
G Model ARTICLE IN PRESS - modena.units.itmodena.units.it/Literature General Documents/1-s2.0... ·...
10
Please cite this article in press as: E. Laurini, et al., MoDeNa Nanotools: An integrated multiscale simulation workflow to predict thermophysical properties of thermoplastic polyurethanes, J. Comput. Sci. (2015), http://dx.doi.org/10.1016/j.jocs.2015.11.006 ARTICLE IN PRESS G Model JOCS-433; No. of Pages 10 Journal of Computational Science xxx (2015) xxx–xxx Contents lists available at ScienceDirect Journal of Computational Science journa l h om epage: www.elsevier.com/locate/jocs MoDeNa Nanotools: An integrated multiscale simulation workflow to predict thermophysical properties of thermoplastic polyurethanes Erik Laurini a,∗ , Paola Posocco a , Maurizio Fermeglia a , Sabrina Pricl a,b a Molecular Simulation Engineering (MOSE) Laboratory, University of Trieste, DI3, Piazzale Europa 1 34127, Trieste, Italy b National Interuniversity Consortium for Material Science and Technology (INSTM), Research Unit MOSE-DEA, University of Trieste, Trieste, Italy a r t i c l e i n f o Article history: Received 16 July 2015 Received in revised form 5 November 2015 Accepted 22 November 2015 Available online xxx Keywords: Molecular simulations Computational recipes Thermoplastic polyurethanes Software integration a b s t r a c t In this work we describe and assess the performance of Nanotools, a feature of the MoDena software we are currently developing in the framework of a granted EU project devoted to the implementation of a multi- scale modeling environment for nanomaterials and systems by design. Specifically, Nanotools integrates multi-step computational procedures based on atomistic molecular dynamics and Monte Carlo simula- tions for the estimation of major thermophysical properties of thermoplastic polyurethanes (TPUs). The predicted results obtained with Nanotools for density, thermal conductivity, surface tension, gas perme- ability, and Young modulus are in good agreement with the relevant experimental data, thus paving the way for the use of Nanotools in the current design of new TPUs for advanced applications. © 2015 Elsevier B.V. All rights reserved. 1. Introduction Nanotechnology and related production processes play an increasingly important role in our modern society [1]. The ulti- mate, macroscopic performance of nanotechnology products is determined by the material properties at each scale–from nano to micro. These properties can, in turn, be affected by the choice of the production conditions and ingredients [2]. Materials science and engineering is a field that has probably benefited most by the introduction of nanotechnologies, whilst the chemical industry, in particular high-performance product sectors like the pharma- ceutical industry, is somewhat lagging behind. Likely, the lack of integrating the different specialized areas, both in terms of knowl- edge and computational tools, currently represents for these big industrial realities a formidable hurdle. The ultimate goal of the MoDeNa project, funded in 2014 by the European Community within the 7th European Framework Program under the Call NMP (Nanosciences, nanotechnologies, Materials and new Production technologies), is indeed to control macroscopic material proper- ties and ultimate product performances by mastering the behavior of materials and ingredients at all length scales. In this scenario, the MoDeNa project aims at developing, demonstrating and assessing ∗ Corresponding author. Tel.: +39 40 5583440. E-mail address: [email protected] (E. Laurini). an easy-to-use multiscale simulation suite under an open-source licensing scheme that delivers models with feasible computational loads for the design and production of specific industrial products, i.e., polyurethanes (PUs). Specifically, the MoDeNa project focus on (i) the development of high-fidelity models to predict properties and behaviors of new nanomaterials at different scales – from quan- tum mechanics to finite element calculations – (ii) the development of a multiscale modeling software framework that integrates these models across the scales, and (iii) the development of reference standards and standardized methods for the representation, stor- age, and communication of models and data. The general challenges for achieving innovations in nanoma- terials technology are the limited atomically precise production capabilities that exist today, together with an incomplete under- standing of thermodynamic and kinetic processes at the nanoscale. This is even truer at the mesoscale, that is, the interval of time and length scales reaching up to seconds and to hundreds of nanometers, respectively. According to the dogma of nanotech- nology, structural features and relaxation phenomena taking place in this time-scale domain ultimately define the properties on the macroscale where production, utilization, and exploitation of the product take place [2]. Thus, for the optimization of the produc- tion process and the achievement of the maximum quality of the end product, it is essential to understand all molecular mechanisms taking place at each scale and how these reflect on the final product performance. http://dx.doi.org/10.1016/j.jocs.2015.11.006 1877-7503/© 2015 Elsevier B.V. All rights reserved.
Transcript of G Model ARTICLE IN PRESS - modena.units.itmodena.units.it/Literature General Documents/1-s2.0... ·...

J
Mp
Ea
b
a
ARRAA
KMCTS
1
imdtoaiicieiMw(ttoM
h1
ARTICLE IN PRESSG ModelOCS-433; No. of Pages 10
Journal of Computational Science xxx (2015) xxx–xxx
Contents lists available at ScienceDirect
Journal of Computational Science
journa l h om epage: www.elsev ier .com/ locate / jocs
oDeNa Nanotools: An integrated multiscale simulation workflow toredict thermophysical properties of thermoplastic polyurethanes
rik Laurinia,∗, Paola Posoccoa, Maurizio Fermegliaa, Sabrina Pricl a,b
Molecular Simulation Engineering (MOSE) Laboratory, University of Trieste, DI3, Piazzale Europa 1 34127, Trieste, ItalyNational Interuniversity Consortium for Material Science and Technology (INSTM), Research Unit MOSE-DEA, University of Trieste, Trieste, Italy
r t i c l e i n f o
rticle history:eceived 16 July 2015eceived in revised form 5 November 2015ccepted 22 November 2015
a b s t r a c t
In this work we describe and assess the performance of Nanotools, a feature of the MoDena software we arecurrently developing in the framework of a granted EU project devoted to the implementation of a multi-scale modeling environment for nanomaterials and systems by design. Specifically, Nanotools integratesmulti-step computational procedures based on atomistic molecular dynamics and Monte Carlo simula-
vailable online xxx
eywords:olecular simulations
omputational recipeshermoplastic polyurethanesoftware integration
tions for the estimation of major thermophysical properties of thermoplastic polyurethanes (TPUs). Thepredicted results obtained with Nanotools for density, thermal conductivity, surface tension, gas perme-ability, and Young modulus are in good agreement with the relevant experimental data, thus paving theway for the use of Nanotools in the current design of new TPUs for advanced applications.
© 2015 Elsevier B.V. All rights reserved.
. Introduction
Nanotechnology and related production processes play anncreasingly important role in our modern society [1]. The ulti-
ate, macroscopic performance of nanotechnology products isetermined by the material properties at each scale–from nanoo micro. These properties can, in turn, be affected by the choicef the production conditions and ingredients [2]. Materials sciencend engineering is a field that has probably benefited most by thentroduction of nanotechnologies, whilst the chemical industry,n particular high-performance product sectors like the pharma-eutical industry, is somewhat lagging behind. Likely, the lack ofntegrating the different specialized areas, both in terms of knowl-dge and computational tools, currently represents for these bigndustrial realities a formidable hurdle. The ultimate goal of the
oDeNa project, funded in 2014 by the European Communityithin the 7th European Framework Program under the Call NMP
Nanosciences, nanotechnologies, Materials and new Productionechnologies), is indeed to control macroscopic material proper-
Please cite this article in press as: E. Laurini, et al., MoDeNa Nanotthermophysical properties of thermoplastic polyurethanes, J. Comput
ies and ultimate product performances by mastering the behaviorf materials and ingredients at all length scales. In this scenario, theoDeNa project aims at developing, demonstrating and assessing
∗ Corresponding author. Tel.: +39 40 5583440.E-mail address: [email protected] (E. Laurini).
ttp://dx.doi.org/10.1016/j.jocs.2015.11.006877-7503/© 2015 Elsevier B.V. All rights reserved.
an easy-to-use multiscale simulation suite under an open-sourcelicensing scheme that delivers models with feasible computationalloads for the design and production of specific industrial products,i.e., polyurethanes (PUs). Specifically, the MoDeNa project focus on(i) the development of high-fidelity models to predict propertiesand behaviors of new nanomaterials at different scales – from quan-tum mechanics to finite element calculations – (ii) the developmentof a multiscale modeling software framework that integrates thesemodels across the scales, and (iii) the development of referencestandards and standardized methods for the representation, stor-age, and communication of models and data.
The general challenges for achieving innovations in nanoma-terials technology are the limited atomically precise productioncapabilities that exist today, together with an incomplete under-standing of thermodynamic and kinetic processes at the nanoscale.This is even truer at the mesoscale, that is, the interval of timeand length scales reaching up to seconds and to hundreds ofnanometers, respectively. According to the dogma of nanotech-nology, structural features and relaxation phenomena taking placein this time-scale domain ultimately define the properties on themacroscale where production, utilization, and exploitation of theproduct take place [2]. Thus, for the optimization of the produc-
ools: An integrated multiscale simulation workflow to predict. Sci. (2015), http://dx.doi.org/10.1016/j.jocs.2015.11.006
tion process and the achievement of the maximum quality of theend product, it is essential to understand all molecular mechanismstaking place at each scale and how these reflect on the final productperformance.

ARTICLE IN PRESSG ModelJOCS-433; No. of Pages 10
2 E. Laurini et al. / Journal of Computational Science xxx (2015) xxx–xxx
oject
ntactoIcpi–cnaoqcsbfispfbc
itpeaEftbApno
Nmmppmtptr
Fig. 1. The MoDena pr
These wide-ranging challenges are well reflected in the MoDeNaanomaterial case study, namely the production and manufac-uring of compact thermoplastic polyurethane (TPU) materialsnd low density polyurethane (PU) foams. Both systems areharacterized by the presence of nano-, meso-, and microstruc-ures, which, in turn, are crucial for the application propertiesf the final material. Scale interactions are everywhere in PUs.ndeed, TPUs can be seen as generally nanophase separated di-blockopolymers, being constituted by a so-called hard (crystalline)hase (HF) – prototypical components being 4,4′-methyl-diphenyl
socyanate (4,4′MDI) and the chain extender 1,4 butandiol (BDO) interspersed within a soft (amorphous) phase (SF) – mostommonly a polyester, or polyether block. In case of the alter-ative MoDeNa material case, i.e., low-density open-cell foam,nother nanophase separation leads to an early solidificationf the foam closed-cell membranes. These, under the subse-uent action of a blowing agent, expand till rupture therebyreating the final foam open-cell structure. Since the nanophaseeparation is the basis for the solidification process of the mem-rane, it also controls cell growth, membrane rupture, andnally the morphology of the open cell foam. Accordingly, theound prediction, understanding, and control of nanophase mor-hology and/or phase separation in TPUs and PUs are keysor further improving the ultimate, macroscopic properties ofoth type of materials, while benefitting from vastly reducedosts.
The choice of TPUs and PUs as case study in the MoDeNa projects motivated by the applicability of these polymeric systems andhe possible impact of this research on the industrial productionrocesses over the coming years. The global market for PU wasstimated at 13,650 Ktons in 2010 (of which 5000 Ktons in Europe)nd is expected to reach 18 Mtons by 2016 (of which 6 Mtons inurope), growing at a compound annual growth rate (CAGR) of 4.7%rom 2011 to 2016. In terms of revenue, the market was estimatedo be worth US$ 33 million in 2010 and is expected to reach US$ 56illion by 2016, growing at a CAGR of 6.8% from 2011 to 2016 [3].ccording to BASF – a leader European chemical industry, a majorroducer of TPUs and PUs, and partner of the MoDeNa project –anoengineered TPU materials will replace the current generationf TPUs over the years to come.
Within MoDeNa, our group specifically developed the MoDeNaanotools feature, that is, an integrated and automated series ofolecular simulation procedures [4–8] to predict the major ther-ophysical properties of TPUs, and its integration in the general
roject workflow. Conceptually, our aim is to provide (i) com-utational recipes for determining fundamental thermophysicalaterial property data (i.e., density, thermal conductivity, surface
Please cite this article in press as: E. Laurini, et al., MoDeNa Nanotthermophysical properties of thermoplastic polyurethanes, J. Comput
ension, gas diffusivity, solubility and permeability, and mechanicalroperties) for TPUs, also termed exact models; (ii) the implemen-ation of recipes and surrogate models in Nanotools, and (iii) theelevant integration within the MoDeNa software.
modeling framework.
2. Theory and computational methodologies
2.1. The MoDeNa software
The concept of MoDeNa is an interconnected multiscale mod-eling software platform. Four scales are linked together by thisframework namely: nano-, meso-, micro-, and macroscale. Asstated in the introduction, this unifying software platform will ulti-mately allow for enhanced product and process design across allthese scales. As shown in Fig. 1, the modeling framework is inti-mately coupled with the software framework; thus, the softwareframework will facilitate and greatly enhance all material model-ing activities. The orchestrator, in turn, will link the modeling acrossthe scales, which is a necessary condition to obtain an integralapproach, in contrast to a series of disconnected phases calcula-tions.
Multiscale coupling requires the exchange of informationbetween software instances developed for specific scales in aconsistent way. In order to achieve this, generating consistentrepresentations for models and data is necessary. The MoDeNaframework handles the communication across scales through Com-putational Code/Tool (CCT) and adaptors. CCT perform simulationsby executing applications for a given set of inputs while adaptorshandle the communication with the MoDeNa software framework.Both are application specific.
The software framework consists of an orchestrator, a databaseand an interface library. The orchestrator is based on FireWorks [9],and constitutes the backbone of the software framework in that itschedules simulations and property estimation operations, whichmake up the workflow of the overall simulation. It is very muchlike a dynamic workflow engine, in which the different applica-tions are “orchestrated” to obtain, analyze, and pass informationto other operations. The NoSQL database MongoDB [10] is usedto store the state of the workflow as well as all associated datasuch as simulation parameters, data used for parameter estimation,and meta-data. Finally, the interface library consists in a high-levelpython module providing access to the database as well regressionanalysis capabilities.
2.2. Nanotools
According to the Nanotools concept, as conceived and imple-mented in the MoDeNa framework, computational recipes basedon low scale (i.e., atomistic-level) simulations were conceived,implemented, and tested within Nanotools to estimate major ther-mophysical properties (i.e., density, thermal conductivity, surfacetension, gas diffusivity, solubility and permeability, and mechani-
ools: An integrated multiscale simulation workflow to predict. Sci. (2015), http://dx.doi.org/10.1016/j.jocs.2015.11.006
cal properties) of different polyether- and polyester-based TPUs.Exploiting the MoDeNa workflow, these computational recipescould be fully or partially automatized to calculate the abovemen-tioned polymer properties, as shown in Fig. 2.

ARTICLE ING ModelJOCS-433; No. of Pages 10
E. Laurini et al. / Journal of Computatio
F
mddalNeMPupwmM
raap
aPfirnoadectsmdpt
2
2
ucpvBswu
ig. 2. Nanotools concept and its implementation within the MoDeNa workflow.
Fully automatized procedures (FAP) could be developed for ther-ophysical properties such as density and thermal conductivity. In
etails, the first step of each FAP was the development of a set ofedicated bash shell scripts to initialize the environmental variablesnd writing the perl scripts required to start the different molecu-ar dynamics (MD)/Monte Carlo (MC) based computational recipes.ext, the perl files perform the relevant computational recipes andxtract the value of the property of interest from the generatedD/MC simulations. All initial simulation recipe parameters (T, V,
, molecular structures,. . .) are configured using a specific config-ration file. The entire computational workflow for each materialroperty is ultimately created and orchestrated by a python script,hich exploits the FireWorks library and, through a php file, ulti-ately uploads the calculated material property value onto theongoDB database.On the other hand, given the underlying theories, the process of
ecipe automatization for the other material properties mentionedbove was hardly to conceive. Therefore, we developed partiallyutomatized procedure (PAP) for the calculation of mechanicalroperties, gas permeability, and surface tension.
A prototypical example of a property for which a fully autom-tized procedure is hardly feasible is gas permeability (P). In fact,
is notoriously defined as the product of the gas diffusion coef-cient D and the gas solubility S. The obtainment of D and Sequires user intervention, as data fitting and extrapolation areecessary (vide infra for more details). However, some sub-partsf these recipes could still be automatized, and for those autom-tization was achieved using the same file scheme and workflowescribed above for the FAPs. For instance, the generation of differ-nt isotherms required to estimate gas solubility S in a TPU matrixould be easily automatized and stored in the database. However,he successive extrapolation of the ratio of the cell loading and pres-ure P to zero pressure to obtain the value of S requires further dataanipulation, which cannot be bluntly automatized. In this case,
ata are automatically transferred to a datasheet, thereby post-rocessed, and the final results are again automatically transferredo MongoDB.
.3. Nanotools computational recipes
.3.1. General detailsAll energy minimizations and molecular dynamics (MD) sim-
lations were performed in parallel on 24 CPUs of our MOSE20luste11, using the Forcite modulus of the Materials Studio suite ofrograms (MS, v. 7.0, Accelrys Inc., San Diego, CA, USA), which pro-ides high performance via numerous algorithmic optimizations.
Please cite this article in press as: E. Laurini, et al., MoDeNa Nanotthermophysical properties of thermoplastic polyurethanes, J. Comput
ulk MD simulations of TPUs were carried out using a con-tant pressure and temperature (NPT) ensemble at 1 bar pressureith periodic boundary conditions (PBCs); all other simulationssed a constant volume and temperature (NVT) ensemble again
PRESSnal Science xxx (2015) xxx–xxx 3
under periodic boundary conditions. The COMPASS force field[11] was used throughout the entire work. The Berendsen ther-mostat/barostat [12] with coupling time constants �T = 0.1 s and�P = 0.5 ps, was used for temperature and pressure control duringthe MD run. Short-range intermolecular interactions of full atom-istic polymer models were accounted for by the Lennard-Jones9-6 potential function with geometric mean combination rules.Long-range Coulombic interactions of polymer models were han-dled with the particle mesh Ewald (PME) [13] method (accuracy0.001 kcal/mol). The cut-off distance for truncation of all non-bonding interactions was set equal to half of the simulation celllength. In all MD simulations, integration of Newton’s equations ofmotion was achieved using the velocity Verlet algorithm with anintegration time step of 1 fs, if not otherwise states. The length ofeach MD simulation and the number of frames collected along eachMD trajectory varied depending on the specific system and the rel-evant property to be estimated. Therefore, the details will be givenin the specific sections below.
2.3.2. Molecular model building and force field validationThe model structures of the TPU components, i.e., 4,4′MDI, 1,4-
butandiol, and the different polyether or polyether blocks werebuilt from scratch using the Visualizer tool of MS. Each molecu-lar structure was optimized via force field energy minimizationprotocols based on the subsequent use of steepest descent, con-jugate gradient, and Newton–Raphson methods, until the energyderivative was less than 10−4 kcal/(mol A). The performance of theall-atom COMPASS force fields for the simulation of TPU polymerswas ensured by determining (i) the main geometrical features ofthe 4,4′MDI and 1,4-BDO molecules and comparing them with boththe results from ab initio calculations and crystallographic infor-mation, and (ii) a set of thermophysical properties which were alsocompared with the corresponding available experimental data. Thispart is reported in full in the Supporting Information.
Starting from the optimized monomer structures, all TPU chainswere built using composition and length corresponding to theavailable experimental systems (see Section 3 for each systemdetails) by means of the Amorphous Cell modulus of MS. Specifically,10 independent configurations of each TPU chain were constructedusing the Rotational Isomeric State (RIS) method [14] at the desiredtemperature. For each of these chains, a three-dimensional (3D)simulation box with PBCs was built, filled with three copies of thechain, and the resulting cell was relaxed by energy minimization.Accordingly, 10 different 3D simulation boxes were obtained foreach RIS generated TPU chain, and the computational recipe forthe determination of a given property was applied to each of theseboxes. Thus, each resulting property is expressed as the averagevalue over 10 MD runs. Since in all computational recipes the opti-mized cell used to predict TPU density values was employed as astarting point, the cell optimization procedure will be given in nextsection.
2.3.3. Nanotools computational recipe for the determination ofdensity (�) of TPUs
For the determination of TPU density as a function of tem-perature, the following multi-step MD-based fully automatedprocedure (FAP) was developed within Nanotools and integrated inthe MoDeNa workflow. First, for each considered TPU 10 3D simu-lation cells were built at 298 K as described in Section 2.3.2, startingby imposing an initial density value of 0.5 g/cm3. The resultingcell configurations were optimized via the following refinementprotocol: (i) initial system energy relaxation (ii) brief MD simu-
ools: An integrated multiscale simulation workflow to predict. Sci. (2015), http://dx.doi.org/10.1016/j.jocs.2015.11.006
lation run (0.5 ns) in the constant volume–constant temperature(i.e., the canonical or NVT) ensemble at 298 K, and (iii) final energyminimization via combined conjugate gradient/Newton–Raphsonmethods using 10−3 kcal/(mol A) as the convergence criterion for

ING ModelJ
4 utatio
tmtrsbapiwoplgamtsortef
ttos
2t
Tptt
�
wtftb
∣∣wfa
Eikavts
firbftt
ARTICLEOCS-433; No. of Pages 10
E. Laurini et al. / Journal of Comp
he energy gradient. All steps were performed using the Forciteodulus of MS. The relaxed simulation boxes were then subjected
o MD runs in the isochoric–isothermal (NPT) ensemble for 2.5 ns toeach realistic density values at the desired temperature and pres-ure (e.g., 298 K and 1 atm). The simulation cell were further relaxedy carrying out an NPT-based MD simulation annealing protocols follows: each system was first heated from the desired tem-erature T (e.g., 298 K) to the temperature T + 100 K (e.g., 398 K) in
ncrements of 20 K, and then cooled back to the initial temperatureith the same temperature variation scheme. The total duration
f the annealing process was 0.2 ns. At the end of the annealingrocess, each cell was again energy minimized, according to the fol-
owing protocol: first, a combination of steepest descent/conjugateradient methods was applied until the energy derivative on anytom decreased to 1000 kcal/mol. Next, Newton–Raphson mini-ization was performed until the energy derivative became less
han 0.1 kcal/mol. For the final equilibrium density determination,tarting from the well-relaxed simulation cells obtained at the endf the simulated annealing process further 1 ns NPT MD simulationun was performed for data acquisition at the desired tempera-ure. The first 0.5 ns were discarded and only the last 0.5 ns of thequilibrated NPT MD trajectory were retained for data analysis. MDrames were saved every 5 ps, for a total number of frames of 100.
The entire sequence described above, from TPU chain genera-ion via RIS to data production NPT runs, was repeated to estimatehe density values for all TPU systems in the temperature rangef interest (i.e., 298–423 K). All data generated were collected andtored in the MongoDB database.
.3.4. Nanotools computational recipe for the determination ofhermal conductivity (�) of TPUs
The developed recipe for calculating the thermal conductivity ofPUs was based on a non-equilibrium molecular dynamics (NEMD)rocedure, in which an energy flux is imposed on the system, andhe resulting temperature gradient is measured [15]. By definition,he thermal conductivity is then simply given by:
= − J
dT/dz(1)
here, J is the energy flux in the z-direction, and dT/dz is theemperature gradient. Since the direction of the flux is oppositerom the gradient, the thermal conductivity is positive. At everyime interval �t, the flux is imposed by exchanging, an energy �Eetween two fixed layers in the system as:
J∣∣ = 1
2A
�E
�t(2)
here, A is the area perpendicular to the flux direction, and theactor 2 is due to the adopted periodic boundary conditions, sincen amount �E/2 flows in or out either sides of the layer.
Our recipes specifically implements the so-called Reverse Nonquilibrium Molecular Dynamics (RNEMD) method [16], accord-ng to which the energy exchange is carried out by exchanging theinetic energy of two particles: the hottest particle in the cold layernd the coldest particle in the hot layer. The energy �E is thereforeariable and needs averaging over many exchanges. Importantly,he method conserves total linear momentum and energy of theystem.
Specifically, starting from the NPT equilibrated TPU cells usedor density calculations (see Section 2.3.3) each cell was extendedn the z-direction, corresponding to the direction of the flux, at aatio of 1:3. The system was divided into 40 layers, and then equili-
Please cite this article in press as: E. Laurini, et al., MoDeNa Nanotthermophysical properties of thermoplastic polyurethanes, J. Comput
rated under NVT MD simulation at constant temperature of 500 Kor 0.5 ns, using again the COMPASS force field [11], the Berendsenhermostat [12], and a time step of 5 fs. After each cell optimiza-ion, RNEMD simulations were carried out with velocity exchange
PRESSnal Science xxx (2015) xxx–xxx
performed every 250 steps, that is, every 1.25 ps. After a transientperiod of 500 exchanges (625 ps), a production stage was run over1000 exchanges (1.25 ns). The resulting flux and gradient were thencollected, and the corresponding thermal conductivity was finallycalculated.
2.3.5. Nanotools computational recipe for the determination ofmechanical properties for TPUs
Any small volume element of an amorphous material in atomicscale can be characterized by a unique distribution of matter withinit and consequently displays unique properties that sometimes canbe pretty far from the macroscopic properties of the sample. Itmeans that a macroscopically homogeneous amorphous materialcan be viewed as heterogeneous at the nanoscale. Thus, the macro-scopic sample can be partitioned into many small elements and,taking an average of the properties of the individual elements, themacroscopic properties of the material under investigation can bepredicted. The aim of the present computational recipe is to be ableto distinguish between properties of very similar materials, e.g.,TPU differing in soft segment (SS)/hard segment (HS) molar ratiosor, at equal molar ratio, differing in chemical composition. This lastcondition may involve, sometimes, very small chemical differencesuch as one–CH2- group in the SS component. Accordingly, to derivestatistical meaningful variations of the elastic constants for TPUmaterials, a large number of nanoscopic elements are to be consid-ered and the relevant mechanical properties are to be expressed asaverages over the relevant ensembles.
The isothermal elastic constants are defined as the elementsof the 4th rank tensor of the second derivatives of the Helmholtzenergy per unit volume, equivalent to the first derivative of thestress with respect to strain, i.e.,:
Ciklm = 1V
∂2A
∂�ik∂�lm
∣∣∣∣T,�ik,�lm
= ∂�ik
∂εlm
∣∣∣∣T,εlm
(3)
For small deformations, the stress � and the strain ε are relatedby the following relationships:
�lm = Clmnkεnk (4)
εlm = Slmnk�nk (5)
where, C denotes the stiffness coefficients and S the compliancecoefficient of the materials. Since C and S are both symmetric, therecan be at most 21 elastic constants. For highly isotropic amorphousmaterials the number of independent constants reduces to justtwo—the so-called Lamé constants � and . Written in terms ofthe Lamé constants, the stiffness matrix takes on the form:⎡⎢⎢⎢⎢⎢⎢⎣
� + 2 0 0 0 0 0
0 � + 2 0 0 0 0
0 0 � + 2 0 0 0
0 0 0 0 0
0 0 0 0 0
0 0 0 0 0
⎤⎥⎥⎥⎥⎥⎥⎦
(6)
The more familiar elastic constants (or mechanical moduli) canbe written in terms of the Lamé constants as:
Bulk modulus = K = � + 23
(7)
Shear modulus = G = (8)(3� + 2
)
ools: An integrated multiscale simulation workflow to predict. Sci. (2015), http://dx.doi.org/10.1016/j.jocs.2015.11.006
Young modulus = E = � +
(9)
Poisson’s ratio = v = �
2 (� + )(10)

ING ModelJ
utatio
ittatopimtb
cntthsm1saTmd
scmttrgiwie1faitdFarfadp
2g
c
P
w(fipfAi
ARTICLEOCS-433; No. of Pages 10
E. Laurini et al. / Journal of Comp
For the computational recipe adopted to estimate the mechan-cal properties of TPUs the static approach to the estimation ofhe Lamé constants was adopted. According to the recipe, threeensile and three shear deformations of a small magnitude werepplied to the simulation cells containing the TPU of interest in thehree spatial directions, with subsequent energy minimization. Thebtained stress tensor was then used to calculate stiffness and com-liance matrices Cij and Sij of the simulation cells. Finally, assuming
sotropic symmetry of the model, these stiffness and compliancesatrices were used to calculate the Lamé constants from which, in
urn, the Young, shear, and bulk moduli, and Poisson’s ratio coulde easily obtained by means of Eqs. (6)–(10).
As mentioned above, because of its high computational effi-iency, this methodology allowed the elastic constants of a largeumber of nanoscopically small volume elements to be analyzed,hus giving narrow bounds of the material properties. Such sta-istical treatment allowed mimicking the effect of nanoscopiceterogeneities that are always present in real macroscopicamples, partially overreaching the size/time limitation of, e.g.,olecular dynamics-based procedures. For this specific purpose,
00 topologically distinct simulation cells for each TPU were con-idered and the relevant mechanical properties were presented asverages over the 100 values obtained from each single calculation.o the best of our knowledge, we are not aware of any all-atomolecular modeling that treated such a great number of statistical
ata to calculate elastic constants.Operationally, the procedure can be divided in the following
teps: for each TPU model the relevant dense periodic packing wasreated at density values lower than the target density (as deter-ined in Section 2.3.3). The periodic cell length parameters were
hen decreased in small increments, with local energy minimiza-ion carried out after each change until the target density waseached. The building process was repeated 100 times for eachiven system to provide a meaningful sampling of different start-ng points for the subsequent dynamics. Each cell building process
as followed by short (20 ps) MD simulations first in the canon-cal (NVT) ensemble and then in the isothermal–isobaric (NPT)nsemble to stabilize temperature and pressure near 298 K and
atm. Visual inspection of the packed models and a check of theree-volume ensured that there were no obvious indications ofnisotropic conditions. For the calculations of the relevant mechan-cal properties, each optimized cell resulting from the last frame ofhe NPT dynamics was subjected to three tensile and three sheareformations of a small magnitude (max strain amplitude 0.003).our steps of cell geometry relaxation were carried out after thepplication of each deformation. The corresponding values of theesulting mechanical properties were finally collected and storedor final ensemble averaging. The computational recipe describedbove can be repeated in any desired temperature range, if theependence of the mechanical property of the material on tem-erature is requires.
.3.6. Nanotools computational recipe for the determination ofas permeability (P), diffusivity (D) and solubility (S) of TPUs
The permeability coefficient of a gas within a polymeric matrixan be generally expressed as:
= D × S (11)
here, P is the gas permeability coefficient, expressed in Barrer1 Barrer = 10−10 cm3 (STP) cm cm−2 s−1 cmHg−1), D is the gas dif-usion coefficient (in cm2/s), and S the gas solubility, expressedn cm3 cm−3 of polymer cm Hg−1). D reflects the dynamics of the
Please cite this article in press as: E. Laurini, et al., MoDeNa Nanotthermophysical properties of thermoplastic polyurethanes, J. Comput
enetrant gas/polymer matrix while S represent a thermodynamicactor depending on the penetrant gas/polymer matrix interaction.ccording to the proposed recipe [17], atomistic molecular dynam-
cs and Grand Canonical Monte Carlo (GCMC) simulations were
PRESSnal Science xxx (2015) xxx–xxx 5
performed to estimate the gas diffusion coefficients and gas solu-bility in TPUs, respectively. The theoretical background underlyingthe computational procedures is briefly described below.
2.3.6.1. Gas diffusion coefficients. The most popular method to cal-culate the diffusion coefficient from atomistic MD simulations isbased on Einstein’s relationship:
D = 16t
limt→∞
[r (t) − r (0)]2 (12)
where, the angular brackets denote the ensemble average of themean square displacement (MSD) of the inserted gas moleculeswithin a molecular trajectory. Accordingly, the limiting slope ofthe MSD as a function of time can be used to estimate the diffusioncoefficient of a molecule undergoing random Brownian motion inthree dimensions. It should be noted that the Einstein relationshipassumes a random walk for the penetrant gas. In short times, aregime of anomalous diffusion may occur, in which the diffusingparticles may diffuse slowly, and the MSD would be proportionalto tx, with x < 1. Or, if during its motion a gas molecule encoun-ters no other molecules, traveling ballistically, then the distancethat it travels would be proportional to the time interval and theMSD increases quadratically with t. However, for simulation timeslong enough, (i.e., in the hydrodynamic limit), the diffusing par-ticles transit from anomalous to Einstein diffusion, so the Fickianregime occurs, in which the MSD is indeed proportional to tx withx ≈ 1.
A further consideration is important at this point. The use ofEinstein equation implies that the diffusion coefficient D, being aconstant, is independent of the diffusing gas concentration. Indeed,constant D values have been reported for the diffusion of light gaseswith low critical temperatures in several polymers. Also, such gasesare only barely soluble in polymers, even at elevated pressures.Accordingly in a computational experiment for the determinationof D, it can be safely assume the penetrant gas concentration in thepolymeric matrices of interest to be very low. This can be accountedfor by placing a small number of each gas molecules per each sim-ulation cell.
2.3.6.2. Gas solubility. Computationally, the widest used procedureto estimate gas solubility in a polymeric matrix relies on the GCMCmethod [18], which is based on the calculation of the energy changebetween two successive molecular configurations. A standard wayto perform such calculations involves creation and destruction ofgas molecules, which interact with the potential field generated bythe atoms of comprising the polymeric matrix, which is assumedrigid. Molecular creation and destruction attempts are made atpoint chosen randomly within the matrix (i.e., in the simulationbox) with a volume V or, in other word, with density �. In the GCMCensemble, the chemical potential of the bulk phase, which can bewritten as a function of temperature T and fugacity f as:
= kBT ln
(f3
kBT
)(13)
in which kB is Boltzmann’s constant and is the thermal de Brogliewavelength. At low pressure values, bulk gases can be assumed toobey the ideal gas law, and fugacity can be replaced by gas pres-sure P. This leads to the following expressions for the probabilityacceptance criteria for molecular creation pc and destruction pd,respectively:
pc = min
[1,
(PV
)exp
(−�Ep
)](14)
ools: An integrated multiscale simulation workflow to predict. Sci. (2015), http://dx.doi.org/10.1016/j.jocs.2015.11.006
(N + 1) kBT kBT
pd = min
[1,
(NkBT
PV
)exp
(−�Ep
kBT
)](15)

ARTICLE IN PRESSG ModelJOCS-433; No. of Pages 10
6 E. Laurini et al. / Journal of Computational Science xxx (2015) xxx–xxx
Fig. 3. Left: Structure and composition [19] of the TPUs considered as validation data set for the Nanotools density estimation. Composition acronyms and definitions:PTMG poly(tetramethylene ether) glycol; PPG, polypropylene glycol; PEG, polyethylene glycol; PEA, polyethylene adipate; BD, polybutadiene diol; BAN, poly(butadiene-co-acrylonitrile) diamine; 2,4TDI, 2,4-toluene diisocyanate; BHBP, 4,4-bis(2-hydroxytheoxy) biphenyl. 570, 650, 1000, 1200, and 2000 are the molecular weights of the TPUsoft phase (SF). Ch. Ext. stands for chain extender. The last column refers to the relative composition of the HF and SF. Right: Comparison of MD predicted vs. experimentaldensity data. The average % absolute error is 2.2.
Fig. 4. Left: Structure and composition [21,22] of the TPUs considered as validation data set for the Nanotools Young modulus prediction. Composition acronyms anddefinitions: PTMG poly(tetramethylene ether) glycol; PPG, polypropylene glycol; PCL, polycaprolactone glycol; HMADEG, poly(diethylene glycol hydromuconate); HMDATEG,poly(tetraethylene glycol hydromuconate); ADPDEG, (polydiethylene glycol adipate); DCDAOD, Poly(1,8-octanediol 4,5-dimethylcyclohex-4-ene cis-1,2-dicarboxylate);D ylate)B olecuc D pre
wtedaf
p
2TiDtjd1mrtmfMp
2tbt
CDATEG, Poly(tetraethylene glycol 4,5-dimethylcyclohex-4-ene cis-1,2-dicarboxDO, 1,4-butandiol. 1500, 1960, 2000, 2760, 3000, 3223, 3370, and 3410 are the molumn refers to the relative composition of the HF and SF. Right: Comparison of M
here, N is the number of molecules in the simulation box beforehe molecular move attempt and �Ep is the change in the potentialnergy associated with the attempt. In addition to the creation andestruction attempts, which can be assumed to have equal prob-bility, molecular rotations, and displacements are also accountedor, each of which is associated with a probability p given by:
= min
[1, exp
(−�Ep
kBT
)](16)
.3.6.3. Computational recipe for determining gas permeability inPUs. The initial polymer matrix simulation cells used for perform-ng the MD and GCMC simulations required to obtain the values of
and S, and hence P, for a series of gases in different TPUs wereaken from the last frame of the equilibrated production MD tra-ectories utilized for determining the corresponding equilibriumensity value (see Section 2.3.3). For each investigated polymer,0 cells for each TPU were again employed. Subsequently, threeolecules of gas were inserted into each cell. The energy of the
esulting systems was then minimized keeping all atoms other thanhose of the gas fixed. Next, the positions constraint on the polymer
atrix was removed, and 0.5 ns of NVT MD simulations were per-ormed at the desired temperature (e.g., 298 K), during which the
SD of the gas molecules was determined from the equilibratedortion of the corresponding MD trajectory.
Please cite this article in press as: E. Laurini, et al., MoDeNa Nanotthermophysical properties of thermoplastic polyurethanes, J. Comput
.3.6.4. Computational recipe for gas diffusion coefficient determina-ion. The value of D for each gas in each TPU matrix was calculatedy the slope of the graph of MSD vs. time in the region wherehe gas diffusion behavior followed Fick’s law. The criterion of
; 4,4′MDI, 4,4′-methyl diphenyl diisocyanate; HDI, hexamethylene diisocyanate;lar weights of the TPU soft phase (SF). Ch. Ext. stands for chain extender. The lastdicted vs. experimental Young modulus data. Data standard deviation is 2.9.
calculation of the diffusion coefficient by this method is that theslope of log (MSD) vs. log (t) approaches unity. In other words, inthe region of validity of the Einstein equation, the MSD vs. t datawere fitted to a linear function, the slope of which yielded the gasdiffusion coefficient D according to Eq. (12). For all systems consid-ered, the slope of the MSD vs. t graphs for all gases approached unitywell below 0.3 ns; accordingly, the simulation conditions adoptedand, consequently, the calculated diffusion coefficients obtainedby the proposed recipe could be considered reliable. The recipe canbe repeated in any desired temperature if the behavior of D as afunction of T is to be determined.
2.3.6.5. Computational recipe for gas solubility determination. Start-ing from gas-loaded equilibrated simulation cells employed for thedetermination of D, sorption isotherms over a pressure range of0–10 atm were obtained for each polymer/gas couple. At each pres-sure, 2,500,000 steps of GCMC calculations were performed, theinitial equilibration period being set to 500,000 steps. The totalpotential energy of the system Ep of the polymer matrix and thepenetrant gas was obtained as the sum of the interaction ener-gies between the gas and the matrix (EpGM) and that between thegas molecules (EpGG). The calculation of �Ep in the present recipewas preceded by a check for overlapping interaction sites. If a pairof interaction sites was closer than half the sum of the van derWaals radii for the two sites, the sites were considered overlap-ping. Accordingly, any attempt to create, displace, rotate, or destroy
ools: An integrated multiscale simulation workflow to predict. Sci. (2015), http://dx.doi.org/10.1016/j.jocs.2015.11.006
a molecule accompanying an overlapping of sites was discarded.If the sites were not overlapping, �Ep was evaluated to deter-mine whether to accept the attempt or not in accordance with theappropriate acceptance probability (Eqs. (14)–(16)). This procedure

ARTICLE IN PRESSG ModelJOCS-433; No. of Pages 10
E. Laurini et al. / Journal of Computational Science xxx (2015) xxx–xxx 7
Fig. 5. Top left: Structure and composition [19] of the TPUs considered as validation data set for the Nanotools gas diffusivity and solubility computational recipes. Compositionacronyms and definitions: PTMG poly(tetramethylene ether) glycol; PPG, polypropylene glycol; PEG, polyethylene glycol; PEA, polyethylene adipate; BD, polybutadiene diol;BAN, poly(butadiene-co-acrylonitrile) diamine; 2,4TDI, 2,4-toluene diisocyanate; BHBP, 4,4-bis(2-hydroxytheoxy) biphenyl. 570, 650, 1000, 1200, and 2000 are the molecularweights of the TPU soft phase (SF). Ch. Ext. stands for chain extender. The last column refers to the relative composition of the HF and SF. Top right: Comparison of MDp dictedv xperimd
cstptpo
2s
icrael
d
redicted vs. experimental oxygen diffusion data. Center left: comparison of MD pres. experimental oxygen solubility data. Bottom: Comparison of MD predicted vs. eiffusion, N2 diffusion, O2 solubility, and N2 solubility, respectively.
orresponds to imposing impenetrable hard cores to interactionites and, therefore, the non-Coulombic part of the pairwise poten-ial is the Lennard-Jones potential supplemented by the hard sphereotential. Each gas solubility was finally calculated by extrapolatinghe ratio of the cell loading and the pressure P to zero pressure. Therocedure can be repeated at any temperature should the behaviorf S vs. T be determined.
.3.7. Nanotools computational recipe for the determination ofurface tension (�) of TPUs
The surface tension � is related to the disruption of molecularnteractions when new interfaces are created. For free surfaces, �an be calculated as the excess energy at the surface of a mate-ial compared to the bulk, i.e., the Gibbs definition, which can bedapted to the case of polymer films as follows. A fundamental
Please cite this article in press as: E. Laurini, et al., MoDeNa Nanotthermophysical properties of thermoplastic polyurethanes, J. Comput
quation for the film phase in energy representation can be formu-ated as follows:
U = TdS − PdV + dn + �da (17)
vs. experimental nitrogen diffusion data. Center right: Comparison of MD predictedental nitrogen solubility data. Average % absolute errors are 5, 8, 46, and 41 for O2
where, T, S, V, , n, � , and a stand for temperature, entropy, volume,chemical potential, number of moles, total surface area (both sides),and surface tension, respectively. If the Helmholtz free energy A isintroduced as the first Legendre transform of the internal energy Uwith respect to entropy S, then:
dA = −SdT − PdV + dn + �da (18)
Accordingly, the surface tension � can be expressed as:
� = ∂A
∂a
∣∣∣∣T,V,n
= ∂U
∂a
∣∣∣∣T,V,n
− T∂S
∂a
∣∣∣∣T,V,n
= �U + �S (19)
Furthermore, by the Maxwell relation, the entropic contributionto � , �S, can be shown to be equal to:
ools: An integrated multiscale simulation workflow to predict. Sci. (2015), http://dx.doi.org/10.1016/j.jocs.2015.11.006
�S = T∂�
∂T
∣∣∣∣T,V,a
(20)

ING ModelJ
8 utatio
s
�
tbt
c
�
icottbmUewaob
�
Efitrt
t[tadficbwmjttUgrtwtsrt
3
3
tp
ARTICLEOCS-433; No. of Pages 10
E. Laurini et al. / Journal of Comp
o that a rearrangement of the above equations yields:
U = ∂U
∂a
∣∣∣∣T,V,n
= � + T∂S
∂a
∣∣∣∣T,V,n
≈ � − Td�
dT(21)
Very accurate ∂U∂a
∣∣T,V,n
values can be obtained by calculating
he internal energy difference between a thin film model and aulk liquid model with the same number of molecule at the sameemperature. Accordingly, for such a configuration ∂U
∂a
∣∣T,V,n
can be
alculated using the following expression:
U = U2D − U3D
2LxLy(22)
n which 2LxLy denotes the total surface area of the two surfacesreated on both sides of the bulk cell, Lx and Ly being the dimensionsf the film in the x and y direction, respectively (whereas, Lz ishe direction normal to the film surface). U2D and U3D representhe average internal energy of the model system in the film andulk states and, as such, can be calculated from the correspondingolecular dynamics simulations under the same NVT conditions.nder the perspective of the theory presented above, the internalnergy contribution to the surface tension �U of each TPU systemas then calculated from the corresponding difference in the MD
verage energy between the thin film and the MD average energyf the corresponding bulk cell, divided by the surface area, as giveny Eq. (22):
U = U2D − U3D
A≈ � (23)
However, we reasoned that the use of a flat area (i.e., 2LxLy inq. (22)) for the calculation of �U was not appropriate for the thinlm model as the surface of such model is rough. Thus, in ordero properly account for amorphous polymer surface roughness, weeplaced the flat area term 2LxLy with the corresponding value ofhe solvent accessible surface area A, as shown in Eq. (22).
The computational recipe for calculating the surface tensions ofhe different TPUs polymers is then based on the following steps6]. Starting from the equilibrated bulk models of the TPUs usedo determine density values, in order to generate a surface withppropriate dimensions the periodic cell was replicated in x and yirections to yield a final cell with the adequate dimensions. Thinlms of each TPU polymer were subsequently generated from theorresponding bulk model cells by elongating one of the periodicoundary conditions until the parent chains no longer interactedith their images along that coordinate. Each equilibrated bulkodel and thin film cells obtained as described above were sub-
ected to 400 ps of NVT MD simulation at 298 K. Of the entirerajectory, only the last 200 ps were used in the subsequent analysiso extract the corresponding component of the potential energies3D and U2D. Finally, the A parameter for each given polymer wasenerated by the center of a solvent molecule probe, modeled as aigid sphere, when being rolled over the van der Waals surface ofhe compound molecular model; obviously, different surface areasould be obtained using probes with different radii. Accordingly,
he van der Waals radius of one molecule of water (1.4 A) was cho-en as the probe radius for water. The computational recipe can beepeated in any desired temperature range if the �(T) behavior iso be determined.
. Results and discussion
.1. Nanotools density prediction of TPUs
Please cite this article in press as: E. Laurini, et al., MoDeNa Nanotthermophysical properties of thermoplastic polyurethanes, J. Comput
In order to check the validity of the Nanotools automated compu-ational recipe for the prediction of density values for thermoplasticolyurethane systems, a set of 10 different polyether-based TPUs
PRESSnal Science xxx (2015) xxx–xxx
(Fig. 3, left panel) synthesized and characterized by Wolinska-Grabczyk and Jankowski [19], was considered. The right panel inFig. 3 shows the comparison between the density values estimatedwith the Nanotools automatized procedure described in Section2.3.3 and the corresponding experimental values at room temper-ature [19]. As it can be can be seen from this image, the agreementachieved using the Nanotools MD-based FAP is very good, consid-ering the underlying complexity of the simulated materials.
3.2. Nanotools prediction of TPU thermal conductivity
Notwithstanding heat transport is a fundamental material prop-erty, experimental data of � for thermoplastic polyurethanes arenot very common. However, the Nanotools � values predicted fortwo commercial TPUs, namely Elastollan 1195A (BASF, Germany)and Pellethane 2102-90A (Lubrizol, USA) are in agreement with thecorresponding experimental data. In fact, the calculated � value forElastollan is 0.202 W/(m K) while the reported experimental valueis 0.208 W/(m K) (BASF, personal communication); for Pellethane,the predicted value of � is 0.261 W/(m K), which well compareswith the experimental thermal conductivity of 0.258 W/(m K)[20].
3.3. Nanotools prediction of TPU Young modulus
The performance of the Nanotools computational recipe forthe estimation of the mechanical properties of thermoplasticpolyurethanes was tested taking a series of 9 TPUs featuring mainlyMDI as the HF, either polyester or polyether blocks as the SF,and 1,4-butanodiol as eventual chain extender, as synthesized andcharacterized by Yilgor et al. [21] and Pierce et al. [22]. Fig. 4 shows acomparison between the Young modulus values estimated with theNanotools procedure described in Section 2.3.5 and the correspond-ing experimental values. As we see, considering the complexity ofthe quantity being calculated, the agreement achieved using theNanotools MD-based computational recipe is good.
3.4. Nanotools prediction of gas diffusivity, solubility andpermeability in TPUs
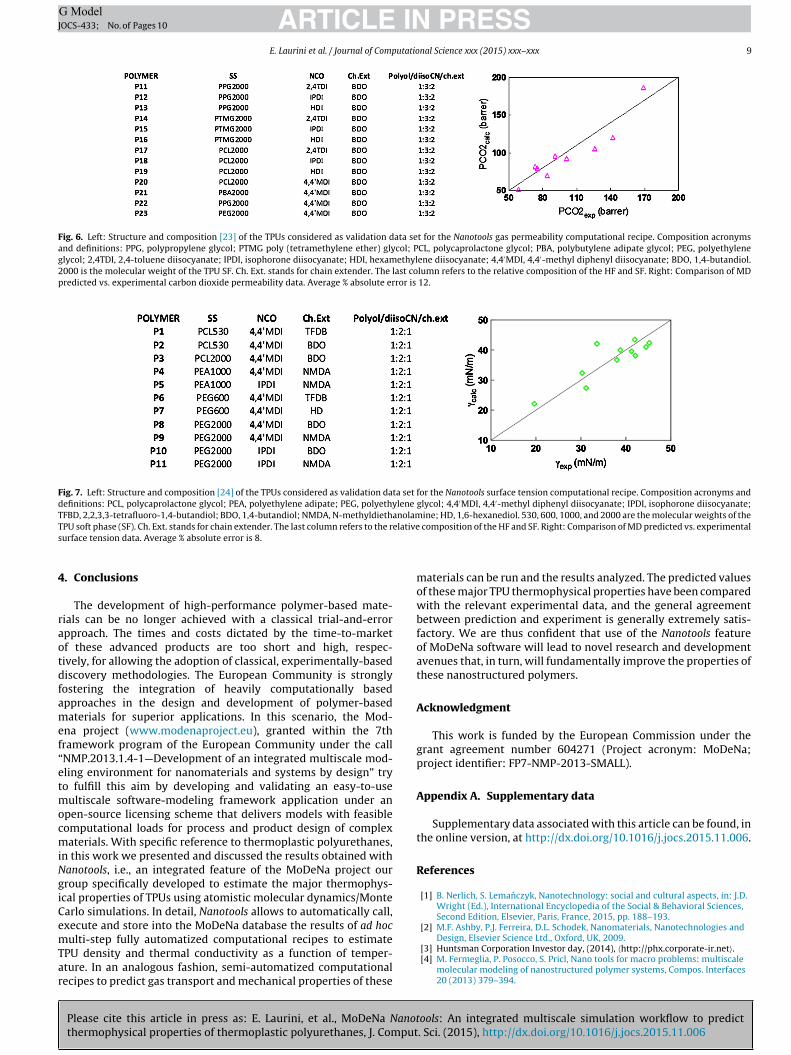
The transport (diffusivity D and permeability P) and thermody-namic (solubility, S) properties of gas in different TPU matrices wereestimated using the Nanotools computational recipes described inSection 2.3.6. Simulation predictions were then validated against aset of 23 experimental data relative to both polyether and polyesterTPUs characterized by Wolinska-Grabczyk and Jankowski [19] andSadeghi et al. [23].
Figs. 5 and 6 show examples of comparison between D, S, andP values estimated with the Nanotools procedure described in Sec-tion 2.3.6 and the corresponding experimental values the differentpolyether/polyester-based TPUs considered [19,23]. As can be seenfrom these figures, the agreement achieved using the NanotoolsMD/MC-based computational recipes is more than satisfactory.
3.5. Nanotools prediction of surface tension of TPUs
The surface tension of different polyester/polyether TUPs, assynthesized and studied by Krol and Krol [24], was predicted usingthe Nanotools computational recipe developed in Section 2.3.7.
ools: An integrated multiscale simulation workflow to predict. Sci. (2015), http://dx.doi.org/10.1016/j.jocs.2015.11.006
Fig. 7 shows the comparison between predicted and experimen-tal surface tension values. As can be seen from these figures, theagreement achieved using the Nanotools MD-based computationalrecipes is very good.
ARTICLE IN PRESSG ModelJOCS-433; No. of Pages 10
E. Laurini et al. / Journal of Computational Science xxx (2015) xxx–xxx 9
Fig. 6. Left: Structure and composition [23] of the TPUs considered as validation data set for the Nanotools gas permeability computational recipe. Composition acronymsand definitions: PPG, polypropylene glycol; PTMG poly (tetramethylene ether) glycol; PCL, polycaprolactone glycol; PBA, polybutylene adipate glycol; PEG, polyethyleneglycol; 2,4TDI, 2,4-toluene diisocyanate; IPDI, isophorone diisocyanate; HDI, hexamethylene diisocyanate; 4,4′MDI, 4,4′-methyl diphenyl diisocyanate; BDO, 1,4-butandiol.2000 is the molecular weight of the TPU SF. Ch. Ext. stands for chain extender. The last column refers to the relative composition of the HF and SF. Right: Comparison of MDpredicted vs. experimental carbon dioxide permeability data. Average % absolute error is 12.
Fig. 7. Left: Structure and composition [24] of the TPUs considered as validation data set for the Nanotools surface tension computational recipe. Composition acronyms anddefinitions: PCL, polycaprolactone glycol; PEA, polyethylene adipate; PEG, polyethylene glycol; 4,4′MDI, 4,4′-methyl diphenyl diisocyanate; IPDI, isophorone diisocyanate;TFBD, 2,2,3,3-tetrafluoro-1,4-butandiol; BDO, 1,4-butandiol; NMDA, N-methyldiethanolamine; HD, 1,6-hexanediol. 530, 600, 1000, and 2000 are the molecular weights of theT elatives
4
raotdfamef“etmocmiNgiCemTar
PU soft phase (SF). Ch. Ext. stands for chain extender. The last column refers to the rurface tension data. Average % absolute error is 8.
. Conclusions
The development of high-performance polymer-based mate-ials can be no longer achieved with a classical trial-and-errorpproach. The times and costs dictated by the time-to-marketf these advanced products are too short and high, respec-ively, for allowing the adoption of classical, experimentally-basediscovery methodologies. The European Community is stronglyostering the integration of heavily computationally basedpproaches in the design and development of polymer-basedaterials for superior applications. In this scenario, the Mod-
na project (www.modenaproject.eu), granted within the 7thramework program of the European Community under the callNMP.2013.1.4-1—Development of an integrated multiscale mod-ling environment for nanomaterials and systems by design” tryo fulfill this aim by developing and validating an easy-to-use
ultiscale software-modeling framework application under anpen-source licensing scheme that delivers models with feasibleomputational loads for process and product design of complexaterials. With specific reference to thermoplastic polyurethanes,
n this work we presented and discussed the results obtained withanotools, i.e., an integrated feature of the MoDeNa project ourroup specifically developed to estimate the major thermophys-cal properties of TPUs using atomistic molecular dynamics/Montearlo simulations. In detail, Nanotools allows to automatically call,xecute and store into the MoDeNa database the results of ad hoc
Please cite this article in press as: E. Laurini, et al., MoDeNa Nanotthermophysical properties of thermoplastic polyurethanes, J. Comput
ulti-step fully automatized computational recipes to estimatePU density and thermal conductivity as a function of temper-ture. In an analogous fashion, semi-automatized computationalecipes to predict gas transport and mechanical properties of these
composition of the HF and SF. Right: Comparison of MD predicted vs. experimental
materials can be run and the results analyzed. The predicted valuesof these major TPU thermophysical properties have been comparedwith the relevant experimental data, and the general agreementbetween prediction and experiment is generally extremely satis-factory. We are thus confident that use of the Nanotools featureof MoDeNa software will lead to novel research and developmentavenues that, in turn, will fundamentally improve the properties ofthese nanostructured polymers.
Acknowledgment
This work is funded by the European Commission under thegrant agreement number 604271 (Project acronym: MoDeNa;project identifier: FP7-NMP-2013-SMALL).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.jocs.2015.11.006.
References
[1] B. Nerlich, S. Lemanczyk, Nanotechnology: social and cultural aspects, in: J.D.Wright (Ed.), International Encyclopedia of the Social & Behavioral Sciences,Second Edition, Elsevier, Paris, France, 2015, pp. 188–193.
[2] M.F. Ashby, P.J. Ferreira, D.L. Schodek, Nanomaterials, Nanotechnologies and
ools: An integrated multiscale simulation workflow to predict. Sci. (2015), http://dx.doi.org/10.1016/j.jocs.2015.11.006
Design, Elsevier Science Ltd., Oxford, UK, 2009.[3] Huntsman Corporation Investor day, (2014), 〈http://phx.corporate-ir.net〉.[4] M. Fermeglia, P. Posocco, S. Pricl, Nano tools for macro problems: multiscale
molecular modeling of nanostructured polymer systems, Compos. Interfaces20 (2013) 379–394.

ING ModelJ
1 utatio
[[
[
[
[
[
[
[
[
[
[
[
[
[
[
ARTICLEOCS-433; No. of Pages 10
0 E. Laurini et al. / Journal of Comp
[5] Z. Posel, P. Posocco, M. Fermegla, M. Lisal, S. Pricl, Modeling hierarchicallystructured nanoparticle/diblock copolymer systems, Soft Matter 9 (2013)2936–2946.
[6] D.R. Nieto, F. Santese, R. Toth, P. Posocco, M. Fermeglia, Simple, fast, andaccurate in silico estimations of contact angle, surface tension, and work ofadhesion of water and oil nanodroplets on amorphous polypropylenesurfaces, ACS Appl. Mater. Interfaces 4 (2012) 2855–2859.
[7] R. Toth, F. Santese, S.P. Pereira, D.R. Nieto, M. Fermeglia, S. Pricl, P. Posocco,Size and shape matter! A multiscale molecular simulation approach topolymer nanocomposites, J. Mater. Chem. 22 (2012) 5398–5409.
[8] P. Posocco, Z. Posel, M. Fermeglia, M. Lisal, S. Pricl, A molecular simulationapproach to the prediction of the morphology of self-assembled nanoparticlesin diblock copolymers, J. Mater. Chem. 20 (2010) 10511–10520.
[9] A. Jain, S.P. Ong, W. Chen, B. Medasani, X. Qu, M. Kocher, M. Brafman, G.Petretto, G.M. Rignanese, G. Hautier, D. Gunter, K.A. Persson, FireWorks: adynamic workflow system designed for high-throughput applications,Concurr. Computat.: Pract. Exper. (2015), http://dx.doi.org/10.1002/cpe.350(in press).
10] https://www.mongodb.org/.11] H. Sun, COMPASS: an ab initio force-field optimized for condensed-phase
applications overview with details on alkane and benzene compounds, J.Phys. Chem. B 102 (1998) 7338–7364.
12] H.J.C. Berendsen, J.P.M. Postma, W.F. van Gunsteren, A. DiNola, J.R. Haak,Molecular-dynamics with coupling to an external bath, J. Chem. Phys. 81(1984) 3684–3690.
13] A. Toukmaji, C. Sagui, J. Board, T. Darden, Efficient particle mesh Ewald basedapproach to fixed and induced dipolar interaction, J. Chem. Phys. 113 (2000)10913–13927.
14] C.A. Helfer, W.L. Mattice, in: J.E. Mark (Ed.), The Rotational Isomeric StateModel in Physical Properties of Polymers Handbook, Springer Verlag, NewYork, USA, 2007, pp. 43–57 (Chapter 3).
15] G. Ciccotti, R. Kapral, A. Sergi, in: Y. Sidney (Ed.), “Non equilibrium MolecularDynamics” in Handbook of Materials Modeling, Springer Verlag, New York,USA, 2005, pp. 745–761 (Chapter 2.17).
16] F. Müller-Plathe, P. Bordat, Reverse non-equilibrium molecular dynamics,Lect. Notes Phys. 640 (2004) 310–326.
17] S. Pricl, M. Fermeglia, Atomistic molecular dynamics simulations of gasdiffusion and solubility in rubbery amorphous hydrocarbon polymers, Chem.Eng. Commun. 190 (2003) 1267–1292.
18] P.G. Lithoxoos, D.L. Peristeras, C.G. Boulougouris, G.I. Economou, Monte Carlosimulation of carbon monoxide, carbon dioxide and methane adsorption onactivated carbon, Mol. Phys. 110 (2012) 1153–1160.
19] A. Wolinska-Grabczyk, A. Jankowski, Gas transport properties of segmentedpolyurethanes varying in the kind of soft segments, Sep. Purif. Technol. 57(2007) 413–417.
20] D. Marchant, J.H. Koo, R.L. Blanski, E.H. Weber, P.N. Ruth, A. Lee, C.E. Schlaeffer,Flammability and thermophysical characterization of thermoplasticelastomer nanocomposites, ACS Natl. Meet. Book Abstr. 228 (2004) 1–19.
21] I. Yilgor, E. Yilgor, G.L. Wilkes, Critical parameters in designing segmentedpolyurethanes and their effect on morphology and properties: acomprehensive review, Polymer 58 (2015) A1–A36.
22] B.F. Pierce, A.H. Brown, V.V. Sheares, Thermoplastic poly (ester urethane)swith novel soft segments, Macromolecules 41 (2008) 3866–3873.
Please cite this article in press as: E. Laurini, et al., MoDeNa Nanotthermophysical properties of thermoplastic polyurethanes, J. Comput
23] M. Sadeghi, M.A. Semsarzadeh, M. Barikani, B. Ghalei, Study on themorphology and gas permeation property of polyurethane membranes, J.Membr. Sci. 385–386 (2011) 76–85.
24] P. Kroll, B. Kroll, Surface free energy of polyurethane coatings with improvedhydrophobicity, Colloid Polym. Sci. 290 (2012) 879–893.
PRESSnal Science xxx (2015) xxx–xxx
Erik Laurini is assistant professor of Chemical Engineering, Molecular Simulations,and Molecular Simulation Techniques for the Pharmaceutical Sciences at the Univer-sity of Trieste, Italy. The main research activities are focused on (i) computer-assisteddrug design and development for targeted therapies and personalized medicine, (ii)study, correlation and prediction of the structure/property relationships for viralproperties, oncogenes, onco-suppressors, and pathologically mutant proteins. He isthe author of more than 40 scientific publications on ISI peer-reviewed internationaljournals and over 50 conference proceedings in the field of molecular modeling andcomputer simulation of complex systems.
Paola Posocco is assistant professor of Chemical Engineering. She is graduated inChemical Engineering at the University of Trieste in 2005. In the same year, shehad an education grant in Plast-Optic Research Center (UD), in the field of plasticmaterials and optoelectronic devices for the automotive. From July 2006 she startedher collaboration with Molecular Simulation Engineering Laboratory (MOSE), activewithin the University of Trieste in the area of molecular modeling of nanostructuredand nanomaterials for life and material science. Her research activity is mainlyfocused on the multiscale modeling of nanostructured complex bio/non bio sys-tems. She is the author of more than 100 scientific publications and conferenceproceedings.
Maurizio Fermeglia is full professor at the Engineering Faculty of the Universityof Trieste, where he holds the course in Chemical and Biochemical Reaction Engi-neering and Data Base Design. From 2006 to 2012 he was appointed as head ofthe Department of Industrial Engineering & Information and, from 201o to 2012 heserved as president of the Research board of the University of Trieste. At present,he is the director of the PhD School of Nanotechnology at the University of Tri-este. Professor Fermeglia research interests are focused on: multiscale modeling formaterials design and life science, phase equilibrium and applied thermodynamics,process simulation, molecular simulation, nanotechnology, and nanomedicine. Pro-fessor Fermeglia currently holds, among others, the following positions: memberof the EFCE working party on thermodynamics and transport properties, IUPAC fel-lows, member of the AIChE, member of GRICU and AIDIC. Maurizio Fermeglia haspublished more than 170 peer reviewed journal articles and book chapters and hasgiven more than 180 conference presentations as invited plenary, keynote, and oralcommunications. He also gives invited lectures seminars at numerous companiesand academic institutions, and organized several workshops on topics related toprocess engineering, nanotechnology and multiscale modeling. At present, he is therector of University of Trieste.
Sabrina Pricl is associate professor of Chemical Engineering, Molecular Simula-tions, and Molecular Simulation Techniques for the Pharmaceutical Sciences at theUniversity of Trieste, Italy. She is also a member of the Scientific Board of thePhD School in Nanotechnology of the University of Trieste. PI and co-PI of sev-eral national and international projects in the field of nano-biotechnology (e.g.,AIRC; EU-FP, Era-NET,.), she is also a member of various international scientificsocieties (e.g., American Chemical Society, American Association of Chemical Engi-neers, American Association for Cancer Research, European Association for researchand Treatment of Cancer, European Society of Nanomedicine, and the AmericanSociety for Nanomedicine). She regularly holds courses and seminars at nationaland international Universities and Scientific Institutions and conferences on top-ics as, including biomacromolecular structure and properties, thermodynamics andmolecular modeling and simulation. The main research activities are focused on (i)computer-assisted drug design and development for targeted therapies and per-
ools: An integrated multiscale simulation workflow to predict. Sci. (2015), http://dx.doi.org/10.1016/j.jocs.2015.11.006
sonalized medicine, (ii) study, correlation and prediction of the structure/propertyrelationships for viral properties, oncogenes, onco-suppressors, and pathologicallymutant proteins. She is the author of more than 180 scientific publications on ISIpeer-reviewed international journals and over 250 conference proceedings in thefield of rheology, molecular modeling and computer simulation of complex systems.


















