
FROM STRUCTURE TO FUNCTION IN PROTEINS: A …
275
FROM STRUCTURE TO FUNCTION IN PROTEINS: A COMPUTATIONAL STUDY A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy in the Faculty of Life Sciences 2010 Tracey Bray Faculty of Life Sciences
Transcript of FROM STRUCTURE TO FUNCTION IN PROTEINS: A …

FROM STRUCTURE TO FUNCTION IN
PROTEINS: A COMPUTATIONAL STUDY
A thesis submitted to the University of Manchester for the degree of
Doctor of Philosophy in the Faculty of Life Sciences
2010
Tracey Bray
Faculty of Life Sciences

2
List of Contents ABSTRACT ............................................................................................................................................. 14
DECLARATION ..................................................................................................................................... 15
COPYRIGHT .......................................................................................................................................... 16
ACKNOWLEDGEMENTS .................................................................................................................... 17
THE AUTHOR ........................................................................................................................................ 18
CHAPTER 1: INTRODUCTION....................................................................................................... 19
1.1 PROTEINS AND THEIR ROLE IN BIOLOGY ....................................................................................... 19
1.1.1 Enzymes ............................................................................................................................. 20
1.1.1.1 Enzyme Kinetics ..........................................................................................................................23
1.1.1.2 Enzyme Functions ........................................................................................................................25
1.2 COMPUTATIONALLY DETERMINING PROTEIN FUNCTION............................................................... 30
1.2.1 Defining Protein Function ................................................................................................. 32
1.2.1.1 Classification Schemes.................................................................................................................32
1.2.2 Functional Transfer Based on Homology.......................................................................... 35
1.2.2.1 Sequence Similarity......................................................................................................................38
1.2.2.2 Structural Similarity .....................................................................................................................39
1.2.2.3 Dynamic Similarity ......................................................................................................................40
1.2.3 Predicting Protein Function in the Absence of Sequence or Structural Similarity............ 41
1.2.3.1 Sequence Motifs...........................................................................................................................41
1.2.3.2 Functional Sites ............................................................................................................................44
1.2.3.3 Genomic Context..........................................................................................................................47
1.2.3.4 Protein-Protein Interactions..........................................................................................................49
1.2.3.5 Subcellular Localisation ...............................................................................................................50
1.2.3.6 Structural Features........................................................................................................................51
1.3 THESIS STRUCTURE...................................................................................................................... 54
1.4 REFERENCES ................................................................................................................................ 55
CHAPTER 2: SEQUENCE AND STRUCTURAL FEATURES OF ENZYMES BY EC CLASS
63
2.1 INTRODUCTION ............................................................................................................................ 63
2.2 METHODS..................................................................................................................................... 66
2.2.1 Dataset Creation................................................................................................................ 66
2.2.2 Defining Active Site Residues ............................................................................................ 67
2.2.3 Calculating Features ......................................................................................................... 68
2.2.4 Culling Redundancy in Features. ...................................................................................... 69
2.2.5 Statistical Analysis............................................................................................................. 69
2.2.6 Rotamer Calculations. ....................................................................................................... 71
2.3 RESULTS AND DISCUSSION........................................................................................................... 72

3
2.3.1 Dataset and Active Site Definition. .................................................................................... 72
2.3.2 Overall Description of Features. ....................................................................................... 76
2.3.3 Unique Descriptive Features. ............................................................................................ 83
2.3.4 Differences in Structure Sizes due to Different Oligomeric State Preferences .................. 86
2.3.4.1 Lyases and Hydrolases in Metabolic Networks............................................................................89
2.3.5 Active-site Non-polarity in Oxidoreductases ..................................................................... 92
2.3.6 Active-site Aspartic Acid Content in Oxidoreductases ...................................................... 94
2.3.6.1 Rotamers ......................................................................................................................................95
2.3.6.2 Hydrogen Bonding .......................................................................................................................96
2.4 CONCLUSIONS .............................................................................................................................. 98
2.5 REFERENCES .............................................................................................................................. 101
CHAPTER 3: FUNCTIONAL SITE IDENTIFICATION IN PROTEINS .................................. 104
3.1 INTRODUCTION: COMPUTATIONAL APPROACHES FOR THE PREDICTION OF FUNCTIONAL SITES .. 105
3.2 METHODS: BENCHMARKING THE ACCURACY OF FUNCTIONAL SITE IDENTIFICATION TOOLS .... 109
3.2.1 Selection of Prediction Methods ...................................................................................... 109
3.2.2 Creation of Test Sets ........................................................................................................ 114
3.2.3 Obtaining and Unifying Functional Site Predictions....................................................... 117
3.3 METHODS: SITESIDENTIFY WEBSERVER .................................................................................... 120
3.3.1 Functional Site Prediction Methods ................................................................................ 120
3.3.2 SitesIdentify Workflow ..................................................................................................... 122
3.3.3 SitesIdentify Usage .......................................................................................................... 122
3.4 RESULTS: BENCHMARKING THE ACCURACY OF FUNCTIONAL SITE IDENTIFICATION TOOLS ...... 124
3.4.1 Recall Accuracy Rates for Real Sites ............................................................................... 124
3.4.2 Crescendo ........................................................................................................................ 126
3.4.3 PASS ................................................................................................................................ 130
3.4.4 Fuzzy Oil Drop ................................................................................................................ 134
3.4.5 QSiteFinder...................................................................................................................... 139
3.4.6 PDBSiteScan.................................................................................................................... 143
3.4.7 Consurf ............................................................................................................................ 147
3.4.8 Thematics......................................................................................................................... 151
3.4.9 SitesIdentify(GM) – Geometry-based............................................................................... 154
3.4.10 SitesIdentify(ConsGM) – Conservation and geometry-based ..................................... 158
3.4.11 All Methods ................................................................................................................. 161
3.5 RESULTS: SITESIDENTIFY WEB-SERVER ..................................................................................... 165
3.6 DISCUSSION ............................................................................................................................... 170
3.7 REFERENCES .............................................................................................................................. 172
CHAPTER 4: PREDICTING EC CLASS FROM ENZYME STRUCTURE.............................. 176
4.1 INTRODUCTION .......................................................................................................................... 176
4.1.1 Machine Learning Theory ............................................................................................... 178
4.1.1.1 Support Vector Machines ...........................................................................................................180

4
4.2 METHODS................................................................................................................................... 186
4.2.1 Dataset Creation.............................................................................................................. 186
4.2.2 Defining Active Site Residues .......................................................................................... 187
4.2.2.1 Dataset 4.1..................................................................................................................................187
4.2.2.2 Dataset 4.2..................................................................................................................................187
4.2.3 Calculating Features. ...................................................................................................... 187
4.2.3.1 Structural Features......................................................................................................................188
4.2.3.2 Sequence Features ......................................................................................................................189
4.2.4 Prediction Methods.......................................................................................................... 190
4.2.4.1 Functional Classification where the Active Site is Known.........................................................190
4.2.4.2 Functional Prediction where the Active Site is not Known ........................................................192
4.3 PREDICTING EC CLASS FOR ENZYMES WITH KNOWN ACTIVE SITE LOCATION .......................... 194
4.4 PREDICTING EC CLASS FOR ENZYMES WITH PREDICTED ACTIVE SITE LOCATIONS.................... 197
4.5 CONCLUSIONS ............................................................................................................................ 202
4.6 REFERENCES .............................................................................................................................. 203
CHAPTER 5: GAUSSIAN NETWORK MODELING OF OLIGOMERIC PROTEINS .......... 205
5.1 INTRODUCTION .......................................................................................................................... 205
5.1.1 Cooperativity in Oligomeric Enzymes ............................................................................. 205
5.1.2 Application of Normal Mode Analysis to the Study of Proteins....................................... 210
5.1.3 Normal Mode Analysis and the Gaussian Network Model .............................................. 213
5.2 METHODS................................................................................................................................... 217
5.2.1 Dataset Creation for the Cooperativity Analysis ............................................................. 217
5.2.2 Dataset Creation for the Active Site Correlation Analysis .............................................. 221
5.2.3 Dataset Creation for the Structural Environment Correlation Analysis ......................... 222
5.2.4 Calculation of Residue Motion Correlation..................................................................... 225
5.3 CORRELATED RESIDUE MOTIONS IN COOPERATIVE OLIGOMERIC ENZYMES ............................. 228
5.3.1 Analysis of Residue Correlations in Co-operative and Non Cooperative Enzymes......... 229
5.3.2 Discussion of Cooperative Enzyme Analysis ................................................................... 238
5.4 CORRELATION OF RESIDUE MOTIONS IN ENZYME ACTIVE SITE REGIONS.................................. 240
5.4.1 Analysis of Residue Correlations in Enzyme Active Sites ................................................ 240
5.4.2 Discussion of Correlation of Motion in Active Sites ........................................................ 245
5.5 PATTERNS OF CORRELATION OF RESIDUE MOTION AS A STRUCTURAL FEATURE OF OLIGOMERIC
PROTEINS............................................................................................................................................. 246
5.5.1 Differences in Residue Motion Correlation According to Structural Environment......... 247
5.5.1.1 Are Residues with High Dynamic Coupling to their Equivalent Residues more Evolutionarily
Conserved?.................................................................................................................................................258
5.5.2 Discussion of Correlation of Motion According to Structural Environment ................... 260
5.6 CONCLUSIONS ............................................................................................................................ 263
5.7 REFERENCES .............................................................................................................................. 266
CHAPTER 6: CONCLUSIONS ....................................................................................................... 272
Final word count : 65,147

5
List of Figures
Figure 1.1 A schematic representation of how an enzyme increases the rate of reaction by
lowering the energy barrier in order for the reaction to proceed. ........................................... 21
Figure 1.2 Schematic illustration of the concerted and sequential models for cooperative
substrate binding............................................................................................................................. 22
Figure 1.3 A simplified representation of a mechanism for single-substrate enzyme
reactions. .......................................................................................................................................... 23
Figure 1.4 A Michaelis-Menton graph showing the maximum velocity at saturation (Vmax),
and the Michaelis-Menton constant (Km).................................................................................... 24
Figure 1.5 A plot showing the difference in change in reaction rate with the concentration
between cooperative and non-cooperative enzymes. ................................................................ 25
Figure 1.6 A simple example of a redox reaction. ..................................................................... 27
Figure 1.7 A simple example of a transferase reaction.............................................................. 27
Figure 1.8 A schematic equation for the hydrolysis reaction. .................................................. 27
Figure 1.9 The proportion of each top EC class in the PDB. ................................................. 29
Figure 1.10 The rise in the number of structures deposited into the PDB since 1986. ....... 31
Figure 1.11 The accuracy of function annotation with varying sequence identity................ 37
Figure 1.12 Schematic diagram of a generic approach for constructing sequence motifs... 42
Figure 1.13 A schematic representation of the Rosetta Stone method of assigning protein
function. ........................................................................................................................................... 48
Figure 1.14 The 52 structural features used to classify into enzyme/non-enzyme from a
previous study by Dobson and Doig .......................................................................................... 53
Figure 2.1 A flow diagram showing how the dataset is culled from the original 880 CSA
literature entries to the dataset of 294 unique non-redundant enzymes................................. 73
Figure 2.2 The percentage coverage of CSA residues by varying active site criteria
thresholds for (a) surface area and (b) distance from centroid. ............................................... 75
Figure 2.3 The median aromatic proportion of the active site for each EC class................. 79
Figure 2.4 Amino acids that showed significant differences between the six EC classes in
either the active site, surface residues or the total protein........................................................ 79
Figure 2.5 The median value of significantly different charge-related features for each EC
class. .................................................................................................................................................. 80

6
Figure 2.6 The median proportion of the total surface area that belongs to the active site
for each EC class. ........................................................................................................................... 80
Figure 2.7 The median value of significantly different size-related features for each EC
class. .................................................................................................................................................. 80
Figure 2.8 The median value of the total sum of B factors for each EC class. ..................... 81
Figure 2.9 The percentage of each EC class on each oligomeric status catergory................ 81
Figure 2.10 The median amino acid composition of the total protein for amino acids
showing significant differences between the EC classes........................................................... 81
Figure 2.11 The median amino acid composition of the protein surface for amino acids
showing significant differences between the EC classes........................................................... 82
Figure 2.12 The median amino acid composition of the active site for amino acids showing
significant differences between the EC classes .......................................................................... 82
Figure 2.13 A network diagram showing the significantly different features (as nodes)
connected by lines where there is a probable correlation (the R value is more than 0.195,
the critical R value at the 5% significance level)......................................................................... 84
Figure 2.14 The median proportion of the total protein that is either helix or non-helix and
non-sheet for each EC class. ......................................................................................................... 85
Figure 2.15 The percentage of oligomers that have single sub-unit or shared sub-unit active
sites in each class............................................................................................................................. 89
Figure 2.16 The observed number of enzymes divided by the expected number of enzymes
in each class for all choke points and the 50% most loaded choke points in the
Saccharomyces cerevisiae metabolic network. .................................................................................... 91
Figure 2.17 The observed number of enzymes divided by the expected number of enzymes
in each class for the 25% most loaded enzymes (incoming and outgoing) from the yeast
metabolic network. ......................................................................................................................... 92
Figure 2.18 The distribution of active site non-polar proportions for the cofactor-binding
and non-cofactor-binding oxidoreductases. ............................................................................... 93
Figure 2.19 The percentage of enzymes in each set that prefer aspartic acid as an active site
residue (there is a higher proportion of active site ASP than GLU), prefer glutamic acid as
an active site residue (there is a higher proportion of active site GLU than ASP), and where
there are equal amounts of aspartic and glutamic acid in the active sites............................... 94
Figure 2.20 The percentage of accessible rotamers available to all active site ASP and GLU
in each class. .................................................................................................................................... 96
Figure 2.21 The underlying distribution for the number of hydrogen bonds per ASP or
GLU in the active site for EC1..................................................................................................... 97

7
Figure 3.1 The asymmetric unit structure of 1daa. .................................................................. 119
Figure 3.2 Distribution of annotated residues recall rates in real sites. ................................ 125
Figure 3.3 The distribution of absolute recall rates per protein for Crescendo in A) the
enzyme set and B)the non-enzyme set. ..................................................................................... 128
Figure 3.4 The cumulative percentage of distances between Crescendo-predicted and real
centroids within the two sets. ..................................................................................................... 129
Figure 3.5 Diagram taken from Brady and Stouten, 2000 showing how the PASS method
defines buried volume.................................................................................................................. 130
Figure 3.6 The distribution of absolute recall rates per protein for PASS in A) the enzyme
set and B) the non-enzyme set.................................................................................................... 132
Figure 3.7 The cumulative percentage of distances between PASS-predicted and real
centroids within the two sets. ..................................................................................................... 133
Figure 3.8 The distribution of absolute recall rates per protein for FOD in A) the enzyme
set and B) the non-enzyme set.................................................................................................... 137
Figure 3.9 The cumulative percentage of distances between FOD-predicted and real
centroids within the two sets. ..................................................................................................... 138
Figure 3.10 The distribution of absolute recall rates per protein for QSiteFinder in A) the
enzyme set and B) the non-enzyme set. .................................................................................... 141
Figure 3.11 The cumulative percentage of distances between QSiteFinder-predicted and
real centroids within the two sets. .............................................................................................. 142
Figure 3.12 The distribution of absolute recall rates per protein for PDBSiteScan in A) the
enzyme set and B) the non-enzyme set. .................................................................................... 145
Figure 3.13 The cumulative percentage of distances between PDBSiteScan-predicted and
real centroids within the two sets. .............................................................................................. 146
Figure 3.14 The distribution of absolute recall rates per protein for Consurf in A) the
enzyme set and B) the non-enzyme set. .................................................................................... 149
Figure 3.15 The cumulative percentage of distances between Consurf-predicted and real
centroids within the two sets. ..................................................................................................... 150
Figure 3.16 The distribution of absolute recall rates per protein for Thematics in the
enzyme set...................................................................................................................................... 152
Figure 3.17 The cumulative percentage of distances between Thematics-predicted and real
centroids within the enzyme set. ................................................................................................ 153
Figure 3.18 The distribution of absolute recall rates per protein for SitesIdentify(GM) in A)
the enzyme set and B) the non-enzyme set. ............................................................................. 156

8
Figure 3.19 The cumulative percentage of distances between SitesIdentify(GM) predicted
and real centroids within the enzyme and non-enzyme set.................................................... 157
Figure 3.20 The distribution of absolute recall rates per protein for SitesIdentify(ConsGM)
in A) the enzyme set and B) the non-enzyme set. ................................................................... 160
Figure 3.21 The cumulative percentage of distances between SitesIdentify(ConsGM)
predicted and real centroids within the enzyme and non-enzyme set. ................................. 161
Figure 3.22 Comparison of distances between the real centroids and the predicted
centroids in the enzyme dataset for each method. .................................................................. 162
Figure 3.23 Comparison of distances between the real centroids and the predicted
centroids in the non-enzyme dataset for each method. .......................................................... 162
Figure 3.24 Comparison of distances between the real centroid and the predicted centroid
for Consurf and SitesIdentify(ConsGM) run on the first chain of the enzyme structures.
......................................................................................................................................................... 164
Figure 3.25 Screenshot for SitesIdentify showing the required user input fields............... 166
Figure 3.26 Screenshot of an example results output for SitesIdentify. .............................. 167
Figure 3.27 An example of highlighted residues in an alternative predicted site. ............... 168
Figure 3.28 An example of differential site prediction between asymmetric and biological
unit structures................................................................................................................................ 169
Figure 4.1 A schematic diagram representing the classification of two groups of data by an
SVM model.................................................................................................................................... 181
Figure 4.2 A schematic diagram representing how the transformation of data into a higher-
dimensional space by using kernel functions can allow the separation of the data by a linear
function. ......................................................................................................................................... 182
Figure 4.3 An example of a decision tree that can be followed to classify into multiple
groups using binary classifications. ............................................................................................ 183
Figure 4.4 A schematic diagram showing how varying the error penalty parameter, C can
identify a hyperplane that achieves a high accuracy on test data. .......................................... 185
Figure 4.5 A schematic representation of the vector comparison method used to predict
the EC class of enzymes with known active sites. ................................................................... 191
Figure 4.6 Accuracies achieved using the top n-ranked features in the prediction model.195
Figure 4.7 Prediction accuracies achieved using a default grid search method for the best C
and γ parameters. A) Shows the accuracies on a 2D plot and B) shows this in 3D.......... 198
Figure 4.8 Accuracies achieved using the top-ranked features with 10-fold cross-validation
on the training set. ........................................................................................................................ 199

9
Figure 5.1 Example reaction rate (v/Vmax) vs. substrate concentration ([S]) for a non-
cooperative (A), a positively cooperative (B) and a negatively cooperate enzyme (C). ...... 207
Figure 5.2 A protein structure (lysine–arginine–ornithine binding protein; top) shown as an
elastic network............................................................................................................................... 214
Figure 5.3 A schematic representation of the basic terms used in the Gaussian network
model. ............................................................................................................................................. 216
Figure 5.4 An example of a protein structure (1ji7) with the interface residues highlighted.
......................................................................................................................................................... 223
Figure 5.5 An example cross-correlation matrix for 1D3V (Manganese Metalloenzyme
Arginase), which is a homo-trimer. ............................................................................................ 226
Figure 5.6 The biological unit structure for 1D3V coloured according to each residue’s
cc_equiv score. .............................................................................................................................. 228
Figure 5.7 Positively cooperative enzyme structures............................................................... 232
Figure 5.8 Negatively cooperative enzyme structures. ............................................................ 233
Figure 5.9 Non-cooperative enzyme structures. ...................................................................... 234
Figure 5.10 Distribution of all residue’s cc_equiv values for positively, negatively and non-
cooperative enzymes. ................................................................................................................... 238
Figure 5.11 The distribution of scaled cc_equiv values for site and non-site residues for all
enzymes in the dataset. ................................................................................................................ 242
Figure 5.12 The cumulative percentage of cc_equiv values for all site and non-site residues
in each set....................................................................................................................................... 242
Figure 5.13 The distribution of scaled cc_equiv scores for each structural environment
over all residues in the dataset. ................................................................................................... 248
Figure 5.14 The distribution of Spearman’s rank correlation coefficients between cc_equiv
values and distance between equivalent residues for individual proteins in the dataset. ... 252
Figure 5.15 An example of a protein in the dataset (1h16) where the closest equivalent
residues in the interface have the highest dynamic coupling and the rest of the interface
residues are less-coupled in comparison. .................................................................................. 253
Figure 5.16 The distribution of scaled cc_equiv values for the closest pair of equivalent
residues in each protein. .............................................................................................................. 254
Figure 5.17 The distribution of scaled distances between the most highly-correlated
equivalent pair in each protein.................................................................................................... 254
Figure 5.18 The distribution of Spearman’s correlation coefficients between cc_within and
cc_equiv values for all proteins in the set. ................................................................................ 255

10
Figure 5.19 The distribution of Spearman’s correlation coefficients between cc_within and
cc_equiv values derived from GNM calculations on both the oligomer and the individual
subunits for all proteins in the set. ............................................................................................. 256
Figure 5.20 An example of a protein (1cq3) with residues coloured by cc_equiv value (A),
cc_within values derived using GNM calculations on the oligomer (B), and cc_within
values derived from GMN calculations on the individual monomers(C). ........................... 257
Figure 5.21 The distribution of Spearman’s correlation coefficients for the relationship
between conservation and degree of correlation of motion between equivalent residues for
each protein in the set. ................................................................................................................. 259
Figure 5.22 The cross-correlation matrix for a homo-trimer (1d3v), which shows obvious
patterns of correlation between residues within subunits but less definition between
residues in different subunits. ..................................................................................................... 262

11
List of Tables Table 1.1 A table showing how the coverage of classification schemes varies per protein. 35
Table 1.2 The main primary sequence databases with their URL and relevant reference. .. 38
Table 1.3 Examples of structure comparison programs with their URL and reference. ..... 39
Table 1.4 A list of sequence motif resources.............................................................................. 43
Table 1.5 Functional/active/binding site residue databases and comparison tools available
via the web. ...................................................................................................................................... 45
Table 2.1 PDB codes for each enzyme in the dataset. .............................................................. 74
Table 2.2 List of all features calculated for each enzyme.......................................................... 77
Table 2.3 The p-value (adjusted for the false discovery rate), the EC class that had the
highest mean or median value and the EC class with the lowest mean or median value for
all features that showed a significant difference between EC classes (p<0.05)..................... 78
Table 2.4 The correlation between total leucine and proline composition and the secondary
structure environments that they are typically associated with. ............................................... 84
Table 2.5 Subcellular location annotation (where available) for each EC class..................... 88
Table 2.6 Number of enzymes that are bound to cofactors and those that are not............. 93
Table 2.7 Average number of hydrogen bonds per aspartic acid/glutamic acid split by
active-site residues and non-active-site residues ........................................................................ 97
Table 3.1 The seven tools used in this analysis along with the broad category of their
method. Each method is described in more detail in their relevant section below. .......... 110
Table 3.2: Functional site prediction tools not included in the comparison analysis.
Reasons for non-inclusion in the analysis are further explained below:............................... 113
Table 3.3 The PDB codes for the 237 structures in the enzyme dataset ............................. 115
Table 3.4 The PDB codes for the 13 structures in the non-enzyme dataset. ...................... 116
Table 3.5 Annotated residues recalled by the site definition criteria..................................... 125
Table 3.6 The functional site prediction accuracy results for Crescendo............................. 127
Table 3.7 The functional site prediction accuracy results for PASS. .................................... 131
Table 3.8 The functional site prediction accuracy results for FOD...................................... 136
Table 3.9 The functional site prediction accuracy results for QSiteFinder.......................... 140
Table 3.10 The functional site prediction accuracy results for PDBSiteScan...................... 144
Table 3.11 The functional site prediction accuracy results for Consurf. .............................. 148
Table 3.12 The functional site prediction accuracy results for Thematics. .......................... 152
Table 3.13 The functional site prediction accuracy results for SitesIdentify (Uniform charge
method) .......................................................................................................................................... 155

12
Table 3.14 The functional site prediction accuracy results for SitesIdentify(ConsGM). ... 159
Table 3.15 The absolute and relative recall rates achieved for the enzyme dataset along with
the average distance between real and predicted centroids for each method...................... 163
Table 3.16 The absolute and relative recall rates achieved for the non-enzyme dataset along
with the average distance between real and predicted centroids for each method. ............ 163
Table 4.1 Features used in the EC class prediction methods................................................. 188
Table 4.2 Features that are removed in the EC class prediction method where the active
site location is known................................................................................................................... 196
Table 4.3 The number of enzyme structures in each class in Dataset 4.2............................ 197
Table 4.4 The 10 lowest ranked features that were removed from the dataset to train the
final model. .................................................................................................................................... 199
Table 4.5 The number of predictions of each class made by the model without class
weightings. ..................................................................................................................................... 200
Table 4.6 The number of predictions of each class made by the model with class
weightings. ..................................................................................................................................... 201
Table 5.1 Dataset 5.1: A list of enzymes with annotated Hill coefficients and a structure
deposited in the PDB for the same organism. ......................................................................... 220
Table 5.2 Dataset 5.2. A list of 114 non redundant homo-oligomeric enzyme PDB
structures with a literature-based active site information obtained from the CSA. ............ 221
Table 5.3 Dataset 5.3: A list of 636 non-redundant homo-oligomeric PDB structures. ... 224
Table 5.4 The average equivalent residue cross-correlation (cc_equiv) scores for site and
non-site residues for cooperative and non-cooperative enzymes.......................................... 235
Table 5.5 The Spearman’s rank correlation coefficient for the comparison between distance
from active site centroid and cc_equiv for each enzyme........................................................ 237
Table 5.6 Average scaled cc_equiv values for pooled residues from enzymes within each
set. ................................................................................................................................................... 237
Table 5.7. Site correlation vs. non-site correlation results for individual enzymes within the
set. ................................................................................................................................................... 241
Table 5.8 The Spearman’s rank correlation coefficient for the relationship between distance
from active site centroid and cc_equiv for all enzymes in the set. ........................................ 243
Table 5.9 Table showing the breakdown of Spearman’s rank correlation coefficients
between distance from active site centroid and cc_equiv value for individual enzymes in
the dataset. ..................................................................................................................................... 243
Table 5.10 The number of proteins in the whole dataset that have either a lower or higher
average active site B-factor than non-site residues, split by significance.............................. 244

13
Table 5.11 The number of proteins in the whole dataset that have either a negative or
positive correlation between B-factor and cc_equiv, split by significance. .......................... 244
Table 5.12 Number of proteins where the active site residues are significantly more
correlated than non-site residues that have either higher or lower average site B-factors in
comparison to the rest of the protein, split by significance. .................................................. 244
Table 5.13 Number of proteins where the active site residues are significantly more
correlated than non-site residues that have either a positive or negative relationship
between cc_equiv and B-factor, split by significance. ............................................................. 244
Table 5.14 The mean scaled cc_equiv values for each structural environment for pooled
residues from all proteins in the set. .......................................................................................... 248
Table 5.15. Pairwise comparison of average cc_equiv values for each structural
environment. ................................................................................................................................. 249
Table 5.16 The number of proteins in the Dataset 2.3 that have either a negative or
positive correlation between B-factor and cc_equiv, split by significance. .......................... 250
Table 5.17 The average scaled B-factors for each structural environment over all residues
in the set. ........................................................................................................................................ 250
Table 5.18 Pairwise comparison of average scaled B-factors for each structural
environment. ................................................................................................................................. 250
Table 5.19 The average scaled distance between equivalent residues for each structural
environment over all residues in the set. ................................................................................... 250
Table 5.20 The number of proteins in the Dataset 2.3 that have either a negative or
positive correlation between the distance between equivalent residues and cc_equiv, split
by significance. .............................................................................................................................. 252
Table 5.21 The average scaled conservation score for each structural environment. ........ 258
List of Equations
Equation 1.1 The Hill equation. ................................................................................................... 25
Equation 2.1 The calculation of the FDR-adjusted p-value (P(FDR))........................................ 70
Equation 3.1 The equation for the conservation score of residue x, which is used to weight
the uniform charge. ...................................................................................................................... 121
Equation 5.1 The Hill equation. ................................................................................................. 206
Equation 5.2 The correlation between fluctuations for residues i and j. .............................. 216

14
Abstract
Name: Tracey Bray University: The University of Manchester Degree: Doctor of Philosophy Thesis title: From structure to function in proteins: A computational study The study of proteins and their function is key to understanding how the cell works in normal and disease states. Historically, the study of protein function was limited to biochemical characterisation, but as computing power and the number of available protein sequences and structures increased this allowed the relationship between sequence, structure and function to be explored. As the number of sequences and structures grows beyond the capacity for experimental groups to study them, computational approaches to inferring function become more important. Enzymes make up approximately half of the known protein sequences and structures, and most of the work in this thesis focuses on the relationship between the sequence, structure and function in enzymes. Firstly, the differences in sequence and structural features between enzymes of the six main functional classes are explored. Features that exhibited the most significant differences between the six classes were further studied to explore their link with function. This study suggested reasons as to why groups of functionally similar but non-homologous enzymes share similar sequence and structural features. A computational tool to predict EC class was then developed in an attempt to exploit the differences in these features. In order to calculate features relating to a particular active site to be used in the EC class prediction method, it was first necessary to predict the active site location. A comprehensive analysis of currently-available functional site prediction tools identified an approach previously developed by this group as amongst the best-performing methods. Here, a tool was created to deliver this approach via a publicly-available web-server, which was subsequently used in the attempt to predict EC class. The study of differences in sequence and structural features between classes revealed differences in oligomeric status between functions. High-order oligomers were linked to an increase in metabolic control in the lyases, possibly via mechanisms such as cooperativity. To further test this idea, it was necessary to be able to computationally identify oligomeric enzymes that act cooperatively. Since no such method currently exists, the degree of coupling of dynamic fluctuations between subunits was explored as a possible way of detecting cooperativity. Whilst this was unsuccessful, the study highlighted the existence of a pattern of correlated motions that were conserved over a wide range of non-homologous and functionally diverse proteins. These observations shed further light on the link between sequence, structure and function and highlight the functional importance of dynamics in protein structures.

15
Declaration
No portion of the work referred to in this thesis has been submitted in support of an
application for another degree or qualification of this or any other university or other
institute of learning.

16
Copyright
i. The author of this thesis (including any appendices and/or schedules to this thesis)
owns certain copyright or related rights in it (the “Copyright”) and s/he has given
The University of Manchester certain rights to use such Copyright, including for
administrative purposes.
ii. Copies of this thesis, either in full or in extracts and whether in hard or electronic
copy, may be made only in accordance with the Copyright, Designs and Patents
Act 1988 (as amended) and regulations issued under it or, where appropriate, in
accordance with licensing agreements which the University has from time to time.
This page must form part of any such copies made.
iii. The ownership of certain Copyright, patents, designs, trade marks and other
intellectual property (the “Intellectual Property”) and any reproductions of
copyright works in the thesis, for example graphs and tables (“Reproductions”),
which may be described in this thesis, may not be owned by the author and may be
owned by third parties. Such Intellectual Property and Reproductions cannot and
must not be made available for use without the prior written permission of the
owner(s) of the relevant Intellectual Property and/or Reproductions.
iv. Further information on the conditions under which disclosure, publication and
commercialisation of this thesis, the Copyright and any Intellectual Property
and/or Reproductions described in it may take place is available in the University
IP Policy (see
http://www.campus.manchester.ac.uk/medialibrary/policies/intellectual-
property.pdf), in any relevant Thesis restriction declarations deposited in the
University Library, The University Library’s regulations (see
http://www.manchester.ac.uk/library/aboutus/regulations) and in The
University’s policy on presentation of Theses

17
Acknowledgements
I would firstly like to thank my two supervisors, Prof Andrew Doig and Dr Jim Warwicker,
for not only giving me the opportunity to work on this project, but for their continuous
support, advice and unrivalled expertise throughout the past four years. I would also like
to thank the members of their groups, Tala Bakheet, Salim Bougouffa, Pedro Chan,
Andrew Cawley, Richard Greaves, Myra Kinalwa-Nalule and James Kitchen, for
generously sharing their skills, knowledge and opinions. I also owe thanks to other
members of the bioinformatics groups (past and present), such as Jennifer Bradford, John
Pinney and Julian Selley, for their technical support and expert advice. I am extremely
grateful to the BBSRC for funding this research.
I would also like to thank my family and friends who have supported, counseled and
encouraged me throughout this time. I am forever indebted to my parents for their
encouragement and unwavering belief. It was their dream to see me go to university and it
is to them that I owe this achievement. Lastly, I am enormously grateful for the patience,
encouragement and support from my husband, Paul, who has smoothed the world in order
to make this possible.

18
The Author
Prior to this PhD, I completed a BSc (Hons) in Biological and Computational Science
(Bioinformatics) at the University of Manchester. This was a 4 year program that
incorporated a 12 month placement in industry, which I spent at Amgen in Cambridge.
During my placement I worked as a biostatistical programmer on the analysis of a phase
III clinical trial on a colorectal cancer therapy. I also spent a 3 month period as a database
curator in the WormBase team during a summer placement at the Wellcome Trust Sanger
Institute in Cambridge.

19
Chapter 1: Introduction
1.1 Proteins and their role in biology
Proteins, made from polymers of amino acids, are involved in almost every biological
process within a cell. They come in a wide variety of structural arrangements and perform
a broad range of roles, such as structural proteins, enzymes and signaling proteins.
Enzymes act as a catalyst to speed up metabolic reactions and are often globular in
structure, whilst structural proteins like collagen or fibrillin tend to form fibrous structures
that play a supportive role in the cell. Receptor proteins transfer signals, typically in and
out or cells or organelles, and contain a transmembrane portion that traverses the cell or
organelle membrane. These interact with signaling proteins and a range of other effectors
to transmit signals throughout the cell and come in a wide range of structures.
The study of proteins and their function is key to understanding how the cell functions and
how to exploit their properties in order to treat diseases. Historically, the study of protein
function was limited to biochemical characterisation, but the advent of sequencing
methods meant that the underlying amino acid sequence of proteins could be obtained.
Structural determination methods, such as X-ray crystallography and later Nuclear
Magnetic Resonance (NMR) allowed the visualisation of the three-dimensional structure of
a protein. The increase in use of these technologies has made it possible to examine and
compare structural and sequence attributes in order to study protein function and
evolution.
This increase in data has driven the production of large databases, such as Uniprot1 and the
Protein Data Bank2 (PDB), that enable the storage, organisation and retrival of the huge
amounts of protein sequence and structural data. Comparative studies of the data stored in
these databases have provided much information about how proteins and their structures
have evolved. Enzymes are the single largest class of proteins contained in sequence and
structural databases and make up approximately half of the protein structures deposited in
the PDB and half of the protein sequences deposited in UniProt. Enzymes are one of the
most well-studied group of proteins and information, not only in terms of sequence and
structure, but in terms of their biochemical data. Mechanism and biochemical information,
is widely available via a number of well-annotated databases (i.e. MACIE3, CSA4, KEGG5,

20
BRENDA6). The majority of the work in this thesis focuses on the relationship between
the sequence, structure and function in enzymes.
1.1.1 Enzymes
Enzymes speed up the rate of a reaction by lowering the activation energy required for the
reaction to occur. They are highly specific to the substrates that they bind and the
reactions that they carry out. Early research suggested that the specificity of substrate
binding occurs via a “lock and key” mechanism7, where the predefined geometric shape of
the active site perfectly complemented the shape of the substrate, therefore allowing a
perfect fit. This explanation was widely held until the late 1950s when Daniel Koshland
proposed that enzymes exhibit flexibility in their active site structure in reaction to the
bound substrate so that the transition state conformation can be stabilised8.
The increase in speed in reaction is usually achieved by an enzyme either stabilizing the
transition state of the enzyme or substrate or providing an alternative reaction path
through the production of intermediates. Some enzymes also bind cofactors, which
interact with the substrate in order to allow the reaction. The enzyme brings the substrate
and cofactor into close proximity by binding them in the active site. This increases the
speed of interaction between the substrate and cofactor over what would occur by normal
diffusion and therefore increases the rate of reaction.
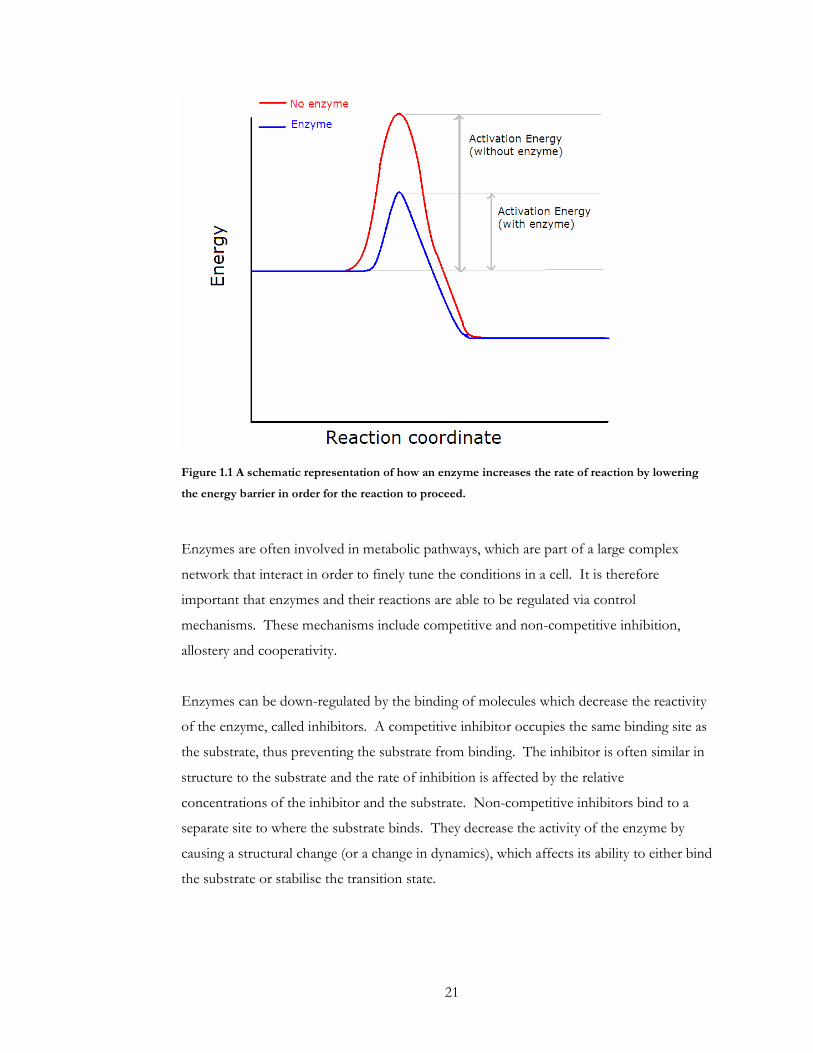
21
Figure 1.1 A schematic representation of how an enzyme increases the rate of reaction by lowering
the energy barrier in order for the reaction to proceed.
Enzymes are often involved in metabolic pathways, which are part of a large complex
network that interact in order to finely tune the conditions in a cell. It is therefore
important that enzymes and their reactions are able to be regulated via control
mechanisms. These mechanisms include competitive and non-competitive inhibition,
allostery and cooperativity.
Enzymes can be down-regulated by the binding of molecules which decrease the reactivity
of the enzyme, called inhibitors. A competitive inhibitor occupies the same binding site as
the substrate, thus preventing the substrate from binding. The inhibitor is often similar in
structure to the substrate and the rate of inhibition is affected by the relative
concentrations of the inhibitor and the substrate. Non-competitive inhibitors bind to a
separate site to where the substrate binds. They decrease the activity of the enzyme by
causing a structural change (or a change in dynamics), which affects its ability to either bind
the substrate or stabilise the transition state.
22
The binding of an effector at a site distal to the binding site that affects the rate of the
enzyme reaction is also termed allosteric regulation. In contrast to non-competitive
inhibition, allosteric effectors can also up-regulate an enzyme by changing the structure or
dynamics to favour the formation of the transition state. Usually the allosteric modulator
is heterotrophic (i.e. different from the enzyme’s substrate) but enzymes can be regulated
by their own substrate (homotrophic allostery). A special case of this is cooperativity.
Cooperativity occurs in a multimeric enzyme where the binding of the enzyme substrate
into the binding site on one subunit increases the affinity for the substrate in binding sites
on other subunits. Negative cooperativity can also occur where substrate binding on one
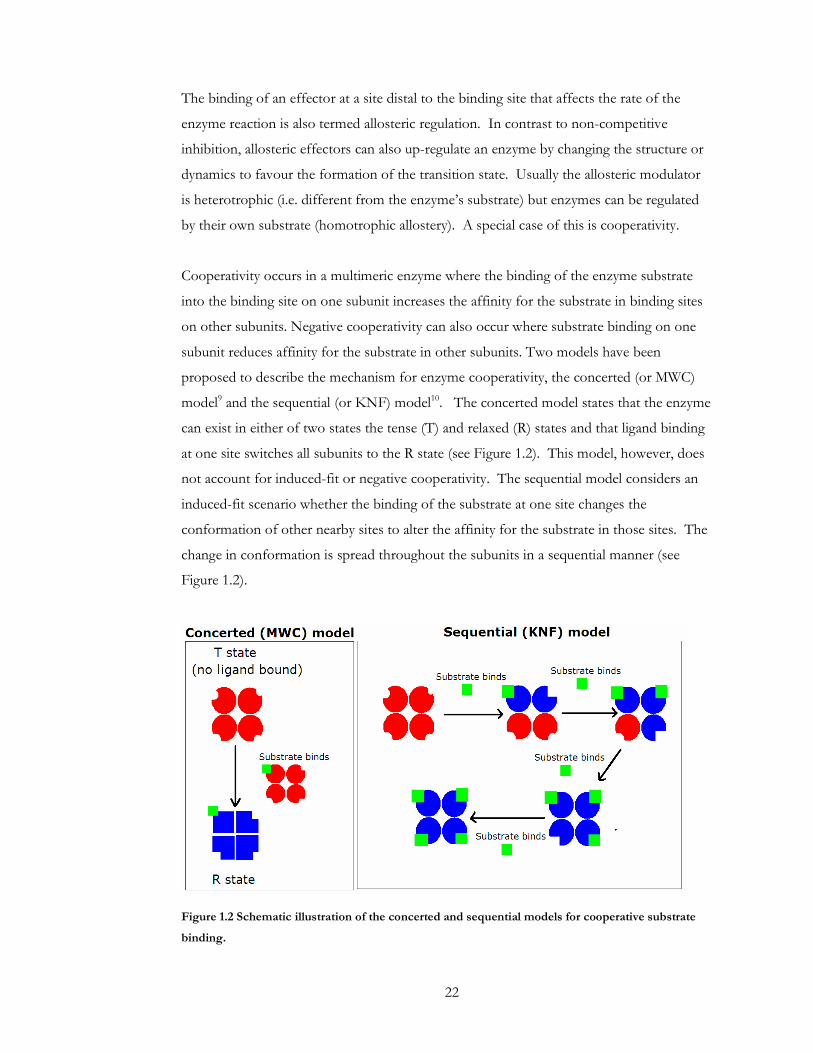
subunit reduces affinity for the substrate in other subunits. Two models have been
proposed to describe the mechanism for enzyme cooperativity, the concerted (or MWC)
model9 and the sequential (or KNF) model10. The concerted model states that the enzyme
can exist in either of two states the tense (T) and relaxed (R) states and that ligand binding
at one site switches all subunits to the R state (see Figure 1.2). This model, however, does
not account for induced-fit or negative cooperativity. The sequential model considers an
induced-fit scenario whether the binding of the substrate at one site changes the
conformation of other nearby sites to alter the affinity for the substrate in those sites. The
change in conformation is spread throughout the subunits in a sequential manner (see
Figure 1.2).
Figure 1.2 Schematic illustration of the concerted and sequential models for cooperative substrate
binding.

23
1.1.1.1 Enzyme Kinetics
The kinetics of non-cooperative enzymes with a single substrate can usually be described
by the Michaelis-Menton model11. This states that a substrate (S) binds with an enzyme (E)
to form an enzyme-substrate complex (ES), which undergoes catalysis and produces the
product (P) as shown in Figure 1.3. Where the substrate concentration is high, the rate of
the reaction is limited by the number of enzymes (or number of active sites) available to
form complexes with the substrate. Initially, therefore, the increase in the rate of reaction
is high as substrates diffuse quickly into active sites. As more enzyme active sites become
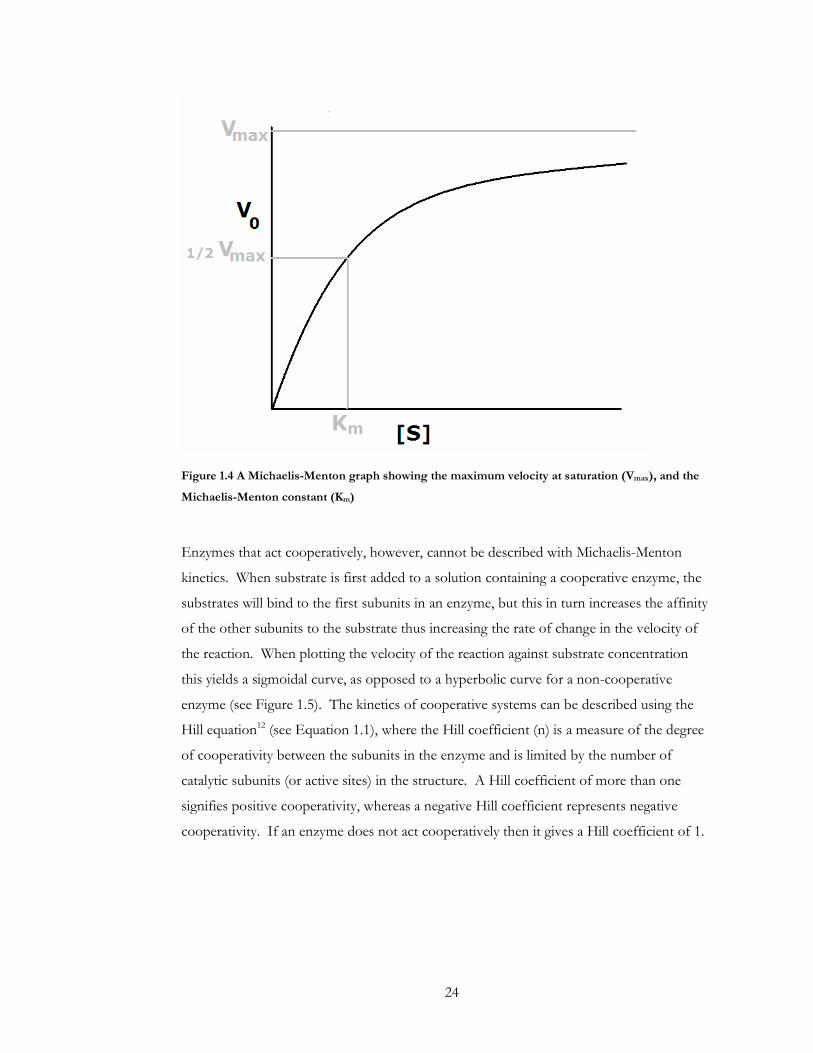
occupied the increase in the rate slows until the maximum speed of the reaction (Vmax) is
reached. An important measure of an enzyme’s kinetics is its Michealis-Menton constant
(Km), which is the concentration of the substrate required for the reaction rate to reach half
its maximum velocity (see Figure 1.4). The efficiency of an enzyme can also be measured
by dividing Kcat by Km, termed the specificity constant and is useful for comparing the
kinetics of different enzymes.
Figure 1.3 A simplified representation of a mechanism for single-substrate enzyme reactions.
k1 is the rate constant for substrate binding, k-1 is the rate constant for dissociation and kcat is rate
constant for the catalytic step (or combination of steps) involved in converting the substrate into the
product. It can also be thought of as the number of substrates that the enzyme can convert in one
second.
24
Figure 1.4 A Michaelis-Menton graph showing the maximum velocity at saturation (Vmax), and the
Michaelis-Menton constant (Km)
Enzymes that act cooperatively, however, cannot be described with Michaelis-Menton
kinetics. When substrate is first added to a solution containing a cooperative enzyme, the
substrates will bind to the first subunits in an enzyme, but this in turn increases the affinity
of the other subunits to the substrate thus increasing the rate of change in the velocity of
the reaction. When plotting the velocity of the reaction against substrate concentration
this yields a sigmoidal curve, as opposed to a hyperbolic curve for a non-cooperative
enzyme (see Figure 1.5). The kinetics of cooperative systems can be described using the
Hill equation12 (see Equation 1.1), where the Hill coefficient (n) is a measure of the degree
of cooperativity between the subunits in the enzyme and is limited by the number of
catalytic subunits (or active sites) in the structure. A Hill coefficient of more than one
signifies positive cooperativity, whereas a negative Hill coefficient represents negative
cooperativity. If an enzyme does not act cooperatively then it gives a Hill coefficient of 1.

25
Figure 1.5 A plot showing the difference in change in reaction rate with the concentration between
cooperative and non-cooperative enzymes.
Equation 1.1 The Hill equation.
The Hill coefficient is denoted by n, Kd is the equilibrium dissociation constant, [L] is the
concentration of the ligand and θ is the fraction of binding sites that are occupied by substrate.
1.1.1.2 Enzyme Functions
The importance of enzymes in biological and evolutionary terms is evident in that all living
organisms contain enzymes. They are also practically important and its estimated that half
of all drug targets are classed as enzymes13,14. Whilst enzymes participate in the reaction
they are not considered as reactants as the enzyme remains chemically the same at the end
of the reaction. They contain highly specific active sites that dictate not only chemical
specificity but stereo- and regiospecificity. Catalytic residues use a wide variety of
mechanisms to catalyse each enzyme’s reaction, amongst which the most common are
stabilisation of intermediates, usually via electrostatic interactions and proton-shuttling
events15. Whilst enzymes involved in similar cellular functions can catalyse their reactions
via different intermediate steps, they can demonstrate propensity for certain reaction

26
mechanisms. Oxidoreductases, for example, tend to carry out their reactions by shuffling
electrons around their active site, whilst the transferase mechanisms tend to involve
nucleophillic addition and substitution15.
Molecular functions of enzymes are usually characterised by an E.C number, given
according to the Enzyme Commision (EC) classification scheme by the International
Union of Biochemistry and Molecular Biology (IUBMB)16. This is a hierarchical scheme
that represents individual enzymes by a four-digit number according the reaction it
catalyses. The EC number is given in the format a.b.c.d, where a represents one of the six
main classes, b denotes the sub-class, c represents the sub-subclass and d is the serial
number of the enzyme within the class (and usually translates to the substrate specificity).

27
The six main classifications of enzymes are;
1. Oxidoreductases (EC1)
This class of enzymes is involved in oxidation-reduction reactions where one
species is oxidised in order to reduce another. Oxidoreductases facilitate the
transfer of electrons from the reductant to the oxidant as is shown in Figure 1.6.
There are a further 22 subclasses of oxidoreductase that are differentiated by the
chemical group that they react on.
A- + B ���� A + B-
Figure 1.6 A simple example of a redox reaction.
2. Transferases (EC2)
These enzymes are involved in reactions where a chemical group (rather than
electrons in the case of oxidoreductases) are transferred from a donor species to an
acceptor species (see Figure 1.7). Enzymes called kinases transfer a phosphate
group (usually fron ATP) to other donor molecules. Protein kinases transfer a
phosphate group specifically onto proteins and have important roles in regulation
and signaling.
A-X + B ���� A + B-X
Figure 1.7 A simple example of a transferase reaction.
3. Hydrolases (EC3)
This is class with the largest amount of structural and sequence information (both
in terms of redundant and non-redundant sequences and structures, see Figure 1.9).
Their small size makes them easy targets for determination of their sequence and
structure, and therefore hydrolases were amongst the most popular early candidates
for structural determination. These enzymes catalyse hydrolysis reactions, where a
substrate is divided apart by the addition of water. One part of the substrate
accepts the proton and the other accepts the hydroxyl group as shown in Figure
1.8. There are 13 subclasses of hydrolases, which act on different chemical bonds.
A-B + H2O ���� A-H + B-OH
Figure 1.8 A schematic equation for the hydrolysis reaction.

28
4. Lyases (EC4)
Like hydrolases, lyases break a chemical bond on their substrate to form two
molecules. Lyases, however, do not cleave the chemical bond by oxidation or
hydrolysis and act on bonds such as C-C, C-N and C-O. Lyase reactions usually
result in the elimination of a species from the substrate and the formation of a
double bond or ring structure in the remaining molecule. There are 7 subclasses of
lyases, depending on what kind of bond is cleaved.
5. Isomerases (EC5)
Isomerases catalyse the reaction that changes a substrate to a chemically identical,
but structurally different isomer. This can take form as a structural isomer, where
the chemical formula is the same but the bonds rearranged to form a different
structure, or a stereo-isomer where the structure is the same but the arrangement
of the groups in 3D space is different. There are 6 subclasses of isomerases, which
depend on the method of isomerisation. Three of these subclasses reflect reactions
that are catalysed by oxidoreductases, transferases and lyases, but are carried out
within the substrate (instead of on a second molecule) to create a single structurally
different product.
6. Ligases (EC6)
This is by far the smallest class of enzymes, perhaps as they have an energetically
difficult task. Ligases create a chemical bond which joins two chemical substrates,
often by hydrolysing a group from one or both of the substrate molecules. For
example, DNA ligase forms a phosphodiester bond between the 3' nucleotide and
the 5' phosphate group in a discontinuous strand of DNA and is involved in DNA
replication and repair.

29
Hydrolases (EC3) are the most abundant class of enzyme in sequence and structural
databases (even when accounting for the overrepresentation from redundant
sequences/structures, see Figure 1.9). There are small numbers of isomerase (EC5) and
ligase (EC6) structures in the PDB, however when duplicate structures are removed the
relative proportion of isomerases increases whilst the proportion of ligases remains low
(see Figure 1.9).
Figure 1.9 The proportion of each top EC class in the PDB.
The number of structures annotated is represented in panel A, whereas panel B represents the
number of non-redundant structures (i.e. do not contain subunits from the same SCOP
superfamily). This is also representative of the spread of enzyme functions seen in sequence
databases.
The relationship between EC classification and levels of sequence and structural similarity
is complicated. It has been shown that beyond the traditional function annotation
threshold of 40% sequence identity, EC number is widely conserved between proteins17,18.
Another study by Rost19, however, showed that EC classification is only fully conserved in
30% of enzyme pairs that exhibit more than 50% sequence identity. It is also unclear as to
how well EC classification is conserved in structurally similar proteins. In a study of 167
homologous structural CATH superfamilies17 it was shown that almost half contained
enzymes that had differing EC classifications. Whilst most of these differences were in the
fourth digit, 22 of the superfamilies had EC numbers that differed at all levels. Similarly,
there is evidence of structural differences within EC classifications. Approximately 8.5%
(185) of the total number of EC nodes (full four-digit numbers) in the classification
scheme contain two or more enzymes that are structurally unrelated20. There is therefore
evidence that enzyme function has evolved via both divergent and convergent evolution.

30
1.2 Computationally determining protein function
Knowledge of protein function is fundamental to elucidating the exact mechanisms of
biological process within the cell. Understanding these processes is important in
developing therapeutic agents and identifying drug targets. Biochemical studies of a
protein’s function can be lengthy, expensive and sometimes fruitless and therefore
computational methods have been developed to try to predict a proteins function without
experimentation. The most common approach for this is by inferring function from a
similar protein of known function. Similar proteins are identified based either on the
degree of similarity between their sequences or three-dimensional structures.
As it has been observed that evolution is more tightly constrained for the structure of a
protein than it is for its sequence 21, structural information is increasingly being used to
identify a protein’s function. Due to the wealth of functional information held in these
structures and the recognition that the protein structures available only represented a
proportion of the total fold space thought to exist, there has been a change in the way
protein structures are solved.
Traditionally a protein’s structure was solved once the protein’s function had been
characterised with a view to understanding the exact mechanisms of its function. The
structural genomics initiatives have reversed that practice22 and many structures are now
being produced for proteins that have little functional characterisation in order to provide
insight into its biochemical function.
This has created a huge surge in the number of protein structures being deposited into the
Protein Data Bank1 (PDB). Over the past 5 years the number of structures in the PDB has
risen from 16,466 to just over 66,000 (see Figure 1.10). There is however a limited capacity
of laboratories to experimentally study each of these proteins and as a result there has been
an increase in the number of protein structures in the PDB with an ‘unknown function’
annotation from 19 to over 1500 in the last 5 years.
1 http://www.rcsb.org/pdb/

31
Due to the drive to produce structures for proteins that inhabit fold space not represented
in the current set, some of the structures produced may not exhibit similarity to another
functionally annotated protein. This is one of the reasons for the increase in the number
of proteins that cannot be assigned a function by similarity. There is therefore a need for
new methods to predict function without transfer of annotation via similarity.
0
10000
20000
30000
40000
50000
60000
70000
1986 1988 1990 1992 1994 1996 1998 2000 2002 2004 2006 2008 2010
Year
Nu
mb
er
of
str
uctu
res in t
he
PD
B
Figure 1.10 The rise in the number of structures deposited into the PDB since 1986.

32
1.2.1 Defining Protein Function
Defining what is meant by protein function is fundamental to the task of protein function
prediction. There is much ambiguity in the definition of protein function due to the fact
that it depends upon the context in which it is used. This has resulted in a range of
biological classification schemes which could differentially annotate a given protein.
The main source of confusion over the definition of protein function is due to the multi-
dimensional nature of the way function can be thought of. For example, trypsin can be
classified according to its biochemical function (peptide bond hydrolysis), molecular
function (a proteolytic enzyme), cellular role (protein degradation) or physiological role
(e.g. digestion). It could be even further complicated by considering cellular location or
regulatory roles.
Another issue when classifying protein function is that often proteins exhibit multiple
functions. The average number of experimentally verified functions for proteins in the
Gene Ontology Annotation project (GOA) is 1.35 23, showing that proteins have a
tendency to carry out more than one function. Multi-functionality may be inherent in its
role (for example, the lac repressor has a role in both carbohydrate metabolism and
osmoprotection) or circumstantial (RNA polymerase enzyme function can considered to
be different at the various stages of the transcription cycle, because the reactions it
catalyses are very different).
1.2.1.1 Classification Schemes
One of the first attempts to classify proteins with regards to their function was the Enzyme
Commission (EC) classification scheme24, which was first developed in 1955. As detailed
above, the EC classification scheme consists of six principle classes of enzymes, which are
then further broken down into 3 further levels with respect to reaction mechanisms,
reactants and products and lastly specificities. Each of these categories (and subsequent
sub-categories) is associated with numerical values, thus each classification of an enzyme
can be represented by a number in the format a.b.c.d.

33
The main advantage of the EC classification is its controlled vocabulary which lends itself
to computational analysis because of its numerical representation. Whilst the EC
classification is simple and well established, it does have properties that make it
problematic for use in bioinformatics analyses. Firstly, the classes are inconsistently
defined by using substrates, transferred groups and acceptor residues in different ways.
Secondly, enzymes are classified based on the overall reaction that they catalyse. The
reaction may consist of multiple sub-reactions catalysed by the enzyme but the
classification number will only represent the overall reaction mechanism. Another point
worth noting is that EC numbers are associated with the reaction catalysed not the protein.
Therefore enzymes that have similar EC numbers are not always evolutionarily similar or
take part in a similar cellular role.
Whilst useful, the EC numbering system only applies to enzymes and therefore other
classification methods have been developed to cover a wider range of protein functions.
The first more comprehensive classification scheme to cover products of a whole genome
was developed by Riley, who proposed a classification scheme based on the physiological
function for products of the E.Coli genome25. An updated version of this scheme was
implemented in GenProtEC 26,27. This classification scheme became the basis for others
such as TIGR28 and SubtiList29.
In the early days a series of classification schemes developed for a species specific database
such as Saccharomyces Genome Database (SGD)30 and Yeast Protein Database (YPD)31, that
contain species specific derivatives. Whilst these classification schemes only apply to
specific species, some of these schemes have been expanded to include other organisms.
For example, MIPS/PENDANT started out as a yeast specific classification but has been
modified to include many other species32.
Instead of focusing on a specific species or type of protein, some classification schemes
focus around specific types of functions. The Kyoto Ontology, which is implemented in
the KEGG PATHWAY database5 and the What Is There (WIT, now renamed as
ERGO)33 mainly address regulation and metabolic pathways. Other schemes may classify
based on another aspect of function such as location, for example YPL.db34, or molecular
interactions35-37.

34
Most of these classification schemes can be thought of as trees, whereby progression along
the tree from top to bottom represents increasingly specific functions. There is, however,
a move away from the traditional tree structure towards a more complex organisation. The
Gene Ontology (GO) classification scheme38 is one of the first classification schemes to
move away from this simple tree structure. The GO scheme seeks to remove some of the
ambiguity that exists in other databases by classifying function based on three different
areas; molecular function, biological process and localisation to cellular components. This
increase in complexity over tree-based schemes presents ease of use and navigation issues,
and lends itself less to computational analyses.
When approaching the task of predicting protein function, it is important to consider what
is meant by function and therefore which classification scheme to use. The coverage of
proteins in functional classification schemes can vary depending on the individual protein
(see Table 1.1). Due to lack of coverage and consistency in how function is described
using different classification schemes, the development of a single unified scheme would
be desirable. Whilst there are some efforts being made into the development of a unified
classification scheme39, there is a lack of a clear consensus on how to tackle this problem.

35
Protein ID Coverage of functional annotation schemes (%)
Annotation
CT080 15 Late transcription unit B hypothetical Protein
CT094 25 tRNA pseudouridine synthase CT313 30 Transaldolase EC 2.2.1.2 CT664 25 FHA domain-containing protein
Table 1.1 A table showing how the coverage of classification schemes varies per protein.
Adapted from Ouzounis et al.40. Examples of four randomly selected proteins from the Chlamydia
trachomatis serovar D genome sequence41 and their annotations. The coverage of consistent
annotations for the 20 functional classification schemes is shown as a percentage. The 20
classification schemes analysed are listed in the original publication40.
1.2.2 Functional Transfer Based on Homology
Functional annotation of proteins by biological experimentation is generally slow and
expensive in terms of time and resources and therefore computational techniques are often
used. The most widely used technique takes advantage of structural or sequence
similarities between proteins that have evolved from a common ancestor and therefore
may exhibit similar functionality.
Although the transfer of functional annotation by homology is very powerful, it has
limitations and has been blamed as one of the main sources of error for incorrect
functional annotation in current databases42,43. One such limitation is that there may be a
lack of an accurately annotated homologue in the databases from which to transfer
functional information. It has been estimated that ~25% of newly sequenced genes have
no annotated homologue44 and the ability to detect homologues decreases as the sequence
similarity threshold (and therefore the accuracy of annotation transfer) is raised (see Figure
1.11).
A second problem faced by this method is the level of similarity required to enable
accurate transfer of functional information is unclear. Indeed, there are even differences in
the level of sequence identity required to transfer different types of functional annotation

36
(see Figure 1.11). Several groups studied the relationship between sequence identity and
functional conservation and, despite using different approaches, agree that below 50%
identity functions diverge very quickly17,18,45,46. Rost, however, argued that these types of
simple pairwise comparison studies might be misleading due to database bias and suggested
that annotation transfer cannot be reliably employed below 70% sequence identity19. Tian
and Skolnick later carried out an analysis similar to Rost, which also took into account
database bias47. This suggested that a 40% threshold can still be used as a confident
threshold for functional transfer.
Ultimately, however, protein function can differ on a small number of changes in amino
acid composition and therefore even when transferring annotation for highly similar
proteins, errors can still occur. For example, the acidic endochitinase WIN6.2b precursor
sequence exhibits 94% sequence identity with DNA topoisomerase II (β-isozyme) despite
them having different functions (EC numbers 3.2.1.14 and 5.99.1.3 respectively)19.
One of the biggest dangers of this method is the prospect that it may propagate any
existing errors in the database, thus amplifying the effects of one incorrect annotation
across a potentially large number of annotations. A study by Jones et al. suggests that
almost half of all annotations assigned by sequence similarity in the GOSeqLite database
are erroneous48. Once an incorrect annotation has been transferred there is the
opportunity for the amount of annotation errors to spread rapidly throughout the database.

37
Figure 1.11 The accuracy of function annotation with varying sequence identity (adapted from Rost
et al.19)
(A) Accuracy of annotation transfer according to percentage sequence identity. The black line
indicates subcellular location annotation and the purple line indicates enzymatic function
annotation. (B) The power of transfer of annotation according to the sequence identity threshold
used for annotation transfer. The arrows represent points in the curve where the error margin is 10%
(i.e. 10% of the annotations are incorrect).

38
1.2.2.1 Sequence Similarity
Assessing sequence similarity to evaluate whether two proteins are homologs and therefore
are likely to share common functionality is the most widely used method of predicting
function. Even with the rapidly growing number of protein structures, the amount of
structural data is far out-weighed by the availability of sequence data and therefore
sequence data is still favoured for use in comparative studies.
Various tools exist to assess the level of similarity between sequences such as BLAST49,
PSI-BLAST50 and FASTA51, which compare a given sequence to sequences deposited in
the major sequence databases (see Table 1.2).
Database URL Reference
DDBJ (DNA Data
Bank of Japan)
http://www.ddbj.nig.ac.jp/ 52
GenBank http://www.ncbi.nlm.nih.gov/Genbank/index.ht
ml
53
EMBL Nucleotide
DB
http://www.ebi.ac.uk/embl/index.html 54
Table 1.2 The main primary sequence databases with their URL and relevant reference.
Kellar et al. highlighted the hazards of basing annotations on sequence similarity alone,
without considering the protein structure55. They suggested that CbiT, which is involved in
vitamin B12 biosynthesis, is a methyltransferase based on structural similarities between the
crystal structure of CbiT and other methyltransferases. This was then later confirmed by
experimentation. However, CbiT was previously annotated as a decarboxylase based upon
sequences similarities with other decarboxylases.

39
1.2.2.2 Structural Similarity
Transferring functional annotation based on structural similarity is often more reliable than
sequence comparison alone. This is mainly due to the fact that protein structure is more
conserved than sequence56, thus allowing homology to be detected even when the
sequence similarity is low. It is worth noting that structurally related proteins are not
always homologous (i.e. evolving from a common ancestor). Some structural similarities
may have occurred due to convergent evolution to an energetically favourable fold.
There are a number of algorithms available for searching for structural similarity between
proteins (see Table 1.3). The tools shown in Table 1.3 were analysed for their sensitivity
and specificity in identifying homologs and topologs (proteins with similar topology that
may or may not be homologous)57. In agreement with earlier studies58,59, Sierk and
Pearson found that using automatic pairwise structure comparison it was very difficult to
distinguish between non-homologous topologs and true homologs. They found that the
best performing algorithm was Dali, which was capable of predicting 840 of the 1120
homologous pairs in their test set. At this coverage level they suggested that 500-700 non-
homologous topologs would also be included in the predictions. This shows that
functional annotation transferred on structural similarity alone may be incorrect due to the
reference protein not being a true homolog.
Name URL Reference
SSM http://www.ebi.ac.uk/msd-srv/ssm/ 60
Cathedral http://www.cathdb.info/cgi-
bin/CathedralServer.pl
61
CE http://cl.sdsc.edu/ 62
VAST http://www.ncbi.nlm.nih.gov/Structure/VAST/
vast.shtml
63
Matras http://biunit.aist-nara.ac.jp/matras/ 64
Table 1.3 Examples of structure comparison programs with their URL and reference.
Another limitation of annotation transfer based on structural similarity is that some of the
new structures emerging from structural genomics projects have no highly similar
structures in the PDB. This is because the aims of structural genomics projects are to

40
obtain structures for regions of protein fold space that are currently under-represented in
the PDB.
1.2.2.3 Dynamic Similarity
The mechanisms of most protein functions require some sort of dynamic motion to elicit
their effect, either by small fluctuations in residue side chains or large-scale conformational
changes. The increase in computational power has allowed the estimation of protein
motion by approaches such as molecular dynamics and also by more simplified approaches
based on normal mode analysis. Since it is widely accepted that proteins with similar
sequence or structure are likely to have similar function, it may also carry that similarity in
dynamics can be used to transfer annotation.
Early attempts at aligning the dynamics of proteins relied on first creating a structural or
sequence alignment to create reference points from which to compare dynamics65,66.
Further studies have proposed other methods that either loosely constrain the alignment
by structure67 or attempt to align the dynamics without prior structural alignment at all68.
The study of the dynamic similarity between families of proteins has shown that similarities
in dynamic behaviour can be detected66 and can allow clustering within these families that
is reflective, not only of clusters of structural similarity, but of clusters of similar
mechanisms or functions68,69. In a study of representative protease structures from
different folds, the dynamic properties were shown to be strongly conserved, particularly
around their functional site, despite their lack of similarity in structure and sequence70.
This suggests that convergent evolution may also act to produce dynamics that are essential
for a particular function in the same way as is thought of for structure.
The results of these studies suggest that functional annotation may be able to be
transferred by detection of similar dynamic properties. However, a study of the dynamic
similarity between representative enzymes from the main functional and structural classes
showed that dynamics are inconsistently conserved between members of the same
functional class67,70. It was, however, possible to detect a subset of homologous protein
pairs by dynamic alignment alone that would not have been detected using usual structural
or sequence comparison thresholds.

41
1.2.3 Predicting Protein Function in the Absence of Sequence or
Structural Similarity
It is estimated that 25% of known sequences show no homology to any annotated
sequence with a further 37% exhibiting levels of sequence similarity that may give rise to
unreliable annotation by automatic transfer44. This provides a large set of proteins for
which homology based methods fail. There has been much recent effort in developing
new methods to predict protein function without the traditional global alignment followed
by functional transfer. These methods use a wide variety of properties and methods using
both sequence and structural attributes. As no single approach is 100% accurate, it is
becoming increasingly important to combine approaches. This integrated approach to
function prediction is implemented in several servers such as ProKnow 71 and ProFunc 72.
1.2.3.1 Sequence Motifs
Even in the absence of overall sequence similarity, a common motif (an isolated sequence
pattern) or fingerprint (a number of sequence patterns) are often observed in proteins that
carry out the same function. Whilst the construction of sequence motifs often involves the
detection of homologs (see Figure 1.12), sequence motifs can be used without having to
infer homology by whole sequence similarity.
There are several motif databases, each with their own search tools (see Table 1.4).
Sequence motifs from PROSITE73 and PRINTS74 are presented in the integrated protein
annotation database INTERPRO75 in an attempt to combine the strengths of motifs with
other annotation methods. Machine learning techniques have also been applied to
functional annotation using sequence motifs. One example is the Anagram server76, which
uses the protomotifs (subtle amino acid patterns) as features of different functional classes
in SWISS-PROT to train a support vector machine algorithm to predict functional
classification.

42
Figure 1.12 Schematic diagram of a generic approach for constructing sequence motifs
43
Whilst sequence motifs are often quite powerful tools to predict function in the absence of
significant sequence similarity, caution needs to be taken when interpreting a match.
Because of the short length of some motifs, a match may occur due to chance and not due
to functional similarity. Databases such as PRINTS try to combat this by using
fingerprints (a series of motifs) to identify a match. A match can be more confidently
identified as functionally similar if it matches all the motifs of the fingerprint with the
correct juxtaposition.
Name Description URL Reference
PROSITE A database of protein
families and domains. It
consists of biologically
significant sites, patterns
and profiles given as
regular expressions.
http://www.expasy.org/prosite
73
PRINTS A database (including
scanning tools) containing
fingerprints representative
of protein families
http://www.bioinf.manchester.
ac.uk/dbbrowser/PRINTS
74
BLOCKS Includes motif making,
retrieving and scanning
tools. BLOCKS also
searches PRINTS
http://blocks.fhcrc.org/ 77
Table 1.4 A list of sequence motif resources
There are also a number of inherent sources of error in the method of constructing motifs.
For example, construction of motifs relies on the formation of a multiple sequence
alignment of functionally related homologs. As discussed above, there is no 100% accurate
automated method of doing this without costly and timely manual intervention. Secondly,
some methods, such as the one used by PRINTS, employ reiterative cycles of searching the
database for additional homologs using the motif formed from the previous cycle. If a
non-homologous sequence is introduced to the alignment by chance it has the opportunity
to influence the construction of a sub-optimal motif.

44
1.2.3.2 Functional Sites
Analysing the features of functional sites and using them to predict function seems logical
since it is the part of the protein which is arguably most important to protein function.
Whilst the rest of the protein may have roles such as stabilising or trafficking the protein,
the functional site contains the most information about the specific function of the protein
and therefore may be the most useful part to study in order to assign function.
It is important to point out that there is ambiguity in what is meant by a ‘functional site’.
In some cases a binding site may be considered to be a protein’s functional site, especially
in cases where enzymes bind their substrate in their catalytic site. However, proteins such
as G- protein coupled receptors bind a ligand on their extracellular C-terminal end and
elicit their response on their intracellular C-terminal domain. In this case, the site that
actually elicits its function is separate from its ligand binding site. Indeed, both the ligand
binding site and the G-protein coupled site could be considered to be functional sites.
Enzymes lend themselves to binding site analysis since they have well defined active sites.
It is also worth noting that some proteins, such as structural proteins, have no obvious
functional site.

45
Name Description URL Reference
PdbFun A database compiling annotated residues from the PDB. Contains binding site residues using the HETERO groups from the PDB and catalytic residues from CATRES
http://pdbfun.uniroma2.it/
78
PDBSiteScan/PDBSite
PDBSite is a database containing functional sites extracted from PDB using the SITE records and of an additional set containing the protein interaction sites inferred from the contact residues in heterocomplexes. PDBSiteScan provides structural alignment with known functional sites stored in PDBSite.
http://wwwmgs.bionet.nsc.ru/mgs/gnw/pdbsitescan/
79
PINTS A server that can compare a protein structure against a database of patterns or a structural pattern against a database of protein structures
http://www.russell.embl.de/pints/
80
PROCAT/TESS/Jess/Catalytic Site Atlas
The Catalytic Site Atlas (CSA) is a database documenting enzyme active sites and catalytic residues in enzymes of 3D structure.
http://www.ebi.ac.uk/thornton-srv/databases/CSA/
81
pvSOAR Detects surface similarities in protein structures. It allows a user to search a protein surface pattern derived from a pocket or a void against all known surface patterns from the CASTp (Computed Atlas of Surface Topology of proteins) database
http://pvsoar.bioengr.uic.edu/
82
SPASM/RIGOR A server that takes PDB style residue coordinates of a motif or template and searches them against known motifs based in a database derived from the PDB
http://portray.bmc.uu.se/cgi-bin/spasm/scripts/spasm.pl
83
SuMo Screens the PDB for protein structures that match a binding site in a given protein structure. It uses its own heuristics for defining ligand binding sites
http://sumo-pbil.ibcp.fr/
84
Table 1.5 Functional/active/binding site residue databases and comparison tools available via the
web.

46
In a similar way to homology-based annotation, information about active sites can be used
to transfer annotation. There are many resources that store structural and sequence
information about known active sites of proteins with well characterised function (see
Table 1.5). A protein of unknown function can be compared to these resources to identify
any similarities with known active sites. One such tool, the Structure-Function Linkage
Database85 organises enzymes into groups defined by highly conserved residues in the
active site that are thought to be related to the reaction that members of that group
mediate. An uncharacterized protein can then be assigned to a reaction group based on
whether it possesses these specific active site residues in the corresponding locations.
Unlike transfer of annotation via homology, such methods do not rely on finding a match
with significant overall sequence or structural similarity. A match can be detected even if
the less evolutionarily constrained parts of the protein have diverged, making them
undetectable to traditional homology searches. Indeed, this method can find proteins with
similar function that have evolved through convergent evolution to some favourable active
site formation.
Other methods of predicting protein function by using active site information do not rely
on finding similarity between existing active sites on other characterised proteins. Instead,
they identify functional sites using the geometry (SARIG86), chemistry (WEBFEATURE87,
THEMATICS88) or electrostatic properties89,90 of a site. These properties may be
associated with a function and therefore can be used to predict the functional class of
given protein. An approach to finding a functional site using electrostatic properties90
illustrated that they were also able to use these properties to discriminate between enzymes
and non-enzymes.
There are other methods for finding functional sites that require alignment with known
homologs such as Evolutionary Trace (ET). ET identifies and orders amino acids
variations in a diverging phylogenetic tree 91. The least varied (most conserved) amino
acids have been shown to correlate well with functional sites and thus ET has been used in
several methods of functional site detection 92-95. One of the problems with this method,
however, is that residues may be conserved for reasons other than to maintain the active
site, such as for the preservation of a favourable structure. Cheng et al. have developed a
method that predicts sequence profiles expected purely under structural constraints and

47
then uses them to predict whether an observed conservation pattern is due to structural or
evolutionary constraints 96. In a similar approach Cheliah et al., construct profiles of
conservation that are expected under functional and structural constrains and use this to
identify regions of the protein that are conserved for functional reasons95.
1.2.3.3 Genomic Context
The ability to predict the function of a protein by considering its genomic context is based
upon four theories. Firstly, functionally similar proteins often evolve in a similar manner.
The measure of a gene’s presence or absence from a set of genomes over an evolutionary
period is termed phylogenetic profiling 97. Function is predicted by matching the
phylogenetic profiles of the unknown protein to those which are known. Barker and Pagel
predicted functional associations by mapping absence/presence data of gene pairs over 15
species’ genomes 98.
Secondly, genes of functionally related proteins may exist in an order which is conserved in
a number of genomes (the Gene Neighbour method)99,100. Thirdly, functionally related
genes may exist as part of an operon101, and lastly, they may fuse to form a novel single
gene in another genome (the Rosetta Stone method 102, see Figure 1.13). The disadvantage
with the latter approach is that it relies on one of the fused domains to have a known
function. If both fused genes are uncharacterised then very little can be inferred.

48
Figure 1.13 A schematic representation of the Rosetta Stone method of assigning protein function.
Coloured blocks represent genes, with sections of the same colours representing sequence
similarity. Sequence A is from an uncharacterised protein, which is found to have high sequence
similarity to an isolated section (in red) of a gene in sequence B in another genome, although it may
not show significant overall sequence similarity to be considered to be homologous. The other non-
similar section (in blue) of sequence B has a high level of sequence similarity to another sequence,
C. The protein of sequence C is functionally characterised and therefore it can be inferred that
protein A is functionally similar to C since they appear to have fused to form protein B.
There are several tools that utilise these basic concepts to predict function. Phydbac103
uses phylogenetic profiles, chromosomal proximity and the Rosetta Stone method to
predict function using GO terms. Another tool, SNAP104, adds to these approaches by
constructing graphs of similarity versus neighbourhood (proximity) for co-located and
homologous genes of bacterial genomes. Functionally related genes are thought to exhibit
similar graphs.
Although there has been some success using these methods, they tend to work better in
prokaryotes, especially the operon and gene-ordering based methods. Gene order-based
functional prediction seems to be almost impossible for eukaryotes as they apparently lack
functional gene clusters.

49
1.2.3.4 Protein-Protein Interactions
It is often observed that proteins carry out their function as a group of proteins that
physically interact with each other. For this reason function can be inferred of an
uncharacterised protein if it is shown, or predicted, to interact with a protein of known
function. Attempts have been made to map GO terms to uncharacterised proteins using
protein-protein interactions with reasonable success 105-108.
There are many databases holding experimentally derived protein-protein interaction
information36,109,110, however it is unlikely that when trying to annotate a hypothetical
protein there will be any experimental interaction data available for that protein. Thus,
protein-protein interaction prediction methods are needed to be able to predict any
interaction and therefore a possible shared function with another protein.
There have been many different approaches to predicting protein-protein interactions.
Pazos et al. have developed a method, which uses correlated mutation analysis to predict
true protein-protein interactions and suggests likely regions for the interaction interface111.
The same group also proposed a method that uses similarity between the evolutionary
distance between the sequence of the proposed interacting pairs112. Another approach
looks at unusually exposed amino acids as an interaction site predictive feature113.
As with the Rosetta stone method, the major drawback of this method is the fact that to be
able to predict a function for a protein, the predicted interaction partner has to have a
known function.

50
1.2.3.5 Subcellular Localisation
It is possible to use subcellular location as a feature to use for predicting protein function
as it is reasonable to assume that proteins must be co-localised within the same subcellular
compartment in order to cooperate in a shared function. Also certain functions are
indicative of subcellular localization (i.e. DNA ligases are often found in the nucleus).
Proteins need to be transported from where they are made to the location where they carry
out their function. In order for the cell to successfully traffic these proteins they contain
sorting signals in their amino acid sequence 114,115. This prompted studies which revealed
that localisation correlates with total amino acid composition and sequence motifs (signal
sequences). This has led to the development of a number of methods based around the
analysis of amino acid composition116,117. Phylogenetic profiling has also been used to
predict localisation118, but it has been less successful than using amino acid composition.
Some tools have attempted to combine the amino acid composition approach with other
methods such as searching databases of known signal sequences119 or analysing expression
levels120. One other approach by Drawid and Gerstein121 was to use a diverse range of 30
features in a Bayesian probabilistic approach, which updates a protein’s probability that it is
found in a given subcellular component.

51
1.2.3.6 Structural Features
Since structure is more highly conserved than sequence, analysing structural features may
give clues to a protein’s function. Not only can structural information be used as a
comparative tool to transfer annotation by homology but also as a direct tool to predict
function.
In a similar way to sequence motifs, structural motifs or patterns are used to identify a
protein with a similar function. These can be residue based motifs 122 or motifs based on
geometric and chemical similarity 123. As a proteins function is strongly associated with a
‘functional site’, for many proteins such as enzymes or binding proteins the best structural
motif classifiers are their active site or binding site. However, there are some tools that
attempt to predict function by using 3D templates not solely limited to information from
functional sites124. Espadaler et al. have developed a method to look at the properties of
loop regions as predictors of function without defining the functional site125.
Rather than use short 3D motifs, some approaches use more global structural features to
define function. From a study of structural features of the proteases, Stawiski et al. found
that they exhibit similar characteristics such as smaller than average surface areas and
higher Cα densities, regardless of whether or not they were evolutionarily related126. They
also showed different secondary structure content to the non-proteases. By using these
features in a machine learning approach they were able to define a set of structural
classifiers that could predict whether a protein is a protease or non-protease with an
accuracy of over 86%. In a later study, Stawiski et al. also reported structural features that
are characteristic of the O-glycosidases such as distinctive electrostatic properties of the
proteins surface, despite differences in the overall fold127.

52
Whilst the above studies are limited to a subset of protein functions, attempts have been
made to use similar structural features to classify proteins into more generic subsets.
Dobson and Doig used 52 simple structural features (see Figure 1.14) in a support vector
machine-learning algorithm to distinguish between enzymes and non-enzymes 128. These
52 features were culled to a set of 36 (the bold items in Figure 1.14) best performing
features, which increased the predictive accuracy from 77% to 80%. A later study129
attempted to classify the enzyme predictions further into the top EC classification number
with an overall accuracy of 35% with the top ranked prediction, increasing to 60% with the
top two ranked predictions. These studies however, focused on global structural features
(for example, overall size or amino acid composition) and since the active site of an
enzyme is more closely associated with its function, it is hypothesised that these methods
could be improved upon by including structural features specifically of an enzymes active
site.
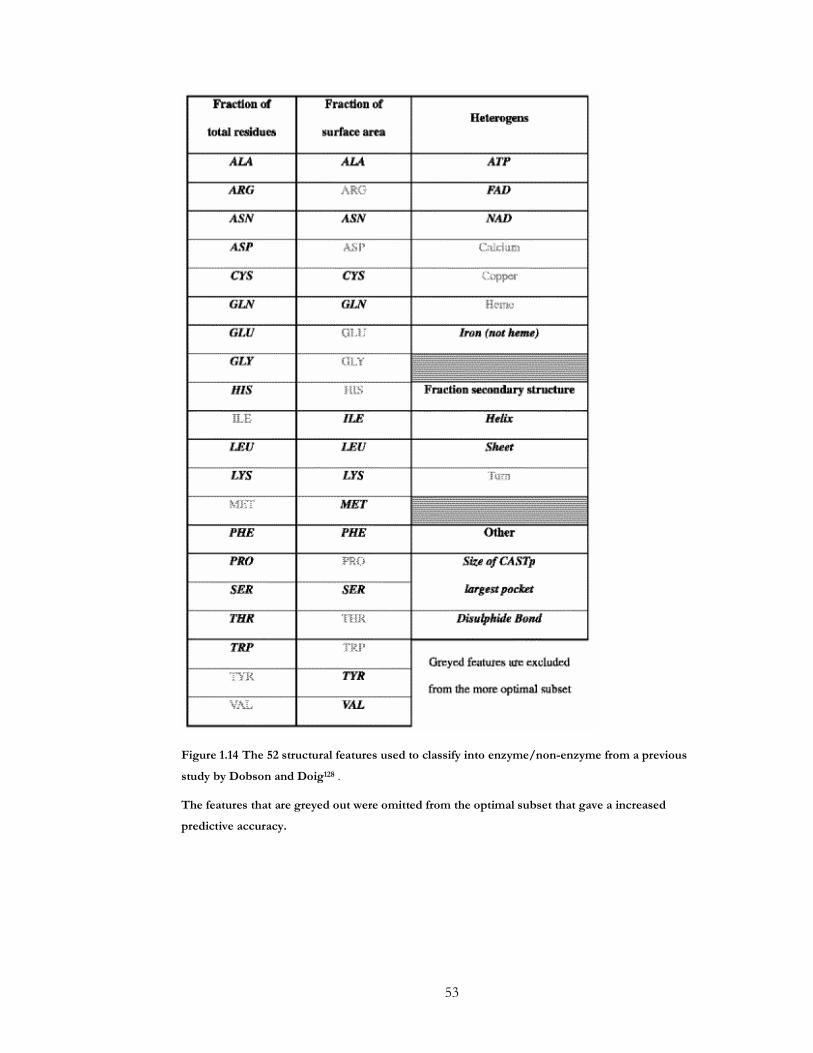
53
Figure 1.14 The 52 structural features used to classify into enzyme/non-enzyme from a previous
study by Dobson and Doig128 .
The features that are greyed out were omitted from the optimal subset that gave a increased
predictive accuracy.

54
1.3 Thesis Structure
The initial aim of this work was to continue the work by Dobson and Doig129 in looking at
the relationship between sequence and structural features of enzymes and their function.
Previous work focused on producing a computational method to predict the functional class
(top EC classification) of an enzyme based on these features. The machine learning method
used in this work, however, made the interpretation of the exact relationships between the
features and the enzyme function difficult. In Chapter 2 of this thesis, a study of the
differences in structural and sequence features of a non-redundant set of enzymes and their
active sites explores the structure-function relationship and further investigates those
features that differ the most between functions.
Furthermore, improvement to the performance of the previous functional prediction
method was attempted by the inclusion of active-site specific features (Chapter 4). In order
for this tool to be applicable to proteins with little or no characterisation, it was necessary to
predict the location of the functional site on the enzyme. Chapter 3 details a comprehensive
benchmark study of current publicly-available software for the prediction of functional sites
and also presents the creation of a webserver to deliver a previously published method by
this group90,130.
In order to further study one of the main findings in Chapter 2, a method to detect
cooperativity in enzymes by assessing the communication in dynamics between residues in
different subunits of oligomers was attempted in Chapter 5. This chapter also goes on to
further investigate the dynamic properties of oligomers in general.
The work contained in Chapter 2 (and some in Chapter 4) has been published as an article in
J. Mol. Biol131. The work in Chapter 3 has been published as an article in BMC
Bioinformatics132 and the work in Chapter 5 is written as a manuscript and currently in
review.

55
1.4 References
1. Apweiler R MM, O'Donovan C, Magrane M, Alam-Faruque Y, Antunes R, Barrell D, Bely B, Bingley M, Binns D, Bower L, Browne P, Chan WM, Dimmer E, Eberhardt R, Fedotov A, Foulger R, Garavelli J, Huntley R, Jacobsen J, Kleen M, Laiho K, Leinonen R, Legge D, Lin Q, Liu W, Luo J, Orchard S, Patient S, Poggioli D, Pruess M, Corbett M, di Martino G, Donnelly M, van Rensburg P, Bairoch A, Bougueleret L, Xenarios I, Altairac S, Auchincloss A, Argoud-Puy G, Axelsen K, Baratin D, Blatter MC, Boeckmann B, Bolleman J, Bollondi L, Boutet E, Quintaje SB, Breuza L, Bridge A, deCastro E, Ciapina L, Coral D, Coudert E, Cusin I, Delbard G, Doche M, Dornevil D, Roggli PD, Duvaud S, Estreicher A, Famiglietti L, Feuermann M, Gehant S, Farriol-Mathis N, Ferro S, Gasteiger E, Gateau A, Gerritsen V, Gos A, Gruaz-Gumowski N, Hinz U, Hulo C, Hulo N, James J, Jimenez S, Jungo F, Kappler T, Keller G, Lachaize C, Lane-Guermonprez L, Langendijk-Genevaux P, Lara V, Lemercier P, Lieberherr D, de Oliveira Lima T, Mangold V, Martin X, Masson P, Moinat M, Morgat A, Mottaz A, Paesano S, Pedruzzi I, Pilbout S, Pillet V, Poux S, Pozzato M, Redaschi N, Rivoire C, Roechert B, Schneider M, Sigrist C, Sonesson K, Staehli S, Stanley E, Stutz A, Sundaram S, Tognolli M, Verbregue L, Veuthey AL, Yip L, Zuletta L, Wu C, Arighi C, Arminski L, Barker W, Chen C, Chen Y, Hu ZZ, Huang H, Mazumder R, McGarvey P, Natale DA, Nchoutmboube J, Petrova N, Subramanian N, Suzek BE, Ugochukwu U, Vasudevan S, Vinayaka CR, Yeh LS, Zhang J. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res 2010;38(Database issue):D142-148.
2. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res 2000;28(1):235-242.
3. Holliday GL, Bartlett GJ, Almonacid DE, O'Boyle NM, Murray-Rust P, Thornton JM, Mitchell JB. MACiE: a database of enzyme reaction mechanisms. Bioinformatics 2005;21(23):4315-4316.
4. Bartlett GJ, Porter CT, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. J Mol Biol 2002;324(1):105-121.
5. Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 2006;34(Database issue):D354-357.
6. Barthelmes J, Ebeling C, Chang A, Schomburg I, Schomburg D. BRENDA, AMENDA and FRENDA: the enzyme information system in 2007. Nucleic Acids Res 2007;35(Database issue):D511-514.
7. Fischer E. Einfluss der Configuration auf die Wirkung der Enzyme. Ber Dt Chem Ges 1894(27):2985–2993.
8. Koshland DE. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci U S A 1958;44(2):98-104.
9. Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: a Plausible Model. J Mol Biol 1965;12:88-118.
10. Koshland DE, Jr., Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 1966;5(1):365-385.
11. Michaelis L. MM. Die Kinetik der Invertinwirkung. Biochem Z 1913(49):333-369. 12. Hill AV. The possible effects of the aggregation of the molecules of hemoglobin on
its dissociation curves. J Physiol 1910;40:iv-vii.

56
13. Zheng CJ, Han LY, Yap CW, Ji ZL, Cao ZW, Chen YZ. Therapeutic targets: progress of their exploration and investigation of their characteristics. Pharmacol Rev 2006;58(2):259-279.
14. Bakheet TM, Doig AJ. Properties and identification of human protein drug targets. Bioinformatics 2009;25(4):451-457.
15. Holliday GL, Mitchell JB, Thornton JM. Understanding the functional roles of amino acid residues in enzyme catalysis. J Mol Biol 2009;390(3):560-577.
16. Barrett AJC, C. R.; Liebecq, C.; Moss, G. P.; Saenger, W.; Sharon, N.; Tipton, K. F.; Vnetianer, P.; Vliegenthart, V. F. G. Enzyme Nomenclature. San Diego, CA: Academic Press; 1992.
17. Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol 2001;307(4):1113-1143.
18. Wilson CA, Kreychman J, Gerstein M. Assessing annotation transfer for genomics: quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J Mol Biol 2000;297(1):233-249.
19. Rost B. Enzyme function less conserved than anticipated. J Mol Biol 2002;318(2):595-608.
20. Omelchenko MV, Galperin MY, Wolf YI, Koonin EV. Non-homologous isofunctional enzymes: A systematic analysis of alternative solutions in enzyme evolution. Biol Direct;5:31.
21. Thornton JM, Todd AE, Milburn D, Borkakoti N, Orengo CA. From structure to function: approaches and limitations. Nat Struct Biol 2000;7 Suppl:991-994.
22. Burley SK. An overview of structural genomics. Nat Struct Biol 2000;7 Suppl:932-934.
23. Eisner R, Poulin B, Szafron D, Lu P, Greiner R. Improving Protein Function Prediction using the Hierarchical Structure of the Gene Ontology. Computational Intelligence in Bioinformatics and Computational Biology, 2005 CIBCB '05 Proceedings of the 2005 IEEE Symposium 2005:1-10.
24. Bairoch A. The ENZYME database in 2000. Nucleic Acids Res 2000;28(1):304-305. 25. Riley M. Functions of the gene products of Escherichia coli. Microbiol Rev
1993;57(4):862-952. 26. Serres MH, Goswami S, Riley M. GenProtEC: an updated and improved analysis of
functions of Escherichia coli K-12 proteins. Nucleic Acids Res 2004;32(Database issue):D300-302.
27. Keseler IM, Collado-Vides J, Gama-Castro S, Ingraham J, Paley S, Paulsen IT, Peralta-Gil M, Karp PD. EcoCyc: a comprehensive database resource for Escherichia coli. Nucleic Acids Res 2005;33(Database issue):D334-337.
28. Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, Bult CJ, Kerlavage AR, Sutton G, Kelley JM, Fritchman RD, Weidman JF, Small KV, Sandusky M, Fuhrmann J, Nguyen D, Utterback TR, Saudek DM, Phillips CA, Merrick JM, Tomb JF, Dougherty BA, Bott KF, Hu PC, Lucier TS, Peterson SN, Smith HO, Hutchison CA, 3rd, Venter JC. The minimal gene complement of Mycoplasma genitalium. Science 1995;270(5235):397-403.
29. Moszer I, Jones LM, Moreira S, Fabry C, Danchin A. SubtiList: the reference database for the Bacillus subtilis genome. Nucleic Acids Res 2002;30(1):62-65.
30. Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, Jia Y, Juvik G, Roe T, Schroeder M, Weng S, Botstein D. SGD: Saccharomyces Genome Database. Nucleic Acids Res 1998;26(1):73-79.
31. Hodges PE, McKee AH, Davis BP, Payne WE, Garrels JI. The Yeast Proteome Database (YPD): a model for the organization and presentation of genome-wide functional data. Nucleic Acids Res 1999;27(1):69-73.

57
32. Riley ML, Schmidt T, Wagner C, Mewes HW, Frishman D. The PEDANT genome database in 2005. Nucleic Acids Res 2005;33(Database issue):D308-310.
33. Overbeek R, Larsen N, Walunas T, D'Souza M, Pusch G, Selkov E, Jr., Liolios K, Joukov V, Kaznadzey D, Anderson I, Bhattacharyya A, Burd H, Gardner W, Hanke P, Kapatral V, Mikhailova N, Vasieva O, Osterman A, Vonstein V, Fonstein M, Ivanova N, Kyrpides N. The ERGO genome analysis and discovery system. Nucleic Acids Res 2003;31(1):164-171.
34. Habeler G, Natter K, Thallinger GG, Crawford ME, Kohlwein SD, Trajanoski Z. YPL.db: the Yeast Protein Localization database. Nucleic Acids Res 2002;30(1):80-83.
35. Chatr-Aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, Cesareni G. MINT: the Molecular INTeraction database. Nucleic Acids Res 2006.
36. Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res 2004;32(Database issue):D449-451.
37. Alfarano C, Andrade CE, Anthony K, Bahroos N, Bajec M, Bantoft K, Betel D, Bobechko B, Boutilier K, Burgess E, Buzadzija K, Cavero R, D'Abreo C, Donaldson I, Dorairajoo D, Dumontier MJ, Dumontier MR, Earles V, Farrall R, Feldman H, Garderman E, Gong Y, Gonzaga R, Grytsan V, Gryz E, Gu V, Haldorsen E, Halupa A, Haw R, Hrvojic A, Hurrell L, Isserlin R, Jack F, Juma F, Khan A, Kon T, Konopinsky S, Le V, Lee E, Ling S, Magidin M, Moniakis J, Montojo J, Moore S, Muskat B, Ng I, Paraiso JP, Parker B, Pintilie G, Pirone R, Salama JJ, Sgro S, Shan T, Shu Y, Siew J, Skinner D, Snyder K, Stasiuk R, Strumpf D, Tuekam B, Tao S, Wang Z, White M, Willis R, Wolting C, Wong S, Wrong A, Xin C, Yao R, Yates B, Zhang S, Zheng K, Pawson T, Ouellette BF, Hogue CW. The Biomolecular Interaction Network Database and related tools 2005 update. Nucleic Acids Res 2005;33(Database issue):D418-424.
38. Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 2004;32(Database issue):D258-261.
39. Rison SC, Hodgman TC, Thornton JM. Comparison of functional annotation schemes for genomes. Funct Integr Genomics 2000;1(1):56-69.
40. Ouzounis CA, Coulson RM, Enright AJ, Kunin V, Pereira-Leal JB. Classification schemes for protein structure and function. Nat Rev Genet 2003;4(7):508-519.
41. Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 1998;282(5389):754-759.
42. Bork P, Koonin EV. Predicting functions from protein sequences--where are the bottlenecks? Nat Genet 1998;18(4):313-318.
43. Iliopoulos I, Tsoka S, Andrade MA, Enright AJ, Carroll M, Poullet P, Promponas V, Liakopoulos T, Palaios G, Pasquier C, Hamodrakas S, Tamames J, Yagnik AT, Tramontano A, Devos D, Blaschke C, Valencia A, Brett D, Martin D, Leroy C, Rigoutsos I, Sander C, Ouzounis CA. Evaluation of annotation strategies using an entire genome sequence. Bioinformatics 2003;19(6):717-726.

58
44. Ofran Y, Punta M, Schneider R, Rost B. Beyond annotation transfer by homology: novel protein-function prediction methods to assist drug discovery. Drug Discov Today 2005;10(21):1475-1482.
45. Devos D, Valencia A. Practical limits of function prediction. Proteins 2000;41(1):98-107.
46. Pawlowski K, Jaroszewski L, Rychlewski L, Godzik A. Sensitive sequence comparison as protein function predictor. Pac Symp Biocomput 2000:42-53.
47. Tian W, Skolnick J. How well is enzyme function conserved as a function of pairwise sequence identity? J Mol Biol 2003;333(4):863-882.
48. Jones CE, Brown AL, Baumann U. Estimating the annotation error rate of curated GO database sequence annotations. BMC Bioinformatics 2007;8:170.
49. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990;215(3):403-410.
50. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25(17):3389-3402.
51. Pearson WR. Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol 1990;183:63-98.
52. Okubo K, Sugawara H, Gojobori T, Tateno Y. DDBJ in preparation for overview of research activities behind data submissions. Nucleic Acids Res 2006;34(Database issue):D6-9.
53. Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res 2006;34(Database issue):D16-20.
54. Cochrane G, Aldebert P, Althorpe N, Andersson M, Baker W, Baldwin A, Bates K, Bhattacharyya S, Browne P, van den Broek A, Castro M, Duggan K, Eberhardt R, Faruque N, Gamble J, Kanz C, Kulikova T, Lee C, Leinonen R, Lin Q, Lombard V, Lopez R, McHale M, McWilliam H, Mukherjee G, Nardone F, Pastor MP, Sobhany S, Stoehr P, Tzouvara K, Vaughan R, Wu D, Zhu W, Apweiler R. EMBL Nucleotide Sequence Database: developments in 2005. Nucleic Acids Res 2006;34(Database issue):D10-15.
55. Keller JP, Smith PM, Benach J, Christendat D, deTitta GT, Hunt JF. The crystal structure of MT0146/CbiT suggests that the putative precorrin-8w decarboxylase is a methyltransferase. Structure 2002;10(11):1475-1487.
56. Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. Embo J 1986;5(4):823-826.
57. Sierk ML, Pearson WR. Sensitivity and selectivity in protein structure comparison. Protein Sci 2004;13(3):773-785.
58. Matsuo Y, Bryant SH. Identification of homologous core structures. Proteins 1999;35(1):70-79.
59. Russell RB, Saqi MA, Sayle RA, Bates PA, Sternberg MJ. Recognition of analogous and homologous protein folds: analysis of sequence and structure conservation. J Mol Biol 1997;269(3):423-439.
60. Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 2004;60(Pt 12 Pt 1):2256-2268.
61. Redfern OC, Harrison A, Dallman T, Pearl FM, Orengo CA. CATHEDRAL: a fast and effective algorithm to predict folds and domain boundaries from multidomain protein structures. PLoS Comput Biol 2007;3(11):e232.
62. Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng 1998;11(9):739-747.
63. Madej T, Gibrat JF, Bryant SH. Threading a database of protein cores. Proteins 1995;23(3):356-369.

59
64. Kawabata T, Nishikawa K. Protein structure comparison using the markov transition model of evolution. Proteins 2000;41(1):108-122.
65. Pang A, Arinaminpathy Y, Sansom MS, Biggin PC. Comparative molecular dynamics--similar folds and similar motions? Proteins 2005;61(4):809-822.
66. Maguid S, Fernandez-Alberti S, Ferrelli L, Echave J. Exploring the common dynamics of homologous proteins. Application to the globin family. Biophys J 2005;89(1):3-13.
67. Zen A, Carnevale V, Lesk AM, Micheletti C. Correspondences between low-energy modes in enzymes: dynamics-based alignment of enzymatic functional families. Protein Sci 2008;17(5):918-929.
68. Munz M, Lyngso R, Hein J, Biggin PC. Dynamics based alignment of proteins: an alternative approach to quantify dynamic similarity. BMC Bioinformatics 2010;11:188.
69. Capozzi F, Luchinat C, Micheletti C, Pontiggia F. Essential dynamics of helices provide a functional classification of EF-hand proteins. J Proteome Res 2007;6(11):4245-4255.
70. Carnevale V, Raugei S, Micheletti C, Carloni P. Convergent dynamics in the protease enzymatic superfamily. J Am Chem Soc 2006;128(30):9766-9772.
71. Pal D, Eisenberg D. Inference of protein function from protein structure. Structure 2005;13(1):121-130.
72. Laskowski RA, Watson JD, Thornton JM. ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res 2005;33(Web Server issue):W89-93.
73. Hulo N, Bairoch A, Bulliard V, Cerutti L, De Castro E, Langendijk-Genevaux PS, Pagni M, Sigrist CJ. The PROSITE database. Nucleic Acids Res 2006;34(Database issue):D227-230.
74. Attwood TK, Bradley P, Flower DR, Gaulton A, Maudling N, Mitchell AL, Moulton G, Nordle A, Paine K, Taylor P, Uddin A, Zygouri C. PRINTS and its automatic supplement, prePRINTS. Nucleic Acids Res 2003;31(1):400-402.
75. Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bradley P, Bork P, Bucher P, Cerutti L, Copley R, Courcelle E, Das U, Durbin R, Fleischmann W, Gough J, Haft D, Harte N, Hulo N, Kahn D, Kanapin A, Krestyaninova M, Lonsdale D, Lopez R, Letunic I, Madera M, Maslen J, McDowall J, Mitchell A, Nikolskaya AN, Orchard S, Pagni M, Ponting CP, Quevillon E, Selengut J, Sigrist CJ, Silventoinen V, Studholme DJ, Vaughan R, Wu CH. InterPro, progress and status in 2005. Nucleic Acids Res 2005;33(Database issue):D201-205.
76. Perez AJ, Thode G, Trelles O. AnaGram: protein function assignment. Bioinformatics 2004;20(2):291-292.
77. Henikoff JG, Greene EA, Pietrokovski S, Henikoff S. Increased coverage of protein families with the blocks database servers. Nucleic Acids Res 2000;28(1):228-230.
78. Ausiello G, Zanzoni A, Peluso D, Via A, Helmer-Citterich M. pdbFun: mass selection and fast comparison of annotated PDB residues. Nucleic Acids Res 2005;33(Web Server issue):W133-137.
79. Ivanisenko VA, Pintus SS, Grigorovich DA, Kolchanov NA. PDBSite: a database of the 3D structure of protein functional sites. Nucleic Acids Res 2005;33(Database issue):D183-187.
80. Stark A, Russell RB. Annotation in three dimensions. PINTS: Patterns in Non-homologous Tertiary Structures. Nucleic Acids Res 2003;31(13):3341-3344.
81. Porter CT, Bartlett GJ, Thornton JM. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res 2004;32(Database issue):D129-133.
82. Binkowski TA, Adamian L, Liang J. Inferring functional relationships of proteins from local sequence and spatial surface patterns. J Mol Biol 2003;332(2):505-526.

60
83. Kleywegt GJ. Recognition of spatial motifs in protein structures. J Mol Biol 1999;285(4):1887-1897.
84. Jambon M, Andrieu O, Combet C, Deleage G, Delfaud F, Geourjon C. The SuMo server: 3D search for protein functional sites. Bioinformatics 2005;21(20):3929-3930.
85. Pegg SC, Brown SD, Ojha S, Seffernick J, Meng EC, Morris JH, Chang PJ, Huang CC, Ferrin TE, Babbitt PC. Leveraging enzyme structure-function relationships for functional inference and experimental design: the structure-function linkage database. Biochemistry 2006;45(8):2545-2555.
86. Amitai G, Shemesh A, Sitbon E, Shklar M, Netanely D, Venger I, Pietrokovski S. Network analysis of protein structures identifies functional residues. J Mol Biol 2004;344(4):1135-1146.
87. Wei L, Altman RB. Recognizing protein binding sites using statistical descriptions of their 3D environments. Pac Symp Biocomput 1998:497-508.
88. Ko J, Murga LF, Wei Y, Ondrechen MJ. Prediction of active sites for protein structures from computed chemical properties. Bioinformatics 2005;21 Suppl 1:i258-265.
89. Elcock AH. Prediction of functionally important residues based solely on the computed energetics of protein structure. J Mol Biol 2001;312(4):885-896.
90. Bate P, Warwicker J. Enzyme/non-enzyme discrimination and prediction of enzyme active site location using charge-based methods. J Mol Biol 2004;340(2):263-276.
91. Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol 1996;257(2):342-358.
92. Berezin C, Glaser F, Rosenberg J, Paz I, Pupko T, Fariselli P, Casadio R, Ben-Tal N. ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics 2004;20(8):1322-1324.
93. Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003;19(1):163-164.
94. Nimrod G, Glaser F, Steinberg D, Ben-Tal N, Pupko T. In silico identification of functional regions in proteins. Bioinformatics 2005;21 Suppl 1:i328-337.
95. Chelliah V, Chen L, Blundell TL, Lovell SC. Distinguishing structural and functional restraints in evolution in order to identify interaction sites. J Mol Biol 2004;342(5):1487-1504.
96. Cheng G, Qian B, Samudrala R, Baker D. Improvement in protein functional site prediction by distinguishing structural and functional constraints on protein family evolution using computational design. Nucleic Acids Res 2005;33(18):5861-5867.
97. Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci U S A 1999;96(8):4285-4288.
98. Barker D, Pagel M. Predicting functional gene links from phylogenetic-statistical analyses of whole genomes. PLoS Comput Biol 2005;1(1):e3.
99. Dandekar T, Snel B, Huynen M, Bork P. Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci 1998;23(9):324-328.
100. Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N. Use of contiguity on the chromosome to predict functional coupling. In Silico Biol 1999;1(2):93-108.
101. Salgado H, Moreno-Hagelsieb G, Smith TF, Collado-Vides J. Operons in Escherichia coli: genomic analyses and predictions. Proc Natl Acad Sci U S A 2000;97(12):6652-6657.
102. Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA. Protein interaction maps for complete genomes based on gene fusion events. Nature 1999;402(6757):86-90.
103. Enault F, Suhre K, Claverie JM. Phydbac "Gene Function Predictor": a gene annotation tool based on genomic context analysis. BMC Bioinformatics 2005;6:247.

61
104. Kolesov G, Mewes HW, Frishman D. SNAPping up functionally related genes based on context information: a colinearity-free approach. J Mol Biol 2001;311(4):639-656.
105. Deng M, Tu Z, Sun F, Chen T. Mapping Gene Ontology to proteins based on protein-protein interaction data. Bioinformatics 2004;20(6):895-902.
106. Kirac M, Ozsoyoglu G, Yang J. Annotating proteins by mining protein interaction networks. Bioinformatics 2006;22(14):e260-270.
107. Samanta MP, Liang S. Predicting protein functions from redundancies in large-scale protein interaction networks. Proc Natl Acad Sci U S A 2003;100(22):12579-12583.
108. Vazquez A, Flammini A, Maritan A, Vespignani A. Global protein function prediction from protein-protein interaction networks. Nat Biotechnol 2003;21(6):697-700.
109. Kerrien S, Alam-Faruque Y, Aranda B, Bancarz I, Bridge A, Derow C, Dimmer E, Feuermann M, Friedrichsen A, Huntley R, Kohler C, Khadake J, Leroy C, Liban A, Lieftink C, Montecchi-Palazzi L, Orchard S, Risse J, Robbe K, Roechert B, Thorneycroft D, Zhang Y, Apweiler R, Hermjakob H. IntAct--open source resource for molecular interaction data. Nucleic Acids Res 2007;35(Database issue):D561-565.
110. Zanzoni A, Montecchi-Palazzi L, Quondam M, Ausiello G, Helmer-Citterich M, Cesareni G. MINT: a Molecular INTeraction database. FEBS Lett 2002;513(1):135-140.
111. Pazos F, Valencia A. In silico two-hybrid system for the selection of physically interacting protein pairs. Proteins 2002;47(2):219-227.
112. Pazos F, Valencia A. Similarity of phylogenetic trees as indicator of protein-protein interaction. Protein Eng 2001;14(9):609-614.
113. Hoskins J, Lovell S, Blundell TL. An algorithm for predicting protein-protein interaction sites: Abnormally exposed amino acid residues and secondary structure elements. Protein Sci 2006;15(5):1017-1029.
114. Mattaj IW, Englmeier L. Nucleocytoplasmic transport: the soluble phase. Annu Rev Biochem 1998;67:265-306.
115. Schatz G, Dobberstein B. Common principles of protein translocation across membranes. Science 1996;271(5255):1519-1526.
116. Hua S, Sun Z. Support vector machine approach for protein subcellular localization prediction. Bioinformatics 2001;17(8):721-728.
117. Reczko M, Hatzigerrorgiou A. Prediction of the subcellular localization of eukaryotic proteins using sequence signals and composition. Proteomics 2004;4(6):1591-1596.
118. Marcotte EM, Xenarios I, van Der Bliek AM, Eisenberg D. Localizing proteins in the cell from their phylogenetic profiles. Proc Natl Acad Sci U S A 2000;97(22):12115-12120.
119. Nakai K, Horton P. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci 1999;24(1):34-36.
120. Drawid A, Jansen R, Gerstein M. Genome-wide analysis relating expression level with protein subcellular localization. Trends Genet 2000;16(10):426-430.
121. Drawid A, Gerstein M. A Bayesian system integrating expression data with sequence patterns for localizing proteins: comprehensive application to the yeast genome. J Mol Biol 2000;301(4):1059-1075.
122. Laskowski RA, Watson JD, Thornton JM. Protein function prediction using local 3D templates. J Mol Biol 2005;351(3):614-626.
123. Chen BY, Bryant DH, Fofanov VY, Kristensen DM, Cruess AE, Kimmel M, Lichtarge O, Kavraki LE. Cavity-aware motifs reduce false positives in protein function prediction. Comput Syst Bioinformatics Conf 2006:311-323.
124. Polacco BJ, Babbitt PC. Automated discovery of 3D motifs for protein function annotation. Bioinformatics 2006;22(6):723-730.

62
125. Espadaler J, Querol E, Aviles FX, Oliva B. Identification of function-associated loop motifs and application to protein function prediction. Bioinformatics 2006;22(18):2237-2243.
126. Stawiski EW, Baucom AE, Lohr SC, Gregoret LM. Predicting protein function from structure: unique structural features of proteases. Proc Natl Acad Sci U S A 2000;97(8):3954-3958.
127. Stawiski EW, Mandel-Gutfreund Y, Lowenthal AC, Gregoret LM. Progress in predicting protein function from structure: unique features of O-glycosidases. Pac Symp Biocomput 2002:637-648.
128. Dobson PD, Doig AJ. Distinguishing enzyme structures from non-enzymes without alignments. J Mol Biol 2003;330(4):771-783.
129. Dobson PD, Doig AJ. Predicting enzyme class from protein structure without alignments. J Mol Biol 2005;345(1):187-199.
130. Greaves R, Warwicker J. Active site identification through geometry-based and sequence profile-based calculations: burial of catalytic clefts. J Mol Biol 2005;349(3):547-557.
131. Bray T, Doig AJ, Warwicker J. Sequence and structural features of enzymes and their active sites by EC class. J Mol Biol 2009;386(5):1423-1436.
132. Bray T, Chan P, Bougouffa S, Greaves R, Doig AJ, Warwicker J. SitesIdentify: a protein functional site prediction tool. BMC Bioinformatics 2009;10:379.

63
Chapter 2: Sequence and structural features
of enzymes by EC class
In this chapter, simple sequence and structural features, both of the whole protein and
specifically of the active site, are analysed for differences over the six EC classes. This
systematic study of enzymes, and their active sites in particular, aims to increase
understanding of how the structure of an enzyme relates to its functional role. Features
analysed include amino acid compositions, secondary structure content, charge fractions,
average hydrophobicity score, B-factors, average isoelectric point, and surface area, both for
the total enzyme and the active site region. The features that differ significantly in frequency
between the 6 classes cluster into major groupings. Exploration of these groups sheds new
light on the relationship between protein structure and function, for example suggesting an
association between enzyme oligomeric status and position within metabolic networks.
The content of this chapter (along with some of the work from Chapter 4) was published as
an article in Journal of Molecular Biology1. The author of this thesis was the first author of
this paper, alongside the author’s two PhD supervisors.
2.1 Introduction
Over the last 10 years the number of protein structures available in the Protein Data Bank
(PDB2) has increased more than five-fold. A large and growing number have no functional
annotation, partly due to the recent efforts of structural genomics initiatives. Experimental
functional characterisation is time consuming and expensive, hence the requirement for
improved computational techniques to assign function. The most commonly used methods
rely on the transfer of annotation from a characterised homologue, identified by sequence or
structural similarity. The transfer of functional information via sequence or structural
similarity has a number of known limitations and has been blamed as one of the main
sources of error for incorrect functional annotation in current databases.3; 4
The EC classification scheme5 has traditionally been used to define the function of an
enzyme. The scheme is a hierarchical organization of enzyme reactions into six main classes
(oxidoreductases, transferases, hydrolases, lyases, isomerases, and ligases), which are then

64
split by a further 3 hierarchical levels. Each reaction is represented numerically in the format
a.b.c.d, where a is one of the six main classes and d corresponds to an individual reaction.
Enzyme information databases such as BRENDA6 and ENZYME7 use the EC
classification, whilst other databases classify enzymes based on evolutionary similarity8 or
reaction mechanism.9
It is difficult to predict the function of enzymes by transferring annotation via homology for
a number of reasons. Fewer than 30% of enzymes pairs that shared at least 50% sequence
identity actually share the same EC classification number. It has also been noted that
structural similarity does not always correspond to catalytic similarity. In an analysis of 167
homologous CATH10 superfamilies, almost half contained enzymes with differing catalytic
functions (denoted by differing EC classification numbers). Whilst many of these enzymes
differed only in their final digit, 22 superfamilies contained enzyme functions that were not
conserved at any level.11
The annotation of function via homology is further complicated for enzymes due to
convergent evolution. Several studies have reported cases of the same catalytic function
evolving independently.12; 13 George et al.11 found 105 cases where the same EC number was
allocated to enzymes that displayed no detectable sequence similarity. Furthermore, 34 of
these EC numbers represented enzymes that have entirely different structural folds,
indicative of convergent evolution. For these cases, functional similarity would not be
recognised by sequence or structural comparison methods.
Enzymes of similar function, whether or not they are evolutionarily related, have been
shown to exhibit shared sequence and structural characteristics. Understanding the link
between these characteristics and protein function is important in the development of
methods to predict and understand protein function. From a study of structural features of
the proteases,14 Stawiski et al. found that they exhibit similar characteristics such as smaller
than average surface areas and higher Cα densities, regardless of whether or not they were
evolutionarily related. They also showed different secondary structure content relative to the
non-proteases. By using these features in a machine learning approach they were able to
define a set of structural classifiers that could predict whether a protein is a protease or non-
protease with an accuracy of over 86%. In a later study,15 Stawiski et al. also reported
structural features that are characteristic of the O-glycosidases such as distinctive electrostatic
properties of the proteins surface, despite differences in the overall fold.

65
It has been shown that simple protein structural features, such as secondary structure
content and amino acid surface fractions, were of value in predicting the top EC class for an
enzyme.16 The machine learning algorithm used in this study found that the utility of
features in predicting EC class differed depending on the class being predicted. Some
unexpected observations were uncovered, such as unusually high tryptophan usage on the
surface of hydrolases. However, due to the complexities of how features combine in
machine learning methods it was difficult to deconstruct the exact relationships between
features and enzyme class.
Whilst such features have performed well in predicting enzyme function, more useful
features are likely to relate to the region of the enzyme that is directly involved in catalysis.
Structural templates made from active site geometry have shown utility in detecting other
enzymes of similar function.17 This shows that, even in the absence of homology, features
of active sites may provide more functional information than available from the whole
protein alone.
The properties of enzyme active sites and the specific residues involved in catalysis have
been well studied. An analysis of a set of 178 enzyme active sites with catalytic residues
annotated from the literature showed how properties such as amino acid identity, secondary
structure state and B-factor differed for catalytic residues.18 Whilst these properties may be
useful for identifying active-site residues, the features were not used to differentiate between
different enzyme functions. Similarly, numerous other studies have used features ranging
from geometry-based features19; 20 to electrostatic21; 22 and chemical features23; 24 to identify
enzyme active sites. However, as yet, these features have not been used to distinguish active
sites of different EC classes.

66
2.2 Methods
2.2.1 Dataset Creation
In order to calculate the features in this analysis the enzymes in the dataset needed an
annotated EC number, a structure deposited in the PDB and a known catalytic site location.
The dataset was therefore created from enzymes contained in the Catalytic Site Atlas,25 a
database of catalytic residues annotated from literature or from comparison to closely related
enzymes. Due to the need to accurately locate each structure’s active site, only the enzymes
that have residues annotated from literature were used. These were then split into the top
six classes of the EC hierarchy based on their primary EC class annotation. If an enzyme
had more than one EC annotation with different top EC classes then the enzyme was
represented in each of the class sets for which it has an annotation.
In some of the enzymes, the annotated catalytic residues were not in spatial proximity to
each other. Often this was caused by residues on separate chains (and in separate models of
the biological unit file) forming sites at their interface. The CSA annotates residues with
chain identifiers but does not differentiate between separate models of the biological unit
file. Where possible these annotations were updated, however in a number of enzymes the
annotated catalytic residues were not in spatial proximity to each other at all and these
enzymes were rejected from the set.
In order to reduce bias towards over-representation of sets of closely related enzymes, it
was important to cull the structures in each class for redundancy. Due to the features being
mostly structure-based, it was more relevant in this study to cull by structural similarity
rather than sequence similarity.

67
Firstly, as the features should be calculated on the structure that is likely to exist in the cell,
any enzyme that did not have a biological unit structure file was omitted. In order to ensure
that the most accurate and reliable structures were favoured, the PDB structures listed for
the enzymes in the CSA were ranked by their AEROSPACI score.26 The AEROSPACI
score is a numerical representation of the quality of a PDB structure. No structure was
included in the set with an AEROSPACI score of less than 0.3, which would represent
structures of a reasonable quality and omits structures with aberrant comments, such as
“misfolded” or “mistraced” in their entry in SCOP.27
The constituent SCOP domains of each of the remaining enzymes were then identified for
each structure. The culling process centered on the principle that no two enzymes within an
EC class should have an active site domain (the domain that contains the active site) from
the same superfamily. Within each functional class the domain superfamilies from the top
AEROSPACI ranking structure were searched for in the subsequent ranked structures. If
they were found to match another enzyme’s catalytic domain, then the lower ranking enzyme
was removed from the list. This process was continued iteratively until the bottom of the
list of remaining enzymes. This was carried out for each functional class, hence producing
non-redundant sets of enzyme structures for each EC class.
2.2.2 Defining Active Site Residues
The catalytic residue information for each enzyme in the datasets was obtained from the
CSA (version 2.2.1). The coordinate of the β carbon atom (or α carbon for Glycine) of each
catalytic residue was taken from the PDB biological unit file as the residue’s reference
coordinate. A central point was then calculated by taking the geometric average of the
reference coordinates for the catalytic residues for each protein. This was termed the
centroid.
To find the residues within the active site, residues were extracted from the PDB file that
had at least one atom within 10Å of the centroid. Residues were then further selected if they
exhibited more than or equal to 5Å2 solvent accessible surface area. Solvent accessible
surface area was calculated using an in-house program called SACALC (Jim Warwicker),
which calculates the solvent accessible surface area by rolling a solvent probe (1.4Å) around

68
the surface of the protein and calculating the area accessible to the probe. These residues
were then considered to be active site residues.
2.2.3 Calculating Features
Active site amino acid compositions were calculated by dividing the number of each residue
type in the active site residues by the total number of residues in the active site. Total amino
acid composition and surface amino acid composition were calculated similarly, either using
all residues or only those with at least 5Å2 surface area respectively.
The polarity/charge fractions were calculated by dividing the number of residues from each
group (in either the total biological unit or the active site) by number of residues in the
biological unit or active site.
Secondary structure states for each residue were taken from the secondary structure
annotation from the PDB file, which is generated by a program that incorporates DSSP28
and Promotif.29 Average hydrophobicity values were obtained by dividing the sum of the
Kyte & Doolittle30 values for each residue in the protein (or in the active site) by the number
of residues in the protein (or in the active site). The polar amino acids contained the
positively charged (R, H, K), negatively charged (D, E) and uncharged amino acids (N, Q, S,
T). The non-polar amino acids were represented by the aromatic amino acids (F, W) and
non-polar amino acids (G, A, V, L, I, P, M). Cysteine and Tyrosine were not included as they
can be either polar or non-polar depending on the pH of the environment.
The isoelectric point (pI) of a protein is the pH at which the protein has a net electrical
charge of zero. The pI of each enzyme was calculated by the Pepstats program, which is part
of the EMBOSS package of applications.31

69
2.2.4 Culling Redundancy in Features.
Some features are obviously highly dependent on each other and should not be considered
separately, such as the proportions of polar residues and non polar residues. Groups of
features that correlated strongly with each other can therefore be represented by just one
feature. Pearson correlation coefficients were calculated for all possible pairs of significant
features and those that had a coefficient of at least 0.5 were considered to correlate strongly.
In order to retain the most descriptive features, the significant features were ranked
according to their p-value. Each feature in the list was compared to the top-ranking feature
and removed if they correlated strongly. This procedure was performed iteratively,
comparing the remaining features in the list to the next highest-ranked feature, until the
bottom of the list. This method produced a set of significantly-different features that are not
strongly correlated with each other.
2.2.5 Statistical Analysis.
The use of the appropriate statistical test to analyse the difference between EC classes
depends on how the data are distributed. In order to test whether the data were normally
distributed or not over the six EC classes, a Kolmogorov-Smirnov test was performed for
each feature.
If the values for a feature were distributed normally, the differences between the EC classes
were be analysed using the One-Way ANOVA, with exception of categorical data such as
oligomeric status. This test evaluates the equality of data over three or more groups. A
significant p-value would indicate that at least one group’s mean is significantly different to
the others.
If data for a feature were not normally distributed, the non-parametric version of the One-
Way ANOVA, the Kruskal-Wallis test, was used. Again, this tests for equality of the data
between three or more groups. However, rather than comparing the group means of the
raw data as the ANOVA does, the Kruskal-Wallis test ranks the data and then compares the
distributions of the ranked data. It was therefore more appropriate to show mean values on

70
histograms for features that were normally distributed and median values for features that
were not.
This study involves the statistical testing of a large number of hypotheses at the 5%
confidence level, which is likely to lead to a number of false positive results (features that
have a p-value of less than 0.05, but do not show real differences in values between the six
classes). There are a number of statistical procedures that attempt to address this problem
and reduce the number of false positives by adjusting the p-value. These include the
Bonferroni correction32, the Holm-Bonferroni correction33, and the Benjamini and
Hochberg34 method for controlling the false discovery rate (FDR).
The Bonferroni and the Holm-Bonferroni procedures are very stringent and focus on
reducing the probability of rejecting even one true null hypothesis (the family-wise error
rate). The penalty for this reliability is that these procedures lack power and are likely to
accept a large number of null hypotheses that are not correct. The likelihood of rejecting a
true hypothesis by the Bonferroni procedure has been a source for some criticism.35 The
FDR has been suggested as a more suitable method to overcome some of the problems with
the Bonferroni procedure.36; 37; 38 Briefly, this method involves ranking each experiment result
by its p-value (in ascending order), then creating a new FDR-adjusted p-value accounding to
the formula shown in Equation 2.1. If this FDR-adjusted p-value is below the significant
threshold (0.05 is used here) then the null hypothesis for that experiment can be rejected.
FDR is a much more powerful and less restrictive method, although the cost is an increased
likelihood of false positive results.
Equation 2.1 The calculation of the FDR-adjusted p-value (P(FDR)).
i is the ordered rank position of the experiment, n is the total number of experiments and Pi is the
original unadjusted p-value for that experiment.

71
In this study, it is appropriate to use a more powerful method in order to give a good
coverage of probable true results, rather than ensure that every result is true at the expense
of many false negatives. The p-values obtained from the hypothesis tests have therefore
been adjusted using the Benjamini and Hochburg method for controlling the false discovery
rate.34
2.2.6 Rotamer Calculations.
In order to calculate the relative flexibilities of aspartic acid and glutamic acid side chains we
used a mean-field program39 developed from earlier work.40 This uses pairwise packing of
rotamers to derive probabilities for rotamers within a fixed sidechain, according to an
allowed van der Waals tolerance in the packing. This tolerance was set at 0.8Å, in keeping
with earlier work.39 Rotamers with zero probability are inaccessible, given the surrounding
mainchain and sidechains. The remaining (non-zero probability) rotamers are then
compared to the number of rotamers in the dictionary to assess the conformational freedom
of a sidechain. These calculations are made for Asp and Glu residues to compare their
flexibility.

72
2.3 Results and Discussion
2.3.1 Dataset and Active Site Definition.
The dataset was created using the criteria outlined in 2.2.1, and contains 294 unique enzymes
from a starting set of 880 (see Figure 2.1 and Table 2.1). Redundancy was culled by
structural similarity, ensuring no two proteins within a class share a domain where at least
one contains the active site, from a common SCOP27 superfamily. This produced a dataset
where the maximum sequence identity between two pairs is 24.1% and the average sequence
identity between enzymes in the set is 11.4%. For each of these enzymes, active site residues
were defined as residues that had at least one atom within 10Å of the centroid calculated
over CSA residues, and at least 5Å2 of solvent accessible surface area. A radius of 10Å
returns almost 95% of the catalytic residues (Figure 2.2), and beyond this the number of
CSA residues returned diminishes rapidly in comparison with other residues. A trade-off is
also required for the solvent accessibility threshold, where we look to exclude buried
residues within the active site radius. A 5Å2 solvent accessible surface area returns over 75%
of CSA residues (Figure 2.2).
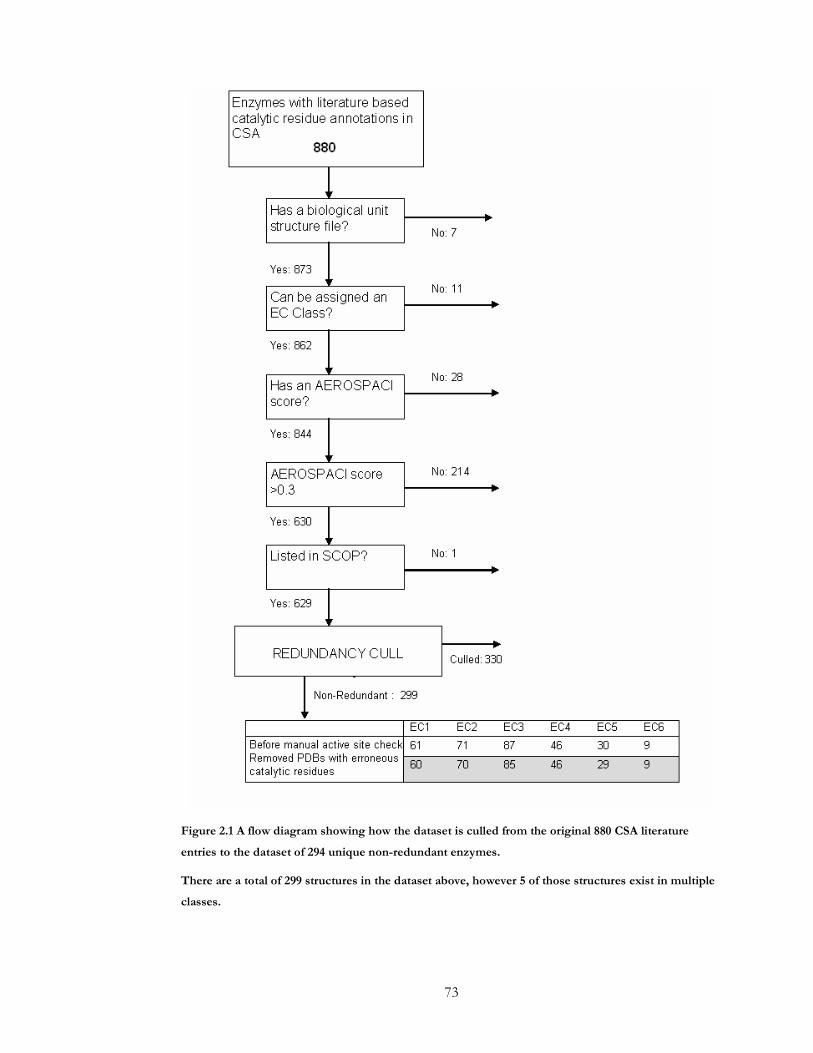
73
Figure 2.1 A flow diagram showing how the dataset is culled from the original 880 CSA literature
entries to the dataset of 294 unique non-redundant enzymes.
There are a total of 299 structures in the dataset above, however 5 of those structures exist in multiple
classes.

74
EC PDB EC PDB EC PDB EC PDB EC PDB EC PDB
1 1a05
1 1a4i
1 1a8q
1 1akd
1 1aop
1 1b5t
1 1bou
1 1bt1
1 1c0k
1 1c9u
1 1d3g
1 1d4a
1 1dhf
1 1do6
1 1dqa
1 1dqs
1 1dve
1 1fnb
1 1g72
1 1g79
1 1gcu
1 1gp1
1 1gpj
1 1gqg
1 1h2r
1 1hfe
1 1i19
1 1jnr
1 1l1d
1 1l1l
1 1l6p
1 1lci
1 1ljl
1 1luc
1 1mrq
1 1ndo
1 1ni4
1 1nid
1 1nir
1 1nml
1 1o04
1 1o9i
1 1oac
1 1opm
1 1qje
1 1qv0
1 1s3i
1 1sox
1 1ti6
1 1vie
1 1vlb
1 1yve
1 2bbk
1 2cpo
1 2jcw
1 2toh
1 3mdd
1 3nos
1 7atj
2 1aj0
2 1al6
2 1bg0
2 1brw
2 1c2t
2 1c3j
2 1cg6
2 1cgk
2 1cqq
2 1cs1
2 1cwy
2 1d0s
2 1d8c
2 1d8d
2 1daa
2 1dqs
2 1e19
2 1e2a
2 1ecf
2 1eh6
2 1ez1
2 1f75
2 1f7l
2 1f8x
2 1foa
2 1g24
2 1g6t
2 1g8f
2 1gpr
2 1h3i
2 1h54
2 1hiv
2 1hka
2 1hxq
2 1hy3
2 1ig8
2 1ir3
2 1iu4
2 1j53
2 1jdw
2 1jm6
2 1jms
2 1k30
2 1lij
2 1mla
2 1moq
2 1nsp
2 1oas
2 1oe8
2 1oj4
2 1onr
2 1oyg
2 1p4n
2 1p4r
2 1pfk
2 1pud
2 1qd1
2 1qpr
2 1rhs
2 1ro7
2 1trk
2 1tys
2 1uam
2 1un1
2 1vid
2 2tdt
2 2tps
2 2ypn
2 3cla
3 135l
3 1a2t
3 1a4i
3 1a79
3 1abr
3 1ah7
3 1ako
3 1apy
3 40391
3 1bol
3 1bp2
3 1bs4
3 1bs9
3 1bwp
3 1cd5
3 1cev
3 1chm
3 1czf
3 1d1q
3 1d2t
3 1d8h
3 1dl2
3 1dmu
3 1dup
3 1e7l
3 1eb6
3 1ef0
3 1eug
3 1fy2
3 1hdh
3 1hzf
3 1itx
3 1j79
3 1j7g
3 1jh6
3 1jhf
3 1js4
3 1k32
3 1k82
3 1kaz
3 1lam
3 1lba
3 1lbu
3 1m21
3 1m6k
3 1mqw
3 1mud
3 1nf9
3 1nln
3 1nlu
3 1nsf
3 1nww
3 1p4r
3 1pa9
3 1pgs
3 1pyl
3 1q3q
3 1qaz
3 1qcn
3 1qd6
3 1qgx
3 1qh5
3 1qq5
3 1qtn
3 1qum
3 1qz9
3 1r16
3 1r4f
3 1s95
3 1ssx
3 1tml
3 1uaq
3 1uf7
3 1v0y
3 1vas
3 2acy
3 40270
3 2eng
3 2nlr
3 2pth
3 3eca
3 5fit
4 1aw8
4 1b66
4 1b93
4 1bfd
4 1bix
4 1c3c
4 1c82
4 1ca2
4 1cl1
4 1db3
4 1dco
4 1dio
4 1dnp
4 1dqs
4 1dw9
4 1dxe
4 1ecm
4 1et0
4 1fgh
4 1fro
4 1fua
4 1hrk
4 1i6p
4 1i7q
4 1mka
4 1mvn
4 1nhx
4 1p1x
4 1pii
4 1pix
4 1ps1
4 1pya
4 1qd1
4 1qj4
4 1qrg
4 1r6w
4 1r76
4 1rbl
4 1ru4
4 1sll
4 1uqr
4 1uro
4 2abk
4 2ahj
4 7odc
5 1bd0
5 1cb7
5 1d6o
5 1dbf
5 1e3v
5 1ecl
5 1ecm
5 1eej
5 1f2v
5 1f6d
5 1jfl
5 1k0w
5 1k4t
5 1lvh
5 1m53
5 1m9c
5 1muc
5 1n20
5 1nn4
5 1o98
5 1otg
5 1p5d
5 1pii
5 1pym
5 1qhf
5 1snn
5 1tph
5 2sqc
5 2xis
6 12as
6 1a4i
6 1dae
6 1gsa
6 1j09
6 1kp2
6 1p3d
6 1qmh
6 1v25
Table 2.1 PDB codes for each enzyme in the dataset.

75
(a)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0 2 4 6 8 10 12 14 16 18
Distance (Angstroms)
Cum
ula
tive P
erc
enta
ge
(b)
0
20
40
60
80
100
0 5 10 15 20 25 30 35 40Solvent Accessible Surface Area Threshold
(Angstrom2)
Pe
rce
nta
ge
of
CS
A
resid
ue
s c
ove
red
Figure 2.2 The percentage coverage of CSA residues by varying active site criteria thresholds for (a)
surface area and (b) distance from centroid.

76
2.3.2 Overall Description of Features.
The total set of features that were analysed is shown in Table 2.2. Features that were shown
to be significantly different (have a Benjamini and Hochberg adjusted p-value of less than
0.05) over the six EC classes are shaded. Table 2.3 shows the features with significant
differences over the six EC classes, their Kruskal-Wallis/ANOVA p-value, and the class
with the highest and lowest mean or median values for that feature. The highest/lowest
mean values are used where the distribution of the data is normal and the median is used
where the data are non-normally distributed. Where the highest or lowest class is the ligases
(EC6), the next highest or lowest class is actually given in the table and denoted with an
asterisk. There are only a small number of ligases (9) in the dataset and their mean/medians
are more influenced by extreme values than other classes.
As an example of features with significant differences we looked at active site aromatic
residue content (Figure 2.3) and amino acid compositions (Figure 2.4). Oxidoreductases
(EC1) and hydrolases (EC3) had the highest active site aromatic proportions. The high
aromatic active site proportion seen in the hydrolases may be influenced by those that bind
proteins as a substrate, since it has been observed that protein-protein interfaces often
contain high proportions of aromatic residues. 41; 42 There was, however, no significant
difference between the active site aromatic proportions observed in hydrolases that bind
other proteins as a substrate and those that do not. The median active site aromatic content
for EC6 showed no aromatic residues in the active site, although this is difficult to interpret
because of the very small class size.
Most amino acids have significantly different composition values over the six EC classes for
one or more of the location categories; active site, surface, total (Figure 2.4). No amino acids
had significantly different proportions between the six classes in all three sets. Distributions
for all significant features between EC classes are shown in Figure 2.5 to Figure 2.12.

77
Attribute Structural Features
Active site proportion helix Active site proportion sheet Active site proportion turn Active site proportion non-helix
and non-sheet Active site total B-factor Active site average atomic B-
factor Active site surface area Relative active site surface area Active site non-polar proportion Active site aromatic proportion Active site negative proportion Active-site polar proportion Active site mean hydrophobicity
score Active site positive proportion Active site mean isoelectric point Average total atomic B-factor Relative average active site
atomic B-factor Number of chains Total proportion of helix Total proportion of beta sheet Total proportion of turn Total proportion of non-helix and
non-sheet
Sequence Features
Total negative proportion Total polar proportion Total non-polar proportion Total aromatic proportion Total positive proportion Total mean hydrophobicity score Total mean Isolelectric point Proportion of low complexity
sequence
Size-associated Features
Total surface area Number of residues in the
biological unit Number of chains Length of sequence Total B-factor
Amino Acid Compositions Active site ALA
Active site ARG
Active site ASN
Active site ASP
Active site CYS
Active site GLN
Active site GLU
Active site GLY
Active site HIS
Active site ILE
Active site LEU
Active site LYS
Active site MET
Active site PHE
Active site PRO
Active site SER
Active site THR
Active site TRP
Active site TYR
Active site VAL
Surface ALA
Surface ARG
Surface ASN
Surface ASP
Surface CYS
Surface GLN
Surface GLU
Surface GLY
Surface HIS
Surface ILE
Surface LEU
Surface LYS
Surface MET
Surface PRO
Surface SER
Surface THR
Surface TRP
Surface TYR
Surface VAL
Total ALA
Total ARG
Total ASN
Total ASP
Total CYS
Total GLN
Total GLU
Total GLY
Total HIS
Total ILE
Total LEU
Total LYS
Total MET
Total PHE
Total PRO
Total SER
Total THR
Total TRP
Total TYR
Total VAL
Table 2.2 List of all features calculated for each enzyme.
Features that showed significant differences between the six classes are shaded.
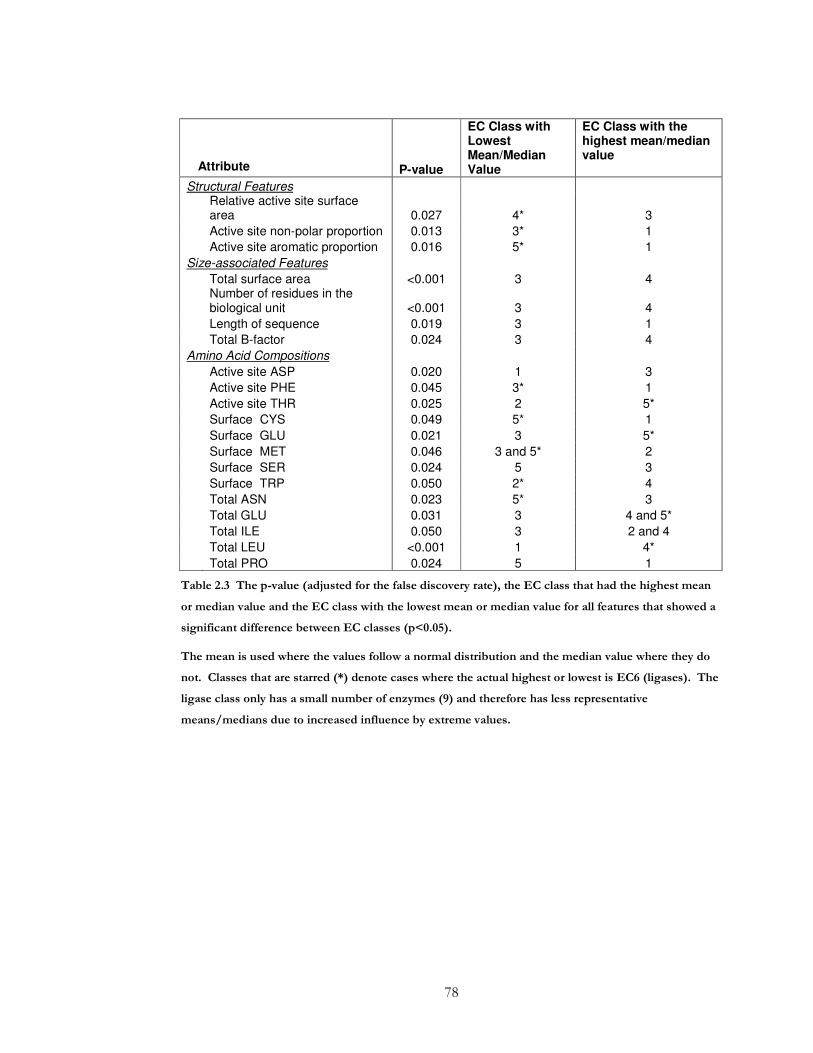
78
Attribute P-value
EC Class with Lowest Mean/Median Value
EC Class with the highest mean/median value
Structural Features
Relative active site surface area 0.027 4* 3
Active site non-polar proportion 0.013 3* 1
Active site aromatic proportion 0.016 5* 1
Size-associated Features
Total surface area <0.001 3 4
Number of residues in the biological unit <0.001 3 4
Length of sequence 0.019 3 1
Total B-factor 0.024 3 4
Amino Acid Compositions Active site ASP 0.020 1 3
Active site PHE 0.045 3* 1
Active site THR 0.025 2 5*
Surface CYS 0.049 5* 1
Surface GLU 0.021 3 5*
Surface MET 0.046 3 and 5* 2
Surface SER 0.024 5 3
Surface TRP 0.050 2* 4
Total ASN 0.023 5* 3
Total GLU 0.031 3 4 and 5*
Total ILE 0.050 3 2 and 4
Total LEU <0.001 1 4*
Total PRO 0.024 5 1
Table 2.3 The p-value (adjusted for the false discovery rate), the EC class that had the highest mean
or median value and the EC class with the lowest mean or median value for all features that showed a
significant difference between EC classes (p<0.05).
The mean is used where the values follow a normal distribution and the median value where they do
not. Classes that are starred (*) denote cases where the actual highest or lowest is EC6 (ligases). The
ligase class only has a small number of enzymes (9) and therefore has less representative
means/medians due to increased influence by extreme values.

79
0.00
0.02
0.04
0.06
0.08
0.10
0.12
EC1 EC2 EC3 EC4 EC5 EC6
Me
dia
n A
rom
atic P
rop
ort
ion
of
Active
Site
Figure 2.3 The median aromatic proportion of the active site for each EC class.
Figure 2.4 Amino acids that showed significant differences between the six EC classes in either the
active site, surface residues or the total protein.
A shaded box indicates a false discovery rate adjusted p-value of less than 0.05.

80
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
Active Site Non-PolarProportion
Active Site AromaticProportion
Pro
port
ion o
f active s
ite
Figure 2.5 The median value of significantly different charge-related features for each EC class.
0.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
0.040
0.045
0.050
Relative Active Site Surface Area
Pro
po
rtio
n o
f active s
ite
Figure 2.6 The median proportion of the total surface area that belongs to the active site for each EC
class.
0
100
200
300
400
500
600
700
800
Sequence Length Number of Residues in theBiological Unit
Num
ber
of
Resid
ues
Figure 2.7 The median value of significantly different size-related features for each EC class.
Key
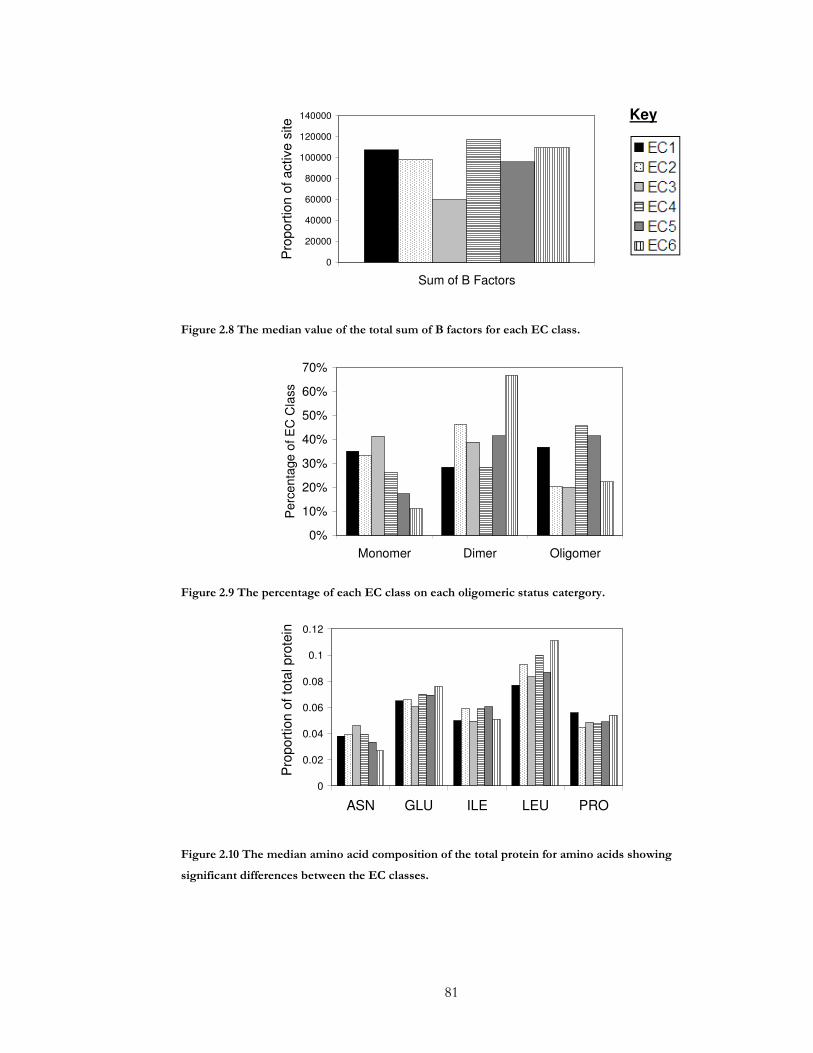
81
0
20000
40000
60000
80000
100000
120000
140000
Sum of B FactorsP
ropo
rtio
n o
f active s
ite
Figure 2.8 The median value of the total sum of B factors for each EC class.
0%
10%
20%
30%
40%
50%
60%
70%
Monomer Dimer Oligomer
Pe
rce
nta
ge
of E
C C
lass
Figure 2.9 The percentage of each EC class on each oligomeric status catergory.
0
0.02
0.04
0.06
0.08
0.1
0.12
ASN GLU ILE LEU PRO
Pro
port
ion
of to
tal pro
tein
Figure 2.10 The median amino acid composition of the total protein for amino acids showing
significant differences between the EC classes.
Key
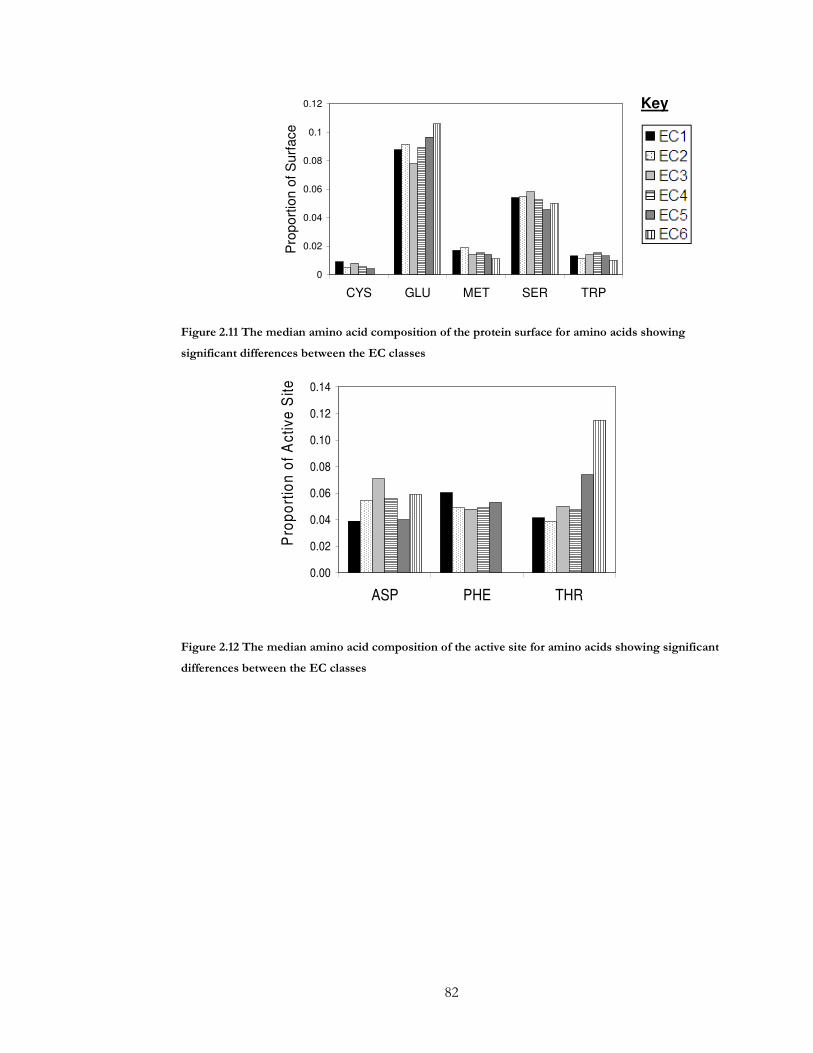
82
0
0.02
0.04
0.06
0.08
0.1
0.12
CYS GLU MET SER TRPP
rop
ort
ion
of
Surf
ace
Figure 2.11 The median amino acid composition of the protein surface for amino acids showing
significant differences between the EC classes
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
ASP PHE THR
Pro
po
rtio
n o
f A
cti
ve
Sit
e
Figure 2.12 The median amino acid composition of the active site for amino acids showing significant
differences between the EC classes
Key

83
2.3.3 Unique Descriptive Features.
At this stage it was decided to look at correlations between features, since groups of features
that correlate strongly with each other can be reduced to a single representative feature
(Figure 2.13). The nodes, which represent features, are connected where there is a probable
correlation between them (i.e. Pearson’s correlation coefficient, R, exceeds the critical value
at the 5% significance level). The critical value of R, 0.195 (given from a table of critical
values of R), is very low due to the large number of features in this study. Whilst a
correlation is likely to exist with this R value, it would not denote a strong correlation,
therefore we have defined a strong correlation as R >=0.5 (shown by the darker edges in
Figure 2.13).
In order to retain the features that are most significant, the features were ranked by the p-
value for the differences between functional groups. Features were then chosen that did not
correlate strongly with any higher-ranking features. In Figure 2.13 the features retained are
shaded in light grey and those that were discarded are shaded in dark grey. It can be seen
that no retained features correlate strongly with any other retained features. The features
appear to cluster into three main groups: the size-associated features in the lower left part of
the network, features relating to active-site non-polarity in the upper right, and total and
surface amino acid proportions to the upper left.
Of the top 5 most significantly different features, two are the total amino acid proportions
for Leu and Pro. Secondary structure preferences for the six EC classes were investigated,
since leucine has a high propensity for helix, and proline a low propensity for sheet and
helix.43; 44 For several, but not all, EC classes we find correlations between total proportions
of either leucine or proline, and secondary structure that are in line with their overall
propensities for helix and sheet (Table 2.4). This, however, does not translate to a significant
difference in secondary structure over the six EC classes (Figure 2.14). It is therefore
difficult to assess the extent to which variation in secondary structure content could be
responsible for the differences in leucine and proline compositions between EC classes.

84
Figure 2.13 A network diagram showing the significantly different features (as nodes) connected
by lines where there is a probable correlation (the R value is more than 0.195, the critical R value
at the 5% significance level).
The darker lines represent a strong correlation, where R is at least 0.5. The features that are
shaded dark grey are the ones that were discarded and those shaded light grey were retained for
further analysis.
Total Leucine vs. Total Helix Content Total Proline vs. Total Non-helix and
Non-sheet
Pearson's correlation coefficient (p value)
Pearson's correlation coefficient (p value)
EC1 0.471 (0.000) 0.086 (0.000)
EC2 0.342 (0.002) -0.061 (0.002)
EC3 0.336 (0.001) -0.085 (0.000)
EC4 0.531 (0.000) 0.009 (0.258)
EC5 0.451 (0.007) 0.208 (0.058)
EC6 -0.008 (0.354) -0.236 (0.498)
Table 2.4 The correlation between total leucine and proline composition and the secondary
structure environments that they are typically associated with.
The Pearson’s correlation coefficient is shown along with the significance associated with this
correlation for the number of proteins in each class.
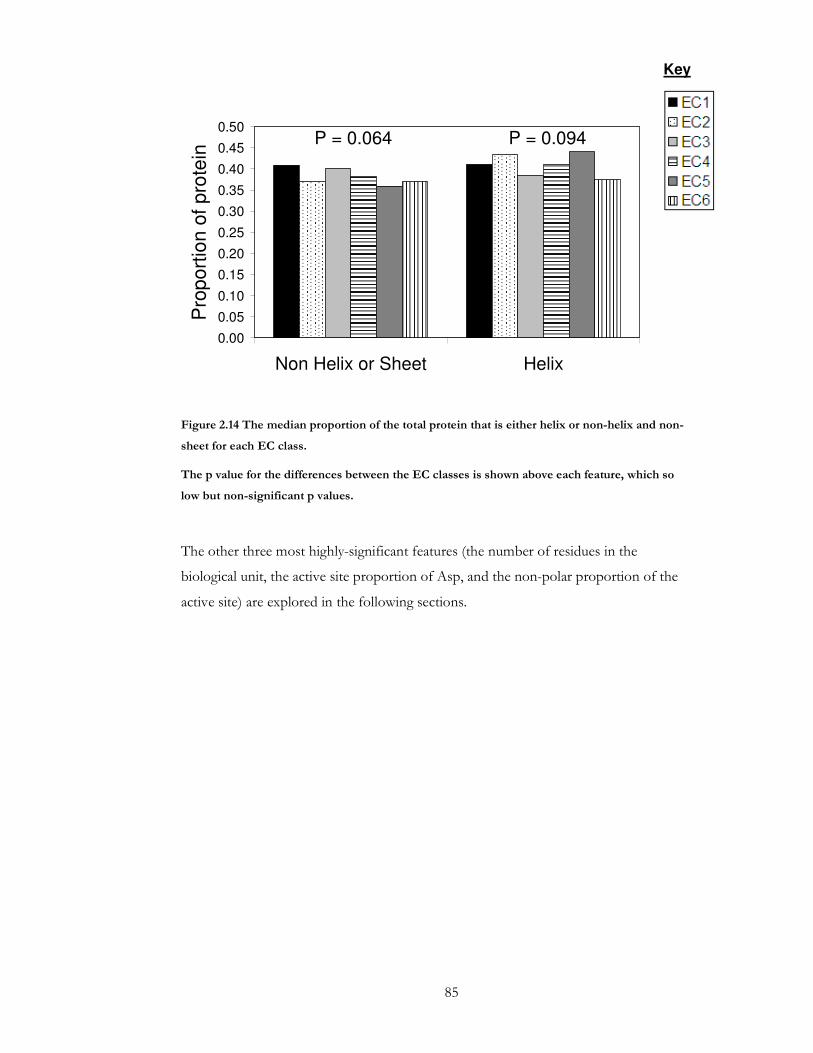
85
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
Non Helix or Sheet Helix
Pro
po
rtio
n o
f p
rote
in
P = 0.064 P = 0.094
Figure 2.14 The median proportion of the total protein that is either helix or non-helix and non-
sheet for each EC class.
The p value for the differences between the EC classes is shown above each feature, which so
low but non-significant p values.
The other three most highly-significant features (the number of residues in the
biological unit, the active site proportion of Asp, and the non-polar proportion of the
active site) are explored in the following sections.
Key

86
2.3.4 Differences in Structure Sizes due to Different Oligomeric
State Preferences
All but one of the size-related features were found to show significant differences
between the six classes. These features correlated strongly with each other, apart from
sequence length (see Figure 2.13), and the number of residues in the biological unit was
chosen as the representative feature for further detailed analysis. Figure 2.7 shows the
differences in sequence length and the number of residues in the biological unit PDB
file. The sequence length is the number of residues in the sequence of each distinct
chain in the PDB file (duplicate chains are not counted twice), whereas the number of
residues in the biological unit counts residues in duplicate chains. The number of
residues in the biological unit is on average larger than the number of residues in the
sequence due to the oligomerisation of protein chains in biologically functional units.
It could be expected that since the oxidoreductases have the largest sequence lengths,
they would also have the largest number of residues in the biological unit. The lyases,
however, actually have the largest number of residues in the biological unit, due to a
preference for higher order oligomers compared to oxidoreductases (Figure 2.9).
The hydrolases (EC3) and lyases (EC4) were the only classes that had significantly
different proportions of monomers, dimers and oligomers to the other classes (p-value
= 0.001 and 0.024, respectively). Lyases have the largest percentages of enzymes that
form oligomers and the lowest proportion of enzymes that form monomers, whereas
hydrolases tend to exist as monomers and have the lowest proportion of oligomers of
all the classes (see Figure 2.9).
Generally, hydrolases have the simplest task of the six classes since hydrolysis is usually
an energetically favourable reaction and therefore they may not require the
complication of forming higher order oligomers. There are also other functional
advantages to enzymes existing as monomers, for example stability at low
concentrations and rapid diffusion. Rapid diffusion is particularly relevant for
extracellular hydrolases, for passage through the cell membranes to their site of
action.24 Subcellular location annotation is only available for 21 of the 85 hydrolases
(see Table 2.5). Two of these hydrolases are annotated as extracellular (secreted), both

87
of which are monomers. All 9 extracellular enzymes in the total set are monomeric,
which suggests that extracellular enzymes prefer to exist as monomers. There is,
however, not enough information to reveal the influence of extracellular location on
the preference of EC3 to exist as a monomer.
Conversely, the lyases have the highest proportion of oligomers and the lowest
proportion of monomers (see Figure 2.9). There are stability benefits to proteins
forming large complexes due an increase in the number of internal interactions
enabling a lower surface to volume ratio. Furthermore, multimeric complexes,
particularly homo-oligomers, are a genetically economical way of producing large
proteins and the subunit based assembly allows for an extra step in error control
whereby defective subunits can be discarded.45 One functional advantage of
multimeric enzymes is the opportunity for increased catalytic control by cooperativity
between active sites in individual subunits or by allosteric action between the subunits.
For the multimeric protein structures in our dataset, we assessed whether the active site
of the enzyme was close to or at a subunit boundary as a means of estimating how
many of the enzymes may have their action regulated by the formation of the
multimeric complex. If the active site amino acids (defined in 2.2.2) from a single site
in an enzyme come from separate chains then the active site is defined as ‘shared’. If
they are all from the same chain then the site is defined as ‘single’.
In all EC classes a larger proportion of dimers have single active sites than shared
active sites (60% have a single active site, whereas 40% have a shared active site). This
is opposite to oligomers with three or more chains, which are more likely to have
shared active sites than single (40% have a single active site, whereas 60% have a
shared active site). When broken down into EC class it is evident that the class with
the largest number of oligomers (EC4) is also the class that has the largest percentage
of oligomers having shared active sites (see Figure 2.15). This suggests that the over-
representation of oligomers in this class can be attributed to the formation of the active
site by multiple subunits.

88
Subcellular Location EC1 EC2 EC3 EC4 EC5 EC6 All
Cytoplasm 8 (30.77%) 15 (60.00%) 12 (57.14%) 9 (64.29%) 6 (75.00%) 7 (100.00%) 57 (56.44%) Mitochondria 4 (15.38%) 4 (16.00%) 0 (0.00%) 2 (14.29%) 0 (0.00%) 0 (0.00%) 10 (9.90%)
Secreted 4 (15.38%) 1 (4.00%) 2 (9.52%) 2 (14.29%) 0 (0.00%) 0 (0.00%) 9 (8.91%)
Periplasm 5 (19.23%) 0 (0.00%) 1 (4.76%) 0 (0.00%) 1 (12.50%) 0 (0.00%) 7 (6.93%)
Nucleus 0 (0.00%) 2 (8.00%) 3 (14.29%) 1 (7.14%) 0 (0.00%) 0 (0.00%) 6 (5.94%)
Membrane 2 (7.69%) 1 (4.00%) 1 (4.76%) 0 (0.00%) 1 (12.50%) 0 (0.00%) 5 (4.95%)
Peroxisome 2 (7.69%) 0 (0.00%) 0 (0.00%) 0 (0.00%) 0 (0.00%) 0 (0.00%) 2 (1.98%)
Chloroplast 1 (3.85%) 1 (4.00%) 0 (0.00%) 0 (0.00%) 0 (0.00%) 0 (0.00%) 2 (1.98%)
Endoplasmic Reticulum 0 (0.00%) 0 (0.00%) 1 (4.76%) 0 (0.00%) 0 (0.00%) 0 (0.00%) 1 (0.99%)
Lysosome 0 (0.00%) 0 (0.00%) 1 (4.76%) 0 (0.00%) 0 (0.00%) 0 (0.00%) 1 (0.99%)
Golgi Apparatus 0 (0.00%) 1 (4.00%) 0 (0.00%) 0 (0.00%) 0 (0.00%) 0 (0.00%) 1 (0.99%)
26 (100%) 25 (100%) 21 (100%) 14 (100%) 8 (100%) 7 (100%) 101 (100%)
Table 2.5 Subcellular location annotation (where available) for each EC class.
The percentages are based on the number in the class with subcellular location annotation. Approximately a third of the total set (101 out of 294) have subcellular
location information.

0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
EC1 EC2 EC3 EC4 EC5 EC6
Pe
rcenta
ge o
f O
ligom
ers
in the
C
lass
Single
Shared
Figure 2.15 The percentage of oligomers that have single sub-unit or shared sub-unit active sites
in each class.
The hydrolases, which have the highest monomer and lowest oligomer proportions, is
the only class where the oligomers prefer to have single subunit active sites (excluding
EC6, which only has 2 oligomers). In contrast to the lyases, when hydrolases do form
oligomers it does not appear to be for functional reasons associated with their active
site and may be in an attempt to overcome the stability disadvantages of small proteins
(hydrolases have the smallest average number of residues in the biological unit).
2.3.4.1 Lyases and Hydrolases in Metabolic Networks
Metabolic networks represent how enzymes are linked to each other via their reactions.
Enzymes in the network that are highly connected to other enzymes (involved in
catalytic reactions with multiple different enzymes) or are at critical points in the
network through which many reaction pathways flow, are important to the stability of
the network. These enzymes therefore have to be highly regulated and their catalytic
rate tightly controlled. Cooperativity and allosteric interaction of active sites can be
used as a further level of control of enzyme action and therefore enzymes whose active
sites communicate in this way may be found at highly regulated points in a metabolic
network.

90
The Pathway Hunter tool46 holds information about metabolic networks for a given
organism, with the nodes representing enzymes or metabolites and the connections
representing their reactions. It also gives statistical and quantative information relating
to the importance of the enzymes in the networks. Traditionally, important enzymes in
a metabolic network would be identified by the number of connections to other
enzymes. Raman and Schomburg, however, proposed other measures of importance in
networks, namely choke points and load points.47 Load points are a measure of the
enzyme’s importance in a network. They are calculated by dividing the number of
metabolic pathways that pass through a node (the shortest route between two
metabolites is assumed) by the number of incoming or outgoing connections. This is
then divided by the average load value for the whole network. Choke points are used
to identify biochemical lethality in the network, where a choke point is defined as an
enzyme that uniquely produces or consumes a particular metabolite.
The distribution of enzymes over the six EC classes in a list of enzymes defined as
choke points in the Saccharomyces cerevisiae metabolic network was calculated using the
Pathway Hunter tool. The expected number of enzymes in each EC class was
calculated using the background distribution from all enzymes in the Saccharomyces
cerevisiae genome. The observed number of enzymes in each class in sets of defined
nodes (either choke points or load points) was divided by the expected number. The
class with the highest percentage of oligomers, the lyases (EC4), was significantly
overrepresented in the list of choke points (Figure 2.16). Similarly, the class with the
lowest percentage of oligomers, the hydrolases (EC3) was significantly
underrepresented. These results were even more marked when only considering the
top 50% of choke points with the highest incoming load value. This was also repeated
for the 25% most loaded (incoming and outgoing) enzymes and similar results were
obtained (Figure 2.17). We suggest that the more important enzymes in a network are
more likely to be oligomeric.
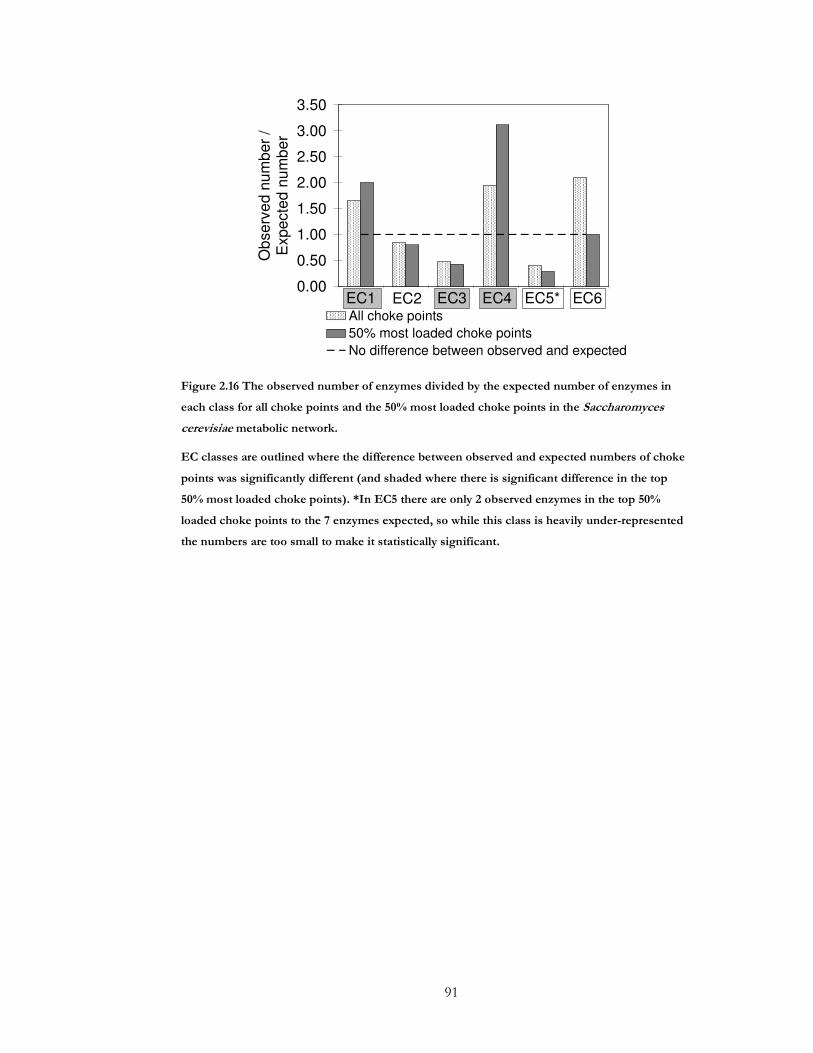
91
0.00
0.50
1.00
1.50
2.00
2.50
3.00
3.50
1 2 3 4 5 6
Ob
serv
ed n
um
ber
/E
xpecte
d n
um
ber
All choke points
50% most loaded choke points
No difference between observed and expected
EC1 EC2 EC3 EC4 EC5* EC6
Figure 2.16 The observed number of enzymes divided by the expected number of enzymes in
each class for all choke points and the 50% most loaded choke points in the Saccharomyces
cerevisiae metabolic network.
EC classes are outlined where the difference between observed and expected numbers of choke
points was significantly different (and shaded where there is significant difference in the top
50% most loaded choke points). *In EC5 there are only 2 observed enzymes in the top 50%
loaded choke points to the 7 enzymes expected, so while this class is heavily under-represented
the numbers are too small to make it statistically significant.
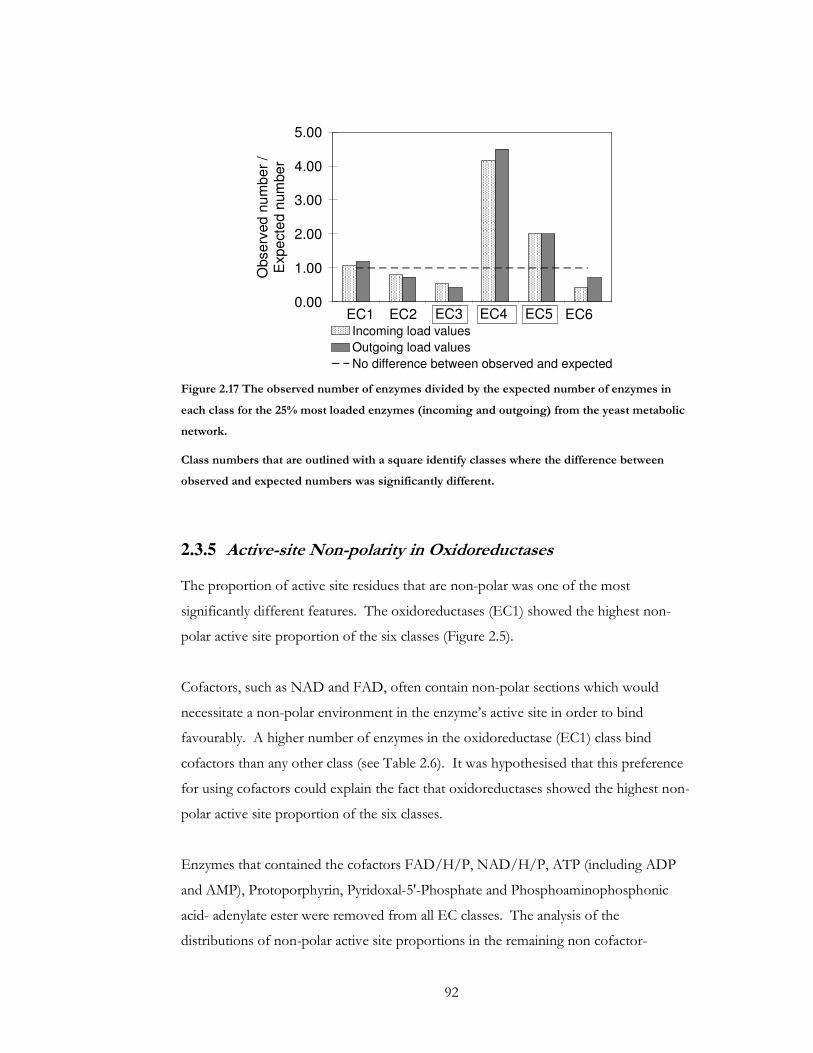
92
0.00
1.00
2.00
3.00
4.00
5.00
1 2 3 4 5 6
Obse
rved
nu
mb
er
/E
xp
ecte
d n
um
be
r
Incoming load values
Outgoing load values
No difference between observed and expected
EC1 EC2 EC3 EC4 EC5 EC6
Figure 2.17 The observed number of enzymes divided by the expected number of enzymes in
each class for the 25% most loaded enzymes (incoming and outgoing) from the yeast metabolic
network.
Class numbers that are outlined with a square identify classes where the difference between
observed and expected numbers was significantly different.
2.3.5 Active-site Non-polarity in Oxidoreductases
The proportion of active site residues that are non-polar was one of the most
significantly different features. The oxidoreductases (EC1) showed the highest non-
polar active site proportion of the six classes (Figure 2.5).
Cofactors, such as NAD and FAD, often contain non-polar sections which would
necessitate a non-polar environment in the enzyme’s active site in order to bind
favourably. A higher number of enzymes in the oxidoreductase (EC1) class bind
cofactors than any other class (see Table 2.6). It was hypothesised that this preference
for using cofactors could explain the fact that oxidoreductases showed the highest non-
polar active site proportion of the six classes.
Enzymes that contained the cofactors FAD/H/P, NAD/H/P, ATP (including ADP
and AMP), Protoporphyrin, Pyridoxal-5'-Phosphate and Phosphoaminophosphonic
acid- adenylate ester were removed from all EC classes. The analysis of the
distributions of non-polar active site proportions in the remaining non cofactor-
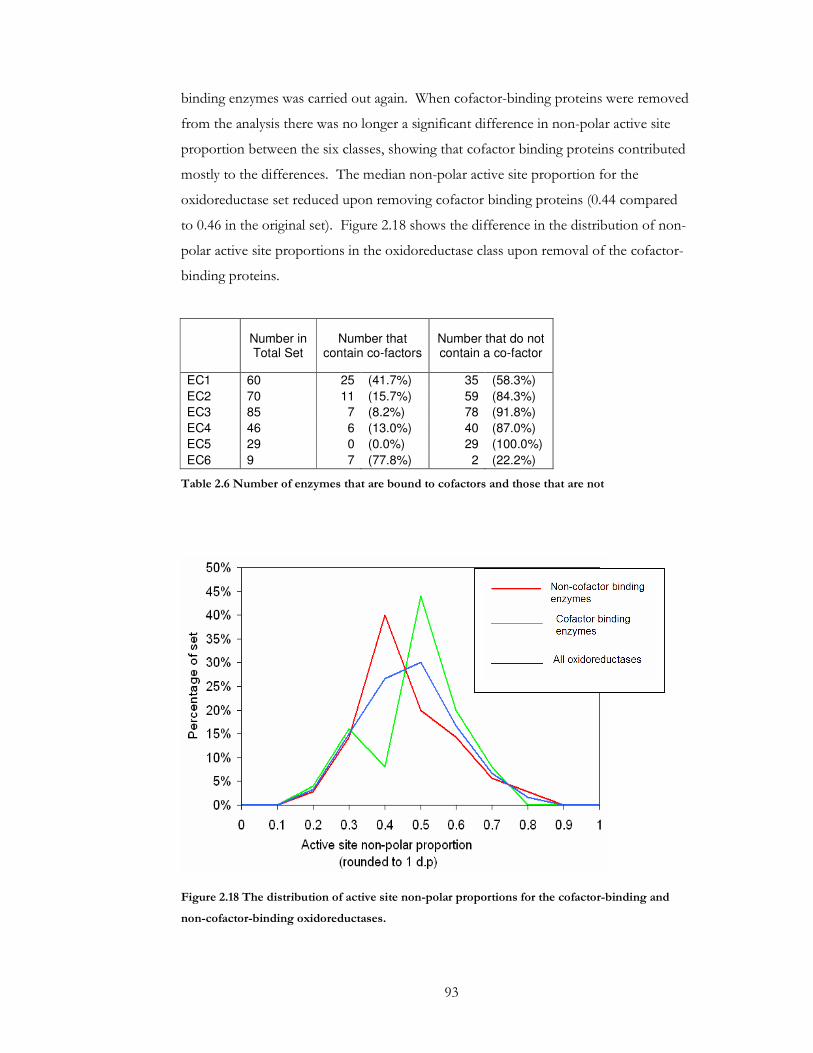
93
binding enzymes was carried out again. When cofactor-binding proteins were removed
from the analysis there was no longer a significant difference in non-polar active site
proportion between the six classes, showing that cofactor binding proteins contributed
mostly to the differences. The median non-polar active site proportion for the
oxidoreductase set reduced upon removing cofactor binding proteins (0.44 compared
to 0.46 in the original set). Figure 2.18 shows the difference in the distribution of non-
polar active site proportions in the oxidoreductase class upon removal of the cofactor-
binding proteins.
Number in Total Set
Number that contain co-factors
Number that do not contain a co-factor
EC1 60 25 (41.7%) 35 (58.3%)
EC2 70 11 (15.7%) 59 (84.3%)
EC3 85 7 (8.2%) 78 (91.8%)
EC4 46 6 (13.0%) 40 (87.0%)
EC5 29 0 (0.0%) 29 (100.0%)
EC6 9 7 (77.8%) 2 (22.2%)
Table 2.6 Number of enzymes that are bound to cofactors and those that are not
Figure 2.18 The distribution of active site non-polar proportions for the cofactor-binding and
non-cofactor-binding oxidoreductases.
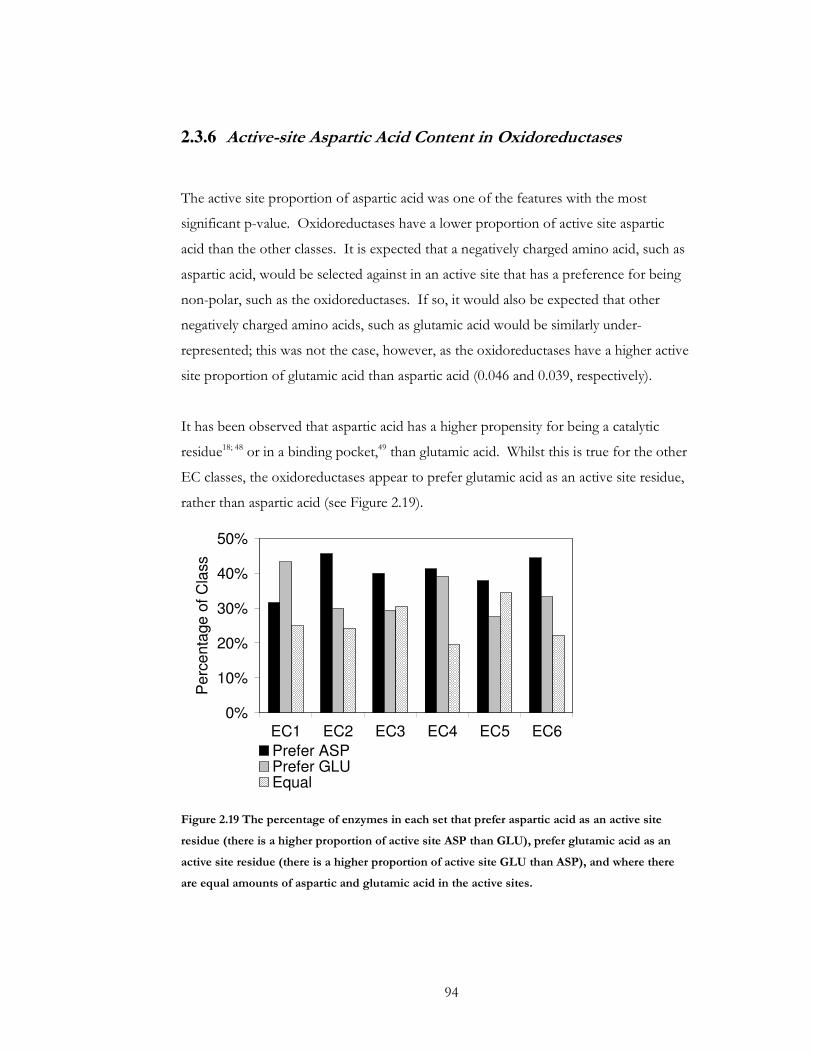
94
2.3.6 Active-site Aspartic Acid Content in Oxidoreductases
The active site proportion of aspartic acid was one of the features with the most
significant p-value. Oxidoreductases have a lower proportion of active site aspartic
acid than the other classes. It is expected that a negatively charged amino acid, such as
aspartic acid, would be selected against in an active site that has a preference for being
non-polar, such as the oxidoreductases. If so, it would also be expected that other
negatively charged amino acids, such as glutamic acid would be similarly under-
represented; this was not the case, however, as the oxidoreductases have a higher active
site proportion of glutamic acid than aspartic acid (0.046 and 0.039, respectively).
It has been observed that aspartic acid has a higher propensity for being a catalytic
residue18; 48 or in a binding pocket,49 than glutamic acid. Whilst this is true for the other
EC classes, the oxidoreductases appear to prefer glutamic acid as an active site residue,
rather than aspartic acid (see Figure 2.19).
0%
10%
20%
30%
40%
50%
EC1 EC2 EC3 EC4 EC5 EC6
Perc
enta
ge o
f C
lass
Prefer ASPPrefer GLUEqual
Figure 2.19 The percentage of enzymes in each set that prefer aspartic acid as an active site
residue (there is a higher proportion of active site ASP than GLU), prefer glutamic acid as an
active site residue (there is a higher proportion of active site GLU than ASP), and where there
are equal amounts of aspartic and glutamic acid in the active sites.

95
Unlike the effect on active site non-polarity, removing cofactor-binding proteins from
the analysis did not remove the differences in active site aspartic acid composition
between the classes (p = 0.018). The preference for active site glutamic acid over
aspartic acid was still shown for oxidoreductases in the non cofactor-binding set (34%
prefer ASP, whereas 45% prefer GLU). This suggests that the preference for glutamic
acid over aspartic acid in the active sites of oxidoreductases is not due to the fact that
they bind cofactors more often.
2.3.6.1 Rotamers
Glutamic acid has an extra methylene group compared with aspartic acid. We
hypothesised that the preferred usage of glutamic acid over aspartic acid in the
oxidoreductases may be related to increased flexibility in glutamic side chains due to
the longer side chain length, thus being more adapted to transferring protons and
compensating charge around the active site during oxidation/reduction.
We calculated the number of allowed amino acid sidechain rotamers available to each
of the aspartic and glutamic acid side chains in the active sites of the proteins in each
class (see 2.2.6) and divided them by the maximum number of rotamers possible for
that side chain.
On average however, the percentage of rotamers accessible of the maximum available
was larger for aspartic acid than glutamic acid by ~15% for all classes (see Figure 2.20).
There was no significant difference in the percentage of available rotamers allowed
between oxidoreductases and any of the other classes for either aspartic acid (p = 0.35)
or glutamic acid (p = 0.50).
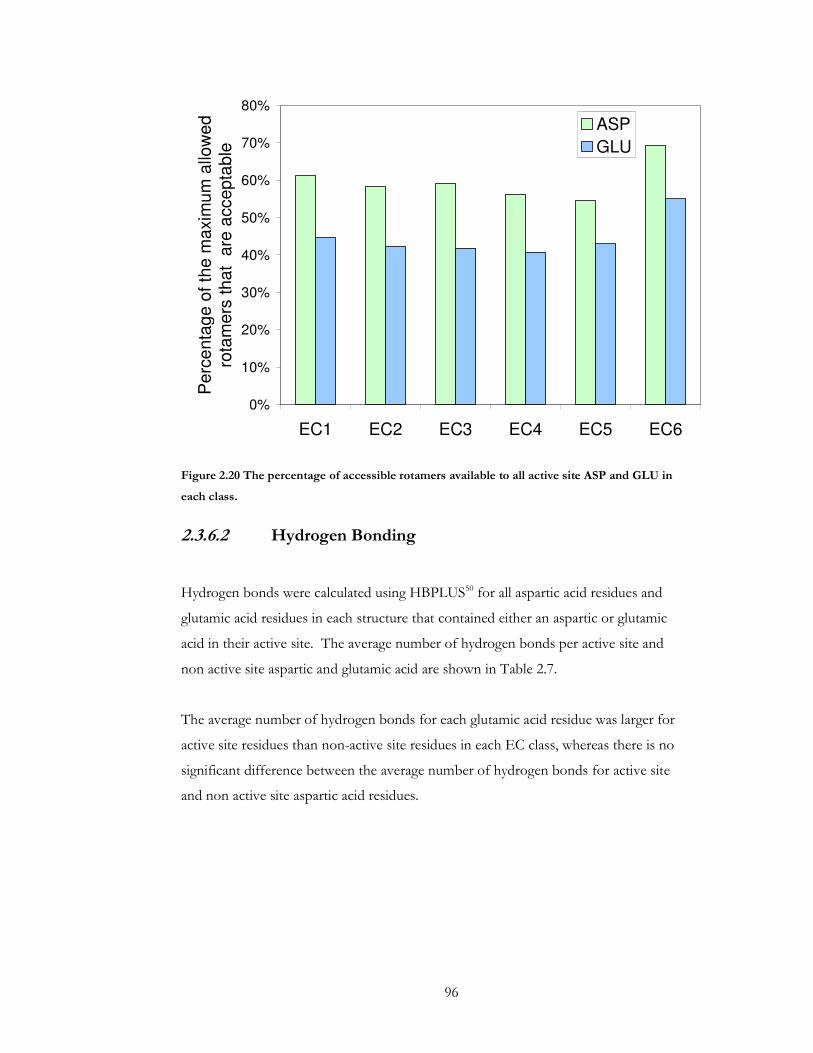
96
0%
10%
20%
30%
40%
50%
60%
70%
80%
EC1 EC2 EC3 EC4 EC5 EC6
Perc
enta
ge o
f th
e m
axim
um
allo
wed
ro
tam
ers
that
are
accepta
ble
ASP
GLU
Figure 2.20 The percentage of accessible rotamers available to all active site ASP and GLU in
each class.
2.3.6.2 Hydrogen Bonding
Hydrogen bonds were calculated using HBPLUS50 for all aspartic acid residues and
glutamic acid residues in each structure that contained either an aspartic or glutamic
acid in their active site. The average number of hydrogen bonds per active site and
non active site aspartic and glutamic acid are shown in Table 2.7.
The average number of hydrogen bonds for each glutamic acid residue was larger for
active site residues than non-active site residues in each EC class, whereas there is no
significant difference between the average number of hydrogen bonds for active site
and non active site aspartic acid residues.

97
EC1 is the only class for which the average number of hydrogen bonds per active site
glutamic acid is significantly larger than average number of hydrogen bonds per active
site aspartic acid. This class also has the largest average number of hydrogen bonds per
active site glutamic acid of all the six classes. This may be related to the fact that
oxidoreductases are the only class to prefer glutamic acid over aspartic acid in their
active site. Figure 2.1 shows the difference in distribution of number of hydrogen
bonds per active site residue between aspartic acid and glutamic acid in EC1.
All Active site Non active site
EC
Average hydrogen bonds per ASP
Average hydrogen bonds per GLU
Average hydrogen bonds per ASP
Average hydrogen bonds per GLU
Average hydrogen bonds per ASP
Average hydrogen bonds per GLU
1 2.95 2.72 2.72 3.32 2.95 2.70
2 2.84 2.52 2.88 2.85 2.84 2.51
3 2.97 2.55 2.85 2.92 2.97 2.53
4 2.93 2.57 2.62 2.96 2.94 2.55
5 2.68 2.54 2.95 3.03 2.67 2.52
6 2.99 2.56 2.20 2.88 3.06 2.54
Table 2.7 Average number of hydrogen bonds per aspartic acid/glutamic acid split by active-
site residues and non-active-site residues
0%
5%
10%
15%
20%
1 2 3 4 5 6 7 8 9Number of hydrogen bonds per
residue
Perc
enta
ge o
f active s
ite
AS
P &
GLU
in E
C1
Asp
Glu
Figure 2.21 The underlying distribution for the number of hydrogen bonds per ASP or GLU in
the active site for EC1.

98
2.4 Conclusions
Previous studies of the properties of enzyme active site residues,18; 49 have focused on
the difference between catalytic or binding pocket residues and other residues, rather
than between active sites of different functions. Other studies14; 15 have shown
differences in structural properties between proteins of different functions, though
these have tended to focus on specific individual functions against all other functions.
To our knowledge, this is the first systematic study of the differences in sequence and
structural features of the six main functional classes of non-evolutionarily related
enzymes and their active sites.
Previous work by Dobson and Doig,16 has shown that global structural features can be
used to distinguish between enzyme functions. The use of non-transparent machine
learning methods in this work made the interpretation of the relationships between
these features and the different functions difficult. Here, we systematically evaluate the
relationship between global attributes and the six main functional classes, as well as
adding active site features. We find numerous features show significant differences
between proteins in the six main classes, and have investigated the relationship
between the most significantly different features and enzyme function, following a
clustering procedure.
Here it is shown that an enzyme’s oligomerisation status differs between the six EC
classes with hydrolases having a significantly larger proportion of monomers and a
lower proportion of oligomers than the other classes. Lyases have a significantly
higher proportion of enzymes existing as oligomers (3 or more chains). The lyases also
have the highest number of oligomers that have active sites located at, or very close to,
subunit interfaces. It was hypothesised that lyases may prefer to have structures that
allow communication between active sites in order to achieve a higher level of
regulation. This was supported by evidence that lyases are indeed over-represented in
comparison to the other classes in the most biochemically important points in the yeast
metabolic network. Conversely, the hydrolases, which contained the lowest proportion
of oligomers, were significantly underrepresented in these highly controlled network
positions.

99
The proportion of the enzyme’s active site that is non-polar differed significantly
between the functional classes. The oxidoreductases showed the highest active site
non-polar proportion, which was found to be related to the oxidoreductase’s
preference for binding cofactors. It may be advantageous for enzymes that bind
cofactors to have non-polar active sites in order to accommodate the non-polar regions
contained in the cofactors. Enzymes that were found to bind cofactors in their crystal
structure were removed from the analyses and a significant difference was no longer
found in the active site non-polarity between the functional classes.
Oxidoreductases also showed unusually low Asp usage in their active sites. This is
unrelated to cofactor-binding, as differences in active site Asp proportions remain
when the cofactor-binding enzymes are removed from the analysis. Indeed, the under-
representation of Asp residues in oxidoreductase active sites was not mirrored by the
other negatively charged residue, Glu. Despite it being reported that Asp is more often
found as an active site residue than Glu,18; 48;49 the oxidoreductases exhibit a preference
for Glu over Asp in their active sites. The other EC classes show the expected
preference for Asp over Glu. We have shown that this possibly relates to active site
glutamic acid residues making a significantly higher average number of hydrogen bonds
in oxidoreductases than any other class. Oxidoreductase was also the only class in
which the active site glutamic acid made significantly more hydrogen bonds per residue
than the active site aspartic acid residues. A study of sidechain rotameric freedom
showed little difference between Asp and Glu, but how rotamers are related to
hydrogen bonding networks remains to be established. An obvious feature of
oxidoreductases is that they require electron transfer, often compensated by proton
movement. It is possible that the preference for Glu over Asp relates to an adaptation
related to charge transfer, but in a complex manner that remains to be established.
Indeed, subsequent to this work being published, a survey of the information
contained in the catalytic mechanism database, MACiE9; 51, revealed that Glu is used as
a catalytic residue in oxidoreductases more often than Asp48. It was also noted that the
most common annotated mechanistic function of catalytic residues in oxidoreductases
is “proton shuffling” and that Glu has a much higher likelihood of acting as a general

100
acid/base and taking part in proton shuffling in oxidoreductases (and all other classes
apart from the ligases) than Asp.
It is discussed here how three of the significantly different features directly may relate
to the enzyme’s catalytic action. There may however be complications in relating every
feature directly to a common function for the top EC class due to the way that the
enzymes are classified in the EC classification. EC classifies enzymes by the overall
reaction which they catalyse, hence enzymes that use similar mechanistic steps to
catalyse different reactions, will be grouped in different classes. Mandalate racemase
(EC5), galactonate dehydratase (EC4) and carboxyphosphonoenolpyruvate synthase
(EC2) have diverse overall reactions, and as such are classified in different EC classes.
They do however, share a common mechanistic step; abstracting the α-proton of a
carboxylic acid to form an enolic intermediate.52 Similarly, enzymes in the same top
EC class are unlikely to share the same complete mechanism. It has been shown that
little mechanistic similarity is common within enzymes at the class level of the EC
hierarchy.53
Structural features, particularly of the active site, may relate to these mechanistic steps
rather than the overall reaction. If a structural feature does relate to a common step
involved in the catalysis of diverse overall reactions, grouping enzymes by their top EC
class would not reveal significant differences in this feature. It therefore does not
mean that features that lack significant differences between EC classes do not relate to
the enzyme’s function.
This systematic study of novel differences in structural features between enzyme
function sheds new light on the relationship between protein structure and function.
This may aid the development of further methods to predict protein function from
structure without the use of alignments and in enzyme design.

101
2.5 References
1. Bray, T., Doig, A. J. & Warwicker, J. (2009). Sequence and structural features of enzymes and their active sites by EC class. J Mol Biol 386, 1423-36.
2. Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). The Protein Data Bank. Nucleic Acids Res 28, 235-42.
3. Bork, P. & Koonin, E. V. (1998). Predicting functions from protein sequences--where are the bottlenecks? Nat Genet 18, 313-8.
4. Iliopoulos, I., Tsoka, S., Andrade, M. A., Enright, A. J., Carroll, M., Poullet, P., Promponas, V., Liakopoulos, T., Palaios, G., Pasquier, C., Hamodrakas, S., Tamames, J., Yagnik, A. T., Tramontano, A., Devos, D., Blaschke, C., Valencia, A., Brett, D., Martin, D., Leroy, C., Rigoutsos, I., Sander, C. & Ouzounis, C. A. (2003). Evaluation of annotation strategies using an entire genome sequence. Bioinformatics 19, 717-26.
5. Barrett, A. J., Canter, C. R., Liebecq, C., Moss, G. P., Saenger, W., Sharon, N., Tipton, K. F., Vnetianer, P. & Vliegenthart, V. F. G. (1992). Enzyme Nomenclature, Academic Press, San Diego, CA.
6. Barthelmes, J., Ebeling, C., Chang, A., Schomburg, I. & Schomburg, D. (2007). BRENDA, AMENDA and FRENDA: the enzyme information system in 2007. Nucleic Acids Res 35, D511-4.
7. Bairoch, A. (2000). The ENZYME database in 2000. Nucleic Acids Res 28, 304-5.
8. Pegg, S. C., Brown, S. D., Ojha, S., Seffernick, J., Meng, E. C., Morris, J. H., Chang, P. J., Huang, C. C., Ferrin, T. E. & Babbitt, P. C. (2006). Leveraging enzyme structure-function relationships for functional inference and experimental design: the structure-function linkage database. Biochemistry 45, 2545-55.
9. Holliday, G. L., Almonacid, D. E., Bartlett, G. J., O'Boyle, N. M., Torrance, J. W., Murray-Rust, P., Mitchell, J. B. & Thornton, J. M. (2007). MACiE (Mechanism, Annotation and Classification in Enzymes): novel tools for searching catalytic mechanisms. Nucleic Acids Res 35, D515-20.
10. Pearl, F. M., Bennett, C. F., Bray, J. E., Harrison, A. P., Martin, N., Shepherd, A., Sillitoe, I., Thornton, J. & Orengo, C. A. (2003). The CATH database: an extended protein family resource for structural and functional genomics. Nucleic Acids Res 31, 452-5.
11. Todd, A. E., Orengo, C. A. & Thornton, J. M. (2001). Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol 307, 1113-43.
12. George, R. A., Spriggs, R. V., Thornton, J. M., Al-Lazikani, B. & Swindells, M. B. (2004). SCOPEC: a database of protein catalytic domains. Bioinformatics 20 Suppl 1, i130-6.
13. Hegyi, H. & Gerstein, M. (1999). The relationship between protein structure and function: a comprehensive survey with application to the yeast genome. J Mol Biol 288, 147-64.
14. Stawiski, E. W., Baucom, A. E., Lohr, S. C. & Gregoret, L. M. (2000). Predicting protein function from structure: unique structural features of proteases. Proc Natl Acad Sci U S A 97, 3954-8.

102
15. Stawiski, E. W., Mandel-Gutfreund, Y., Lowenthal, A. C. & Gregoret, L. M. (2002). Progress in predicting protein function from structure: unique features of O-glycosidases. Pac Symp Biocomput, 637-48.
16. Dobson, P. D. & Doig, A. J. (2005). Predicting enzyme class from protein structure without alignments. J Mol Biol 345, 187-99.
17. Laskowski, R. A., Watson, J. D. & Thornton, J. M. (2005). Protein function prediction using local 3D templates. J Mol Biol 351, 614-26.
18. Bartlett, G. J., Porter, C. T., Borkakoti, N. & Thornton, J. M. (2002). Analysis of catalytic residues in enzyme active sites. J Mol Biol 324, 105-21.
19. Amitai, G., Shemesh, A., Sitbon, E., Shklar, M., Netanely, D., Venger, I. & Pietrokovski, S. (2004). Network analysis of protein structures identifies functional residues. J Mol Biol 344, 1135-46.
20. Goyal, K., Mohanty, D. & Mande, S. C. (2007). PAR-3D: a server to predict protein active site residues. Nucleic Acids Res 35, W503-5.
21. Bate, P. & Warwicker, J. (2004). Enzyme/non-enzyme discrimination and prediction of enzyme active site location using charge-based methods. J Mol Biol 340, 263-76.
22. Elcock, A. H. (2001). Prediction of functionally important residues based solely on the computed energetics of protein structure. J Mol Biol 312, 885-96.
23. Ko, J., Murga, L. F., Wei, Y. & Ondrechen, M. J. (2005). Prediction of active sites for protein structures from computed chemical properties. Bioinformatics 21 Suppl 1, i258-65.
24. Wei, L. & Altman, R. B. (1998). Recognizing protein binding sites using statistical descriptions of their 3D environments. Pac Symp Biocomput, 497-508.
25. Porter, C. T., Bartlett, G. J. & Thornton, J. M. (2004). The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res 32, D129-33.
26. Chandonia, J. M., Hon, G., Walker, N. S., Lo Conte, L., Koehl, P., Levitt, M. & Brenner, S. E. (2004). The ASTRAL Compendium in 2004. Nucleic Acids Res 32, D189-92.
27. Murzin, A. G., Brenner, S. E., Hubbard, T. & Chothia, C. (1995). SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247, 536-40.
28. Kabsch, W. & Sander, C. (1983). Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577-637.
29. Hutchinson, E. G. & Thornton, J. M. (1996). PROMOTIF--a program to identify and analyze structural motifs in proteins. Protein Sci 5, 212-20.
30. Kyte, J. & Doolittle, R. F. (1982). A simple method for displaying the hydropathic character of a protein. J Mol Biol 157, 105-32.
31. Rice, P., Longden, I. & Bleasby, A. (2000). EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16, 276-7.
32. Miller, R. G. (1981). Simultaneous Statistical Inference. 2nd edit, Springer-Verlag, New York.
33. Holm, S. (1979). A simple sequentially rejective multiple test procedure. Scand. J. Statist. 6, 65--70.
34. Benjamini, Y. & Hochburg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B. Methodological 57.

103
35. Perneger, T. V. (1998). What's wrong with Bonferroni adjustments. Bmj 316, 1236-8.
36. Farcomeni, A. (2008). A review of modern multiple hypothesis testing, with particular attention to the false discovery proportion. Stat Methods Med Res 17, 347-88.
37. Levin, B. (1996). On the Holm, Simes, and Hochberg multiple test procedures. Am J Public Health 86, 628-9.
38. Curran-Everett, D. (2000). Multiple comparisons: philosophies and illustrations. Am J Physiol Regul Integr Comp Physiol 279, R1-8.
39. Cole, C. & Warwicker, J. (2002). Side-chain conformational entropy at protein-protein interfaces. Protein Sci 11, 2860-70.
40. Koehl, P. & Delarue, M. (1994). Application of a self-consistent mean field theory to predict protein side-chains conformation and estimate their conformational entropy. J Mol Biol 239, 249-75.
41. Jones, S. & Thornton, J. M. (1996). Principles of protein-protein interactions. Proc Natl Acad Sci U S A 93, 13-20.
42. Bogan, A. A. & Thorn, K. S. (1998). Anatomy of hot spots in protein interfaces. J Mol Biol 280, 1-9.
43. Chou, P. Y. & Fasman, G. D. (1974). Conformational parameters for amino acids in helical, beta-sheet, and random coil regions calculated from proteins. Biochemistry 13, 211-22.
44. Pace, C. N. & Scholtz, J. M. (1998). A helix propensity scale based on experimental studies of peptides and proteins. Biophys J 75, 422-7.
45. Goodsell, D. S. & Olson, A. J. (2000). Structural symmetry and protein function. Annu Rev Biophys Biomol Struct 29, 105-53.
46. Rahman, S. A., Advani, P., Schunk, R., Schrader, R. & Schomburg, D. (2005). Metabolic pathway analysis web service (Pathway Hunter Tool at CUBIC). Bioinformatics 21, 1189-93.
47. Rahman, S. A. & Schomburg, D. (2006). Observing local and global properties of metabolic pathways: 'load points' and 'choke points' in the metabolic networks. Bioinformatics 22, 1767-74.
48. Holliday, G. L., Mitchell, J. B. & Thornton, J. M. (2009). Understanding the functional roles of amino acid residues in enzyme catalysis. J Mol Biol 390, 560-77.
49. Tseng, Y. Y. & Liang, J. (2007). Predicting enzyme functional surfaces and locating key residues automatically from structures. Ann Biomed Eng 35, 1037-42.
50. McDonald, I. K. & Thornton, J. M. (1994). Satisfying hydrogen bonding potential in proteins. J Mol Biol 238, 777-93.
51. Holliday, G. L., Bartlett, G. J., Almonacid, D. E., O'Boyle, N. M., Murray-Rust, P., Thornton, J. M. & Mitchell, J. B. (2005). MACiE: a database of enzyme reaction mechanisms. Bioinformatics 21, 4315-6.
52. Babbitt, P. C., Hasson, M. S., Wedekind, J. E., Palmer, D. R., Barrett, W. C., Reed, G. H., Rayment, I., Ringe, D., Kenyon, G. L. & Gerlt, J. A. (1996). The enolase superfamily: a general strategy for enzyme-catalyzed abstraction of the alpha-protons of carboxylic acids. Biochemistry 35, 16489-501.
53. O'Boyle, N. M., Holliday, G. L., Almonacid, D. E. & Mitchell, J. B. (2007). Using reaction mechanism to measure enzyme similarity. J Mol Biol 368, 1484-99.

104
Chapter 3: Functional site identification in
proteins
Further work in this thesis attempts to predict enzyme function based on features of
the whole protein and those specifically relating to the active site. Using features of
functional sites to predict function seems logical since it is the part of the protein
which is arguably most important to the protein’s function. Whilst other parts of the
protein may have roles such as stabilising or trafficking, the functional site should
contain more detailed information about the specific function of the protein.
Active site information is very unlikely to be known for enzymes of unknown function,
therefore in order to calculate the features relating to active sites in the function
prediction method (see Chapter 4) the location of the active site must first be
identified. In this chapter current methods for the prediction of functional site
location are analysed and the SitesIdentify webserver, which delivers two of these
methods, is presented. The best-performing tool will then be used to identify active
sites on enzymes with no functional site annotation in order to calculate active-site
features for use in a functional prediction method (Chapter 4).
The content of the chapter was published in BMC Bioinformatics1. The author of this
thesis was the first author of this paper, and the contributions of the other non-
supervisor authors are detailed below.
Pedro Chan: Supplied some of the code to provide the webserver with basic
functionality such as integrity checking of input and automatic email notifications. He
also wrote the code to annotate structures with their conservation score.
Salim Bougouffa: The webserver layout and style template is based on another of this
group’s webtools produced by Salim.
Richard Greaves: The original author of the method behind SitesIdentify. The paper
reporting this method has been referenced where appropriate.

105
3.1 Introduction: Computational approaches for the
prediction of functional sites
Efforts, primarily by structural genomics groups, have provided a rapidly growing
number of protein structures with little or no functional annotation. This has caused
new interest in the relationship between structure and function and has increased focus
on ways to elucidate a protein’s function from its structure rather than solely from
sequence. In order to investigate the role of a protein using its structure, it is useful to
be able to identify the portion of the protein that is most closely involved with its
function.
It is first important to point out that there is ambiguity in what is meant by a
‘functional site’. In enzymes, the functional site is generally considered to be its active
site, which is where the enzyme’s ligand is bound. Other proteins, such as G protein-
coupled receptors, bind a ligand on their extracellular N terminal end and elicit their
response via their intracellular C terminal G protein binding domain. In this case, the
site which actually elicits its function is separate from the ligand binding site. Indeed,
both the ligand binding site and the G protein coupled site could be considered to be
functional sites. It is also worth noting that some proteins, such as structural proteins
have no obvious discrete functional site. Enzymes lend themselves to functional site
analysis since they have well defined functional sites that are defined by their catalytic
residues.
There are currently several computational approaches that predict functional sites
which use either structural or sequence information. The most widely used methods
rely on sequence information in order to predict functionally important residues, due to
the greater availability of sequence data as opposed to structural data for
uncharacterised proteins. Sequence based methods mainly centre around the concept
of functionally important residues being more highly conserved through evolution and
identify the most conserved residues by comparing positions in a multiple sequence
alignment with homologous proteins. Some methods use only sequence conservation
information in making predictions 2; 3, whilst others also include additional computed
sequence features4, or structural properties predicted from sequence such as predicted
secondary structure and solvent accessible surface area 5; 6, particularly in order to

106
distinguish between residues conserved for function and those conserved for structure 7; 8. Many methods focus on predicting catalytic residues in enzyme active sites, but
measures of sequence conservation have also been successfully used to predict residues
in contact with a ligand 2; 6; 9 or in contact with other proteins, although sequence
conservation has been shown to perform less well as a predictive feature in the latter
cases 2; 10.
Whilst there are a large number of sequence-based methods available, there are also a
growing number of methods that predict functional sites based on structural
information. These methods fall into two main categories: those that identify structural
similarities and transfer annotation from a protein with a known functional site and
those that predict functional sites by non-homology related structural features such as
geometrical or electrostatic properties 6; 11; 12.
There are many resources that store structural and sequence information about
proteins with known active sites, such as PdbFun 13, CSA 14, PDBSite 15 and ProSite 16.
A protein of unknown active site location can be compared to these resources (CSS 17
scans the CSA and PDBSiteScan 18 scans PDBSite), or to databases derived specifically
for the prediction method, to identify any structural similarities with known active sites 19; 20; 21; 22; 23; 24; 25; 26. While these methods often produce accurate results, they assume the
existence of a functionally annotated homologue of similar active site structure in their
respective databases. As one of the aims of structural genomics initiatives is to obtain
structures for proteins that occupy remote fold space, these methods may be of limited
use for such proteins.
In this situation, ab initio methods that do not rely on the existence of a functionally
characterised homolog may be of more value. A wide range of structural properties
have been used, showing that the relationship between a protein’s structure and its
function is affected by many structural characteristics. A study of catalytic residues and
their properties 27 showed that they have a low propensity to exist in helix or sheet
secondary structure, have a higher propensity to be a charged residue and exhibit lower
B-factor values than non-catalytic residues. A number of methods have used these
characteristics to predict residues involved in catalysis 28; 29. Bartlett et al. noted that
catalytic residues tend to line the surface of large surface clefts, yet remain relatively

107
buried within the protein geometry. It was also observed in a study of 67 single-chain
enzymes that 83% of enzyme active sites are found in the largest surface cleft 30,
resulting in methods to predict active sites by finding surface clefts 31; 32.
Previous work by this group 33 attempted to identify functional sites by locating peak
electrostatic potentials near to the surface of a protein resulting from the interaction of
charged residues that are under electrostatic strain. The greatest functional site
prediction accuracy, however, was obtained by applying a uniform charge weighting
across the protein rather than using actual charges. This uniform charge weighting
essentially acts as a cleft-finding algorithm and will predict the most buried surface
cleft. This gave a prediction accuracy of 77%, where a successful prediction is when
the peak potential was within 5% of the protein surface from the real active site centre.
Other studies have successfully used electrostatics calculations to predict active site and
ligand-binding site residues 34; 35; 36; 37; 38. Elcock identified residues that had destabilising
effects on the stability of the protein using continuum electrostatics methods and
found that these correlated with residues involved in protein functionality 34. This
method, however, was not tested on a large experimentally annotated dataset and so it
is hard to interpret the degree of accuracy it achieved. Another approach predicts
enzyme active sites by identifying residues with unusually-shaped titration curves 36; 39 as
well as predicting enzyme function 40. Other chemistry-based approaches, such as
identifying residues that are unusually hydrophobic for their position in a structure
have also been successful 41.
Other ab initio methods use the degree of connectivity of residues to predict those
involved in function. A number of methods assess the closeness centrality of residues 42; 43; 44, whilst one study found that catalytic residues are more likely to exist in close
proximity to the molecular centroid 45.
Perhaps the best accuracies can be achieved by combining structural approaches and
sequence conservation. Residues may be evolutionarily conserved due to structural as
well as functional constraints and a number of studies have attempted to distinguish
these two factors by considering the degree of conservation and the residue’s structural
environment 7; 46. Mapping the degree of evolutionary conservation onto the structure

108
is useful in identifying clusters of conserved residues in the structure that may indicate
a functional site 47; 48. Combining the types of structural information used in ab initio
structural methods with sequence conservation can be effective 11; 12; 35; 49; 50.
Despite the success of the large number of varied approaches, only a relatively small
subset of these methods are currently available either via a software package or a web-
server. Tools report various levels of accuracy that are difficult for a user to compare
due to their separate test datasets, outputs and reporting methods. Here we present a
user-friendly functional site prediction tool, SitesIdentify, based on previously
published work by this group 11; 33. This is made publicly available via a web-server
(www.manchester.ac.uk/bioinformatics/sitesidentify)51, and is compared to other
accessible tools in a comparison of performance on two common datasets.

109
3.2 Methods: Benchmarking the Accuracy of Functional
Site Identification Tools
3.2.1 Selection of Prediction Methods
There have been many functional site prediction techniques published in the literature,
however for this analysis it was essential to have access to the method in order to apply
it to a common dataset. Only published techniques with the method available for
download or via a webserver were therefore applicable to this study.
In order to be included in this analysis, a method had to adhere to the following
criteria:
• The method must require no prior knowledge about the active site.
• It produces output that identifies the active site either by a coordinate location,
the identities of catalytic residues or identities of residues found in the binding
site.
• It produces results within a reasonable time scale. The method should return
results for a test protein with 330 residues in 10 minutes or less.
• It does not simply access known annotation about the test protein.
The applications that met these criteria are listed in Table 3.1. Other applications that
were considered but were not included in this study, along with the reason for not
including them, are listed in Table 3.2.
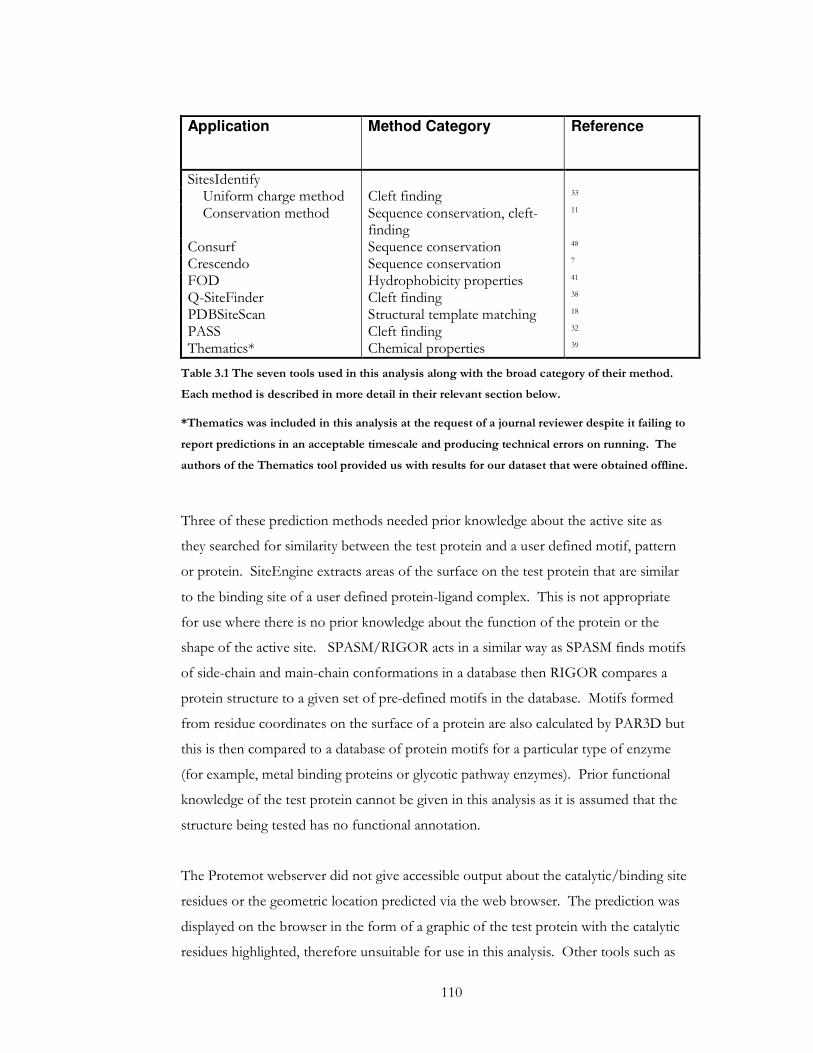
110
Application
Method Category Reference
SitesIdentify Uniform charge method Cleft finding 33 Conservation method Sequence conservation, cleft-
finding 11
Consurf Sequence conservation 48 Crescendo Sequence conservation 7 FOD Hydrophobicity properties 41 Q-SiteFinder Cleft finding 38 PDBSiteScan Structural template matching 18 PASS Cleft finding 32 Thematics* Chemical properties 39
Table 3.1 The seven tools used in this analysis along with the broad category of their method.
Each method is described in more detail in their relevant section below.
*Thematics was included in this analysis at the request of a journal reviewer despite it failing to
report predictions in an acceptable timescale and producing technical errors on running. The
authors of the Thematics tool provided us with results for our dataset that were obtained offline.
Three of these prediction methods needed prior knowledge about the active site as
they searched for similarity between the test protein and a user defined motif, pattern
or protein. SiteEngine extracts areas of the surface on the test protein that are similar
to the binding site of a user defined protein-ligand complex. This is not appropriate
for use where there is no prior knowledge about the function of the protein or the
shape of the active site. SPASM/RIGOR acts in a similar way as SPASM finds motifs
of side-chain and main-chain conformations in a database then RIGOR compares a
protein structure to a given set of pre-defined motifs in the database. Motifs formed
from residue coordinates on the surface of a protein are also calculated by PAR3D but
this is then compared to a database of protein motifs for a particular type of enzyme
(for example, metal binding proteins or glycotic pathway enzymes). Prior functional
knowledge of the test protein cannot be given in this analysis as it is assumed that the
structure being tested has no functional annotation.
The Protemot webserver did not give accessible output about the catalytic/binding site
residues or the geometric location predicted via the web browser. The prediction was
displayed on the browser in the form of a graphic of the test protein with the catalytic
residues highlighted, therefore unsuitable for use in this analysis. Other tools such as

111
Crescendo and FOD did not explicitly give residue identities or geometric locations but
gave per-residue scores related to their measures. Predictions were then made by
taking a set of top-scoring residues from the output (see the description of each
method below for the criteria).
Due to the large number of proteins used in the test set (237 enzymes), the time taken
to compute predictions is important to this analysis. Five of the methods took
inappropriate amounts of time to compute results, these usually included methods that
had some conservation-related aspect to them. FrPred uses conservation information
and was problematic to run as it could only handle proteins with less than 500 residues.
Even proteins with a length less than this, however, took a large amount of time to
run. PinUP also uses conservation scoring in its method and thus took unsuitable
lengths of time to compute. SuMo compares the test proteins to all ligand binding sites
in the PDB database. It gives the option of using only part of the test protein and/or a
subset of PDB files to search against, however, using the whole protein and
exhaustively searching against the whole PDB database takes a prolonged amount of
time.
PDBSiteScan, whilst used in this analysis, transfers annotation from similar proteins in
the PDB. As this test set contains proteins with known active site information it is
likely to retrieve information from the test protein in the PDB database. In this
analysis, a prediction from PDBSiteScan has been removed if the information was
obtained from the same structure as the test structure. Another method, CSS, scans
the CSA in order to compare the test protein to proteins with annotation in the CSA.
It was deemed unsuitable for this analysis since the test set is derived from the CSA
and so the top active site prediction for each protein in the test set was annotation
transferred from itself.

112
The remaining six prediction methods (CrPred, FSPS, MFS, PINTS, PvSOAR and
SARIG) could not be included in the analysis for technical reasons. CrPred’s
predictions were pre-computed for specific datasets of structurally related proteins.
This was deemed unsuitable for the methods later use in a function prediction method.
The web server was inaccessible for PvSOAR, whilst the PINTS web server gave
errors and the web page containing SARIGs output also gave errors. The web server
for MFS was unreliable and often gave error messages, whilst the command-line
download option did not give a single prediction program but a list of standalone
feature calculation programs with no instruction as to order of running or
interpretation of output.

113
Name Reference publication Reason for non-inclusion in analysis
CrPred Zhang et al., (2008) Technical reasons. CSS Torrence et al. (2005) Scans test set. FrPred Fischer et al. (2007) Processing time.
(could only process <500 residues)
Functional Site Prediction Server (FSPS)
Cheng et al. (2005) Technical reasons.
MFS Wang et al. (2008) Technical reasons. Par3D Goyal et al. (2007) Prior knowledge needed PINTS Stark and Russell (2003) Technical reasons. PinUP Liang et al. (2006) Processing time. Protemot Chang et al. (2006) Cannot process results PvSOAR Binkowski et al. (2004) Technical reasons. SARIG Amitai et al. (2004) Technical reasons. SiteEngine Shulman-Peleg et al. (2005) Prior knowledge needed SPASM/RIGOR Kleywegt (2005) Prior knowledge needed SuMo Jambon et al. (2005) Processing time.
Table 3.2: Functional site prediction tools not included in the comparison analysis. Reasons
for non-inclusion in the analysis are further explained below:
Technical reasons. Web-servers that produced errors on attempting to submit a protein or
accessing results pages were not included.
Prior knowledge needed. These prediction methods needed prior knowledge about the active
site as they search for similarity between the test protein and a user defined motif, pattern or
protein.
Cannot process results. The results are not given in a form that can be automatically processed
(in this case the prediction was displayed as a graphic of the test protein with the catalytic
residues highlighted).
Processing time. Due to the large number of proteins used in the test set (237), the time taken
to compute predictions is important to this analysis. A tool was excluded if results were not
returned within 10 minutes for an example test protein of 330 residues (PDBID: 12as).
Scans test set. CSS scans the CSA in order to compare the test protein to proteins with
annotation in the CSA. It was deemed unsuitable for this analysis since the test set is derived
from the CSA.

114
3.2.2 Creation of Test Sets
Two datasets are used in this analysis; an enzyme set and a non-enzyme set. As
mentioned previously, enzymes are usually used to test functional site predictors due to
their functional sites being easily defined by the location of their catalytic residues. The
primary dataset that these methods are tested on is the enzyme dataset, however to
assess each method’s applicability to other types of proteins a non-enzyme dataset was
also gathered.
The enzyme dataset was gathered from the 880 literature annotated entries in version
2.2.1 of the Catalytic Sites Atlas (CSA) database14. In order to reduce bias towards
methods that are particularly good at classifying a particular enzyme (and its related
homologues) it was important to remove redundancy from this dataset. The set of 880
proteins were culled for redundancy on the basis that no two enzymes shared an active
site-containing domain from the same SCOP superfamily with any other protein of a
lesser structural quality. This procedure is described in more detail in Chapter 2,
however in this analysis the enzymes were not split into EC classes.
The resultant dataset contained 237 enzymes, each having one or more annotated
active sites (see Table 3.3). This gave a total of 747 catalytic residues with an average
of 3.2 catalytic residues per site per protein.

115
1ssx 1h2r 1dxe 1gpr 2plc 1b65 1eb6 1gcu 1c3c 1f7l 1wgi 1al6 1p1x 1g6t 1bp2 1c3j 1r16 1abr 1qj4 1dw9 2jcw 1bg0 1pa9 12as 1qv0 2xis 2acy 1sox 1oas 1rbl 1itx 1ru4 1qrg 1qcn 1nsp 1qd6 2nlr 1d3g 1qaz 1gog 1nid 1pya 1nww 1uaq 1e1a 1fua 1m9c 1pfk 1qtn 1s95 1cg6 1d6o 1gpj 1nir 1o9i 1r6w 1uro 1d0s 1eh6 1n20 1e7l 1qq5 1tys 1bs4 1e2a 1nn4 2tps 1moq 1tml 1b93 1bou 1mvn 2pth 1lam 1j79 1apy 1a05 1mqw 1vlb 1jnr 1foa 1a4i 2cpo 1ef0 1qje 1dl2 1chd 1a2t 1yve 1cd5 1fy2 1cs1 1r51 1mrq 1nml 1r4f 1dbf 1aop 1pgs 1l1d 1lci 1q3q 1nlu 1v0y 1p4n 1kp2 1f75 1ndo 1rhs 1qgx 1oe8 1jhf 1bol 1f8x 1hdh 1eug 1lbu 1jdw 1aj0 1dhf 2eng 7odc 1jh6 1i6p 1otg 1cev 1c0k 1uqr 1j53 1chm 1k4t 3nos 135l 1qhf 1j09 1akd 1fro 3mdd 7atj 1qd1 1g72 1oj4 1f2v 2toh 1pyl 1p3d 1dup 1oac 1dmu 1d8h 1nln 1o04 1d4a 1hxq 1c2t 1a79 1nf9 1gqg 1cgk 1vid 1bwp 1vas 1dj0 1d1q 2sqc 1pii 3eca 1jms 1oyg 1ako 1tph 1js4 2ypn 1dve 1mla 2bbk 1pud 1do6 1uam 1dqa 1m6k 2apr 1m21 1aug 1lij 1c9u 1e19 2ahj 1l1l 1a95 1lba 1b5t 1qh5 1j7g 1g24 1nhx 1k82 1qpr 1dci 1i19 1daa 1hrk 1h3i 1dco 1p5d 1dae 1uf7 1g79 1ez1 1r76 1ah7 1rhc 1ro7 1fgh 1dqs 1bt1 1snn 3cla 1mka 1ecm 1dnp 1jm6 1l6p 1mud 1k30 1ecl 1dio 1hka 1kaz 1jfl 1d8c 1ca3 1hfe 1fnb 1ir3 1d2t 1brw Table 3.3 The PDB codes for the 237 structures in the enzyme dataset

116
As in previous work (see Chapter 2)52, active site residues were defined by taking
residues that had >5Å2 solvent accessible surface area (SASA) and had at least one
atom within a 10Å radius of a point defined by taking the geometric average of the Cβ
coordinates (Cα for glycine) of the annotated catalytic residues for that protein.
It is difficult to construct datasets of well-definied functional sites for non-enzymes
since “functional sites” are defined differently depending on the function of the
protein. For example, a GPCR can be thought of as having multiple functional sites
(the G-protein binding site and the ligand binding site) and structural proteins, such as
fibrilin don’t have any self-contained functional site. The dataset containing non-
enzymes was formed by taking the non-enzymes from the dataset used by Laurie and
Jackson38 to test their functional site predictor, Q-SiteFinder. Of the 134 proteins
listed in their publication, 31 were non-enzymes. These were then put through the
same culling procedure as for the enzyme test set, which resulted in 13 remaining
proteins (see Table 3.4). The functional site residues were defined in a similar way to
the active site residues in the enzyme set, however for this dataset the annotated
catalytic residues from the CSA are replaced with the residues that are listed as being
within van der Waals contact or hydrogen-bonded to the ligand (as listed by the
PDBeMotif Ligand Environment database53).
1lic
1abe
1mrk
1eta
1lst
1srj
1wap
1nco
1a71*
1tyl
1igj
1ctr
2plv
Table 3.4 The PDB codes for the 13 structures in the non-enzyme dataset.
*1a71 replaces 1slt from the Laurie and Jackson paper as 1slt had no ligand bound in the PDB
structure.

117
3.2.3 Obtaining and Unifying Functional Site Predictions
The predictions for each method were obtained by automatically running the dataset
through each webserver and capturing the output via a perl-CGI script for
QSiteFinder, Consurf, PDBSiteScan and FOD. Thematics and Crescendo were unable
to be run automatically online for large datasets and therefore the results were provided
by their respective groups from offline runs. Due to availability of source code, results
from SitesIdentify and PASS were obtained from running the code locally. The
asymmetric unit structure file was supplied to each method on the basis that a newly
solved structure of a protein of unknown function would be unlikely to have any clear
indication of its true in vivo quaternary structure. Some methods can only deal with one
chain and where this is the case only the first chain in the file has been passed to the
method.
SitesIdentify and PASS give predictions by specifying PDB geometry coordinates
relating to the centre of the active site, which is used directly as the centroid for further
defining active site residues. For methods that give output in the form of a number of
predicted residues (whether that be catalytic only in the case of enzymes or a set of site
environment residues) the centroid is calculated by averaging the Cβ atom coordinates
(Cα for glycine). The standardised predicted residues used to assess the prediction
accuracy of each method are defined as those that have at least one atom within a 10Å
radius of this centroid and have a SASA of 5Å2 or more. This provides standardised
output that can be fairly compared between the different methods.
Prediction accuracy can be measured in a number of different ways; the most simple is
measuring the linear distance between the real site centroid and the predicted centroid.
This measure may, however, be misleading due to the geometry of the functional site.
The predicted and real centroid may be some distance apart whilst the environment
around each centroid may contain most of the same residues, therefore identifying the
same site.
It is possibly more important to consider how many of the biologically active residues
(i.e. catalytic in the case of enzymes and ligand-binding in non-enzymes) are recalled as

118
predicted site residues by each method. It is important to note however, that in the
enzyme dataset the active site residues defined using the CSA generated centroid do
not recall 100% of the CSA annotated catalytic residues (see Figure 3.2 and Table 3.5).
It is therefore unfair to evaluate a method by its absolute CSA/ligand-binding residue
recall rate (termed the absolute recall rate). It is more realistic to compare its
CSA/ligand-binding residue recall rate to the recall rate achieved by the real centroid
(termed the relative recall rate).
In the previous analysis of structural and sequence features of active sites (Chapter 2),
many features were found that differed significantly between enzyme functions. These
features were calculated on the active site residues defined by the CSA generated
centroid. The best performing method from this analysis will go on to be used to
predict active site residue sets of unknown proteins in order to calculate values for
features in the same way as in the previous study. The number of residues that are
shared between the real active site residue set and the predicted active site residue set is
therefore of interest. It should not be taken as a definite measure of a method’s
accuracy as it has limited relevance outside of the use of this study.
It is also important to consider which site on a protein is being predicted. There may
be more than one genuine active site on a given structure, particularly where there are
symmetrical chains. The CSA annotation deals with this by providing a number of site
entries per protein. When a site is predicted by a method, it is assessed to see which
real site listed in the CSA the predicted site is closest to. It would be unfair to compare
a prediction simply to the first site in the CSA if it happened to be on an opposite
symmetrical chain as this would produce a falsely erroneous result (see Figure 3.1).
Similarly in the non-enzyme dataset, where there is more than one identical site there
will be multiple bound ligands in the PDB file. Ligand-binding residues for these
multiple ligands are split into their respective sites.

119
Figure 3.1 The asymmetric unit structure of 1daa.
Chain A is shown in yellow and chain B is shown in blue. The CSA annotated residues for its
two separate sites are shown in green with the active site centroid coordinates predicted shown
in red. It can be seen that if the predicted coordinates are compared to the first site in the CSA,
it would give a poor prediction. In reality the predicted coordinates are very close to the second
site given in the CSA and therefore should be compared to the centroid of the second site
instead of the first. This demonstrates the importance of comparing predictions to all CSA
annotated sites.

120
3.3 Methods: SitesIdentify Webserver
3.3.1 Functional Site Prediction Methods
SitesIdentify can predict functional site location by two separate approaches, which
have been published previously11; 33 The first method essentially identifies buried clefts
on the surface of the protein via electrostatics calculations33. In a previous publication
by Bate and Warwicker, a number of electrostatic properties were used to attempt to
identify active sites in enzymes. A 2Å grid was placed over the protein and the
electrostatic potential from the atoms contained within the neighboring grid volumes is
calculated for each second nearest grid point to the protein’s surface. The electrostatic
potential was calculated firstly by assigning charges to all ionisable residues based on
model pKas at pH 5.5, pH 7 and irrespective of pH, and secondly by applying a
uniform charge density to all Cα atoms from all non-hydrogen residues. The electric
potential is calculated at each grid point using finite difference Poisson-Boltzmann
(FDPB) calculations and the grid point that had the greatest peak potential was
predicted to be the active site centre coordinate.
The peak-potential calculations from applying estimated charges from pKa didn’t,
however, perform as well as the simpler uniform charge density method. The peak
potential from the uniform charge method therefore essentially identifies the most
buried cleft on the protein surface. Here, this method is termed SitesIdentify(GM),
where GM stands for geometric.
The second method of SitesIdentify presented here is based on the uniform charge
method but the charges are weighted with normalised conservation scores that reflect
the amino acid/sterochemical diversity and the gap occurrence of that residue (see
Equation 3.1). Close homologues are found by running the sequence through PSI-
BLAST with an E value cut-off of 1e-20 and then a profile is created from which the
conservation score is calculated. The peak potential on the grid is then calculated by
FDPB calculation by using the conservation-weighted charges on each residue. This

121
method of Sitesidentify is called here, SitesIdentify(ConsGM) as it essentially identifies
the most conserved buried cleft.
Once the coordinates of the grid point with the peak potential (from both SitesIdentify
methods) has been identified, a sphere of a user-defined radius (the default is 10Å) is
drawn around the coordinate and residues that have at least one atom within the
sphere and exist on the surface of the protein (having at least 5Å2 of SASA) are
identified as predicted site residues.
Equation 3.1 The equation for the conservation score of residue x, which is used to weight the
uniform charge.
t is the normalised symbol diversity, r is the normalised stereochemical diversity (based on the
BLOSUM-62 matrix) and g the gap cost. Each of these terms are weighted by integral values
ranging between 0 and 5 (α, β and γ), the values for which are defined as those giving the best
predictive performance in the original publication11.

122
3.3.2 SitesIdentify Workflow
Upon submission of a job, SitesIdentify starts a number of programs depending on
which method the user requested. If the conservation approach is selected, the in-
house Conserved Residue Colouring program(CRC) is run first, which identifies
homologues by running the sequence contained in the SEQRES records in the PDB
file through PSI-BLAST 54. PSI-BLAST is run for one iteration (in default settings) on
the non-redundant database with an E-value cut-off for inclusion of sequences of 1e-
20. A profile file containing the conservation scores for each residue is produced.
SitesIdentify uses the conservation scores as charge weightings on a single atom for
each amino acid (Cβ or Cα for glycine), and calculates the location of the peak potential
as described above 11.
If no homologue can be identified for a protein using CRC then the method
automatically switches to only charge-based calculations. If the conservation method is
not selected then the CRC program is omitted and the location of the peak potential is
calculated using the uniform charge-weighting method 33. A sphere of user-supplied
radius is drawn around the predicted centroid coordinates and residues are selected that
have at least one atom within that sphere and also exhibit more than 5Å2 of solvent-
accessible surface area (SASA) as calculated using the Lee and Richards method 55. This
list of residues represents the predicted functional site, which is given on the results
page as a text list and also highlighted on the PDB structure using Jmol 56 .
3.3.3 SitesIdentify Usage
SitesIdentify is available for use via a web browser and is freely accessible without
license or an account registration. The main web page allows a user to enter either a
pre-existing PDB structure ID (and whether to use the biological unit or the
asymmetric unit) or upload a structure file, the radius around the predicted site to use,
the method to use and an email address so that a user can be notified and emailed the
results link upon job completion.

123
If a user has submitted their own structure file then this is validated to ensure that
contains an acceptable PDB-format structure, the rules for which are given in the user
guide available from the website. The file must be less than 2MB in size and contain
only text. It also must contain at least SEQRES and ATOM records and be spaced
exactly as the standard PDB format. If the user-supplied information is invalid (non-
existent PDB ID or invalid email address) then the job is not initialised and the user
informed of the incorrect information via the browser. Upon successful completion of
a job the web-server directs the user to the results page and also sends an email to the
user at the address specified with a link to the results page.

124
3.4 Results: Benchmarking the Accuracy of Functional Site
Identification Tools
3.4.1 Recall Accuracy Rates for Real Sites
The criteria for the definition of functional site residues (see 3.2.2) recalled 544 of the
total 747 catalytic residues in the enzyme dataset and 52 of the 80 (65%) ligand-binding
residues in the non-enzyme dataset (see Table 3.5). The average recall rate per protein
was 76.1% in the enzyme set and 71.6% in the non-enzyme set. Figure 3.2 shows how
the recall rates were distributed for the 237 proteins in the enzyme set and the 13
proteins in the non-enzyme set. Surprisingly, 15 (6.3%) enzymes recalled no catalytic
residues with the definition criteria. This was due to their annotated residues having
less than 5Å2 solvent accessible surface area. It is known that catalytic residues may
not exist on the surface of an enzyme active site27, however it was deemed unsuitable to
allow residues underneath the surface to be classed as active site residues as it would
introduce too many non-catalytic and non-binding residues into the selection.
An example of an enzyme in the set where the active site definition criteria did not
recall any of the CSA annotated residues is 1O9I, a manganese catalase. It has one
residue annotated as catalytic in the CSA, a glutamic acid at position 178. It doesn’t
meet the active site definition criteria as GLU 178 only has 1.5Å2 of solvent accessible
surface area.
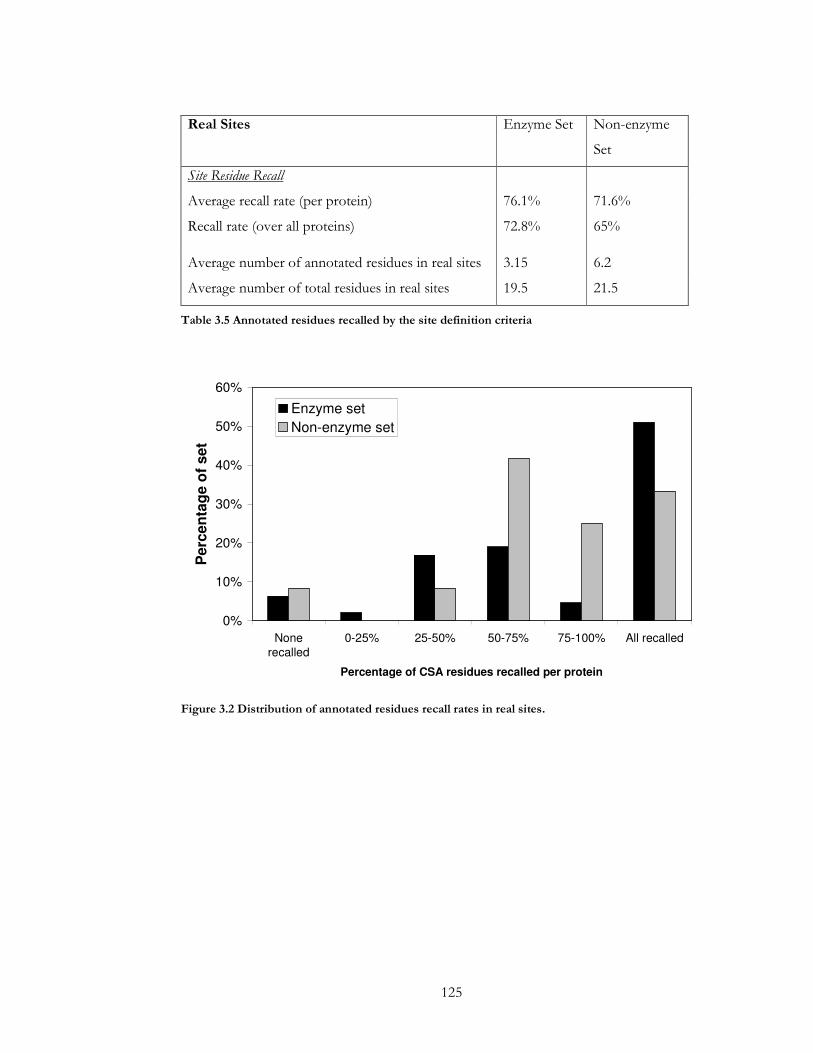
125
Real Sites Enzyme Set Non-enzyme
Set
Site Residue Recall
Average recall rate (per protein) 76.1% 71.6%
Recall rate (over all proteins) 72.8% 65%
Average number of annotated residues in real sites 3.15 6.2
Average number of total residues in real sites 19.5 21.5
Table 3.5 Annotated residues recalled by the site definition criteria
0%
10%
20%
30%
40%
50%
60%
None
recalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of CSA residues recalled per protein
Per
cen
tag
e o
f se
t
Enzyme set
Non-enzyme set
Figure 3.2 Distribution of annotated residues recall rates in real sites.

126
3.4.2 Crescendo
Method Description
Crescendo seeks to identify active sites by identifying clusters of residues that have
higher than usual evolutionary constraint. Residues under evolutionary constraint were
identified by three measures; 1) whether there was a higher degree of evolutionary
conservation than expected at a position, 2) whether environment specific substitution
tables made weak predictions of the amino acid substitution patterns, and 3) residues
that have spatially conserved positions when structures of proteins within the same
family are superimposed. The method is able to distinguish between residues that are
conserved for structural or functional regions.
In the reference paper7, the scores calculated by these measures are overlaid onto the
protein structure and clusters of high scoring residues are identified and predicted as
functional site locations. The method provided in the webserver however, omits this
stage of the process and returns restraint scores on a per residue basis for the whole
protein. A user can then load this PDB format file into a molecular graphics program
to visually scan for clusters of highly conserved residues. Since this analysis calls for
the automatic identification of functional sites without visual inspection, site residues
are identified by taking a sample of the top-scoring residues. The average number of
functional residues per protein over the three test sets that the authors used was 6.8.
In our testing dataset there is an average of 3.2 literature annotated catalytic residues
per enzyme, which is similar to the average number of 3.5 annotated catalytic residues
per protein quoted in the original CSA analysis27. As a compromise between these
figures, the top 5 scoring residues given in the crescendo output were taken as the
predicted functional residues.

127
Prediction Accuracy
Predictions were obtained successfully for each of the 237 proteins in the enzyme set
and each of the 13 proteins in the non-enzyme set. Crescendo only evaluates one
chain so the first chain identifier in the PDB was used. Despite this limitation
Crescendo performed well (see Table 3.6), achieving a relative recall rate of 63.8% for
the enzyme set and 65.8% for the non-enzyme set. The distribution of distances
between predicted centroids and real centroids is shown in Figure 3.4.
In the enzyme set Crescendo performed better than the CSA defined active site for
two structures, 1e2a and 1f8x. The CSA defined centroid recalled 2 out of the 4
annotated residues for 1e2a whilst Crescendo recalled 3 out of 4. Crescendo recalled
all of the 4 annotated residues for 1f8x, whilst the CSA generated centroid only recalled
3. Crescendo recalled more annotated residues than the real centroid for 6 of the 13
proteins in the non-enzyme set.
Crescendo Enzyme Set Non-enzyme
Set
Site Residue Recall
Absolute recall rate 46.9% 44.2%
Relative recall rate 63.8% 65.8%
Distance between real and predicted centroids
Average distance (Ǻ) 10.3 11.8
Minimum distance (Ǻ) 1.0 2.9
Maximum distance (Ǻ) 28.4 33.8
Site residues shared between real and predicted sites
Average number of residues in real sites 19.6 21.5
Average number of residues in predicted sites 18.3 21.2
Average number of site residues shared 7.1 9.1
Average percentage of site residue shared per protein 35.7% 44.3%
Table 3.6 The functional site prediction accuracy results for Crescendo.
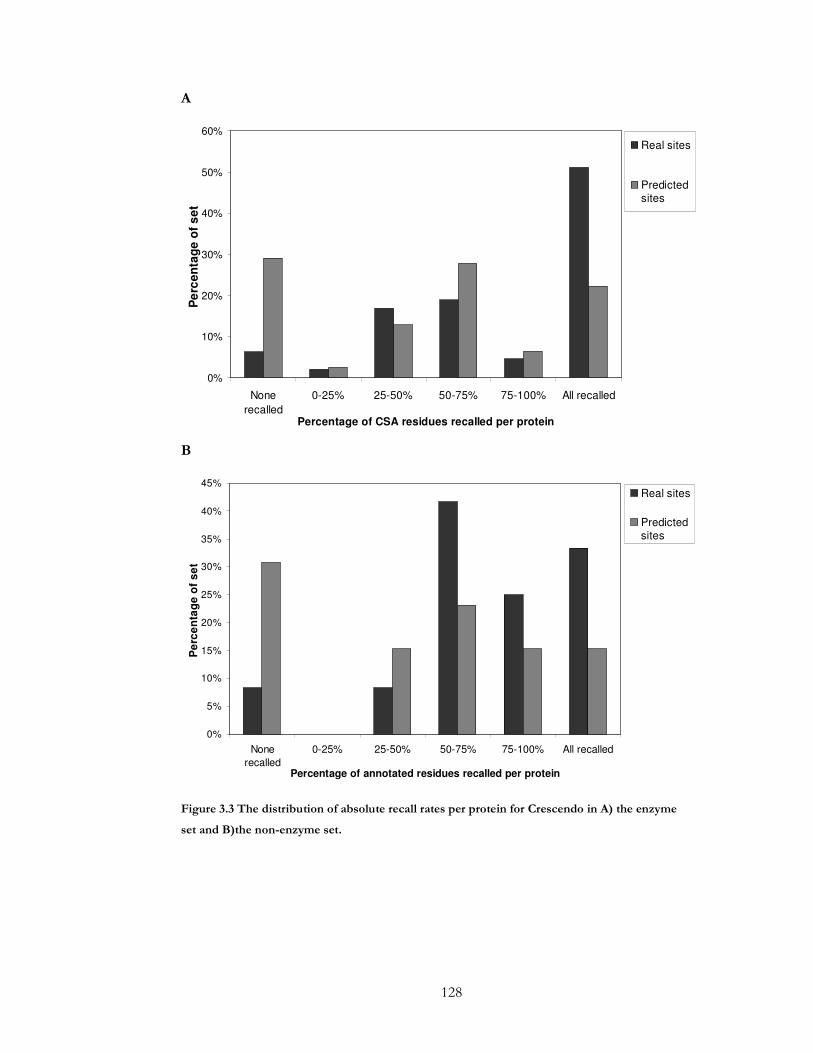
128
A
0%
10%
20%
30%
40%
50%
60%
None
recalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of CSA residues recalled per protein
Per
cen
tag
e o
f se
t
Real sites
Predictedsites
B
0%
5%
10%
15%
20%
25%
30%
35%
40%
45%
None
recalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of annotated residues recalled per protein
Per
cen
tag
e o
f se
t
Real sites
Predictedsites
Figure 3.3 The distribution of absolute recall rates per protein for Crescendo in A) the enzyme
set and B)the non-enzyme set.

129
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 5 10 15 20 25 30 35 40 45 50
Distance between predicted and real centroid (rounded to nearest Ångstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
Enzymes
Non-enzymes
Figure 3.4 The cumulative percentage of distances between Crescendo-predicted and real
centroids within the two sets.

130
3.4.3 PASS
Method description
PASS (Putative Active Site Spheres) is essentially a geometric cleft-finding method. It
characterises regions of buried volume by iteratively coating the surface of a protein
with probe spheres until all cavity space is filled (see Figure 3.5). The ASPs (Active Site
Points) are the centres of a spherical representation of the cavities found. The shape,
volume and burial depth determines whether a cavity is predicted to be an active site
cleft and the ASP for that cleft is returned as the active site prediction coordinate.
Figure 3.5 Diagram taken from Brady and Stouten, 200032 showing how the PASS method
defines buried volume.

131
Prediction accuracy
PASS did not run successfully for 9 proteins in the enzyme dataset and for one protein
in the non-enzyme set. For two further structures in the enzyme set, 1bs4 and 1qdl,
and one in the non-enzyme set (1eta) the centroid coordinates given were not within
the coordinate limits of the protein structure and thus no residues could be found
within the 10Ǻ of the centroid predicted. PASS achieved an average relative recall rate
of 49.3% for enzymes and 44.1% for non-enzymes.
Putative Active Sites with Spheres (PASS) Enzyme Set Non-enzyme
Set
Site Residue Recall
Absolute recall rate 36.6% 37.5%
Relative recall rate 49.3% 44.1%
Distance between real and predicted centroids
Average distance (Ǻ) 14.8 17.4
Minimum distance (Ǻ) 1.6 3.8
Maximum distance (Ǻ) 44.3 63.9
Site residues shared between real and predicted sites
Average number of residues in real sites 19.4 21.3
Average number of residues in predicted sites 22.2 22.5
Average number of site residues shared 4.3 8.7
Average percentage of site residue shared per protein 21.7% 33.8%
Table 3.7 The functional site prediction accuracy results for PASS.
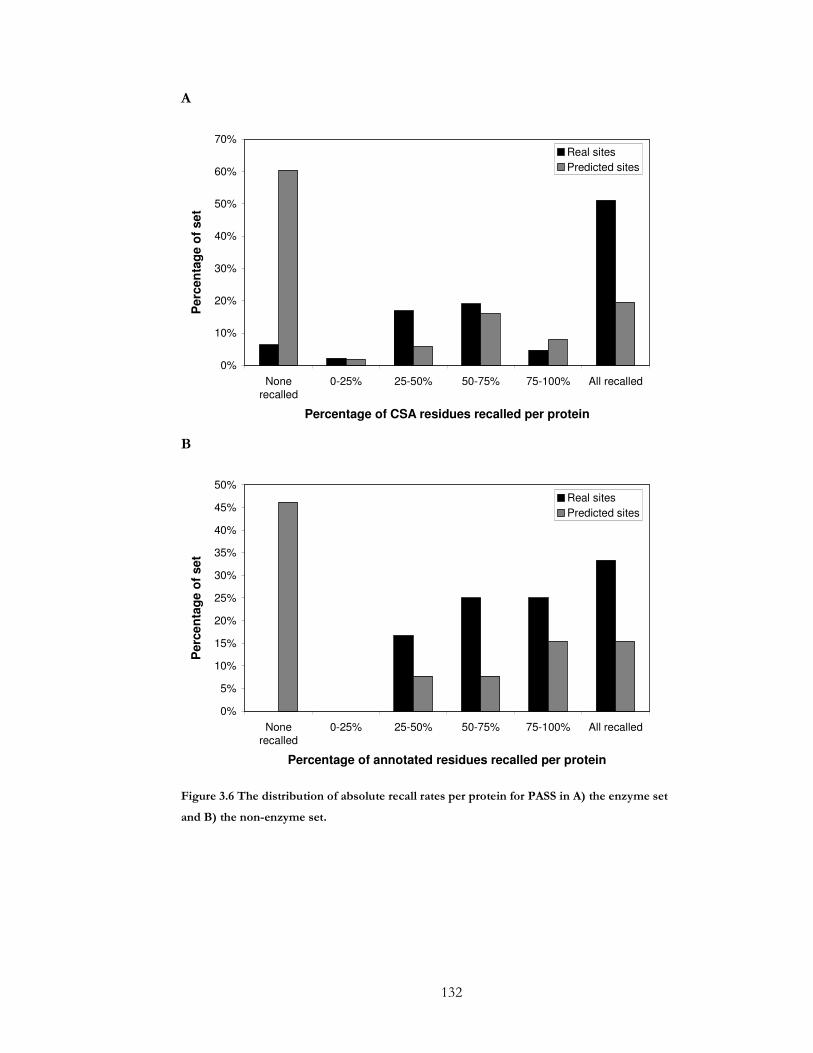
132
A
0%
10%
20%
30%
40%
50%
60%
70%
Nonerecalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of CSA residues recalled per protein
Per
cen
tag
e o
f se
t
Real sites
Predicted sites
B
0%
5%
10%
15%
20%
25%
30%
35%
40%
45%
50%
Nonerecalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of annotated residues recalled per protein
Per
cen
tag
e o
f se
t
Real sites
Predicted sites
Figure 3.6 The distribution of absolute recall rates per protein for PASS in A) the enzyme set
and B) the non-enzyme set.
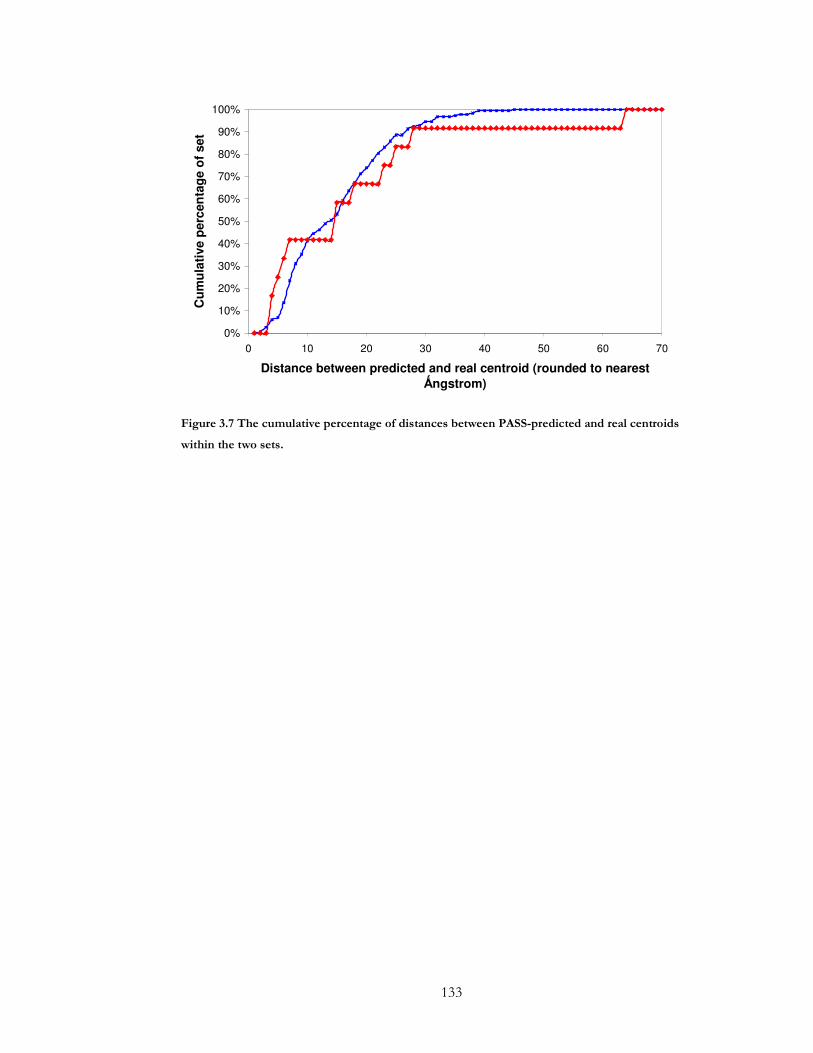
133
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 10 20 30 40 50 60 70
Distance between predicted and real centroid (rounded to nearest Ǻngstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
Figure 3.7 The cumulative percentage of distances between PASS-predicted and real centroids
within the two sets.

134
3.4.4 Fuzzy Oil Drop
Method description
It has long been observed that residues in the solvent-inaccessible core of a protein are
more likely to be more hydrophobic than the residues existing on, or close to, the
surface of a protein57. Residues involved in binding have been shown to frequently
exhibit levels of hydrophobicity that are unusual for their position within the protein
structure58. This prediction method predicts residues as forming a functional site
where there is a large difference between the expected hydrophobicity of a residue at
that position and the observed hydrophobicity value.
The hydrophobicity force field calculated in this method is based on the assumption
that the theoretical hydrophobicity in proteins follows a 3D Gaussian distribution. The
expected hydrophobicity of a residue is determined by a residues relative position to
the theoretically most hydrophobic point in the protein. The observed hydrophobicity
value is calculated by assessing the hydrophobicity characteristics of the sidechains
within the protein model and the residues position in relation to those side chains.
They have shown that this model does not produce significantly different results to
more commonly used scales such as Eisenberg59 and Kyte and Doolittle60 scales.
The hydrophobic deficiency score for each residue is the difference between the
expected hydrophobicity and the observed hydrophobicity. The highest scoring
residues are then predicted to be involved in functional sites. The residues were then
ranked by descending score and the minimum score of top 5% residues were then
predicted as functional site residues as the reference paper instructs.

135
Prediction accuracy
As with Crescendo, the FOD method can only calculate predictions of a single
polypeptide chain and so the first chain in the PDB file was used. Predictions were
obtained successfully for all 237 proteins in the enzyme set, giving relative recall rate of
56.1% and for 12 of the 13 non-enzymes giving a relative recall rate of 33.3% (Table
3.8). There were, however, some issues in the output that meant a slightly altered
analysis was required.
One issue was that the residue numbering in their output was different to the
numbering of residues in the PDB. The output of this method includes a modified
PDB file that uses their own numbering scheme and so centroids were calculated from
this PDB file instead of the standard PDB file. The PDB file supplied in the output
occasionally misses atom coordinates for some of their predicted active site residues.
Where the Cβ atom coordinates for a predicted residue were unavailable in the output
PDB file, the coordinates for the next available atom in that residue were used in order
to calculate the centroid.
The second issue was that the hydrophobic deficiency score applied to each residue
was truncated to 2 decimal places in the output in order for it to be accommodated
into the temperature factor column of the PDB format file. This produced degeneracy
in the scoring system since multiple residues could be assigned the same score.
Multiple residues having the same score created a problem when attempting to cut off
the top 5% scoring residues to predict as active site residues. The boundary residue for
the top 5% often lay within a group of residues with the same score and therefore
discriminating which of these residues to consider a prediction is somewhat arbitrary.
The output for the method also gives the raw (un-normalised) hydrophobic deficiency
score for each residue. In order to avoid the above problem, the residues with the top
5% of raw hydrophobic deficiency values (rather than normalised, as reported in their
publication) were predicted as active site residues.
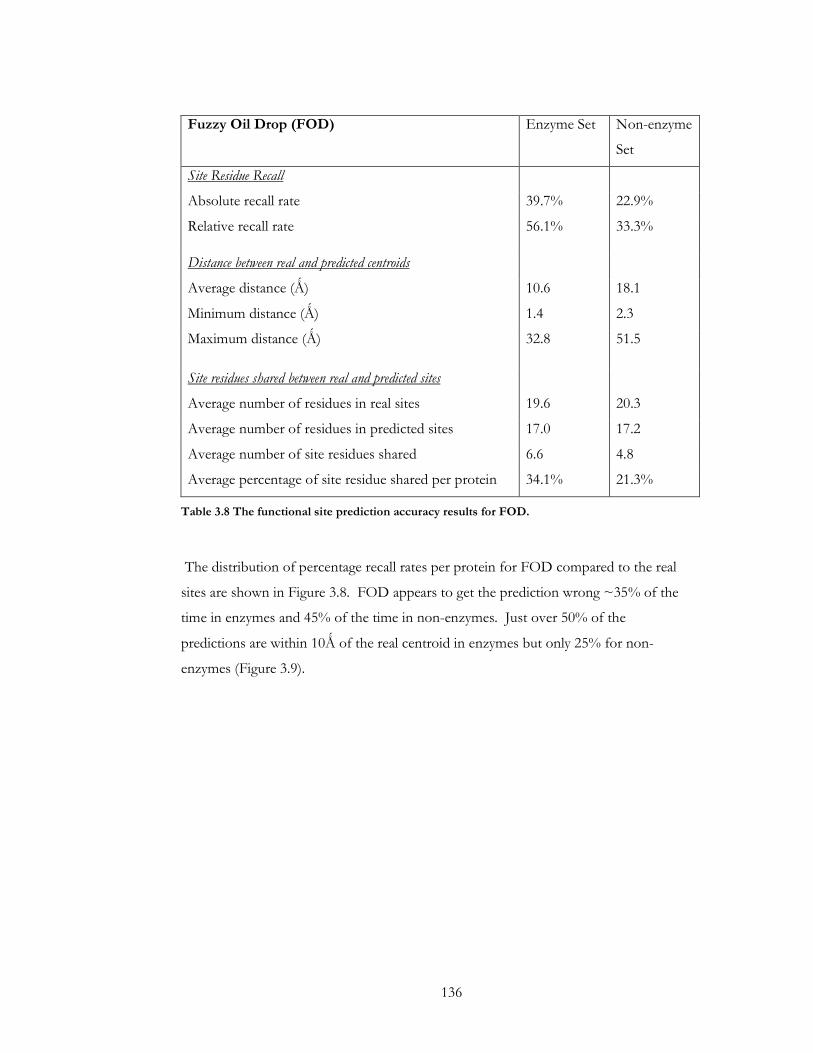
136
Fuzzy Oil Drop (FOD) Enzyme Set Non-enzyme
Set
Site Residue Recall
Absolute recall rate 39.7% 22.9%
Relative recall rate 56.1% 33.3%
Distance between real and predicted centroids
Average distance (Ǻ) 10.6 18.1
Minimum distance (Ǻ) 1.4 2.3
Maximum distance (Ǻ) 32.8 51.5
Site residues shared between real and predicted sites
Average number of residues in real sites 19.6 20.3
Average number of residues in predicted sites 17.0 17.2
Average number of site residues shared 6.6 4.8
Average percentage of site residue shared per protein 34.1% 21.3%
Table 3.8 The functional site prediction accuracy results for FOD.
The distribution of percentage recall rates per protein for FOD compared to the real
sites are shown in Figure 3.8. FOD appears to get the prediction wrong ~35% of the
time in enzymes and 45% of the time in non-enzymes. Just over 50% of the
predictions are within 10Ǻ of the real centroid in enzymes but only 25% for non-
enzymes (Figure 3.9).
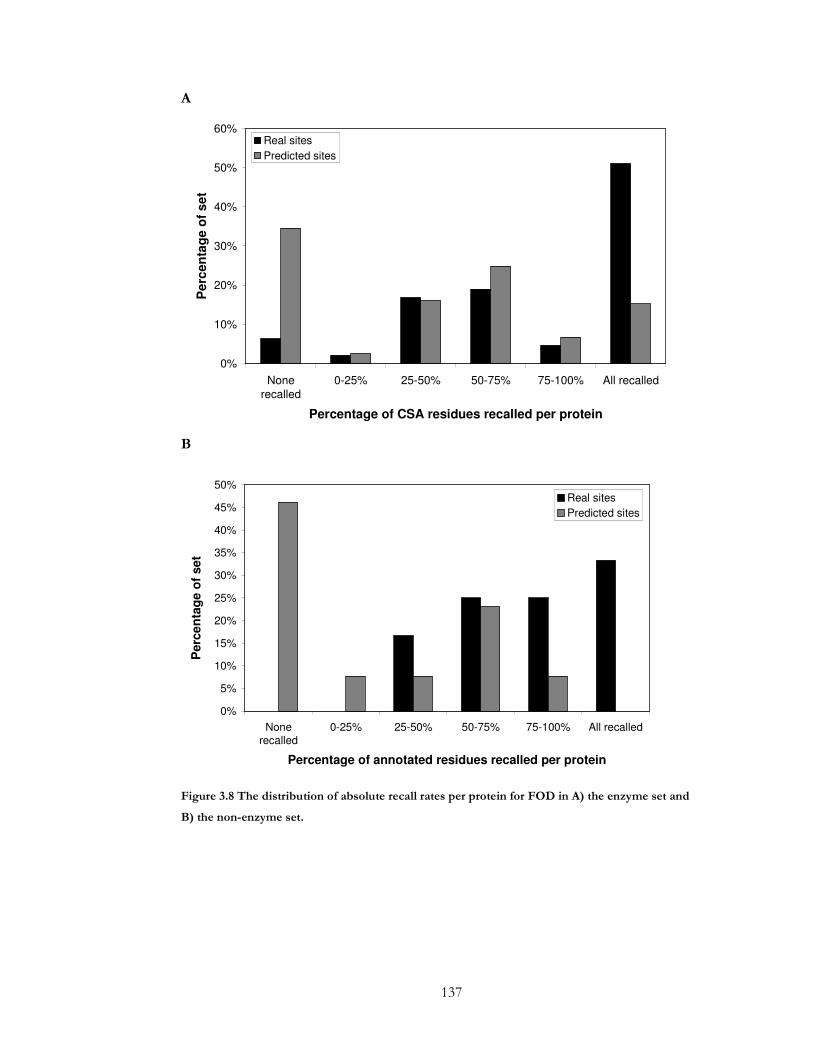
137
A
0%
10%
20%
30%
40%
50%
60%
None
recalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of CSA residues recalled per protein
Per
cen
tag
e o
f se
t
Real sites
Predicted sites
B
0%
5%
10%
15%
20%
25%
30%
35%
40%
45%
50%
Nonerecalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of annotated residues recalled per protein
Per
cen
tag
e o
f se
t
Real sites
Predicted sites
Figure 3.8 The distribution of absolute recall rates per protein for FOD in A) the enzyme set and
B) the non-enzyme set.
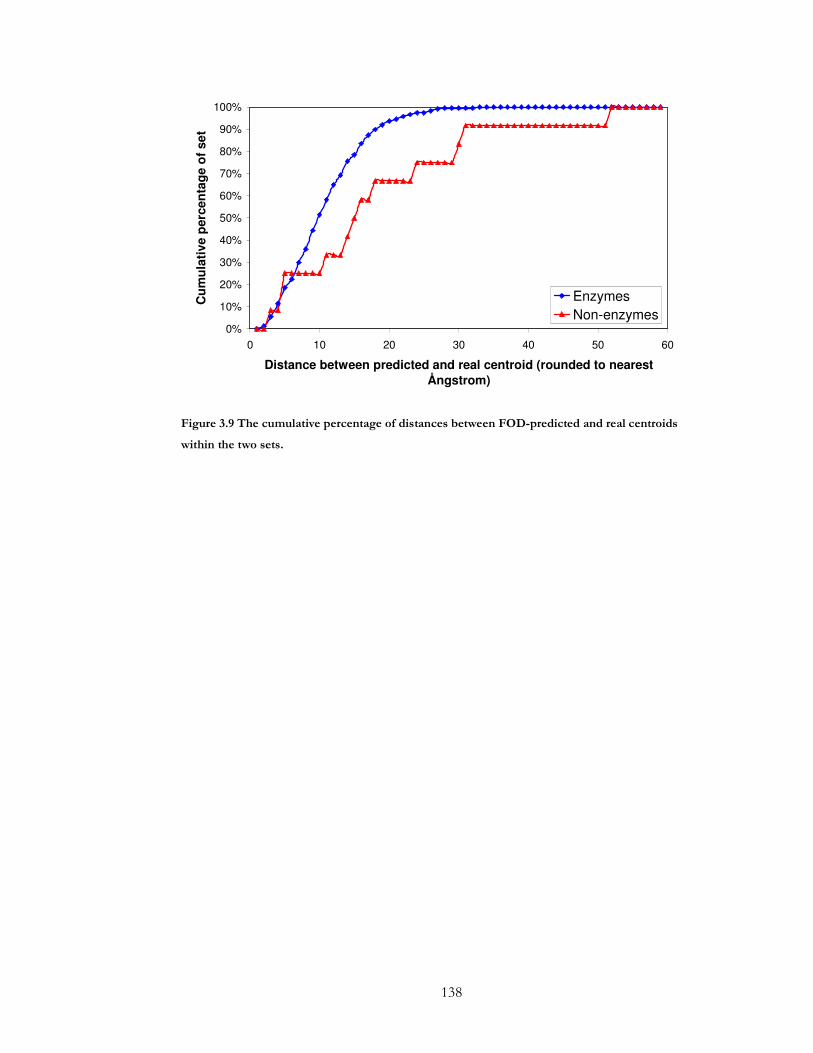
138
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 10 20 30 40 50 60
Distance between predicted and real centroid (rounded to nearest Ångstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
Enzymes
Non-enzymes
Figure 3.9 The cumulative percentage of distances between FOD-predicted and real centroids
within the two sets.

139
3.4.5 QSiteFinder
Method Description
QSiteFinder finds clefts on the proteins surface and then ranks them according to their
interaction energy between the protein and a van der Waals probe. Non-bonded
interaction energies are calculated by placing a 0.9Å 3D grid over the whole protein
and then evaluating the interaction energy between the protein and a methyl group at
each point on the grid. The positions of the probes on the grid that gave the best
interaction energies were then spatially clustered to identify groups of close probes.
These clusters are then assigned a single interaction energy based on the energies of
their member probes. The clusters are then ranked by their representative interaction
energy and the highest ranked cluster is predicted as the functional site. Protein residue
atoms that are in contact with the predicted site are given as the predicted functional
site residues. The output gives a list of ranked sites but only the top-ranking site will
be used for this analysis.
Prediction Accuracy
QSiteFinder gives output for multiple site predictions ranked in order of the likelihood
of being a real active site. For this analysis the first ranked predicted site is taken. For
each site, a set of atoms predicted as existing in the active site is given. Not all atoms
for a residue are predicted, but for consistentcy with other methods, if an atom of a
residue is given in the prediction the coordinates of the Cβ atom of that residue, even if
it is not given in the prediction, are used to calculate the centroid.
QSiteFinder can only process structures with less than 10,000 atoms and this excluded
24 proteins from the enzyme set and one from the non-enzyme set. For the remainder
of the enzyme dataset QSiteFinder achieved relative recall rate of 53.0% and 54.0% for
the non-enzyme set (see Table 3.9). Q-SiteFinder performed better than the CSA-
generated centroid for 1qd6 in the enzyme set. The QSiteFinder-generate centroid
recalled all 3 of the CSA annotated residues as opposed to the two recalled by the CSA-
generated centroid.

140
The distribution of percentage recall rates per protein for QSiteFinder compared to the
real sites are shown in Figure 3.10. QSiteFinder appears to get the prediction wrong
40% of the time in enzymes and ~30% of the time in non-enzymes. Just over 45% of
the predictions are within 10Ǻ of the real centroid in enzymes but almost 60% for non-
enzymes (Figure 3.11).
QSiteFinder Enzyme Set Non-enzyme
Set
Site Residue Recall
Absolute recall rate 40.1% 33.6%
Relative recall rate 53.0% 54.0%
Distance between real and predicted centroids
Average distance (Ǻ) 13.0 12.5
Minimum distance (Ǻ) 1.7 2.3
Maximum distance (Ǻ) 39.5 30.6
Site residues shared between real and predicted sites
Average number of residues in real sites 22.3 23.4
Average number of residues in predicted sites 19.8 21.8
Average number of site residues shared 5.9 8.9
Average percentage of site residue shared per protein 29.0% 41.4%
Table 3.9 The functional site prediction accuracy results for QSiteFinder
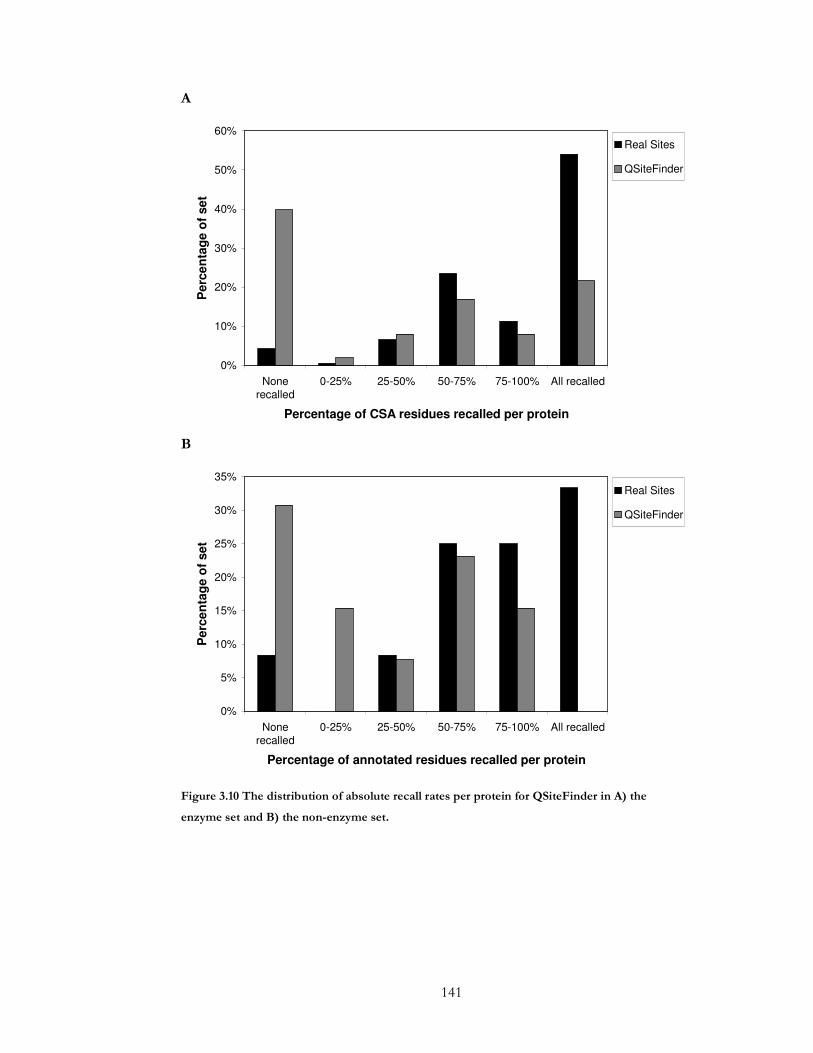
141
A
0%
10%
20%
30%
40%
50%
60%
Nonerecalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of CSA residues recalled per protein
Per
cen
tag
e o
f se
t
Real Sites
QSiteFinder
B
0%
5%
10%
15%
20%
25%
30%
35%
Nonerecalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of annotated residues recalled per protein
Per
cen
tag
e o
f se
t
Real Sites
QSiteFinder
Figure 3.10 The distribution of absolute recall rates per protein for QSiteFinder in A) the
enzyme set and B) the non-enzyme set.

142
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 10 20 30 40 50 60
Distance between predicted and real centroid (rounded to nearest Ångstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
Enzymes
Non-enzymes
Figure 3.11 The cumulative percentage of distances between QSiteFinder-predicted and real
centroids within the two sets.

143
3.4.6 PDBSiteScan
Method description
PDBSiteScan takes 3D fragments of a protein structure and compares them to 3D
structure fragments of known active sites. The known active sites structures are held in
a collection called PDBSite that is formed from annotation in the PDB SITE field and
also REMARK 800 fields. PDBSite stores several types of ‘functional’ sites including
protein-protein interactions and posttranslational modification sites. In the enzyme
analysis only the sites marked as “active sites” were searched.
The alignment of the site template and the test protein are performed using CE61
(Combinatorial Extension) and the N, C and Cα atoms are used to define the
orientation of the residue. For a template to match a 3D fragment the maximum
distance mismatch (MDM), the sum of the Cartesian distances between each atom in
the template and the fragment, has to be less than the user defined cut-off. In this
analysis the default setting of 2Å was used.
Prediction accuracy
PDBSiteScan was problematic to run and gave errors for 53 of the 237 structures in
the enzyme dataset. This included 19 structures where the protein found during the
scan of the PDB was the same as the query structure, 18 structures where the scan
could not find any similar proteins and a further 16 where PDBSiteScan did not run
due to other technical errors. It performed relatively poorly on the remaining enzyme
structures, giving a relative recall rate of 38.4% (see Table 3.10). PDBSiteScan could
not produce results for three of the non-enzyme structures and only produced a
relative recall rate of 23.5% for the non-enzyme set.

144
PDBSiteScan Enzyme Set Non-enzyme
Set
Site Residue Recall
Absolute recall rate 28.1% 11.4%
Relative recall rate 38.4% 23.5%
Distance between real and predicted centroids
Average distance (Ǻ) 15.5 19.2
Minimum distance (Ǻ) 0.1 7.9
Maximum distance (Ǻ) 49.1 41.2
Site residues shared between real and predicted sites
Average number of residues in real sites 22.3 21.8
Average number of residues in predicted sites 23.2 17.7
Average number of site residues shared 4.6 2.9
Average percentage of site residues shared per protein 23.2% 15.0%
Table 3.10 The functional site prediction accuracy results for PDBSiteScan
The distribution of recall rates is shown in Figure 3.12 and the distance between
predicted and real site centroids is shown in Figure 3.13. PDBSiteScan appears to get
the prediction wrong over half of the time in both sets and with some degree of
accuracy the remaining time (Figure 3.12). Approximately only 30% and 20% of the
predictions are within 10Ǻ of the real enzyme and non-enzyme centroids, respectively
(Figure 3.13).
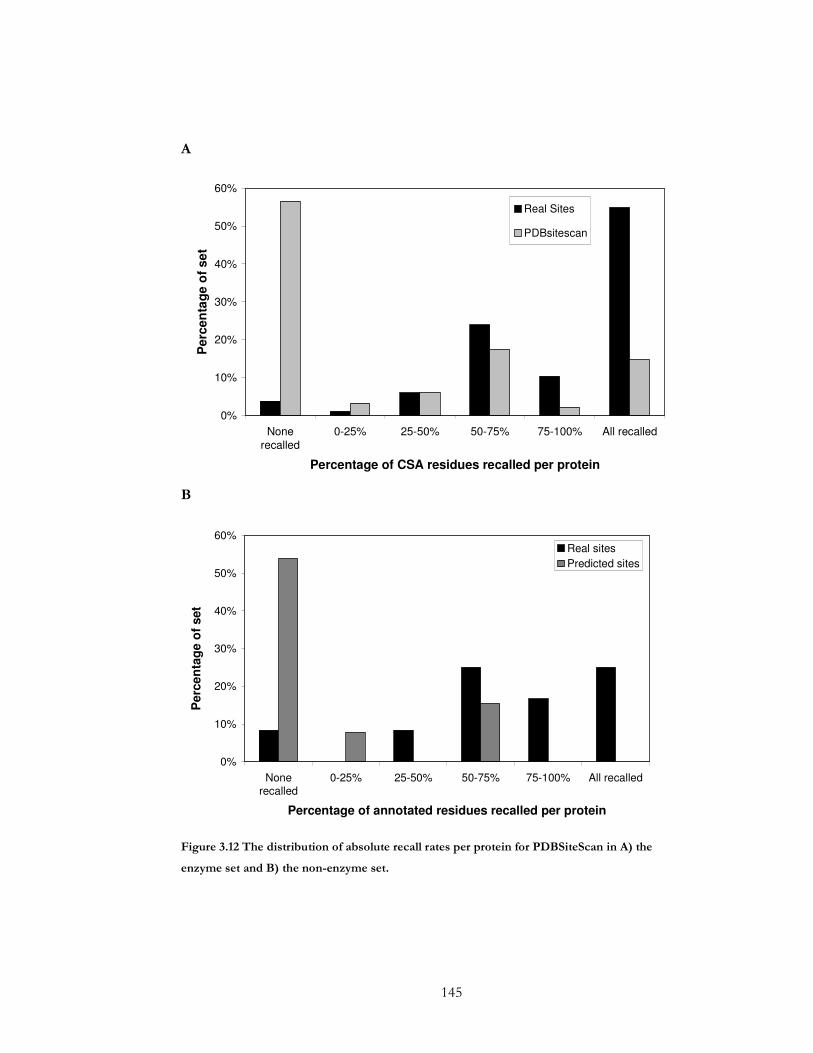
145
A
0%
10%
20%
30%
40%
50%
60%
None
recalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of CSA residues recalled per protein
Per
cen
tag
e o
f se
tReal Sites
PDBsitescan
B
0%
10%
20%
30%
40%
50%
60%
Nonerecalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of annotated residues recalled per protein
Per
cen
tag
e o
f se
t
Real sites
Predicted sites
Figure 3.12 The distribution of absolute recall rates per protein for PDBSiteScan in A) the
enzyme set and B) the non-enzyme set.

146
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 10 20 30 40 50
Distance between predicted and real centroid (rounded to nearest Ǻngstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
Figure 3.13 The cumulative percentage of distances between PDBSiteScan-predicted and real
centroids within the two sets.

147
3.4.7 Consurf
Method description
Consurf calculates the degree of evolutionary conservation for each residue in a
structure and assigns them an integer score from 1 to 9, with 9 being the most
conserved residues. A graphical representation of the structure is then coloured
according to these residue conservation scores, which allows visual identification of
highly conserved patches, which are predicted to be functional sites.
Prediction Accuracy
Consurf only accepts one chain as input to the method and so the first chain from the
structure file was used to obtain predictions. Where there are multiple copies of this
chain in the structure the predicted site annotation from the input chain is copied
across to all other identical chains. The primary output of this method is via a graphic
of the protein structure coloured according to the residues’ degree of conservation.
Visual inspection of this coloured structure allows the identification of surface patches
of highly conserved residues. This analysis however, requires output to be
automatically evaluated and therefore residues were taken as predicted site residues
when they were assigned the top conservation score (9).
Consurf did not produce output for 4 of the 237 proteins in the enzyme set and 3 of
the 13 proteins in the non-enzyme dataset. Despite only being able to predict sites for
one chain of a protein, Consurf achieved an average relative recall rate of 78.2% for
enzymes and 52.1% for non-enzymes (see Table 3.11). Around 70% of proteins had
predicted centroids within 10Å of the real centroid for both the enzyme and non-
enzyme set (see Figure 3.15).

148
Consurf Enzyme Set Non-enzyme
Set
Site Residue Recall
Absolute recall rate 58.6% 36.3%
Relative recall rate 78.2% 52.1%
Distance between real and predicted centroids
Average distance (Ǻ) 8.2 13.0
Minimum distance (Ǻ) 0.5 3.6
Maximum distance (Ǻ) 28.6 37.8
Site residues shared between real and predicted sites
Average number of residues in real sites 17.3 20.3
Average number of residues in predicted sites 19.6 21.5
Average number of site residues shared 8.7 7.9
Average percentage of site residue shared per protein 44.5% 35.8%
Table 3.11 The functional site prediction accuracy results for Consurf.
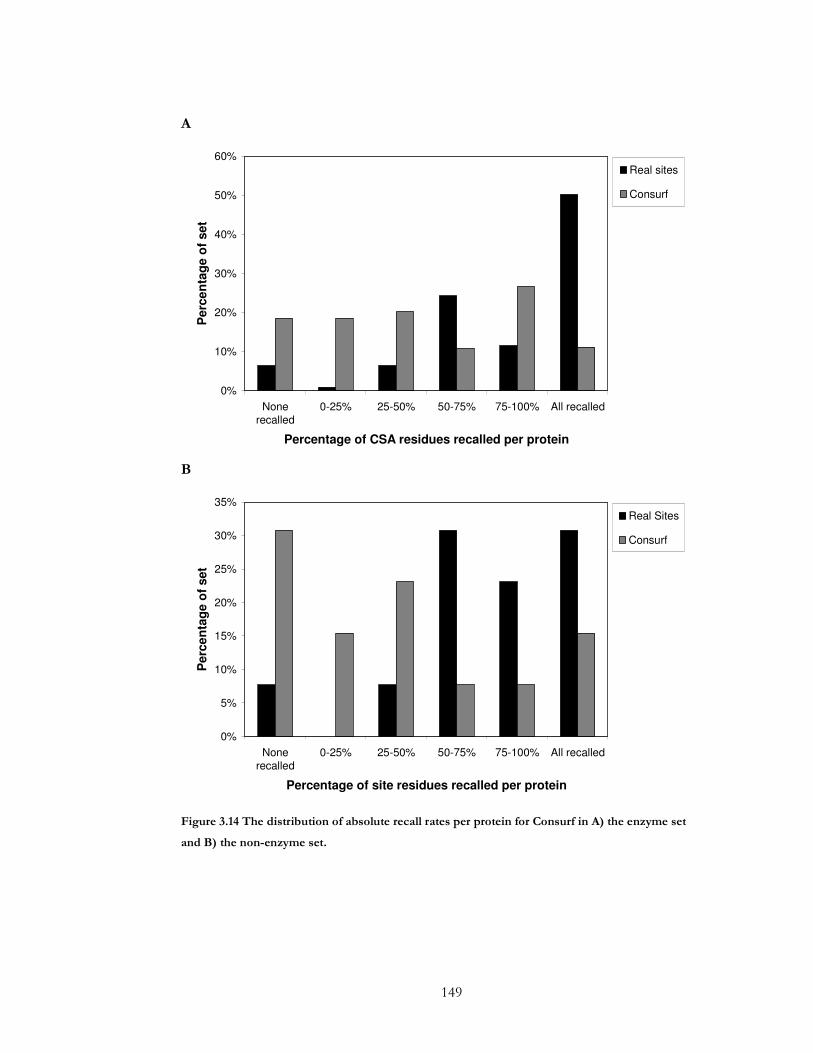
149
A
0%
10%
20%
30%
40%
50%
60%
Nonerecalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of CSA residues recalled per protein
Per
cen
tag
e o
f se
t
Real sites
Consurf
B
0%
5%
10%
15%
20%
25%
30%
35%
Nonerecalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of site residues recalled per protein
Per
cen
tag
e o
f se
t
Real Sites
Consurf
Figure 3.14 The distribution of absolute recall rates per protein for Consurf in A) the enzyme set
and B) the non-enzyme set.
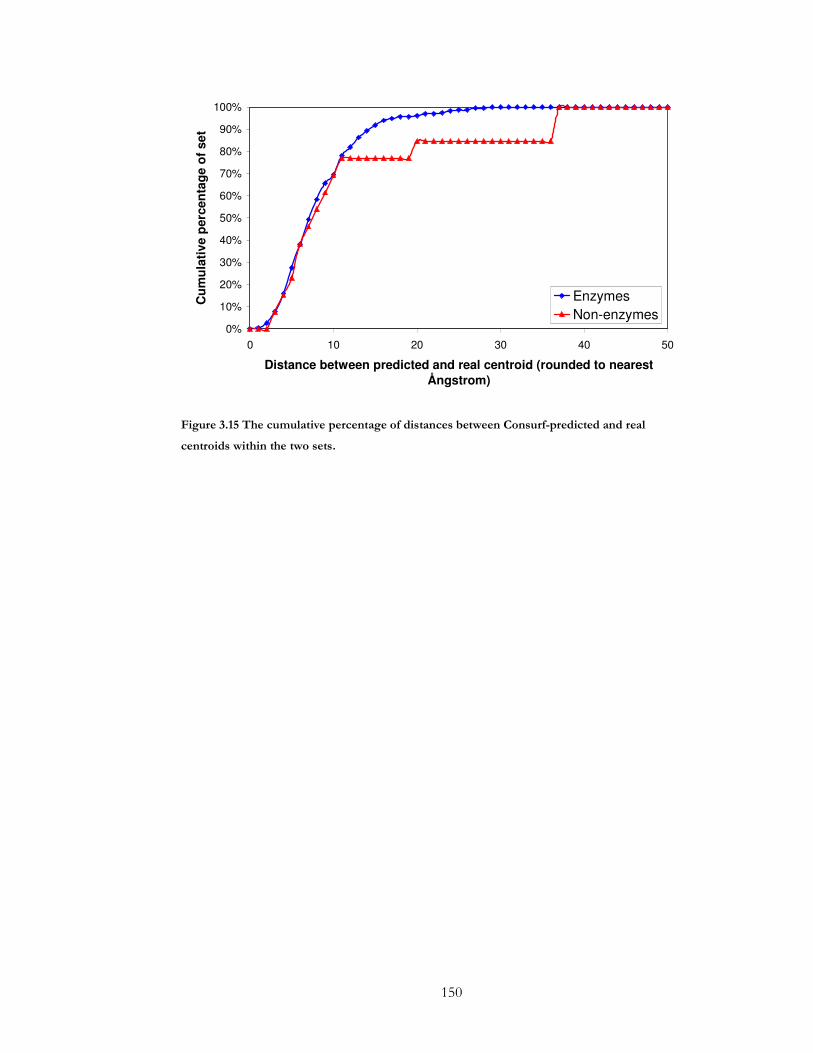
150
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 10 20 30 40 50
Distance between predicted and real centroid (rounded to nearest Ångstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
Enzymes
Non-enzymes
Figure 3.15 The cumulative percentage of distances between Consurf-predicted and real
centroids within the two sets.

151
3.4.8 Thematics
Method description
Thematics identifies ionisable residues with unusually perturbed titrations curves.
Active sites are predicted where two or more of these ionisable residues form a cluster
in 3D space. This method is only applicable to enzyme active sites, and therefore isn’t
assessed for the non-enzyme set.
Prediction accuracy
Thematics was unable to produce output for 25 of the 237 proteins in the enzyme set
and since Thematics is developed as an active-site predictor for enzymes, it was not
used n the non-enzyme dataset. Thematics achieved an average relative recall rate of
48.9% for enzymes (see Table 3.11 ) with around 40% of the predicted centroids being
within 10Å of the real centroid (see Figure 3.17).
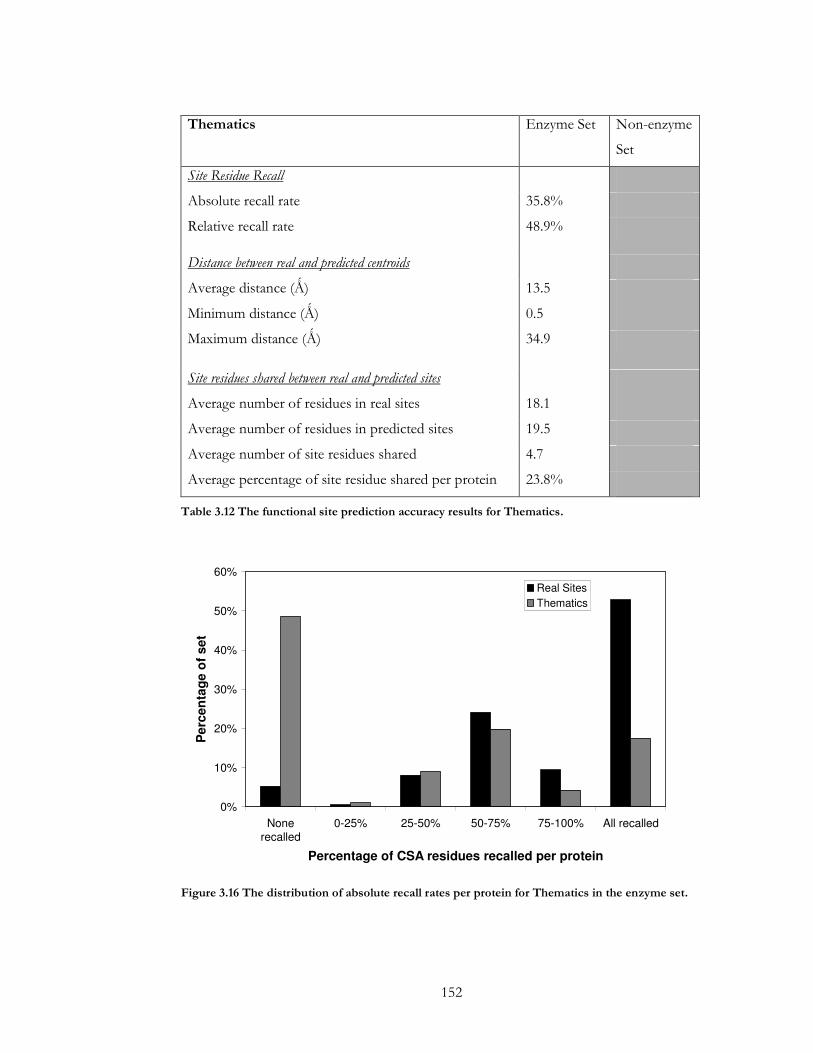
152
Thematics Enzyme Set Non-enzyme
Set
Site Residue Recall
Absolute recall rate 35.8%
Relative recall rate 48.9%
Distance between real and predicted centroids
Average distance (Ǻ) 13.5
Minimum distance (Ǻ) 0.5
Maximum distance (Ǻ) 34.9
Site residues shared between real and predicted sites
Average number of residues in real sites 18.1
Average number of residues in predicted sites 19.5
Average number of site residues shared 4.7
Average percentage of site residue shared per protein 23.8%
Table 3.12 The functional site prediction accuracy results for Thematics.
0%
10%
20%
30%
40%
50%
60%
Nonerecalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of CSA residues recalled per protein
Per
cen
tag
e o
f se
t
Real Sites
Thematics
Figure 3.16 The distribution of absolute recall rates per protein for Thematics in the enzyme set.
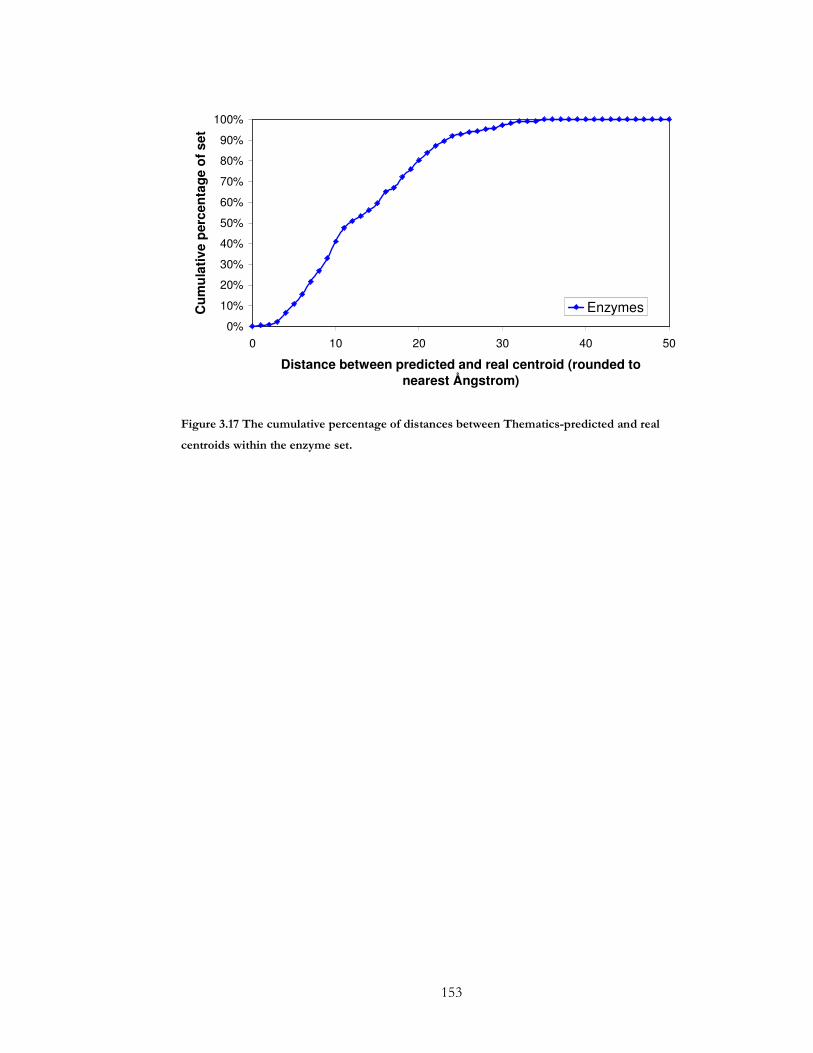
153
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 10 20 30 40 50
Distance between predicted and real centroid (rounded to nearest Ångstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
Enzymes
Figure 3.17 The cumulative percentage of distances between Thematics-predicted and real
centroids within the enzyme set.

154
3.4.9 SitesIdentify(GM) – Geometry-based
Method Description
The method behind SitesIdenitfy(GM) is explained in detail in 3.3.1 but in brief, a 2Å
grid is placed over the protein structure and a uniform charge is applied to each non-
hydrogen atom. The electrostatic potential is calculated using Finite Difference
Poisson-Boltzmann calculations with no dielectric boundary and the peak potential is
predicted as the centroid of the functional site.
Prediction Accuracy
SitesIdentify(GM) did not run successfully for two structures, 10o4 and 1rbl, but did
successfully produce output for the other 235. It achieved an average relative
percentage accuracy per protein of 63.0% for enzymes yet a higher accuracy of 69.5%
for non-enzymes (see Table 3.13). For 4 of the structures in the enzyme dataset, 1snn,
2sqc, 1qd6 and 1mvn, the SitesIdentify-generated centroid recalled one more CSA
annotated residue than the CSA-generated centroid.
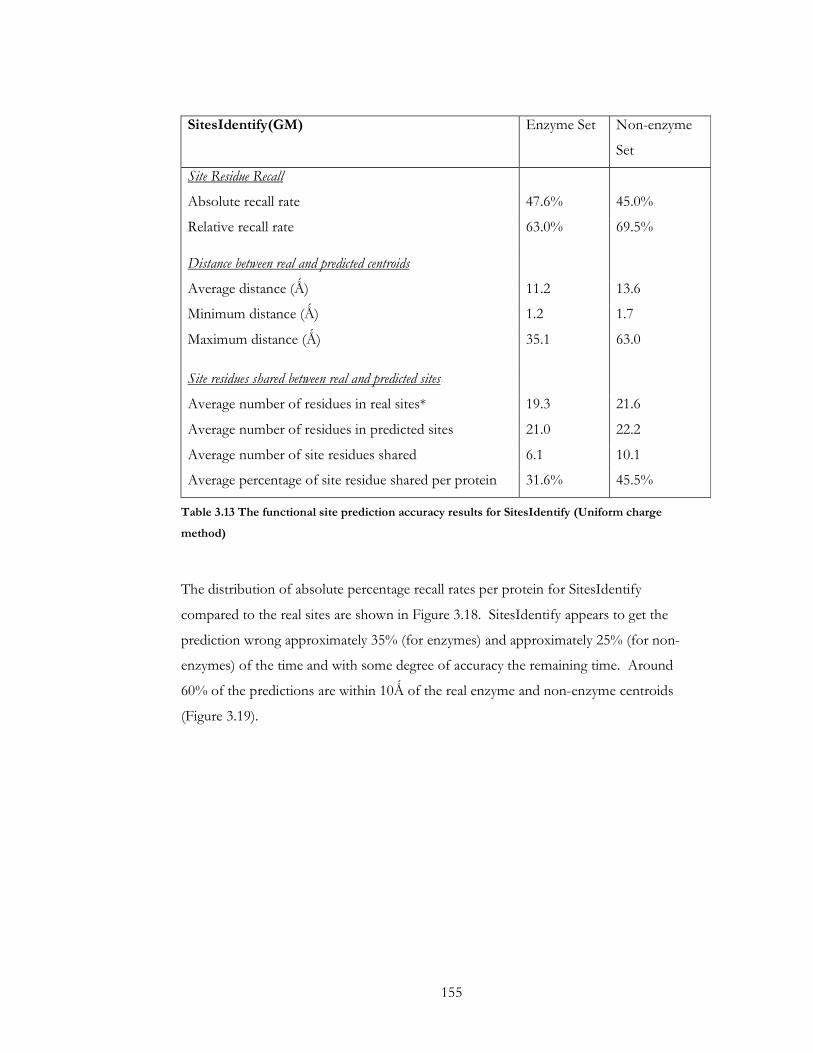
155
SitesIdentify(GM) Enzyme Set Non-enzyme
Set
Site Residue Recall
Absolute recall rate 47.6% 45.0%
Relative recall rate 63.0% 69.5%
Distance between real and predicted centroids
Average distance (Ǻ) 11.2 13.6
Minimum distance (Ǻ) 1.2 1.7
Maximum distance (Ǻ) 35.1 63.0
Site residues shared between real and predicted sites
Average number of residues in real sites* 19.3 21.6
Average number of residues in predicted sites 21.0 22.2
Average number of site residues shared 6.1 10.1
Average percentage of site residue shared per protein 31.6% 45.5%
Table 3.13 The functional site prediction accuracy results for SitesIdentify (Uniform charge
method)
The distribution of absolute percentage recall rates per protein for SitesIdentify
compared to the real sites are shown in Figure 3.18. SitesIdentify appears to get the
prediction wrong approximately 35% (for enzymes) and approximately 25% (for non-
enzymes) of the time and with some degree of accuracy the remaining time. Around
60% of the predictions are within 10Ǻ of the real enzyme and non-enzyme centroids
(Figure 3.19).

156
0%
10%
20%
30%
40%
50%
60%
None
recalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of CSA residues recalled per protein
Per
cen
tag
e o
f se
t
Real Sites
SitesIdentify
0%
5%
10%
15%
20%
25%
30%
35%
40%
45%
None
recalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of site residues recalled per protein
Per
cen
tag
e o
f se
t
Real Sites
SitesIdentify
Figure 3.18 The distribution of absolute recall rates per protein for SitesIdentify(GM) in A) the
enzyme set and B) the non-enzyme set.

157
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 10 20 30 40 50 60 70
Distance between predicted and real centroid (rounded to nearest Ångstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
Enzymes
Non-enzymes
Figure 3.19 The cumulative percentage of distances between SitesIdentify(GM) predicted and
real centroids within the enzyme and non-enzyme set.

158
3.4.10 SitesIdentify(ConsGM) – Conservation and geometry-based
Method description
The second method11, SitesIdentify (ConsGM), combines the electrostatics method
used in SitesIdentify(GM) with sequence conservation information. Close homologues
are found by running the sequence through PSI-BLAST with an E value cut-off of 1e-
20. A normalised conservation score is calculated for each residue based on the amino
acid and stereochemical diversity and the gap occurrence at that position,
C(x)=(1−t(x))α(1−r(x))β(1−g(x))γ, where t is the normalised symbol diversity, r is the
normalised stereochemical diversity (based on the BLOSUM-62 matrix) and g the gap
cost. Each of these terms are weighted by integral values ranging between 0 and 5 (α, β
and γ), the values for which are defined as those giving the best predictive performance
in the original publication11. The peak potential is then calculated in the same way as
the first method, but now with a single central atom in each amino acid weighted with
the conservation scores. This method is described in more detail in 3.3.1.
Prediction accuracy
SitesIdentify(ConsGM) did not run successfully for the same two structures as
SitesIdentify(GM), 10o4 and 1rbl. It achieved an average relative percentage accuracy
per protein of 74.7% for enzymes and 62.2% for non-enzymes (see Table 3.14).
The distribution of absolute percentage recall rates per protein for SitesIdentify
compared to the real sites are shown in Figure 3.20. SitesIdentify appears to get the
prediction wrong approximately 25% (for enzymes) and approximately 30% (for non-
enzymes) of the time and with some degree of accuracy the remaining time. Around
60% of the predictions are within 10Ǻ of the real enzyme centroids and approximately
55% are within 10Ǻ of the non-enzyme centroids (Figure 3.21).
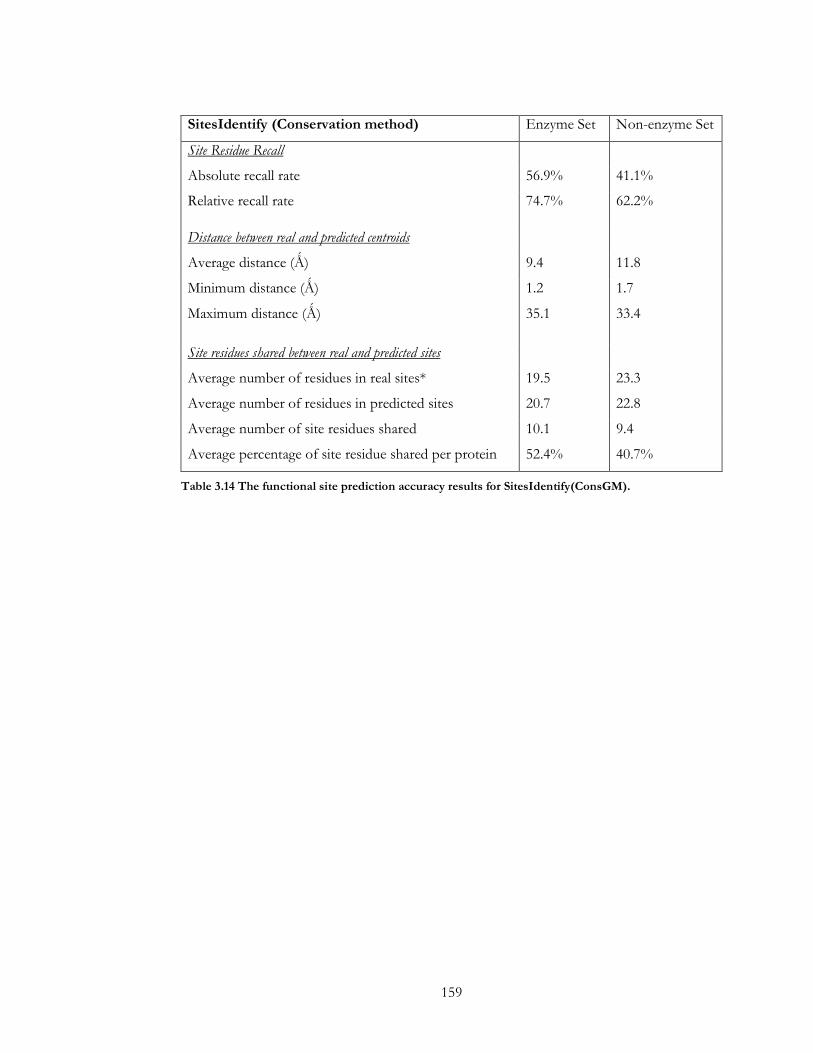
159
SitesIdentify (Conservation method) Enzyme Set Non-enzyme Set
Site Residue Recall
Absolute recall rate 56.9% 41.1%
Relative recall rate 74.7% 62.2%
Distance between real and predicted centroids
Average distance (Ǻ) 9.4 11.8
Minimum distance (Ǻ) 1.2 1.7
Maximum distance (Ǻ) 35.1 33.4
Site residues shared between real and predicted sites
Average number of residues in real sites* 19.5 23.3
Average number of residues in predicted sites 20.7 22.8
Average number of site residues shared 10.1 9.4
Average percentage of site residue shared per protein 52.4% 40.7%
Table 3.14 The functional site prediction accuracy results for SitesIdentify(ConsGM).
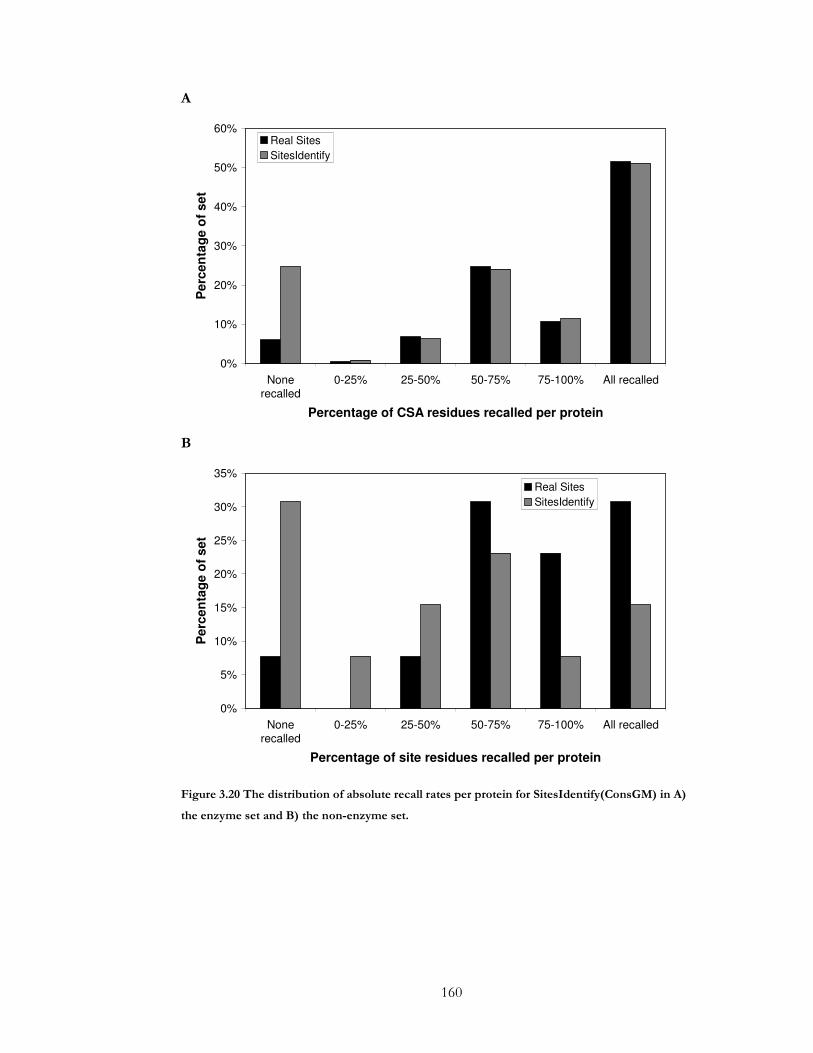
160
A
0%
10%
20%
30%
40%
50%
60%
Nonerecalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of CSA residues recalled per protein
Per
cen
tag
e o
f se
t
Real Sites
SitesIdentify
B
0%
5%
10%
15%
20%
25%
30%
35%
Nonerecalled
0-25% 25-50% 50-75% 75-100% All recalled
Percentage of site residues recalled per protein
Per
cen
tag
e o
f se
t
Real Sites
SitesIdentify
Figure 3.20 The distribution of absolute recall rates per protein for SitesIdentify(ConsGM) in A)
the enzyme set and B) the non-enzyme set.

161
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 10 20 30 40 50
Distance between predicted and real centroid (rounded to nearest Ångstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
Enzymes
Non-enzymes
Figure 3.21 The cumulative percentage of distances between SitesIdentify(ConsGM) predicted
and real centroids within the enzyme and non-enzyme set.
3.4.11 All Methods
The absolute and relative recall rates of all methods are shown in Table 3.15 for the
enzyme set and Table 3.16 for the non-enzymes set. A comparison of the cumulative
percentages of the distances between predicted and real centroids between all methods
are shown in Figure 3.22 for enzymes and Figure 3.23 for non-enzymes. These show
that Consurf achieves the highest relative recall rate for enzymes with
SitesIdentify(GM) achieving the highest relative recall rate for non-enzymes.

162
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 5 10 15 20 25 30 35 40 45 50
Distance between predicted and real centroid (rounded to nearest Angstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
SitesIdentify (Geometry)SitesIdentify (Geometry + Conservation)FODQsitefinderCrescendoPDBsitescanPASSThematicsConsurf
Figure 3.22 Comparison of distances between the real centroids and the predicted centroids in
the enzyme dataset for each method.
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 10 20 30 40 50 60
Distance between predicted and real centroid (rounded to nearest Ǻngstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
SitesIdenitfy (Uniform charge method)
SitesIdentify (Conservation Method)
FOD
QSiteFinder
Crescendo
PDBSiteScan
PASS
Consurf
Figure 3.23 Comparison of distances between the real centroids and the predicted centroids in
the non-enzyme dataset for each method.

163
Method Absolute Recall Rate
Relative Recall Rate
Average Distance between Predicted and Real Centroid
(Å) SitesIdentify SitesIdentify(GM) 47.6% 63.0% 11.2 SitesIdentify(ConsGM) 56.9% 74.7% 9.4 Consurf 58.6% 78.2% 8.2 Crescendo 46.9% 63.8% 10.3 FOD 39.7% 56.1% 10.6 QSiteFinder 40.1% 53.0% 13.0 PDBSiteScan 28.1% 38.4% 15.5 PASS 36.6% 49.3% 14.8 Thematics 35.8% 48.9% 13.5
Table 3.15 The absolute and relative recall rates achieved for the enzyme dataset along with the
average distance between real and predicted centroids for each method.
Method Absolute Recall Rate
Relative Recall Rate
Average Distance between Predicted and
Real Centroid (Å) SitesIdentify SitesIdentify(GM) 45.0% 69.1% 13.6 SitesIdentify(ConsGM) 41.0% 62.2% 11.8 Consurf 36.3% 52.1% 13.1 Crescendo 44.2% 65.8% 11.8 FOD 22.9% 33.7% 18.1 QSiteFinder 33.6% 54.0% 12.5 PDBSiteScan 11.4% 23.5% 19.5 PASS 37.5% 47.1% 17.4
Table 3.16 The absolute and relative recall rates achieved for the non-enzyme dataset along with
the average distance between real and predicted centroids for each method.
Whilst achieving a slightly lower recall accuracy than Consurf, SitesIdentify(ConsGM)
also performs well for the enzyme dataset. Consurf, however, only makes predictions
for one chain of a structure and whilst this may be a limitation of the method, on this
dataset it had an advantageous effect on the recall accuracy for Consurf. Residues in a
structure can be conserved for either functional or structural reasons, and residues that
form a subunit interface may exhibit similar levels of conservation to functional
residues. Since SitesIdentify(ConsGM) identifies clusters of highly conserved residues,
it could be distracted from the functional site by a cluster of conserved residues at the
interface between two chains. Consurf would not be able to detect a cluster of
conserved residues between two chains as it only evaluates the degree of conservation
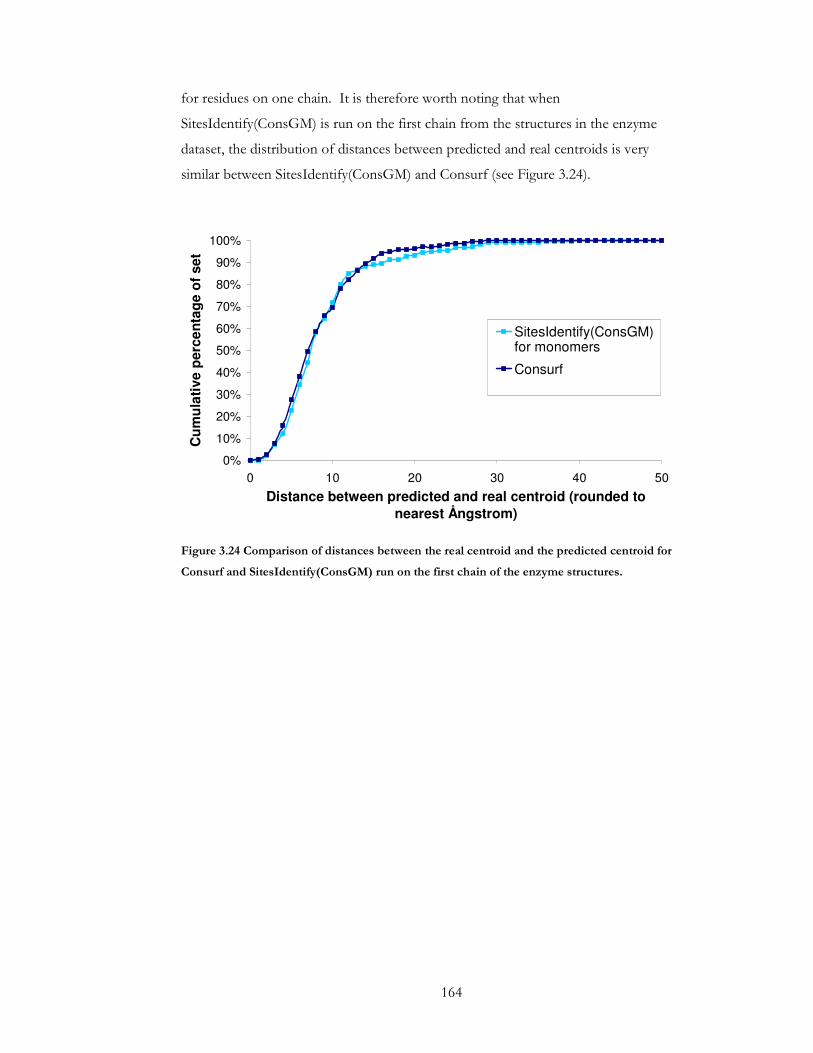
164
for residues on one chain. It is therefore worth noting that when
SitesIdentify(ConsGM) is run on the first chain from the structures in the enzyme
dataset, the distribution of distances between predicted and real centroids is very
similar between SitesIdentify(ConsGM) and Consurf (see Figure 3.24).
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
0 10 20 30 40 50
Distance between predicted and real centroid (rounded to nearest Ångstrom)
Cu
mu
lati
ve p
erce
nta
ge
of
set
SitesIdentify(ConsGM)for monomers
Consurf
Figure 3.24 Comparison of distances between the real centroid and the predicted centroid for
Consurf and SitesIdentify(ConsGM) run on the first chain of the enzyme structures.

165
3.5 Results: SitesIdentify web-server
SitesIdentify is available to run for single protein entries at
www.manchester.ac.uk/bioinformatics/sitesidentify/ or can be downloaded to run
offline for multiple proteins. It requires some basic user-input via a web-browser (see
Figure 3.25). Once this information is validated a new job is initiated. The average
calculation time per protein is approximately 6 minutes when using the method
including conservation information and approximately 2 minutes if only using charge-
based calculations. If the protein takes longer than 45 minutes to produce results,
which may occur for very large proteins, the job is terminated and the user is notified
by email.
Upon completion of a job an email is sent to the user at the address specified, which
provides a link to the results page. The results page displays a Jmol applet illustrating
the protein structure with the predicted site residues highlighted, a text list of the
predicted residues and a link to a text file containing the predicted residue information
(see Figure 3.26 for an example). The methods used in SitesIdentify can distinguish
between enzyme and non-enzyme with a high degree of accuracy 33 and so an
enzyme/non-enzyme prediction is also given along with the functional site prediction.

166
Figure 3.25 Screenshot for SitesIdentify showing the required user input fields.
A user can either input a pre-existing PDB code and whether to use the asymmetric or
biological unit structure or upload their own PDB-style structure file. All fields are compulsory.

167
Figure 3.26 Screenshot of an example results output for SitesIdentify.
The output for 1j2c (rat heme oxygenase-1) when submitted using the geometry-based method
and a 10Ǻ radius. The list of active site residues is truncated for display purposes.

168
SitesIdentify only gives a prediction for a single functional site as it makes predictions
based on the single highest peak potential. In oligomeric structures, however, the same
site may be present in multiple subunits and so where there is a similar site in other
chains SitesIdentify identifies it as another possible site. These residues are highlighted
in purple on the protein structure (see Figure 3).
Figure 3.27 An example of highlighted residues in an alternative predicted site.
The biological unit structure for 2af4 (phosphotransacetylase) is a homodimer and identical
active sites are present on both chains. SitesIdentify identifies only one site (in red), but the
annotation is transformed onto the other chain in order to identify the other active site (shown
in purple).
Where a user inputs a pre-existing PDB ID to SitesIdentify, the option to use either the
asymmetric unit or the biological unit structure is given. Where the real functional site
is formed in or near subunit boundaries in the biological unit, running SitesIdentify on
the asymmetric unit may fail to give the correct prediction. Some biological units,
however, may give a false prediction particularly where there is an internal void formed
by a cyclical arrangement of subunits. Such voids tend to be well-buried, more so than
the real surface clefts, and the residues on the edges of these voids may be

169
evolutionarily conserved in order to retain the quaternary structure. These voids are
therefore sometimes incorrectly selected as predicted functional sites. Where a
biological unit has an internal void it would be useful to also run SitesIdentify on the
asymmetric unit. For example, running the asymmetric unit for 1B6T through the
SitesIdentify server locates the functional site in the correct location, however the site
is predicted incorrectly for the biological unit as the void formed in the centre of the
molecule (see Figure 3.28).
Figure 3.28 An example of differential site prediction between asymmetric and biological unit
structures.
The active site predicted for the asymmetric unit of 1b6t (phosphopantetheine
adenylyltransferase) is reasonably close to the bound ligand shown in part A. The biological
unit is formed by a cyclical arrangement of the asymmetric unit and when SitesIdentify is run
on this structure it incorrectly identifies the central void as the enzyme active site (part B).

170
3.6 Discussion
Both Consurf and SitesIdentify(ConsGM) are based around predicting conserved
residues as functional site residues but whilst Consurf appears to perform slightly
better overall, it could not produce predictions for three of the proteins in the set
(1C3J, 1DMU and 1PGS) as it was unable to identify enough homologues.
SitesIdentify(ConsGM) uses both a combination of residue conservation information
with an electrostatics-based cleft-finding algorithm and so still gives predictions where
there is little or no conservation information available. SitesIdentify was able to recall
100% of the annotated catalytic residues for the three proteins in this set for which
Consurf did not make any prediction. SitesIdentify, therefore, is likely to give better
predictions for structures from uncharacterised families, such as those being generated
by structural genomics initiatives.
In general, most methods perform better for predicting the functional sites of enzymes
than non-enzymes. The best-performing methods for the enzyme set, Consurf and
SitesIdentify(ConsGM), which both use conservation as a predictive feature are
overtaken by a non-conservation-based approach, SitesIdentify(GM) for the non-
enayme set. Residue conservation is known to be less indicative of functionality for
non-enzymes than for enzymes9,60,62. A study of four non-enzyme families by Magliery
et al. found that rather than binding sites being conserved, they showed a higher degree
of variation than the rest of the protein 9. This may explain why some conservation-
based methods, including those tested here (SitesIdentify and Consurf) and those not50;
62, report better accuracies in predicting functional sites of enzymes than non-enzymes.
It is therefore useful to the user if analysing a protein of unknown function to predict
whether the structure is an enzyme or non-enzyme when choosing which method of
SitesIdentify to use. Indeed, the webserver implementation of both SitesIdentify
methods includes an enzyme/non-enzyme prediction in the results output in order to
allow the user to select which method is likely to give the best functional site
prediction.
PDBSiteScan achieved the lowest absolute and relative recall rates (28.1% and 38.4%,
respectively) and also the largest average distance between predicted and real active-site

171
centroids (15.5Å). PDBSiteScan scans the query protein against proteins of known
annotation. In this analysis the test set consists of enzymes with known annotation and
therefore it was necessary to reject predictions that simply accessed the annotation of
any of these test proteins. As the number of proteins with well-characterised active site
information is limited, removing these proteins from the set that PDBSiteScan
compares to will obviously reduce the prediction power of the method. If tested on
proteins outside of this set (i.e. proteins with uncharacterised functional sites) the
prediction accuracy may increase.
Q-SiteFinder identifies energetically favourable methyl binding sites by calculating the
interaction energy between the protein and a methyl probe and then ranking clusters of
probes by their total interaction energy. Similar to the electrostatics-based method of
SitesIdentify, Q-SiteFinder is essentially a cleft-finding algorithm. Despite similar
approaches the uniform charge method of SitesIdentify achieves a 10% higher relative
recall rate than Q-SiteFinder. Both Q-SiteFinder and SitesIdentify performed better
than the other cleft-finding method, PASS, which also selects for cleft depth. Since
SitesIdentify implicitly detects the atom density around a cleft rather than the cleft
geometry itself, it suggests that this may be a contributing factor to the increased
accuracy over PASS.
It is interesting that whilst SitesIdentify(GM) and Crescendo use very different
approaches they give very similar accuracies on the enzyme dataset, suggesting that
both conservation and geometrical information are equally useful in identifying
functional sites. The combination of both of these approaches in
SitesIdentify(ConsGM) further improves the accuracy achieved by either one alone.
Both of the SitesIdentify methods are delivered as a publicly-available tool via a
webserver at www.manchester.ac.uk/bioinformatics/sitesidentify.

172
3.7 References
1. Bray, T., Chan, P., Bougouffa, S., Greaves, R., Doig, A. J. & Warwicker, J. (2009). SitesIdentify: a protein functional site prediction tool. BMC Bioinformatics 10, 379.
2. Capra, J. A. & Singh, M. (2007). Predicting functionally important residues from sequence conservation. Bioinformatics 23, 1875-82.
3. Manning, J. R., Jefferson, E. R. & Barton, G. J. (2008). The contrasting properties of conservation and correlated phylogeny in protein functional residue prediction. BMC Bioinformatics 9, 51.
4. Zhang, T., Zhang, H., Chen, K., Shen, S., Ruan, J. & Kurgan, L. (2008). Accurate sequence-based prediction of catalytic residues. Bioinformatics 24, 2329-38.
5. Fischer, J. D., Mayer, C. E. & Soding, J. (2008). Prediction of protein functional residues from sequence by probability density estimation. Bioinformatics 24, 613-20.
6. Liang, S., Zhang, C., Liu, S. & Zhou, Y. (2006). Protein binding site prediction using an empirical scoring function. Nucleic Acids Res 34, 3698-707.
7. Chelliah, V., Chen, L., Blundell, T. L. & Lovell, S. C. (2004). Distinguishing structural and functional restraints in evolution in order to identify interaction sites. J Mol Biol 342, 1487-504.
8. Berezin, C., Glaser, F., Rosenberg, J., Paz, I., Pupko, T., Fariselli, P., Casadio, R. & Ben-Tal, N. (2004). ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics 20, 1322-4.
9. Magliery, T. J. & Regan, L. (2005). Sequence variation in ligand binding sites in proteins. BMC Bioinformatics 6, 240.
10. Caffrey, D. R., Somaroo, S., Hughes, J. D., Mintseris, J. & Huang, E. S. (2004). Are protein-protein interfaces more conserved in sequence than the rest of the protein surface? Protein Sci 13, 190-202.
11. Greaves, R. & Warwicker, J. (2005). Active site identification through geometry-based and sequence profile-based calculations: burial of catalytic clefts. J Mol Biol 349, 547-57.
12. Wang, K., Horst, J. A., Cheng, G., Nickle, D. C. & Samudrala, R. (2008). Protein meta-functional signatures from combining sequence, structure, evolution, and amino acid property information. PLoS Comput Biol 4, e1000181.
13. Ausiello, G., Zanzoni, A., Peluso, D., Via, A. & Helmer-Citterich, M. (2005). pdbFun: mass selection and fast comparison of annotated PDB residues. Nucleic Acids Res 33, W133-7.
14. Porter, C. T., Bartlett, G. J. & Thornton, J. M. (2004). The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res 32, D129-33.
15. Ivanisenko, V. A., Pintus, S. S., Grigorovich, D. A. & Kolchanov, N. A. (2005). PDBSite: a database of the 3D structure of protein functional sites. Nucleic Acids Res 33, D183-7.

173
16. Hulo, N., Bairoch, A., Bulliard, V., Cerutti, L., Cuche, B. A., de Castro, E., Lachaize, C., Langendijk-Genevaux, P. S. & Sigrist, C. J. (2008). The 20 years of PROSITE. Nucleic Acids Res 36, D245-9.
17. Torrance, J. W., Bartlett, G. J., Porter, C. T. & Thornton, J. M. (2005). Using a library of structural templates to recognise catalytic sites and explore their evolution in homologous families. J Mol Biol 347, 565-81.
18. Ivanisenko, V. A., Pintus, S. S., Grigorovich, D. A. & Kolchanov, N. A. (2004). PDBSiteScan: a program for searching for active, binding and posttranslational modification sites in the 3D structures of proteins. Nucleic Acids Res 32, W549-54.
19. Binkowski, T. A., Freeman, P. & Liang, J. (2004). pvSOAR: detecting similar surface patterns of pocket and void surfaces of amino acid residues on proteins. Nucleic Acids Res 32, W555-8.
20. Chang, D. T., Weng, Y. Z., Lin, J. H., Hwang, M. J. & Oyang, Y. J. (2006). Protemot: prediction of protein binding sites with automatically extracted geometrical templates. Nucleic Acids Res 34, W303-9.
21. Jambon, M., Andrieu, O., Combet, C., Deleage, G., Delfaud, F. & Geourjon, C. (2005). The SuMo server: 3D search for protein functional sites. Bioinformatics 21, 3929-30.
22. Kleywegt, G. J. (1999). Recognition of spatial motifs in protein structures. J Mol Biol 285, 1887-97.
23. Shulman-Peleg, A., Nussinov, R. & Wolfson, H. J. (2005). SiteEngines: recognition and comparison of binding sites and protein-protein interfaces. Nucleic Acids Res 33, W337-41.
24. Stark, A. & Russell, R. B. (2003). Annotation in three dimensions. PINTS: Patterns in Non-homologous Tertiary Structures. Nucleic Acids Res 31, 3341-4.
25. Kristensen, D. M., Chen, B. Y., Fofanov, V. Y., Ward, R. M., Lisewski, A. M., Kimmel, M., Kavraki, L. E. & Lichtarge, O. (2006). Recurrent use of evolutionary importance for functional annotation of proteins based on local structural similarity. Protein Sci 15, 1530-6.
26. Goyal, K., Mohanty, D. & Mande, S. C. (2007). PAR-3D: a server to predict protein active site residues. Nucleic Acids Res 35, W503-5.
27. Bartlett, G. J., Porter, C. T., Borkakoti, N. & Thornton, J. M. (2002). Analysis of catalytic residues in enzyme active sites. J Mol Biol 324, 105-21.
28. Tseng, Y. Y. & Liang, J. (2007). Predicting enzyme functional surfaces and locating key residues automatically from structures. Ann Biomed Eng 35, 1037-42.
29. Tang, Y. R., Sheng, Z. Y., Chen, Y. Z. & Zhang, Z. (2008). An improved prediction of catalytic residues in enzyme structures. Protein Eng Des Sel 21, 295-302.
30. Laskowski, R. A., Luscombe, N. M., Swindells, M. B. & Thornton, J. M. (1996). Protein clefts in molecular recognition and function. Protein Sci 5, 2438-52.
31. Gutteridge, A., Bartlett, G. J. & Thornton, J. M. (2003). Using a neural network and spatial clustering to predict the location of active sites in enzymes. J Mol Biol 330, 719-34.
32. Brady, G. P., Jr. & Stouten, P. F. (2000). Fast prediction and visualization of protein binding pockets with PASS. J Comput Aided Mol Des 14, 383-401.
33. Bate, P. & Warwicker, J. (2004). Enzyme/non-enzyme discrimination and prediction of enzyme active site location using charge-based methods. J Mol Biol 340, 263-76.

174
34. Elcock, A. H. (2001). Prediction of functionally important residues based solely on the computed energetics of protein structure. J Mol Biol 312, 885-96.
35. Ota, M., Kinoshita, K. & Nishikawa, K. (2003). Prediction of catalytic residues in enzymes based on known tertiary structure, stability profile, and sequence conservation. J Mol Biol 327, 1053-64.
36. Tong, W., Williams, R. J., Wei, Y., Murga, L. F., Ko, J. & Ondrechen, M. J. (2008). Enhanced performance in prediction of protein active sites with THEMATICS and support vector machines. Protein Sci 17, 333-41.
37. Dessailly, B. H., Lensink, M. F. & Wodak, S. J. (2007). Relating destabilizing regions to known functional sites in proteins. BMC Bioinformatics 8, 141.
38. Laurie, A. T. & Jackson, R. M. (2005). Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 21, 1908-16.
39. Wei, Y., Ko, J., Murga, L. F. & Ondrechen, M. J. (2007). Selective prediction of interaction sites in protein structures with THEMATICS. BMC Bioinformatics 8, 119.
40. Ondrechen, M. J., Clifton, J. G. & Ringe, D. (2001). THEMATICS: a simple computational predictor of enzyme function from structure. Proc Natl Acad Sci U S A 98, 12473-8.
41. Brylinski, M., Prymula, K., Jurkowski, W., Kochanczyk, M., Stawowczyk, E., Konieczny, L. & Roterman, I. (2007). Prediction of functional sites based on the fuzzy oil drop model. PLoS Comput Biol 3, e94.
42. Amitai, G., Shemesh, A., Sitbon, E., Shklar, M., Netanely, D., Venger, I. & Pietrokovski, S. (2004). Network analysis of protein structures identifies functional residues. J Mol Biol 344, 1135-46.
43. del Sol, A., Fujihashi, H., Amoros, D. & Nussinov, R. (2006). Residue centrality, functionally important residues, and active site shape: analysis of enzyme and non-enzyme families. Protein Sci 15, 2120-8.
44. Chea, E. & Livesay, D. R. (2007). How accurate and statistically robust are catalytic site predictions based on closeness centrality? BMC Bioinformatics 8, 153.
45. Ben-Shimon, A. & Eisenstein, M. (2005). Looking at enzymes from the inside out: the proximity of catalytic residues to the molecular centroid can be used for detection of active sites and enzyme-ligand interfaces. J Mol Biol 351, 309-26.
46. Cheng, G., Qian, B., Samudrala, R. & Baker, D. (2005). Improvement in protein functional site prediction by distinguishing structural and functional constraints on protein family evolution using computational design. Nucleic Acids Res 33, 5861-7.
47. Landgraf, R., Xenarios, I. & Eisenberg, D. (2001). Three-dimensional cluster analysis identifies interfaces and functional residue clusters in proteins. J Mol Biol 307, 1487-502.
48. Landau, M., Mayrose, I., Rosenberg, Y., Glaser, F., Martz, E., Pupko, T. & Ben-Tal, N. (2005). ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res 33, W299-302.
49. Thibert, B., Bredesen, D. E. & del Rio, G. (2005). Improved prediction of critical residues for protein function based on network and phylogenetic analyses. BMC Bioinformatics 6, 213.
50. Glaser, F., Morris, R. J., Najmanovich, R. J., Laskowski, R. A. & Thornton, J. M. (2006). A method for localizing ligand binding pockets in protein structures. Proteins 62, 479-88.
51. SitesIdentify.

175
52. Bray, T., Doig, A. J. & Warwicker, J. (2009). Sequence and structural features of enzymes and their active sites by EC class. J Mol Biol 386, 1423-36.
53. Golovin, A. & Henrick, K. (2008). MSDmotif: exploring protein sites and motifs. BMC Bioinformatics 9, 312.
54. Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389-402.
55. Lee, B. & Richards, F. M. (1971). The interpretation of protein structures: estimation of static accessibility. J Mol Biol 55, 379-400.
56. Stawiski, E. W., Mandel-Gutfreund, Y., Lowenthal, A. C. & Gregoret, L. M. (2002). Progress in predicting protein function from structure: unique features of O-glycosidases. Pac Symp Biocomput, 637-48.
57. Rose, G. D. & Roy, S. (1980). Hydrophobic basis of packing in globular proteins. Proc Natl Acad Sci U S A 77, 4643-7.
58. Jones, S. & Thornton, J. M. (1997). Analysis of protein-protein interaction sites using surface patches. J Mol Biol 272, 121-32.
59. Eisenberg, D., Schwarz, E., Komaromy, M. & Wall, R. (1984). Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol 179, 125-42.
60. Kyte, J. & Doolittle, R. F. (1982). A simple method for displaying the hydropathic character of a protein. J Mol Biol 157, 105-32.
61. Shindyalov, I. N. & Bourne, P. E. (1998). Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng 11, 739-47.
62. Burgoyne, N. J. & Jackson, R. M. (2006). Predicting protein interaction sites: binding hot-spots in protein-protein and protein-ligand interfaces. Bioinformatics 22, 1335-42.

176
Chapter 4: Predicting EC class from
enzyme structure
The aim of this work is to be able to predict protein function from structural and
sequence features without transferring functional annotation from homologous
proteins. Here, this relates to the prediction of top EC classes from the structural
features of a group of structurally non-homologous enzymes. Two prediction models
are presented in this chapter, one developed on a set of enzymes with known active site
locations and another on a larger set of enzymes that may, or may not, have known
active site locations. The former prediction method was included, along with the work
in Chapter 2, in a published article in J. Mol. Biol1.
4.1 Introduction
The importance of being able to predict protein function from structure (or sequence)
is reflected in the growing number of structures in the PDB2 that have little or no
annotation. It is estimated that between 1% and 3% of the structures in the PDB3,4
and approximately 40% of the sequences in GenBank3 have an ‘unknown function’
annotation. These are conservative estimates since many proteins are tentatively
assigned functions, or vague annotation, based on similarity to others. The rate at
which proteins structures are being solved is higher than the capacity for experimental
groups to characterise them. This has led to the rise in the number of protein
structures lacking functional annotation and presents a requirement for automatic
annotation methods.
Traditional methods to automatically predict protein function have centered around
transferring annotation from a characterised homologue. It is, however, one of the
aims of structural genomics initiatives to resolve proteins that may occupy more
remote areas of protein fold space. This has increased the number of structures for
which a functionally annotated homologue cannot be reliably identified. This has
produced a subset of functionally uncharacterized proteins for which tradition

177
similarity-based methods fail and therefore the need for methods that do not rely on
identifying a homologue has arisen.
A number of approaches exist to predict protein function from sequence or structure
in the absence of homology. Sequence features, such as hydrophobicity, polarity and
polarisability, have been used to predict classes of metal binding proteins5, lipid binding
proteins6, RNA binding proteins7 and enzyme8 and other function classifications9
without the inference of homology to other proteins.
Other approaches have included structural features to predict protein function. Within
a study of structural features of the proteases, Stawiski et al. found that they exhibit
similar characteristics such as smaller than average surface areas and higher Cα
densities, regardless of whether or not they were evolutionarily related10. They also
showed different secondary structure content to the non-proteases. By using these
features in a machine learning approach they were able to define a set of structural
classifiers that could predict whether a protein is a protease or non-protease with an
accuracy of over 86%. In a later study, Stawiski et al. also reported structural features
that are characteristic of the O-glycosidases such as distinctive electrostatic properties
of the proteins surface, despite differences in the overall fold11.
Previous work 12 has shown that the use of structural features of proteins in a machine
learning method can predict the top EC classification of an enzyme. It is hypothesised
that to further improve these methods, structural features specifically relating to a
protein’s functional site, such as active site specific amino acid compositions and
secondary structure content, may increase the prediction power of this method

178
4.1.1 Machine Learning Theory
Machine learning is a computational method that exploits relationships between
variables in datasets to make predictions about further outcomes. These outcomes, for
example, can be associations (i.e. predicting whether a customer X is likely to buy
product B based on them having bought product A) or classifications (predicting the
socio-economic group of customer X based on the features of their purchases). Most
machine learning methods take advantage of the underlying probability distributions of
feature values and can identify complex patterns in large datasets that are not easily
detectable without the use of such methods.
A vast number of machine learning approaches have been proposed, which fall broadly
into a number of categories; supervised, unsupervised, semi-supervised and
reinforcement learning. Supervised learning occurs when the input data for the
training set is labeled with the correct outcomes. The machine learning method then
derives classification rules based on the observed relationships between the feature
values and the true classification. These rules are then used to predict an outcome
based upon an unlabeled test case. In contrast, unsupervised machine learning
methods do not require the input training data to be labeled with the correct outcome.
Instead, feature values are used to cluster the data into sets of similar cases with some
optimum separation. The rules used to cluster this data are then used to identify a
cluster given a new test case.
Semi-supervised learning is a compromise between the two above methods, where the
training data contains a mixture of labeled and unlabeled data. The labeled data can be
used to seed or guide the clustering algorithm used to separate the unlabeled data.
Reinforcement machine learning methods assess the outcomes of sets of action
sequences to generate a new sequence of actions that are likely to yield a given
outcome, regardless of the outcome of individual actions. This type of machine
learning is often used on game-playing type applications where there are multiple
routes to a successful outcome with complex dependencies between actions.

179
The work described in this chapter lends itself to supervised learning since a large
number of enzyme structures are available with known classification labels (EC
numbers). There are a number of issues and limitations attached to using supervised
learning methods. Firstly, the size of the dataset is important to the outcome of
supervised learning methods. Only very simple classification functions can be learnt
from a small dataset and so complex classification functions require large datasets. The
ability of a method to find the correct classification function therefore depends on the
availability of suitable data.
The number of input variables (features) in the training (and test) dataset also has a
large impact on the accuracy of supervised learning models. Large numbers of features
increase the dimensionality that the method must use to find a suitable classifier and so
increase the complexity of the problem. The optimal classifier tends to be formed
from only a subset of the total features. Incorporating irrelevant features into a model
makes it too detailed to be accurate on more general datasets and can be prevented by
removing features from the dataset using a feature selection algorithm.
It is important in all supervised learning methods that both the training set and the test
set are non-redundant, both within the sets and across the sets. If the same, or highly
similar, cases exist in the training set then the classifier will be skewed towards
classifying on the feature values from the overrepresented set of cases despite this bias
not being present in the test population. Similarly, if redundancy exists within the test
set then the accuracy will be either artificially enhanced or reduced depending on
whether that set of redundant cases are predicted correctly by the model or not. If
redundancy exists between the test and the training set then it will result in a classifier
that matches test cases to the training cases rather than forming a classifier that is
representative of the features that describe the classes.
The degree of heterogeneity in the data also can affect supervised learning methods.
Some supervised learning methods find it difficult to handle data where values of
features are on different scales to each other (or the dataset contains both continuous
and discrete data). Attributes that have small absolute values will exhibit smaller
absolute variance in their values and may be overshadowed by a higher absolute, but
smaller relative, variance in an attribute with a larger absolute value. Decision trees

180
(where a classifier is obtained using a series of binary rules to split the data) can handle
heterogeneity in the data well, but other methods such as linear regression and support
vector machines need to have each of their features scaled in a consistent manner
(usually -1 to 1 or 0 to 1) to reduce this bias.
There are a wide range of algorithms available that use supervised learning methods,
however a support vector machine (SVM) approach is used in the second prediction
method in this analysis. SVMs have been widely used in bioinformatics prediction
problems with success5-9,12,13 and are capable of high prediction accuracies.
4.1.1.1 Support Vector Machines
The SVM model can be thought of as representing a set of points in high dimensional
space, which are orientated according to their attributes. The model attempts to
identify a hyperplane that separates these points into their labeled classifications. The
optimum hyperplane is the one that separates the data into each class with the largest
distance between the hyperplane and the nearest points in each class (see Figure 4.1 for
a schematic representation). The ability for this method to find the optimum solution
quickly, by finding the hyperplane that divides the groups with the largest gap (i.e.
maximizing the margin between them) is one of the major advantages of this method.
Other approaches may identify a solution that separates the data, but not necessarily
the optimum and most general classifier and hence are susceptible to finding local
minima.

181
Figure 4.1 A schematic diagram representing the classification of two groups of data by an SVM
model.
Whilst the hyperplane A separates the data correctly the distance between the nearest points to
hyperplane A in each group (represented by grey dotted lines) is smaller than that of an
alternative hyperplane, B. The nearest points to the hyperplane (those that delimit the
maximum margin between the points and the hyperplane) are termed the support vectors.
Figure 4.1 represents a linear classifier, where cases can be separated by a linear
function. On real-world problems it is not often possible to separate the data using a
linear function and a technique called the kernel trick needs to be used. This maps the
points to a higher-dimensional space in order to achieve a separation by a hyperplane
with a linear function (see Figure 4.2).

182
Figure 4.2 A schematic diagram representing how the transformation of data into a higher-
dimensional space by using kernel functions can allow the separation of the data by a linear
function.
SVMs are binary classifiers and so need to be modified in order to classify into more
than two groups. One way of doing this is by conducting binary classifications
A
B

183
between all pairs of classes, which is termed one-versus-one classification. A model is
built for the binary classification between all pairs and then the test case is evaluated by
each model, assigning a vote to the class predicted by each model. The class with the
most votes is the group that the test case is classified into. Another method is one-
versus-all where a model is built for each class versus all other classes (i.e. EC1 vs.
EC2-6). The classification with the highest function output (the largest classifier
margin) is the final predicted class. Another way to predict from multiple classes is by
constructing a series of decision trees that compare individual classes and/or groups of
classes until a prediction of an individual class is reached (see Figure 4.3). The
structure of the decision tree can be decided upon by either unsupervised clustering
methods or evaluating a range of trees on a validation set.
Figure 4.3 An example of a decision tree that can be followed to classify into multiple groups
using binary classifications.
In order to achieve the best accuracy possible on a test set, it is necessary to find the
best parameters to use for the SVM. One of the most useful parameters to alter is the
error penalty C. This controls how incorrect classifications are tolerated in training the
classification model. The model that correctly predicts all cases in a training set may
not give a high accuracy on the test set as it may be overly specific in describing the
training data. Where there is noise in the dataset or there are cases that are difficult to
separate, it may be more beneficial to allow misclassification in training the model in
order to correctly classify the majority of test cases (see Figure 4.4). A high value of C

184
increases the penalty cost of creating a model that misclassifies examples and therefore
may produce a model that overfits the data. If the penalty is too low then the model
may misclassify too many examples and produce a model which is not meaningful.
Where non-linear kernel functions are used to generate the model, it may be useful to
optimise the values of kernel parameters. The γ parameter in the radial basis function
(RBF), a function whose value depends only on the distance from origin, specifies the
flexibility of the function. A high value of γ enables the kernel to closely fit the
separation of the training data and therefore may cause overfitting, whereas a small
value of γ may over generalise the model. Optimal values for each of these parameters
vary according to the classification problem and should be searched in order to identify
the best parameter values.
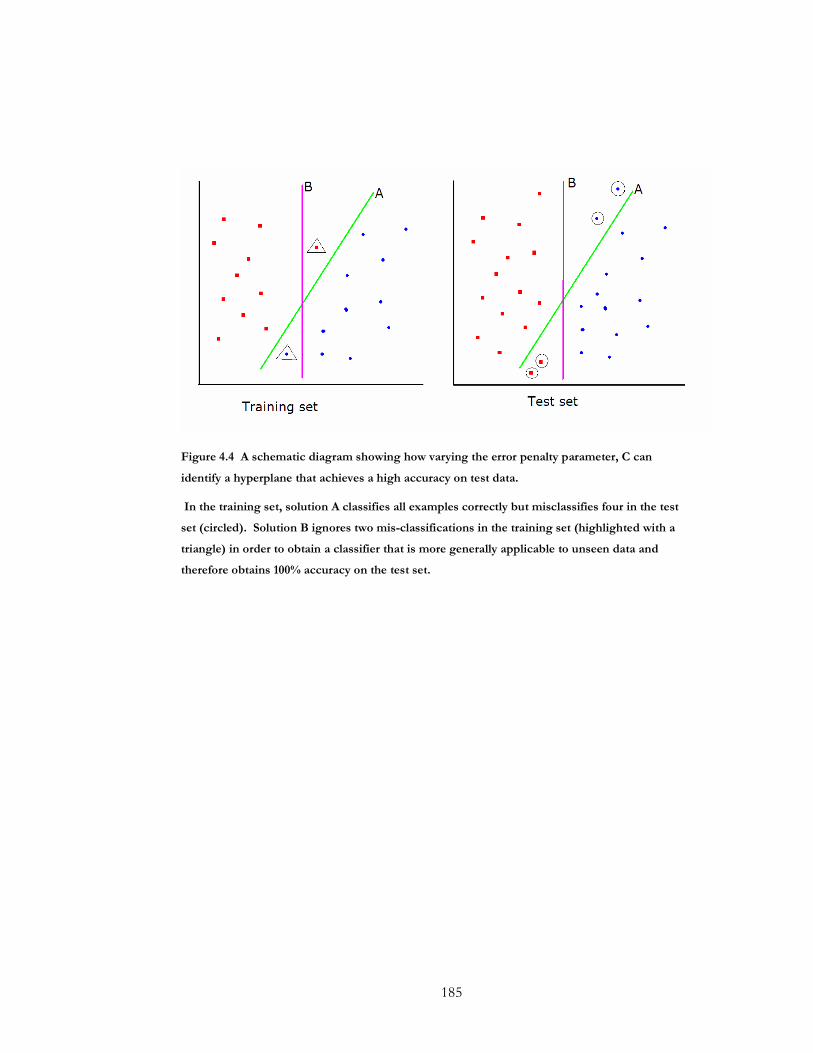
185
Figure 4.4 A schematic diagram showing how varying the error penalty parameter, C can
identify a hyperplane that achieves a high accuracy on test data.
In the training set, solution A classifies all examples correctly but misclassifies four in the test
set (circled). Solution B ignores two mis-classifications in the training set (highlighted with a
triangle) in order to obtain a classifier that is more generally applicable to unseen data and
therefore obtains 100% accuracy on the test set.

186
4.2 Methods
4.2.1 Dataset Creation
There are two datasets used in this chapter. Firstly, a prediction method is created that
uses features of known active sites and hence uses a dataset of enzymes with known
active sites (Dataset 4.1). Secondly, a prediction method is developed for enzymes
where the location of the active site is not known, and so enzymes without known
active site locations are introduced to the dataset (Dataset 4.2).
For the first method, the same dataset as in Chapter 2 was used. The creation of the
dataset is explained in detail in 2.2.1 and 2.3.1. In brief, the redundancy cull involves
ranking each enzyme in each class by descending AEROSPACI14 score and removing
any lower-ranked enzymes that share a domain from the same SCOP15 superfamily as
the domain containing the active site.
Dataset 4.2 was created in a similar manner as Dataset 4.1, however instead of using
enzymes that have known active site locations in the Catalytic Sites Atlas (CSA16)
Dataset 4.2 originates from all enzymes in the PDB that have a biological unit file. If
the PDB file contains an EC annotation then the top EC number is assigned to the
PDB. In the case where there is more than one EC classification for one PDB file the
enzyme is added to all classes for which there is annotation. If there is no EC number
in the PDB file, its corresponding Uniprot17 entry is searched for EC annotation. The
protein is assumed to be a non-enzyme if there is no EC annotation in either the PDB
file or the Uniprot entry. These non-enzymes are then checked manually to identify
any obvious omissions in EC number allocation (here, there were 105 cases of enzymes
that had to be manually annotated with an EC number). If the words “putative”,
“hypothetical”, “predicted”, “similarity”, “unknown” or “not known” were found in
the function comment line of the Uniprot entry then the corresponding PDB was
discarded as its function may not be certain.

187
4.2.2 Defining Active Site Residues
In order to include active site features to use in the prediction method, the active site
residues must first be defined. For Dataset 4.1 the location of the catalytic residues
(and hence the active site) are known but for Dataset 4.2 the active site residues must
be predicted.
4.2.2.1 Dataset 4.1
The methods for defining active site features for Dataset 4.1 are described in detail in
2.2.2. Briefly, the geometric average is calculated from the coordinates of the Cβ atom
(or Cα for glycine) of the catalytic residues listed in the CSA. This is termed the
centroid. All residues that have at least one atom within a 10Å radius of the centroid
and that have at least 5Å2 of solvent-accessible surface area (SASA) are defined as
active site residues.
4.2.2.2 Dataset 4.2
As the location of the active site is not known for all enzymes in this set, its location is
predicted using the SitesIdentify(ConsGM) method described in 3.3.1 and 3.4.10. This
prediction method gives XYZ coordinates relating to the central point in the predicted
active site of a PDB structure. In the same way as for Dataset 4.1, active site residues
are defined as any residue which has at least one atom within a 10Å radius of this
centroid and have at least 5Å2 of SASA.
4.2.3 Calculating Features.
The list of features that are calculated for these prediction methods are given in Table
4.1. For features listed as “Total” the feature is calculated using the whole sequence or
structure (the biological unit structure) and active site features are calculated using only
the active site residues as defined above. For sequence features the sequence given in
the SEQRES entry in the PDB file is used.

188
Feature Active site (AS), Total (T), Surface (S)
Structural features
Surface area AS/T Relative active site surface area AS Secondary structure content AS/T Average atomic B-factor Relative active site B-factor
AS/T AS
Oligomeric status (number of chains) T Number of residues in the biological unit T Molecular weight T
Sequence features
Sequence length T Amino acid composition (for all 20 amino acids) AS/T/S Polar/Non-polar/Negative/Aromatic/Positive proportions
AS/T
Average hydrophobicity (Kyte Doolittle score) AS/T Average isoelectric point (pI) AS/T Low complexity regions* T Table 4.1 Features used in the EC class prediction methods.
* Low sequence complexity was recorded in the form of three features; a binary feature that
recorded whether a low complexity region was identified or not and, if so, the number of low
complexity regions and the total length of the low complexity sequence(s).
4.2.3.1 Structural Features
Solvent-accessible surface area was calculated by an in-house program SACALC (J.
Warwicker), which rolls a solvent probe around the surface of the proteins to estimate
the amount of surface area (Å2) that is accessible to the probe. The relative active site
surface area is the proportion of the total surface area that is contributed to by the
active site residues. Secondary structure states for each residue were taken from the
secondary structure annotation from the PDB file, which is generated by a program
that incorporates DSSP18 and Promotif19. The average atomic B-factor is calculated by
averaging the atomic B-factors over all atoms in the protein (or active site). The
relative atomic B-factor for the active site is calculated by dividing the active site
average B-factor by the total average B-factor. The molecular weight of each enzyme
was calculated by the Pepstats program, which is part of the EMBOSS package of
applications20.

189
4.2.3.2 Sequence Features
Active site amino acid compositions were calculated by dividing the number of each
residue type in the active site residues by the total number of residues in the active site.
Total amino acid composition and surface amino acid composition were calculated
similarly, either using all residues or only those with at least 5Å2 surface area
respectively.
The polarity/charge fractions were calculated by dividing the number of residues from
each group (in either the total biological unit or the active site) by number of residues
in the biological unit or active site.
Average hydrophobicity values were obtained by dividing the sum of the Kyte &
Doolittle21 values for each residue in the protein (or in the active site) by the number of
residues in the protein (or in the active site). The polar amino acids contained the
positively charged (R, H, K), negatively charged (D, E) and uncharged amino acids (N,
Q, S, T). The non-polar amino acids were represented by the aromatic amino acids (F,
W) and non-polar amino acids (G, A, V, L, I, P, M). Cysteine and Tyrosine were not
included as they can be either polar or non-polar depending on the pH of the
environment.
The isoelectric point (pI) of a protein is the pH at which the protein has a net electrical
charge of zero. The pI of each enzyme was calculated by the Pepstats program, which
is part of the EMBOSS package of applications20. Low complexity regions were
predicted using SEG, a program that identifies low complexity regions in sequences22.

190
4.2.4 Prediction Methods
4.2.4.1 Functional Classification where the Active Site is
Known.
The classification tool is built around the principle of comparing a vector of the feature
values for each protein to vectors of average values for each functional class. Variation
in features with small values, such as the active site tyrosine proportion, cannot be
compared to the variation achievable for features with larger values, such as surface
area. The values for each feature were therefore normalised on a scale from 0 to 1 to
reduce the bias caused by features with larger absolute values. The minimum value for
a feature over the whole set is set to 0 and the maximum set to 1 and values in between
are linearly scaled accordingly.
In order to avoid bias by allowing the classification tool to use information from the
test enzyme in the class-average vector, a leave-one-out analysis was carried out. Each
enzyme was iteratively removed from the set and the class-average vectors were
formed from the class average of each feature for the remaining enzymes. The vector
formed from feature values for the test protein was then compared to each class
average vector and the angle between them calculated with a scalar product (see Figure
4.5). The closest class-average vector to the test enzyme vector (the pair giving the
smallest angle between them), represented the functional class predicted for the test
enzyme.
To reduce the effects of overfitting by using all features in the prediction model, each
feature’s contribution to the accuracy was evaluated. The accuracy achieved using all
features was obtained, then each feature was removed individually and the effect on the
accuracy observed. If the accuracy decreased when a feature was removed then the
feature was deemed to contribute positively to the prediction model and vice versa.
Features were then ranked by their individual contribution to the prediction model and
the prediction accuracy was iteratively assessed using increasing top n-ranked features.
The top n-ranked features that gave the highest accuracy were used as the features for
the final prediction model.

191
Figure 4.5 A schematic representation of the vector comparison method used to predict the EC
class of enzymes with known active sites.
The cosine of the angle between two vectors can be obtained by the scalar product of the
vectors (a and b, where a is the test enzyme and b is the class average vector). The class vector
that gives the smallest angle θ (or the largest cosine θ) between itself and the test protein vector
is the class predicted.

192
4.2.4.2 Functional Prediction where the Active Site is not
Known
The classification method used to predict the enzyme class of enzymes without a
known active site location was created using a support vector machine learning package
called LIBSVM23. Firstly each feature in the set was scaled between 0 and 1, where the
lowest value observed for that feature was set to 0 and the highest value was set to 1.
The dataset (Dataset 4.2) was then randomly split into a training and a test set, which
contains 625 (90%) and 70 (10%) enzymes, respectively.
The default kernel used in LIBSVM is the radial basis function (RBF), which is used in
this analysis as it can handle classifiers that are not linearly associated with each group.
LIBSVM also has an internal function to handle multi-class classification problems,
which is based on a one-against-one algorithm. Each class is compared against all
other classes and a model for the binary classification between each pair is constructed.
Each test case is then evaluated by each model and a vote is assigned to the class
predicted by each of the models. The class receiving the maximum number of votes is
the one assigned to that test case.
Two of the most influential factors when training a machine learning algorithm are the
parameters used and the features used. As discussed in the introduction, the error
penalty parameter, C, and the kernel parameter, γ, can be varied to identify the values
that give the optimum prediction performance. LIBSVM provides a grid-searching
algorithm that searches a range of C and γ values (the default is log2-5 to log213 for C
and log2-15 to log23 for γ). The performance of each pair of parameters was evaluated
using 10-fold cross-validation on the training set and the parameter values giving the
best accuracy were recorded.
The dataset used in this analysis is unbalanced in that the class sizes are very different
(for example the largest class, EC3, has over 5 times as many enzymes as EC6). This
imbalance in class sizes can result in the predictions being dominated by the largest
classes. LIBSVM provides an option to weight the error penalty for each class in order
to penalise predictions from larger classes more than those from smaller classes in

193
order to balance the predictions. Here, the error penalty parameter C is inversely
weighted by the ratios of the class sizes relative to the largest.
The second biggest contributor to the effectiveness of a machine learning model is the
features used. Using too many features in a machine learning method can be
detrimental for two reasons; 1) features that do not contain information relating to the
classes can distract the model from using more meaningful features and 2) using too
many features can lead to overfitting. Overfitting occurs when a very detailed model is
built and optimised for a training set using a large number of features. This detailed
model will give good accuracy on cross-validation evaluation on the dataset, since it
accurately describes the data within it. When this detailed model is used on data
outside of this training set the intricacies that described the training data well may
hinder the model. Removing features from the model will increase the ability for the
model to generalise and therefore produce better accuracy on unseen data.
A backward-pruning method was used here to remove features that negatively
impacted on the prediction model in the same way as described in 4.2.4.1. The features
were ranked according to their usefulness to the model and the number of top-ranked
features that gave the best cross-validation prediction accuracy on the dataset were
retained. These features, along with the optimum parameters values found in the grid
search were used to create a model on the training dataset, which was then used to
classify the unseen data in the test set.

194
4.3 Predicting EC class for Enzymes with Known Active
Site Location
A leave-one-out classification of each enzyme in the dataset into a top EC class using
all features achieved an accuracy of 29.1%. Each feature was then individually
removed and the effect on the prediction accuracy was observed. If the accuracy
decreases on removal of a feature it is deemed to have contributed positively to the
prediction model. The features were then ranked according to how much they had
contributed to the prediction accuracy. The prediction accuracy was assessed using an
increasing number of top n-ranked features. The highest accuracy (33.1%) was
achieved using the 74 top-ranked features (see Figure 4.6). The features that were
removed in the final prediction method and their rank are listed in Table 4.2. To assess
how much active site features contributed to the model over whole-protein features,
the active site features were removed from the model. The resulting prediction
accuracy was 26.1%.
A prediction accuracy of 16.7% would be expected if each enzyme were randomly
assigned to a class. The features used here therefore contain information that is able to
classify enzymes into their top EC class with an accuracy of 16.4% better than random.
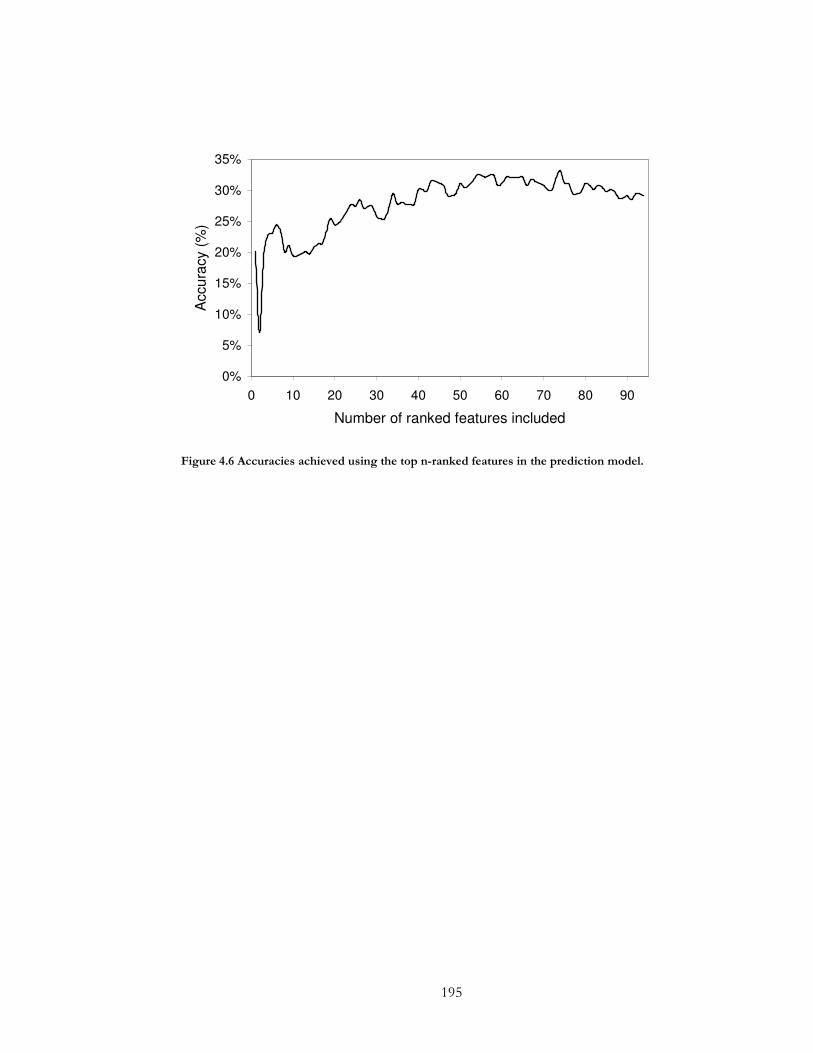
195
0%
5%
10%
15%
20%
25%
30%
35%
0 10 20 30 40 50 60 70 80 90
Number of ranked features included
Accu
racy (
%)
Figure 4.6 Accuracies achieved using the top n-ranked features in the prediction model.

196
Feature Rank
Amino acid compositions Active site GLU 89 Active site HIS 90 Active site LEU 94 Active site LYS 77 Active site THR 93 Active site VAL 82 Surface ALA 79 Surface CYS 75 Surface VAL 76 Total ASN 78 Total HIS 81 Total LEU 91 Other features Active site polar proportion 80 Active site beta sheet proportion 86 Average active site B-factor 83 Total negative proportion 85 Average hydrophobicity score 87 Proportion of non helix or sheet 92 Isoelectric point 84 Proportion of structure annotated as turn 88 Table 4.2 Features that are removed in the EC class prediction method where the active site
location is known.

197
4.4 Predicting EC class for Enzymes with Predicted Active
Site Locations
The methodology for creating Dataset 4.2 described in 4.2.1 produced a dataset of 695
enzymes. Table 4.3 shows the number of enzymes in each EC class. This was split into
a training set of 625 structures and a test set of 70 structures. The distribution of
structures between the classes in the test set is representative of the class sizes in the
total dataset.
The best values for C and γ parameters were searched for by using the grid-searching
algorithm supplied in the LIBSVM package. Using parameters that were not weighted
by the class sizes, a maximum 10-fold cross-validation prediction accuracy of 38.2%
was achieved using 0.5 for both the C and the γ parameter. When class size
weightings were used (EC1 = 2.4, EC2= 1.3, EC3 = 1, EC4 = 2.8, EC5 = 3.9, EC6 =
5.5), the best prediction accuracy achieved was 36.0% (C = 8 and γ = 0.5) using a
coarse-grained grid search of the default range of C and γ parameter values (see Figure
4.7). A further grid search was then performed using a scale of 2-1 to 28 for C and 2-5 to
25 for γ. The optimal prediction accuracy remained at 36.0% with C = 8 and γ = 0.5.
EC class Number of enzyme structures
1 97 2 181 3 231 4 84 5 60 6 42
Table 4.3 The number of enzyme structures in each class in Dataset 4.2

198
Figure 4.7 Prediction accuracies achieved using a default grid search method for the best C and
γ parameters. A) Shows the accuracies on a 2D plot and B) shows this in 3D.
A
B

199
To reduce the effects of overfitting, features were removed that negatively contributed
to the model accuracy. The accuracies achieved using the top n-ranked features are
shown in Figure 4.8. The best prediction accuracy (39.0%) was achieved with the top
91 features. The final prediction model was then trained using a value of 8.0 for the C
and 0.5 the γ parameter and removing the 10 lowest ranked features (see Table 4.4)
from the training set.
32%
33%
34%
35%
36%
37%
38%
39%
40%
0 10 20 30 40 50 60 70 80 90 100
Feature rank
Accu
racy (
%)
Figure 4.8 Accuracies achieved using the top-ranked features with 10-fold cross-validation on
the training set.
The red line shows the minimum number of top-ranked features (91) needed to achieve the
maximum accuracy (39.0%).
Rank Feature 92 Total PRO
93 Proportion of active site B-sheet
94 Molecular weight
95 Surface PRO
96 Active site THR
97 Active site GLY
98 Active site ALA
99 Active site VAL
100 Presence of low complexity regions
101 Active site PRO
Table 4.4 The 10 lowest ranked features that were removed from the dataset to train the final
model.

200
The final prediction model using the optimized parameters and the 91 top-ranked
features was run on the testing dataset, which resulted in 32.9% accuracy (23 correct
class predictions out of 70). Despite the class weightings for the C parameter, the
larger classes had the best prediction accuracy. The best model achieved without class
weightings (the top-ranked 91 features and values of 0.5 for both parameters) resulted
in a higher accuracy of 34.3%. These predictions were, however, dominated by the
two largest classes (EC2 and EC3) and no cases were predicted as EC4, 5 or 6 and only
three cases were predicted as EC1 (see Table 4.5). Introducing weightings for the C
parameter reduced the overall accuracy achieved but the predictions it made were
slightly more balanced between the classes (see Figure 4.7).
Due to the lack of balance in the accuracy achieved between the classes, this method is
of limited use in real-world function prediction problems. Assuming that the accuracy
expected by random class choice is the sum of the squares of the class sizes divided by
the size of the dataset, the accuracy expected by random is 22.3%. Whilst the accuracy
achieved by this prediction method is 12% higher than this, it is still below that of the
percentage accuracy achieved by predicting all test cases as the largest class, EC3
(37%). Even predicting the second largest class, EC2, for all test cases would achieve a
prediction accuracy better than random selection (27%).
Actual EC Classification
Predicted EC Classification 1 2 3 4 5 6
Total (% correct)
1 2 0 1 0 0 0 3 (66.7%)
2 1 7 10 3 4 0 25 (28.0%)
3 6 12 15 6 2 1 42 (35.7%)
Total (% correct)
9 (22.2%)
19 (36.8%)
26 (57.7%)
9 (0%)
6 (0%)
1 (0%)
Table 4.5 The number of predictions of each class made by the model without class weightings.
The correct predictions are highlighted in green. No predictions were made by the model for
EC4-6.
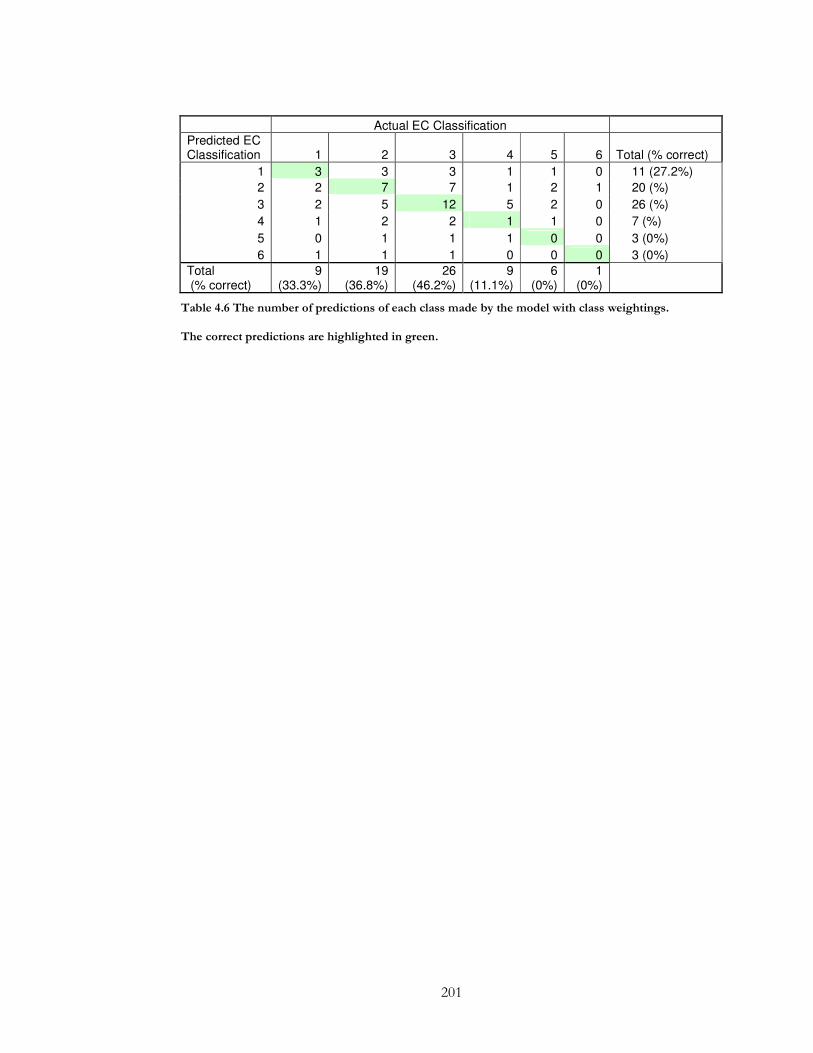
201
Actual EC Classification Predicted EC Classification 1 2 3 4 5 6 Total (% correct)
1 3 3 3 1 1 0 11 (27.2%)
2 2 7 7 1 2 1 20 (%)
3 2 5 12 5 2 0 26 (%)
4 1 2 2 1 1 0 7 (%)
5 0 1 1 1 0 0 3 (0%)
6 1 1 1 0 0 0 3 (0%)
Total (% correct)
9 (33.3%)
19 (36.8%)
26 (46.2%)
9 (11.1%)
6 (0%)
1 (0%)
Table 4.6 The number of predictions of each class made by the model with class weightings.
The correct predictions are highlighted in green.

202
4.5 Conclusions
The two prediction models presented in this chapter show that the information
contained in the features used in the model can be used to predict the top class of an
enzyme with better accuracy than random. The machine learning method, however, is
less able to deal with issues surrounding differing class sizes and results in imbalanced
prediction dominated by the largest classes.
The work in Chapter 2 attempts to deconstruct the relationships between differences in
features and the six functional classes on an individual basis. This showed trends in
individual features that were significantly different between the six EC classes for 20
features, of which three were explored further in relation to their functional
importance. The EC class prediction tools here attempt to quantify the usefulness of
these features in relation to predicting the top EC class of enzymes.
Of the 20 features that showed significant differences between EC classes listed in
Chapter 2, 6 of them were not used in the prediction model using known active sites
(prediction model 1) and one feature was not used in the machine learning method
(prediction model 2).
The active site aromatic proportion and the active site phenylalanine proportion were
not included in prediction model 1, but both strongly correlate with the active site
tryptophan proportion (from Chapter 2), which was included. Similarly, the active site
non-polar proportion was not included in prediction model 1 whilst active site
hydrophobicity, which correlated strongly with the active site non-polar proportion
(from Chapter 2), was included. The other three significant features not included in
prediction model 1, relative active site surface area, sequence length (which both
strongly correlated with each other) and total isoleucine proportion, were not strongly
correlated with other features that were included in the model. Only one of the
significantly different features (total proline composition) from the analysis in Chapter
2 was not used in the machine learning method (prediction model 2).

203
4.6 References
1. Bray T, Doig AJ, Warwicker J. Sequence and structural features of enzymes and their active sites by EC class. J Mol Biol 2009;386(5):1423-1436.
2. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res 2000;28(1):235-242.
3. Friedberg I, Jambon M, Godzik A. New avenues in protein function prediction. Protein Sci 2006;15(6):1527-1529.
4. Doppelt O, Moriaud F, Bornot A, de Brevern AG. Functional annotation strategy for protein structures. Bioinformation 2007;1(9):357-359.
5. Lin HH, Han LY, Zhang HL, Zheng CJ, Xie B, Cao ZW, Chen YZ. Prediction of the functional class of metal-binding proteins from sequence derived physicochemical properties by support vector machine approach. BMC Bioinformatics 2006;7 Suppl 5:S13.
6. Lin HH, Han LY, Zhang HL, Zheng CJ, Xie B, Chen YZ. Prediction of the functional class of lipid binding proteins from sequence-derived properties irrespective of sequence similarity. J Lipid Res 2006;47(4):824-831.
7. Han LY, Cai CZ, Lo SL, Chung MC, Chen YZ. Prediction of RNA-binding proteins from primary sequence by a support vector machine approach. Rna 2004;10(3):355-368.
8. Cai CZ, Han LY, Ji ZL, Chen YZ. Enzyme family classification by support vector machines. Proteins 2004;55(1):66-76.
9. Cai CZ, Han LY, Ji ZL, Chen X, Chen YZ. SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res 2003;31(13):3692-3697.
10. Stawiski EW, Baucom AE, Lohr SC, Gregoret LM. Predicting protein function from structure: unique structural features of proteases. Proc Natl Acad Sci U S A 2000;97(8):3954-3958.
11. Stawiski EW, Mandel-Gutfreund Y, Lowenthal AC, Gregoret LM. Progress in predicting protein function from structure: unique features of O-glycosidases. Pac Symp Biocomput 2002:637-648.
12. Dobson PD, Doig AJ. Predicting enzyme class from protein structure without alignments. J Mol Biol 2005;345(1):187-199.
13. Dobson PD, Doig AJ. Distinguishing enzyme structures from non-enzymes without alignments. J Mol Biol 2003;330(4):771-783.
14. Chandonia JM, Hon G, Walker NS, Lo Conte L, Koehl P, Levitt M, Brenner SE. The ASTRAL Compendium in 2004. Nucleic Acids Res 2004;32(Database issue):D189-192.
15. Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 1995;247(4):536-540.
16. Porter CT, Bartlett GJ, Thornton JM. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res 2004;32(Database issue):D129-133.
17. Apweiler R MM, O'Donovan C, Magrane M, Alam-Faruque Y, Antunes R, Barrell D, Bely B, Bingley M, Binns D, Bower L, Browne P, Chan WM, Dimmer E, Eberhardt R, Fedotov A, Foulger R, Garavelli J, Huntley R, Jacobsen J, Kleen M, Laiho K, Leinonen R, Legge D, Lin Q, Liu W, Luo J, Orchard S, Patient S, Poggioli D, Pruess M, Corbett M, di Martino G, Donnelly

204
M, van Rensburg P, Bairoch A, Bougueleret L, Xenarios I, Altairac S, Auchincloss A, Argoud-Puy G, Axelsen K, Baratin D, Blatter MC, Boeckmann B, Bolleman J, Bollondi L, Boutet E, Quintaje SB, Breuza L, Bridge A, deCastro E, Ciapina L, Coral D, Coudert E, Cusin I, Delbard G, Doche M, Dornevil D, Roggli PD, Duvaud S, Estreicher A, Famiglietti L, Feuermann M, Gehant S, Farriol-Mathis N, Ferro S, Gasteiger E, Gateau A, Gerritsen V, Gos A, Gruaz-Gumowski N, Hinz U, Hulo C, Hulo N, James J, Jimenez S, Jungo F, Kappler T, Keller G, Lachaize C, Lane-Guermonprez L, Langendijk-Genevaux P, Lara V, Lemercier P, Lieberherr D, de Oliveira Lima T, Mangold V, Martin X, Masson P, Moinat M, Morgat A, Mottaz A, Paesano S, Pedruzzi I, Pilbout S, Pillet V, Poux S, Pozzato M, Redaschi N, Rivoire C, Roechert B, Schneider M, Sigrist C, Sonesson K, Staehli S, Stanley E, Stutz A, Sundaram S, Tognolli M, Verbregue L, Veuthey AL, Yip L, Zuletta L, Wu C, Arighi C, Arminski L, Barker W, Chen C, Chen Y, Hu ZZ, Huang H, Mazumder R, McGarvey P, Natale DA, Nchoutmboube J, Petrova N, Subramanian N, Suzek BE, Ugochukwu U, Vasudevan S, Vinayaka CR, Yeh LS, Zhang J. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res 2010;38(Database issue):D142-148.
18. Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983;22(12):2577-2637.
19. Hutchinson EG, Thornton JM. PROMOTIF--a program to identify and analyze structural motifs in proteins. Protein Sci 1996;5(2):212-220.
20. Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 2000;16(6):276-277.
21. Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982;157(1):105-132.
22. Wootton JC, Federhen S. Analysis of compositionally biased regions in sequence databases. Methods Enzymol 1996;266:554-571.
23. Chih-Chung Chang C-JL. LIBSVM : a library for support vector machines. Software available at http://wwwcsientuedutw/~cjlin/libsvm 2001.

205
Chapter 5: Gaussian Network Modeling of
Oligomeric Proteins
One of the observations resulting from an analysis of differences in structural and
sequence features between EC classes in Chapter 2 was that lyases (EC4) tend to prefer
to exist as oligomers and conversely hydrolases (EC3) preferred to exist as monomers.
The preference for different oligomeric statuses in these functions was linked to the
function’s over/under-representation at highly loaded points in metabolic networks. It
was suggested that some metabolically important enzymes may have evolved to exist as
oligomers in order to enable them to be regulated via mechanisms such as
cooperativity.
Due to the difficulty in obtaining biochemical data for a large amount of structures in
order to further investigate this theory, a method to detect cooperative action from
enzyme structure was required. Currently no such computational method exists for
this purpose and this chapter begins with an attempt to address this. It goes on to
further investigate the patterns of coupled motions in protein structures in terms of
enzyme active sites and as a feature of oligomeric structures in general.
5.1 Introduction
5.1.1 Cooperativity in Oligomeric Enzymes
Cooperativity, in relation to oligomeric enzymes, describes enzymes where the affinity
of binding of a ligand at one binding site induces a change in the rate of binding of the
ligand at further sites on the enzyme. Bohr et al. first noticed that the oxygen binding
curve for hemoglobin was sigmoidal and suggested that the binding of the first oxygen
molecule made it easier for subsequent oxygen molecules to bind1.

206
Many enzymes, especially monomeric enzymes, have a constant affinity for binding of
their substrate as the substrate concentration increases until the enzyme approaches
saturation and the maximum rate of reaction is reached (termed Vmax). Figure 5.1a
shows how the rate of reaction varies with the concentration of the substrate for a
non-cooperative enzyme.
For cooperative enzymes, binding of a substrate molecule changes the affinity for
binding subsequent substrate molecules at other sites. This alters the rate of change in
the speed of the reaction as the substrate concentration is increased. The reaction rate
vs. substrate concentration curve for a positively cooperative enzyme would therefore
be sigmoidal (see Figure 5.1b).
In contrast to positively cooperative enzymes, substrate binding in negatively
cooperative enzymes reduces the affinity for the enzyme to bind further substrate
molecules. As the concentration of substrate increases the rate of increase of the
reaction rate diminishes. This produces a plot with a slope that is less steep than for a
non-cooperative protein (see Figure 5.1c).
A common measure of enzyme cooperativity is the Hill coefficient, which was
proposed by A. V. Hill in 1910 to explain the sigmoidal oxygen binding plot for
hemoglobin2. It describes the fraction of the enzyme saturated by substrate as a
function of the substrate concentration (derived from the Hill equation shown in
Equation 5.1). Enzymes for which there is no evidence of cooperativity have a Hill
coefficient of 1; a coefficient of more than 1 indicates a positively cooperative enzyme
and less than 1 signifies a negatively cooperative enzyme. The upper limit of an
enzyme’s Hill coefficient is the number of sites, and therefore subunits, that the
enzyme has.
Equation 5.1 The Hill equation.
The Hill coefficient is denoted by n, Kd is the equilibrium dissociation constant, [L] is the
concentration of the ligand and θ is the fraction of binding sites that are occupied by substrate.

207
A
B
C
Figure 5.1 Example reaction rate (v/Vmax) vs.
substrate concentration ([S]) for a non-
cooperative (A), a positively cooperative (B)
and a negatively cooperate enzyme (C).
The grey line in figure shows the curve expected
from a non-cooperative enzymes in comparison
to the negative cooperative black line.

208
The response coefficient, RS, was proposed by Koshland et al. as a measure of an
enzyme’s sensitivity3. It measures the difference between the concentrations of
substrate required at 10% and 90% of the maximum reaction rate. For enzymes that
follow Michaelis-Menten kinetics the response coefficient RS equals 81, but for
positively cooperative enzymes the change in substrate required to increase the reaction
rate from 10% to 90% of Vmax is much less. An enzyme with a hill coefficient of 2.5,
for example, has a response coefficient of only 5. This allows the enzyme to react with
greater sensitivity to small changes in substrate concentrations.
Sensitivity to changes in substrate concentrations is important in many key biological
processes and can act as a further control mechanism for biochemically important
enzymes, particularly where substrate concentrations are low. For example, in cases
where defective hemoglobin lacks the ability to bind oxygen cooperatively in humans,
it causes cyanosis (a blue pigmentation of the skin due to oxygen saturation typically
dropping below 85%), which can have devastating effects4. Cooperativity can be used
as a mechanism to change the reaction kinetics of an enzyme without changing the
critical arrangement of the enzyme active site5. The arrangement of the oxygen-
binding site in hemoglobin is critical to its function and is highly conserved over
different organisms. Changes in environmental constraints for an organism (for
example between the amphibious environment of the frog and the aqueous
environment of the tadpole) dictate different binding kinetics, which can be altered by
varying the subunit interactions without disturbing the critical binding-site residues6.
As the response to substrate concentration is damped for negatively cooperative
enzymes the change in substrate concentration required to increase the reaction rate
from 10% to 90% of Vmax is often significantly larger than a non-cooperative enzyme.
This effectively increases the range of substrate concentrations over which the enzyme
is active. This is particularly advantageous to enzymes for which it is critical to
maintain some level of reaction in stressful situations where usually-plentiful
metabolites are in short supply5. There is also a suggestion that branch-point enzymes
are likely to act cooperatively in order to ensure that the multiple pathways that rely on
the products of that branch point enzyme are not inhibited by an excess of one
substrate5.

209
A further level of control for cooperative enzymes may exist due to their oligomeric
status. Errors in transcription or translation rarely have a dramatic effect on an
enzyme’s activity unless the mutation occurs at the enzyme’s binding site. A single
residue mutation may have little or no effect on a non-cooperative, monomeric enzyme
outside of these functionally critical areas. Even in a non-cooperative oligomer,
without the need for interaction between cooperatively-acting subunits, a mutation that
disturbs the oligomeric interfaces may have little phenotypic effect. As mentioned
above for hemoglobin, mutations that affect the subunit interface and therefore the
ability to act cooperatively can have damaging effects on the rate of binding. A larger
proportion of a cooperative enzyme’s residues is therefore functionally constrained
than for a non-cooperative enzyme and provides further support for cooperativity as a
mechanism for tight catalytic control in metabolic systems.
Unfortunately the amount and quality of cooperativity data for enzymes is patchy and
inconsistent. To evaluate the extent of cooperativity as a metabolic control mechanism
in tightly regulated enzymes it is necessary to be able to identify enzymes that are (or
are not) cooperative without lengthy and detailed biochemical experiment on individual
enzymes. Currently there is no computational approach to distinguish oligomeric
enzymes that are likely to act cooperatively and those that are not. As outlined below,
computational analyses of structural dynamics on an individual basis have been able to
characterise allosteric mechanisms and here it is assessed whether general dynamic
properties, such as the degree of correlation of motion over oligomer subunits, are
indicative of enzyme cooperativity. In particular, it is questioned whether the
communication between active sites on cooperative proteins is mediated or observable
through coupled motion in dynamic fluctuations between residues.

210
5.1.2 Application of Normal Mode Analysis to the Study of Proteins
It is well documented that a protein’s function is not entirely coded in its DNA or
protein sequence7; 8; 9; 10 and despite the increasing availability of protein structures
thanks to structural genomics initiatives, the gap between sequence and function
cannot yet be accounted for by structure alone. Since biological activity in proteins is
often accompanied with a change in protein structural conformation, perhaps these
conformational changes contain further functional information that is missing from the
sequence and structure alone. The study of protein dynamics has become increasingly
popular over the last 20 years and has growing recognition as an important additional
step in the sequence, structure and function paradigm.
Despite computing power having increased dramatically since the first protein
structures were produced, modeling these conformational changes over relevant
timescales is still difficult due to the total number of conformations available to a
protein in solution. Functional proteins in native state conditions however, tend to
sample conformations in equilibrium around their folded state. These subsets of
available conformations are termed microstates and modeling their vibrational modes is
a fast and efficient way of approximating them. Approaches such as Normal Mode
Analysis (NMA) have been used to successfully model proteins dynamics for over 25
years11 and have become even more popular as computational capacity increases.
This increase in popularity has prompted a number of simple models based on NMA
to assess large-scale protein dynamics quickly and efficiently12; 13; 14; 15; 16. These methods
are particularly useful in modeling dynamics in large systems, for which more detailed
methods such as molecular dynamics are too computationally expensive to be feasible.
Despite the coarse-grained nature of these methods, they have been found to give
remarkably similar results to complex methods such as molecular dynamics12; 17; 18. It is
also surprising that the resolution of the model has little effect on the results of
modeling dynamic motion of a protein in this way, indeed it has been shown that
motion can be modeled sufficiently accurately in the absence of crystal structure
coordinates by using electron density maps obtained from cryo-electon microscopy or

211
X-ray diffraction19. This shows the robustness and generality of this method as an
estimate of protein motion.
These simplified NMA approaches, have been able to successfully model the
machinery and conformational dynamics of several large protein systems including
RNA polymerase20, HIV reverse transcriptase21, GroEL-GroES22, F1 ATPase23, and
aspartate transcarbamylase24. The different sets of modes (i.e. frequencies) have been
shown to contain information on different functional properties of their dynamics.
The slowest (lowest frequency) modes are highly cooperative and transmit signal across
large distances throughout the protein structure. These frequencies are most
commonly linked to the functional conformational change in proteins and residues
with low fluctuation magnitudes in low frequency modes have been shown to be
indicative of hinge-bending regions22; 24; 25; 26; 27. Fluctuations in high frequency modes
are highly localised and tend to form pockets of local fluctuations in tightly packed
regions. Peaks in these high-frequency mode fluctuations are indicative of residues
important for protein folding25.
Such elastic network-based methods have shown to be particularly useful in elucidating
conformational changes relating specifically to allosteric mechanisms in proteins28; 29.
Ming and Wall were able to detect communication between the regulator site and the
active site in the allosteric enzyme, bovine trypsinogen by observing that both sites
exhibited similarly large changes in conformational distribution upon binding of the
regulator ligand29. Whilst the mechanisms of allosteric regulation have been extensively
characterised for a variety of individual proteins by the analysis of their dynamic
properties, homotropic cooperativity is less well-studied in the same way.
In addition to characterising the functional dynamics of individual systems, similar
methods have been used in a wide range of other roles. Structural domains within
crystal structures are able to be automatically delimited via analysis of the degree of
coupling of motion between residues30. Structural domains have generally highly
connected (and therefore highly-coupled) contacts between residues within the subunit
whilst maintaining only weak coupling between residues of separate domains. Another
useful application of modeling dynamic fluctuation via elastic network models is to
identify residues that are important for protein folding. Bahar et al.31 found that the

212
magnitude of fluctuations correlated with the resistance of residues to undergo
hydrogen-deuterium exchange and it was suggested that the more protected residues
are more critical to the folding process. Similar studies have also found that residues
with peak fluctuation displacements in the fast modes are critical to folding32 and have
gone on to specifically predict folding cores17.
The simplicity, robustness and speed of these models make them attractive to large-
scale analysis of protein dynamics and their success in elucidating functional
conformational change in proteins makes them an attractive choice for this analysis.
Amongst the simplified models based on NMA, the Gaussian Network Model (GNM),
described in further detail in 5.1.3, is one of the most commonly used and has been
shown to give good correlation between theoretical and experimental data33. Many
software applications and webservers are available to model the dynamics of a given
protein structure34; 35; 36 and thus it is not necessary to create a new method as part of
this work.
Since it has been shown that conformational change on binding correlates with
dynamic motion intrinsic in the native folded protein37, and it has been suggested that
communication can be transmitted between distant sites without large-scale
conformational changes38, it was hypothesised that cooperativity can be detected from
dynamic fluctuations in a protein’s native folded structure. It is reasonable to imagine
that the cooperative action between multiple active sites on separate subunits may be
communicated via dynamic motion intrinsic in the structure and that it may result in
correlated fluctuations between the active site residues.

213
5.1.3 Normal Mode Analysis and the Gaussian Network Model
A protein can be imagined as a system of oscillating nodes, the normal modes of which
are the patterns of motion where all parts move sinusoidally. This can occur at
different frequencies for different systems and such frequencies are called natural
frequencies. Normal Mode Analysis (NMA) assumes that near the energy minimum of
a system, forces act like springs, which can be approximated by atomic force-fields
taken from molecular dynamics simulations. One of the major computational hurdles
of NMA is minimizing the energy of the system before the normal modes can be
analysed. The computational overhead of this step has led to the application of NMA
to more simplified models, such as the elastic network model.
One such application of this approach is Gaussian Network Modeling (GNM)12. It is
based on the above principles but rather than having to minimize the energy of a
system, the normal modes are evaluated on a simple elastic network of nodes that
represent the protein structure (see Figure 5.2). Each residue is represented by a node,
which corresponds to its alpha carbon coordinates and a network topology is built by
connecting nodes within a given cutoff distance. An all-atom model can also be
constructed by representing each atom with a node, but this increases the
computational overhead beyond any advantage that the increased resolution provides.
Indeed, it has been shown that coarse-grained models give comparably representative
results to all-atom models39.
The connections between nodes in this elastic network are modeled as springs,
enabling the nodes to move with Gaussian motion around their coordinates with
varying frequencies according to each mode. In GNM the fluctuations are assumed to
be isotropic, which means that the fluctuation is uniform in all directions. This is
where GNM differs to NMA applied to elastic networks (also termed Anisotropic
Network Modeling [ANM]) as the latter do not assume fluctuations are isotropic. In
the context of this analysis the degree of similarity in fluctuations between residues is
important rather than the overall direction of the movement and therefore GNM is

214
preferred for this analysis. GNM results have also been shown to give higher
agreement to experimental B-factors than ANM33.
Figure 5.2 A protein structure (lysine–arginine–ornithine binding protein; top) shown as an
elastic network.
The nodes represent residues and the lines represent the elastic connections between them.
Picture taken from Tama and Sanejouand, 2001.15

215
Of interest in this analysis is the degree of similarity between the fluctuations of pairs
of residues. In GNM this information is derived in the following way:
• If the equilibrium position vectors of a residue i as Ri0 and the position of the
node in this vector at any one time as Ri, the fluctuations around the starting
point can be described as ∆Ri = Ri – Ri0
• The fluctuations of the other residue, j, in the pair are defined as above (∆Rj).
• The difference vector between these two residue fluctuations is described as
∆Rij = ∆Ri – ∆Rj (see Figure 5.3 for a schematic representation).
• The correlation of fluctuations for i and j is given by the dot product of their
fluctuation vectors (see Equation 5.2) using the statistical mechanical
probabilities defined by the GNM method12. The overall correlation between
the two residues is obtained by summing over all non-zero modes.
There are many webservers and software applications that can perform normal mode
analysis for a given protein, but most deliver the anisotropic NMA/elastic network
method35; 36. As mentioned above, the isotropic GNM approach was deemed more
suitable for this analysis. The Bahar group have produced a webserver, oGNM34, that
performs GNM analysis of a given protein structure. In addition to mode shapes and
mean square fluctuations (displacements), it also provides a cross-correlation matrix for
all residues versus all residues, which forms the basis of the data used in this chapter.
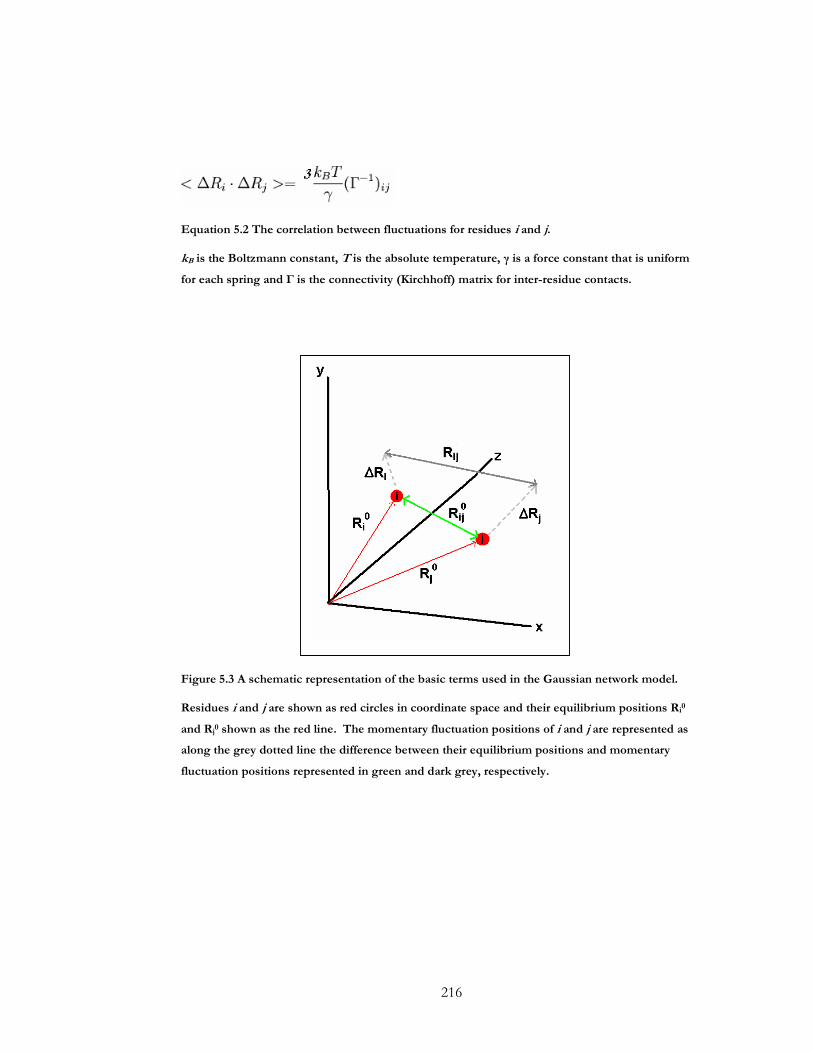
216
Equation 5.2 The correlation between fluctuations for residues i and j.
kB is the Boltzmann constant, T is the absolute temperature, γ is a force constant that is uniform
for each spring and Г is the connectivity (Kirchhoff) matrix for inter-residue contacts.
Figure 5.3 A schematic representation of the basic terms used in the Gaussian network model.
Residues i and j are shown as red circles in coordinate space and their equilibrium positions Ri0
and Rj0 shown as the red line. The momentary fluctuation positions of i and j are represented as
along the grey dotted line the difference between their equilibrium positions and momentary
fluctuation positions represented in green and dark grey, respectively.

217
5.2 Methods
5.2.1 Dataset Creation for the Cooperativity Analysis
To test the hypothesis that the active sites of cooperative enzymes exhibit increased
correlation of motion over non-site residues than non-cooperative enzymes it was
necessary to collect a collection of structures of known cooperative enzymes. There is
currently no database that holds large-scale information about cooperative enzymes.
Enzyme databases such as BRENDA40 and SABIO-RK41 annotate entries with their
hill coefficients where this is available but the number of enzymes with this
information given is relatively small.
It is further necessary for enzymes with known hill coefficient data to have a known
structure deposited in the PDB. It is also important to ensure that the enzyme
structure is from the same organism as the hill coefficient is reported for as it has been
shown that cooperativity can vary for the same protein between different organisms42.
A review by Koshland and Hamadani5 attempted to estimate the comparative
proportions of negatively cooperative and positively cooperative enzymes in nature by
surveying the literature from 1980-1990. In doing so they produced small datasets of
positive, negative and non-cooperative enzymes. Many of these enzymes, however,
could not be associated with a PDB structure from the same organism. A benchmark
set of allosteric protein structures was reported by Daily and Gray43, however allosteric
proteins are not necessarily cooperative and only a subset of the enzymes they list are
cooperative.
The most productive source of cooperative enzyme information was the database,
SABIO RK41. This is a database containing information about biochemical reactions,
alongside their kinetic equations and associated parameters. The reaction pathways and
enzyme annotation in SABIO RK is obtained from KEGG44 and reaction kinetics are
annotated by manual literature curation. This focus on reaction kinetics using literature

218
curation results in a larger number of entries for which there is Hill coefficient
information than BRENDA or KEGG.
In addition to the above data sources, a literature search was performed in order to
find papers for individual enzymes that report the hill coefficients of ligand binding.
Since a null set (i.e. non-cooperative proteins) was needed, enzymes were also recorded
where a hill coefficient of 1 was reported, in addition to negatively and positively
cooperative enzymes. Enzymes were matched to PDB structures via their Uniprot
entry and where more than one PDB structure exists for the enzyme, preference was
given to the highest resolution structure where the ligand that the hill coefficient relates
to is present. Table 5.1 shows the resultant dataset (named Dataset 5.1) that was
obtained from these sources.
This analysis also required the location of the active site to be known for each of the
proteins in the set. Since each of these proteins has been relatively well-studied in
order to report detailed biochemical parameters, the active sites for each of the
proteins is very likely to already be known. If the PDB file contained the enzyme’s
ligand then active site residues were defined as those that had any atom within 3Å of
the bound ligand. If the PDB structure did not contain a bound ligand, either the
residues listed in the SITE records in the PDB file (if present) or in that structure’s
entry in the CSA were used.
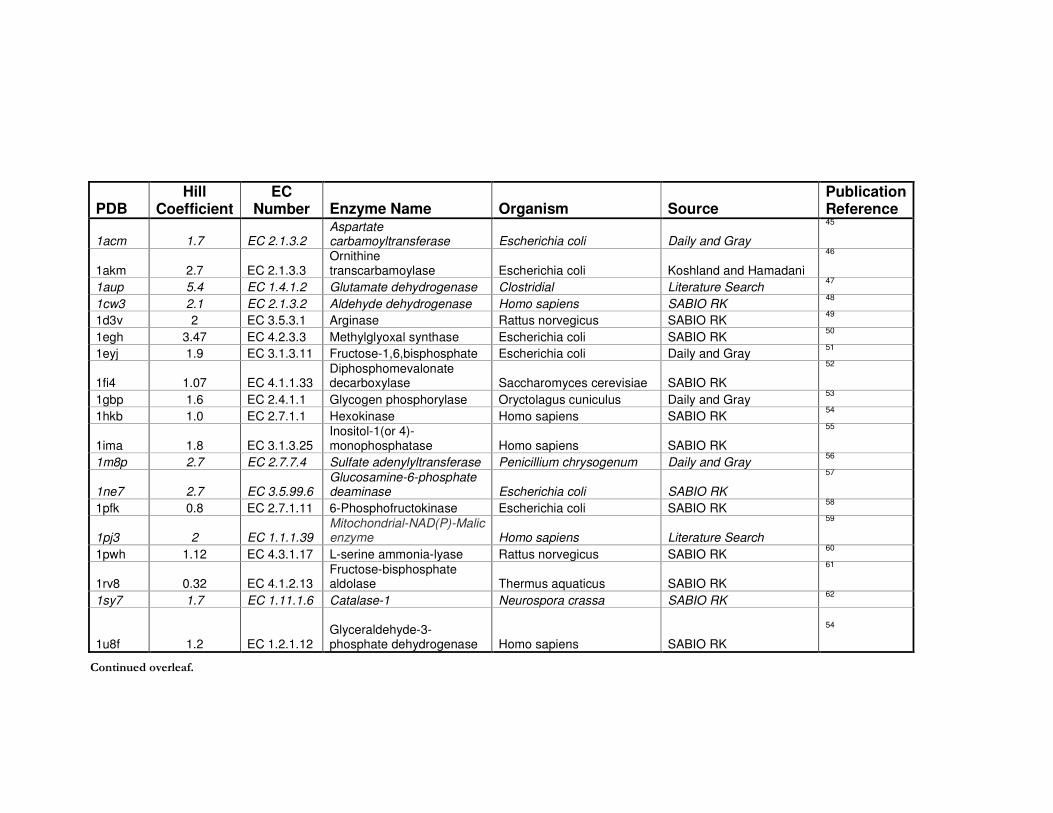
PDB Hill
Coefficient EC
Number Enzyme Name Organism Source Publication Reference
1acm 1.7 EC 2.1.3.2 Aspartate carbamoyltransferase Escherichia coli Daily and Gray
45
1akm 2.7 EC 2.1.3.3 Ornithine transcarbamoylase Escherichia coli Koshland and Hamadani
46
1aup 5.4 EC 1.4.1.2 Glutamate dehydrogenase Clostridial Literature Search 47
1cw3 2.1 EC 2.1.3.2 Aldehyde dehydrogenase Homo sapiens SABIO RK 48
1d3v 2 EC 3.5.3.1 Arginase Rattus norvegicus SABIO RK 49
1egh 3.47 EC 4.2.3.3 Methylglyoxal synthase Escherichia coli SABIO RK 50
1eyj 1.9 EC 3.1.3.11 Fructose-1,6,bisphosphate Escherichia coli Daily and Gray 51
1fi4 1.07 EC 4.1.1.33 Diphosphomevalonate decarboxylase Saccharomyces cerevisiae SABIO RK
52
1gbp 1.6 EC 2.4.1.1 Glycogen phosphorylase Oryctolagus cuniculus Daily and Gray 53
1hkb 1.0 EC 2.7.1.1 Hexokinase Homo sapiens SABIO RK 54
1ima 1.8 EC 3.1.3.25 Inositol-1(or 4)-monophosphatase Homo sapiens SABIO RK
55
1m8p 2.7 EC 2.7.7.4 Sulfate adenylyltransferase Penicillium chrysogenum Daily and Gray 56
1ne7 2.7 EC 3.5.99.6 Glucosamine-6-phosphate deaminase Escherichia coli SABIO RK
57
1pfk 0.8 EC 2.7.1.11 6-Phosphofructokinase Escherichia coli SABIO RK 58
1pj3 2 EC 1.1.1.39 Mitochondrial-NAD(P)-Malic enzyme Homo sapiens Literature Search
59
1pwh 1.12 EC 4.3.1.17 L-serine ammonia-lyase Rattus norvegicus SABIO RK 60
1rv8 0.32 EC 4.1.2.13 Fructose-bisphosphate aldolase Thermus aquaticus SABIO RK
61
1sy7 1.7 EC 1.11.1.6 Catalase-1 Neurospora crassa SABIO RK 62
1u8f 1.2 EC 1.2.1.12 Glyceraldehyde-3-phosphate dehydrogenase Homo sapiens SABIO RK
54
Continued overleaf.

220
PDB
Hill Coefficient EC Number Enzyme Name Organism Source
Publication Reference
1vgv 1.8 EC 5.3.1.14 UDP-N-acetylglucosamine 2-epimerase Escherichia coli SABIO RK
63
1xbt 0.8 EC 2.7.1.21 Thymidine kinase Homo sapiens SABIO RK 64
1xge 1.57 EC 3.5.2.3 Dihydroorotase Escherichia coli Literature Search 65
1xva 2.3 EC 2.2.1.20 Glycine Methyltransferase Rattus norvegicus Koshland and Hamadani 66
1xz8 1.0 EC 2.4.2.9 uracil phosphoribosyltransferase Bacillus caldolyticus Daily and Gray
67
1y3i 1.4 EC 2.7.1.23 NAD+ kinase Mycobacterium tuberculosis BRENDA 68
2bz0 1.3 EC 3.5.4.25 GTP cyclohydrolase II Escherichia coli BRENDA 69
2csm 1.6 EC 5.4.99.5 Chorismate mutase Saccharomyces cerevisiae Daily and Gray 70
2hbq 1.5 EC 3.4.2.36 Caspase I Homo sapiens Daily and Gray 71
2hgs 0.8 EC 6.3.2.3 Glutathione synthase Homo sapiens SABIO RK 72
2hxd 1.9 EC 3.5.4.30 dCTP deaminase Methanococcus jannaschii SABIO RK 73
2jlc 1.0 EC 2.5.1.64
2-Succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate Escherichia coli SABIO RK
74
2pah 1.6 EC 1.14.16.1 Phenylalanine 4-monooxygenase Homo sapiens SABIO RK
75
Table 5.1 Dataset 5.1: A list of enzymes with annotated Hill coefficients and a structure deposited in the PDB for the same organism.
Enzymes shown in italics are later deleted from the dataset for technical reasons.

5.2.2 Dataset Creation for the Active Site Correlation Analysis
In order to test the hypothesis that active site residues are generally more coupled than
non-active site residues, a test set of homo-oligomeric enzymes with known structures
and annotated active sites was needed. The non-redundant set of enzymes with
literature active site annotation from the Catalytic Sites Atlas (CSA)76 that was used to
test active site predictions in Chapter 5: was used again here (details of creation of this
dataset are given in 3.2.2). Only the structures that contained 2 or more identical
chains were applicable to this analysis and those proteins with more than 9 chains were
discarded due to the technical reasons discussed in 5.2.4.
The resultant dataset (Dataset 5.2, shown in Table 5.2) contains 114 non redundant
homo-oligomeric enzyme structures with a literature-annotated active site. The active
site residues for these proteins were defined in the same way as described in 2.2.2.
1a4i 1n20 1f75 1e2a 1d2t
1b5t 1nir 1f7l 1f8x 1dco
1c9u 1nww 1fua 1fro 1dxe
1cd5 1qrg 1gpj 1hrk 1ez2
1cgk 1r16 1gpr 1jfl 1j80
1cs1 1r51 1j7g 1nn5 1jm7
1d8h 1wgi 1kp2 1oe9 1m6k
1dhf 1yve 1mka 1otg 1moq
1dqa 12as 1nid 1qh6 1mvn
1e7l 1a05 1nsp 1qpr 1nf10
1ecm 1a79 1snn 1qq6 1o9i
1ef0 1a95 1sox 1r4f 1oac
1g79 1b65 1tys 1r6w 1oas
1gqg 1b93 1uro 1uf8 1pfk
1hxq 1brw 3cla 2jcw 1qj5
1i6p 1cg6 3eca 2toh 1rhc
1ir3 1d4a 3mdd 7odc 1ro8
1jdw 1daa 1c2t 1al7 1tph
1jhf 1dci 1cev 1bwp 1uam
1mqw 1do6 1dae 1c0k 1uaq
1o04 1dqs 1dbf 1c3c 2xis
1qd1 1dup 1dj0 1chm 3nos
1qhf 1f2v 1e19 1d0s
Table 5.2 Dataset 5.2. A list of 114 non redundant homo-oligomeric enzyme PDB structures
with a literature-based active site information obtained from the CSA.

222
5.2.3 Dataset Creation for the Structural Environment Correlation
Analysis
In contrast to Datasets 2.1 and 2.2, this analysis concentrates on the structural
environment of residues it was not necessary for the proteins in this set to have any
biochemical data or functional site annotation. Homo-oligomeric structures were
extracted from the entire PDB where there was a biological unit file available and it
contained less than 9 chains and/or 1000 residues (due to technical reasons discussed
in 5.2.4). Some of the structures contain very short chains that are unlikely to form full
subunits and thus structures were rejected that had a chain length of less than 50
residues. To ensure that bias is reduced towards proteins that are over-represented in
the PDB, the dataset was culled for redundancy in the same way as described in 3.2.2.
In some of the structure files contained in this dataset, the residue-numbering is
inconsistent with the standard PDB format. The vast majority of files differentiate
chains by their chain identifier field and provide the same residue numbering for
equivalent residues on different chains. The residue numbering for different chains
was inconsistent in 34 of the remaining files and there was no consensus in the scheme
that was employed to number them. For example, residue 1 from chain A in 2dlb was
numbered ‘1001' yet residue 1 from chain B was numbered ‘2001’, whilst in 1p60
residues from chain A were numbered from 3-158 and residues from chain B were
numbered from 198 onwards. Another problem with some files was that chains
identifiers were denoted using numbers (rather than the traditional letters). This
creates problems when trying to uniquely identify a residue; residue 10 from the first
chain would traditionally be identified as 10A, whereas if the first chain is given the
identifier ‘1’, then it becomes residue 101, which becomes confusing. It is important to
be able to accurately identify equivalent residues in this analysis in order to assess the
degree of coupling between them, therefore PDB structures with inconsistent residue
numbering were deleted from the dataset. The resultant dataset (Dataset 5.3, shown in
Table 5.3) contains 636 proteins.
As this analysis tests the hypothesis that the degree of coupling of a residue’s
fluctuation with its equivalent residue on another chain depends on its structural
environment, residues in each structure need to be assigned to the “interface”,
“surface” or “core”. A residue is defined as being in an interface when any of its atoms

223
are within 5Å from any other atom from another chain (see Figure 5.4 for an example
structure with the interface highlighted using this definition). Surface residues are
defined as those that have at least 5Å2 of solvent-accessible surface area (S-ASA, which
is calculated using an in-house program, SACALC, as described in 2.2.2). All other
residues are defined as “core” residues.
Figure 5.4 An example of a protein structure (1ji7) with the interface residues highlighted.
Residues are coloured according to their chain identity (Red atoms are hetero-atoms) and
space-filled residues are defined as being in the interface.

224
1a12 1dk8 1gmw 1jke 1n0e 1ox0 1r7l 1u7g 1ut7 1xko 2bgx 2g3p 2j0n 2phn 3c70
1a1x 1dmh 1gpr 1jkx 1n0q 1p0w 1r8h 1u7k 1utg 1xm3 2bko 2g7s 2j6b 2pi8 3c9u
1a3a 1dov 1gut 1jlt 1n1c 1p32 1rcq 1uae 1vke 1xs0 2bm5 2g8l 2j73 2pif 3ce1
1a8o 1dqa 1gve 1jly 1n3l 1p35 1reg 1uan 1vki 1xsv 2brj 2g8o 2j7j 2pju 3chb
1aap 1dqe 1gxj 1jm0 1n55 1p5f 1rfx 1uc2 1vkm 1xub 2bw4 2gbo 2j80 2pk7 3cla
1aqt 1dto 1gxr 1jr8 1n69 1p9h 1rfy 1ucr 1vkp 1xv2 2bz1 2gec 2j8w 2pk8 3cmb
1at0 1duv 1gxu 1k04 1n7v 1pc6 1rge 1ueh 1vky 1xvh 2c9v 2gf4 2jb2 2pl7 3cw9
1aua 1dys 1gyo 1k4i 1n7z 1pfo 1ris 1ufy 1vm0 1xvs 2car 2gfq 2jhf 2pmr 3d03
1awd 1e0b 1gyy 1k4z 1n93 1pm4 1ro7 1uku 1vmh 1y0g 2cc0 2ghv 2jl1 2pq7 3d3r
1ayi 1e19 1h16 1k51 1nc7 1ppr 1rqp 1uq5 1vp6 1y12 2ccb 2gj4 2lig 2pr1 3dwr
1ayo 1e58 1h34 1kic 1nco 1ppy 1rw0 1usg 1vp7 1y2i 2ccv 2gjv 2nlv 2ps1 3eeq
1b2p 1eaj 1h8g 1kjn 1nfp 1psr 1rwz 1usm 1vp8 1y6x 2cg6 2glk 2nml 2pt0 3eip
1b4b 1ecw 1h99 1kjq 1njh 1ptq 1ry9 1uty 1vpb 1y71 2chh 2glz 2nr5 2pw4 3erj
1b5t 1edh 1hf8 1kpt 1nki 1puc 1rya 1uuj 1vps 1y7m 2cjt 2gom 2nrk 2pw6 3lyn
1b79 1ejb 1hqs 1kqp 1nlq 1pvm 1rzl 1uv7 1vq3 1y9i 2cn4 2gsv 2ntk 2pzz 3sdh
1bdo 1ek9 1hta 1kut 1nms 1q2h 1s0p 1uw1 1vr0 1yer 2cu6 2gud 2nw8 2q03 3ssi
1bgf 1ekq 1hx6 1kzq 1no4 1q2o 1s2e 1uwk 1vr7 1yki 2d00 2guk 2o35 2q3t 4bcl
1bjt 1ekr 1hz4 1l3p 1nog 1q6o 1s7m 1uww 1vz0 1ylx 2d8d 2gum 2o3i 2q4o 5hpg
1ble 1el6 1i40 1l4i 1nqd 1q7e 1s7z 1ux5 1vzy 1yox 2dek 2gx9 2o70 2qif 5rub
1byi 1eqt 1i4u 1l5o 1ns5 1q8r 1s98 1uz3 1w23 1yoz 2dm9 2h1t 2o7m 2qii 8rsa
1c02 1es9 1i6p 1l8d 1nvj 1q9u 1sed 1v5v 1w53 1ypq 2dsy 2h2n 2oa5 2qsw
1c0p 1euw 1ig3 1lfa 1nxj 1qc7 1sei 1v6p 1w7c 1z0p 2e2a 2hft 2ob5 2qzg
1c5e 1ext 1ihk 1lkt 1nxm 1qcz 1sf8 1v7l 1wc9 1z41 2e2r 2hh6 2od0 2rcf
1c9o 1eyv 1ijy 1lm5 1nxu 1qh4 1sg4 1v7z 1whi 1zed 2e50 2hmz 2odk 2rde
1cbk 1ezg 1iom 1ln0 1o0w 1qhd 1sj1 1v8c 1who 1zei 2efm 2hng 2oee 2rfr
1cby 1ezj 1iro 1lnd 1o22 1qhv 1skz 1v8d 1wlg 1zjc 2elc 2hqv 2oez 2rh2
1cku 1f08 1itv 1m0k 1o3u 1qi9 1sqj 1v8h 1wm3 1zke 2ewh 2hqx 2oik 2rl8
1cq3 1f1m 1iu8 1m1f 1o5h 1qks 1su8 1v8q 1wmg 1zkp 2f01 2huh 2okf 2rsl
1cru 1f46 1ixb 1m1l 1o6a 1ql0 1szq 1v96 1wo8 1zps 2f22 2hzb 2oku 2uu8
1ctf 1f7l 1iyb 1m2d 1o75 1qlm 1t0a 1v9y 1wpn 1zq7 2f48 2i52 2ook 2uui
1cun 1f86 1izm 1m4j 1o7j 1qre 1tej 1vd6 1wq8 1zro 2f4l 2i71 2opl 2uzq
1cxq 1f8e 1j2r 1m4z 1o7k 1qsd 1tfe 1vdd 1wud 1zso 2f5t 2i8d 2ou3 2v41
1cy9 1few 1j31 1m5w 1o8b 1qu1 1thw 1vdw 1wvf 2a2l 2f62 2i9i 2ouf 2vg1
1d1j 1fjj 1j8b 1m65 1o9i 1qve 1to6 1ve1 1wwh 2a9s 2f6s 2i9x 2ox7 2vgx
1d3y 1fqt 1i12 1mby 1ocy 1qw2 1tu1 1vgg 1wzd 2aeb 2f7f 2iba 2oy9 2vpa
1d5f 1ftr 1i2k 1mg7 1of8 1qwg 1tul 1vh4 1x2i 2aib 2f9h 2idl 2oyn 2vvp
1d7d 1fu1 1j8u 1mkf 1ofz 1qx4 1tv8 1vh6 1x6m 2aj7 2fb5 2ie7 2p02 2yx4
1dcs 1fx2 1j98 1mo1 1oi2 1qxm 1twd 1vhd 1x99 2arc 2fbl 2iim 2p12 2zgw
1ddt 1flg 1j9j 1mqi 1ojr 1qxo 1tx2 1vhw 1x9i 2axw 2fef 2ikb 2p3y 3bbb
1dg6 1fn9 1jd0 1msc 1oki 1r0m 1tyx 1vi6 1x9z 2b0a 2ffg 2ilk 2p62 3bex
1di6 1fx8 1jfl 1mvl 1oms 1r0v 1tzp 1vjl 1xeq 2b3n 2fgq 2in5 2p6v 3bge
1dj0 1g8e 1jg5 1mvo 1osy 1r29 1u07 1vjq 1xfs 2b5a 2fn0 2iqq 2p8i 3bpd
1dj8 1ggx 1jhg 1mw5 1ou0 1r3s 1u2m 1vk8 1xg7 2b82 2fyx 2it9 2p90 3byp
1djt 1gml 1ji7 1mwq 1ova 1r4c 1u6k 1vka 1xi3 2bay 2fzt 2ivy 2peq 3byq
Table 5.3 Dataset 5.3: A list of 636 non-redundant homo-oligomeric PDB structures.

225
5.2.4 Calculation of Residue Motion Correlation
As mentioned above, a number of webtools and downloadable applications exist to
calculate protein normal modes. It is therefore out of the scope of this project to
create further software for the calculation of normal modes and thus a pre-existing
tool, oGNM34, was used.
This analysis seeks to assess whether residue fluctuations are more correlated over
different subunits in different situations. The output provided by oGNM includes
residue ‘cross-correlation’ values, which is a measure of how correlated a residues
movement is with another according to their average fluctuations from all modes. An
outline of the theory behind GNM is explained in section 5.1.3 and the cross-
correlations are calculated as shown in Equation 5.2.
This produces a matrix of normalised cross-correlation values for all residues against all
residues in a protein (see Figure 5.5 for an example). Residues have a cross-correlation
value of 1 if their fluctuations are perfectly coupled in the same direction. Residues
that are not correlated have a cross-correlation value of 0, while residues with a cross-
correlation value of -1 are perfectly coupled but in the opposite direction. For the
purpose of this work, the degree of coupling was of interest rather than the direction
and so cross-correlations were converted to represent only their magnitude.

226
Figure 5.5 An example cross-correlation matrix for 1D3V (Manganese Metalloenzyme
Arginase), which is a homo-trimer.
Here red sections show the most correlated residue pairs, whilst the darkest blue are the most
anti-correlated residue pairs. Residue pairs within the same subunit show higher correlations
than inter-subunit residue pairs, therefore it can be seen that this structure is trimeric.
In order to obtain oGNM results for a large number of proteins, the Bahar group
kindly provided the oGNM source code to enable it to be run offline. It was also
modified to allow analysis of larger systems (up to 2000 nodes) than is possible online
(there is a 500 node limit on the oGNM webserver for cross-correlation results). It is,
however, computationally expensive to run oGNM for large proteins, even for systems
within this limit, therefore oGNM could not produce results for a number of proteins
in Dataset 5.1 (see italicised entries in Table 5.1) and in Dataset 5.2 and 2.3 proteins
were restricted to those with less than 9 chains and/or 1000 residues.
The underlying network of residues in oGNM is created by connecting two residues
where their alpha carbons are within a given cut-off distance. The cut-off distance was
set at 7.3Å for these analyses as was shown to give the optimum correlation between
theoretically-derived mean square fluctuations using GNM and the experimentally-
derived B-factors33. Each residue was represented by a single node instead of three in
order to increase the size of protein that the method was valid for.

227
Each residue is then assigned a cross-correlation value that represents the residue’s
degree of coupling to its equivalent residues on the opposite chain(s). For each residue
this equivalent residue cross-correlation (cc_equiv) score is calculated by taking the
average of that residue’s cross-correlation with each equivalent residue on each of the
other chains. It is then possible to colour protein structures according to each residue’s
cc_equiv score as is shown in Figure 5.6. The cc_equiv score is multiplied by 100 in
order to allow colouring by cc_equiv as a replacement of the B-factor in PDB files.
Where analyses dictate that multiple proteins cc_equiv scores be pooled, the cc_equiv
scores are normalised for each protein to make the highest cc_equiv residue score in
that protein equal to one and the lowest equal to zero. This allows fair comparison
between differences in residues over proteins that have cc_equiv scores on different
scales.

228
Figure 5.6 The biological unit structure for 1D3V coloured according to each residue’s cc_equiv
score.
Residues coloured red are the most correlated residues in that structure, whereas dark blue are
the least correlated. The space-filled black atoms represent the ligand.
5.3 Correlated Residue Motions in Cooperative Oligomeric
Enzymes
The following work addresses whether active sites on the separate subunits in
cooperative enzymes have a higher degree of coupling between their dynamic
fluctuations than the active sites of non-cooperative enzymes. Cross-correlation scores
between each residue and their equivalent residue on the opposite chain (cc_equiv)
were calculated for all residues in proteins in Dataset 5.1 as discussed in 5.2. The
degree of correlation of equivalent residue fluctuations was compared for cooperative
and non-cooperative proteins and the results are shown below.

229
5.3.1 Analysis of Residue Correlations in Co-operative and Non
Cooperative Enzymes.
There are 17 positively cooperative, 4 negatively cooperative and 4 non-cooperative
proteins in the dataset for this analysis (Dataset 5.1). The average cc_equiv score for
site and non-site residues and the level of significance of the difference between them
(the Mann-Whitney p-value) is shown in Table 5.4. Where sites have a different
average cc_equiv (due to small changes in site residue annotation or symmetry) the
mean value is taken for all sites. The structure of each enzyme in the dataset is
coloured by each residues degree of cross-correlation with their equivalent residue
(positively cooperative enzymes are shown in Figure 5.7, negatively cooperative
enzymes are shown in Figure 5.8 and non-cooperative enzymes are shown in Figure
5.9).

230
Positively cooperative enzymes
1akm
1d3v
1egh
1eyj
1gbp
1ima
Continued overleaf.

231
1pwh
1u8f
1vgv
1xge
1xva
1y3i
Continued overleaf.

232
2bz0
2csm
2hbq
2hxd
2pah
Figure 5.7 Positively cooperative enzyme
structures.
Each residue is coloured by its cc_equiv value,
dark blue residues represent those with the
lowest cc_equiv and red residues are those
with the highest cc_equiv. Residues that are
space-filled in 3D represent active site
residues, typically for structures with no bound
ligand. Space-filled atoms shown in black are
bound ligands or metal ions, where present in
the PDB file, representing the location of the
active site.

233
Negatively cooperative enzymes
1pfk
1rv8
1xbt
2hgs
Figure 5.8 Negatively cooperative enzyme structures.
Each residue is coloured by its cc_equiv value, dark blue residues represent those with the
lowest cc_equiv and red residues are those with the highest cc_equiv. Space-filled atoms shown
in black are bound ligands or metal ions, where present in the PDB file, representing the
location of the active site.
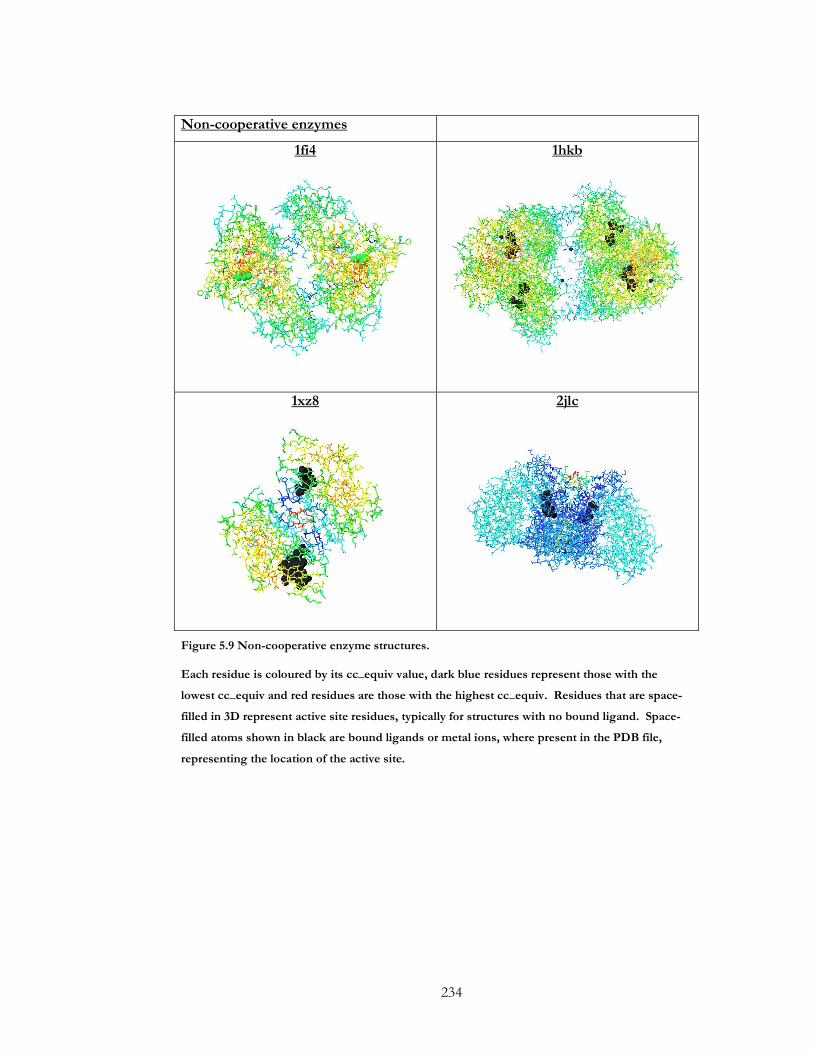
234
Non-cooperative enzymes
1fi4
1hkb
1xz8
2jlc
Figure 5.9 Non-cooperative enzyme structures.
Each residue is coloured by its cc_equiv value, dark blue residues represent those with the
lowest cc_equiv and red residues are those with the highest cc_equiv. Residues that are space-
filled in 3D represent active site residues, typically for structures with no bound ligand. Space-
filled atoms shown in black are bound ligands or metal ions, where present in the PDB file,
representing the location of the active site.

235
PDB Hill Coefficient Non-site average
cc_equiv Site average
cc_equiv Mann-Whitney
p-value
Positively cooperative 1egh 3.47 7.46 7.54 0.271
1akm 2.7 9.06 10.45 0.176
1xva 2.3 11.57 11.94 0.226
1d3v 2 11.16 13.31 0.023 1eyj 1.9 8.41 13.27 <0.001 2hxd 1.9 3.33 2.09 0.005 1ima 1.8 12.02 13.64 0.129
1vgv 1.8 18.89 18.59 0.956
1gpb 1.6 12.71 20.80 0.001 2csm 1.6 11.29 17.57 0.001 2pah 1.6 17.50 20.42 0.001 1xge 1.57 18.33 21.99 0.001 2hbq 1.5 9.54 11.74 0.028 1y3i 1.4 7.23 5.63 0.004 2bz0 1.3 12.83 8.15 0.002 1u8f 1.2 7.28 7.71 0.14
1pwh 1.12 18.35 21.44 <0.001
Negatively-cooperative 1pfk 0.8 5.77 5.36 0.316
1xbt 0.8 8.12 5.86 <0.001 2hgs 0.8 16.41 18.03 0.129
1rv8 0.32 15.60 20.82 0.059 Non-cooperative
1fi4* 1.07 21.37 23.44 0.602
1hkb 1 23.49 29.35 <0.001 1xz8 1 18.83 13.96 0.03 2jlc 1 8.16 5.19 0.024
Table 5.4 The average equivalent residue cross-correlation (cc_equiv) scores for site and non-
site residues for cooperative and non-cooperative enzymes.
The p-value (from Mann-Whitney tests) for the significance of the difference between the site
and non-site cc_equiv scores is also given. Enzymes shown in bold had a significant difference
between site and non-site cc_equiv values. * 1fi4 has a reported Hill coefficient of slightly more
than 1, but it is not significantly different than 1 and therefore has been defined as non-
cooperative.
The number of site residues defined by the criteria set out in 5.2.1 defines a relatively
small number of residues per site, particularly in cases where site residues are taken
from the CSA. The CSA annotates residues known to be involved in catalysis and so
other residues in the environment of the active site, but not involved in catalysis, are
not taken into account. Active sites that are in, or close to, highly correlated regions of

236
the structure yet have uncorrelated catalytic residues would therefore not show any
difference between active site and non-active site correlation. The centroid of the
active site was calculated from the site residues in the same way as described in 2.2.2
and the Spearman’s rank correlation coefficient between the distance from the active-
site centroid and the cc_equiv value was evaluated for each protein. The Spearman’s
rank correlation coefficient and its associated significance value for each enzyme are
given in Table 5.5.
Overall the majority (18 out of 25) of proteins has either a higher average active-site
cc_equiv value than the non-site residues or a negative correlation between cc_equiv
and the distance from the active site. Only 9 of the 17 cases where the average active
site cc_equiv is larger than non-active site cc_equiv values, however, are significant.
Enzymes for which their active sites show a significantly increased amount of
correlation in their dynamics exist in both the cooperative and non-cooperative sets.
Similarly, strong significant negative correlations exist between distance of a residue
from the active site centroid and its cc_equiv value for both cooperative and non-
cooperative enzymes.
Each enzyme has a different background distribution of cc_equiv values and so it
would be misleading to pool all cc_equiv values for site and non-site residues from
different proteins. The cc_equiv values were therefore scaled from 0 to 1 within each
enzyme before pooling residues from difference enzymes. Table 5.6 shows the mean
scaled cc_equiv values for all site and non-site residues in the cooperative (positive and
negative) and non-cooperative set. There is a non-significant increase in dynamic
correlation for site residues over non-site residues for cooperative enzymes, whereas
the increase is much larger (and significant) for non-cooperative enzymes.
Furthermore, the distribution of scaled cc_equiv values for site and non-site residues is
different for cooperative and non-cooperative proteins. Figure 5.10 shows that this
increase in dynamic correlation values over all residues for non-cooperative proteins is
also reflected in the raw cc_equiv values.

237
PDB Hill Coefficient Spearman's Rank Correlation Coefficient P-value
Positively cooperative
1egh 3.47 0.048 0.149
1akm 2.7 -0.070 0.029
1xva 2.3 -0.263 <0.001
1d3v 2 -0.361 <0.001
1eyj 1.9 -0.544 <0.001
2hxd 1.9 0.163 <0.001
1ima 1.8 -0.262 <0.001
1vgv 1.8 0.001 0.969
1gpb 1.6 -0.659 <0.001
2csm 1.6 -0.631 <0.001
2pah 1.6 -0.426 <0.001
1xge 1.57 -0.354 <0.001
2hbq 1.5 -0.366 <0.001
1y3i 1.4 0.172 <0.001
2bz0 1.3 0.349 <0.001
1u8f 1.2 -0.055 0.045
1pwh 1.12 -0.367 <0.001
Negatively-cooperative
1pfk 0.8 0.023 0.402
1xbt 0.8 -0.182 <0.001
2hgs 0.8 -0.378 <0.001
1rv8 0.32 -0.598 <0.001
Non-cooperative
1fi4* 1.07 -0.742 <0.001
1hkb 1 -0.663 <0.001
1xz8 1 0.113 0.042
2jlc 1 0.241 <0.001
Table 5.5 The Spearman’s rank correlation coefficient for the comparison between distance
from active site centroid and cc_equiv for each enzyme.
The level of significance associated with this correlation coefficient is also given. Residues in
enzymes that are given in red show no significant correlation between the distance from the
active site centroid and cc_equiv.
Cooperative (both positive and negative)
Non-cooperative
P-value (Cooperative vs. Non-cooperative)
Site residues 0.394 0.500
0.001
Non-site residues 0.388 0.440
<0.001
P-value (Site vs. Non-site) 0.436 0.018
Table 5.6 Average scaled cc_equiv values for pooled residues from enzymes within each set.
Mann-Whitney p-values are given for the differences in scaled cc_equiv values in each category.
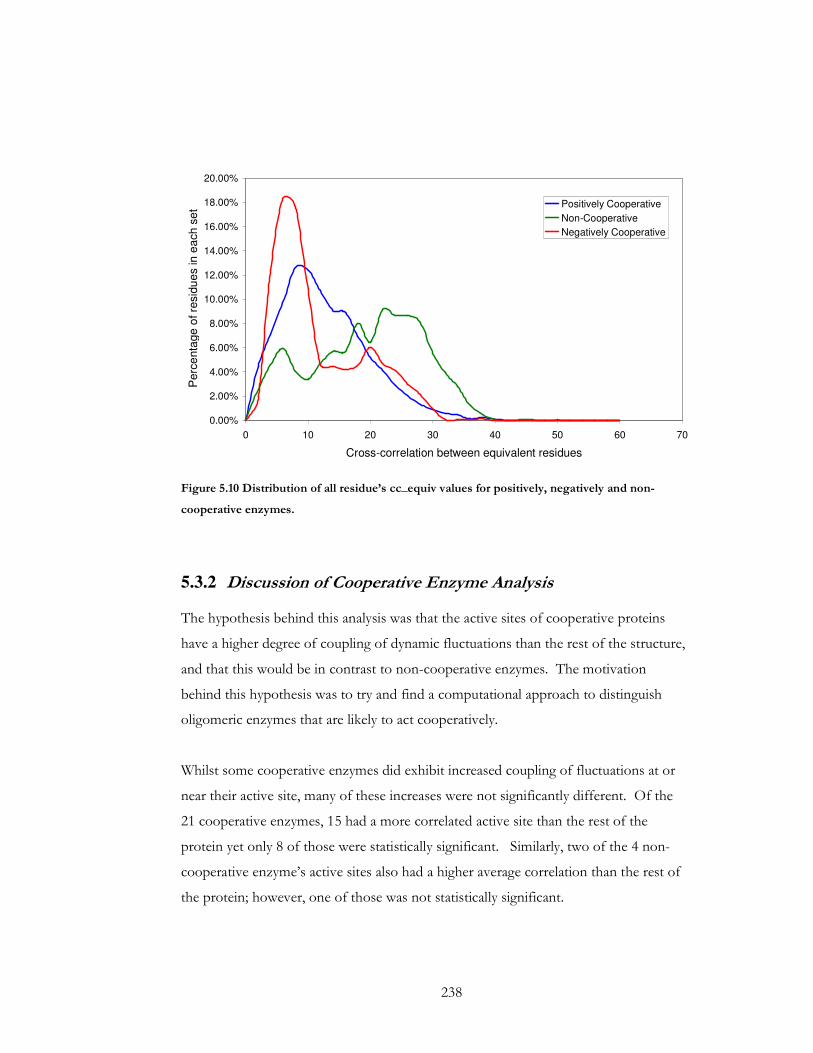
238
0.00%
2.00%
4.00%
6.00%
8.00%
10.00%
12.00%
14.00%
16.00%
18.00%
20.00%
0 10 20 30 40 50 60 70
Cross-correlation between equivalent residues
Perc
enta
ge
of re
sid
ue
s in
ea
ch s
et Positively Cooperative
Non-Cooperative
Negatively Cooperative
Figure 5.10 Distribution of all residue’s cc_equiv values for positively, negatively and non-
cooperative enzymes.
5.3.2 Discussion of Cooperative Enzyme Analysis
The hypothesis behind this analysis was that the active sites of cooperative proteins
have a higher degree of coupling of dynamic fluctuations than the rest of the structure,
and that this would be in contrast to non-cooperative enzymes. The motivation
behind this hypothesis was to try and find a computational approach to distinguish
oligomeric enzymes that are likely to act cooperatively.
Whilst some cooperative enzymes did exhibit increased coupling of fluctuations at or
near their active site, many of these increases were not significantly different. Of the
21 cooperative enzymes, 15 had a more correlated active site than the rest of the
protein yet only 8 of those were statistically significant. Similarly, two of the 4 non-
cooperative enzyme’s active sites also had a higher average correlation than the rest of
the protein; however, one of those was not statistically significant.

239
When the scaled cross-correlation values are pooled for cooperative and non-
cooperative proteins there is no significant difference between site and non-site
correlation for cooperative proteins, whereas non-cooperative proteins do seem to
exhibit higher site correlation values. Even without differentiating between site and
non-site residues, residues in non-cooperative enzymes seem to be more highly
correlated as a whole than in cooperative enzymes.
A major issue with this study is the small size of the dataset available. It is impossible
to identify any real trends in a dataset this small, particularly for non-cooperative
enzymes, of which there are only 4. The limitations of finding enzymes where there is
not only experimental evidence to support it acting cooperatively (or not) but also a
good quality structure from the same organism as the biochemical data is derived, is
prohibitive to forming a large dataset.
The pooled results (Figure 5.10 and Table 5.6), show that cc_equiv values tend to be
higher for site and non-site residues in non-cooperative proteins. This, however, may
be artificially skewed by two large non-cooperative enzymes in the small dataset. Out
of the 4 non-cooperative enzymes, two (1xz8 and 2jlc) have relatively small cc_equiv
values and have significantly less-correlated active sites than the rest of the protein,
whereas the remaining two (1fi4 and 1hkb) have relatively high cc_equiv values with
only one having a significantly higher-correlated active site than the rest of the protein.
It is therefore surprising that when the residues from the 4 enzymes are pooled they
show both a larger increase in active-site correlation over non-site residues and a higher
degree of correlation in general than non-cooperative proteins. This is due to the large
number of residues in the most highly-coupled enzyme, 1hkb (1834 residues),
dominating over the contributions from 1xz8, 2jlc and 1fi4 (which have 358, 1154 and
832 residues, respectively). This illustrates why it would be misleading to draw solid
conclusions from a dataset of this size.
The results from this limited dataset show that highly-correlated residue dynamic
fluctuations between active sites on different chains of oligomeric enzymes have not
been able to computationally identifying enzymes likely to act in a cooperative manner.
Further work is necessary to identify systematic links between computed correlations
and enzyme cooperativity/Hill coefficients.

240
5.4 Correlation of Residue Motions in Enzyme Active Site
Regions.
The results from the analysis of dynamic coupling in a very small set of cooperative
and non-cooperative enzymes show that the majority have either a higher average
active-site cc_equiv value than the non-site residues or a negative correlation between
cc_equiv and the distance from the active site. There was little distinction in this trend
between cooperative and non-cooperative enzymes and the small dataset size
prevented any solid conclusions to be reached. The suggestion from these results that
dynamic coupling may be a general feature of active sites in oligomeric enzymes,
regardless of their cooperative action, prompted a more extensive study of dynamic
coupling in a larger set of oligomeric enzymes with known active sites. A larger dataset
of oligomeric enzymes with known active sites was compiled as described in 5.2.2 and
the results are shown below.
5.4.1 Analysis of Residue Correlations in Enzyme Active Sites
The dataset described in 5.2.2 (Dataset 2.2) contains 114 homo-oligomeric enzymes
with literature annotated active site information. A similar analysis was carried out as
detailed in 5.3 for Dataset 2.2, this time looking to see whether active site residues were
significantly more highly-coupled with their equivalent residues on opposite chains
than non-active site residues for the whole set.
Due to the large number of proteins in this set results are mostly shown for pooled
data rather than per individual protein. Residue cc_equiv values were scaled between 0
and 1 within each enzyme, which allows fair comparison between differences in site
and non-site residues over enzymes with different background cc_equiv values. The
mean scaled cc_equiv value for pooled non-site residues is 0.362 and for site residues is
0.373. The Mann-Whitney p-value for the difference between non-site and site
cc_equiv values is <0.001 and the 95% confidence intervals are 0.360/0.363 and
0.367/0.379, respectively. This shows that active sites residues are significantly more

241
correlated than non-site residues, but by a very small margin. Figure 5.11 shows the
distribution of scaled cc_equiv values for site and non-site residues in the dataset and
Figure 5.12 shows the cumulative percentage of cc_equiv values for all site and non-
site residues. Whilst there is a significant increase in correlation for site residues over
non-site residues in the pooled dataset, when each enzyme is evaluated individually this
trend is only seen in just over a quarter of the enzymes (see Table 5.7).
As in the previous analysis (detailed in 5.3), the distance of each residue from the active
site centroid and its cc_equiv value was compared. The number of residues in all 114
enzymes is too large to show a clear representation of this data on a plot. The
Spearman’s rank correlation coefficient (and its associated p-value) between cc_equiv
and distance from active site centroid for pooled and scaled data is shown in Table 5.8
(the distance from active site centroid for each residue was also scaled between 0 and 1
in the same way as for cc_equiv). This shows a significant but weak negative
correlation between distance from active-site centroid and cc_equiv.
Table 5.9 shows how this relationship varies for individual enzymes within the set. A
larger number of enzymes have a significant negative correlation between distance
from active-site centroid and cc_equiv than have significantly higher active site
correlations vs. non-site correlations (75 and 31, respectively).
Site correlation > Non-site correlation?
Yes No
Significant 31 23
Non Significant 32 28
Table 5.7. Site correlation vs. non-site correlation results for individual enzymes within the set.
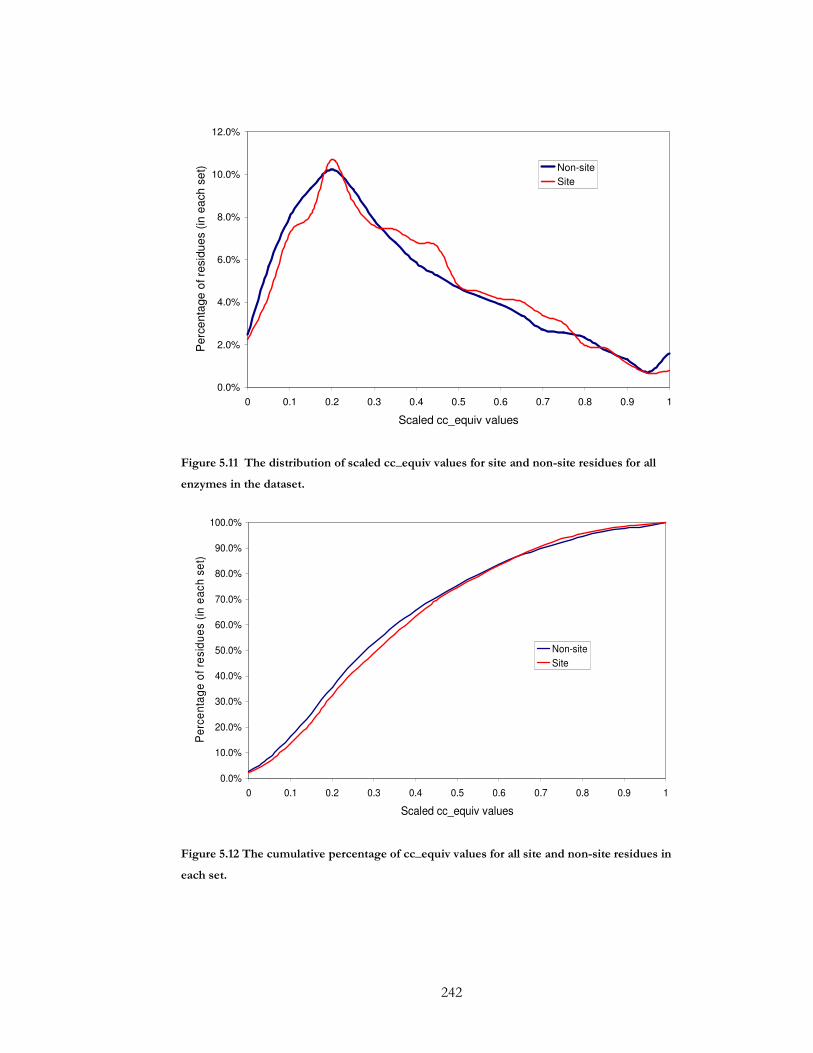
242
0.0%
2.0%
4.0%
6.0%
8.0%
10.0%
12.0%
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Scaled cc_equiv values
Perc
enta
ge
of re
sid
ue
s (
in e
ach
se
t) Non-site
Site
Figure 5.11 The distribution of scaled cc_equiv values for site and non-site residues for all
enzymes in the dataset.
0.0%
10.0%
20.0%
30.0%
40.0%
50.0%
60.0%
70.0%
80.0%
90.0%
100.0%
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Scaled cc_equiv values
Pe
rce
nta
ge
of
resid
ue
s (
in e
ach
se
t)
Non-site
Site
Figure 5.12 The cumulative percentage of cc_equiv values for all site and non-site residues in
each set.

243
Spearman's rank correlation coefficient P-value
-0.071 >0.001
Table 5.8 The Spearman’s rank correlation coefficient for the relationship between distance
from active site centroid and cc_equiv for all enzymes in the set.
Spearman's rank correlation coefficient
Negative Positive
Significant 75 25
Non-significant 8 6
Table 5.9 Table showing the breakdown of Spearman’s rank correlation coefficients between
distance from active site centroid and cc_equiv value for individual enzymes in the dataset.
A low B-factor (which approximates to the degree of structural constraint of a residue)
is a well documented feature of catalytic residues77; 78 and similarly the magnitude of
normal mode fluctuations has also been shown to be indicative of catalytic residues79.
It is a reasonable argument that residues in a constrained environment have a higher
probability of having more highly correlated motion with each other than those with
more freedom. If active site residues (which contain catalytic residues) tend to be more
constrained than other residues then this may explain their slight increase in fluctuation
correlation.
In this dataset only around half of the enzymes (58 out of 114) have a significantly
lower average B-factor for active site residues than non-site residues (see Table 5.10).
The Spearman’s rank correlation coefficient is significantly negative for the relationship
between cc_equiv and B-factor for 65 of the 114 enzymes in the dataset, indicating that
the hypothesis that more constrained residues are likely to be more highly correlated is
only supported for approximately half of the set (see Figure 5.11).
It is furthermore not the case that the enzymes where there are significantly higher
cc_equiv values in the active site (31 out of 114) are more likely to show significantly
lower B-factors or significantly negative relationships between cc_equiv and B-factor.
Table 5.12 shows that, as for the whole set, only around half of the enzymes where
active site residues are significantly more correlated than non-site residues have

244
significantly lower active-site B-factors and two-thirds have significant negative
relationships between cc_equiv and B-factor.
Number of proteins in the set
Higher average active site
B-factor than non-site Lower average active site
B-factor than non-site
Significant 18 58
Non-significant 9 29
Table 5.10 The number of proteins in the whole dataset that have either a lower or higher
average active site B-factor than non-site residues, split by significance.
Number of proteins in the set
Negative correlation between
B-factor and cc_equiv Positive correlation between
B-factor and cc_equiv
Significant 65 17
Non-significant 19 13
Table 5.11 The number of proteins in the whole dataset that have either a negative or positive
correlation between B-factor and cc_equiv, split by significance.
Number of proteins where the active site residues are
significantly more correlated than non-site residues
Higher average active site
B-factor than non-site Lower average active site
B-factor than non-site
Significant 6 16
Non-significant 2 7
Table 5.12 Number of proteins where the active site residues are significantly more correlated
than non-site residues that have either higher or lower average site B-factors in comparison to
the rest of the protein, split by significance.
Number of proteins where the active site residues are
significantly more correlated than non-site residues
Negative correlation between
B-factor and cc_equiv Positive correlation between
B-factor and cc_equiv
Significant 20 4
Non-significant 5 2
Table 5.13 Number of proteins where the active site residues are significantly more correlated
than non-site residues that have either a positive or negative relationship between cc_equiv and
B-factor, split by significance.

245
5.4.2 Discussion of Correlation of Motion in Active Sites
It was the hypothesis that active site residues are at or near to parts of the enzyme
structure which have high correlation of residue fluctuations with their equivalent
residues in opposite chains. When scaled cc_equiv values are pooled for all enzymes in
the set there is a significant but small increase in cc_equiv values for site residues over
non-site residues. This trend however, only holds true for 31 of the 114 enzymes in
the set on an individual basis.
The motivation behind the hypothesis was to support identification of active sites in
oligomeric enzymes; however the very small overall difference in cc_equiv values over
the total dataset and the inconsistent pattern of active-site correlation values on an
individual basis suggests that it is not a promising candidate for use in characterising
active sites.
In contrast to the previous analysis, a problem in interpreting the results of this analysis
is due to the large number of data points (i.e. the total number of residues from all 114
enzymes) used to evaluate significance. Due to the large number of residues, very weak
correlation values are statistically significant. The Spearman’s rank correlation
coefficient (rho) for the relationship between B-factor and cc_equiv over the whole set
is very weakly negative (-0.071), which does not demonstrate a strong relationship
between these two features. Due to the large number of data points in the sample, the
threshold for rho to be significant is very low and therefore the above relationship is
statistically significant even though it is very weak.
Catalytic residues have been shown to have low B-factors78 and smaller magnitude of
normal mode fluctuations79 than other residues. It is a reasonable assumption that if a
residue is structurally constrained and the amount of space that its fluctuations can
sample is limited then it has a higher probability of being correlated with its equivalent
residues on the opposite chain. If this was true then it could explain the slight increase
in correlation between equivalent residues in active sites over non-active sites. Over all
residues in all enzymes in the dataset there was only a very weak (but significant)
negative correlation between cc_equiv and B-factor and only 65 out of 114 enzymes

246
showed a significant negative correlation on an individual basis. Of the 31 enzymes
that did show a significantly more correlated active site than the rest of the protein,
only half of these showed a significant negative relationship between cc_equiv and B-
factor, which suggests that for at least half of the cases where active sites are more
dynamically coupled that it is not the structural constraint of those residues that is
driving it.
5.5 Patterns of Correlation of Residue Motion as a
Structural Feature of Oligomeric Proteins
Correlations between residues within subunit structures have been shown to be of use
in characterising functional dynamics of protein structures80; 81; 82 but less is known
about how correlations across subunits affects the function of oligomers. Bai et al.,
observed that the overall degree of dynamic coupling was increased when a functional
dimer was considered in its oligomeric state rather than by considering the monomer
subunits separately83. This suggests that the oligomeric state of a protein has a
functional effect on its dynamic properties.
In the analyses in 5.3 and 5.4 the functional significance of these cross-subunit
fluctuation correlations was investigated. The first analysis failed to reach a solid
conclusion about whether active sites of cooperative enzymes have coupled motions
between subunits that are distinguishable from those of non-cooperative active sites.
Similarly, the previous analysis suggested only a slight increase in active site fluctuation
correlations between subunits in oligomers in general.
If the degree of coupling of a given residue with its equivalent residue on another
subunit isn’t associated with any functional or structural purpose for that enzyme, it
might be expected that either there is no variation in the degree of coupling between
different residues or that the variation is distributed in a random manor within the
structure. It is interesting, however, that the structures of enzymes in both analyses
tend to exhibit a broadly similar non-random architecture of coupling of residue
fluctuations.
The structures of oligomers coloured according to their per-residue cc_equiv value (see
Figure 5.7 to Figure 5.9 for enzymes from Dataset 2.1, for example) show a smooth,

247
ordered distribution of varying cross-subunit residue correlations over the protein
structure. The degree of correlation between equivalent residues appears from many of
these examples to organize itself into a common architecture. Highly-correlated
residues appear to assemble inside the subunit cores, with the surface of the subunit
exhibiting moderate cross-correlations and the subunit interfaces typically being the
least correlated.
If the assumption that tightly packed residues have a higher probability of their
fluctuations being coupled because of their limited freedom is true then it is perhaps
unsurprising that the cores of the subunits tend to be highly correlated with each other.
Given this hypothesis, however, the least packed residues- those on the surface of the
subunits- would be expected to show the lowest degree of coupling. It is therefore
interesting that surface residues appear to be more correlated than those in the
interface, even though such residues experience more structural constraint. It would
also be reasonable to assume that equivalent pairs that are the closest in the 3D
structure (as are the residues in the interface) would have higher-coupled motion than
those connected by longer range distances and so, again, it is surprising that interface
residues appear to be among the least correlated.
These observations are based on visual inspection of the relatively limited set of
enzymes in the previous analyses. To further investigate these observations, a new
dataset was created to include a wider range of homo-oligomeric proteins including
non-enzymes. The pattern of variation of cross-correlations within these structures
was evaluated quantitatively and the results are shown below.
5.5.1 Differences in Residue Motion Correlation According to
Structural Environment
The dataset used in this analysis (described in 5.2.3) contains 636 homo-oligomeric
proteins. As in previous analyses, the cc_equiv values were scaled from 0 to 1 within
each protein to enable residues from all proteins to be pooled. A structural
environment status (interface, core or surface) was assigned to each residue based on
the rules described in 5.2.3. The mean scaled cc_equiv values for each structural
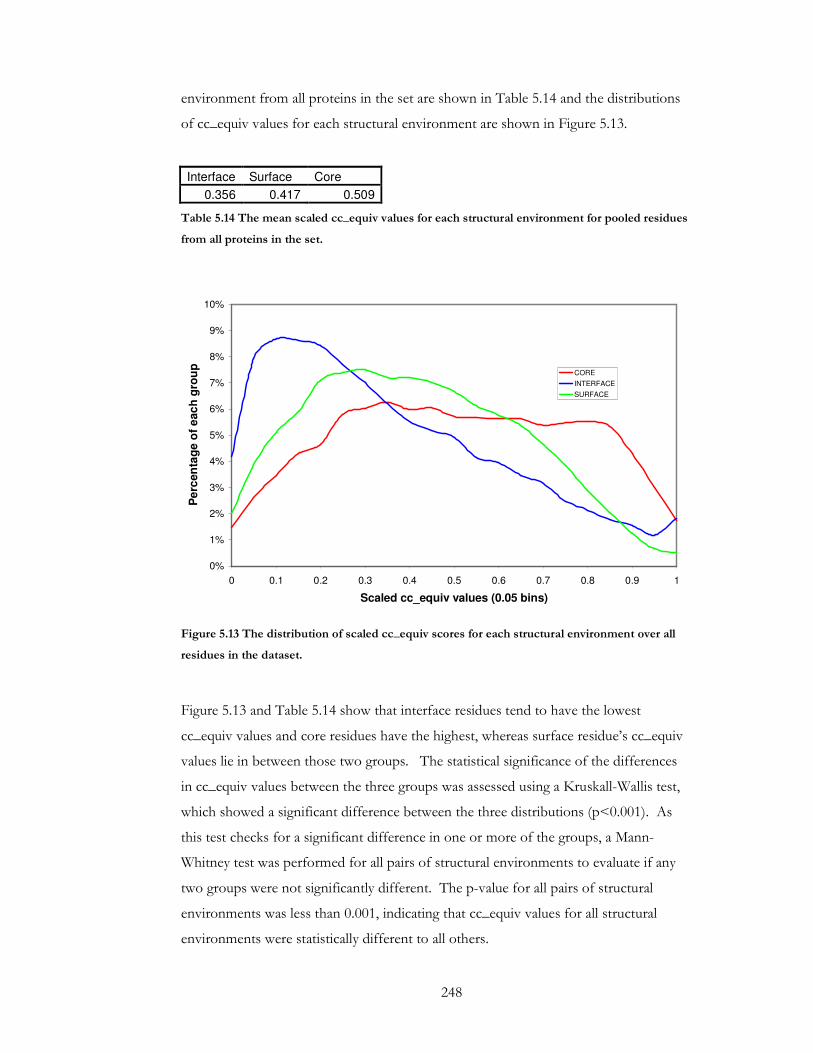
248
environment from all proteins in the set are shown in Table 5.14 and the distributions
of cc_equiv values for each structural environment are shown in Figure 5.13.
Interface Surface Core
0.356 0.417 0.509
Table 5.14 The mean scaled cc_equiv values for each structural environment for pooled residues
from all proteins in the set.
0%
1%
2%
3%
4%
5%
6%
7%
8%
9%
10%
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Scaled cc_equiv values (0.05 bins)
Per
cen
tag
e o
f ea
ch g
rou
p
CORE
INTERFACE
SURFACE
Figure 5.13 The distribution of scaled cc_equiv scores for each structural environment over all
residues in the dataset.
Figure 5.13 and Table 5.14 show that interface residues tend to have the lowest
cc_equiv values and core residues have the highest, whereas surface residue’s cc_equiv
values lie in between those two groups. The statistical significance of the differences
in cc_equiv values between the three groups was assessed using a Kruskall-Wallis test,
which showed a significant difference between the three distributions (p<0.001). As
this test checks for a significant difference in one or more of the groups, a Mann-
Whitney test was performed for all pairs of structural environments to evaluate if any
two groups were not significantly different. The p-value for all pairs of structural
environments was less than 0.001, indicating that cc_equiv values for all structural
environments were statistically different to all others.

249
On an individual basis the majority of proteins (419) have lower average cc_equiv for
their interface residues than their surface and core residues, where the core residues
have the highest average cc_equiv. The core residues have the highest average
cc_equiv in 83% (529) of the dataset, and the interface has the lowest cc_equiv in 68%
of the proteins. The variation of cc_equiv values over the three structural
environments for individual proteins is shown in Table 5.15.
Interface Surface Core
Interface 436 (392) 540 (490)
Surface 230 (142) 603 (533)
Core 96 (45) 33(6)
Table 5.15. Pairwise comparison of average cc_equiv values for each structural environment.
The value given is the number of proteins in the set where the average cc_equiv value for the
environment in the row is lower than the environment in the column. The figure in brackets is
the number of these cases where the difference is statistically significant.
The Spearman’s rank correlation coefficient for the relationship between the cc_equiv
score and the B-factor in this dataset shows a very weak negative correlation of -0.066
(p-value < 0.001). Table 5.16 shows the number of proteins in the dataset that have
either a negative or positive correlation between cc_equiv and B-factor (split by
significance).
The B-factors were scaled from 0 to 1 within each file to allow their distributions to be
compared over the whole set. The average scaled B-factor values for each structural
environment are shown in Table 5.17. The difference between B-factors in these three
environments is statistically significant for all pairs of environments (Mann-Whitney
p<0.001). The surface residues have the overall highest average B-factor and the core
residues have the lowest. Table 5.18 shows how differences in average B-factors for
each structural environment break down for individual proteins in the dataset. The
core residues have the highest average B-factors in over 90% of the proteins in the set,
and 88% have lower average B-factors for the interface than the surface.

250
Number of proteins in the set
Negative correlation between
B-factor and cc_equiv Positive correlation between
B-factor and cc_equiv
Significant 414 61
Non-significant 96 65
Table 5.16 The number of proteins in the Dataset 2.3 that have either a negative or positive
correlation between B-factor and cc_equiv, split by significance.
Interface Surface Core
0.222 0.317 0.143
Table 5.17 The average scaled B-factors for each structural environment over all residues in the
set.
Core Surface Interface
Core 636 (615) 580 (406)
Surface 0 (0) 74 (25)
Interface 56 (21) 562 (453)
Table 5.18 Pairwise comparison of average scaled B-factors for each structural environment.
The value given is the number of proteins in the set where the average scaled B-factor for the
environment in the row is lower than the environment in the column. The figure in brackets is
the number of these cases where the difference is statistically significant.
As mentioned previously, it is reasonable to assume that the closer the equivalent
residues are to each other in the structure the more correlated their motions will be.
The distance between Cβ atoms (Cα for glycine) for equivalent residues on 2 separate
chains was calculated and the distances scaled from 0 to 1 within each protein. The
average scaled distances for each structural environment are shown in Table 5.19,
which shows that interface residues are on average the closest to each other (on an
individual basis this is true for over 90% of the set). This is unsurprising since
interface residues are defined as being close to residues on other chains and the
symmetry of oligomers often puts equivalent residues at the interface between chains.
Interface Surface Core
0.318 0.545 0.452
Table 5.19 The average scaled distance between equivalent residues for each structural
environment over all residues in the set.

251
It is surprising, however, that despite interface residues being the closest to their
equivalent residues they have on average the least correlated motion. Over all residues
from all proteins in the set there is a significant positive correlation between the
distance of a residue from its equivalent residue on the opposite chain and the degree
of dynamic coupling between them (Spearman’s rank correlation coefficient of 0.237
with a p-value of less than 0.001). Table 5.20 and Figure 5.14 show how the
correlation between cc_equiv and distance between equivalent residues differs for
individual proteins in the set. Almost 95% (603) of the proteins in the set show either
a positive or a non-significantly negative correlation between the degree of motion
correlation and distance between equivalent residues (Table 5.20).
Whilst in general the closer a residue is to its equivalent residue does not necessarily
translate into a higher degree of dynamic coupling, where two equivalent residues are
directly adjacent to each other in the protein structure they are often highly correlated.
These highly correlated residues are often isolated within the interface, with the other
surrounding interface residues still being weakly coupled (see Figure 5.15 for an
example). The closest pair of equivalent residues is the most correlated in 111 (17%)
of the 636 proteins in the set and the distribution of scaled cc_equiv values for the
closest pair of equivalent residues is shown in Figure 5.16. It should be noted that
cc_equiv values have been rounded to the nearest 0.05 in order to plot the data in this
figure and thus an extra 11 residue pairs have had their scaled cc_equiv rounded up to
1. Despite the closest equivalent pair having the highest cc_equiv value, 61 of these
111 still have a significantly lower degree of coupling for their interface residues than
the rest of the protein.
Similarly, the distribution of scaled distances between equivalent pairs that have the
largest cc_equiv is shown in Figure 5.17. An extra 73 residue pairs have a scaled
distance that is rounded down to 0, indicating that, whilst they are not the closest
residue pairs, they are one of the closest. This shows that for 71% (452) of the
proteins in the set, the highest-correlated pair is not one of the closest residue pairs in
the structure. The highest-correlated residue pair is one of the closest residues (the
scaled distance between them rounds to 0) in 184 proteins, yet in over 75% of these
(140) the interface residues are still significantly less-coupled than the rest of the
protein.

252
Number of proteins in the set
Positive correlation between distance between equivalent
residues and cc_equiv
Negative correlation between distance between equivalent
residues and cc_equiv
Significant 418 87
Non-significant 155 33
Table 5.20 The number of proteins in the Dataset 2.3 that have either a negative or positive
correlation between the distance between equivalent residues and cc_equiv, split by
significance.
Figure 5.14 The distribution of Spearman’s rank correlation coefficients between cc_equiv
values and distance between equivalent residues for individual proteins in the dataset.
0
10
20
30
40
50
60
70
-1 -0.8 -0.6 -0.4 -0.2 0 0.2 0.4 0.6 0.8
Spearman's rank correlation coefficient between scaled cc_equiv values and distance between equivalent residues
Nu
mb
er
of
pro
tein
s

253
Figure 5.15 An example of a protein in the dataset (1h16) where the closest equivalent residues
in the interface have the highest dynamic coupling and the rest of the interface residues are less-
coupled in comparison.
Red residues have the highest cc_equiv value and dark blue have the lowest.
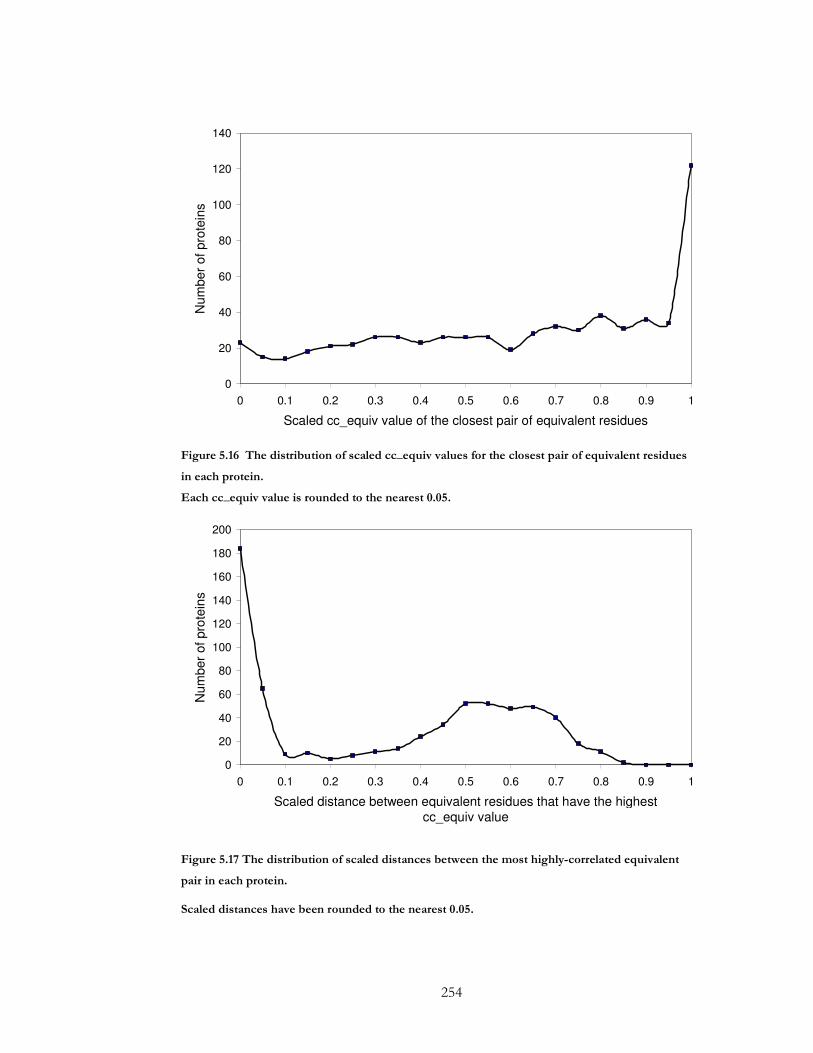
254
0
20
40
60
80
100
120
140
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Scaled cc_equiv value of the closest pair of equivalent residues
Num
ber
of pro
tein
s
Figure 5.16 The distribution of scaled cc_equiv values for the closest pair of equivalent residues
in each protein.
Each cc_equiv value is rounded to the nearest 0.05.
0
20
40
60
80
100
120
140
160
180
200
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Scaled distance between equivalent residues that have the highest cc_equiv value
Num
ber
of pro
tein
s
Figure 5.17 The distribution of scaled distances between the most highly-correlated equivalent
pair in each protein.
Scaled distances have been rounded to the nearest 0.05.

255
To investigate the extent to which the oligomeric status affects this common
architecture of correlated motion, the degree of correlation across subunits was
compared to the pattern of correlation of residue motion within subunits. The degree
of dynamic correlation of a residue to the all other residues within a single subunit was
estimated by averaging the correlation between a given residue and all other residues in
the subunit (termed the cc_within value). The distribution of Spearman’s correlation
coefficients between cc_equiv and cc_within values for each protein in the dataset is
shown in Figure 5.18. It shows that the patterns of variation of cc_equiv and
cc_within values over the structure are similar for most proteins.
0
20
40
60
80
100
120
140
-1.000 -0.800 -0.600 -0.400 -0.200 0.000 0.200 0.400 0.600 0.800 1.000
Sprearman's rank correlation coefficient between cc_within and
cc_equiv value
Num
ber
of pro
tein
s
Figure 5.18 The distribution of Spearman’s correlation coefficients between cc_within and
cc_equiv values for all proteins in the set.
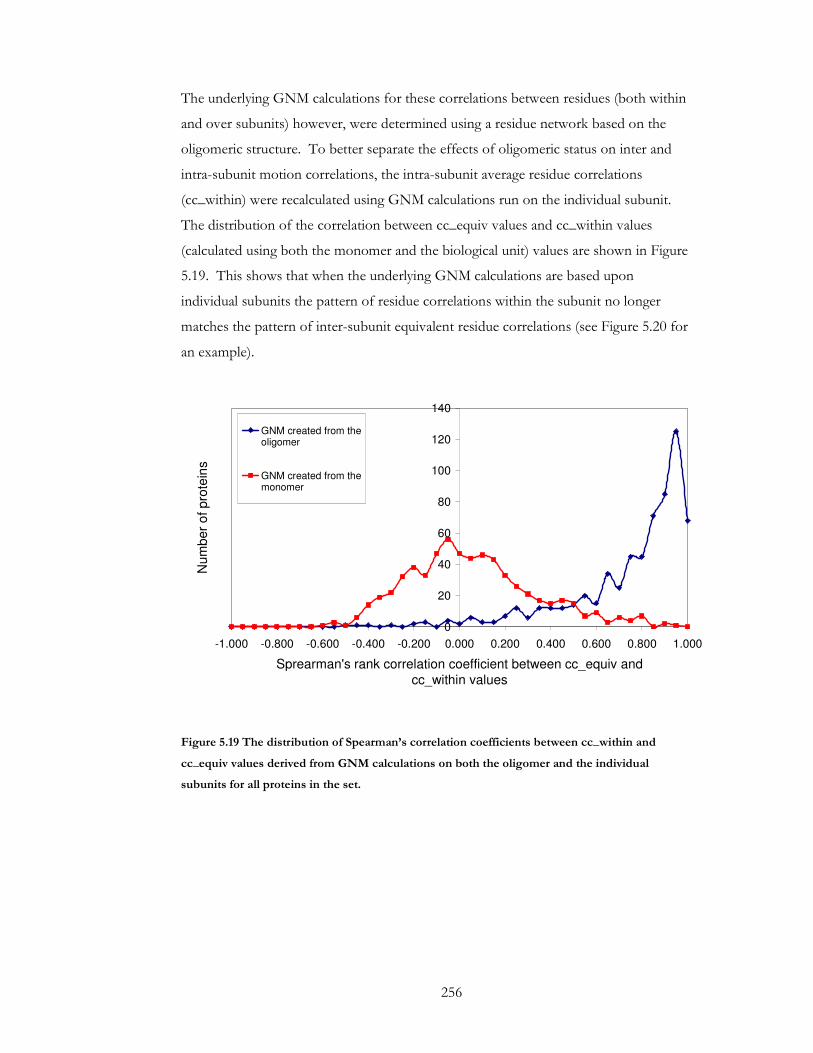
256
The underlying GNM calculations for these correlations between residues (both within
and over subunits) however, were determined using a residue network based on the
oligomeric structure. To better separate the effects of oligomeric status on inter and
intra-subunit motion correlations, the intra-subunit average residue correlations
(cc_within) were recalculated using GNM calculations run on the individual subunit.
The distribution of the correlation between cc_equiv values and cc_within values
(calculated using both the monomer and the biological unit) values are shown in Figure
5.19. This shows that when the underlying GNM calculations are based upon
individual subunits the pattern of residue correlations within the subunit no longer
matches the pattern of inter-subunit equivalent residue correlations (see Figure 5.20 for
an example).
0
20
40
60
80
100
120
140
-1.000 -0.800 -0.600 -0.400 -0.200 0.000 0.200 0.400 0.600 0.800 1.000
Sprearman's rank correlation coefficient between cc_equiv and
cc_within values
Nu
mb
er
of p
rote
ins
GNM created from theoligomer
GNM created from themonomer
Figure 5.19 The distribution of Spearman’s correlation coefficients between cc_within and
cc_equiv values derived from GNM calculations on both the oligomer and the individual
subunits for all proteins in the set.

257
A
B
C
Figure 5.20 An example of a protein (1cq3) with residues coloured by cc_equiv value (A),
cc_within values derived using GNM calculations on the oligomer (B), and cc_within values
derived from GMN calculations on the individual monomers(C).

258
5.5.1.1 Are Residues with High Dynamic Coupling to their
Equivalent Residues more Evolutionarily Conserved?
The degree of evolutionary conservation was mapped onto each protein structure by
assigning each residue a conservation score (the methods for which are given in 3.3.1).
The conservation score was normalised from 0 to 1 within each protein structure. The
Spearman’s correlation coefficient between a residue’s normalised conservation score
and its cc_equiv value is plotted for each protein in the set (a conservation profile
could not be produced by PSI-BLAST for 21 of the proteins). Over all proteins the
correlation between conservation and the degree of dynamic coupling between
equivalent residues was 0.098 (p<0.001), which shows that there is only a weak positive
relationship between dynamic coupling and evolutionary conservation. Whilst it is true
that the core residues are generally the most conserved (and also have the highest
average cc_equiv values) the surface residues are the least conserved but do not have
the lowest average cc_equiv value (Table 5.21). Interface residues have the lowest
average cc_equiv value yet are evolutionarily conserved to a higher degree, therefore
only a weak positive relationship between conservation and degree of correlation of
motion exists.
Interface Surface Core
0.339 0.258 0.385
Table 5.21 The average scaled conservation score for each structural environment.
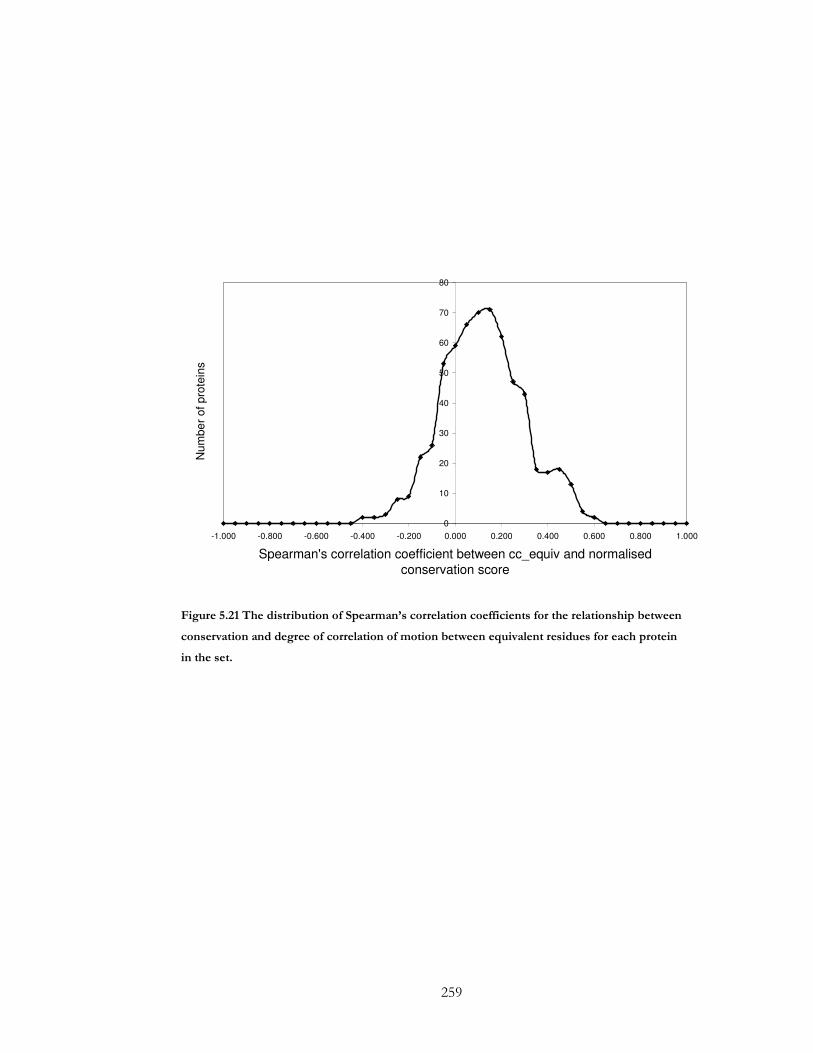
259
0
10
20
30
40
50
60
70
80
-1.000 -0.800 -0.600 -0.400 -0.200 0.000 0.200 0.400 0.600 0.800 1.000
Spearman's correlation coefficient between cc_equiv and normalised
conservation score
Num
be
r of pro
tein
s
Figure 5.21 The distribution of Spearman’s correlation coefficients for the relationship between
conservation and degree of correlation of motion between equivalent residues for each protein
in the set.

260
5.5.2 Discussion of Correlation of Motion According to Structural
Environment
As discussed previously, the degree of correlation of motion between pairs of residues
within subunits has been able to identify structural regions that relate to a protein’s
function80; 81; 82 but little is known about how much functional information is contained
in the correlation of motion between subunits. Cross-correlation matrices reveal
striking correlation patterns within subunits but these analyses show variation and order
in correlations across subunits, which is not obvious from these matrices (see Figure
5.22). If correlation of motion between subunits has no role in the structure or
function of a protein then it could be expected to either not vary, or vary in a random
pattern over the protein structure. This analysis shows that not only does the degree of
coupling over subunits vary for residues within a structure, but they seem to form a
common architecture where subunit cores are highly correlated, and subunit interfaces
are the lowest correlated.
It is possible that differences in the degree of dynamic coupling between equivalent
pairs are purely consequences of the constraints that their different environments place
on them. One such constraint, the degree of structural freedom of a residue’s side
chain (approximated by a residue’s B-factor), was investigated in the above analysis. If
the degree of structural constraint that a side chain experienced was solely responsible
for the variation in residue coupling then it would be expected that there would be a
strong negative correlation between B-factor and degree of coupling for proteins in the
set. It was shown, however, that only a very weak negative relationship existed
between these two variables (Spearman’s rank correlation coefficient = -0.066).
This small negative relationship was driven by core residues (which have the lowest
average B-factor) having the highest average degree of dynamic coupling. This was
however, contradicted by surface residues having the highest average B-factor but not
showing the smallest level of coupling. Despite experiencing a greater degree of
structural constraint than surface residues, the interface residues actually showed

261
significantly less correlation between equivalent residues than surface residues (and
core residues).
It was also investigated whether the distance between equivalent residue pairs was
driving the variation in the degree of correlation between them. Whilst the residue pair
with the highest cross-correlation was one of the closest (their scaled distance rounded
down to 0) in 184 proteins, 140 of these still showed a significantly lower degree of
coupling in their interface than the rest of the protein. If it is true that high dynamic
coupling is driven by residue pairs being closer in proximity, then a negative
relationship between the cc_equiv values and the distance between equivalent residues
would be expected. The relationship between these two factors however, was positive
(Spearman’s rank correlation coefficient = 0.237, p<0.001) and the closest residues (the
interface residues) did not have the highest average cross-correlation (which belonged
to the core residues).
These results suggest that there may be some structural or functional reasons for the
patterns of coupling of dynamic fluctuations over subunits in oligomers, which are not
merely consequences of their proximity to each other or their degree of structural
constraint. It is not entirely obvious that the degree of coupling of residues across
separate subunits should form an ordered arrangement over the protein structure, or
moreover that there should be a common architecture of this arrangement across
evolutionarily unrelated proteins. Furthermore, it was also shown that this
architecture is produced by patterns of motion specific to the oligomer and is distinct
from patterns of motion from within each monomer.
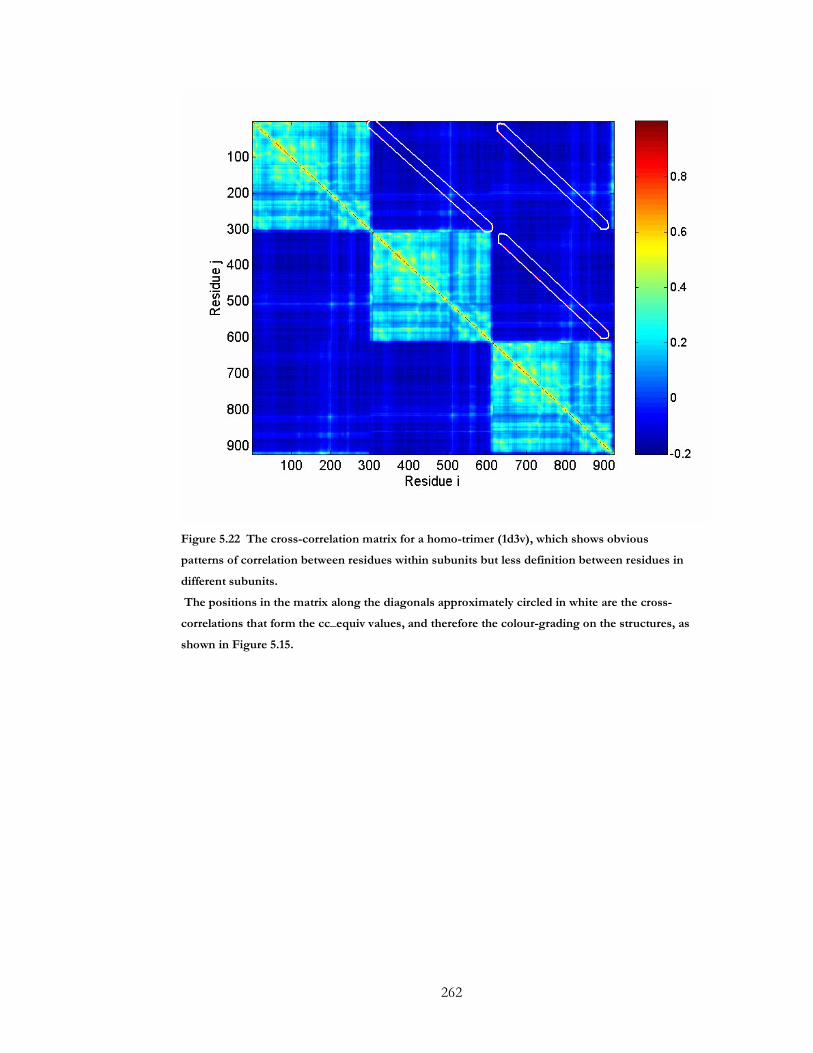
262
Figure 5.22 The cross-correlation matrix for a homo-trimer (1d3v), which shows obvious
patterns of correlation between residues within subunits but less definition between residues in
different subunits.
The positions in the matrix along the diagonals approximately circled in white are the cross-
correlations that form the cc_equiv values, and therefore the colour-grading on the structures, as
shown in Figure 5.15.

263
5.6 Conclusions
The initial aim of the work in this chapter was to assess whether dynamic coupling
between residues in the multiple active sites of cooperative proteins is distinguishable
from those in non-cooperative proteins. The intention of this work was to guide an
attempt to create a method to computationally identify enzymes which are likely to act
cooperatively from their structure. The results showed that the residues in active sites
of cooperative proteins are no more dynamically coupled than those in non-
cooperative proteins and thus, at present, is not able to successfully distinguish
cooperative and non-cooperative enzymes.
A number of observations whilst carrying out this work led to further analyses; firstly,
that active site regions appeared to be situated close to highly-coupled sections of the
enzyme structure and secondly, that the distribution of the degree of coupling between
equivalent residues on separate chains appeared to vary in an ordered manner over the
enzyme structure. These two observations were investigated further analyses on large
sets of non-redundant homo-oligomeric proteins.
The first of these analyses, which focused on the coupling of active site regions,
showed that over the whole dataset there is a statistically significant but very small
increase in the dynamic coupling of active site residues over non-active site residues in
homo-oligomeric enzymes. On an individual basis however, proteins within the set
were equally likely to have active sites with decreased coupling at their active sites than
increased coupling. Even for proteins with a significant increase in coupling in their
active sites, the magnitude of the difference is inadequate to distinguish active sites
residues from non-site residues.
The final analysis investigated the observation that oligomeric proteins, regardless of
the evolutionary relatedness, seemed to show a common architecture of the pattern of
coupling between equivalent residues over subunits of homo-oligomeric proteins. It
has been previously shown that similar architectures of protein folds exhibit similar
dynamics84 but here it is shown that dynamic coupling between subunits in homo-
oligomers is broadly conserved over a wide range of non-homologous proteins.

264
On a large, non-redundant set of homo-oligomeric proteins (including non-enzymes)
it was shown that the interface residues have the lowest distribution of cross-subunit
coupling, the core has the highest and the surface correlations fall in between. It is
perhaps surprising that interface residues are less correlated than others, particularly
due to their proximity to each other. It was also shown that the degree of constraint
was not responsible for the pattern of coupling, specifically as interfaces are generally
more tightly packed than surface residues yet they exhibit less coupling between them
than surface residues. Correlation of motion between residues within a subunit have
been shown to contain information relating to the protein’s function80; 81; 82 but little is
known about how residue motions are correlated over separate subunits and whether
these motions are important to the protein’s structure and function. A very weak
positive correlation was found between the evolutionary constraint of a residue and the
degree of correlation of motion to its equivalent residues, suggesting that evolution
does not necessarily act to preserve the most highly correlated motions over subunits
in the same way as has been suggested within subunits. The fact that the degree of
coupling over separate subunits varies in a smooth and ordered fashion over non-
homologous protein structures from a wide range of functions suggest that this pattern
of dynamics is functionally important to oligomeric proteins. It was shown that this
common architecture of dynamic coupling between residues is not intrinsic in the
monomer alone as it was altered in an inconsistent manner when the influence of the
oligomeric status was removed from the estimation of residue dynamics.
It is still unclear exactly what functional significance these cross-subunit dynamic
correlations have for oligomeric proteins. Perhaps the most plausible functionality that
correlated motion between subunits might bestow is communicating structural change
between distal residues for the purposes of cooperativity. An analysis of this concept
was attempted in this chapter and the results were inconclusive, especially due to the
availability of only a small number of biochemically-annotated structures. It is
possible that, due to the wide range of functions for which this pattern of coupling is
displayed, that the coupling is a structural feature of oligomeric proteins rather than
being associated with a particular function.

265
The dynamic coupling between interface residues is arguably the most important to the
viability of the oligomeric structural arrangement. Residue pairs on the surface, or even
in the core, can sample a variety of motion combinations without jeopardising the
overall quaternary structure. If, for example, a pair of interface residues was to move
in a correlated manor but in opposite directions, this could create a solvent-accessible
pocket in the subunit interface. The creation of a solvent-accessible pocket in an
interface would reduce the interaction energy between the two subunits and in turn,
potentially destabilise the quaternary structure. It is, therefore, reasonable to imagine
that the coupling between residue dynamics at the interface is selected to be more
chaotic and disorganised, and therefore less-correlated, to avoid creating solvent-
accessible space in the subunit interface, which would be detrimental to the
preservation of the quaternary structure.

266
5.7 References
1. Bohr C., H. K. A., Krogh A. (1904). Ueber einen in biologischer Beziehung wichtigen Einfluss, den die Kohlensäurespannung des Blutes auf dessen Sauerstoffbindung übt. Skand. Arch. Physiol 16, 402-412.
2. Hill, A. V. (1910). The possible effects of the aggregation of the molecules of hemoglobin on its dissociation curves. J. Physiol 40, iv-vii.
3. Goldbeter, A. & Koshland, D. E., Jr. (1981). An amplified sensitivity arising from covalent modification in biological systems. Proc Natl Acad Sci U S A 78, 6840-4.
4. Perutz, M. F. & Lehmann, H. (1968). Molecular pathology of human haemoglobin. Nature 219, 902-9.
5. Koshland, D. E., Jr. & Hamadani, K. (2002). Proteomics and models for enzyme cooperativity. In J Biol Chem, Vol. 277, pp. 46841-4.
6. Perutz, M. F. & Brunori, M. (1982). Stereochemistry of cooperative effects in fish an amphibian haemoglobins. Nature 299, 421-6.
7. Todd, A. E., Orengo, C. A. & Thornton, J. M. (2001). Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol 307, 1113-43.
8. Thornton, J. M., Todd, A. E., Milburn, D., Borkakoti, N. & Orengo, C. A. (2000). From structure to function: approaches and limitations. Nat Struct Biol 7 Suppl, 991-4.
9. Bork, P. & Koonin, E. V. (1998). Predicting functions from protein sequences--where are the bottlenecks? Nat Genet 18, 313-8.
10. Iliopoulos, I., Tsoka, S., Andrade, M. A., Enright, A. J., Carroll, M., Poullet, P., Promponas, V., Liakopoulos, T., Palaios, G., Pasquier, C., Hamodrakas, S., Tamames, J., Yagnik, A. T., Tramontano, A., Devos, D., Blaschke, C., Valencia, A., Brett, D., Martin, D., Leroy, C., Rigoutsos, I., Sander, C. & Ouzounis, C. A. (2003). Evaluation of annotation strategies using an entire genome sequence. Bioinformatics 19, 717-26.
11. Go, N., Noguti, T. & Nishikawa, T. (1983). Dynamics of a small globular protein in terms of low-frequency vibrational modes. Proc Natl Acad Sci U S A 80, 3696-700.
12. Bahar, I., Atilgan, A. R. & Erman, B. (1997). Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold Des 2, 173-81.
13. Tirion, M. M. (1996). Large Amplitude Elastic Motions in Proteins from a Single-Parameter, Atomic Analysis. Phys Rev Lett 77, 1905-1908.
14. Atilgan, A. R., Durell, S. R., Jernigan, R. L., Demirel, M. C., Keskin, O. & Bahar, I. (2001). Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J 80, 505-15.
15. Tama, F. & Sanejouand, Y. H. (2001). Conformational change of proteins arising from normal mode calculations. Protein Eng 14, 1-6.
16. Li, G. & Cui, Q. (2002). A coarse-grained normal mode approach for macromolecules: an efficient implementation and application to Ca(2+)-ATPase. Biophys J 83, 2457-74.
17. Micheletti, C., Carloni, P. & Maritan, A. (2004). Accurate and efficient description of protein vibrational dynamics: comparing molecular dynamics and Gaussian models. Proteins 55, 635-45.

267
18. Doruker, P., Atilgan, A. R. & Bahar, I. (2000). Dynamics of proteins predicted by molecular dynamics simulations and analytical approaches: application to alpha-amylase inhibitor. Proteins 40, 512-24.
19. Ming, D., Kong, Y., Lambert, M. A., Huang, Z. & Ma, J. (2002). How to describe protein motion without amino acid sequence and atomic coordinates. Proc Natl Acad Sci U S A 99, 8620-5.
20. Van Wynsberghe, A., Li, G. & Cui, Q. (2004). Normal-mode analysis suggests protein flexibility modulation throughout RNA polymerase's functional cycle. Biochemistry 43, 13083-96.
21. Temiz, N. A. & Bahar, I. (2002). Inhibitor binding alters the directions of domain motions in HIV-1 reverse transcriptase. Proteins 49, 61-70.
22. Keskin, O., Bahar, I., Flatow, D., Covell, D. G. & Jernigan, R. L. (2002). Molecular mechanisms of chaperonin GroEL-GroES function. Biochemistry 41, 491-501.
23. Cui, Q., Li, G., Ma, J. & Karplus, M. (2004). A normal mode analysis of structural plasticity in the biomolecular motor F(1)-ATPase. J Mol Biol 340, 345-72.
24. Thomas, A., Hinsen, K., Field, M. J. & Perahia, D. (1999). Tertiary and quaternary conformational changes in aspartate transcarbamylase: a normal mode study. Proteins 34, 96-112.
25. Bahar, A. R. A., M.C. Demirel and B. Erman,. (1998). Dynamics of folded proteins: significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett., 2733–2736.
26. Jernigan, R. L., Demirel, M.C., and Bahar, I. (1999). Relating structure to function through the dominant slow modes of motion of DNA topoisomerase II. Int. J. Quant. Chem.
27. Wang, Y., Rader, A. J., Bahar, I. & Jernigan, R. L. (2004). Global ribosome motions revealed with elastic network model. J Struct Biol 147, 302-14.
28. Xu, C., Tobi, D. & Bahar, I. (2003). Allosteric changes in protein structure computed by a simple mechanical model: hemoglobin T<-->R2 transition. J Mol Biol 333, 153-68.
29. Ming, D. & Wall, M. E. (2005). Allostery in a coarse-grained model of protein dynamics. Phys Rev Lett 95, 198103.
30. Kundu, S., Sorensen, D. C. & Phillips, G. N., Jr. (2004). Automatic domain decomposition of proteins by a Gaussian Network Model. Proteins 57, 725-33.
31. Bahar, I., Wallqvist, A., Covell, D. G. & Jernigan, R. L. (1998). Correlation between native-state hydrogen exchange and cooperative residue fluctuations from a simple model. Biochemistry 37, 1067-75.
32. Micheletti, C., Lattanzi, G. & Maritan, A. (2002). Elastic properties of proteins: insight on the folding process and evolutionary selection of native structures. J Mol Biol 321, 909-21.
33. Kundu, S., Melton, J. S., Sorensen, D. C. & Phillips, G. N., Jr. (2002). Dynamics of proteins in crystals: comparison of experiment with simple models. Biophys J 83, 723-32.
34. Yang, L. W., Rader, A. J., Liu, X., Jursa, C. J., Chen, S. C., Karimi, H. A. & Bahar, I. (2006). oGNM: online computation of structural dynamics using the Gaussian Network Model. Nucleic Acids Res 34, W24-31.
35. Suhre, K. & Sanejouand, Y. H. (2004). ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res 32, W610-4.

268
36. Hollup, S. M., Salensminde, G. & Reuter, N. (2005). WEBnm@: a web application for normal mode analyses of proteins. BMC Bioinformatics 6, 52.
37. Tobi, D. & Bahar, I. (2005). Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc Natl Acad Sci U S A 102, 18908-13.
38. Cooper, A. & Dryden, D. T. (1984). Allostery without conformational change. A plausible model. Eur Biophys J 11, 103-9.
39. Tama, F., Gadea, F. X., Marques, O. & Sanejouand, Y. H. (2000). Building-block approach for determining low-frequency normal modes of macromolecules. Proteins 41, 1-7.
40. Barthelmes, J., Ebeling, C., Chang, A., Schomburg, I. & Schomburg, D. (2007). BRENDA, AMENDA and FRENDA: the enzyme information system in 2007. Nucleic Acids Res 35, D511-4.
41. Wittig U., G., M., Kania, R., Krebs, O., Mir, S., Weidemann, A., Anstein, S., Saric, J. and Rojas, I. (2006). SABIO-RK: Integration and Curation of Reaction Kinetics Data. In Data Integration in the Life Sciences, Vol. 4075, pp. 94-103. Springer Berlin / Heidelberg.
42. Milo, R., Hou, J. H., Springer, M., Brenner, M. P. & Kirschner, M. W. (2007). The relationship between evolutionary and physiological variation in hemoglobin. Proc Natl Acad Sci U S A 104, 16998-7003.
43. Daily, M. D. & Gray, J. J. (2007). Local motions in a benchmark of allosteric proteins. Proteins 67, 385-99.
44. Kanehisa, M., Goto, S., Hattori, M., Aoki-Kinoshita, K. F., Itoh, M., Kawashima, S., Katayama, T., Araki, M. & Hirakawa, M. (2006). From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34, D354-7.
45. Yang, Y. R. & Schachman, H. K. (1993). In vivo formation of active aspartate transcarbamoylase from complementing fragments of the catalytic polypeptide chains. Protein Sci 2, 1013-23.
46. Kuo, L. C., Lipscomb, W. N. & Kantrowitz, E. R. (1982). Zn(II)-induced cooperativity of Escherichia coli ornithine transcarbamoylase. Proc Natl Acad Sci U S A 79, 2250-4.
47. Wang, X. G. & Engel, P. C. (1995). Positive cooperativity with Hill coefficients of up to 6 in the glutamate concentration dependence of steady-state reaction rates measured with clostridial glutamate dehydrogenase and the mutant A163G at high pH. Biochemistry 34, 11417-22.
48. Kikonyogo, A., Abriola, D. P., Dryjanski, M. & Pietruszko, R. (1999). Mechanism of inhibition of aldehyde dehydrogenase by citral, a retinoid antagonist. Eur J Biochem 262, 704-12.
49. Maggini, S., Stoecklin-Tschan, F. B., Morikofer-Zwez, S. & Walter, P. (1992). New kinetic parameters for rat liver arginase measured at near-physiological steady-state concentrations of arginine and Mn2+. Biochem J 283 ( Pt 3), 653-60.
50. Saadat, D. & Harrison, D. H. (1998). Identification of catalytic bases in the active site of Escherichia coli methylglyoxal synthase: cloning, expression, and functional characterization of conserved aspartic acid residues. Biochemistry 37, 10074-86.
51. Nelson, S. W., Honzatko, R. B. & Fromm, H. J. (2004). Origin of cooperativity in the activation of fructose-1,6-bisphosphatase by Mg2+. J Biol Chem 279, 18481-7.

269
52. Krepkiy, D. & Miziorko, H. M. (2004). Identification of active site residues in mevalonate diphosphate decarboxylase: implications for a family of phosphotransferases. Protein Sci 13, 1875-81.
53. Sergienko, E. A. & Srivastava, D. K. (1997). Kinetic mechanism of the glycogen-phosphorylase-catalysed reaction in the direction of glycogen synthesis: co-operative interactions of AMP and glucose 1-phosphate during catalysis. Biochem J 328 ( Pt 1), 83-91.
54. Dunaway, G. A., Jr. & Smith, E. C. (1971). A comparative study of some of the enzymes involved in glucose metabolism of human diploid and SV40-transformed human diploid cells. Cancer Res 31, 1418-21.
55. Ganzhorn, A. J., Lepage, P., Pelton, P. D., Strasser, F., Vincendon, P. & Rondeau, J. M. (1996). The contribution of lysine-36 to catalysis by human myo-inositol monophosphatase. Biochemistry 35, 10957-66.
56. Foster, B. A., Thomas, S. M., Mahr, J. A., Renosto, F., Patel, H. C. & Segel, I. H. (1994). Cloning and sequencing of ATP sulfurylase from Penicillium chrysogenum. Identification of a likely allosteric domain. J Biol Chem 269, 19777-86.
57. Calcagno, M., Campos, P. J., Mulliert, G. & Suastegui, J. (1984). Purification, molecular and kinetic properties of glucosamine-6-phosphate isomerase (deaminase) from Escherichia coli. Biochim Biophys Acta 787, 165-73.
58. Auzat, I., Le Bras, G. & Garel, J. R. (1994). The cooperativity and allosteric inhibition of Escherichia coli phosphofructokinase depend on the interaction between threonine-125 and ATP. Proc Natl Acad Sci U S A 91, 5242-6.
59. Hsieh, J. Y., Chen, S. H. & Hung, H. C. (2009). Functional roles of the tetramer organization of malic enzyme. J Biol Chem 284, 18096-105.
60. Lopez-Flores, I., Barroso, J. B., Valderrama, R., Esteban, F. J., Martinez-Lara, E., Luque, F., Peinado, M. A., Ogawa, H., Lupianez, J. A. & Peragon, J. (2005). Serine dehydratase expression decreases in rat livers injured by chronic thioacetamide ingestion. Mol Cell Biochem 268, 33-43.
61. Sauve, V. & Sygusch, J. (2001). Molecular cloning, expression, purification, and characterization of fructose-1,6-bisphosphate aldolase from Thermus aquaticus. Protein Expr Purif 21, 293-302.
62. Diaz, A., Munoz-Clares, R. A., Rangel, P., Valdes, V. J. & Hansberg, W. (2005). Functional and structural analysis of catalase oxidized by singlet oxygen. Biochimie 87, 205-14.
63. Samuel, J. & Tanner, M. E. (2004). Active site mutants of the "non-hydrolyzing" UDP-N-acetylglucosamine 2-epimerase from Escherichia coli. Biochim Biophys Acta 1700, 85-91.
64. Frederiksen, H., Berenstein, D. & Munch-Petersen, B. (2004). Effect of valine 106 on structure-function relation of cytosolic human thymidine kinase. Kinetic properties and oligomerization pattern of nine substitution mutants of V106. Eur J Biochem 271, 2248-56.
65. Lee, M., Chan, C. W., Mitchell Guss, J., Christopherson, R. I. & Maher, M. J. (2005). Dihydroorotase from Escherichia coli: loop movement and cooperativity between subunits. J Mol Biol 348, 523-33.
66. Konishi, K. & Fujioka, M. (1988). Rat liver glycine methyltransferase. Cooperative binding of S-adenosylmethionine and loss of cooperativity by removal of a short NH2-terminal segment. J Biol Chem 263, 13381-5.
67. Chander, P., Halbig, K. M., Miller, J. K., Fields, C. J., Bonner, H. K., Grabner, G. K., Switzer, R. L. & Smith, J. L. (2005). Structure of the nucleotide complex

270
of PyrR, the pyr attenuation protein from Bacillus caldolyticus, suggests dual regulation by pyrimidine and purine nucleotides. J Bacteriol 187, 1773-82.
68. Raffaelli, N., Finaurini, L., Mazzola, F., Pucci, L., Sorci, L., Amici, A. & Magni, G. (2004). Characterization of Mycobacterium tuberculosis NAD kinase: functional analysis of the full-length enzyme by site-directed mutagenesis. Biochemistry 43, 7610-7.
69. Ritz, H., Schramek, N., Bracher, A., Herz, S., Eisenreich, W., Richter, G. & Bacher, A. (2001). Biosynthesis of riboflavin: studies on the mechanism of GTP cyclohydrolase II. J Biol Chem 276, 22273-7.
70. Schnappauf, G., Strater, N., Lipscomb, W. N. & Braus, G. H. (1997). A glutamate residue in the catalytic center of the yeast chorismate mutase restricts enzyme activity to acidic conditions. Proc Natl Acad Sci U S A 94, 8491-6.
71. Scheer, J. M., Romanowski, M. J. & Wells, J. A. (2006). A common allosteric site and mechanism in caspases. Proc Natl Acad Sci U S A 103, 7595-600.
72. Njalsson, R., Carlsson, K., Bhansali, V., Luo, J. L., Nilsson, L., Ladenstein, R., Anderson, M., Larsson, A. & Norgren, S. (2004). Human hereditary glutathione synthetase deficiency: kinetic properties of mutant enzymes. Biochem J 381, 489-94.
73. Bjornberg, O., Neuhard, J. & Nyman, P. O. (2003). A bifunctional dCTP deaminase-dUTP nucleotidohydrolase from the hyperthermophilic archaeon Methanocaldococcus jannaschii. J Biol Chem 278, 20667-72.
74. Bhasin, M., Billinsky, J. L. & Palmer, D. R. (2003). Steady-state kinetics and molecular evolution of Escherichia coli MenD [(1R,6R)-2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate synthase], an anomalous thiamin diphosphate-dependent decarboxylase-carboligase. Biochemistry 42, 13496-504.
75. Bjorgo, E., de Carvalho, R. M. & Flatmark, T. (2001). A comparison of kinetic and regulatory properties of the tetrameric and dimeric forms of wild-type and Thr427-->Pro mutant human phenylalanine hydroxylase: contribution of the flexible hinge region Asp425-Gln429 to the tetramerization and cooperative substrate binding. Eur J Biochem 268, 997-1005.
76. Porter, C. T., Bartlett, G. J. & Thornton, J. M. (2004). The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res 32, D129-33.
77. Tseng, Y. Y. & Liang, J. (2007). Predicting enzyme functional surfaces and locating key residues automatically from structures. Ann Biomed Eng 35, 1037-42.
78. Bartlett, G. J., Porter, C. T., Borkakoti, N. & Thornton, J. M. (2002). Analysis of catalytic residues in enzyme active sites. J Mol Biol 324, 105-21.
79. Yang, L. W. & Bahar, I. (2005). Coupling between catalytic site and collective dynamics: a requirement for mechanochemical activity of enzymes. Structure 13, 893-904.
80. Ma, W., Tang, C. & Lai, L. (2005). Specificity of trypsin and chymotrypsin: loop-motion-controlled dynamic correlation as a determinant. Biophys J 89, 1183-93.
81. Keskin, O., Durell, S. R., Bahar, I., Jernigan, R. L. & Covell, D. G. (2002). Relating molecular flexibility to function: a case study of tubulin. Biophys J 83, 663-80.
82. Bahar, I., Erman, B., Jernigan, R. L., Atilgan, A. R. & Covell, D. G. (1999). Collective motions in HIV-1 reverse transcriptase: examination of flexibility and enzyme function. J Mol Biol 285, 1023-37.

271
83. Bai, H., Ma, W., Liu, S. & Lai, L. (2008). Dynamic property is a key determinant for protein-protein interactions. Proteins 70, 1323-31.
84. Keskin, O., Jernigan, R. L. & Bahar, I. (2000). Proteins with similar architecture exhibit similar large-scale dynamic behavior. Biophys J 78, 2093-106.

272
Chapter 6: Conclusions
The work included in this thesis broadly addresses aspects of how structure relates to
function in proteins (for most of the thesis this specifically relates to enzymes). The
initial aim of the project was to improve prediction of EC class from structural and
sequence features of enzymes by including active-site specific features. In working
towards this aim, the general relationships between these features and the functions of
each EC class were explored in Chapter 2. This involved identifying features that
differ significantly between the six EC classes and further analysing those that showed
the most significant differences.
Three features that showed significant differences between EC classes were
investigated further; these included the proportion of non-polar residues in the active
site, the proportion of aspartic acid in the active site and the number of residues in the
biological unit (which relates to the oligomeric status). The proportion of the active
site composed of non-polar residues was one of the most significantly-different
features between the six classes. The oxidoreductases (EC1) exhibited the highest
proportion of non-polar residues in the active sites. Oxidoreductases are the most
likely group of enzymes to elicit their function by binding a cofactor and these
cofactors often contain large non-polar groups. Upon removing the cofactor-binding
proteins from the whole dataset the composition of the active site that is non-polar was
reduced for the oxidoreductases and there was no longer a significant different
between the six classes.
The active site composition of aspartic acid was also one of the most significantly-
different features between the six EC classes. The aspartic acid active site composition
was the lowest for the oxidoreductases. The reduction in aspartic acid composition for
oxidoreductases was compensated by a preference for glutamic acid. This was
surprising since aspartic acid is preferred as an active site (and catalytic) residue over
glutamic acid, which is seen in the other five classes. Glutamic acid has different
hydrogen-bonding properties to aspartic acid and it was shown that Glu residues form
significantly more hydrogen bonds in oxidoreductases, than in other classes and also
significantly more than aspartic acid. It was suggested that Glu is preferred to Asp in

273
order to form hydrogen bond networks in the active site that play a role in proton
shuffling, the most common catalytic mechanism of the oxidoreductases.
Further work to investigate this hypothesis would include structural bioinformatics and
experimentation. For example, the static calculations of hydrogen-bonding reported in
this thesis could be extended with calculations of alternate rotameric forms in networks
of sidechains, particularly where pathways for proton transfer have been suggested in
the literature. The aim would be to examine whether swapping Asp for Glu impedes
these pathways through restriction of alternate hydrogen-bonded networks. The same
question could be asked experimentally, with mutagenesis and substitution of Glu and
Asp sidechains, alongside a read-out of catalytic activity.
Lastly, the number of residues in the biological unit structures differed significantly
between the functional classes. The lyases (EC4) had the largest number of residues in
the biological unit, but not the largest average/median sequence length. This suggested
that the differences in size were due to differences in oligomeric status. Indeed, lyases
were the class most likely to form high-order oligomers (three or more chains). It was
also found that lyases were also more likely to active sites at, or near to, subunit
boundaries. It was suggested that they form high-order oligomers to allow finely-tuned
control of their action since lyases were also found to be over-represented at important
points in metabolic networks. Conversely, the hydrolases were the smallest and
preferred to exist as monomers and were also under-represented at important points in
metabolic networks.
Lyases may prefer to exist in high-order oligomers in order to allow cooperative action
between subunits in order to elicit a high level of control over their catalytic action.
Since biochemical data is incomplete for much of the dataset, a method to
computationally identify oligomeric enzymes that act cooperatively was needed.
Chapter 5 starts by analysing the degree of coupling of residue motion between
oligomers in an attempt to distinguish oligomers that act cooperatively than those that
do not. Whilst this method was unable to distinguish between cooperative and non-
cooperative oligomers, it was observed that the pattern of correlation between residue
motion is broadly conserved over a large number of oligomers. This was further

274
identified in a large, functionally diverse, non-redundant set of oligomeric protein
structures.
Since the role of protein dynamics in function is receiving increasing attention, it will
be important to investigate further the observation of a common pattern of correlated
motion at interfaces. In purely computational terms, there is scope for more complex
analysis of the normal modes than reported in this thesis, for example examining the
role of individual lower frequency modes, which are generally expected to play
important roles in functional properties.
In order to address the original aim of this thesis, to improve EC class prediction by
the addition of sequence and structural features of the active site region, it was
necessary to use an active site prediction method in order to identify active sites in
proteins that may have no functional annotation. Many computational tools have been
developed to predict functional sites of proteins and it was considered out of the scope
of this project to develop one. Chapter 3 contains a thorough benchmark analysis of
current publicly-available functional site prediction tools in order to identify the best-
performing method to be used in subsequent function prediction methods. We found
that, alongside another tool (Consurf) a previous method developed by the Warwicker
group (SitesIdentify) predicted enzyme active sites and functional sites of non-enzymes
with the highest accuracy. This method was not previously publicly-available and so
Chapter 3 also presents the creation of a web-server to deliver the SitesIdentify method
via the web.
Lastly, structural and sequence features of enzymes, including those relating to their
active sites, were used to create a method to predict the top EC class of an enzyme
without the transfer of information via homology. The first attempt used a vector
comparison method on enzymes with known active sites. Whilst this did not achieve a
high level of accuracy it was more than expected by chance, which indicates that the
features used in the model held information that was indicative of the top EC function
in enzymes. It was a further improvement on increase in accuracy than obtained by
previous attempts by this group using a similar method without including active site
features. This suggested that features specific to the active site increase the model’s
ability to predict function. A further approach in this chapter used a larger set of
enzymes with predicted active site locations to calculate features. Prediction models

275
were made using a machine learning approach (SVMs) and achieved a similar level of
accuracy.
The levels of accuracy and lack of balance in the EC class prediction methods limit
their use in real-world function prediction problems. In order to increase prediction
accuracy then it is possible that the model needs to be updated to include alternative
prediction features such as electrostatic profiles, particularly of active sites. Other
machine learning approaches, such as Random forests may display better utility for this
particular prediction problem. Random forests are a collection of decision trees, where
the prediction is the most popular output from individual trees. Trees are constructed
using a random subset of variables at each branch point. The advantages of such a
method are that it can handle large number of input features and can give indications
of the level of importance of these in making the predictions.
Predicting EC class is obviously only applicable to enzymes, and it becomes necessary
to therefore predict beforehand whether the protein is an enzyme or non-enzyme.
This introduces a further level of error in predicting the correct function of the protein.
It also does not aid in predicting the functions of non-enzymes. It would, therefore,
also be useful to construct prediction models based on classification schemes that do
not only apply to enzymes, such as the Gene Ontology (GO). Despite the limitation of
the applicability of this EC class prediction model, it gives a quantitative indication of
how well the differences in features described in Chapter 2 are predictive of EC class.
Even without prediction, there is still much to be learnt from understanding how
structural and sequence features relate to functional class and why evolutionary diverse
but functionally similar proteins can exhibit similar features.


















