
treatment of enamel hypoplasia in a patient with Usher syndrome

PEDIATRIC DENTISTRY/Copyright © 1986 byThe American Acaciemy of Pediatric Dentistry
Volume 8 Number 3
Focal dermal hypoplasia syndrome (Goltz syndrome):the first dental case report
Steven D. Ureles, DMD, MS Howard L. Needleman, DMD
AbstractThe first dental case report of a patient with focal dermal
hypoplasia syndrome (FDHS) is presented. A review of signs andsymptoms of FDHS is presented along with newly reported oralfindings. This patient apparently is the first person with FDHS tohave an absent sternum and the fifteenth person reported withconfirmed osteopathia striata.
Carious lesions can be difficult to restore in FDHS due to asevere enamel hypoplasia and large pulp horns associated withthis syndrome. Orofacial swellings in FDHS can be either odon-togenic in origin or related to an infected skin lesion as presentedin this case report.
Focal dermal hypoplasia syndrome (FDHS), also
known as Goltz syndrome, is a rare congenital me-soectodermal disorder which was first described in1962.I This syndrome is characterized by cutaneousdefects consisting of thinning of the skin, herniationsof adipose tissue in the form of yellowish papulesand abnormal skin pigmentations. Patients with FDHSalso may present with skeletal, dental, ocular, hair,and nail anomalies.2-6
The purpose of this report is to: (1) present the firstcase report of FDHS in the dental literature; (2) re-view histopathology of the dermal lesions; (3) doc-ument the oral manifestations of FDHS in the liter-ature; and (4) present new oral findings associatedwith this syndrome.
Literature ReviewIn 1983 Hall4 conducted a review of the literature
and documented 125 reported cases of FDHS in theUnited States, Europe, Japan, and Mexico. AlthoughFDHS generally has been considered incompatiblewith survival of the male fetus,3 12% (15/125) of Hall’sreported cases were living males. Some familial re-lationships have been reported, yet the etiology andtransmission methods still are unknown.
In 1970 Goltz suggested that FDHS is an inheriteddisease since 5 of 33 reported cases of FDHS hadrelatives with anomalies associated with the syn-drome.2
The most favored genetic hypothesis is that FDHSis caused by an X-linked dominant gene of variableexpressivity with a pronounced lethality inmales.2,3,6,7-t° Alternately, an X-linked autosomaltranslocation or environmental teratogen might beresponsible. 11 To date, karyotype examinations haveprovided no indication of chromosome alterations.4,12
The first case reports in the literature of what ap-pear to be FDHS were reported by Jessner in 1921and 1928.13,14 The first patient, however, who was his-tologically confirmed to have what is now known asFDHS was reported by Lieberman in 193525 He de-scribed a 14-year-old female with hypoplasia of theteeth, wart-like lesions on the maxillary mucosa andanal orifice, papillomas on the gingiva, generalizederuptions of macular, annular, and linear areas ofcutaneous atrophy, telangiectasia, and pigmentationof the skin. The patterns of cutaneous atrophy canbe reticular, cribriform, punctate, or linear in shape,with reddish to tan pigmentations or areas of hypo-pigmentation,t,2,t4 Microscopic evaluation of theseskin lesions revealed normal epidermis with varyingareas of partial to complete replacement of the con-nective tissue in the dermis by adipose tissue. Lie-berman called this finding "lipomatosis" of the cu-tis25
In 1941 Cole et al. 16 reported a case of a 26-year-old female whose clinical and histologic presenta-tion was consistent with FDHS. This patient exhib-ited microdontia and hypoplasia of the upper rightmolars and premolars, syndactyly of several toes andfingers, and a missing toe with an aplastic metatarsalbone. The patient’s skin lesions were consistent withFDHS both histologically and clinically. Histologic
PEDIATRIC DENTISTRY: September 1986/Vol. 8 No. 3 239

evaluation of patients with FDHS reveals a hypo-plastic dermis with thin, sparse collagen bundles andcomplete absence of the elastic fibers with vascularhyperplasia and ectasia,5-17 which clinically producetelangiectactic atrophy. The dermis exhibits abun-dant quantities of adipose tissue which herniates andproduces yellowish and brown papules, nodules, andplaques of the skin.18 Several authors have ques-tioned whether adipose tissue near the epidermis isthe result of the overgrowth of fat or the underde-velopment of collagen. Either or both of these couldresult from the profound mesodermal or ectodermaldysplasia which these patients manifest in a varietyof ways.2'19
In 1978 Happle20 suggested that the migration of 2populations of dermal cells might explain the linearstreaking of the hypoplastic areas of skin. Accordingto the Lyon hypothesis,21 only 1 X-chromosome isactive in any mammalian cell. Since this is a matterof chance in each cell, 2 populations of dermal cellscould arise, resulting in pigmented areas being mixedwith normal areas.7
Osteopathia striata is a recent documented findingwhich may be a diagnostic feature of FDHS. Thisradiographic presentation, unique to this syndrome,consists of fine linear areas of dense bone arrangedparallel to the long axes of bones originating at ar-ticular surfaces and extending for short distances intothe diaphyses.6-22'24 This finding first was reported byLarregue et al. in 197225 who found a longitudinalstriation in the metaphysis of long bones in 9 of 11patients. This finding has been confirmed since byseveral authors, and in 1979 Knockaert and Dequeker6
described in the literature the fourteenth known caseof osteopathia striata. They stressed the importanceof a radiological survey of the skeleton for differen-tial diagnosis of FDHS and related syndromes. Thismorphologically variable presentation also may beexplained by the Lyon hypothesis.7 23'26
In 1983 Hall noted 34 of his 125 cases presentedwith oral manifestations.4 The most commonly re-ported oral manifestations enumerated by Hall aredysplasia of teeth, papillomas, enamel defects withcaries, oligodontia and malocclusion with irregularspacing, microdontia, gingival hypertrophy, higharched palate, hypoplasia of the mandible, defect inthe alveolar ridge, notching of incisors, and mediancleft of tongue. In addition to these findings, earlyloss of teeth,6-8-9-12-27-30 high frenum attachment,14-17-28
midline deviation,17-31 and ectopic eruption32 also havebeen reported.
Case ReportMedical History
JB is a Caucasian female born August 15, 1978, tononconsanguinous parents. She was the product of
FIG 1. Hand and wrist radiograph illustrating congeni-tally missing radius, 4 metacarpal bones, and absent thumbon the left upper limb, and 2 metacarpal bones with thumbpresent on the right hand.
a 41-week gestation of a gravida 5/para 5 mother andweighed 2.6 kg. The pregnancy was normal and themother denied having taken medications during herpregnancy. The family history was negative for anycongenital anomalies.
The child was noted at birth to have atrophic skinlesions with an erythematous and bullous-type der-matitis producing streaking of the face, arms, andabdomen. Multiple congenital anomalies were pres-ent and consisted of low-set ears, midline cleft of thechest, dermal hypoplasia, nail dystrophy, bilateralsyndactyly and oligodactyly forming a lobster clawdeformity of both hands and feet, shortened extrem-ities, and spina bifida. In addition, the left eye wasmicroophthalmic with a purulent discharge and pre-sented with an iris coloboma. The right eyelid alsohad a coloboma appearing as a punched out lesionand without lashes in its upper-middle portion. Theoral cavity was remarkable for a slightly high-archedpalate and a long pointed tongue which deviated tothe right. There was a visible cardiac pulsation in themidline, but the heart had a normal si and s2 splitwithout a murmur or gallop. A neurologic exami-nation, including a CAT scan and electroencephalo-gram revealed no neurologic abnormalities. Hermental status exam was age appropriate with no signof mental deficiency. A roentgenographic survey re-vealed vertebral anomalies, abnormalities of bothupper and lower limbs, as well as the absence of asternum. The left upper limb had a congenitallymissing radius, short forearm, 4 metacarpal bones,and an absent thumb. The right upper limb had anormal radius and ulna with only 2 metacarpal bonesand a thumb (Fig 1).
Shortly after birth JB underwent surgical closureof a supraumbilical defect of the abdomen and tho-
240 FOCAL DERMAL HYPOPLASIA SYNDROME: Ureles and Needleman

racic closure of the anterior chest wall defect at TheChildren's Hospital, Boston. JB was discharged at age3 weeks with a head circumference and weight inthe tenth percentile.
FIG 2. Radiograph ofdistal femur with ar-row demarcating ra-diopaque lines of cal-cifications extendingfrom the articulatingsurfaces suggestingosteopathia striata.
At age 2, calcified striations in the distal femoralmetaphysis compatible with osteopathia striata werenoted (Fig 2). She was admitted for surgical place-ment of a pin in her left hand and correction of the
FIG 3. Extraoral photograph of JB illustrating diffuse ery-thematous cribriform scar tissue over her cheeks and chinand the triangular appearance of the mandible. Note thecoloboma of the right eyelid and the scarring about the leftmicroophthalmic eye which results in a closed eyelid atrest.
FIG 4. Intraoral photograph illustrating significant notch-ing of the alveolar ridge in the maxillary left lateral incisorarea and the mandibular midline. Minor alveolar notchingis evident in the maxillary midline and mandibular leftlateral incisor. Note the presence of gemination of themandibular right primary incisor and fusion of the max-illary right primary central and lateral incisors.
syndactyly between her fingers to allow for bettergrasping of objects and self-feeding. At age 3, sheunderwent a cystoscopy with a bilateral ureteralreimplantation to correct pyelonephritic changes inboth kidneys resulting from chronic urinary tract in-fections.
Dental FindingsJB presented to the dental clinic at The Children's
Hospital at age 5 years for an initial clinical and ra-diographic evaluation. The extraoral examination re-vealed diffuse erythematous cribriform scar tissueover the cheeks and chin. Her chin was pointed, giv-ing the mandible a triangular appearance (Fig 3). In-traorally, she presented with significant notching ofthe alveolar ridge in the maxillary left lateral incisorarea and the mandibular midline. Minor alveolarnotching was evident in the maxillary midline andthe mandibular left lateral incisor area. She had ahigh maxillary and mandibular labial frenum attach-ment and a generalized gingivitis (Fig 4).
The masticatory oral mucosa was normal except foran unusual striated pattern of the palatal mucosa.The primary dentition exhibited several morpholog-ical abnormalities: microdontia of the mandibular leftprimary central incisor, gemination of the mandib-ular right primary incisor, fusion of the maxillaryright primary central and lateral incisors, and mul-berry-like molars and maxillary left primary canine(Figs 5a, b). The dentition exhibited numerous smallto moderate carious lesions. Multiple chronic drain-ing fistulas were noted. The radiographic survey re-vealed agenesis of the following permanent teeth:maxillary right first and second molars, second pre-
PEDIATRIC DENTISTRY: September 1986/Vol. 8 No. 3 241
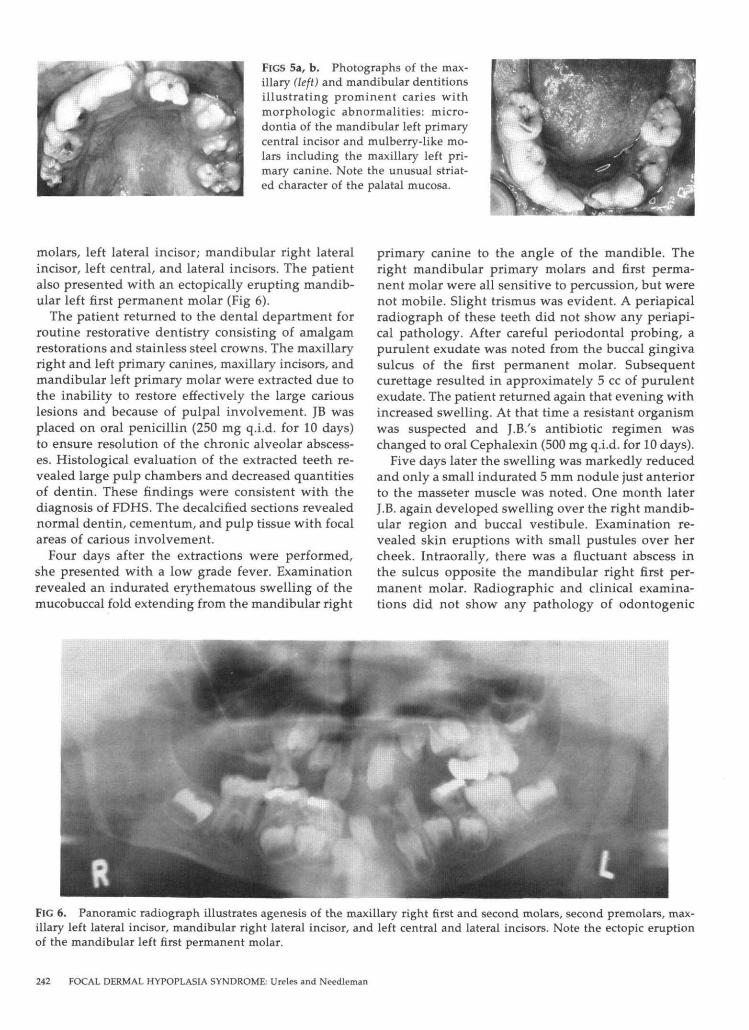
FIGS 5a, b. Photographs of the max-illary (left) and mandibular dentitionsillustrating prominent caries withmorphologic abnormalities: micro-dontia of the mandibular left primarycentral incisor and mulberry-like mo-lars including the maxillary left pri-mary canine. Note the unusual striat-ed character of the palatal mucosa.
molars, left lateral incisor; mandibular right lateralincisor, left central, and lateral incisors. The patientalso presented with an ectopically erupting mandib-ular left first permanent molar (Fig 6).
The patient returned to the dental department forroutine restorative dentistry consisting of amalgamrestorations and stainless steel crowns. The maxillaryright and left primary canines, maxillary incisors, andmandibular left primary molar were extracted due tothe inability to restore effectively the large cariouslesions and because of pulpal involvement. JB wasplaced on oral penicillin (250 mg q.i.d. for 10 days)to ensure resolution of the chronic alveolar abscess-es. Histological evaluation of the extracted teeth re-vealed large pulp chambers and decreased quantitiesof dentin. These findings were consistent with thediagnosis of FDHS. The decalcified sections revealednormal dentin, cementum, and pulp tissue with focalareas of carious involvement.
Four days after the extractions were performed,she presented with a low grade fever. Examinationrevealed an indurated erythematous swelling of themucobuccal fold extending from the mandibular right
primary canine to the angle of the mandible. Theright mandibular primary molars and first perma-nent molar were all sensitive to percussion, but werenot mobile. Slight trismus was evident. A periapicalradiograph of these teeth did not show any periapi-cal pathology. After careful periodontal probing, apurulent exudate was noted from the buccal gingivasulcus of the first permanent molar. Subsequentcurettage resulted in approximately 5 cc of purulentexudate. The patient returned again that evening withincreased swelling. At that time a resistant organismwas suspected and J.B.'s antibiotic regimen waschanged to oral Cephalexin (500 mg q.i.d. for 10 days).
Five days later the swelling was markedly reducedand only a small indurated 5 mm nodule just anteriorto the masseter muscle was noted. One month laterJ.B. again developed swelling over the right mandib-ular region and buccal vestibule. Examination re-vealed skin eruptions with small pustules over hercheek. Intraorally, there was a fluctuant abscess inthe sulcus opposite the mandibular right first per-manent molar. Radiographic and clinical examina-tions did not show any pathology of odontogenic
FIG 6. Panoramic radiograph illustrates agenesis of the maxillary right first and second molars, second premolars, max-illary left lateral incisor, mandibular right lateral incisor, and left central and lateral incisors. Note the ectopic eruptionof the mandibular left first permanent molar.
242 FOCAL DERMAL HYPOPLASIA SYNDROME: Ureles and Needleman

origin. An incision and drainage was performed andthe area was decompressed. A culture was taken andsent to the lab for identification. The patient wasagain placed on oral Cephalexin since this was aneffective antibiotic during her previous infective ep-isode.
The laboratory culture was positive for staphlococ-cus aureus, streptococcus viridans, alpha hemolyticstreptococcus, and haemophilus para influenza sug-gesting skin and respiratory flora. On follow-up ex-amination, the intraoral swelling and the skin erup-tions over her cheek had completely resolved.
DiscussionAlthough FDHS has not been reported previously
in the dental literature, this mesoectodermal disor-der has many significant dental anomalies. After re-viewing the literature, a retabulation of Hall’s studyalong with cases not included in that report whichthe author has collated, reveals that the occurrenceof oral anomalies associated with FDHS is more fre-quent than previously reported. ]B presented withmany of these oral anomalies. Her pointed mandible,ectopically erupting molars, oligodontia of the max-illary right molars, notching of the alveolar ridge,irregular spacing of the dentition, high frenum at-tachments, and midline deviation all contributed toher dental malocclusion. The morphological abnor-malities in the size and shape of her dentition andpoor dexterity of her deformed hands made plaquecontrol difficult, resulting in a generalized gingivitisand high caries rate.
Although "notched incisors" is an anomaly fre-quently associated with FDHS, according to the lit-erature no further clinical delineation of this anom-aly has been made, i.e., a Hutchinson’s incisor, fusion,or gemination. JB presented with fusion of the max-illary left primary central and lateral incisor. Thisfinding is based on the appearance of a bifid crown,2 pulp canals, and a congenitally missing tooth con-firmed by radiographic analysis. 33 The clinical andradiographic findings of the bifid crown and 1 pulpcanal suggested that the mandibular right primaryincisor is geminated and that agenesis of the man-dibular left incisor exists. Although missing and mal-formed teeth occur in FDHS, other conditions, suchas Down’s syndrome, anhidrotic ectodermal dyspla-sia, chondroectodermal dysplasia, incontinentia pig-menti, and Rieger’s syndrome should be consideredwhen making a differential diagnosis.2~
The notching of the alveolus appears to mimicanomalies often associated with the cleft lip and pal-ate patients. In the areas where notching occurred inthe alveolar ridge, ]B exhibited an obvious distur-bance of the dental lamina resulting in the anomaliesof agenesis, fusion, gemination, and/or microdontia
of the primary and permanent dentition. It is alsointeresting to note that the palatal mucosa is remark-able for a striated character which is similar in ap-pearance to the linear streaking of the hypoplasticareas of the skin. Orofacial infections in FDHS canbe either odontogenic in origin or related to an in-fected skin lesion. One can only speculate that therecurrent buccal space swellings noted in J.B. mighthave originated as a staphlococcus aureus skin infec-tion which left a scarred residual lymph node in theanterior masseteric region. Presumably, the masse-teric node became reinfected during dental manip-ulation and the tract led to the buccal sulcus.
SummaryA report of focal dermal hypoplasia syndrome
(FDHS) is presented with a review of the literature,signs and symptoms and newly reported oral find-ings of fusion, and a striated character of the palatalmucosa. JB is the first person with FDHS to have anabsent sternum, and is now the fifteenth person withconfirmed osteopathia striata.
Although there is often a complicated medical his-tory associated with children who present withFDHS, there is no contraindication to dental treat-ment. Due to the severe enamel hypoplasia and largepulp horns, carious lesions can be difficult to restore.Differential diagnosis of facial swellings should in-clude infections of both dental origin and facial der-mal lesions. The restorability of the teeth is depen-dent on tooth morphology and the extent of pulpalinvolvement. Prevention should be emphasizedthrough dietary control, fluoride supplementation,and special consideration in plaque control. Sincemany of these children are of normal intelligence,esthetics becomes an important consideration inmasking their physical handicap. Further consider-ations in dental treatment should emphasize spacemaintenance and/or replacement of missing teeth foresthetics and guidance of the developing occlusionwhich eventually will require full corrective ortho-dontics.
The authors wish to thank Drs. Leonard B. Kaban and Gary S.Lindner for their review of this manuscript and Patricia Kennedyfor preparation of this manuscript.
At the time of this writing, Dr. Ureles was chief resident, Depart-
ment of Dentistry, The Children’s Hospital in Boston, and Dr.
Needleman is assistant clinical professor, pediatric dentistry, Har-vard School of Dental Medicine, and associate dentist in chief at
The Children’s Hospital. Reprint requests should be sent to: Dr.
Steven D. Ureles, Dept. of Dentistry, The Children’s Hospital, 300
Longwood Ave., Boston, MA 02115.
1. Goltz RW, Peterson WC, Gorlin RJ, Ravitis HG: Focal dermal
hypoplasia. Arch Derm 86:708-17, 1962.
PEDIATRIC DENTISTRY: September 1986/Vol. 8 No. 3 243

2. Goltz RW, Henderson RR, Hitch JM, Ott JE: Focal dermal hy-poplasia syndrome. Arch Derm 101:1-11, 1970.
3. Gottlieb SK, Fisher BK, Violin GA: Focal dermal hypoplasia--a nine-year follow-up study. Arch Derm 108:551-53, 1973.
4. Hall EH, Terezhalmy GT: Focal dermal hypoplasia syndrome.J Am Acad Derm 9:443-51, 1983.
5. Holden JD, Akers WA: Goltz’s syndrome: focal dermal hypo-plasia: a combined me:~oectodermal dysplasia. Am J Dis Child114:292-300, 1967.
6. Knockaert D, Dequeker J: Osteopathia striata and focal dermalhypoplasia. Skeletal Radiol 4:223-27, 1979.
7. Burgdorf WHC, Dick GF, Soderberg MD, Goltz RW: Focal der-mal hypoplasia in a father and daughter. J Am Acad Derm 4:273-77, 1981.
8. Fjellner BO: Focal dermal hypoplasia in a 46 XY male. Int JDerm 18:812-15, 1979.
9. Mair IWS, Hoy C: Focal dermal hypoplasia. J Laryngol Otol85:853-56, 1971.
10. Ruiz-Maldonado R, Carnevale A, Tamayo L, Milonas D, Man-tiel E: Focal dermal hypoplasia. Clin Genet 6:36-45, 1974.
I1. Walbaum R, Samaille G, Dehaene P: Syndrome de Goltz chezun garcon. Pediatrie 25:911-20, 1970.
12. Warburg M: Focal dermal hypoplasia ocular and general man-ifestations with a survey of the literature. Acta Ophthalmol48:525-36, 1970.
13. Jessner M: Breslauer dermatologische vereingung. Arch DermSyph 133:48, 1921.
14. Gorlin RJ, Meskin LH, Peterson WC, Goltz RW: Focal dermalhypoplasia syndrome. Acta Dermatovener 43:421-40, 1963.
15. Lieberman S: Atrophoderma linearis maculosa et papilloma-tosis congenitalis. Acta Dermatovener 16:476-84, 1935.
16. Cole HN, Driver JR, Giffen HK, Norris CB, Stroud G: Ectoder-mal and mesodermal dysplasia with osseous involvement. ArchDerm Syph 44:773-88, 1941.
17. Zergollern L, Laktic N, Schmutzer L: Focal dermal hypoplasia.Dermatologica 148:240-46, 1974.
18. Ishibashi A, Kurihara Y: Goltz’s syndrome: focal dermal dys-plasia syndrome (focal dermal hypoplasia). Dermatologica 144:156-67, 1972.
19. Tsuji T: Focal dermal hypoplasia syndrome. An electron mi-croscopical study of the skin lesions. J Cutan Pathol 9:271-81,1982.
20. Happle R: Genetische interpretation streifenformiger haut-anomalien. Hautarzt 29:357-63, 1978.
21. Sutton HE: An Introduction to Human Genetics, 2nd ed. NewYork; Holt, Rinehart and Winston, 1975 pp 50-52.
22. Behrman RE, Vaughan VC: Nelson Textbook of Pediatrics, 12thed. Philadelphia; WB Saunders Co, 1983.
23. Happle R, Lenz W: Striation of bones in focal dermal hypo-plasia: manifestation of functional mosaicism? Br J Derm 96:133-38, 1977.
24. Prentice FM, Mackie RM: A case of focal dermal hypoplasia.Clin Exp Derm 7:149-53, 1982.
25. Larregue M, Maroteaux P, Michel Y, Faure C: L’osteopathiastriee symptome radiologique de l’hypoplasie dermique enaires. Ann Radiol 15:287-95, 1972.
26. Happle R, Lenz W: Striation of bones in focal dermal hypo-plasia: manifestation of functional mosaicism? Br J Derm 96:133-38, 1977.
27. Ferrara A: Goltz syndrome. Am J Dis Child 123:263, 1972.28. Howell JB: Nevus angiolipomatosus vs. focal dermal hypopla-
sia. Arch Derm 92:238-48, 1965.29. Toro-Sola MA, Kistenmacher ML, Punnett HH: Focal dermal
hypoplasia syndrome in a male. Clin Genet 7:325-27, 1975.30. Sommer A, Eaton AP, Kontras SB: Goltz’s syndrome. Ohio State
Med J 70:721-24, 1974.31. Becker SW, Lindsay DG: A case for diagnosis: Thomson’s
poikiloderma; ectodermal and mesodermal dysplasia with os-seous involvement. Rothmund’s syndrome? Arch Derm 77:620-21, 1958.
32. Daly JF: Focal dermal hypoplasia. Cutis 4:1354-59, 1968.33. Levitas TC: Gemination, fusion, twinning, and concrescence.
J Dent Child 32:93-100, 1965.34. Wesley RK, Baker JD, Golnick AL: Rieger’s syndrome: (oligo-
dontia and primary mesodermal dysgenesis of the iris) clinicalfeatures and report of an isolated case. J Pediatr OphthalmolStrabismus 15:67-70, 1978.
244 FOCAL DERMAL HYPOPLASIA SYNDROME: Ureles and Needleman














![Case Report Goldenhar Syndrome - Peertechz · range from malocclusion to complete absence of the mandibular . branch and temporomandibular joint [17,18]. ... with hypoplasia of facial](https://static.fdocuments.us/doc/165x107/5f56b5114b0fff1cfd591de4/case-report-goldenhar-syndrome-peertechz-range-from-malocclusion-to-complete-absence.jpg)



