discussion
6.1 General
The patient series consisted of 76 verified Prader-Willi and 47 Angelman patients recruited during the years 1989-2000. The patients were not exclusively from the catchment area of Oulu University Hospital in Northern Finland, but also included individuals from other parts of country. In the years 1989-1994, PWS and AS patients were identified more actively by requesting the institutes for the mentally handicapped, clinical genetics departments and larger paediatric departments of university hospitals to refer PWS and AS patients for the study. In the later years, patients referred to the genetic laboratory and diagnosed for either PWS or AS were also included; all diagnostic cases from Northern Finland were probably included. The series proved to be large enough to allow an analysis of the different types of aetiologic changes. It did not, however, allow estimates of the birth frequencies or prevalence of these syndromes. Although all northern Finnish patients and some of the others were seen at the Department of Clinical Genetics of Oulu University Hospital, the phenotypic evaluation of most patients was based on information obtained from the referring doctors and hospital records.
Most of the Prader-Willi patients (76 %) had a paternal deletion at 15q11-q13, and maternal uniparental disomy accounted for almost all of the other cases (21 %). In a series of 29 Swiss PWS patients described by Robinson et al. (1991), the corresponding percentages were 72% and 28%, and they were also comparable to those reported in the USA (Mascardi et al. 1992), Japan (Mutirangura et al. 1993b) and the United Kingdom (Webb et al. 1995). Furthermore, in the review of Beuten et al. (1993), which covered data from eleven reports published during the years 1988 – 1992, the corresponding figures were 73% (156/214) for deletion and 23% (26/113) for uniparental disomy. Of the present patients, 3% had an imprinting defect as a cause of their Prader-Willi syndrome. Imprinting defects have been estimated to account for approximately 1 % of all PWS cases (Buiting et al. 2003).
In Angelman syndrome, most patients (67.4 %) had a maternal deletion of 15q11-q13, while paternal uniparental disomy was detected in only one patient (2 %). These percentages were comparable to those reported by others (Mutirangura et al. 1993b, Chan

68
et al. 1993, Saitoh et al. 1994a, Smith et al. 1997, Robinson et al. 1998b, Lossie et al. 2001). For example, in the review of Beuten et al (1993) on AS patients from nineteen reports, 70/100 (70%) had a deletion and 5/123 (4%) had uniparental disomy. Imprinting defects were seen in 10.9 % of the present cases (9% of the families) and UBE3A gene mutations in another 10.9 %. In the series of 93 Angelman families described by Lossie et al. (2001), 3/93 (3.2 %) had an imprinting defect and 10/93 (10.8%) a UBE3A gene mutation.
No specific defect causing the Angelman syndrome could be found in four clinically diagnosed patients (8.7%). Similar observations have been made by several research groups (Malzac et al. 1998, Fang et al. 1999), including the study of Lossie et al. (2001), where a genetic defect could not been demonstrated in ten patients (10.8%). It is possible that these cases represent other mutations related to the UBE3A gene or its regulation, or other genes or silencing elements are involved in the pathogenesis of AS. These patients may also represent phenocopies of Angelman syndrome.
6.2 Deletions
6.2.1 Detection of deletions
At the first stage of the laboratory analysis of Prader-Willi and Angelman syndromes, the detection of deletions by high-resolution chromosome analysis was found useful, although it did not detect all the deletions. In our series, the molecularly identified deletions could not be detected cytogenetically in 29% of PWS and 33% of AS patients. In 26% of Swiss PWS patients (Robinson et al. 1991) and in 33%, 30% and 43% of Dutch (Beuten et al.1993), British (Chan et al. 1993) and Japanese (Saitoh et al. 1994a) AS patients, respectively, the deletion could not be seen cytogenetically. Successful identification of deletions by high-resolution chromosome analysis depended on the size of the deletion. In our series, all deletions in the largest category (type I, n=8) could be detected microscopically, while the second largest deletions (type II) could only be seen in 11/16 cases (69 %), and the smallest deletions (types III and IV) remained invisible in ordinary microscopy.
Fluorescence in situ hybridization analysis with 15q11-q13 specific probes was consistent with molecular analysis in selected cases. The D15Z1/SNRPN/PML probe (Vysis) was found useful for the detection of deletions and for excluding structural chromosome 15 rearrangements, since it detected DNA sequences from both the short and the long arm of chromosome 15.
In this study, the use of molecular methods greatly improved the detection of deletions. In the quantitative analysis, the purpose was either to verify or to exclude deletion by estimating the dosages of DNA at the specific fragments. Although the method was very efficient, the measurement and the determination of dosage was laborious, requiring simultaneous or successive hybridizations of the control probe, several Southern blottis, radioactive exposures and high-quality autoradiograms. The

69
simultaneous use of RFLP fragments was useful, as they could be utilized to substantiate, clarify and often simplify the data interpretation in quantitative analysis. This analysis could also be used to confirm the diagnosis by revealing the lack of a parental allele. The informativeness of RFLP analysis was limited, due to the mostly two-allelic nature of the markers spanning the region 15q11-q13. The allele frequencies and polymorphic information content (PIC) values had been shown to vary from 0.29 to 0.49 in northern Finnish (Kokkonen 1992) and from 0.28 to 0.4 in American (Nicholls et al. 1989a) control populations, respectively. In some cases, in addition to Southern blot analysis, polymorphic microsatellite markers were utilized to determine the size and origin of the deletion. All deletions in PWS and AS were detectable by molecular analysis, as verified later by DNA methylation analysis, which made it possible to determine the size of the deletions.
Later, deletion was recognized with the nonpolymorphic hN4HS cDNA probe. This probe detects DNA sequences from the SNRPN gene that has been shown to be deleted in all PWS and AS deletions, except in one case, where the deletion encompassed only the UBE3A gene (Trent et al. 1997). It also detects the SNRPNP1 (6pter-p21) locus that serves as an internal control in DNA dosage measurements, simplifying the quantitative analysis. This probe proved to be effective, as it identified all of the present deletion cases without any need for parental samples.
6.2.2 Size variation of the deletion
In Prader-Willi patients, deletions of four different sizes were found. The most common types were the largest type I and II deletions, comprising altogether 88% of the PWS deletions studied. Corresponding findings have been reported by others, with these deletion types representing 81% - 100% of the deletions detected (Hamabe et al 1991a, Robinson et al. 1991, Christian et al. 1995, Amos-Landgraf et al. 1999). All PWS patients, regardless of the size of deletion, showed clinically typical features. The smallest region of overlap (SRO) of the deletions identified was the region at the loci D15S11 to D15S10 (type IV deletion), which signified the deletions detected in two present PWS patients. According to Buiting et al. (1993), the SRO of deletion in PWS is about 320 kb in size. In addition to the D15S13 locus, it includes the locus for the maternally imprinted polycistronic gene, SNURF-SNRPN (Glenn et al. 1993a, Reed & Leff 1994, Runte et al. 2001), the deficiency of which has been suggested at least to contribute to the PWS phenotype (Nicholls et al. 1999).
All Angelman syndrome patients with deletions, had either a type I or a type II deletion. The presence of a maternal deletion in one patient (PAS-1) without AS placed the critical region for AS in the range from D15S128 to D15S12. The smallest region of overlap of AS deletions has been shown to be located between the loci D15S122 and D15S113 and to be about 300 kb in size (Greger et al. 1994). In addition to the UBE3A gene (Kishino et al. 1997, Matsuura et al. 1997), this region also includes another maternally expressed gene, ATP10C (Meguro et al. 2001b, Herzing et al. 2001), whose role in the aetiology of AS is not known yet.

70
The proximal breakpoints of the type I deletion observed in both Prader-Willi and Angelman patients in our series were located between the centromere and the D15S18 locus and those of type II deletion between the loci D15S18 and D15S9. The distal breakpoints in both deletion types were located between the loci D15S12 and D15S24. The corresponding breakpoints have been described in the literature, indicating that the vast majority of deletions in the proximal 15q cluster at distinct sites (Knoll et al. 1990, Robinson et al. 1991, Kuwano et al. 1992, Robinson et al. 1993, Saitoh et al. 1994a, Christian et al. 1995, Robinson et al. 1998a, Amos-Landgraf et al. 1999, Christian et al. 1999). The common breakpoint regions probably originate from unequal crossing over between the previously identified HERC2 duplicons at 15q11 and 15q13 at either paternal (PWS) or maternal (AS) meiosis (Amos-Landgraft et al. 1999). Both intra- and interchromosomal changes have been observed (Carrozzo et al. 1997, Robinson et al. 1998a). Unequal crossing over between large duplicated genomic segments at the common deletion breakpoints, located adjacent to the alpha-satellite arrays of the centromere, has also been shown to be a mechanism for deletion in several microdeletion syndromes (Shaffer & Lupski 2000). Duplicated sequences near the pericentromeric regions have been suggested to create genomic instability, which may predispose to further chromosomal rearrangements (Christian et al. 1999).
6.3 Uniparental disomy
Uniparental disomy of chromosome 15 (UPD15) was found in fifteen Prader-Willi patients and in one Angelman patient, and it was always maternal in PWS and paternal in AS. The origin of maternal UPD was meiotic in 13 out of 15 cases (87%), of which 90% were due to maternal non-disjunction events in the first meiotic segregation (MI), resulting in heterodisomy. This is consistent with the previous findings showing that maternal UPD was caused by a meiotic error in 90% of cases, of which ∼ 83% had arisen in the first meiotic segregation (Robinson et al. 1998b, Robinson et al. 2000, Robinson 2000). One of the maternal UPD cases showed isodisomy for markers throughout the entire long arm of chromosome 15, suggesting that the additional maternal chromosome arose through a post-zygotic somatic event rather than an error at meiosis. An error at the second meiotic segregation, however, cannot be totally excluded because the distance between the informative markers distal to 15q11-q13 (38 cM) cannot rule out double crossover events. One patient showed isodisomy from D15S10 to D15S87, although the most proximal markers were uninformative of the type of UPD. This case has probably arisen from a somatic error, although a possible non-disjunctional event at the first meiotic segregation cannot be ruled out.
Maternal uniparental disomy of chromosome 15 has mostly been thought to result from postzygotic correction of a trisomic embryo (trisomy rescue) (Robinson et al. 1996, Robinson et al. 2000). Direct evidence of trisomiy rescue as a mechanism of maternal UPD15 has been obtained from the observations of trisomy 15 in chorionic villus samples simultaneously with UPD in the foetus (Purvis-Smith et al. 1992, Cassidy et al. 1992, Christian et al. 1996). Furthermore, indirect evidence of this mechanism has been acquired from significant non-random X-chromosome inactivation (> 90%) in 24%

71
maternal UPD15 patients. This is a consequence of the relatively late origin of the diploid cell line from the trisomic embryo that will result in skewed X-chromosome inactivation (Robinson et al. 1996, 2000).
The UPD15 detected in one Angelman patient was due to a paternal MII error, probably through trisomy rescue, as gamete complementation would require the occurrence of a non-disjunction event in the meiosis of both parents. This is in contrast to most other cases of paternal UPD reported in Angelman syndrome, which have been due to non-disjunction in the maternal meiosis (nullisomic gamete) followed by mitotic duplication of the paternal homology in the monosomic zygote (compensatory UPD or monosomic rescue) (Mutirangura 1993b, Robinson et al. 1993a, Rogan et al. 1993, Robinson et al. 2000, Fridman et al. 2000a). Only three other patients with a paternal MII error have been described earlier (Gyftodiou et al. 1999, Robinson et al. 2000, Fridman et al. 2000c). Seven cases due to a paternal MI error have been reported; five of the patients carried a translocation involving chromosome 15 (Malcolm et al. 1991, Smeets et al. 1992 Smith et al. 1997, Robinson et al. 2000).
DNA markers spanning the length of the long arm of chromosome 15 showed, along with previous data on UPD15 (Robinson et al. 1991, Malcolm et al. 1991, Nicholls et al. 1992, Mascardi et al. 1992, Mutirangura et al. 1993b, Robinson et al. 1993a, 1996 1998b, 2000, Fridman et al. 2000a,b) that, in both syndromes, the uniparental disomy involves the entire chromosome 15 rather than only parts of it. Because four patients with UPD15 scored here as well as patients described by others have shown no additional phenotypic abnormalities compared to patients with a deletion at 15q11-q13 (Robinson et al. 1991, Mascardi et al. 1992, Freeman et al. 1993, Bottani et al. 1994), it is probable that no other regions of chromosome 15 than q11-q13 are subject to genomic imprinting.
The frequency of uniparental disomy is much higher in Prader-Willi syndrome than in Angelman syndrome, as also observed in the present study (21% of PWS and 2 % of AS cases). The reason for this difference is obviously the relatively rare occurrence of meiotic non-disjunction in male compared to female meiosis (Martin et al. 1991, Antonarakis et al.1993b, Hassold et al. 1996). Postspermatogenesis selection of gametes may also be of significance (Robinson et al. 1993a).
The mean maternal age for maternal UPD was 34.5 years, which is much higher than reported for births in Finland in general (mean maternal age 28 years). The mean paternal age was 36.8 years. In the paternal UPD case, both parents were aged 38 years. The mean maternal and paternal ages collected from several countries for maternal UPD15 were 33.2 and 35.3 years, respectively, while those for paternal UPD15 were 33.4 and 39.4 years, respectively (Robinson et al. 2000). In both maternal and paternal UPD15, the apparent advanced parental ages effect can probably be explained by the effect of maternal age on meiotic non-disjunction. The frequency of non-disjunctional events causing aneuploidy has been shown to increase dramatically with maternal age for most chromosomes (Kupke et al. 1989, Hassold et al. 1995, 1996, Antonarakis et al. 1992). For chromosome 15, UPD has been estimated to occur in 1/3400 live births to women aged 40 years or older (Robinson et al. 1998b)

72
6.4 Imprinting defects
An abnormal DNA methylation imprint and biparental inheritance of 15q markers were detected in two Prader-Willi and five Angelman patients, indicating an imprinting defect (ID). A microdeletion of the imprinting centre (IC) was found in one sib pair with Angelman syndrome (AS family IV). In two AS and two PWS patients, heteroduplex and partial sequence analysis did not reveal point mutations of the known IC elements. The PWS and AS patients with an imprinting defect fall into two molecular classes: those with a microdeletion of IC and those with no IC deletion, the former representing 41% of PWS and 26.5% of AS families (table 6). Cases with no deletion have been sporadic, while IC deletion may be hereditary, as it was in our patient series.
In AS family IV, a 15-kb microdeletion of the AS-IC was found in two affected children, the mother, two maternal uncles and the grandfather. The 5.5-kb deletion detected in the large American family (ASO) could be used to narrow down the AS-IC to 1.15 kb (article III). Recently, the AS-IC region has been narrowed down to 880 bp (Buiting et al. 1999a), and deletion of this component alone may be sufficient to prevent the paternal-to-maternal imprint switch in the female germ line.
All the mutations found in the PWS-IC and AS-IC to date have been more than four kilobases in size, and no point mutations or deletions of only a few base pairs in these regions have been detected. Such small changes are apparently not sufficient to prevent imprinting switching, and a larger deletion is needed.
In Angelman patients with IC deletions, the affected chromosome 15 has always been found to be derived from the maternal grandfather, as in our AS family IV (Buiting et al. 1995, Saitoh et al. 1996, article III). Correspondingly, in PWS, the affected chromosome 15 has always been derived from the paternal grandmother.
Table 6. Sporadic and familial imprinting defects in PWS and AS patients
Molecular class
AS Sibs Sporadic cases
PWS Sibs Sporadic cases
IC deletions 7a 2b 10c 4d
No IC deletion detected 25e .. 20f
a Buiting et al. 1995, Saitoh et al. 1996, article III, Buiting et al. 1999 b Buiting et al. 1995, Saitoh et al. 1996 c Sutcliffe et al. 1994, Buiting et al. 1995, Tashima et al. !996, Ohta et al. 1999, Ming et al. 2000, Buiting et al. 2000, McEntagart et al. 2000 d Schuffenhauer et al. 1996, Ohta et al. 1999, Buiting et al. 2000, Bielinska et al. 2000 e Bürger et al. 1997, article II, two additional AS patients described here f Schulze et al. 1997, article II, Ohta et al. 1999, two additional PWS patients described here
Although the mechanism of imprinting defects in non-deletion cases is not known, they represented new mutations in the families. The grandparental origin of the incorrectly imprinted chromosome 15 could be determined in ten non-IC-deletion Angelman cases, including two patients in our series. Three patients, including the patient from AS family III (article II), had the incorrectly imprinted chromosome 15 derived from the maternal grandmother, while in the other AS patients the affected chromosome 15 was derived from the maternal grandfather. In eleven non-IC-deletion Prader-Willi patients, including two of ours as well as patients described by others (Schulze et al. 1997, Ohta et al. 1999),

73
the incorrectly imprinted chromosome was derived from the paternal grandmother. On the basis of the non-reciprocal findings on the grandparental origin of the incorrectly imprinted chromosome in AS and PWS, it was suggested that the paternal imprint might be the default imprint that develops in the paternal germ line if a maternal trans-acting factor is missing and in the maternal germ line of AS patients with an imprinting defect if maternal imprinting fails (article II).
Both affected sibs from the Finnish AS family IV, and all affected Angelman individuals from the other AS family (AS-O) fulfilled the clinical criteria for Angelman syndrome (article III). However, these patients had several features milder than classic AS deletion patients, including a lack of seizures in several individuals and milder seizures in others, absence of microcephaly, larger birth size and more body and muscle mass, better fine and gross motor control, and normal pigmentation. These differences may be due to haploinsufficiency of the genes at 15q11-q13 contributing to the AS phenotype in deletion patients (Minassian et al. 1998, Nicholls 1998) or incomplete imprinting of the UBE3A gene in the brain of ID patients (Albrecht et al. 1997, Rougeulle et al. 1997, Vu & Hoffman 1997), which possible lead to a double dose of paternal allele expression of this gene (article III). This could also be the case in the AS patients with UPD and a milder phenotype. Haploinsufficiency in the Prader-Willi patients with deletion of the paternal 15q11-q13 genes and relaxation of imprinting of either the maternally (maternal UPD patients) or paternally derived chromosome 15 with maternal epigenotype (ID patients) might also explain the milder phenotype observed in many of these patients.
6.5 UBE3A mutations
Of the nine Angelman patients in whom deletion, uniparental disomy and imprinting mutation had been excluded, five (56%) had an UBE3A gene mutation as the cause of their Angelman syndrome. In other corresponding series of sporadic AS patients, the percentages have varied within 5 – 44% (Malzac et al. 1998, Fang et al. 1999, Moncla et al. 1999b, Baumer et al. 1999, van der Ouweland et al. 1999, Russo et al. 2000, Lossie et al. 2001). The proportion of UBE3A mutations among the familial cases of this group has been higher (75%-80%, Malzac et al. 1998, Fang et al. 1999, Lossie et al. 2001).
To identify UBE3A mutations, sequencing of the abnormal products detected by single-strand conformation polymorphism (SSCP) (Kishino et al. 1997, Malzac et al. 1998, Baumer et al. 1999) or direct sequencing of amplified genomic DNA (Matsuura et al. 1997, Fung et al. 1998, Tsai et al. 1998, Fang et al. 1999, Russo et al. 2000) have been used previously. Sequencing of the whole gene is the most sensitive, but relative expensive and laborious method. SSCP is a commonly used mutation scanning technique, but its sensitivity is best with fragments of less than 200 bp, being still only 60%-95% (see Cotton 1993, Eng & Viji 1997). In this study, conformation-sensitive gel electrophoresis (CSGE) was used to screen for mutations. It is a relative simple and fast method reported to detect most mutations in fragments of 200-800 bp (Ganguly et al. 1993, Ganguly & Prockop 1995, Ganguly & Williams 1997). Under improved conditions,

74
its sensitivity and specificity have been reported to be close to 100% in fragments of 200 – 450 bp (Körkkö et al. 1998). CSGE allowed more frequent use of intron primers than SSCP, which made it easier to avoid the processing of the UBE3AP2 pseudogene, which has high sequence homology with UBE3A (Kishino & Wagstaff 1998). Because CSGE is a PCR-based procedure, a deletion or some other sequence variation at the PCR primer binding site or large insertions between the PCR primer sites may go undetected.
The truncating mutations (1930delAG in patient AS1, 3093delAAGA in patients AS2 and AS3) detected were considered to be causative of AS because they are known to lead to catalytically inactive translation products, because of an absence of the extreme carboxyl terminus of the E6-AP protein, the region necessary for the ubiquitination and degradation of the target protein (Huibregtse et al. 1995, Malzac et al. 1998). The mutation 1930delAG has also been described in three patients (Matsuura et al. 1997, Fung et al. 1998, Lossie et al. 2001) and the mutation 3093delAAGA in two other patients (Fung et al. 1998, Lossie et al. 2001). Together, these findings suggest that these sites may be prone to deletions in the UBE3A gene. The other recurrent mutations of the UBE3A gene reported so far consist of a base pair insertion in exon 9 (897insA) (Russo et al. 2000), a 5 bp insertion in exon 16 (3086ins5) (Kishino et al. 1997, Malzac et al. 1998), a C→T transition in exon 9 (2030C→T) (Malzac et al. 1998, Russo et al. 2000) and a deletion of the three nucleotides (2929del3) (Fang et al. 1999, Russo et al. 2000), which all have been found in two patients (Table 7).
The two missense mutations (902A→C inherited from the mother and 975T→C) identified here have not been described earlier. Both are located in exon 9, but outside the functionally important hect domain of the E6-AP protein, where three of the four missense mutations described to date have been found. Instead, these two mutations are located at the p53-binding domain (Huitbregtse et al. 1993b) or the transactivation domain (Nicholls and Knepper, 2001). Although their exact effect on protein structure or function is not known, they could lead to a deficiency in the binding of substrates to be ubiquitinated and degraded. Furthermore, their absence in the northern Finnish control population (200 alleles) supports the view that these DNA changes are not polymorphisms. Polymorphic DNA alterations, on the other hand, are known in the UBE3A gene (Table 7). The A178T polymorphism found in one AS patient and her father and in one control individual has been described previously (Matsuura et al. 1998, Malzac et al. 1998, Fang et al. 1999).
Three of the known six missense mutations have been located in the hect domain and the two mutations found in the present study in the transactivation domain, all leading to typical Angelman syndrome. The missense mutation (648G→A) reported in one patient with a mild phenotype (Matsuura et al.1997) was located outside the three domains known to be involved in ubiquitin-ligase activity, possible leading to a milder form of AS.
Including the point mutations described here, 46 different disease-causing mutations in the coding region of the UBE3A gene have been reported so far (Kishino et al.1997, Matsuura et al. 1997, Malzac et al. 1998, Tsai et al. 1998, Fang et al. 1999, Baumer et al. 1999, Ouweland van der et al. 1999, Russo et al 2000, Lossie et al. 2001) (table 7). Sixteen of these are deletions (14 frameshift mutations, one causing a deletion of one amino acid and one leading to the production of an elongated protein), 15 are insertions (14 frameshift, one producing a protein with one additional amino acid), 13 are point mutations (6 missense, 7 nonsense), one is a splicing mutation (frameshift), and one

75
shows a 26-bp deletion with a 1 bp (A) insertion at position 2230 (frameshift) (Table 7). In addition, one 7 bp deletion in intron 11 has been reported, but the effect of this deletion is not known (Ouweland van der et al. 1999). Approximately 48 % of these mutations are located in exon 9 (n=22), which is the largest exon. Exon 16 has also shown several mutations despite its relative small size (n=10, 22 % of mutations). Other mutations have been distributed as follows: four in the exons 8 and 15 (8.7% each), three in exon 12 (6.5%) and one mutation in the exons 10 and 11 and intron 9 (2.2% each). So far, no mutations have been detected in the non-coding exons or the exons 7, 13 and 14.
Of the 92 known patients with a UBE3A mutation, 57 have been familial (36% of the probands belonging to 20 families) and 35 sporadic (64%). All of the twenty mothers in the familial cases carried the mutation, and one had it in a mosaic form. In the sporadic cases, 6 of the 34 mothers studied (17%) were carriers, two of them mosaics. As to the grandfathers, five in the familial cases were reported to carry the mutation, and one of them was mosaic. Of the sporadic cases where the carrier mother was non-mosaic, one grandfather had the mutation. In most cases, the grandfathers were not studied or reported.
Of the 54 probands whose parents were studied, 28 (52%) had a new mutation, and 26 (48%) had inherited the mutation from the mother. Interestingly, in the families where the children had inherited the mutation from their mother, three of the 26 mothers (11%) were mosaics.
The cause of Angelman syndrome in the remaining four patients remained unknown. Whether these cases represent other mutations related to the UBE3A gene or its regulation or were caused by other possible genes, especially genes involved in the ubiquitin pathway or ones located at 15q11-q13 (e.g. maternally expressed ATP10C), remains to be elucidated.
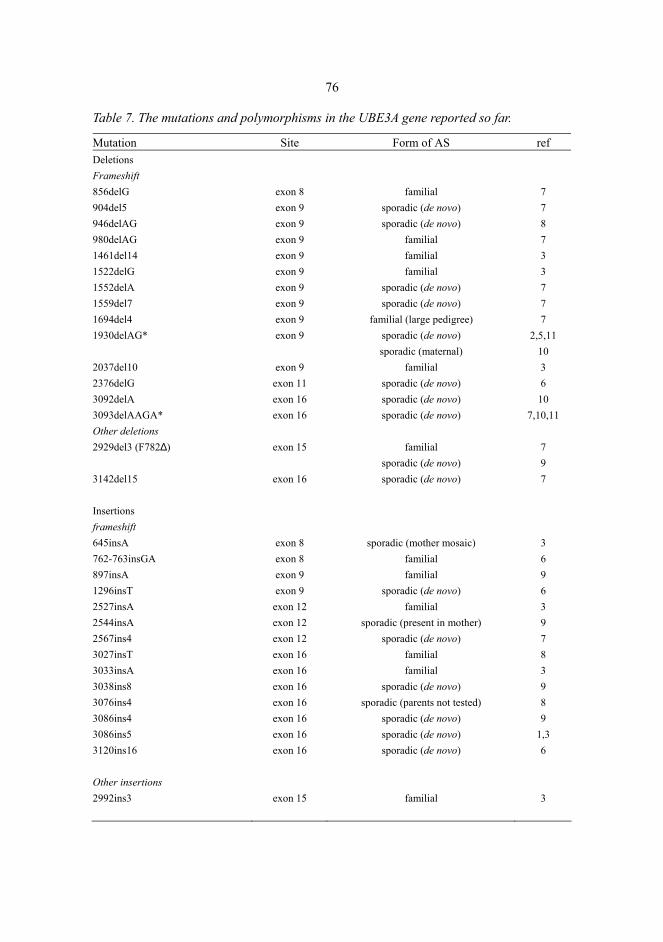
76
Table 7. The mutations and polymorphisms in the UBE3A gene reported so far.
Mutation Site Form of AS ref Deletions Frameshift 856delG 904del5 946delAG 980delAG 1461del14 1522delG 1552delA 1559del7 1694del4 1930delAG* 2037del10 2376delG 3092delA 3093delAAGA* Other deletions 2929del3 (F782∆) 3142del15 Insertions frameshift 645insA 762-763insGA 897insA 1296insT 2527insA 2544insA 2567ins4 3027insT 3033insA 3038ins8 3076ins4 3086ins4 3086ins5 3120ins16 Other insertions 2992ins3
exon 8 exon 9 exon 9 exon 9 exon 9 exon 9 exon 9 exon 9 exon 9 exon 9
exon 9 exon 11 exon 16 exon 16
exon 15
exon 16
exon 8 exon 8 exon 9 exon 9 exon 12 exon 12 exon 12 exon 16 exon 16 exon 16 exon 16 exon 16 exon 16 exon 16
exon 15
familial sporadic (de novo) sporadic (de novo)
familial familial familial
sporadic (de novo) sporadic (de novo)
familial (large pedigree) sporadic (de novo) sporadic (maternal)
familial sporadic (de novo) sporadic (de novo) sporadic (de novo)
familial
sporadic (de novo) sporadic (de novo)
sporadic (mother mosaic) familial familial
sporadic (de novo) familial
sporadic (present in mother) sporadic (de novo)
familial familial
sporadic (de novo) sporadic (parents not tested)
sporadic (de novo) sporadic (de novo) sporadic (de novo)
familial
7 7 8 7 3 3 7 7 7
2,5,11 10 3 6
10 7,10,11
7 9 7 3 6 9 6 3 9 7 8 3 9 8 9
1,3 6 3
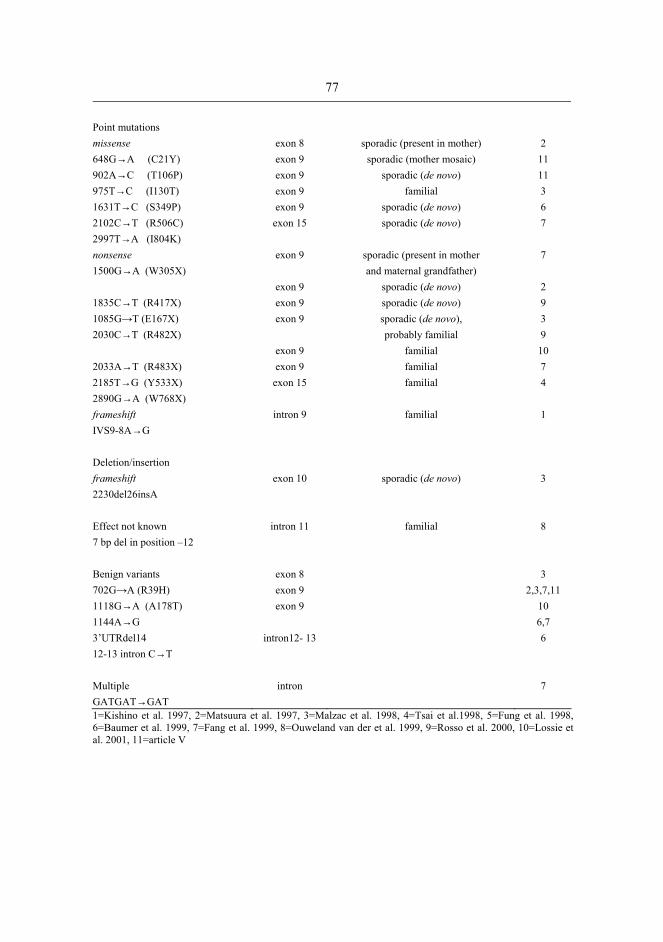
77
Point mutations missense 648G→A (C21Y) 902A→C (T106P) 975T→C (I130T) 1631T→C (S349P) 2102C→T (R506C) 2997T→A (I804K) nonsense 1500G→A (W305X) 1835C→T (R417X) 1085G→T (E167X) 2030C→T (R482X) 2033A→T (R483X) 2185T→G (Y533X) 2890G→A (W768X) frameshift IVS9-8A→G Deletion/insertion frameshift 2230del26insA Effect not known 7 bp del in position –12 Benign variants 702G→A (R39H) 1118G→A (A178T) 1144A→G 3’UTRdel14 12-13 intron C→T Multiple GATGAT→GAT
exon 8 exon 9 exon 9 exon 9 exon 9 exon 15
exon 9
exon 9 exon 9 exon 9
exon 9 exon 9 exon 15
intron 9
exon 10
intron 11
exon 8 exon 9 exon 9
intron12- 13
intron
sporadic (present in mother) sporadic (mother mosaic)
sporadic (de novo) familial
sporadic (de novo) sporadic (de novo)
sporadic (present in mother and maternal grandfather)
sporadic (de novo) sporadic (de novo) sporadic (de novo), probably familial
familial familial familial
familial
sporadic (de novo)
familial
2
11 11 3 6 7 7 2 9 3 9
10 7 4 1 3 8 3
2,3,7,11 10 6,7 6 7
1=Kishino et al. 1997, 2=Matsuura et al. 1997, 3=Malzac et al. 1998, 4=Tsai et al.1998, 5=Fung et al. 1998, 6=Baumer et al. 1999, 7=Fang et al. 1999, 8=Ouweland van der et al. 1999, 9=Rosso et al. 2000, 10=Lossie et al. 2001, 11=article V

78
6.6 Inheritance of defects and recurrence of Prader-Willi and Angelman syndromes
Prader-Willi and Angelman syndromes are caused by genetic mechanisms of several types, as already indicated. Deletions and uniparental disomies with normal parental chromosomes or non-deletion imprinting defects have been considered as de novo genetic errors that occur either pre- or postzygotically. Recurrence of the syndromes has been described only in families with structural chromosomal rearrangements, imprinting defects with a deletion in the imprinting centre (IC) or, in AS, UBE3A mutations.
Of the 84 deletion patients (53 PWS and 31 AS), 82 were sporadic cases representing new mutations with a low recurrence risk. In one family, the mother of one PWS child was a carrier of a Robertsonian translocation between the chromosomes 13 and 15 (der(13;15)(q10;q10)), which was not found in her child and which is not known to increase the risk for deletions. Recurring deletions of 15q11-q13 are extremely rare. So far, only one recurrent maternal, grandpaternally derived very small deletion involving only the loci D15S113, D15S10 and GABRB3 outside the critical region for PWS has been described in affected AS sibs (Saitoh et al. 1992). Chromosomal translocations (Smeets et al 1992, Qumsiyeh et al. 1992, Chan et al. 1993, Reeve et al. 1993, Jauch et al. 1995, Burke et al. 1996, Suzuki et al. 1996, Eliez et al. 1997, Krajewska et al. 1998) or inversions (Webb et al. 1992) involving the chromosome region 15q11-q13 have been reported to lead to deletions responsible for PWS or AS in the offspring.
In the family ASFV, two children with Angelman syndrome had identical large deletions of 15q11-q13 (type I) in the maternal chromosome 15. The mother’s both chromosomes 15 were structurally normal, which ruled out chromosomal rearrangements. The patients and their unaffected brother had inherited an identical, maternally derived haplotype distal to the deletion region, suggesting that all three shared the same grandparental chromosome 15. These findings suggest the presence of germ line mosaicism of del(15)(q11q13) in the mother, which would explain the recurrence of AS in this family. Germ line mosaicism has been reported to explain the unexpected recurrence of many genetic diseases, and it seems to be more frequent than expected (see Zlotogora 1998). In AS, germ line mosaicism for a microdeletion of the imprinting centre (IC) (Saitoh et al. 1996, Gilbert et al. 1997) has already been reported. Germ line mosaicism has also been described in other microdeletion syndromes (CATCH-22 at 22q11 (Hatchwell et al. 1998) and in Williams syndrome at 7q11.23 (Kara-Mostefa et al. 1999)). The present family is the first where the recurrence of a large deletion of 15q11-q13 has been shown to be due to germ line mosaicism. The previous estimated recurrence of less than 1% for deletions in PWS and AS, which was based on theoretical assumptions rather than observations, seems justified after this finding.
Uniparental disomy of chromosome 15 has never been described to recur in Prader-Willi or Angelman families, and all of the 16 cases in the present study were also sporadic. This can probably be explained by the origin of UPD, which is caused by prezygotic non-disjunction followed by postzygotic correction of the monosomic/trisomic zygote, both of which are spontaneous events. It has been shown that, even in UPD15 cases, the meiotic non-disjunction depends on maternal age, but its effect on the recurrence of PWS or AS is probably non-significant in view of the rarity of UPD. The

79
increased risk has been suggested to be related to the UPD in translocation families (Nicholls et al. 1989b, Malcolm et al. 1991, Smeets et al. 1992, Smith et al. 1997, Robinson et al. 2000). Parental gonadal mosaicism for trisomy 15 could theoretically also increase the risk for UPD. To date, as no confirmed recurrences of UPD15 have been reported, a recurrence risk of <1% has been accepted for purposes of genetic counselling.
As to the recurrence of imprinting defects in PWS and AS, the families fall into two categories: IC deletion cases with recurrence risks up to 50% and non-deletion IC cases with a low recurrence risk. In the present study, of the six families with an imprinting defect, one was found to have a microdeletion in the imprinting centre (AS family IV). In addition to the two affected children, this deletion was also found in the mother, the grandfather and two maternal uncles. A similar situation has been described in an even larger family (ASO, article III) with several healthy carriers with IC deletion. These families show that the mutation can be passed through several generations silently by male carriers. No such large pedigrees with IC deletion have been described in PWS so far.
The mechanism for nondeletion imprinting defects has been suggested to be due to a de novo imprint switch failure during parental gametogenesis and/or fertilization. This could explain why no recurrent cases are known and no detectable mutations in these patients have been described. The present two Prader-Willi and two Angelman patients with an imprinting defect without deletion or other observable alterations in the imprinting centre were sporadic. In both PWS patients, the incorrectly imprinted chromosome 15 was derived from the paternal grandmother. In AS family II the affected chromosome 15 was derived from the maternal grandfather and in AS family III from the maternal grandmother. Inheritance of the incorrectly imprinted chromosome from the maternal grandmother in AS appears to be an additional indicator for a low risk of familial recurrence. However, because the cause of incorrect imprinting in non-IC-deletion PWS and AS patients is not yet understood, recurrence of the syndromes in these cases cannot be absolutely excluded.
About half of the cases due to UBE3A mutations have been new mutations, while the other half have been inherited from the mother (Table 7). All the present five patients with UBE3A mutation were sporadic cases in their families. Four of them represented a new mutation with a low risk of recurrence of AS, while the mother in one case was mosaic for the mutation. The recurrence risk in this family depends on the ratio of mutant to normal germ cells. In addition to our family, three other families with either a female or a male mosaic carrier for UBE3A mutation have been described so far (Malzack et al. 1998). Of all carrier mothers known to date, 11% (3/26) are mosaic. In them, the transmission of the mutation to the offspring indicates that the mutation has occurred at a very early embryonic stage before the differentiation of the germinal cells, thus leading to both somatic and germ line mosaicism. It is also plausible that, in these mosaic carriers, the mutation has occurred in the inactive paternal UBE3A allele, as a mutation in the maternal allele could be assumed to lead to phenotypic changes. So far, no AS patients with UBE3A mutation mosaicism have been reported, in whom it could possibly cause a clinically milder form of the syndrome. Patients who are mosaic for deletion of 15q11-q13 (Tekin et al. 2000) or non-IC-deletion imprinting defects (Buiting et al. 2003) have already been described.

80
6.7 Laboratory evaluation of the Prader-Willi and Angelman syndromes
During the past ten years, our understanding of the basic genetic defects underlying the Prader-Willi and Angelman syndromes has allowed increasingly sophisticated laboratory methods to be used in the diagnosis of these syndromes, and laboratory diagnosis has become a necessary tool in resolving the aetiology. Clinical diagnoses of the Prader-Willi and Angelman syndromes remain difficult in many instances because of the individual variations in the phenotype and because the phenotype develops only with age. Knowledge of the basic genetic defect and its possible inheritance is also necessary for genetic counselling.
High-resolution chromosome analysis of the region 15q11-q13, which used to be the only available method, is no longer used, because more accurate molecular methods are available. This method was not accurate enough to detect all deletions, and it also missed uniparental disomies, imprinting defects and other molecular defects, such as UBE3A mutations. Chromosome analysis is, however, still used in, for instance, the evaluation of possible parental chromosomal rearrangements.
Fluorescence in situ hybridization (FISH) analysis with 15q11-q13 specific probes has proved to be a useful and sensitive method in the detection of deletions and cryptic translocations, and many laboratories even use FISH in the primary diagnosis of these two syndromes. Furthermore, in the detection of rare mosaicism of deletion 15q11-q13, FISH analysis can be considered the method of choice. In this study, it was used in specific cases to confirm the presence or absence of a suspected deletion and to exclude a cryptic structural chromosome translocation or inversion, but DNA methylation analysis was found the most useful primary method of diagnosis.
The DNA methylation test has become the method of choice in the primary diagnosis of the Prader-Willi and Angelman syndromes because it detects all cases with a deletion, uniparental disomy or imprinting defects, although additional studies are needed to distinguish between these aetiologies. In this study, of the three probes that detect DNA sequences with different maternal and paternal methylation patterns, the probes KB17 (SNRPN) and PW71B (D15S63) showed abnormal methylation imprinting for all types of deletions, while the small deletions (types III and IV) could not be detected by using the probe DN34 (ZNF127). Furthermore, the analysis of DN34 methylation bands required a comparison of the dosages of the different bands within the sample rather than a simple observation of the absence or presence of either maternal or paternal bands. The probe DN34 proved to be least consistent in a diagnostic assessment. All the three probes proved reliable in the detection of cases with uniparental disomies and imprinting defects. The DNA methylation test with the probe KB17 provided the most reliable means of diagnosing the PWS and AS. It turned out to be sensitive and specific for the detection of both syndromes, it gave easily detectable signals, and it identified all deletions, UPD and ID cases. Furthermore, it is the only probe that can be used in prenatal diagnosis (Kubota et al. 1996b, Slater et al. 1997, Glenn et al. 2000). The probe KB17 did not reveal diagnostically confusing rare deletion polymorphisms, as has been done by the probe PW71B (Buiting et al. 1999), and it did not yield the kind of false positive findings that were observed in one atypical patient (PAS1) in this study, who was studied for early

81
hypotonia and suspected PWS. The DNA methylation test with PW71B, however, showed a missing maternal contribution suggestive of AS, but a small deletion encompassing the loci from D15S13 to D15S63 outside the region critical for AS was found. The deletion was also present in the patient’s healthy mother. The results of the KB17 methylation test were normal, as was also the subsequent development of the child.
To distinguish between the different aetiologies in patients with abnormal methylation patterns, Southern blot hybridization with the hN4HS cDNA probe was used to detect deletions. In addition the SNRPN locus (15q12), it also detected DNA sequences from chromosome 6, which was used as an internal control in quantitative analysis, making the use of another control probe unnecessary. In the diagnostic setup, reference samples from both healthy individuals and deletion patients were used. The test proved to be simple and reliable, and no parental samples were needed.
In the non-deletion cases, DNA marker polymorphism study of the family was needed to identify either uniparental disomy or the presence of an imprinting defect. At the beginning of this study, only RFLP markers within the chromosome region 15q11-q13 were available. Of the nine early UPD cases, RFLP analysis detected six, in three of which only the six-allelic D15S24 locus was informative. Later, the use of highly informative microsatellite markers of chromosome 15 allowed both the detection of all uniparental disomy cases and the assessment of the origin of UPD in them. In the non-UPD cases, it also revealed the biparental inheritance of chromosome 15, which, in these patients, suggested the presence of an imprinting defect. For diagnostic purposes, five different 15q11-q13 specific microsatellite markers (D15S542, D15S11, D15S128, D15S122 and GABRB3) were selected and used simultaneously. In the case of suspected imprinting defects, further DNA methylation analysis with the PW71B and/or DN34 probes was found useful to confirm the presence of an imprinting defect. Deletion analysis of the imprinting centre was performed by using a quantitative Southern blot method with a battery of probes from the IC. The analysis of the IC deletion was done in collaboration with the laboratory of the Institute of Human Genetics, Univeritätsklinikum Essen, Germany, which had long-term experience of research in this field.
In the Angelman cases where the DNA methylation test was normal, conformation-sensitive gel electrophoresis and sequencing were found practical in the detection of UBE3A mutations. It was assumed that, by using modified conditions, the vast majority of mutations could be detected. In this study, sequencing was used to characterize the observed abnormal CSGE products, but it was not applied to the four patients with normal CSGE findings.
In UBE3A analysis, as is the situation with many other genetic diseases, the observed changes could not necessarily be directly interpreted as causal defects of Angelman syndrome. In order to rule out harmless polymorphisms, a control series was needed to check whether any of the observed point mutations could be found in healthy individuals. The observed changes also had to be interpreted in terms of their abnormal effect on protein function, and the literature had to be searched for similar changes to facilitate interpretation. Especially the presence of the mutation in the father, which was the case in patient AS5, was suggestive of polymorphism.
In Prader-Willi syndrome the diagnoses of practically all patients have been confirmed by modern laboratory methods: in the few cases where the methylation test was negative, chromosome analysis has shown the presence of translocation of chromosome 15. As

82
regards Angelman syndrome, however, clinically typical AS patients have been reported - four patients in the present study - where the aetiology of the syndrome has remained unsettled. Further studies will be needed to show whether mutations in the non-coding region of UBE3A or mutations in some other genes of 15q11-q13 or involving the ubiquitin pathway could lead to this syndrome.
The diagnoses of the Prader-Willi and Angelman syndromes require close co-operation between clinicians and laboratories, both for the interpretation of unexpected findings in some instances and for the analysis of the parents to detect the possible hereditary forms or an increased risk of recurrence in the family. Analysis of the parental molecular status was found useful in almost all instances in the present study.

7 Concluding remarks
The major observations and conclusions of this study are as follows:
1. In a Finnish series of Prader-Willi and Angelman patients, the different types of aetiologic genetic defects were found in approximately the same proportions as reported elsewhere. Deletions accounted for 76% of PWS and 67% of AS patients, uniparental disomies for 21% of PWS and 2% of AS patients and imprinting defects for 3% of PWS and for 11% of AS patients, and in Angelman syndrome, UBE3A mutations accounted for 11% of patients. In a minority of AS patients, the aetiology could not be established.
2. All Prader-Willi and most Angelman cases – including all non-IC-deletion imprinting defects and most UBE3A mutation cases – were caused by new mutations and did not carry an increased recurrence risk in the families. The rare inherited cases in this series appeared either familial (IC deletion in one AS family and del(15)(q11q13) in another AS family) or sporadic (UBE3A mutation in one AS family). Interestingly, in a rare case of recurring deletion in Angelman syndrome, the defect was inherited through maternal germ line mosaicism. Although rare, possible inheritance of the syndromes needs to be taken into consideration in genetic counselling.
3. The deletion was paternally derived in Prader-Willi patients and maternally derived in Angelman patients. Although 88% of PWS and all AS deletions were large deletions (type I or II), smaller deletions were also found (type III and IV). In this series, the smallest overlap of the deletion was from locus D15S13 to locus D15S10, being proximal to the GABA receptor genes, for PWS, and from locus D15S128 to locus D15S12, being distal to the D15S63 locus, for AS. The size of the deletion did not seem to have practical clinical consequences. The presence of smaller deletions helped both to define the critical region for these syndromes and to develop diagnostic probes for the detection of deletions.

84
4. Uniparental disomy of chromosome 15, maternal in PWS and paternal in AS, was much more frequent in Prader-Willi syndrome than in Angelman syndrome, in corcondance to other studies. In Prader-Willi syndrome, UPD was mostly due to maternal meiotic non-disjunction, followed by trisomic rescue of the zygote, and it was associated with advanced maternal age (mean age 34.6 years). Most of the maternal UPD cases were heterodisomic (error in the first meiotic segregation), although parts of the chromosome 15 were isodisomic in some cases. In Angelman syndrome, a rare error in the paternal second meiotic segregation was detected as a cause of UPD in one patient.
5. Most of the imprinting defects detected here were non-IC-deletion cases (two PWS and three AS patients), which were thought to be due to a de novo imprinting switch failure during gametogenesis or fertilization. In these and other non-IC deletion patients, the maternally imprinted maternal chromosome region was inherited from the paternal grandmother in Prader-Willi syndrome. In Angelman syndrome, the paternally imprinted maternal chromosome region was inherited from either the maternal grandfather or the maternal grandmother. These observations suggest that a paternal imprint developed either in the maternal germ line or postzygotically, and that the paternal imprint may be the default imprint. In one sib pair with Angelman syndrome, a microdeletion of the imprinting centre was found. IC deletions were shown to occur in multiple unaffected members and to be passed silently through males across several generations. The smallest overlap of imprinting centre deletions in Angelman syndrome was defined to be 1.15 kb.
6. UBE3A mutations were confirmed to represent an important aetiologic group in AS and need to be searched for in patients with normal DNA methylation tests. The mutations 1930delAG and 3093delAAGA detected here have been described concurrently in four cases, suggesting that these sites may be prone to deletions in the UBE3A gene. Also, two novel disease-causing missense mutations (902A→C and 975T→C) were found. Most of the mutations detected here, as also described elsewhere, are clustered to exon 9, which is the largest exon. Because most of the UBE3A mutations have been unique, the possibility of polymorphisms must be taken into consideration in the interpretation of the molecular changes. For diagnostic purposes, CSGE turned out to be a relatively simple, inexpensive and fast mutation screening method, which allowed more frequent use of intron primers than SSCP, making it easier to avoid processing of the UBE3AP2 pseudogene.
7. The genetic diagnoses of the Prader-Willi and Angelman syndromes may be complicated and require molecular analyses at several levels. Although laborious, testing of the parents – and sometimes even other relatives – is necessary both for diagnosis and for the determination of the possible mechanism of inheritance. As the primary diagnostic test of both syndromes, the DNA methylation test proved to be the method of choice. On the basis of its results, further examinations could be designed. In practice, all Prader-Willi patients could be diagnosed by the available molecular methods, while a group of Angelman syndrome patients without an identifiable molecular defect was found. These patients might have a mutation in another gene, e.g. the gene affecting the ubiquitin pathway, and further research is required to resolve the genetic aetiology of these patients.

Reference
Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G & Beaudet AL (1997) Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet 17: 75-78.
American Society of Human Genetics/American College of Mdical Genetics Test and Technology Transfer Committee (1996) Diagnostic testing for Prader-Willi and Angelman syndromes: report of the ASHG/ACMG test and technology transfer committee. Am J Hum Genet 58: 1085-1088.
Amos-Landgraf JM, Ji Y, Gottlieb W, Depinet T, Wandstrat AE, Cassicy SB, Driscoll DJ, Rogan PK, Schwartz & Nicholls RD (1999) Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet 65: 370-386.
Angelman H (1965) Puppet children. A report on three cases. Dev Med Chlid Neurol 7: 681-688. Antonarakis SE, Avramopoulos D, Blouin J-L, Talbot CC & Schinzel AA (1993b) Mitotic (somatic
cell) errors cause trisomy 21 in about 4.5% of cases and are not associated with advanced maternal age. Nature genet 3: 146-150.
Antonarakis SE, Blouin J-L, Maher J, Avramopoulos D, Thomas G & Talbot CC (1993a) Maternal uniparental disomy for human chromosome 14, due to loss of a chromosome 14 from somatic cells with t(13;14) trisomy 14. Am J Hum Genet 52: 1145-1152.
Antonarakis SE, Petersen MB, McInnis MG, Adelsberger PA, Schinzel AA, Binkert F, Pangolas C, Raoul O, Slaugenhaupt SA & Hafez M (1992) The meiotic stage of nondisjunction in trisomy 21: determination by using DNA polymorphisms. Am J Hum Genet 51: 35-40.
Aviv A & Aviv H (1998) Telomeres, hidden mosaicism, loss of heterozygosity and complex genetic traits. Hum Genet 103: 2-4.
Bannister A, Zegerman P, Partridge J, Miska E, Thomas J, Allshire R & Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromosome domain. Nature 410 :120-124.
Barber JCK, Cross IE, Douglas F, Nicholson J, Moore KJ & Browne CE (1998) Neurofibromatosis pseudogene amplification underlines euchromatic cytogenetic duplications and triplications of proximal 15q. Hum Genet 103: 600-607.
Bartolomei MS & Tilgham SM (1997) Genomic imprinting in mammals. Annu Rev Genet 31: 493-525.
Baumer A, Balmer D & Schinzel A (1999) Screening for UBE3A gene mutations in a group of Angelman syndrome patients selected according to non-stringent clinical criteria. Hum Genet 105: 598-602.
Beaudet AL & Jiang Y (2002) A Rheostat Model for a Rapid and Reversible Form of Imprinting-Dependent Evolution. Am J Hum Genet 70: 1389-1397.

86Bettio D, Rizzi N, Giardino D, Gurrieri F, Silvestri G, Grugni G & Larizza L (1997) FISH
characterization of small supernumerary marker chromosomes in two Prader-Willi patients. Am J Med Genet 68: 99-104.
Beuten J, Mangelschots K, Buntix I, et al. (1993) Molecular study of chromosome 15 in 22 patients with Angelman syndrome. Hum Genet 90:489-495.
Bielinska B, Blaydes SM, Buiting K, Yang T, Krajewska-Walasek M, Horsthemke B & Brannan CI (2000) De novo deletions of SNRPN exon 1 in early human and mouse embryos result in a paternal to maternal imprint switch. Nat Genet 25: 74-78.
Boccaccio I, Glatt-Deeley H, Watrin F, Roëckel N, Lalande M & Muscatelli F (1999) The human MAGEL2 gene and its mouse homologue are paternally expressed and mapped to the Prader-Willi region. Hum Mol Genet 8: 2497-2505.
Bottani A, Robinson WP, DeLozier-Blanchet CD, Engel E, Morris MA, Schmitt B, Thun-Hohenstein L & Schinzel A (1994) Angelman syndrome due to paternal uniparental disomy of chromosome 15: a milder phenotype? Am J Med Genet 51: 35-40.
Bower B & Jeavons P (1967) The ‘happy puppet’ syndrome. Arch Dis Child 42:294-298. Brannan CI & Bartolomei MS (1999) Mechanisms of genomic imprinting. Curr Opin Genet & Dev
9: 164-170. Braun R (2001) Packaging paternal chromosomes with protamines. Nat Genet 28: 10-12. Browne CE, Dennis NR, Maher E, Long FL, Nicholson JC, Sillibourne J & Barber CK (1997)
Inherited interstitial duplications of proximal 15q: Genotype-phenotype correlations. Am J Hum Genet 61: 1342-1352.
Buchler EM, Rossier R, Bodis I, Vulliet V, Buchler UK & Stalder G (1963) Chromosomal translocations in a mentally deficient child wioth cryptochidism. Acta Paediat Scand 52: 177-182.
Buckley RH, Dinno N & Weber P (1998) Angelman syndrome: Are the Estimates Too Low? Am J Med Genet 80: 385-390.
Buiting K, Barnicoat A, Lich C, Pembrey M, Malcolm S & Horsthemke B (2001) Disruption of the bipartite imprinting center in a family with Angelman syntrome. Am J Hum Genet 68: 1290-1294.
Buiting K, Dittrich B, Dworniczak B, Lerer I, Abeliovich D, Cottrell S, Temple IK, Harvey JF, Lich C, Groß S & Horsthemke B (1999c) A 28-kb deletion spanning D15S63 (PW71) in five families: a rare neutral variant? Am J Hum Genet 65: 1588-1594.
Buiting K, Dittrich B, Groß, Greger V, Lalande M, Robinson W, Mutirangura A, Ledbetter D & Horsthemke B (1993) Molecular definition of the Prader-Willi syndrome chromosome region and orientation of the SNRPN gene. Hum Mol Genet 2; 1991-1994.
Buiting K, Färber C, Kroisel P, Wagner K, Brueton L, Robertson ME, Lich C & Horsthemke B (2000) Imprintig centre deletions in two PWS families: implications for diagnostic testing and genetic counselling. Clin Genet 58: 284-290.
Buiting K, Groß S, Ji Y, Senger G, Nicholls RD & Horsthemke B (1998) Expressed copies of the MN7 (D15F37) gene family map close to the common deletion breakpoints in the Prader-Willi/Angelman syndromes. Cytogenet Cell Genet 81: 247-253.
Buiting K, Groß S, Lich C, Gillessen-Kaesbach G, El-Maarri O & Horsthemke B (2003) Epimutations in Prader-Willi and Angelman syndrome: A Molecular Study of 136 Patients with an Impritning Defect. Am J Hum Genet 72: 000-000. (Electronic version)
Buiting K, Kaya-Westerloh S & Horsthemke B (1996) A pseudogene for the human ribosomal protein L5 (RPL5P1) maps within an intron of the SNRPN transcription unit on human chrosomsome 15. Cytogenet Cell Genet 75: 224-226.
Buiting K, Korner C, Ulrich B, Wahle E & Horsthemke B (1999b) The human gene for the poly(A)-specific ribonuclease (PARN) maps to 16p13 and has a truncated copy in the Prader-Willi/Angelman syndrome region on 15q11 q13. Cytogenet Cell Genet 87:125-131.
Buiting K, Lich C, Cottrell S, Barnicoat A & Horsthemke B (1999a) A 5-kb imprinting center deletion in a family with Angelman syndrome reduces the shortest region of deletion overlap to 880 bp. Hum Genet 105: 665-666.

87Buiting K, Saitoh S, Groß S, Dittrich B, Schwartz S, Nicholls RD & Horsthemle B (1995) Inherited
microdeletions in the Angelman and Prader-Willi syndromes define an imprinting center on human chromosome 15. Nat Genet 9: 395-400.
Burd L, Vesely B, Martsolf J & Kerbeshian J (1990) Prevalence study of Prader-Willi syndrome in North Dakota. Am J Med Genet 37: 97-99
Burke LW, Wiley JE, Glenn CC, Driscoll DJ, Loud KM, Smith AJ & Kushnick T (1996) Familial cryptic translocation resulting in Angelman syndrome: Implications for imprinting or location of the Angelman gene? Am J Hum Genet 58: 777-784.
Bürger J, Buiting K, Dittrich B, Groß S, Lich C, Sperling K, Horsthemke B & Reis A (1997) Different mechanism and recurrence risks of imprinting defects in Angelman syndrome. Am J Hum Genet 61: 88-93.
Bürger J, Kunze J, Sperling K & Reis A (1996) Phenotypic differences in Angelman syndrome patients: imprinting patients show less frequently microcephaly and hypopigmentation than deletions. Am J Med Genet 66: 221-226.
Butler MG (1990) Prader-Willi Syndrome: Current Understanding of cause and diagnosis. Am J Med Genet 35: 319-332.
Butler MG (1995) High resolution chromosome analysis and fluorescence in situ hybridization in patients referred for Prader-Willi or Angelman syndrome. Am J Med Genet 56: 420-422.
Butler MG, Christian SL, Kubota T & Ledbetter DH (1996) A 5-year-old white girl with Prader-Willi syndrome and a submicroscopic deletion of chromosome 15q11q13. Am J Med Genet 65: 137-141.
Butler MG, Meaney FJ & Palmer CG (1986) Clinical and cytogenetic survey of 39 individuals with Prader-Labhart-Willi syndrome. Am J Med Genet 23: 793-809.
Butler MG & Palmer CG (1983) Parental origin of chromosome 15 deletion in Prader-Willi syndrome (letter). Lancet 1: 1285-1286.
Carrozzo R, Rossi E, Christian SL, Kittikamron K, Livieri C, Corrias A, Pucci L, Fois A, Simi P, Bosio L, Beccaria L, Zuffardi O & Ledbetter DH (1997) Inter- and intrachromosomal rearrangements are both involved in the origin of 15q11-q13 deletions in Prader-Willi syndrome. Am J Hum Genet 61: 228-231.
Cassidy SB (1997a) Prader-Willi syndrome. J Med Genet 34: 917-923. Cassidy SB (1997b) Recurrence risk in Prader-Willi syndrome. Am J Med Genet 28: 59-60. Cassidy SB, Conroy J, Becker L & Schwartz S (1996) Paternal triplication of 15q11-q13 in a
hypotonic, developmentally delayed child without Prader-Willi or Angelman syndrome. Am J Med Genet 62: 206-207.
Cassidy SB, Dykens E & Williams C (2000) Prader-Willi and Angelman syndrome: Sister Imprintes Disorders. Am J Med Genet 97:136-146.
Cassidy SB, Forsythe M, Heeger S, Nicholls RD, Schork N, Benn P & Schwartz S (1997c) Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. Am J Med Genet 68: 433-440.
Cassidy SB, Gainey AJ & Butler MG (1989) Occupational hydrocarbon exposure among fathers of Prader-Willi syndrome patients with and without deletions of 15q. Am J Hum Genet 44: 806-810.
Cassidy SB, Lai L-W, Erickson RP, Magnuson L, Thomas E, Gendron R & Herrmann J (1992) Trisomy 15 with loss of the paternal 15 as a cause of Prader-Willi syndrome due to maternal disomy. Am J Hum Genet 51: 701-708.
Cassidy SB & Schwartz S (1998) Prader-Willi and Angelman syndromes:disorers of genomic imprinting. Medicine 77: 140-151.
Cattanach BM, Barr JA, Evans EP, Burtenshaw M, Beechey CV, Leff SE, Brannan CI, Copeland NG, Jenkins NA & Jones J (1992) A candidate mouse model for Prader-Willi syndrome which shows an absence of Snrpn expression. Nat Genet 2: 270-274.
Cavaille j, Buiting K, Kiefmann M, Lalande M, Brannan CI, Horsthemke B, Bachellerie JP, Brosius J & Huttenhoffer A (2000) Identification of tandemly-repeated C/D snoRNA genes at

88the imprinted human 14q32 domain reminiscent of those at the Prader-Willi/Angelman syndrome region. Hum Mol Genet11: 1527-1538.
Chaillet JR, Knoll JHM, Horsthemke B & Lalande M (1991) The syntenic relationship between the critical deletion region for the Prader-Willi/Angelman Syndromes and proximal mouse chromosome 7. Genomics 11: 773-776.
Chan CT, Clayton-Smith J, Cheng XJ, Buxton J, Webb T, Pembrey ME & Malcom S (1993): Molecular mechanisms in the Angelman syndrome: A survey of 93 patients, J Med Genet 30: 895-902.
Chen K-S, Manian P, Koeuth T, Potocki L, Zhao Q, Chinault C, Lee CC & Rupski JR (1997) Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet 17: 154-163.
Chotai KA & Payne SJ (1998) A rapid, PCR based test for differential molecular diagnosis of Prader-Willi and Angelman syndromes. J Med Genet 35: 472-475.
Christian SL, Fanters JA, Mewborn SK, Huang G & Ledbetter DH (1999) Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum Mol Genet 8: 1025-1037.
Christian SL, Robinson WP, Huang B, Mutirangura A, Line MR, Nakao M, Surti U, Chakravarti A & Ledbetter DH (1995) Molecular characterisation of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am J Hum Genet 57: 40-48.
Christian SL, Smith AC, Macha M, Black SH, Elder FF, Johnson JM, Resta RG, Surti U, Suslak L, Verp MS & Ledbetter DH (1996) Prenatal diagnosis of uniparental disomy 15 following trismoy 15 mosaicism. Prenatal Diag 16: 323-332.
Ciechanover A & Schwartz AL (1998) The ubiquitin-proteasome pathway: The complexity and myriad functions of proteins death. Proc Natl Acad Sci 95: 2727-2730.
Clayton-Smith J, Driscoll DJ, Waters MF, Webb T, Andrews T, Malcolm S, Pembrey ME & Nicholls RD (1993b) Difference in methylation patterns with the D15S9 region of chromosome 15q11-13 in first cousins with Angelman syndrome and Prader-Willi syndrome. Am J Med Genet 47: 683-686.
Clayton-Smith J & Pembrey ME (1992) Angelman syndrome. J Med Genet 29: 412-415. Clayton-Smith J, Webb T, Cheng XJ, Pembrey ME & Malcolm (1993a) Duplication of
chromosome 15 in the region 15q11-13 in a patient with developmental delay and ataxia with similarities to Angelman syndrome. J Med Genet 30: 529-531.
Clayton-Smith J, Webb T, Pembrey ME, Nicholls M & Malcom S (1992) Maternal origin of deletion 15q11-q13 in 25/25 cases of Angelman syndrome. Hum Genet 88: 376-378.
Coerwinkel-Driessen M, Shepens J, Zandvoort van, Oos B van, Mariman E & Wieringa B (1988) NcoI RFLP at the creatine kinase muscle type gene locus (CKMM, chromosome 19). Nucleic Acids Res 16: 8743.
Cohen-Fix O, Peters JM, Kirschner MW & KoshlandD (1996) Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev 10: 3081-3093.
Connerton-Moyer KJ, Nicholls RD, Schwartz S, Driscoll DJ, Hendrickson JE, Williams CA & Pauli RM (1997) Unexpected familial recurrence in Angelman syndrome. Am J Med Genet 70: 253-260.
Conroy JM, Grebe TA, Becker LA, Tsuchiya K, Nicholls RD, Buiting K, Horsthemke B, Cassidy SB & Schwartz S (1997) Balanced translocation 46,XY,t(2;15)(q37.2;q11.2) associated with atypical Prader-Willi syndrome. Am J Hum Genet 61: 388-94.
Cook EH, Lingren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, Lord C & Courchesne E (1997) Autism or atypical autism in maternally but not paernally derived proximal 15q duplication. Am J Hum Genet 60: 928-934.
Cotton RGH (1993) Current methods of mutation detection. Mutat Res 285: 125-144. Cuoco C, Bicocchi MP, Granata D, Mezzano P & Serra G (1990) De novo (15;21) unbalanced
translocation of paternal origin in a girl with Prader-Willi syndrome. Am J Med Genet 37: 62-64.

89DeChiara TM, Robertson EJ & Efstratiadis A (1991) Parental imprinting of the mouse insulin-like
growth factor II gene. Cell 64: 849-859. Delach JA, Rosengren SS, Kaplan L, Greenstein RM; Cassidy SB & Benn PA (1994) Comparison
of high resolution chromosome banding and fluorescence in situ hybridization (FISH) for the laboratory evaluation of Prader-Willi syndrome and Angelman syndrome. Am J Med Genet 52: 85-91.
DeLorey TM, Handroth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD & Olsen RW (1998) Mice lacking the β2 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci 18: 8505-8514.
Dewald G, Stallard R, Bader PI, chen K, Zenger-Hain J, Harris CJ, Higgins R, Hirsch B, Hsu W-T, Johnson E, Kubic V, Kurczynski TW, Malone JM, McCorquodale DJ, Meilinger K, Meisner LF, Moore JW, Schwartz S, Siembieda S, Storto PD, Vance G, Van Tuinen P, Wiktor A & Yung J-F (1996) Toward quality assurance for metaphase FISH: a multi-center experience. Am J Med Genet 64: 539-545.
Dittrich B, Buiting K & Horsthemke B (1996b) PW71 methylation test for Prader-Willi and Angelman syndromes. Am J Med Genet 61: 196-197.
Dittrich B, Buiting K, Korn B, Rickard S, Buxton J, Saitoh S, Nicholls RD, Poustka A, Winterpacht A, Zabel B & Horsthemke B (1996a) Imprint switching on human chromosome 15 may involve alternative transcripts of the SNRPN gene. Nat Genet 14: 163-170.
Dittrich B, Robinson WP, Knoblauch H, Buiting K, Schmidt K, Gillessen-Kaesbach G & Horsthemke B (1992) Molecular diagnosis of the Prader-Willi and Angelman syndromes by detection of parent-of-origin specific DNA methylation in 15q11-q13. Hum Genet 90: 313-315.
Donlon TA (1988) Similar molecular deeltion on chromosome 15q11.2 are encountered in both the Prader-Willi and Angelman syndromes. Hum Genet 80: 322-328.
Donlon TA, Lalande M, Wyman A, Bruns G & Latt SA (1986) Isolation of molecular probes assiciated with the chromosome 15 instability in the Prader-Willi syndrome. Proc Natl Acad Sci USA 83: 4408-4412.
Down JL (1887) Mental Affections of Childhood and Youth. London: Churchill (pub.) pp. 179. Driscoll DJ, Waters MF, Williams CA, Zori RT, Glenn CC, Avidano KM & Nicholls RD (1992) A
DNA methylation imprint, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes. Genomics 13: 918-924.
Driscoll DJ (1994) Genomic imprinting in humans. Molec Genet Med 4: 37-77. Duckett DP, Roberts SH & Davies P (1984) Unbalanced reciprocal translocations in cases of
Prader-Willi syndrome. Hum Genet 67: 156-161. Dupont J-M, Le Tessier D, Rabineau D, Cuisset L, Vasseur C, Jeanpierre M, Delepech M, Pinton
F, Ponsot G & DeNavit M-F (1999) Unexpected Angelman syndrome molecular defect in a girl displaying clinical features of Prader-Willi syndrome. J Med Genet 36: 652-654.
Dutrillaux B & Lejeune J (1971) Sur une novelle technique d’analyse du caryotype humain. C R Acad Sci (D) Paris 272: 2638-2640.
Dutrillaux B & Viegas-pequignot E (1981) High resolution R- and G-banding on the same preparation. Hum Genet 57: 93-95.
Dykens EM, Cassidy SB & King BH (1999) Maladaptive behaviour differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. Am J Ment Retard 104: 67-77.
Ekstrom TJ, Cui H, Li X & Ohlsson R (1995) Promoter-specific IGF2 imprinting status and its plasticity during human liver development. Development 121: 309-316.
Elder FF, Nichols MM, Hood OJ & Harrsion WR (1985) Unbalanced translocation (15;17) (q13;13.3) with apparent Prader-Willi syndrome but without Miller-Dieker syndrome. Am J Med Genet 20: 519-524.
Eliez S, Morris MA, Dahoun-Hadorn S, DeLozier-Blanchet CD, Gos A, Sizonenko P & Antonarakis SE (1997) Familial translocation t(Y;15)(q12;p11) and De Novo deletion of the Prader-Willi syndrome (PWS) critical region on 15q11-q13. Am J Med Genet 70: 222-228.

90El-Maarri O, Buiting K, Peery EG, Kroisel PM, Balaban B, Wagner K, Urman B, Heyd J, Lich C,
Brannan CI, Walter J & Horsthemke B (2001) Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nat Genet 27: 341-344.
Eng C & Vijg J (1997) Genetiv testing: the problems and the promises. Nat Biotechnol 15: 422-426.
Engel E (1993) Uniparental disomy revisited: The first twelve years. Am J Med Genet 46: 670-674. Engel E (1996) Uniparental disomy and genome imprinting: an overview. Acta Geneticae Medicae
et Gemellologiae 45: 19-39. Engel E (1997) Uniparental disomy (UPD). Annales de Genetique 40: 24-34. Engel E (1998) Uniparental disomies in unselected populations. Am J Hum Genet 63: 962-966. Erdel M, Schuffenhauer S, Bucholz B, Barth-Witte U, Köchl S, Utermann B, Duba H-C &
Utermann G (1996) Routine screening for microdeletions by FISH in 77 patients suspected of having Prader-Willi or Angelman syndromes using YAC clone 273A2 (D15S10). Hum Genet 97: 784-793.
Everman DB & Cassidy SB (2000) Genetics of childhood disorders: XII. Genomic imprinting: breaking the rules. J Am Acad Child Adolesc Psychiatry 39: 386-389.
Falls JG, Pulford DJ, Wylie AA & Jirtle RL (1999) Genomic imprinting: implications for human disease. Am J Pathol 154: 635-647.
Fang P, Lev-Lehman E, Tsai T-F, Matsuura T, Benton CS, Sutcliffe JS, Christian SL, Kubota T, Halley DJ, Meijers-Heijboer H, Langlois S, Graham JM, Beuten J, Willems PJ, Ledbetter DH & Beaudet AL (1999) …Hum Mol Genet 8: 129-135.
Feinberg AP (2000) DNA methylation, genomic imprinting, and cancer. Curr Top Microbiol Immunol 249: 87-99.
Fernandez F, Berry C & Mutton D (1987) Prader-Willi syndrome in siblings, due to unbalanced translocation between chromosomes 15 and 22. Arch Dis Child 62: 841-843.
Fournier C, Goto Y, Ballestar E, Delaval K, Hever A, Esteller M & Feil R (2002) Allele-specific histone lysine methylation marks regulatory regions at imprinted mouse genes. The EMBO Journal 21: 6560-6570.
Fraccaro M, Zuffardi O, Buhler EM & Jurik LP (1977) 15/15 translocation in Prader-Willi syndrome. J Med Genet 14: 275-276.
Freeman SB, May KM, Pettay D, Fernhoff PM & Hassold TJ (1993) Paternal uniparental disomy in a child with a balanced 15;15 translocation and Angelman syndrome. Am J Med genet 45(5): 625-630.
Fridman C & Koiffmann CP (2000a) Origin of uniparental disomy 15 in patients with Prader-Willi or Angelman sydnrome. Am J Med Genet 94: 249-253.
Fridman C, Santos M, Ferrari I & Koiffmann CP (2000c) A further Angelman sydnrome patient with UPD15 due to paternal meiosis II nondisjunction. Clin Genet 57: 86-87.
Fridman C,Varela MC, Kok F, Diament A & Koiffmann CP (2000b) Paternal UPD15: Further genetic and clinical studies in four Angelman syndrome patients. Am J Med Genet 92: 322-327.
Fulmer-Smentek SB & Franckie U (2001) Association of acetylated histones with paternally expressed genes in the Prader-Willi deletion region. Hum Mol Genet 10; 645-652.
Fung DCY, Yu B, Cheong KF, Smith A & Trent RJ (1998) UBE3A “mutations” in two unrelated and phenotypically different Angelman syndrome patients. Hum Genet 102: 487-492.
Färber C, Dittrich B, Buiting K & Horstshemke B (1999) The chromosome 15 imprinting centre (IC) region has undergone multiple duplication events and contains an upstream exon of SNRPN that is deleted in all Angelman syndrome patients with an IC microdeletion. Hum Mol Genet 8: 337-343.
Gabriel JM, Gray TA, Stubbs L, Saitoh S, Ohta T & Nicholls RD (1998) Structure and function correlations at the imprinted mouse Snrpn locus. Mamm Genome 9: 788-793.
Gabriel JM, Merchant M, Ohta T, Ji Y, Galdwell RG, Ramsey MJ, Tucker JD, Longnecker R & Nicholls RD (1999) A transgene insertion creating a heritable chromosome deletion mouse model of Prader-Willi and Angelman syndromes. Genetics 96: 9258-9263.

91Gallagher RC, Pils B, ALbalwi M & Francke U (2002) Evidence for the Role of PWCR1/HBII-85
C/D Box Small nucleolar RNAs in Prader-Willi Syndrome. Am J Hum Genet 71: 669-678. Ganguly A & Prockop DJ (1995) Detection of mismatched bases in douple stranded DNA by gel
electrophoresis. Electrophoresis 16: 1830-1835. Ganguly A & Williams C (1997) Detection of mutations in multi-exon genes: Comparison of
conformation sensitive gel electrophoresis and sequencing strategies with respect to cost and time finding mutations. Hum Mutat 9: 339-343.
Gardner JM, Nakatsu Y, Gondon Y, Lee S, Lyon MF, King RA & Brilliant MH (1992) The mouse pink-eyed dilution gene: Association with human Prader-Willi and Angelman syndromes. Science 257: 1121-1124.
Gardner RJM & Sutherland GC (1996) “Chromosome abnormalities and genetic counselling”, Second edition. Oxford University Press, New York, p. 269.
Gérard M, Hernandez L, Wevrick R & Stewart CL (1999) Disruption of the mouse necdin gene results in early post-natal lethality. Nat Genet 23: 199-202.
Gilbert HL, Buxton JL, Chan CTJ, McKay T, Cottrell S, Ramsden S, Winter RM, Pembrey ME & Malcom S (1997) Counselling dilemmas associated with the molecular characterisaton of two Angelman syndrome families. J Med Genet 34: 651-655.
Gillessen-Kaesbach G, Albrecht B, Passarge E & Horsthemke B (1995c) Further patient with Angelman syndrome due to paternal disomy of chromosome 15 and a milder phenotype. Am J Med Genet 56: 328-329.
Gillessen-Kaesbach G, Demuth S, Thiele H, Theile U, Lich C & Horsthemke B (1999) A previously unrecognised phenotype characterised by obesity, muscular hypotonia, and ability to speak in patients with Angelman syndrome caused by an imprinting defect. Eur J Hum Genet 7: 638-644.
Gillessen-Kaesbach G, Gross S, Kaya-Westerloh S, Passarge E & Horsthemke B (1995b) DNA methylation based testing of 450 patients suspected of having Prader-Willi syndrome. J Med Genet 32: 88-92.
Gillessen-Kaesbach G, Robinson W, Lohmann D, Kaya-Westerloh S, Passarge E & Horsthemke B (1995a) Genotype-phenotype correlation in a series of 167 deletion and non-deletion patients with Prader-Willi syndrome. Hum Genet 96: 638-643.
Ginsburg C, Fokstuen S & Schinzel A (2000) The contribution of uniparental disomy to congenital development defects in children born to mothers at advanced childbearing age. Am J Med Genet 95: 454-460.
Glavac D & Dean M (1995) Applications of heteroduplex analysis for mutation detection in disease genes. Hum Mutat 6: 281-287.
Glenn CC, Deng G, Michaelis RC, Tarleton J, Phelan MC, Surh L, Yang TP & Driscoll DJ (2000) DNA methylation analysis with respect to prenatal diagnosis of the Angelman and Prader-Willi syndromes and imprinting. Prenat Diag 20: 300-306.
Glenn CC, Nicholls RD, Robinson WP, Saitoh S, Niikawa N, Schinzel A, Horsthemke B & Driscoll DJ. (1993b): Modification of 15q11-q13 DNA methylation imprints in unique Angelman and Prader-Willi patients. Hum Mol Genet 2: 1377-1382.
Glenn CC, Porter KA, Jong MTC, Nicholls RD & Driscoll DJ (1993a): Functional imprinting and epigenetic modification of the human SNRPN gene. Hum Mol Genet 2; 2001-2005.
Glenn CC, Saitoh S, Jong MTC, Filbrandt MM, Surti U, Driscoll DJ & Nicholls RD (1996): Gene structure, DNA methylation, and imprinted expression of human SNRPN gene. Am J Hum Genet 58: 335-346.
Glotzer M, Murray AW & Kirschner MW (1991) Cyclin is degraded by the ubiquitin pathway. Nature 349: 132-138.
Gray TA, Saitoh S & Nicholls RD (1999) An imprinted, mammalian biscitronic transcript encodes two independent proteins. Genetics 96: 5616-5621.
Greally JM, Gabriel JM, Song L, Zemel S & Nicholls RD (1999) Conserved characteristics of heterochromatin-forming DNA at the 15q11-q13 imprinting center. Proc Natl Acad Sci 96: 14430-14435.

92Greger V, Knoll JH, Wagstaff J, Woolf E, Lieske P, Glatt H, Benn PA, Rosengren SS & Lalande M
(1997) Angelman syndrome associated with an inversion of chromosome 15q11.2-q24.3. Am J Med Genet 60: 574-580.
Greger V, Reis A & Lalande M (1994) The critical region for Angelman syndrome lies between D15S122 and D15S113. Am J Med Genet 53: 396-398.
Gregory CA, Kirkilionis AJ, Greenberg CR, Chudley AE & Hamerton JL (1990) Detection of molecular rearrangements in Prader-Willi syndrome patients by using genomic probes recognizing four loci within the PWCR. Am J Med Genet 35: 536-545.
Grunstein M (1997) Histone acetylation in chromatin structure and transcription. Nature 389: 349-352.
Gunaratne PH, Nakao M, Ledbetter DH, Sutcliffe JS & Chinault AC (1995) Tissue-specific and allele-specific replication timing control in the imprinted human Prader-Willi syndrome region. Genes Dev 9: 808-820.
Gunay-Aygun M, Heeger S, Schwartz S & Cassidy SB (1997) Delayed diagnosis in patients with Prader-Willi syndrome due to maternal uniparental disomy 15. Am J Med Genet 71: 106-110.
Guanti G (1980) A new case of rearrangement of chromosome 15 associated with Prader-Willi syndrome. Clin Genet 17: 423-427.
Gyftodiou J, Karadima G, Pandelia E, Vassilopoulos D & Petersen MB (1999) Angelman syndrome with uniparental disomy due to paternal meiosis II nondisjunction. Clin Genet 55: 483-486.
Halford S, Lindsay E, Nayudu M, Carey AH, Baldini A & Scambler PJ (1993) Low-copy-number repeat sequences flank the DiGeorge/velocardio-facial synrome loci at 22q11. Hum Mol Genet 2: 191-196.
Hall JG (1990) Genomic imprinting: review and relavance to human diseases. Am J Hum Genet 46: 857-873.
Hamabe J, Fukushima Y, Harada N, Abe K, Matsuo N, Nagai T, Yoshioka A, Tonoki H, Tsukino R & Niikawa N (1991a) Molecular study of the Prader-Willi syndrome: deletion, RFLP, and phenotype analyses of 50 patients. Am J Med Genet 41: 54-63.
Hamabe J, Kuroki Y, Imaizumi K, Sugimoto T, Fukushima Y, Yamaguchi A, Izumikawa Y & Niikawa N (1991b) DNA deletion and its parental origin in Angelman syndrome patients. Am J Med Genet 41: 64-68.
Hassold T, Abruzzo M, Adkins K, Griffin D, Merrill M, Millie E, Saker Shen J & Zaragoza M (1996) Human aneuploidy: incidence, origin and etiology. Environ Mol Mutat. 28: 167-175.
Hassold TJ, Merrill M, Adkins K, Freeman S & Sherman S (1995) Recombination and maternal age-dependent nondisjunction: molecular studies of trisomy 16. Am J Hum Genet 57: 867-874.
Hatchwell E, Long F, Wilde J, Crolla J & Temple K (1998) Molecular Confirmaiton of Germ Line Mosaicism for a Submicroscopic Deletion of Chromosome 22q11. Am J Med Genet 78: 103-106.
Hawkley CJ & Smithies A (1976) The Prader-Willi syndrome with a 15 – 15 translocation: case report and review of the litteratue. J Med Genet 13: 152-7.
Hayward B, Kamiya M, Strain L, Moran V, Campell R, Hayashizaki Y & Bonthron D (1998) The human GNAS1 gene is imprinted and encodes distinct paternally and biallelically G proteins. Proc Natl Acad Sci. USA. 95: 10038-10043.
Hershko A & Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67: 425-479. Herzing LBK, Kim SJ, Cook EH & Ledbetter DH (2001) The human aminophospholipid-
transporting ATPase gene ATP10C maps adjacent to UBE3A and exhibits similar imprinted expression. Am J Hum Genet 68: 1501-1505.
Hochstrasser M (1996a) Protein degradation or regulation: Ub the judge. Cell 84: 813-815. Hochstrasser M (1996b) Ubiquitin-dependent protein degradation. Ann Rev Genet 30: 405-439. Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY & Greenberg F
(1993) Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics 91: 398-402.

93Holopainen IE, Metsähonkala E-L, Kokkonen H, Parkkola RK, Manner TE, Någren K & Korpi ER
(2001) Decreased binding of [11C] flumazenil in Angelman syndrome patients with GABAA receptor β3 subunit deletions. Ann Neurol 49: 110-113.
Horstshemke B, Dittrich B & Buiting K (1997) Imprinting mutations on human chromosome 15. Hum Mutat 10: 329-337.
Horsthemke B, Maat-Kievit A, Sleegers E, van den Ouweland A, Buiting K, Lick C, Mollevanger P, Beverstock G, Gillessen-Kaesbach G & Schwanitz G (1996) Familial translocations involving 15q11-q13 can give a rise to interstitial deletions causing Prader-Willi or Angelman syndrome. J Med Genet 33: 848-851.
Huang B, Crolla JA, Christian SL, Wolf-Ledbetter ME, Macha ME, Papenhausen PN & Ledbetter DH (1997) Refined molecular characterization of the breakpoints in small inv dup(15) chromosomes. Hum Genet 99: 11-17.
Huang L, Kinnucan E, Guangli W, Beaudenon S, Howley PM, Huibregtse JM & Pavletich NP (1999) Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science 286: 1321-1326.
Huibregtse JM, Scheffner M, Beaudenon S & Howley PM (1995) A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc Natl Acad Sci 92: 2563-2567.
Huibregtse JM, Scheffner M & Howley PM (1993a): Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol Cell Biol 13: 775-784.
Huibregtse JM, Scheffner M & Howley PM (1993b) Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol Cell Biol 13: 4918-4927.
Hulten M, Hardy G, Gould C, Stergianou R, Waters J & McKeown C (1992) Molecular cytogenetics of Prader-Willi and Angelman syndromes. Lancet 339: 243-244.
Hurst LD & McVean GT (1997) Growth effect of uniparental disomies and the conflict theory of genomic imprinting. Trends Genet 13: 436-443.
Imaizumi K, Takada F, Kuroki Y, Naritomi K, Hamabe J & Niikawa N (1990) Cytogenetic and molecular study of the Angelman syndrome. Am J Med Genet 35: 314-318.
Ishikawa T, Kibe T & Wada Y (1996) Deletion of small nuclear ribonucleoprotein polypeptide N (SNRPN) in Prader-Willi syndrome detected by fluorescence in situ hybridization: Two sibs with the typical phenotype without a cytogenetic deletion in chromosome 15q. Am J Med Genet 62: 350-352.
James RS, Temple IK, Dennis NR & Crolla JA (1995) A search for uniparental disomy in carriers of supernumerary marker chromosomes. Eur J Hum Genet 2: 21-26.
Jauch A, Robson L & Smith A (1995) Investigations with fluorescence in situ hybridization (FISH) demonstrate loss of the telomeres on the reciprocal chromosome in three unbalanced translocations involving chromosome 15 in the Prader-Willi and Angelman syndromes. Hum Genet 96: 345-349.
Jay V, Becker LE, Chan FW & Perry TL Sr. (1991) Puppet-like syndrome of Angelman: a pathologic and neurochemical study. Neurology 41: 416-422.
Jay P, Rougeulle C, Massacrier A, Moncla A, Mattei MG, Malzac P, Roeckel N, Taviaux S, Lefrane JL, Cau P, Berta P, Lalande M & Muscatelli F (1997) The human necdin gene, NDN, is maternally imprinted and located in he Prader-Willi syndrome chromosomal region. Nature Genet 17: 357-361.
Jenuwein T & Allis JF (2001) Translating the histone code. Science 293: 1074-1080. Ji Y, Rebert NA, Joslin JM, Higgins MJ, Schultz RA & Nicholls RD (2000) Structure of the hihgly
conserved HERC2 gene and of multiple partially duplicated paralogs in human. Genome Res 10: 319-329.
Ji Y, Walkowicz MJ, Buiting K, Johnson DK, Tarvin RE, Rinchik EM, Horsthemke B, Stubbs L & Nicholls RD (1999) The ancestral gene for transcribed, low-copy repeats in the Prader-Willi/Angelman region encodes a large protein implicated in protein trafficking, which is

94deficient in mice with neuromuscular and spermiogenic abnormalities. Hum Mol Genet 8: 533-542.
Jiang Y, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Seatt JD & Beaudet AL (1998b) Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21: 799-811.
Jiang Y, Lev-Lehman E, Bressler J, Tsai T-F & Beaudet AL (1999) Genetics of Angelman syndrome. Am J Hum Genet 65: 1-6.
Jiang Y, Tsai T-F, Bressler J & Beaudet AL (1998a) Impriting in Angelman and Prader-Willi syndromes. Curr Opin Genet Dev 8: 334-342.
Jong MTC, Gray TA, Ji Y, Glenn CC, Saitoh S, Driscoll DJ & Nicholls RD (1999) A novel imprinted gene, encoding a RING zing-finger protein, and overlapping antisense transript in the Prader-Willi syndrome critical region. Hum Mol Genet 8: 783-793.
Kaplan LC, Wharton R, Elias E, Mandell F, Donlon T & Latt S (1987) Clinical heterogenity associated with deletions in the long arm of chromosome 15: report of 3 new cases and their possible genetic significance. Am J Med Genet 28: 45-53.
Kara-Mostefa A, Raoul O, Lyonnet S, Amile J, Munnich A, Vekemans M, Magnier S, Ossareh B & Bonnefont J-P (1999) Recurrent Williams-Beuren Syndrome in a Sibship Suggestive of Maternal Germ-Line Mosaicism. Am J Hum Genet 64: 1475-1478.
Kawame H, Gartler SM & Hansen RS (1995) Allele-specific replication timing in imprinted domains: absence of asynchrony at several loci. Hum Mol Genet 4: 2287-2293.
Khan NL & Wood NW (1999) Prader-Willi and Angelman syndromes: update on genetic mechanisms and diagnostic complexities. Curr Opin Neurol 12: 149-154.
Kehrer-Sawatzki H, Schwickardt T, Assum G, Rochhi M & Krone W (1997) A third neurofibromatosis type I (NF1) pseudogene at chromosome 15q11.2. Hum Genet 100: 595-600.
King RA, Wiesner GL, Townsend D & White JG (1993) Hypopigmentation in Angelman syndrome. Am J Med Genet 46: 40-44.
Kishino T, Lalande M & Wagstaff J (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nature Genet 15: 70-73.
Kishino T & Wagstaff J (1998) Genomic oorganization of the UBE3A/E6-AP gene and related pseudogenes. Genomics 47: 101-107.
Kitsberg D, Selig S, Brandeis M, Smon I, Keshet I, et al. (1993) Allele-specific replication timing of imprinted gene regions. Nature 364: 459-463.
Knoll JHM, Chem S-D & Lalande M (1994) Allele specificity of DNA replication timing in the Angelman/Prader-Willi syndrome imprinted chromosomal region. Nature Genet 6: 41-46.
Knoll JH, Glatt KA, Nicholls RD, Malcom S & Lalande M (1991): Chromosome 15 uniparental disomy is not frequent in Angelman syndrome. Am J Hum Genet 48(1): 16-21.
Knoll JHM, Nicholls RD & Lalande M (1989b) On the parental origin of the deletion in Angelman syndrome. Hum Genet 83: 205-206.
Knoll JHM, Nicholls RD, Magenis RE, Glatt K, Graham JM Jr, Kaplan L & Lalande M (1990) Angelman syndrome: three molecular classes identified with chromosome 15q11-q13 specific DNA markers. Am J Hum Genet 47: 149-155.
Knoll JMH, Nichols RD, Magenis RE, Graham JM Jr, Lalande M & Latt SA (1989a) Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in the parent of origin of the deletion. Am J Med Genet 32: 285-290.
Kokkonen H (1992) Prader-Willin oireyhtymään liittyvän kromosomideleetion tunnistaminen molekyyligeneettisin keinoin. Tutkielma. Perinnöllisyystieteen laitos. Oulun yliopisto.
Kosaki K, McGinniss MJ, Veraksa AN, McGinnis WJ & Jones KL (1997) Prader-Willi and Angelman syndromes: diagnosis with a bisulfite-treated methylation-specific PCR method. Am J Med Genet 73: 308-313.
Kotzot D (1999) Abnormal Phenotypes in Uniparental Disomy (UPD): Fundamental Aspects and a Critical Review With Bibliography of UPD Other Than 15. Am J Med Genet 82: 265-274.-587.

95Kotzot D, Schmitt S, Bernasconi F, Robinson WP, Lurie IW, Ilyina H, Mehes K, Hamel BC, Otten
BJ, Hergersberg M, Werder E, Schönle E & Schinzel A (1995) Uniparental disomy 7 in Silver-Russel syndrome and primordial growth retardation. Hum Mol Genet 4: 583-587.
Krajewska WM, Gukowska A, Bielinska B, Goryluk-Kozakiewicz B & Popowska E (1998) A case of Prader-Willi syndrome arising as a result of familial unbalanced translocation t(11;15)(q25;q13). Clin Genet 54: 60-64.
Kubota T, Niikawa N, Jinno Y & Ishimaru T (1994) GABAA receptor beta 3 subunit gene is possibly paternally imprinted in humans. Am J Med Genet 49: 452-453.
Kubota T, Sutcliffe JS, Aradhya S, Gillessen-Kaesbach G, Christian SL, Horsthemke B, Beaudet AL & Ledbetter DH (1996a) Validation studies of SNRPN methylation as a diagnostic test for Prader-Willi syndrome. Am J Med Genet 66: 77-80.
Kubota T, Aradhya S, Macha M, Smith AC, Surh LC, Satish J, Verp MS, Nee HL, Johnson A, Christan SL & Ledbetter DH (1996b) Analysis of parent of origin specific DNA methylation at SNRPN and PW71 in tissues: implication for prenatal diagnosis. J Med Genet 33: 1011-1014.
Kubota T, Das S, Christian SL, Baylin SB, Herman JG & Ledbetter DH (1997) Methylation-specific PCR simplifies imprinting analysis. Nat Genet 16: 16-17.
Kucerova M, Strakova M & Polikova Z (1979) The Prader-Willi syndrome with a 15-3 translocation. J Med Genet 16: 234-235.
Kumar S, Kao WH & Howley PM (1997) Physical interaction between specific E2 and Hect E3 enzymes determines functional cooperativity. J Biol Chem 272: 13548-13554.
Kupke KG & Muller U (1989) Parental origin of the extra chromosome in trisomy 18. Am J Hum Genet 45: 599-605.
Kuslich CD, Kobori JA, Mohapatra G, Gregorio-King C & Donlon TA (1999) Prader-Willi syndrome is caused by disruption of the SNRPN gene. Am J Hum Genet 64: 70-76.
Kuwano A, Mutirangura A, Dittrich B, Buiting K, Horsthemke B, Saitoh S, Niikawa N, Ledbetter SA, Greenberg F, Chinault AC, et al. (1992) Molecular dissection of the Prader-Willi syndrome region (15q11-q13) by YAC cloning and FISH analysis. Hum Mol Genet 1: 417-425.
Körkkö J, Annunen S, Pihlajamaa T, Prockop DJ & Ala-Kokko L (1998) Conformation sensitive gel electrophoresis for simple and accurate detection of mutations: comparison with denaturing gradient gel electrophoresis and nucleotide sequencing. Proc Natl Acad Sci 95: 1681-1685.
Lai W, Erickson RP & Cassidy SB (1993) Clinical correlates of chromosome 15 deletions and maternal disomy in Prader-Willi syndrome. Am J Dis Child 147: 1217-1223.
Lalande M (1996) Parental imprinting and human disease. Annual Reviews of Genetics 30: 173-195.
Lalande M, Dryja TP, Schreck RR, Shipley J, Flint A & Latt SA (1984) Isolation of human chromosome 13-specific DNA sequences cloned from flow sorted chromosomes and potentially linked to the retinoblastoma locus. cancer Genet Cytogenet 13: 283-295.
LaSalle JM & Lalande M (1995) Domain organization of allele-specific replication within the GABRB3 gene cluster requires a biparental 15q11-13 contribution. Nat Genet 9: 386-394.
LaSalle J & Lalande M (1996) Homologous association of oppositely imprinted chromosomal domains. Science 272: 725-728.
Ledbetter DH & Engel E (1995) Uniparental disomy in humans: development of an imprinting map and its implications for prenatal diagnosis. Hum Mol Genet 4: 1757-1764.
Ledbetter DH, Greenberg F, Holm VA & Cassidy SB (1987) Conference report: second annual Prader-Willi syndrome scientific conference. Am J Med Genet 28: 779-790.
Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan B & Crawford JD (1981) Deletion of chromosome 15 as a cause of the Prader-Willi syndrome. New Eng J Med 304: 325-329.
Lee MP, DeBaun MR, Mitsuya K, Galonek HL, Brandenburg S, Oshimura M & Feinberg AP (1999) Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent ofinsulin-like growth factor II imprinting. Proc Natl Acad Sci 96: 5203-5208.

96Lee MP, Hu R, Johnson LA & Feinberg AP (1997) Human KVLQT1 gene shows tissue specific
imprinting and encompasses Beckwith-Wiedemann syndrome chromosomal rearrangements. Nat Genet 15: 181-185
Lee S, Kozlov S, Hernandez L, Chamberlain SJ, Brannan CI, Stewart CL & Wevrick R (2000) Expression and imprinting of MAGEL2 suggest a role in Prader-Willi syndrome and the homologous murine imprinting phenotype. Hum Mol Genet 9: 1813-1819.
Lee ST, Nicholls RD, Schnur RE, Guida LC, Lu-Kuo J, Spinner NB, Zackai EH & Spitz RA (1994) Diverse mutations of the P gene among African-Americans with type II (tyrosinase-positive) oculocutaneous albinism (OCA2). Hum Mol Genet 3: 2047-2051.
Lee S & Wevrick R (2000) Identification of novel imprinted transcripts in the Prader-Willi syndrome and Angelman syndrome deletion region: further evidence for regional imprinting control. Am J Hum Genet 66: 848-858.
Legius E, Marchuck DA, Hall BK, Andersen LB, Wallace MR, Collins FS & Glover TW (1992) NF1-related locus on chromosome 15. Genomics 13: 1316-1318.
Lerchner W & Barlow DP (1997) Paternal repression of the imprinted mouse Igf2r locus occurs during implantation and is stable in all tissues of the postimplantation mouse embryo. Mech Dev 61: 141-149.
Lohmann DR, Brandt B, Höpping W, Passarge E & Horsthemke B (1996) The spectrum of RB1 germline mutations in hereditary retinoblastoma. Am J Hum Genet 58: 940-949.
Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, Hutson A, Nicholls RD, Zori RT, Williams CA & Driscoll DJ (2001) Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet 38: 834-845.
Lubinski M, Zellweger H, Greenswag L, Larson G, Hansmann I & Ledbetter D (1987) Familial Prader-Willi syndrome with apparently normal chromosomes. Am J Med Genet 28: 37-43.
Lupski JR (1998) Charcot-Marie-Tooth disease: lessons in genetic mechanisms. Mol Med 4: 3-11. Lyko F, Buiting K, Horshtemke B & Paro R (1998) Identification of a silencing element in the
human 15q11-q13 imprinting center by using transgenic Drosophila. Genetics 95: 1698-1702. MacDonald H & Wevrick R (1997) The necdin gene is deleted in Prader-Willi syndrome and its
imprinted in human and mouse. Hum Mol Genet 6: 1873-1878. Malcolm S, Clayton-Smith J, Nichols M, Robb S, Webb T, Armour JA, Jeffreys AJ, Pembrey ME.
et al. (1991): Uniparental paternal disomy in Angelman’s syndrome. Lancet 337: 694-697. Malcolm S & Donlon TA (1994) Report of the second international workshop on human
chromosome 15 mapping 1994. Cytogeget Cell Genet 67: 1-22. Malzac P, Webber H, Moncla A, Graham Jr JM, Kukolich M, Williams C, Pagon RA, Ramsdell
LA, Kishino T & Wagstaff J (1998) Mutation analysis of UBE3A in Angelman syndrome patients. Am J Hum Genet 62: 1353-1360.
Mann JR & Lowell-Badge RH (1984) Inviability of parthenogenones is determined by ronuclei, nott egg cytoplasm. Nature 310: 66-67.
Mann MRW & Bartolomei MS (1999) Towards a moleculare understanding of Prader-Willi and Angelman syndromes. Hum Mol Genet 8: 1867-1873.
Mann MRW & Bartolomei MS (2000) Maintaining imprinting. Nat Genet 25: 4-5. Martin RH, Ko E & Rademaker A (1991) Distribution of aneuploidy in human gametes:
comparison between human sperm and oocytes. Am J Med Genet 39: 321-331. Mascardi MJ, Gottlieb W, Rogan PK, Butler MG, Waller DA, Armour JAL, Jeffreys AJ, Ladda RL
& Nicholls RD (1992) The frequency of uniparental disomy in Prader-Willi syndrome. N Engl J Med 326: 1599-1607
Matsuura T, Sutcliffe JS, Fang P, Galjaard SB, Jiang Y, Benton CS, Rommens JM & Beaudet AL (1997) De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet 15: 74-77.
Mattei JF, Mattei MG & Giraud F (1983) Prader-Willi syndrome and chromosome 15: a clinical discussion of 20 cases. Hum Genet 64: 356-362.
McEntagart ME, Webb T, Hardy C & King MD (2000) Familial Prader-Willi syndrome: case report and a literature review. Clin Genet 58: 216-223.

97McGrath J & Solter D (1983) Nuclear transplantationin mouse embryos. J Exp Zool 228: 355-362. McGrath J & Solter D (1984) Completion of mouse embryogenesis requires both the maternal and
paternal genomes. Cell 37: 179-183. Meguro M, Kashiwagi A, Mitsuya K, Nakao M, Kondo I, Saitoh S & Oshimura M (2001b) A novel
maternally expressed gene, ATP10C, encodes a putative aminophospholipid translocase associated with Angelman syndrome. Nat Genet 28: 19-20.
Meguro M, Mitsuya K, Nomura N, Kohda M, Kashiwagi A, Nishigaki R, Yoshioka H, Nakao M, Oishi M & Oshimura (2001a) Large-scale evaluation of imprinting status in the Prader-Willi syndrome region: an imprinted direct repeat cluster resembling small nucleolar RNA genes. Hum Mol Genet 10: 383-394.
Meguro M, Mitsuya K, Sui H, Shigenami K, Kugoh H, Nakao M & Oshimura M (1997) Evidence for uniparental, paternal expression of the human GABAA receptor subunit genes, using microcell-mediated chromosome transfer. Hum Mol Genet 6; 2127-2133.
Michaelis RC, Skinner SA, Lethco BA, Simensen RJ, Donlon TA, Tarleton J & Phelan MC (1995) Deletions involving D15S113 in a mother and son without Angelman syndrome: refinement of the Angelman syndrome critical deletion region. Am J Med Genet 55: 120-126.
Minssian BA, DeLorey TM, Olsen RW, Philippart M, Rronsten Y, Zhang Q, Guerrini R, Ness PV, Livet MO & Delgado-Escueta AV (1998) Angelman syndrome: correlations between syndrome phenotypes and genotypes. Ann Neurol 43: 485-493.
Ming JE, Blagowidow N, Knoll JHM, Rollings L, Fortina P, McDonald-McGinn DM, Spinner NB & Zackai EH (2000) Submicroscopic deletion in cousins with Prader-Willi syndrome causes a grandmatrilineal inheritance pattern: effects of imprinting. Am J Med Genet 92: 19-24.
Mitchell J, Schnizel A, Langlois S, Gillessen-Kaesbach G, Schuffenhauer S, Michaelis R, Abeliovich D, Lerer I, Christian S, Guitart M, McFadden DE & Robinson WP (1996) Comparison of phenotype in uniparental disomy and deletion Prader-Willi syndrome: Sex specific differences. Am J Med Genet 65: 133-136.
Moncla A, Malzac P, Livet M-O, Voelckel M-A, Mancini J, Delaroziere JC, Philif N & Mattei J-F (1999b) Angelman syndrome resulting from UBE3A mutations in 14 patients from eight families: clinical manifestations and genetic counselling. J Med Genet 36: 554-560.
Moncla A, Malzac P, Voelckel M-A, Auquier P, Girardot L, Mattei M-G, Philip N, Mattei J-F, Lalande M & Livet M-O (1999a) Phenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur J Hum Genet 7: 131-139.
Moore T & Haig D (1991) Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet 7: 45-49.
Morison IM & Reeve AE (1998) A catalogue of imprinted genes and parent-of-origin effects in humans and animals. Hum Mol Genet 7: 1599-1609.
Muir WJ (2000) Genetics advances and learning disability. Br J Psychiat 176: 12-19. Muscatelli F, Abrous DN, Massacrier A, Boccaccio I, Moal ML, Cau P & Cremer H (2000)
Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum Mol Genet 9: 3101-3110.
Mutirangura A, Greenberg F, Butler MG, Malcolm S, Nicholls RD, Chakravarti A & Ledbetter DH (1993b) Multiplex PCR of three dinucleotidi repeats in the Prader-Willi/Angelman critical region (15q11-13): molecular diagnosis and mechanism of uniparental disomy. Hum Mol Genet 2: 143-151.
Mutirangura A, Jayakumar A, Sutcliffe JS, Nakao M, McKinney MJ, Buiting K, Horsthemke B, Beaudet AL, Chinault AC & Ledbetter DH (1993a) A complete YAC contig of the Prader-Willi/Angelman chromosome region (15q11-q13) and refined localization of the SNRPN gene. Genomics 18: 546-552.
Nagaoka H, Ozawa K, Matsuda F, Hayashida H, Matsumura R, Haimo M, Shin EY, Fukita Y, Iman T, Anand R, Yokoyama K, Eki T, Soeda E & Honjo T (1994) Recent translocation of variable and diversity segments of human immunoglobulin havy-chain from chromosome 14 to chromosome 15 and 16. Genomics 22: 189-197.

98Nakada Y, Taniura H, Uetsuki T, Inazawa J & Yoshikawa K (1998) The human chromosomal gene
for necdin, a neuronal growth suppressor, in the Prader-Willi syndrome deletion region. Gene 213: 65-72.
Nakao M, Sutcliffe JS, Durtschi B, Mutirangura A, Ledbetter DH & Beaudet AL (1994) Imprinting analysis of three genes in the Prader-Willi/Angelman region: SNRPN, E6-associated protein, and PAR-2 (D15S225E). Hum Mol Genet 3: 309-315.
Nawaz Z, Lonard DM, Smith CL, Lev-Lehman E, Tsai SY, Tsai M-J & O’Malley BW (1999) The Angelman syndrome – associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol Cell Biol 19: 1182-1189.
Nayak A & Browning MD (1999) Presynaptic and postsynaptic mechanism of long-term potentiation. Adv Neurol 79: 645-658.
Nicholls RD (1993) Genomic imprinting and uniparental disomy in Angelman and Prader-Willi syndromes: A review. Am J Med Genet 46: 16-25.
Nicholls RD (1998) Strange bedfellows? Protein degradation and neurological dysfunction. Neuron 21: 647-9.
Nicholls RD (1999) Incriminating gene suspects, Prader-Willi style (news; comment). Nat Genet 23: 132-134.
Nicholls RD (2000) The impact of genomic imprinting for neurobehavioral and developmental disorders. J Clin Invest 105: 413-418.
Nicholls RD, Gottlieb W, Avidano KWilliams CA & Driscoll D (1991) Mouse chromosome mapping of clones of from PWS/AS genetic region. Mouse Genome 89: 254.
Nicholls RD & Knepper JL (2001) Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet 2: 153-175.
Nicholls RD, Knoll JH, Butler MG, Karam S & Lalande M (1989b) Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature 342: 281-285.
Nicholls RD, Knoll JH, Glatt K, Hersh JH, Brewster TD, Graham Jr JM, Wurster-Hill D, Wharton R & Latt SA (1989a) Restriction fragment length polymorphisms within proximal 15q and their use in molecular cytogenetics and the Prader-Willi syndrome. Am J Med Genet 33: 66-77.
Nicholls RD, Ohta T & Gray TA (1999) Genetic abnormalities in Prader-Willi syndrome and lessons from mouse models. Acta Paediatr Suppl 88:99-104.
Nicholls RD, Pai GS, Gottlieb W & Cantu ES (1992) Paternal uniparental disomy of chromosome 15 in a child with Angelman syndrome. Ann Neurol 32: 512-518.
Nicholls RD, Saitoh S & Horsthemke B (1998) Imprinting in Prader-Willi and Angelman syndromes. TIG 14: 194-200.
Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O’Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE & Kouzarides T (2001) Rb targets histone H3 methylation and HP1 to promoters. Nature 412: 561-565.
Niikawa N & Ishikiriyama S (1985) Clinical and cytogenetic studies of the Prader-Willi syndrome: evidence of phenotype-karyotype correlation. HumGenet 69: 22-27.
Ning Y, Roschke A, Christian SL, Lesser J, Sutcliffe JS & Ledbetter DH (1996) Identification of a novel paternally expressed transcript adjacent to snRPN in the Prader-Willi syndrome critical region. Genome Res 6; 742-746.
Orita M, Iwahana H, Kanazawa H, Hayashi K & Sekiya T (1989a) Detection of polumorphisms of human DNA by gel electrophoresis as single-stranded conformation polymorphisms. Proc Natl Acad Sci USA 86: 2766-2770.
Orita M, Suzuki Y, Sekiya T & Hayashi K (1989b) Rapid and sensitive detection of point mutations and DNA polymorphisms using the polymerase chain reaction. Genomics 5: 874-879.
Ouweland van den AM, van der Est MN, Wesby-van Swaay E, Tijmensen TS, Los FJ, van Hemel JO, Hennekam RC, Meijers-Heijboer HJ, Niermeijer MF & Halley DJ (1995) DNA diagnosis of Prader-Willi and Angelman syndromes with the probe PW71 (D15S63). Hum Genet 95: 562-567.

99Pai GS & Thomas GH (1980) A new R-banding technique in clinical cytogenetics. Hum Genet 54:
41-45. Park JP, Moeschler JB, Hani VH, Hawk AB, Belloni DR, Noll WW & Mohandas TK (1998)
Maternal disomy and Prader-Willi syndrome consistent with gamete complementation in a case of familial translocation (3;15) (p25;q11.2). Am J Med Genet 78: 134-139.
Pembrey M, Fennel SJ, Van den Berghe J, Fitchett M, Summer D, Butler L, Clarke C, Griffiths M, Thompson E, Super M & Baraitser M (1989) The assocation of Angelman’s syndrome with deletions within 15q11-13. J Med Genet 26: 73-77.
Pentao L, Wise CA, Chinault AC, Patel PI & Lupski JR (1992) Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nature Genet 2: 292-300.
Perez Jurado LA, Peoples R, Kaplan P, Hamei BCJ & France U (1996) Molecular definition of the chromosome 7 deletion in Williams syndrome and parent-of-origin effects on growth. Am J Hum Genet 59: 781-792.
Petersen MB, Brondum-Nielsen K, Hansen LK & Wulff K (1995) Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: Estimated prevalence rate in Danish county. Am J Med Genet 60: 261-262.
Pettigrew AL, Gollin SM, Greenberg F, Ficcardi VM & Ledbetter DH (1987) Duplication of proximal 15q as a cause of Prader-Willi syndrome. Am J Med Genet 28: 791-802.
Pfeifer K (2000) Mechanisms of genomic imprinting. Am J Hum Genet 67: 777-787. Prader A, Labhart A & Willi H (1956) Ein Syndrom von Adipositas, Kleinwuchs, Kryptorchismus
und Oligophrenic nach Myatonicartigem Zustand im Neugeborenalter. Schweiz Med Wschr 86: 1260-1261.
Purvis-Smith SG, Saville T, Manass S, Yip M-Y, Lam-Po-Tang PRL, Duffy B, Johnston H, Leigh D & McDonald B (1992) Uniparental disomy 15 resulting from “correction” of an initial trisomy 15. Am J Hum Genet 50: 1348-1350.
Prasad C & Wagstaff J (1997) Genotype and phenotyp in Angelman syndrome caused by paternal UPD 15. Am J Med Genet 70: 328-329.
Purandare SM, Breidenbach HH, Li Y, Zhu XL, Sawada S, Neil SM, Brothman A, White R, Cawthon R & Viskochil D (1995) Identification of neurofibromatosis 1 (NF!) homologous loci by direct sequencing, fluorescence in situ hybridization, and PCR amplification of somatic cell hybrids. Genomics 30: 476-485.
Özcelik T, Leff S & Robinson W (1992) Small nuclear ribonucleoprotein ppolypeptide N (SNRPN), an expressed gene in Prader-willi syndrome critical rergion. Nat Genet 2: 265-269.
Qumsiyeh MB, Dalton JD, Gordon PL, Wilroy RS & Tharapel AT (1992) Deletion of chromosome 15pter→q11.2 due to t(Y;15) in a boy with Prader-Willi syndrome. Am J Med Genet 42: 109-111.
Reed ML & Leff SE (1994) Maternal imprinting of human SNRPN, a gene deleted in Prader-Willi syndrome. Nature Genet 6: 163-167.
Reeve A, Norman A, Sinclair P, Whittington-Smith R, Hamey Y, Donnai D & Read A (1993) True telomeric translocation in a baby with the Prader-Willi phenotype. Am J Med Genet 47: 1-6.
Regnier V, Meddeb M, Lecointre G, Richard F, Duverger A, Nguyen VC, Dutrillaux B, Berheim A & Danglot G (1997) Emergence and scattering of multiple neurofibromatosis (NF1)-related sequences during hominoid evolution suggest a process of pericentromeric interchromosomal transposition. Hum Mol Genet 6: 9-16.
Reik W, Cillick A, Norris ML, Barton SC & Surani MA (1987) Genomic imprinting determines methylation of parental alleles in transgenic mice. Nature 328: 248-251.
Reik W & Walter J (2001) Evolution of imprinting mechamisms: the battle of the sexes begins in the zygote. Nat Genet 27: 255-256.
Reis A, Kunze J, Ladanyi L, Enders H, Klien-Vogler U & Niemann G (1993) Exclusion of the GABAA-receptor beta 3 subunit gene as the Angelman’s syndrome gene. Lancet 341: 122-123.

100Reis A, Dittrich B, Greger V, Buiting K, Lalande M, Gillessen-Kaesbach G, Anvert M &
Horsthemke B (1994) Imprinting mutations suggested by abnormal DNA methylation patterns in familial Angelman and Prader-Willi syndromes. Am J Hum Genet 54: 741-747.
Repetto GM, White LM, Bader PJ, Johnson D & Knoll JHM (1998) Interstitial duplications of chromosome region 15q11q13: Clinical and molecular characterization. Am J Med Genet 79: 82-89.
Rich DC, Witkowski CM, Summers KM, Tuinen P van & Ledbetter DH (1988): Highly polymorphic D15S25 (CMW-1) maps to 15pter-q13. Nucleic Acids Res 16: 8740.
Ritchie RJ, Mattei M-G & Lalande M (1998) A large polymorphic repeat in the pericentromeric region of human chromosome 15q contains three partial gene duplications. Hum Mol Genet 7: 1253-1260.
Robinson WP (2000) Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays 22: 452-459.
Robinson WP, Bernasconi F, Mutirangura A, Ledbetter DH, Langlois S, Malcom S, Morris MA & Schinzel AA (1993a) Nondisjunction of chromosome 15: Origin and recombination. Am J Hum genet 53: 740-751.
Robinson WP, Bottani A, Yagang X, Balakrishman J, Binkert F, Machler M, Prader A & Schinzel A (1991) Molecular, cytogenetic and clinical investication of Prader-Willi syndrome patients. Am J Hum Genet 49: 1219-1234.
Robinson WP, Christian SL, Kuchinka BD, Peñaherrera MS, Das S, Schuffenhauer S, Malcolm S, Schinzel AA, Hassold TJ & Ledbetter DH (2000) Somatic segregation error predominantly contribute to the gain or loss of a paternal chromosome leading to uniparental disomy for chromosome 15. Clin Genet 57: 349-358.
Robinson WP, Dutly F, Nicholls RD, Bernasconi F, Peñaherrera M, Michaelis RC, Abeliovich D & Schnizel AA (1998a) The mechanisms involved in formation of deletions and duplications of 15q11-q13. J Med Genet 35: 130-136.
Robinson WP, Kuchinka BD, Bernasconi F, Petersen MB, Schulze A, Brøndum-Nielsen, Chrstian SL, Ledbetter DH, Schinzel AA, Horsthemke B, Schuffenhauer S, Michaelis RC, Langlois S & Hassold TJ (1998b) Maternal meiosis I non-disjunction of chromosome 15: dependence of the maternal age effect on level of recombination. Hum Mol Gen 7: 1011-1019.
Robinson WP, Langlois S, Schuffenhauer S, Horsthemke B, Michaelis RC, Christian S, Ledbetter DH & Schinzel A (1996) Cytogenetic and age-dependent risk factors associated with uniparental disomy 15. Prenatal Diag 16: 837-844.
Robinson WP, Lorda-Sanchez I, Malcolm S, Langlois S, Schuffenhauer S, Knoblauch H, Horsthemke B & Schinzel AA (1993d) Increased parental ages and uniparental disomy 15: a paternal aged effect? Eur J Hum Genet 1: 280-286.
Robinson WP, Spiegel R & Schinzel AA (1993b) Deletion breakpoints associated with the Prader-Willi and Angelman syndromes (15q11-q13) are not sites of high homologous recombination. Hum Genet 91: 181-184.
Robinson WP, Wagstaff J, Bernasconi F, Baccichetti C, Aftifoni L, Franzoni E, Suslak L, Shih L-Y, Aviv H & Schinzel AA (1993c) Uniparental disomy explains the occurrence of the Angelman of Prader-Willi syndrome in patients with an additional small inv dup(15) chromosome. J Med Genet 30: 756-760.
Rogan PK, Lichty TR, Ladda RL, Mascardi MJ, Steele MW, Wenger SL, Malcom S, Driscoll DJ & Nicholls RD (1993) Uniparental disomy in Angelman syndrome: A consequence of paternal meiotic nondisjunction (abstract). Am J Hum Genet 53: 1225.
Rogan PK, Seip JR, White LM, Wenger SL, Steele MW, Sperling MA, Menon R & Knoll JHM (1998) Relaxation of imprinting in Prader-Willi syndrome. Hum Genet 103: 694-701.
Rougeulle C, Cardoso C, Fontes M, Colleaux L & Lalande M (1998a) An imprinted antisens RNA overlaps UBE3A and a second maternally expressed transcript. Nat Genet 19: 15-16.
Rougeulle C, Glatt H & Lalande M (1997) The Angelman syndrome canditate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet 17: 14-15.

101Rougeulle C & Lalande M (1998b) Angelman syndrome: how many genes to remain silent?
Neurogenetics 1: 229-237. Runte M, Huttenhofer A, Gross S, Kiefmann M, Horsthemke B & Buiting K (2001) The IC-
SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet 23: 2687-2700.
Russo D, Cogliati F, Viri M, Cavalleri F, Selicorni A, Turolla L, Belli S, Romeo A & Larizza L (2000) Novel mutations of ubiquitin protein ligase 3A gene in Italian patients with Angelman syndrome. Hum Mutation 15: 387 Online.
Saitoh S, Buiting K, Cassidy SB, Conroy JM, Driscoll DJ, Gabriel JM, Gillessen-Kaesbach G, Glenn CC, Greenswag LR, Horsthemke B, Kondo I, Kuwajima K, Niikawa N, Rogan P, Schwartz S, Seip J, Williams CA & Nicholls RD (1997) Clinical spectrum and molecular diagnosis of Angelman and Prader-Willi syndrome patients with an imprinting mutation. Am J Med Genet 68: 195-206.
Saitoh S, Buiting K, Rogan PK, Buxton JL, Driscoll DJ, Arnemann J, König R, Malcolm S, Horsthemke B & Nicholls RD (1996) Minimal definition of the imprinting center and fixation of a chromosome 15q11-q13 epigenotype by imprinting mutations. Genetics 93: 7811-7815.
Saitoh S, Harada N, Jinno Y, Hashimoto K, Imaizumi K, Kuroki Y, Fukushima Y, Sugimoto T, Renedo M, Wagstaff J, et al. (1994b) Molecular and clinical study of 61 Angelman syndrome patients. Am J Med Genet 52: 158-163.
Saitoh S, Kubota T, Ohta T, Jinno Y, Niikawa N, Subimoto T, Wagstaff J & Lalande M (1992) Familial Angelman syndrome caused by imprinted submicroscopic deletion encompassin GABAA receptor β3-subunit gene. Lancet 339: 366-367.
Saitoh S, Mutirangura A, Kuwano A, Ledbetter DH & Niikawa N (1994a) Isochromosome 15q of maternal origin in two Prader-Willi syndrome patients previously diagnosed erroneously as cytogenetic deletions. Am J Med Genet 50: 64-67.
Saitoh S & Wada T (2000) Parent-of-Origin specific histone acetylation and reactivation of a key imprinted gene locus in Prader-Willi syndrome. Am J Hum Genet 66: 1958-1962.
Salafsky IS, MacGregor SN, Claussen U & Eggeling F (2001) Maternal UPD 20 in an infant from a pregnancy with mosaic trisomy 20. Prenat Diagn 21: 860-863.
Sambrook J, Fritsch EF & Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
Scheffner, M, Werness JM, Huibregtse JM, Levine AJ & Howley PM (1990) The E6 oncoprotein encodes by human papillomavirus types 16 and 18 promotes the degradation of 53. Cell 63: 1129-1136.
Scheffner M, Nuber U & Huibregtse JM (1995) Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 373: 81-83.
Schuffenhauer S, Buchholz T, Stengel-Rutkowski S, Buiting K, Scmidt H & Meitinger (1996) A familial deletion in the Prader-Willi syndrome region including the imprinting control region. Hum Mutation 8: 288-292.
Schulze A, Hansen C, B’gaard P, Blichfeldt S, Petersen MB, Tommerup N & Brindum-Nielsen K (1997) Clinical features and molecular genetic analysis of a boy with Prader-Willi syndrome caused by an imprinting defect. Acta Paediatr 86: 906-910.
Schulze A, Hansen C, Skakkebæk NE, Brøndum-Nielsen K, Ledbetter DH & Tommerup N (1996) Exclusion of SNRPN as a major determinant of Prader-Willi syndrome by a translocation breakpoint. Nature Genet 12: 452-454.
Schumacher A, Buiting K, Zeschnigk M, Doerfler & Horsthemke B (1998) Methylation analysis of the PWS/AS region does not support an enhancer-competition model. Nat Genet 19: 324-325.
Schweizer J, Zynger D & Francke U (1999) In vivo nuclease hypersensitivity studies reveal multiple sites of parental origin-dependent differential chromatin conformation in the 150 kg SNRPN transcription unit. Hum Mol Genet 8: 555-566.
Schwob E, Bohm T, Mendenhall M & Nasmyth K (1994) The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell 79: 233-244.

102Shaffer LG & Lupski JR (2000) Molecular Mechanisms for Constitutional Chromosomal
Rearrangements in Humans. Annu Rev Genet 34: 297-329. Shemer R, Birger Y, Riggs AD & Razin A (1997) Structure of the imprinted mouse Snrpn gene and
establishment of its parental-specific methylation pattern. Genetics 94: 10267-10272. Shemer R, Hershko AY, Perk J, Mostoslavsky R, Tsuberi B, Cedar H, Buiting K & Razin A (2000)
The imprinting box of the Prader-Willi/Angelman syndrome domain. Nat Genet 26: 440-443. Skuse DH, James RS, Bishop DV, Coppin B, Dalton P, Aamodt-Leeper, Bacarese-Hamilton M,
Creswell C, McGurk R & Jacobs PA (1997) Evidence from Turner’s syndrome of an imprinted X-linked locus affecting cognitive function. Nature 387: 705-708.
Slater HR, Vaux C, Pertile M, Burgess T & Petrovic V (1997) Prenatal diagnosis of Prader-Willi syndrome using PW71 methylation analysis – uniparental disomy and the significance of residual trisomy 15. Prenat Diag 17: 109-113.
Smeets DFCM, Hamel BCJ, Nelen MR, Smeets HJM, Bollen JHM, Smith APT, Ropers HH & van Oost B (1992): Prader-Willi syndrome and Angelman syndrome in cousins from a family with a translocation between chromosomes 6 and 15. NEJM 326: 807-811.
Smilinich NJ, Day CD, Fitzpatrick GV, Caldwell GM, Lossie AC, Cooper PR, Smallwood AC, Joyce JA, Schofield PN, Reik W, Nicholls RD, Weksberg R, Driscoll DJ, Maher ER, Shows TB & Higgins MJ (1999) A maternally methylated CpG island in KVLQT1 isassociated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc Natl Acad Sci 96: 8064-8069.
Smith A, Deng Z-M, Beran R, Woodage T & Trent RJ (1994) Familial unbalanced translocation t(8;15)(p23.3;q11) with uniparental disomy in Angelman syndrome. Hum Genet 93: 471-473.
Smith A, Marks R, Haan E, Dixon J & Trent RJ (1997) Clinical features in four patients with Angelman syndrome resulting from paternal uniparental disomy. J Med Genet 34: 426-429.
Smith A, Prasad M, Deng Z-M, Robson L, Woodage T & Trent RJ (1995) Comparison of high resolution cytogenetics, fluoresence in situ hybridisation, and DNA studies to validate the diagnosis of Prader-Willi and Angelman’s syndromes. Arch Dis Child 72: 397-402.
Smith A, Robson L, Neumann A, Mulcahy M, Chabros V, Deng ZM, Woodage T & Trent RJ (1993) Fluorescence in-situ hybridisation and molecular studies used in the characterisation of a Robertsonian translocation (13q15q) in Prader-Willi syndrome. Clin Genet 43: 5-8.
Smith A, Wiles C, Haan E, McCill J, Wallace G, Dixon J, Selby R, Colley A, Marks R & Trent RJ (1996) Clinical features in 27 patients with Angelman syndrome resulting from DNA deletion. J Med Genet 33: 107-112.
Southern EM (1975) Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98: 508-517.
Spence JE, Perciaccante RG, Greig GM, Willard HF, Ledbetter DH, Hejtmancik JF, Pollack MS, O’Brien WE & Beaudet AL (1988) Uniparental disomy as a mechanism for human genetic disease. Am J Hum Genet 42: 217-226.
Spinner NB, Zackai E, Cheng S-D & Knoll JHM (1995) Supernumerary inv dup(15) in a patient with Angelman syndrome and a deletion of 15q11-q13. Am J Med Genet 57: 61-65.
Spritz RA, Bailin T, Nicholls RD, Lee S-T, Park S-K, Mascari MJ & Butler MG (1997) Hypopigmentation in the Prader-Willi syndrome correlates with P gene deletion but not with haplotype of the hemizygous P allele. Am J Med Genet 71: 57-62.
Stalker HJ & Williams CA (1998) Genetic Counseling in Angelman Syndrome: The Challenges of Multiple Causes. Am J Med Genet 77: 54-59.
Stalker HJ, Williams CA & Wagstaff J (1998) Genetic counseling in Angelman syndrome: gonadal mosaicism. Am J Med Genet 78: 482.
Steffenburg U, Hagberg G & Kyllerman M (1996) Characteristics of seizures in a population-based series of mentally retarded children with active epilepsy. Epilepsia. 37: 850-856.
Strakowski SM & Butler MG (1987) Paternal hydrocarbon exposure in Prader-Willi syndrome. Lancet 19: 1458.

103Sun Y, Nicholls RD, Butler MG, Saitoh S, Hainline BE & Palmer CG (1996) Breakage in the
SNRPN locus in a balanced 46,XY,t(15;19) Prader-Willi syndrome patient. Hum Mol Genet 5: 517-524.
Surani MAH & Barton SC (1983) Development of gynogenetic eggs in the mouse: implications for parthenogenetic embryos. Science 222: 1034-1036.
Surani MAH, Barton SC & Norris ML (1984) Development of reconstituted mouse eggs suggest imprinting of the genome during gametogenesis. Nature 308: 548-550.
Surani MAH, Barton SC & Norris ML (1986) Nuclear transplantation in the mouse: heritable differences between parental genomes after activation of the embryonic genome. Cell 45: 127-136
Sutcliffe JS, Han M, Christian SL & Ledbetter DH (1997) Neuronally-expressed necdin gene: an imprinted candidate gene in Prader-Willi syndrome. Lancet 350: 1520-1521.
Sutcliffe JS, Nakao M, Christian S, Orstavik KH, Tommerup N, et al. (1994) Deletions of a differentially methylated CpG island at the SNRPN gene define a putative imprinting control region. Nat Genet 8: 52-58.
Suzuki Y, Sasagawa I, Sawamura T, Ishigooka M, Kaneko H, Kubota Y & Nakada T (1996) Derivative Y chromosome resulting from a t(Y;15)(q12;q11.2) in a boy with Prader-Willi syndrome. International Urology & Nephrology 28: 797-800.
Szabo PE & Mann JR (1995) Allele-specific expression and total expression levels of imprinted genes during early mouse development: implications for imprinting mechanisms. Genes Dev 9: 3097-3108.
Tantravahi U, Nicholls RD, Stroh H, Ringer S, Neve RL, Kaplan L, Wharton R, Wurster-Hill D, Graham Jr JM, Cantú ES, Frias JL, Kousseff BG & Latt SA (1989) Quantitative Calibration and Use of DNA Probes for Investigating Chromosome Abnormalities in the Prader-Willi Syndrome. Am J Med Genet 33: 78-87.
Tekin M, Jackson-Cook C, Buller A, Ferreira-Gonzalez A, Pandya A, Garrett CT & Bodurtha J (2000) Fluorescence in situ hybridization detectable mosaicism for Angelman syndrome with biparental methylation. Am J Med Genet 95: 145-149.
Teshima I, Chadwick D, Chitayat D, Kobayashi J, Ray P, Shuman C, Siegel-Bartelt J, Strasberg P & Weksberg R (1996) FISH detection of chromosome 15 deletions in Prader-Willi and Angelman syndromes. Am J Med Genet 62: 216-223.
Thomas NS, Browne CE, Oley C, Healey S, Crolla JA (1999) Investigation of a cryptic interstitial duplication involving the Prader-Willi/Angelman syndrome critical region. Hum Genet 105: 384-387.
Tilghman SM (1999) The sins of the fathers and mothers: genomic imprinting in mammalian development. Cell 96: 185-193.
Tomlinson IM, Cook GP, Carter NP, Elaswarapu R, Smith S, Walter G, Buluwela L, Rabbitts TH, Winter G (1994) Human immunoglobulin VH and D segments on chromosome 15q11.2 and 16p11.2. Hum Mol Genet 3: 853-860.
Toth-Fejel S, Olson S, Gunter K, Quan F, Wolford J, Popovich BW & Magenis RE (1996) The impact of imprinting: Prader-Willi syndrome resulting from chromosome translocation, recombination, and nondisjunction. Am J Hum Genet 58: 1008-1016.
Tremblay KD, Saam JR, Ingram RS, Tilghman SM & Bartolomei MS (1995) A paternal-specific methylation imprint marks the allels of the mouse H19 gene. Nat Genet 9:407-413.
Trent RJ, Sheffield LJ, Deng Z-M, Kim WS, Nassif NT, Ryce C, Woods CG, Michaelis RC, Tarleton J & Smith A (1997) The elusive Angelman syndrome critical region. J Med Genet 34: 714-718.
Trent RJ, Volpato F, Smith A, Lindeman R, Wong M-K, Warne G & Haan E (1991) Molecular and cytogenetic studies of the Prader-Willi syndrome. J Med Genet 28: 649-654.
Tsai T-F, Armstrong D & Beaudet AL (1999a) Necdin-deficient mice do not show lethality or the obesity and infertility of Prader-Willi syndrome. Nat Genet 22: 15-16.

104Tsai T-F, Jiang Y-H, Bressler J, Armstrong D & Beaudet AL (1999b) Paternal deletion from Snrpn
to Ube3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to Prader-Willi syndrome. Hum Mol Genet 8: 1357-1364.
Tsai T-F, Raas-Rotschild A, Ben-Neriah Z & Beaudet AL (1998) Prenatal diagnosis and carrier detection for a point mutation in UBE3A causing Angelman syndrome. Am J Hum Genet 63: 1561-1563.
Tycko B (1999) Genomic imprinting and cancer. In:Ohlsson R, editor. Genomic impritning: an interdisciplinary approach. Springer-Verlag p 133-169.
Vercesi A, Carvalho MRS, Aguitar MJB & Pena SDJ (1999) Prevalence of Prader-Willi and Angelman syndromes among mentally retarded boys in Brazil. J Med Genet 36: 498.
Vickers S, Dahlitz M, Hardy C, Kilpatrick M & Webb T (1994) A male with a de novo translocation involving loss of 15q11q13 material and Prader-Willi syndrome. J Med Genet 31: 478-481.
Vu TH & Hoffman AR (1994) Promoter-specific imprinting of the human insulin-like growth factor-II gene. Nature 371: 714-717.
Vu TH & Hoffman AR (1997) Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat gebet 17: 12-13.
Vu PK & Sakamoto KM (2000) Ubiquitin-mediated proteolysis and human disease. Mol Genet Metab 71: 261-266.
Wagstaff J, Chaillet JR & Lalande M (1991a) The GABAA receptor β3 subunit gene: Characterization of a Human cDNA from chromosome 15q11q13 and mapping to a region of conserved synteny on mouse chromosome 7. Genomics 11: 1071-1078.
Wagstaff J, Knoll JHM, Fleming J, Kirkness EF, Martin-Gallardo A, Greenberg F, Graham JM, Menninger J, Ward D, Venter JC & Lalande M (1991b) Localization of the gene encoding the GABAA receptor β3 subunit to the Angelman/Prader-Willi region of human chromosome 15. Am J Hum Genet 49: 330-337.
Wagstaff J, Knoll JH, Glatt KA, Shugart YY, Sommer A & Lalande M (1992): Maternal but not paternal transmission of 15q11-q13-linked nondeletion Angelman syndrome leads to phenotypic expression. Nat Genet 1: 291-294.
Wagstaff J, Shugart YY & Lalande M (1993) Linkage analysis in familial Angelman syndrome. Am J Hum Genet 53: 105-112.
Wandstrat AE, Leana-Cox J, Jenkins L & Schwartz S (1998) Molecular cytogenetic evidence for a common breakpoint in the largest inverted duplications of chromosome 15. Am J Hum Genet 62: 925-936.
Watrin F, Roeckel N, Lacroix L, Mignon C, Mattei MG, Disteche C & Muscatelli F (1997) The mouse Necdin gene is expressed from the paternal allele only and lies in the 7C region of the mouse chromosome 7, a region of conserved synteny to the human Prader-Willi syndrome region. Eur J Hum genet 5: 324-332.
Webb T, Clarke D, Hardy CA, Kilpatric MW, Corbert J & Dahilitz M (1995) A clinical, cytogenetic, and molecular study of 40 adults with the Prader-Willi syndrome. J Med Genet 32: 181-185.
Webb T, Clayton-Smith J, Cheng XJ, Knoll JHM, Lalande M, Pembrey ME & Malcom S (1992) Angelman syndrome with a chromosomal inversion 15inv(p11q13) accompanied by a deletion in 15q11-q13. J Med Genet 29: 921-924.
Webber LM & Garson OM (1983) Fluorodeoxyuridine synchronization of bone marrow cultures. Cancer Genet Cytogenet 8: 123-132.
Wenger SL, Sell SL, Painter MF & Steele MW (1997) Inherited unbaanced subteomeric translocation in a child with 8p- and Angelman syndromes. Am J Med Genet 70: 150-154.
Wevrick R & Francke U (1996) Diagnostic test for the Prader-Williy syndrome by SNRPN expression in blood. Lancet 348: 1068-1069.
Wevrick R, Kerns JA & France U (1994) Identification of a novel paternally expressed gene in the Prader-Willi syndrome region. Hum Mol Genet 3: 1877-1882.

105White L & Knoll JHM (1995) Angelman syndrome: validation of molecular cytogenetic analysis of
chromosome 15q11-q13 for deletion detection. Am J Med Genet 56: 101-105. White LM, Rogan PK, Nicholls RD, Wu B-L, Korf B & Knoll JHM (1996) Allele-specific
replication of 15q11-q13 loci: a diagnostic test for detection of uniparental disomy. Am J Hum Genet 59: 423-430.
Wiesner GL, Bendel CM, Olds DP, White JG, Arthur DC, Ball DW & King RA (1987) Hypopigmentation in the prader-Willi syndrome. AmJ Med Genet 40: 431-442.
Williams CA, Angelman H, Claytopn-Smith J, Driscoll DJ, Hendricckson JE, Knoll JHM, Magenis RE, Schinzel A, Wagstaff J, Whidden EM & Zori RT (1995a) Angelman syndrome: Consensus for diagnostic criteria. Am J Med Genet 56: 237-238.
Williams CA & Frias JL (1982) The Angelman (‘happy puppet’) syndrome. Am J Med Genet 11: 453-460.
Williams CA, Zori RT, Hendrickson J, Stalker H, Marum T, Whidden E & Driscoll D (1995b) Angelman syndrome. Curr Probl Pediatr 25: 216-231.
Wirth J, Back E, Hüttenhofer A, Nothwang H-G, Lich C, Groß S, Menzel C, Schinzel A, Kioschis P, Tommerup N, Ropers H-H, Horsthemke B & Buiting K (2001) A translocation breakpoint cluster disrupts the newly defined 3´ end of the SNURF-SNRPN transcription unit on chromosome 15. Hum Mol Genet 10: 201-210.
Woodage T, Deng Z-M, Prasad M, Smart R, Lindeman R, Christian SL, Ledbetter DH, Robson L, Smith A & Trent RJ (1994) A variety of genetic mechanisms are associated with the Prader-Willi syndrome. Am J Med G enet 54: 219-226.
Woodage T, Prasad M, Dixon JW, Selby RE, Romain DR, Columbano-Green LM, Graham D, Rogan PK, Seip JR & Smith A et al. (1994) Bloom syndrome and maternal uniparental disomy for chromosome 15. Am J Hum Genet 55: 74-80.
Working group on Prader-Willi and Angelman syndromes: Diagnostic testing for Prader-Willi and angelman syndromes: Report of the ASHG/ACMG test and technology transfer comittee. Am J Hum Genet 58:1085-1088.
Wutz A, Smrzka OW, Schweifer N, Schellander K, Wagner EF & Barlow DP (1997) Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature 16: 745-749.
Xin Z, Allis CD & Wagstaff J (2001) Parent-specific complementary patterns of histone H3 lysine 9 and H3 lysine 4 methylation at the Prader-Willi syndrome imprinting centre. Am J Hum Genet 69: 1389-1394.
Yamamoto Y, Huibregtse JM & Howley PM (1997) The human E6-AP gene (UBE3A) endoces three potential protein isoforms generated by differential splicing. Genomics 41: 263-266.
Yang T, Adamson TE, Resnick JL, Leff S, Wevrick R, Francke U, Jenkins NA, Copeland NG & Brannan CI (1998) A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nat Genet 19: 25-31.
Yunis JJ (1976) High resolution of human chromosomes. Science 191: 1268-1270. Zackowski JL, Nicholls RD, Gray BA, Bent-Williams A, Gottlieb W, Harris PJ, Waters MF,
Driscoll DJ, Zori RT & Williams CA (1993) Cytogenetic and molecular analysis in Angelman syndrome. Am J Med Genet 46: 7-11.
Zeschnigk M, Lich C, Buiting K, Doerfler W & Horsthemke B (1997) A single-tube PCR test for the diagnosis of Angelman and Prader-Willi syndrome based on allelic methylation differences at the SNRPN locus. Eur J Hum Genet 5: 94-98.
Zlotogora J (1998) Germ line mosaicism. Hum Genet 102:381-386. Zuffardi O, Buhler EM & Fraccaro M (1978) Chromosome 15 and Prader-Willi syndrome. Clin
Genet 14:315-3166. Östravik KH, Tangsrud SE, Kiil R, Hansteen I-L, Steen-Johnsen J, Cassidy SB, Martony A, Anvret
M, Tommerup N & Bröndum-Nielsen K (1992) Prader-Willi syndrome in a brother and sister without cytogenetic or detectable molecular genetic abnormality at chromosome 15q11q13. Am J Med Genet 44: 534-538.

106Özçelik T, Leff S, Robinson W, Donlon T, Lalande M, Sanjines E, Schinzel A & France U (1992)
Small nuclear ribonucleoprotein polypeptide N (SNRPN), an expressed gene in the Prader-Willi syndrome critical region. Nature Genet 2: 265-269.


















