
FORMULATION AND EVALUATION OF TIME CONTROLLED DRUG DELIVERY SYSTEM...
12
INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031 1 | Page Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org FORMULATION AND EVALUATION OF TIME CONTROLLED DRUG DELIVERY SYSTEM OF MONTELUKAST SODIUM Bailpattar Padmaxi * 1 karwa Preeti 1 Patel Kirtan 1 Mondal Md. Sahidullah 2 Pasha Mohamed Irshad 2 1 Nargund college of Pharmacy, Bangalore- 560085, Karnataka 2 MMU College of Pharmacy, Ramanagaram- 562159, Karnataka Abstract: Aim of the present work was to formulate and evaluate an oral, time controlled drug delivery system of Montelukast sodium, based on chronopharmaceutical approach for the treatment of nocturnal asthma. It consisting of a core surrounded by coat layer of different ratios of swellable erodible hydrophilic layer Sodium Alginate and HPMC K4M. The prepared pulsatile tablets were evaluated for the drug content, thickness and in vitro release profile. In vitro release profiles of 6 to 9 hours studies were found to have minimum or no drug release and at the end of 5 hours immediate release was observed. It was observed that lag time increased with increased concentration of HPMC K4M due to no seepage of dissolution fluid into the core. The lag time decreased with the increased concentration of Sodium Alginate resulted in faster and burst release of drug from a core tablets. Higher concentration of HPMC alone was able to maintain the lag time up to 6 h and Sodium alginate for 1 h. The programmable time controlled release has been achieved from a press-coated tablet over a period of 5 hr and burst release was obtained after a lag time, which was consistent with the demands of chronotherapeutic drug delivery. Key words: Time controlled drug delivery; Chronotherapeutics; Montelukast sodium, HPMC K4M; Sodium Alginate INTRODUCTION: For many decades, treatment of an acute disease or a chronic illness has been mostly accomplished by delivering drugs using various pharmaceutical dosage forms, including tablets, capsules, pills, suppositories, creams, ointments, liquids, aerosols, and injectables as carriers. Amongst various routes of drug delivery, oral route is perhaps the most preferred to the patient and the clinician alike. These traditional pharmaceutical products are still commonly seen today in the prescription and over-the-counter drug marketplace. To achieve and maintain the *Corresponding Author Bailpattar Padmaxi
Transcript of FORMULATION AND EVALUATION OF TIME CONTROLLED DRUG DELIVERY SYSTEM...

INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
1 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
FORMULATION AND EVALUATION OF TIME CONTROLLED
DRUG DELIVERY SYSTEM OF MONTELUKAST SODIUM
Bailpattar Padmaxi *1 karwa Preeti 1 Patel Kirtan 1 Mondal Md. Sahidullah 2
Pasha Mohamed Irshad 2
1 Nargund college of Pharmacy, Bangalore- 560085, Karnataka
2MMU College of Pharmacy, Ramanagaram- 562159, Karnataka
Abstract:
Aim of the present work was to formulate and evaluate an oral, time controlled drug delivery
system of Montelukast sodium, based on chronopharmaceutical approach for the treatment of
nocturnal asthma. It consisting of a core surrounded by coat layer of different ratios of
swellable erodible hydrophilic layer Sodium Alginate and HPMC K4M. The prepared
pulsatile tablets were evaluated for the drug content, thickness and in vitro release profile. In
vitro release profiles of 6 to 9 hours studies were found to have minimum or no drug release
and at the end of 5 hours immediate release was observed. It was observed that lag time
increased with increased concentration of HPMC K4M due to no seepage of dissolution fluid
into the core. The lag time decreased with the increased concentration of Sodium Alginate
resulted in faster and burst release of drug from a core tablets. Higher concentration of
HPMC alone was able to maintain the lag time up to 6 h and Sodium alginate for 1 h. The
programmable time controlled release has been achieved from a press-coated tablet over a
period of 5 hr and burst release was obtained after a lag time, which was consistent with the
demands of chronotherapeutic drug delivery.
Key words: Time controlled drug delivery; Chronotherapeutics; Montelukast sodium, HPMC
K4M; Sodium Alginate
INTRODUCTION:
For many decades, treatment of an
acute disease or a chronic illness has been
mostly accomplished by delivering drugs
using various pharmaceutical dosage
forms, including tablets, capsules, pills,
suppositories, creams, ointments, liquids,
aerosols, and injectables as carriers.
Amongst various routes of drug delivery,
oral route is perhaps the most preferred to
the patient and the clinician alike. These
traditional pharmaceutical products are
still commonly seen today in the
prescription and over-the-counter drug
marketplace. To achieve and maintain the
*Corresponding Author
Bailpattar Padmaxi

INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
2 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
drug concentration in the body within the
therapeutic range required for a
medication, it is often necessary to take
this type of drug delivery system several
times a day. [1]
To introduce the concept of
chronopharmaceutics, it is important to
define the concepts of chronobiology and
pharmaceutics. The term "chrono"
basically refers to the observation that
every metabolic event undergoes rhythmic
changes in time. Chronobiology is the
study of biological rhythms and their
mechanisms. Pharmaceutics is an area of
biomedical and pharmaceutical sciences
that deals with the design and evaluation
of pharmaceutical dosage forms (or drug
delivery systems) to assure their safety,
effectiveness, quality and reliability.
The potential benefits of
chronotherapeutics have been
demonstrated in the management of a
number of diseases. In particular there is a
great deal of interest if how chronotherapy
can particularly benefit patients suffering
from allergic rhinitis, rheumatoid arthritis
and related disorders, asthma, cancer,
cardiovascular diseases, and peptic ulcer
disease.[2]
A circadian rhythm is an
endogenously driven roughly 24-hour
cycle in biochemical, physiological, or
behavioral processes. Circadian rhythms
have been widely observed
in plants, animals, fungi and cyanobacteria
. The term "circadian" comes from
the Latin circa, meaning "around",
and diem or dies, meaning "day". The
formal study of biological temporal
rhythms such as daily, tidal, weekly,
seasonal, and annual rhythms is
called chronobiology. Biological rhythms
are defined by a number of characteristics.
Oscillations of shorter duration are termed
‘‘ultradian’’ (more than one cycle per 24
h). Oscillations that are longer than 24 h
are ‘‘infradian’’ (less than one cycle per 24
h) rhythms. Ultradian, circadian, and
infradian rhythms coexist at all levels of
biologic organization. [3]
Several physiological processes
in humans vary in a rhythmic manner, in
synchrony with the internal biological
clock. It represents the overview of most
serious diseases displaying significant
daily variations. Many of circadian
dependent diseases display acute
symptoms in early morning hours or in the
morning at awakening.
Diseases, such as asthmas,
hypertension, peptic ulcer, arthritis, etc,
follow the body's circadian rhythm. For
example, osteoarthritis worsens during the
day and is most bothersome in the
evenings but for people with rheumatoid
arthritis, the pain usually peaks in the
morning and decreases as the day
progresses. Cardiovascular diseases such
as hypertension and angina, and chest pain,
also follow a definite circadian rhythm.
Epidemiologic studies have documented
the heightened morning-time risk of
angina, myocardial infarction, and stroke.
[4]
Through a number of clinical
trials and epidemiological studies, it has
become evident that the levels of diseases
activity of a number of clinical disorders
have a pattern associated with the body’s

INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
3 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
inherent clock set according to circadian
rhythms.
Asthma is a chronic
inflammatory disease of the airways,
characterized by hyper responsiveness to a
variety of stimuli. [5]
The role of circadian
rhythms in the pathogenesis and treatment
of asthma indicates that airway resistance
increases progressively at night in
asthmatic patients. Circadian changes are
seen in normal lung function, which
reaches a low point in the early morning
hours. The worsening of asthma at night,
commonly referred to as nocturnal asthma
(NA).A drug delivery system administered
at bedtime but releasing drug during
morning hours would be ideal in this case.
Nocturnal asthma is a variable
exacerbation of the underlying asthma
condition associated with increases in
symptoms, need for medication, airway
responsiveness, and/or worsening of lung
function. Approximately two-thirds of
asthmatics suffer from nighttime
symptoms. Lung function (e.g., peak
expiratory flow rate or FEV1) is usually
highest at 4 PM and lowest at 4 AM. Many
circadian-dependent factors appear to
contribute to the worsening of nocturnal
asthmatic symptoms. For example, cortisol
(an anti inflammatory substance) levels
were highest at the time of awakening and
lowest in the middle of the night, and
histamine (a mediator of
bronchoconstriction) concentrations
peaked at a level that coincided with the
greatest degree of bronchoconstriction at
4:00 am. [6]
Based on these findings drug
delivery and therapy should be modified to
achieve an effective drug level at the
required time. This can be achieved by
adapting a time controlled or pulsatile drug
delivery system of a suitable drug.
Consequently, the administration of a drug
formulated in such a delivery system, i.e.
taken at bedtime with a programmed start
of drug release in early morning hours,
could offer a more effective therapy than a
typical controlled release drug delivery
system, provided that the most appropriate
drugs are administrated. Pulsatile drug
delivery system is the one type of drug
delivery system, where the delivery device
is capable of releasing drug after
predetermined time-delay (i.e. lag time)
known as pulsatile drug delivery system
(PDDS).[7]
PDDS are gaining lot of interest
and attention these days. These systems
have a peculiar mechanism of delivering
the drug rapidly and completely after a
"lag time," i.e., a period of "no drug
release." Oral pulsatile administration
could be useful for the treatment of certain
diseases, such as asthma, gastric ulcer,
hypertension, ischemic heart disease,
arthritis, etc., which exhibit circadian
rhythms. Pulsatile drug delivery denotes
the capability of a controlled release
preparation to deliver the drug at varying
rates from very low to high over a
desirable time. PDDS is classified into
various systems like enteric coated system,
layered system, sigmoid system, press
coated system, single unit system and
multiple unit system. [8]
Press coated tablets (PCTs)
gained wide interest ‘claiming some

INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
4 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
advantages over regular and (pan-) coated
tablets, such as to protect hygroscopic,
light-sensitive or oxygen-labile drugs from
environmental-atmospheric ill effects or
decomposition of acid-labile drugs by
gastric fluids; to separate incompatible
drugs from each other; to achieve a
sustained release in that the drug in the
core is embedded in waxes or fats
constituting a depot; to protect the gastric
mucosa from irritation by certain drugs by
using enteric coating material in the outer
press-coating granules; or to achieve
intermittent release by incorporating one
portion of drug in the core and the other in
the coat, separated by a film-coat or a
second press-coat. However, common
drawbacks of the press-coating technique
are the multistep processes involved, and
the requirement for reliable and
reproducible central positioning of the core
tablet within press-coated tablet (PCT), a
major challenge for large scale industrial
manufacturing. The lag time of drug
release from PCTs depends upon the
thickness and the composition of the
barrier layer. Generally speaking, the
thicker the barrier layer, the longer the lag
time. The composition of the barrier layer
controls the mechanism of effecting a lag
time. Besides capsule based pulsatile
release systems, formulations, such as
Pulsicap have also been developed. These
systems consist of a water-impermeable or
semi-impermeable capsule half with the
drug formulation contained within the
capsule and sealed by means of a hydrogel
polymer plug. Contact of the dissolution
media or gastrointestinal fluids with the
barrier or the plug results to its removal or
ejection followed by the rapid release of
the drug. [9]
The aim of the present
investigation was to develop and evaluate
an alternative, simple, orally applicable
one pulse drug delivery system based on a
press-coated tablet preparation. It
consisting of a rapidly disintegrating core
tablet press coated by a barrier layer
consisting of varying concentrations of
Sodium Alginate and Hydroxypropyl
methyl cellulose (HPMC K4M). HPMC
and Sodium alginate are used as a rate
controlling polymer. Combination of these
polymers were able to achieve various lag
time.
MATERIALS AND METHOD:
Materials:
Montelukast Sodium - model drug, was
obtained from Micro lab Ltd, Bangalore,
India. Croscarmellose sodium (Ac-Di-Sol),
HPMC K4M (Methocel), Sodium alginate
were gifted from Alembic Pvt. Ltd,
Vadodara, India. Magnesium stearate,
Lactose Monohydrate and Sodium Lauryl
Sulphate (SLS) were gifted from S.D. Fine
chem. Ltd, Mumbai, India. All the other
chemicals and reagents were either
analytical or pharmaceutical grades.
Preformulation Study:
Bulk density, tapped density, Hausner’s
ratio, carr’s index and angle of repose was
perform for polymeric blends.
Drug excipient interaction:
Compatibility of the Drug with the
excipients is determined by subjecting the
physical mixture of the drug and the
polymers of the main formulation to
infrared absorption spectral analysis

INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
5 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
(FTIR). Any changes in chemical
composition of the drug after combining it
with the polymers were investigated with
I.R. spectral analysis.
Procedure: Weighed amount of drug (3
mg) was mixed with 100mg of potassium
bromide (dried at 40-50oC). The mixture
was taken and compressed under 10-ton
pressure in a hydraulic press to form a
transparent pellet. The pellet was scanned
by IR spectrophotometer. Similar
procedure is followed for all relevant
excipients used.
Tablet manufacturing method:
Formulation of core tablet by direct
compression method: [10]
The inner core tablets were prepared by
using direct compression method. The
powder mixtures of Montelukast sodium,
Lactose (lactochem), croscarmellose
sodium (Ac-Di-Sol) ingredients were dry
blended for 20 min. followed by addition
of Magnesium Stearate. The mixtures were
then further blended for 10 min., 100mg of
resultant powder blend was manually
compressed using tablet pushing machine
(Rimek mini press-1) with a 6.3 mm punch
and die to obtain the core tablet. Formula
for formulation of the core tablet was
shown in the Table 1.
Formulation of mixed blend for barrier
layer
The various formulation Compositions
containing sodium alginate and HPMC
K4M were prepared i.e. formulation from
F1 to F8 different compositions were
weighed and dry blended at about 10 min.
and used as press-coating material to
prepare press-coated pulsatile tablets
respectively by direct compression
method.
Precompression including (bulk density,
tapped density, total porosity, Hausner’s
ratio compressibility index, angle of
repose) were done for both core powder
and mixed blend of the barrier layer.
Formula for formulation of the barrier
layer (200 mg) coating was shown in the
Table 2.
Development of Press-coated tablets:
The core tablets were press-coated with
200 mg of mixed Blend. 100 mg of barrier
layer material was weighed and transferred
into a 9.54 mm die then the core tablet was
placed manually at the center. The
remaining 100 mg of the barrier layer
material was added into the die, so that the
core tablet get covered by the barrier layer
completely and compressed by tablet
punching machine (Rimek mini press-1).
The total weight of the tablet would be 300
mg. Formula for formulation of press-
tablets (300 mg) was shown in the Table 3.
Evaluation of press coated tablets: [11]
Core and press-coated tablets were
evaluated for weight variation, thickness,
hardness, friability test, content uniformity
test. These parameters are known as post
compression parameters.
Thickness of the tablets was determined by
Vernier caliper. Pzifer Monsanto tester
was used to determine the hardness of the
tablets. 5 tablets were taken for each test.
Friability was determined by placing 10
tablets in Roche Friabilator, it denotes the
mechanical strength of the tablets. For
determining the content uniformity test, 10

INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
6 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
tablets were taken and crushed, weight
equivalent to 100 mg was taken and
dissolved in SLS. Further dilutions were
made to get the concentration of 10 µg/ml.
the solution was analyzed for Montelukast
content by UV-Spectrophotometer at 345.5
nm by using SLS as a blank.
In vitro release study of core tablets [12]
In vitro release studies of core tablets were
carried out in USP Type II (Paddle Type)
apparatus by using 0.5% of SLS in water
as a dissolution media. Rotated at 50 rpm
and temperature maintained at 37 ± 0.5˚C.
Sample was withdrawn periodically at the
interval of 5 mins and analyzed by UV-
spectrophotometer at 345.5 nm.
In vitro release study of Press-coated
tablets
In vitro dissolution studies were carried
out in USP Type II (Paddle Type)
apparatus. All the conditions were
maintained same as mentioned above.
Sample was withdrawn periodically at the
interval of 1 h until coat ruptures then
samples were withdrawn in ½ h intervals
and analysed by UV-spectrophotometer at
345.5 nm using 0.5% of SLS as a blank.
Swelling index: [13]
Swelling effect on lag time and release
behavior was observed. A known weight
of a tablet was placed in 0.5% of SLS and
allowed to swell for the required period of
time at 37ºC ± 0.5ºC in the dissolution
apparatus (Dissolution Tester USP 2). A
tablet was periodically removed and
blotted with filter paper; then their change
in weight (after correcting for drug loss)
was measured until attainment of
equilibrium. The swelling ratio (SR) was
then calculated using the following
formula:
WF - WI ×100
SR=
WI
Where
SR= swelling ratio,
WF = weight of the tablet after swelling,
WI= initial weight of the tablet.
RESULT AND DISCUSSION:
An absorption maximum was determined
by scanning different concentration of
solution of drug Montelukast sodium. It
was found to be 345.5 nm and method
obeys Beer’s law in concentration range 5
to 25µg/ml, r2 was found to be 0.0097.
Preformulation test of polymeric
blends:
Bulk density, tapped density, Hausner’s
ratio and carr’s index given in the Table 4.
The angle of repose (θ) for all the
formulation blends of Coated was below
30º indicating good flow property.
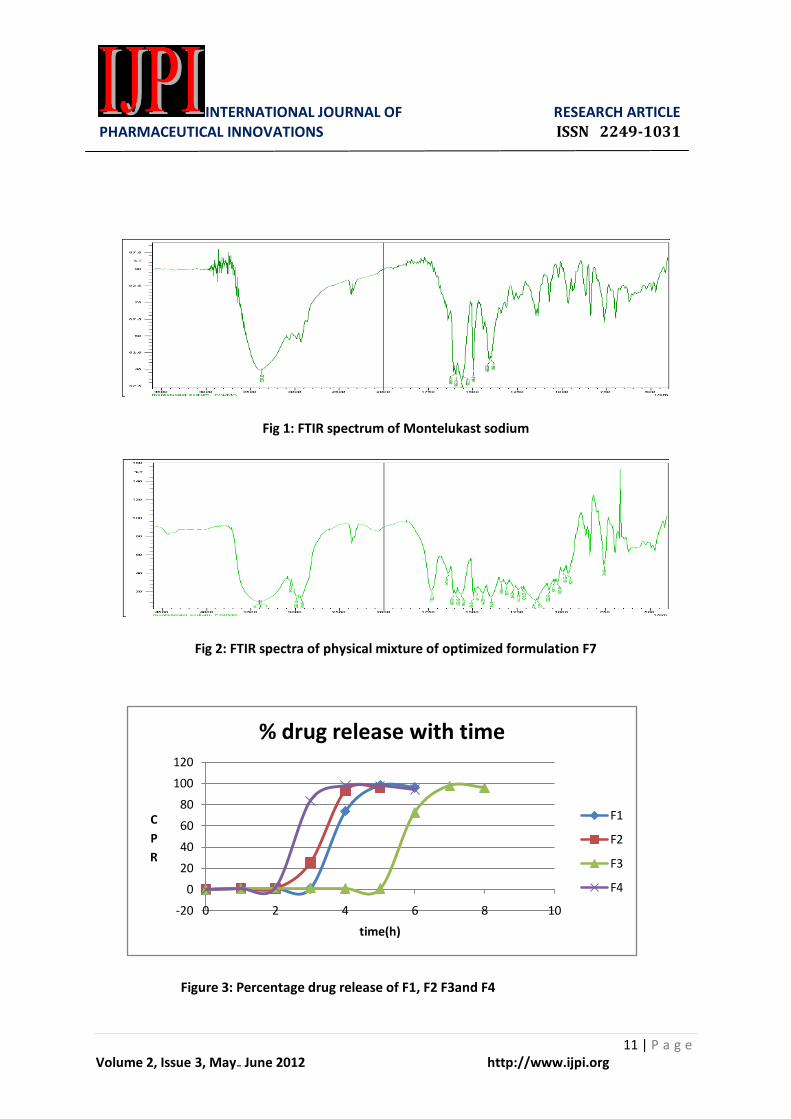
IR spectra of optimized
formulation F7 (25:100) was compared
with IR spectra of the pure drug samples.
No change in the peak occurred with
demonstrated no incompatibility with the
excipients used. FTIR spectrum was
shown in the figure 1 & 2.
Evaluation of press-coated tablets:
Post compression parameters like weight
variation, thickness, hardness, friability are
given in table No 5. Weight variation
(n=20) was found to be 298.89 ± 1.007 to

INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
7 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
300.02 ± 1.741. Thickness (n=5) ranging
from 5.26 ± 0.342 to 5.33 ± 0.031 mm.
Hardness (n=5) was ranging from 5-6
kg/cm2. Friability of press-coated tablets
was found to be 0.177 to 0.289 %,
indicating good mechanical strength. Drug
content of coated tablets were found to be
98.56 ± 0.300 to 99.81 ± 0.312 %
indicating uniform distribution of drug.
Result was shown in the Table 5.
In vitro dissolution of core tablet:
The core tablets shows 70% of drug
release within 5 mins followed by the
maximum release in 15 mins upon contact
with dissolution media.
In vitro dissolution of coated tablets:
From visual observation of in vitro
dissolution studies it become apparent that
upon contact of the tablet with the liquid,
the outer layer consisting of the
hydrophilic polymers, starts to absorb
liquid. As a consequence the polymer
swells and shortly after an expansion of
the layer was noticed. As the time passes
the swelling and the expansion of the top
layer increases creating a considerable
barrier which may delay to some extent the
contact of the bulk liquid with the surface
of the Core drug tablet. The swelling
process of the outer barrier layer could act
as a disintegrating force, which facilitates
firstly the destabilization of the barrier
layer itself. The balance between these two
forces disintegration (sodium alginate) and
swelling (HPMC K4M) controls the
behavior of the outer barrier layer (i.e. the
erosion process of the polymer) and
consequently the performance of the
system. Finally depending on the
properties of each polymer i.e. Sodium
alginate and HPMC K4M, the outer barrier
layer is removed and as result the
dissolution of the Core drug tablet increase
sharply due to increased access of liquid
into the Core of the tablet. HPMC K4M
showed maximum swelling which lasts
longer but the erosion appeared to be
greater after maximum swelling for
Sodium alginate. Thus the greatest bulk
swelling is achieved with both Sodium
alginate and HPMC K4M and after lag
time erosion appeared due to Sodium
alginate and drug is released from Core.
During the course of studies it was
observed that F7 (25:100) showed a lag
time of 5 h and immediate release of drug
was found. Formulation F7 selected as an
optimized formulation as it was meeting
chronotherapeutics system for the
treatment of asthma. Release study of the
prepared formulations was shown in the
Figure 3-4.
Swelling index:
The optimized formulation F 7 were
subjected for the determination of bulk
volume swelling reflecting extent of liquid
uptake and the loss of weight reflecting
erosion on the tablet. Visual observation
indicated that both polymers swelled and
created a viscous gel at the top layer
surface when they are exposed to the
dissolution medium. During the coarse of
the study, a maximum swelling achieved
followed by erosion of the hydrophilic
polymer layer. (Fig- 5)
The barrier layer exhibited slower
liquid uptake and the maximum uptake
swelling (80 %) was achieved at 5 hr.
These indicate that in Montelukast sodium
tablets, drug molecule are released by
diffusion out of the gelatinous layer once

INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
8 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
the polymer outer layer had been fully
hydrated and the liquid molecules come in
contact with the Core tablet. As time
passes, the erosion of the polymer and
progress of the dissolution of the drug
occur almost simultaneously and
termination of drug release coincides
approximately with erosion of the outer
layer.
CONCLUSION:
The novel Time controlled Chronotropic
drug delivery system for oral use was
successfully developed and evaluated. The
formulation consisted of a core tablet
containing a drug Montelukast sodium and
outer layer of combination of swellable,
erodible hydrophilic polymer. The present
study demonstrated that the press-coated
tablets of Montelukast sodium was
successfully developed with desired lag
time of 5 hrs by using combination of
Sodium Alginate and HPMC K4M. It was
concluded that Formulation F7 (25:100)
was the ideal formulation with lag time of
5 hrs followed by burst release of drug and
also meeting all specifications of pre-
compression and post compression
parameters and stability studies.
ACKNOWLEDGEMENT:
We are thankful to wish to Micro lab Ltd,
Bangalore, India for providing the gift
sample of Montelukast sodium. We are
also thankful to the Nargund and M.M.U
college of Pharmacy for providing all
necessary facilities.
REFERENCES:
1. Jain NK. Controlled and novel
drug delivery. 1st ed. New Delhi:
CBS Publishers; 2002.
2. Jha N, Bapat S. Chronobiology and
chronotherapeutics: A review.
Kathmandu university Med. J.
2004; 2(8): 384-388.
3. Sarasija S, Stutie P.
Chronotherapeutics: Emerging role
of biorhythms in optimizing drug
therapy. Indian J. Pharm. Sci.
March-April 2005; 67(2): 135-140.
4. Sangita V. Timing is everything.
Nov. 2009;
http://www.pharmaquality.com/Fea
ture.7ahtm.
5. Smolensky M, Lemmer B,
Reinberg A. Chronobiology and
chronotherapy of allergic rhitis and
bronchial asthma. Advance drug
del. Reviews. 2007; 852-882
6. Gwen SS. Nocturnal asthma:
mechanisms and management. The
Mount Sinai J. Medicine.May
2002; 69(3): 140-147.
7. Reddy J, Jyosthna M, Saleem T,
Chetty C. Pulsatile drug delivery
system: A review. J. Pharm. Sci.
and Res. 2009; 1(4): 109-115.
8. Gothaskar AV, Joshi AM, Joshi
NH. Pulsatile drug delivery
system: A review. Drug
Del.Tech.June 2004; 4(5).
9. Sajan J, Cinu TA, Chacko AJ, Litty
J, Jaseeda T. Chronotherapeutics
and Chronotherapeutics drug
delivery system: A Review.
Tropical J. Pharmaceutical Res.
Oct. 2009; 8(5): 467-475.
INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
9 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
10. Janugade BU, Patil SS, Patil SV,
Lade PD. Formulation and
evaluation of press-coated
Montelukast sodium tablet for
pulsatile drug delivery system. Int.
J. chem. and Tech. Res. July-
sep.2009;1(3): 690-691.
11. Verma S, Valecha V, Singh SK,
Syan N, Mathur P. Development
and evaluation of Montelukast
sodium colon targeted matrix
tablets based on pulsatile approach
for nocturnal asthma.Int.J.
Pharmaceutical sci. Review and
Res. May-June 2011; 8(1): 129-
137.
12. Government of India Ministry of
Health & Family Welfare. Indian
Pharmacopoeia. Delhi:Controller
of Publications. 2007; 3: 1689-
1690.
13. Patel GC, Patel MM. A
comparative in vitro evaluation of
enteropolymers for Pulsatile drug
delivery system. Acta
Pharmaceutica Sci. 2009; 51: 243-
250.
Table 1: Formulation of the core tablet Table 2: Formulation of the barrier layer (200 mg)
SN Sodium alginate (mg)
HPMC K4M (mg)
Lactose (mg)
F1 40 60 100
F2 50 50 100
F3 50 100 50
F4 75 75 50
F5 75 100 25
F6 100 75 25
F7 25 100 75
F8 100 100 0
SN Ingredients Quantity
(mg)
1 Montelukast
Sodium (MKS)
10.4
2 Lactose 82.6
3 Croscarmellose
sodium
10
4 Magnesium stearate 2
Total weight 100 mg

INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
10 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
Table 3: Formulation of press-coated Table 4: Precompression parameters of the polymeric blend
Tablets (300 mg)
Table 5: Post compression parameters of press-coated tablets
Batch Thickness
(mm)
Hardness
(kg/cm2)
Wt. variation Friability
(%)
Drug content
(%)
F1 5.26±0.342 5-6 299.45±2.811 0.231 98.90±0.501
F2 5.33±0.031 5-6 300.01±0.910 0.144 99.81±0.312
F3 5.30±0.024 5-6 300.02±1.741 0.088 99.42±0.402
F4 5.31±0.003 5-6 299.44±1.140 0.289 98.56±0.300
F5 5.29±0.257 5-6 298.89±2.007 0.216 97.32±0.210
F6 5.29±0.269 5-6 300.01±1.119 0.192 98.78±0.251
F7 5.30±0.031 5-6 299.60±1.818 0.177 99.40±0.310
F8 5.31±0.027 5-6 299.55±0.973 0.176 99.12±0.411
BL : CORE
(200:100)
S: H
(RATIO)
F1 40:60
F2 50:50
F3 50:100
F4 75:75
F5 75:100
F6 100:75
F7 25:100
F8 100:100
Batch Bulk
density
(w/v)
Tapped
density
(w/v)
Hausner’s
ratio
Carr’s
index
Angle
of
repose
(θ)
F1 0.384 0.439 1.14 12.53 22.75
F2 0.416 0.480 1.15 13.33 24.14
F3 0.395 0.452 1.14 12.61 25.31
F4 0.367 0.390 1.06 5.90 25.39
F5 0.361 0.409 1.13 11.74 28.11
F6 0.352 0.397 1.13 11.34 26.14
F7 0.413 0.443 1.07 6.77 25.34
F8 0.414 0.477 1.15 6.21 24.34
INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
11 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
Fig 1: FTIR spectrum of Montelukast sodium
Fig 2: FTIR spectra of physical mixture of optimized formulation F7
Figure 3: Percentage drug release of F1, F2 F3and F4
-20
0
20
40
60
80
100
120
0 2 4 6 8 10
C
P
R
time(h)
% drug release with time
F1
F2
F3
F4

INTERNATIONAL JOURNAL OF RESEARCH ARTICLE PHARMACEUTICAL INNOVATIONS ISSN 2249-1031
12 | P a g e Volume 2, Issue 3, May₋ June 2012 http://www.ijpi.org
Figure 4: Percentage drug release of F5, F6, F7 and F8
Figure 5: swelling index of optimized formulation F7
-20
0
20
40
60
80
100
120
0 2 4 6 8 10
C
R
P
time(h)
% drug release with time
F5
F6
F7
F8
0
20
40
60
80
100
0 2 4 6 8
%
w
e
i
g
h
t
g
a
i
n
time (h)
swelling index
F7


















