
Generic expansion of the substrate spectrum of a DNA polymerase by directed evolution
5
LETTERS DNA polymerases recognize their substrates with exceptionally high specificity 1,2 , restricting the use of unnatural nucleotides and the applications they enable. We describe a strategy to expand the substrate range of polymerases. By selecting for the extension of distorting 3′ mismatches, we obtained mutants of Taq DNA polymerase that not only promiscuously extended mismatches, but had acquired a generic ability to process a diverse range of noncanonical substrates while maintaining high catalytic turnover, processivity and fidelity. Unlike the wild-type enzyme, they bypassed blocking lesions such as an abasic site, a thymidine dimer or the base analog 5-nitroindol 3 and performed PCR amplification with complete substitution of all four nucleotide triphosphates with phosphorothioates 4 or the substitution of one with the equivalent fluorescent dye–labeled nucleotide triphosphate. Such ‘unfussy’ polymerases have immediate utility, as we demonstrate by the generation of microarray probes with up to 20-fold brighter fluorescence. Attempts to modify the substrate specificity of polymerases by design, screening or selection have often focused on DNA polymerase I from T. aquaticus (Taq) because of its importance in biotechnology. Successes have included the design of variants with increased accept- ance of dideoxynucleotides (ddNTPs) for sequencing 5 and the identi- fication of variants with increased incorporation of ribonucleotides (NTPs) 6 by screening and phage display 7 with a proximally tethered substrate 7,8 . It would, however, be desirable to engineer polymerases with a more generically expanded substrate spectrum. Polymerase substrate specificity is controled both at the incorporation step and subsequently at the extension step, with 3′ mismatches extended with greatly reduced efficiency 1,9 . Likewise, many unnatural base analogs can be incorporated but then act as terminators 2 . We reasoned that selection for extension of 3′ mismatches might subvert aspects of the stringent geometric control in the polymerase active site and not only allow efficient extension of mismatched primer termini but confer a generic ability to process other noncognate 3′ ends. To select for mismatch extension, we modified the compartment- alized self-replication (CSR) method for the directed evolution of polymerases 10 to include flanking primers bearing the 3′ mismatches A•G (primer•template) and C•C, which were reported to be extended by Taq more than a million times less efficiently than matched ter- mini 9 (Fig. 1a). Starting from two repertoires of randomly mutated Taq genes 10 , efficient mismatch extension evolved readily in just three cycles of CSR. The two mutant Taq polymerases best able to extend 3′ mismatches were chosen for further characterization and named M1 (G84A, D144G, K314R, E520G, F598L, A608V, E742G) and M4 (D58G, R74P,A109T, L245R, R343G, G370D, E520G, N583S, E694K, A743P) (Fig. 1b). Both M1 and M4 not only had greatly increased abil- ity to extend the selected A•G and C•C mismatches, but appeared to have acquired a generic ability to extend 3′ mispaired termini. These included all other strongly disfavored mismatches (such as G•A, A•A, G•G) 9 (Fig. 1c), which wild-type Taq polymerase (wtTaq) is unable to extend in PCR, as reported previously 11 . Single base extension kinetics revealed similar, if slightly lower catalytic rates (V max /K m ) for M1 (17%) and M4 (74%) compared to wtTaq on a matched (G-C) 3′ end. In contrast, M1 (and M4) extended a C•C 3′ mispair 427-fold (and 82-fold) more efficiently than wtTaq (see Supplementary Table 1 online). We first investigated whether the selected polymerases M1 and M4 would be able to bypass common blocking template lesions such as an abasic site and a cis-syn cyclobutane pyrimidine dimer (CPD) as well as the hydrophobic base analog 5-nitroindol (5NI) 3 , which—like many other hydrophobic base analogs—acts as a terminator. Whereas all three lesions arrested DNA replication by wtTaq, both M1 (Fig. 2a,b) and M4 (see Supplementary Fig. 1a online) could completely bypass the lesions. Naturally occurring translesion polymerases are mostly poorly processive 12,13 . We therefore investigated whether the processiv- ity of M1 and M4 was similarly reduced but found that, even at the low- est enzyme concentrations, primer extension and termination probabilities by M1 and M4 closely matched those of wtTaq (see Supplementary Fig. 1b,c online), indicating that both M1 and M4 exhibit processivity equal to (or higher than) that of wtTaq. This is also reflected in the striking proficiency of M1 in long-range PCR. Previously, amplification of DNA targets >5–10 kb required the use of polymerase ‘blends’ comprising, for example, Taq with a small amount of a proofreading polymerase 14 . In contrast, we found that M1 alone, 1 MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 2QH, UK. 2 Section on DNA Replication, Repair and Mutagenesis, Laboratory of Genomic Integrity, National Institutes of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland 20892-2725, USA. 3 BioRobotics, Barton Road, Haslingfield CB3 7LW, UK. 4 Department of Chemistry and Biotechnology, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. Correspondence should be addressed to P.H. ([email protected]). Published online 23 May 2004; doi:10.1038/nbt974 Generic expansion of the substrate spectrum of a DNA polymerase by directed evolution Farid J Ghadessy 1 , Nicola Ramsay 1 , François Boudsocq 2 , David Loakes 1 , Anthony Brown 3 , Shigenori Iwai 4 , Alexandra Vaisman 2 , Roger Woodgate 2 & Philipp Holliger 1 NATURE BIOTECHNOLOGY VOLUME 22 NUMBER 6 JUNE 2004 755 © 2004 Nature Publishing Group http://www.nature.com/naturebiotechnology
Transcript of Generic expansion of the substrate spectrum of a DNA polymerase by directed evolution

L E T T E R S
DNA polymerases recognize their substrates with exceptionallyhigh specificity1,2, restricting the use of unnatural nucleotidesand the applications they enable. We describe a strategy to expand the substrate range of polymerases. By selecting for the extension of distorting 3′ mismatches, we obtainedmutants of Taq DNA polymerase that not only promiscuouslyextended mismatches, but had acquired a generic ability toprocess a diverse range of noncanonical substrates whilemaintaining high catalytic turnover, processivity and fidelity.Unlike the wild-type enzyme, they bypassed blocking lesionssuch as an abasic site, a thymidine dimer or the base analog 5-nitroindol3 and performed PCR amplification withcomplete substitution of all four nucleotide triphosphates with phosphorothioates4 or the substitution of one with theequivalent fluorescent dye–labeled nucleotide triphosphate.Such ‘unfussy’ polymerases have immediate utility, as wedemonstrate by the generation of microarray probes with up to 20-fold brighter fluorescence.
Attempts to modify the substrate specificity of polymerases by design,screening or selection have often focused on DNA polymerase I fromT. aquaticus (Taq) because of its importance in biotechnology.Successes have included the design of variants with increased accept-ance of dideoxynucleotides (ddNTPs) for sequencing5 and the identi-fication of variants with increased incorporation of ribonucleotides(NTPs)6 by screening and phage display7 with a proximally tetheredsubstrate7,8. It would, however, be desirable to engineer polymeraseswith a more generically expanded substrate spectrum. Polymerasesubstrate specificity is controled both at the incorporation step andsubsequently at the extension step, with 3′ mismatches extended withgreatly reduced efficiency1,9. Likewise, many unnatural base analogscan be incorporated but then act as terminators2. We reasoned thatselection for extension of 3′ mismatches might subvert aspects of thestringent geometric control in the polymerase active site and not onlyallow efficient extension of mismatched primer termini but confer ageneric ability to process other noncognate 3′ ends.
To select for mismatch extension, we modified the compartment-alized self-replication (CSR) method for the directed evolution of
polymerases10 to include flanking primers bearing the 3′ mismatchesA•G (primer•template) and C•C, which were reported to be extendedby Taq more than a million times less efficiently than matched ter-mini9 (Fig. 1a). Starting from two repertoires of randomly mutatedTaq genes10, efficient mismatch extension evolved readily in just threecycles of CSR. The two mutant Taq polymerases best able to extend 3′ mismatches were chosen for further characterization and namedM1 (G84A, D144G, K314R, E520G, F598L, A608V, E742G) and M4(D58G, R74P, A109T, L245R, R343G, G370D, E520G, N583S, E694K,A743P) (Fig. 1b). Both M1 and M4 not only had greatly increased abil-ity to extend the selected A•G and C•C mismatches, but appeared tohave acquired a generic ability to extend 3′ mispaired termini. Theseincluded all other strongly disfavored mismatches (such as G•A, A•A,G•G)9 (Fig. 1c), which wild-type Taq polymerase (wtTaq) is unable to extend in PCR, as reported previously11. Single base extensionkinetics revealed similar, if slightly lower catalytic rates (Vmax/Km) forM1 (17%) and M4 (74%) compared to wtTaq on a matched (G-C) 3′ end. In contrast, M1 (and M4) extended a C•C 3′ mispair 427-fold(and 82-fold) more efficiently than wtTaq (see Supplementary Table 1online).
We first investigated whether the selected polymerases M1 and M4would be able to bypass common blocking template lesions such as anabasic site and a cis-syn cyclobutane pyrimidine dimer (CPD) as well asthe hydrophobic base analog 5-nitroindol (5NI)3, which—like manyother hydrophobic base analogs—acts as a terminator. Whereas allthree lesions arrested DNA replication by wtTaq, both M1 (Fig. 2a,b)and M4 (see Supplementary Fig. 1a online) could completely bypassthe lesions. Naturally occurring translesion polymerases are mostlypoorly processive12,13. We therefore investigated whether the processiv-ity of M1 and M4 was similarly reduced but found that, even at the low-est enzyme concentrations, primer extension and terminationprobabilities by M1 and M4 closely matched those of wtTaq (seeSupplementary Fig. 1b,c online), indicating that both M1 and M4exhibit processivity equal to (or higher than) that of wtTaq. This is alsoreflected in the striking proficiency of M1 in long-range PCR.Previously, amplification of DNA targets >5–10 kb required the use ofpolymerase ‘blends’ comprising, for example, Taq with a small amountof a proofreading polymerase14. In contrast, we found that M1 alone,
1MRC Laboratory of Molecular Biology, Hills Road, Cambridge CB2 2QH, UK. 2Section on DNA Replication, Repair and Mutagenesis, Laboratory of Genomic Integrity,National Institutes of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland 20892-2725, USA. 3BioRobotics, Barton Road,Haslingfield CB3 7LW, UK. 4Department of Chemistry and Biotechnology, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. Correspondenceshould be addressed to P.H. ([email protected]).
Published online 23 May 2004; doi:10.1038/nbt974
Generic expansion of the substrate spectrum of a DNA polymerase by directed evolutionFarid J Ghadessy1, Nicola Ramsay1, François Boudsocq2, David Loakes1, Anthony Brown3, Shigenori Iwai4,Alexandra Vaisman2, Roger Woodgate2 & Philipp Holliger1
NATURE BIOTECHNOLOGY VOLUME 22 NUMBER 6 JUNE 2004 755
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy

L E T T E R S
using standard PCR conditions and in the absence of auxiliary poly-merases or other processivity factors, was able to amplify DNA frag-ments >25 kb (see Supplementary Fig. 2 online). It has been speculatedthat the size limitation on amplification products is due to prematuretermination following misincorporation, with mismatch removal bythe proofreading polymerase allowing extension to resume. Our obser-vation that a polymerase (M1) with a generic ability to extend mis-matches amplified targets >25 kb supports this hypothesis.
Next we tested the ability of M1 and M4 to incorporate and repli-cate noncognate nucleotide substrates in PCR. First we examined 7-deaza-dGTP, which is commonly used to facilitate amplification andsequencing of GC-rich regions. When dGTP was completely replacedwith 7-deaza-dGTP for amplification of a 0.4-kb fragment (∼ 70% GC)under standard conditions, no product was obtained with wtTaq,
whereas M1 (and to a lesser extent M4 (not shown)) generated ampli-fication products with good yield (Fig. 2c).
Phosphorothioates (αS dNTPs), in which one of the oxygen atomsin the α phosphate group is replaced by a sulfur atom, are good sub-strates for enzymatic incorporation at partial substitution and havefound many applications4. At full substitution, however, with all fourdNTPs replaced by their αS dNTPs counterparts, they no longer sup-port PCR amplification by wtTaq. M1 (and to a lesser extent M4 (notshown)), however, generated amplification products of up to 2kb offully αS substituted DNA (Fig. 2d).
Other commonly used nucleotide analogs are those bearing bulkyadducts. For example, dye-labeled nucleotides are widely used inapplications such as sequencing or microarrays. Incorporation is gen-erally inefficient and requires high concentrations and prolongedextension times, presumably because of steric crowding effects. InPCR, the effect is potentiated, as both template and product strandsbecome decorated with bulky dye molecules. We tested PCR amp-lification with complete substitution of either dUTP withRhodamine(Rho)-5-dUTP or Biotin(Bio)-16-dUTP or complete sub-stitution of dATP with fluorescein(FITC)-12-dATP. Whereas wtTaqwas unable to generate any amplification products, M1 (but not M4)amplified DNA targets of 0.4 kb with FITC-12-dATP (Fig. 2e) or withmuch reduced efficiency using Rho-5-dUTP (not shown) and DNAfragments up to 2.5 kb in length with Bio-16-dUTP (Fig. 2f). Wecloned and sequenced five of the 100% FITC-12-dATP- substitutedamplification products (corresponding to 590 insertions of FITC-12-dATP). Sequencing revealed no mutations at the sites of incorporation(or elsewhere), indicating incorporation of the dye-modified bases byM1 with a fidelity > 1:500.
Complete substitution of natural nucleotides with their unnaturalcounterparts altered the properties of the resulting amplificationproducts. For example, fully αS substituted DNA was completelyresistant to nuclease digestion (not shown). Decoration of bothstrands of a double stranded DNA fragment with bulky substituents(118 FITC molecules in the 0.4-kb fragment and 838 biotin moleculesin the 2.5-kb fragment) led to a substantial shift in electrophoreticmobility from 0.4 kb to 0.5 kb (FITC) and 2.5 kb to 4.0 kb (biotin))(Fig. 2e,f and Supplementary Fig. 3 online).
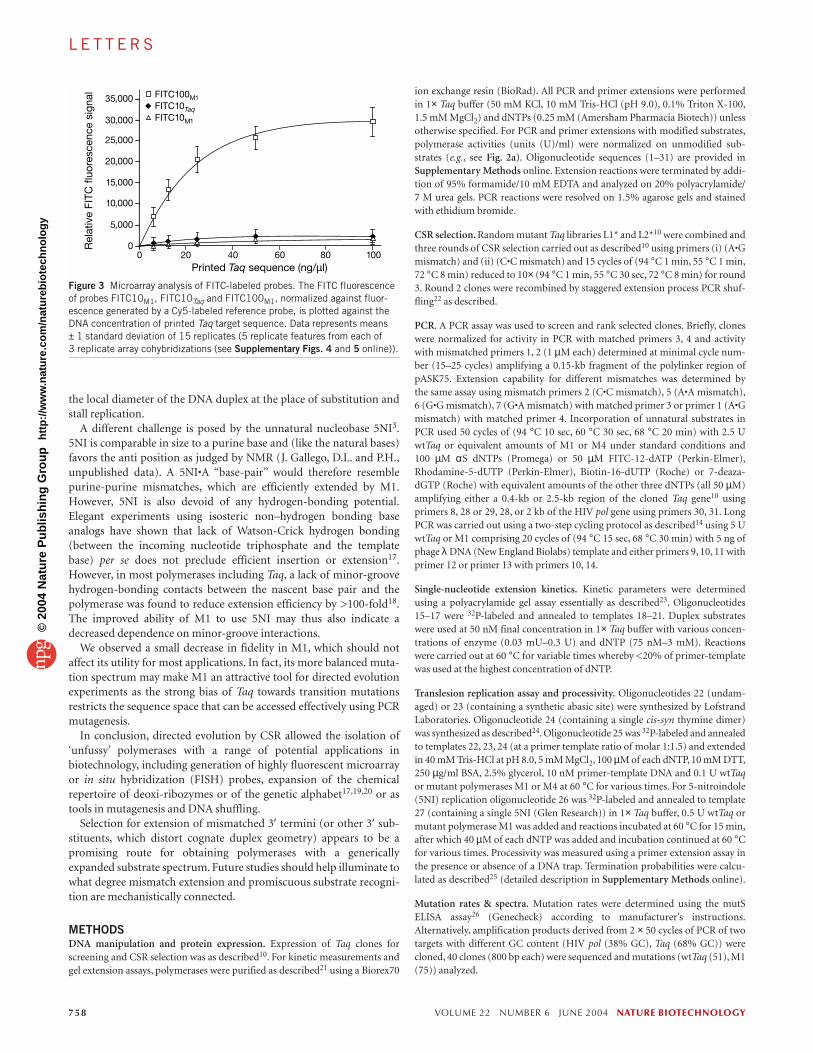
In addition, the 0.4kb fragment, in which all adenosines (dA) onboth strands had been replaced with FITC-12-dAMP (FITC100M1),displayed extremely bright fluorescence. The frequency of fluorophoreincorporation per 1000 nucleotides (FOI) is commonly used to specifythe fluorescence intensity of a probe. FOIs of microarray probes com-monly range from 10 to 50, whereas FITC100M1 has an FOI of 295. Toinvestigate whether such a high level of fluorophore substitutionwould affect hybridization characteristics, we did a series of microar-ray experiments. We compared the fluorescent signal generated byFITC100M1 with equivalent probes generated using either wtTaq orM1 and replacing only 10% of dAMP with FITC-12-dAMP(FITC10Taq, FITC10M1 (FOI=30)). In competitive cohybridizationwith a standard Cy5-labeled probe (Cy5Taq), FITC100M1 hybridizedspecifically only with its cognate Taq polymerase target sequence andnot with any non-cognate control DNA. Hybridization of FITC100M1generated an up to 20-fold higher specific signal than equimolaramounts of the FITC10 probes (Fig. 3) without showing increasedbackground binding (see Supplementary Figs. 4 and 5 online).
Promiscuous mismatch extension might be expected to come at theprice of reduced fidelity, as misincorporation no longer leads to termi-nation. Measurement of the overall mutation rate using both the MutSassay (see Supplementary Fig. 6a online) and direct sequencing ofamplification products, however, indicated only a modestly (1.6-fold)
756 VOLUME 22 NUMBER 6 JUNE 2004 NATURE BIOTECHNOLOGY
A
x
G C
C
pol*
A
G C
C
polx
Pol* Polx
GCGCGC
GCCGC
GCCGA
CGGCG
GGCCA
GGCCG
wtTaq
TP
M4
C A G G A
a
b
c
E520G
D144G
L245R
D58G A109T
R343GG370D
K314R
F598L
N583S
E694K
E742G
A743P
A608V
Figure 1 Mismatch extension. (a) General scheme of CSR selection formismatch extension. Self-replication of the pol gene by the encodedpolymerase requires extension of flanking primers bearing A•G and C•C 3′ mismatches. Only polymerases capable of mismatch extension (Pol*)replicate their own encoding gene (pol*). Black bars denote incorporation of the mismatch into replication products. (b) Mutations in selectedpolymerases M1 (red) and M4 (green) are mapped on a ribbon representa-tion of Taq DNA polymerase (1TAU.pdb27) generated using the programPyMOL (http://www.pymol.org). The DNA template strand is shown inmagenta, the primer strand in yellow. Mutations M1: G84A and M4: R74Pare not visible as residues 69–84 are not resolved. (c) Polymerase activity in PCR for matched versus mismatched 3′ ends (3′ mismatch in bold).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy

L E T T E R S
increased mutation rate for M1 (and M4). However, M1 displays a sig-nificantly altered mutation spectrum compared to wtTaq, with aclearly increased propensity for transversions, in particular G/C toC/G events (see Supplementary Fig. 6b online).
Extension of mismatched 3′ primer termini is a feature of some nat-urally occurring polymerases, notably viral reverse transcriptases, theunusual polB-family polymerase polξ or members of the polY familyof translesion synthesis polymerases12. For these, mismatch extensionforms an integral part of their biological function, permitting error-prone yet processive replication of the viral genome by reverse tran-scriptase or extension of unpaired and distorted primer terminiopposite template strand lesions by polξ and polY polymerases.Unfortunately, these enzymes combine mismatch extension with arange of properties undesirable for biotechnological application suchas low fidelity, poor processivity (in the absence of auxiliary factorssuch as a β sliding clamp)12,13 and slow catalytic turnover. The selectedTaq polymerase mutants M1 and M4 are thus unique in combiningmismatch extension, lesion bypass and relaxed substrate recognitionwith high processivity, catalytic efficiency and fidelity.
In the polY polymerases, the ability to traverse replication blockinglesions is thought to arise from a relaxation of geometric selection due to a more open active site12,15. Preliminary results for M1 and M4
suggest that the observed mutations serve to create a more open act-ive site compared to the wtTaq polymerase (unpublished data).Indeed, modeling showed that a thymine-thymine-CPD cannot beaccommodated into the active site of the wtTaq polymerase withoutmajor steric clashes16.
A more open active site and consequently more relaxed geometricselection may explain the striking ability of M1 and M4 to prom-iscuously extend 3′ mismatches and incorporate and replicate a diverse collection of unnatural substrates (Fig. 2). The relaxation ofgeometric selection may extend beyond the actual nucleotide insertionsite, as efficient mismatch replication, apart from extension, requirestranslocation of the incorporated mismatch through the primer-template duplex binding region. Indeed, mismatches in the primer-template duplex have been found to stall primer extension up to fourbases upstream from the 3′ end1. A relaxation of steric constraintsextending to the DNA duplex binding region may thus be essential forprocessive replication of DNA that is fully or partially substituted withunnatural substrates. For example, at full αS substitution, the signifi-cantly larger volume of the sulfur atoms (in both template and primerstrand) may lead to steric crowding in the DNA duplex binding region.Similarly, in the case of biotin- or dye-labeled bases, decoration ofboth DNA strands with bulky substituents may substantially increase
NATURE BIOTECHNOLOGY VOLUME 22 NUMBER 6 JUNE 2004 757
0.5 kb
wt M1
FITC-12-dATP7-deaza-dGTP
0.5 kb
Biotin-16-dUTP
wt M1
4 kb
Mwt M1 M MM1 wt M1 wt
2.2 kb
0.3 kb
αS-dNTPs
φX λH
0 30"1'2' 5' 0 30"1' 2' 5' 0 5'10'20'30'Undamaged Abasic
wtTaq
CPD
T
TA
XAC
TTA
0 30"1' 2' 5' 030"1' 2' 5' 0 5'10' 20'30'Undamaged Abasic
M1
CPD
TT
A
XAC
TTA
a 2322
2322
23 23
pr
5NIG
TC
G
A
0 10' 20' 30' 40' 0 10' 20' 30' 40'
b wtTaq M1
prprprprprpr
c d e f
Figure 2 Lesion bypass and incorporation of unnatural substrates. Polymerase activity was assayed over time for its ability to extend a radiolabeled primer(pr) annealed to template. To facilitate comparison, the concentration of the enzymes was adjusted to allow for comparable primer extensions on undamagedtemplates. (a) Translesion synthesis activity of wtTaq and M1 on an undamaged template, a template containing an abasic site or a cis-syn cyclobutanethymine-thymine dimer (CPD). The template sequence was identical except for the three bases located immediately downstream of the primer (N1-3). X, abasic site; T-T, CPD. On the template containing an abasic site, wtTaq efficiently inserted a base opposite the lesion, but further extension was negligible.On the CPD-containing template, wtTaq only extended 60% of the primer by one nucleotide (opposite the 3′ T of the CPD). In contrast, M1 was capable ofinsertion opposite the abasic site and lesion bypass with 3.4% (M1) of primers being fully extended. On the CPD template, M1 used >90% of the primer,and extended ∼ 26% to the 5′ T and was capable of completing CPD bypass (19%) upon correct incorporation of two dAMPs (not shown) opposite thethymine-thymine CPD. (b) Bypass activity of wtTaq (wt) and M1 on a template containing 5-nitroindol (5NI). Synthesis by wtTaq essentially terminated at5NI (+1), with <10% extended to the +2 position. In contrast, M1 (and M4 (not shown)), while pausing at +1 and +2 positions, showed up to 50% complete5NI bypass. (c–f) Polymerase activity of wtTaq (wt) and M1 in PCR with complete replacement of (c) dGTP with 7-deaza-dGTP, (d) all four dNTPs with αSdNTPs, (e) dATP with FITC-12-dATP or (f) dTTP with Biotin-16-dUTP. Markers: φX, HaeIII-digested phage φX174 DNA; λH, HindIII-digested phage λ DNA;M, 1-kb DNA ladder (Invitrogen), (for wtTaq and M1 amplification of the same DNA fragments using unmodified dNTPs, see Supplementary Fig. 3 online).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy
L E T T E R S
the local diameter of the DNA duplex at the place of substitution andstall replication.
A different challenge is posed by the unnatural nucleobase 5NI3.5NI is comparable in size to a purine base and (like the natural bases)favors the anti position as judged by NMR (J. Gallego, D.L. and P.H.,unpublished data). A 5NI•A “base-pair” would therefore resemblepurine-purine mismatches, which are efficiently extended by M1.However, 5NI is also devoid of any hydrogen-bonding potential.Elegant experiments using isosteric non–hydrogen bonding baseanalogs have shown that lack of Watson-Crick hydrogen bonding(between the incoming nucleotide triphosphate and the templatebase) per se does not preclude efficient insertion or extension17.However, in most polymerases including Taq, a lack of minor-groovehydrogen-bonding contacts between the nascent base pair and thepolymerase was found to reduce extension efficiency by >100-fold18.The improved ability of M1 to use 5NI may thus also indicate adecreased dependence on minor-groove interactions.
We observed a small decrease in fidelity in M1, which should notaffect its utility for most applications. In fact, its more balanced muta-tion spectrum may make M1 an attractive tool for directed evolutionexperiments as the strong bias of Taq towards transition mutationsrestricts the sequence space that can be accessed effectively using PCRmutagenesis.
In conclusion, directed evolution by CSR allowed the isolation of‘unfussy’ polymerases with a range of potential applications inbiotechnology, including generation of highly fluorescent microarrayor in situ hybridization (FISH) probes, expansion of the chemicalrepertoire of deoxi-ribozymes or of the genetic alphabet17,19,20 or astools in mutagenesis and DNA shuffling.
Selection for extension of mismatched 3′ termini (or other 3′ sub-stituents, which distort cognate duplex geometry) appears to be apromising route for obtaining polymerases with a genericallyexpanded substrate spectrum. Future studies should help illuminate towhat degree mismatch extension and promiscuous substrate recogni-tion are mechanistically connected.
METHODSDNA manipulation and protein expression. Expression of Taq clones forscreening and CSR selection was as described10. For kinetic measurements andgel extension assays, polymerases were purified as described21 using a Biorex70
ion exchange resin (BioRad). All PCR and primer extensions were performed in 1× Taq buffer (50 mM KCl, 10 mM Tris-HCl (pH 9.0), 0.1% Triton X-100,1.5 mM MgCl2) and dNTPs (0.25 mM (Amersham Pharmacia Biotech)) unlessotherwise specified. For PCR and primer extensions with modified substrates,polymerase activities (units (U)/ml) were normalized on unmodified sub-strates (e.g., see Fig. 2a). Oligonucleotide sequences (1–31) are provided inSupplementary Methods online. Extension reactions were terminated by addi-tion of 95% formamide/10 mM EDTA and analyzed on 20% polyacrylamide/7 M urea gels. PCR reactions were resolved on 1.5% agarose gels and stainedwith ethidium bromide.
CSR selection. Random mutant Taq libraries L1* and L2*10 were combined andthree rounds of CSR selection carried out as described10 using primers (i) (A•Gmismatch) and (ii) (C•C mismatch) and 15 cycles of (94 °C 1 min, 55 °C 1 min,72 °C 8 min) reduced to 10× (94 °C 1 min, 55 °C 30 sec, 72 °C 8 min) for round3. Round 2 clones were recombined by staggered extension process PCR shuf-fling22 as described.
PCR. A PCR assay was used to screen and rank selected clones. Briefly, cloneswere normalized for activity in PCR with matched primers 3, 4 and activitywith mismatched primers 1, 2 (1 µM each) determined at minimal cycle num-ber (15–25 cycles) amplifying a 0.15-kb fragment of the polylinker region ofpASK75. Extension capability for different mismatches was determined by the same assay using mismatch primers 2 (C•C mismatch), 5 (A•A mismatch),6 (G•G mismatch), 7 (G•A mismatch) with matched primer 3 or primer 1 (A•Gmismatch) with matched primer 4. Incorporation of unnatural substrates inPCR used 50 cycles of (94 °C 10 sec, 60 °C 30 sec, 68 °C 20 min) with 2.5 UwtTaq or equivalent amounts of M1 or M4 under standard conditions and 100 µM αS dNTPs (Promega) or 50 µM FITC-12-dATP (Perkin-Elmer),Rhodamine-5-dUTP (Perkin-Elmer), Biotin-16-dUTP (Roche) or 7-deaza-dGTP (Roche) with equivalent amounts of the other three dNTPs (all 50 µM)amplifying either a 0.4-kb or 2.5-kb region of the cloned Taq gene10 usingprimers 8, 28 or 29, 28, or 2 kb of the HIV pol gene using primers 30, 31. LongPCR was carried out using a two-step cycling protocol as described14 using 5 UwtTaq or M1 comprising 20 cycles of (94 °C 15 sec, 68 °C 30 min) with 5 ng ofphage λ DNA (New England Biolabs) template and either primers 9, 10, 11 withprimer 12 or primer 13 with primers 10, 14.
Single-nucleotide extension kinetics. Kinetic parameters were determinedusing a polyacrylamide gel assay essentially as described23. Oligonucleotides15–17 were 32P-labeled and annealed to templates 18–21. Duplex substrateswere used at 50 nM final concentration in 1× Taq buffer with various concen-trations of enzyme (0.03 mU–0.3 U) and dNTP (75 nM–3 mM). Reactionswere carried out at 60 °C for variable times whereby <20% of primer-templatewas used at the highest concentration of dNTP.
Translesion replication assay and processivity. Oligonucleotides 22 (undam-aged) or 23 (containing a synthetic abasic site) were synthesized by LofstrandLaboratories. Oligonucleotide 24 (containing a single cis-syn thymine dimer)was synthesized as described24. Oligonucleotide 25 was 32P-labeled and annealedto templates 22, 23, 24 (at a primer template ratio of molar 1:1.5) and extendedin 40 mM Tris-HCl at pH 8.0, 5 mM MgCl2, 100 µM of each dNTP, 10 mM DTT,250 µg/ml BSA, 2.5% glycerol, 10 nM primer-template DNA and 0.1 U wtTaqor mutant polymerases M1 or M4 at 60 °C for various times. For 5-nitroindole(5NI) replication oligonucleotide 26 was 32P-labeled and annealed to template27 (containing a single 5NI (Glen Research)) in 1× Taq buffer, 0.5 U wtTaq ormutant polymerase M1 was added and reactions incubated at 60 °C for 15 min,after which 40 µM of each dNTP was added and incubation continued at 60 °Cfor various times. Processivity was measured using a primer extension assay inthe presence or absence of a DNA trap. Termination probabilities were calcu-lated as described25 (detailed description in Supplementary Methods online).
Mutation rates & spectra. Mutation rates were determined using the mutSELISA assay26 (Genecheck) according to manufacturer’s instructions.Alternatively, amplification products derived from 2 × 50 cycles of PCR of twotargets with different GC content (HIV pol (38% GC), Taq (68% GC)) werecloned, 40 clones (800 bp each) were sequenced and mutations (wtTaq (51), M1(75)) analyzed.
758 VOLUME 22 NUMBER 6 JUNE 2004 NATURE BIOTECHNOLOGY
35,000
30,000
25,000
20,000
15,000
10,000
5,000
00 20 40 60 80 100
Printed Taq sequence (ng/µl)
Rel
ativ
e FI
TC fl
uore
scen
ce s
igna
l FITC100M1FITC10TaqFITC10M1
Figure 3 Microarray analysis of FITC-labeled probes. The FITC fluorescenceof probes FITC10M1, FITC10Taq and FITC100M1, normalized against fluor-escence generated by a Cy5-labeled reference probe, is plotted against theDNA concentration of printed Taq target sequence. Data represents means ± 1 standard deviation of 15 replicates (5 replicate features from each of 3 replicate array cohybridizations (see Supplementary Figs. 4 and 5 online)).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy

L E T T E R S
Array manufacture and hybridization. Targets were prepared by PCR amplifi-cation of a 2.5 kb Taq gene using primers 29, 28 or 2 kb of the HIV polgene using primers 30, 31. Salmon sperm DNA (Invitrogen) was prepared at100 ng/µl in 50% DMSO. FITC and Cy5 probes were prepared by PCR amplifi-cation of 0.4-kb fragment of Taq using primers 8, 28 with either 100%(FITC100M1) or 10% of dATP (FITC10M1, FITC10Taq) replaced by FITC-12-dATP or 10% of dCTP replaced by Cy5-dCTP (Cy5Taq). Cy5 and Cy3 random20 mers (MWG) were used at 250 nM. Targets were purified using PCR purifi-cation kit (Qiagen) and prepared in 50% DMSO and spotted onto GAPSIIaminosilane-coated glass slides (Corning) using a MicroGrid (BioRobotics).Array hybridizations were done according to standard protocols (detaileddescription in Supplementary Methods online).
Note: Supplementary information is available on the Nature Biotechnology website.
ACKNOWLEDGMENTSF.B., A.V. and R.W. were supported by funds from the National Institutes of Healthintramural research program.
COMPETING INTERESTS STATEMENTThe authors declare that they have no competing financial interests.
Received 3 February; accepted 31 March 2004Published online at http://www.nature.com/naturebiotechnology/
1. Kunkel, T.A. & Bebenek, K. DNA replication fidelity. Annu. Rev. Biochem. 69,497–529 (2000).
2. Kool, E.T. Active site tightness and substrate fit in DNA replication. Annu. Rev.Biochem. 71, 191–219 (2002).
3. Loakes, D. Survey and summary: The applications of universal DNA base analogues.Nucleic Acids Res. 29, 2437–2447 (2001).
4. Verma, S. & Eckstein, F. Modified oligonucleotides: synthesis and strategy for users.Annu. Rev. Biochem. 67, 99–134 (1998).
5. Li, Y., Mitaxov, V. & Waksman, G. Structure-based design of Taq DNA polymeraseswith improved properties of dideoxynucleotide incorporation. Proc. Natl. Acad. Sci.USA 96, 9491–9496 (1999).
6. Astatke, M., Ng, K., Grindley, N.D. & Joyce, C.M. A single side chain preventsEscherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonu-cleotides. Proc. Natl. Acad. Sci. USA 95, 3402–3407 (1998).
7. Xia, G. et al. Directed evolution of novel polymerase activities: mutation of a DNApolymerase into an efficient RNA polymerase. Proc. Natl. Acad. Sci. USA 99,6597–6602 (2002).
8. Jestin, J.L., Kristensen, P. & Winter, G. A method for the selection of catalytic activityusing phage display and proximity coupling. Angew. Chem. Int. Ed. 38, 1124–1127(1999).
9. Huang, M.-M., Arnheim, N. & Goodman, M.F. Extension of base mispairs by Taq poly-merase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res.20, 4567–4573 (1992).
10. Ghadessy, F.J., Ong, J.L. & Holliger, P. Directed evolution of polymerase function bycompartmentalized self-replication. Proc. Natl. Acad. Sci. USA 98, 4552–4557(2001).
11. Kwok, S. et al. Effects of primer-template mismatches on the polymerase chain reac-tion: human immunodeficiency virus type 1 model studies. Nucleic Acids Res. 18,999–1005 (1990).
12. Goodman, M.F. Error-prone repair DNA polymerases in prokaryotes and eukaryotes.Annu. Rev. Biochem. 71, 17–50 (2002).
13. Friedberg, E.C., Wagner, R. & Radman, M. Specialized DNA polymerases, cellularsurvival, and the genesis of mutations. Science 296, 1627–1630 (2002).
14. Barnes, W.M. PCR amplification of up to 35-kb DNA with high-fidelity and high-yieldfrom l bacteriophage templates. Proc. Natl. Acad. Sci. USA 91, 2216–2220 (1994).
15. Ling, H., Boudsocq, F., Woodgate, R. & Yang, W. Crystal structure of a Y-family DNApolymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell107, 91–102 (2001).
16. Trincao, J. et al. Structure of the catalytic core of S. cerevisiae DNA polymerase eta:implications for translesion DNA synthesis. Mol. Cell 8, 417–426 (2001).
17. Kool, E.T. Synthetically modified DNAs as substrates for polymerases. Curr. Opin.Chem. Biol. 4, 602–608 (2000).
18. Morales, J.C. & Kool, E.T. Functional hydrogen-bonding map of the minor groovebinding tracks of six DNA polymerases. Biochemistry 39, 12979–12988 (2000).
19. Tae, E.L., Wu, Y., Xia, G., Schultz, P.G. & Romesberg, F.E. Efforts toward expansion ofthe genetic alphabet: replication of DNA with three base pairs. J. Am. Chem. Soc.123, 7439–7440 (2001).
20. Piccirilli, J.A., Krauch, T., Moroney, S.E. & Benner, S.A. Enzymatic incorporation of anew base pair into DNA and RNA extends the genetic alphabet. Nature 343, 33–37(1990).
21. Engelke, D.R., Krikos, A., Bruck, M.E. & Ginsburg, D. Purification of Thermus aquati-cus DNA polymerase expressed in Escherichia coli. Anal. Biochem. 191, 396–400(1990).
22. Zhao, H., Giver, L., Shao, Z., Affholter, J.A. & Arnold, F.H. Molecular evolution bystaggered extension process (StEP) in vitro recombination. Nat. Biotechnol. 16,258–261 (1998).
23. Creighton, S., Bloom, L.B. & Goodman, M.F. Gel fidelity assay measuring nucleotidemisinsertion, exonucleolytic proofreading, and lesion bypass efficiencies. MethodsEnzymol. 262, 232–256 (1995).
24. Murata, T., Iwai, S. & Ohtsuka, E. Synthesis and characterization of a substrate for T4endonuclease V containing a phosphorodithioate linkage at the thymine dimer site.Nucleic Acids Res. 18, 7279–7286 (1990).
25. Kokoska, R.J., McCulloch, S.D. & Kunkel, T.A. The efficiency and specificity ofapurinic/apyrimidinic site bypass by human DNA polymerase eta and Sulfolobus sol-fataricus Dpo4. J. Biol. Chem. 278, 50537–50545 (2003).
26. Debbie, P. et al. Allele identification using immobilized mismatch binding protein:Detection and identification of antibiotic resistant bacteria and determination ofsheep susceptibility to scrapie. Nucleic Acids Res. 25, 4825–4829 (1997).
27. Eom, S.H., Wang, J. & Steitz, T.A. Structure of Taq polymerase with DNA at the poly-merase active site. Nature 382, 278–281 (1996).
NATURE BIOTECHNOLOGY VOLUME 22 NUMBER 6 JUNE 2004 759
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy


















