

Focal Adhesion Proteins Talin-1 and Vinculin Negatively Affect Paxillin Phosphorylation and Limit...
17
Focal Adhesion Proteins Talin-1 and Vinculin Negatively Affect Paxillin Phosphorylation and Limit Retroviral Infection Craig Brown 1,2 , Scott G. Morham 3 , Derek Walsh 2 and Mojgan H. Naghavi 1,4 ⁎ 1 Centre for Research in Infectious Diseases, School of Medicine and Medical Science, University College Dublin, Belfield, Dublin 4, Ireland 2 National Institute for Cellular Biotechnology, Dublin City University, Dublin 9, Ireland 3 Myrexis, Inc., Salt Lake City, UT 84108, USA 4 Department of Biochemistry and Molecular Biophysics, Department of Microbiology and Immunology, Columbia University, New York, NY 10032, USA Received 31 January 2011; received in revised form 30 March 2011; accepted 31 March 2011 Edited by M. F. Summers Keywords: retrovirus; HIV-1; cytoskeleton; focal adhesions; early block Many of the early events in retroviral infection are not well understood, but it is known that the host cytoskeleton and signaling pathways play integral roles in various entry and post-entry processes. Focal adhesion complexes act as sites of integration for both cytoskeletal organization and integrin signaling at the cell surface. Here, we show that talin-1 and vinculin, two interacting proteins that localize in focal adhesions to mediate integrin linkage to the actin cytoskeleton, function during retroviral infection. Transient overexpression of either talin-1 or vinculin reduced the susceptibility of human cells to infection with pseudotyped human immunodeficiency virus type 1 (HIV-1) and Moloney murine leukemia virus. In contrast, transient short interfering RNA-mediated knockdown of talin-1 or vinculin increased infection by pseudotyped HIV- 1 and simian immunodeficiency virus, demonstrating that the endogenous forms of these proteins also impaired retroviral infection. Talin-1 or vinculin overexpression inhibited infection by retroviruses that entered the cell by either fusion or endocytosis, while analysis of HIV-1 DNA synthesis demonstrated that the block occurred early in infection and prior to the initiation of reverse transcription. Both factors retained antiviral activity in the presence of actin or microtubule depolymerizing agents. Finally, talin-1 and vinculin expression was found to negatively influence tyrosine phosphorylation of paxillin, a major focal adhesion scaffolding protein whose transient knockdown decreased pseudotyped HIV-1 infection. Together, these findings demonstrate that talin-1 and vinculin negatively affect tyrosine phosphorylation of paxillin, a novel positive regulator of *Corresponding author. Department of Biochemistry and Molecular Biophysics, Department of Microbiology and Immunology, Columbia University, HHSC 1313, 701 West 168th Street, New York, NY 10032, USA. E-mail address: [email protected]. Abbreviations used: HIV-1, human immunodeficiency virus type 1; M-MuLV, Moloney murine leukemia virus; SIV, simian immunodeficiency virus; VSV-G, vesicular stomatitis virus; MT, microtubule; FAK, focal adhesion kinase; ECM, extracellular matrix; ERM, ezrin/radixin/moesin; ERK, extracellular signal-regulated kinase; siRNA, short interfering RNA; GFP, green fluorescent protein; MSS, minus-strand strong stop; DMSO, dimethyl sulfoxide; FERM, 4.1/ezrin/ radixin/moesin; KS, Kaposi's sarcoma; qPCR, quantitative real-time PCR; TBS-T, Tris-buffered saline and 1% Tween. doi:10.1016/j.jmb.2011.03.076 J. Mol. Biol. (2011) 410, 761–777 Contents lists available at www.sciencedirect.com Journal of Molecular Biology journal homepage: http://ees.elsevier.com.jmb 0022-2836/$ - see front matter. Published by Elsevier Ltd.
-
Upload
craig-brown -
Category
Documents
-
view
214 -
download
0
Transcript of Focal Adhesion Proteins Talin-1 and Vinculin Negatively Affect Paxillin Phosphorylation and Limit...

doi:10.1016/j.jmb.2011.03.076 J. Mol. Biol. (2011) 410, 761–777
Contents lists available at www.sciencedirect.com
Journal of Molecular Biologyj ourna l homepage: ht tp : / /ees .e lsev ie r.com. jmb
Focal Adhesion Proteins Talin-1 and VinculinNegatively Affect Paxillin Phosphorylationand Limit Retroviral Infection
Craig Brown1,2, Scott G. Morham3, DerekWalsh2 andMojgan H. Naghavi1,4⁎1Centre for Research in Infectious Diseases, School of Medicine and Medical Science, University College Dublin,Belfield, Dublin 4, Ireland2National Institute for Cellular Biotechnology, Dublin City University, Dublin 9, Ireland3Myrexis, Inc., Salt Lake City, UT 84108, USA4Department of Biochemistry and Molecular Biophysics, Department of Microbiology and Immunology,Columbia University, New York, NY 10032, USA
Received 31 January 2011;received in revised form30 March 2011;accepted 31 March 2011
Edited by M. F. Summers
Keywords:retrovirus;HIV-1;cytoskeleton;focal adhesions;early block
*Corresponding author. DepartmentImmunology, Columbia University, [email protected] used: HIV-1, huma
simian immunodeficiency virus; VSVextracellular matrix; ERM, ezrin/radRNA; GFP, green fluorescent proteiradixin/moesin; KS, Kaposi's sarcom
0022-2836/$ - see front matter. Publishe
Many of the early events in retroviral infection are not well understood,but it is known that the host cytoskeleton and signaling pathways playintegral roles in various entry and post-entry processes. Focal adhesioncomplexes act as sites of integration for both cytoskeletal organization andintegrin signaling at the cell surface. Here, we show that talin-1 andvinculin, two interacting proteins that localize in focal adhesions tomediate integrin linkage to the actin cytoskeleton, function duringretroviral infection. Transient overexpression of either talin-1 or vinculinreduced the susceptibility of human cells to infection with pseudotypedhuman immunodeficiency virus type 1 (HIV-1) and Moloney murineleukemia virus. In contrast, transient short interfering RNA-mediatedknockdown of talin-1 or vinculin increased infection by pseudotyped HIV-1 and simian immunodeficiency virus, demonstrating that the endogenousforms of these proteins also impaired retroviral infection. Talin-1 orvinculin overexpression inhibited infection by retroviruses that entered thecell by either fusion or endocytosis, while analysis of HIV-1 DNA synthesisdemonstrated that the block occurred early in infection and prior to theinitiation of reverse transcription. Both factors retained antiviral activity inthe presence of actin or microtubule depolymerizing agents. Finally, talin-1and vinculin expression was found to negatively influence tyrosinephosphorylation of paxillin, a major focal adhesion scaffolding proteinwhose transient knockdown decreased pseudotyped HIV-1 infection.Together, these findings demonstrate that talin-1 and vinculin negativelyaffect tyrosine phosphorylation of paxillin, a novel positive regulator of
of Biochemistry and Molecular Biophysics, Department of Microbiology andHSC 1313, 701 West 168th Street, New York, NY 10032, USA. E-mail address:
n immunodeficiency virus type 1; M-MuLV, Moloney murine leukemia virus; SIV,-G, vesicular stomatitis virus; MT, microtubule; FAK, focal adhesion kinase; ECM,ixin/moesin; ERK, extracellular signal-regulated kinase; siRNA, short interferingn; MSS, minus-strand strong stop; DMSO, dimethyl sulfoxide; FERM, 4.1/ezrin/a; qPCR, quantitative real-time PCR; TBS-T, Tris-buffered saline and 1% Tween.
d by Elsevier Ltd.

762 Talin-1 and Vinculin Limit Retroviral Infection
HIV-1 infection, and impose an early block to infection by distinctretroviruses.
Published by Elsevier Ltd.
Introduction
As intracellular parasites, viruses must manipulatemany cellular functions to complete their life cycle,including the activity of their hosts' cytoskeletalnetworks and kinase signaling pathways. The hostcytoskeleton plays a complex role in many stages ofinfection. Many retroviruses surf along the cellsurface using an actin-mediated process to find asuitable site of entry. To enter the cell, they then needto penetrate through the layer of cortical actin that liesbeneath the cell surface and that functions both as afacilitator and as a barrier to the entry of incomingviruses.1–3 Upon entry into the cell, it is known thatviruses use the host cytoskeleton to reach specificcellular locations at the correct stage in the viral lifecycle to facilitate their replication, yet the exacttrafficking mechanisms used are not wellunderstood.4,5 Soon after entry into the cytoplasm,the viral core is uncoated and the viral RNA genomeis reverse transcribed, although the exact order andmechanism(s) involved in these processes remainsomewhat controversial. It is thought that reversetranscription of the human immunodeficiency virustype 1 (HIV-1) genome is completed before nuclearentry and has recently been suggested to occur at thenuclear pore.6,7 It has also been shown that the HIV-1reverse transcription complex associates with thecytoskeleton and that efficient reverse transcription isdependent on functional actin dynamics.8 The pre-integration complex may also associate with actinnetworks, as direct interactions of viral nucleocapsid,reverse transcriptase and integrasewith actin have allbeen described.9–11 Subsequent movement of incom-ing viral cores toward the nucleus exploits the hostcellsmicrotubule (MT) network, a cytoskeletal systemfor the long-range transport of macromolecules.12,13
The processes that govern themovement of incomingcores and the factors involved remain poorly under-stood. Recently, the ezrin/radixin/moesin (ERM)family members moesin and ezrin that mediatelinkage of the plasma membrane to actin filamentswere found to also negatively regulate the formationof a small subset of stable detyrosinatedMTs (termedGlu-MTs) and impose an early post-entry block toinfection by bothmurine leukemia virus andHIV-1 invarious mammalian cells.14,15 In-line with thesefindings, virus-envelope-mediated increases in thelevels of stabilized acetylated MTs have also beensuggested to play a role in HIV-1 cell fusion andinfection.16 In addition, IQGAP1, another regulator ofactin and MT networks, has been shown to directlyinteract with murine leukemia virus matrix protein
and affects both the early and late stages of viralinfection.17 Overexpression of the MT-motor-associated factor FEZ1 blocks the transport ofretroviral DNA into the nucleus after reversetranscription,18 while the kinesin motor proteinKIF4 associates with retroviral Gag and functionsin the outward movement of viral particles.19–21 Assuch, proteins that associate with or regulate actinand/or MT function are likely to play importantroles at a variety of stages in the retroviral life cycle.In addition to the host transport machinery,
retroviruses also hijack critical signaling pathways tofacilitate their replication. HIV-1-envelope-mediatedsignaling events that regulate actin cytoskeletondynamics have recently been suggested to promoteearly post-entry steps of viral infection in immunecells.22,23 HIV-1-envelope-mediated activation ofsignal transduction pathways is also known toregulate intracellular calcium mobilization andPI3K-dependent activation of all three mitogen-activated protein kinase family members (ERK1/2,JNK, and p38) and focal adhesion kinase (FAK).24
FAK phosphorylates paxillin, a key component ofintegrin signaling in focal adhesions, which are largedynamic protein complexes that attach the cytoskel-eton to the extracellular matrix (ECM) and play animportant role in cell signaling and motility.25
Tyrosine phosphorylation of paxillin generates dock-ing sites for diverse cytoskeletal and signalingfactors.26 As focal adhesion proteins are importantfor the transfer of force from the ECM through thehost cytoskeleton and in regulating signaling path-ways from the cell surface, they are good candidatesfor facilitating or obstructing early events in viralinfection. Indeed, FAK activity and focal adhesionassembly have previously been suggested to beinvolved in HIV-1 infection.27–33
In this study, we investigated the effects of talin-1and vinculin, two proteins that localize to focaladhesions, on retroviral infection both by transientoverexpression of these proteins and by shortinterfering RNA (siRNA)-mediated reduction in theexpression of their endogenous forms. We found thatunder both conditions, talin-1 and vinculin hadnegative effects on infection by a variety of pseudo-typed retroviruses in a number of different humancell lines, and these negative effects occurredindependently of the route of viral entry. It wasalso found that the block to infection occurred earlyin the viral life cycle, before the initiation of reversetranscription. While disruption of actin or MTpolymerization did not affect the antiviral activityof talin-1 or vinculin, expression of these proteinswas

763Talin-1 and Vinculin Limit Retroviral Infection
found to downregulate tyrosine phosphorylation ofpaxillin. The negative effects of talin-1 and vinculinon paxillin phosphorylation were specific, as theactivity of extracellular signal-regulated kinase (ERK)remained unaffected by talin-1 or vinculin, whilesiRNAs targeting paxillin reduced pseudotypedHIV-1 infection. As such, our findings identify twocellular factors that have antiretroviral activity andnegatively affect tyrosine phosphorylation of pax-illin, a positive regulator of HIV-1 infection.
Results
Overexpression of talin-1 or vinculin decreasesthe susceptibility of human cells toHIV-1 infection
To examine whether talin-1 expression affectsretrovirus infection, were transfected increasing
Fig. 1. Overexpression of talin-1 inhibits HIV-1 pseudotexpressing full-length talin-1 (pcDNA-HA-talin-1) were transimmunoblotting 48 h post-transfection with anti-talin-1 antibowas confirmed with anti-GAPDH antibodies (a) (lower panel).increasing amounts of plasmid expressing talin-1 were infectewas added to the cells 48 h postinfection, and resistant coloCHME3 (c) and 293A (d) cells transiently transfected with empHIV-1-VSV-luc 48 h post-transfection. The susceptibility of thluciferase activity 48 h postinfection. Similar results were obta
amounts of the full-length talin-1 expression vector(pcDNA-HA-talin-1) or of the control empty vector(pcDNA 3.1+) into HeLa cells (Fig. 1a). Thetransfected cells were infected with HIV-1-puropseudotyped with vesicular stomatitis virus (VSV)-G envelope, to allow entry into CD4-negative HeLacells, followed by puromycin selection 48 h post-infection for 11 days. Puromycin-resistant colonieswere stained and counted to measure the levels ofinfection in each case. Results demonstrated thatoverexpression of talin-1 decreased infection (2.98- to5.62-fold) in a dose-dependent manner (Fig. 1b). Toinvestigate whether the talin-1-mediated decrease ininfection was cell-type dependent, we then tran-siently overexpressed talin-1 in two additionalhuman cell lines: CHME3 microglia cells, a naturaltarget cell for HIV-1, and 293A embryonic kidneycells. Both lines were transfected with either emptyvector control plasmid or pcDNA-HA-talin-1, theninfected with luciferase-expressing HIV-1-VSV-luc
yped virus infection. Increasing amounts of a plasmidfected into HeLa cells, and expression was measured bydies (a) (top panel). Loading of equal amounts of lysates(b) HeLa cells transiently transfected with empty vector ord with HIV-1-VSV-puro 48 h post-transfection. Puromycinnies were stained and counted after 10 days of selection.ty vector or talin-1-expressing plasmid were infected withese cells to infection was then determined by measuringined in at least three independent experiments.

Fig. 2. Overexpression of vinculin inhibits HIV-1 pseudotyped virus infection. Increasing amounts of a plasmidexpressing full-length vinculin (pcDNA-vinculin) were transfected into HeLa cells, and expression was measured byimmunoblotting 48 h post-transfection with anti-vinculin antibodies (a) (top panel). GAPDH was used as a loadingcontrol (a) (lower panel). (b) HeLa cells transiently transfected with empty vector or increasing amounts of plasmidexpressing vinculin were infected with HIV-1-VSV-puro 48 h post-transfection. Puromycin was added to the cells 48 hpostinfection, and resistant colonies were stained and counted after 10 days of selection. CHME3 (c) and 293A (d) cellstransiently transfected with empty vector or vinculin-expressing plasmid were infected with HIV-1-VSV-luc 48 h post-transfection. The susceptibility of these cells to infection was then determined by measuring luciferase activity 48 hpostinfection. Similar results were obtained in at least three independent experiments.
764 Talin-1 and Vinculin Limit Retroviral Infection
pseudotyped virus. Luciferase assays showed that,similar to findings in HeLa cells, talin-1 expressioncaused a decrease in infection in both CHME3(Fig. 1c, 2.28-fold) and 293A (Fig. 1d, 1.84-fold) cells.This demonstrated that the block to infection wasnot cell-type dependent.We then investigated the talin-1 binding partner,
vinculin, to determine what effect this proteinwould have on retroviral infection. In the sameexperimental setup as described above, vinculin wasefficiently overexpressed in HeLa cells (Fig. 2a), andcells were then infected with HIV-1-puro pseudo-typed with VSV-G, followed by puromycin selec-tion, staining, and counting. Similar to results withtalin-1, overexpression of vinculin also decreasedinfection (2.24- to 4.44-fold) in HeLa cells (Fig. 2b).Vinculin also reduced infection by HIV-1-VSV-lucpseudotyped virus when overexpressed in CHME3(2.66-fold) or 293A (2.47-fold) cells, as determinedby luciferase assays (Fig. 2c and d). As such, similar
to its binding partner talin-1, vinculin inhibitedretroviral infection in a number of human cell types,including natural target cells for HIV-1.
Transient knockdown of talin-1 or vinculinincreases the susceptibility of human cellsto HIV-1 infection
Talin-1 and vinculin are ubiquitously expressedproteins, and it does not always follow that endog-enous levels of proteins have the same effect as whenthey are transiently overexpressed. To examine whateffect endogenous talin-1 had on retroviral infection,we used siRNAs to transiently reduce talin-1 levels inHeLa cells. These cells were then infectedwithHIV-1-VSV-luc or HIV-1-VSV-puro for 48 h, followed bymeasurements of luciferase activity or puromycinselection for 11 days, respectively. As shown in Fig. 3aand b, both of the siRNA duplexes targeting talin-1,TLN-B and TLN-C, increased infection (2.46- to 3.85-
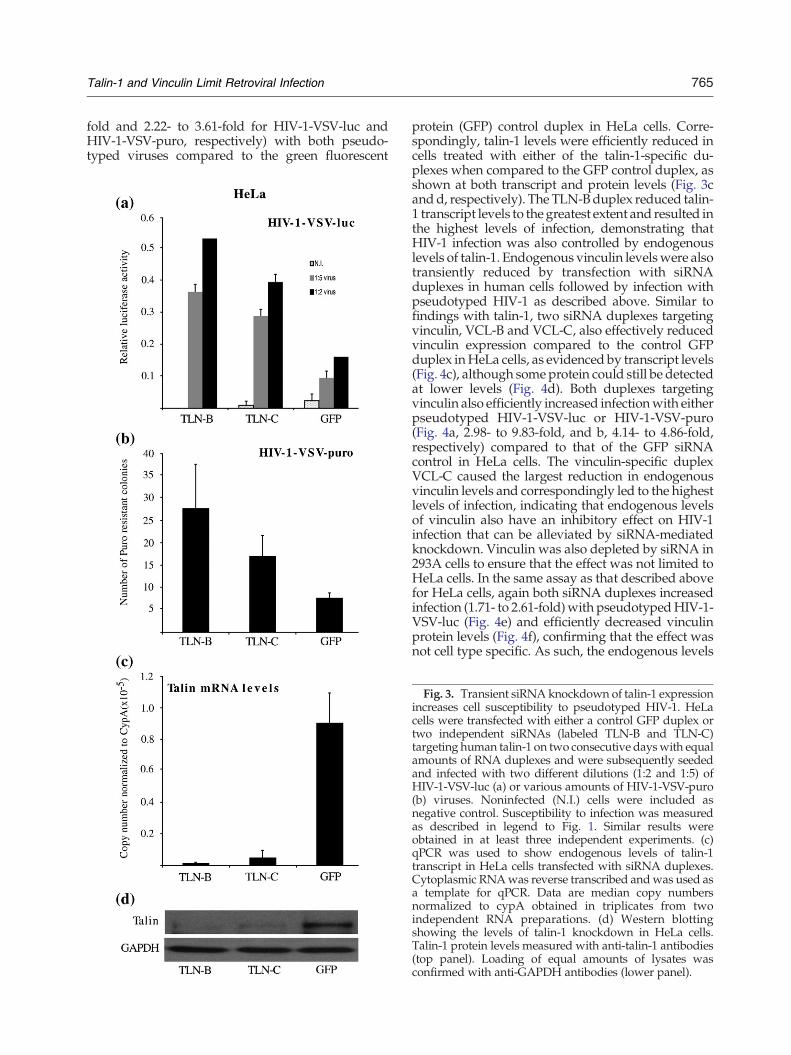
765Talin-1 and Vinculin Limit Retroviral Infection
fold and 2.22- to 3.61-fold for HIV-1-VSV-luc andHIV-1-VSV-puro, respectively) with both pseudo-typed viruses compared to the green fluorescent
protein (GFP) control duplex in HeLa cells. Corre-spondingly, talin-1 levels were efficiently reduced incells treated with either of the talin-1-specific du-plexes when compared to the GFP control duplex, asshown at both transcript and protein levels (Fig. 3cand d, respectively). The TLN-Bduplex reduced talin-1 transcript levels to the greatest extent and resulted inthe highest levels of infection, demonstrating thatHIV-1 infection was also controlled by endogenouslevels of talin-1. Endogenous vinculin levelswere alsotransiently reduced by transfection with siRNAduplexes in human cells followed by infection withpseudotyped HIV-1 as described above. Similar tofindings with talin-1, two siRNA duplexes targetingvinculin, VCL-B and VCL-C, also effectively reducedvinculin expression compared to the control GFPduplex inHeLa cells, as evidenced by transcript levels(Fig. 4c), although some protein could still be detectedat lower levels (Fig. 4d). Both duplexes targetingvinculin also efficiently increased infectionwith eitherpseudotyped HIV-1-VSV-luc or HIV-1-VSV-puro(Fig. 4a, 2.98- to 9.83-fold, and b, 4.14- to 4.86-fold,respectively) compared to that of the GFP siRNAcontrol in HeLa cells. The vinculin-specific duplexVCL-C caused the largest reduction in endogenousvinculin levels and correspondingly led to the highestlevels of infection, indicating that endogenous levelsof vinculin also have an inhibitory effect on HIV-1infection that can be alleviated by siRNA-mediatedknockdown. Vinculin was also depleted by siRNA in293A cells to ensure that the effect was not limited toHeLa cells. In the same assay as that described abovefor HeLa cells, again both siRNA duplexes increasedinfection (1.71- to 2.61-fold)with pseudotypedHIV-1-VSV-luc (Fig. 4e) and efficiently decreased vinculinprotein levels (Fig. 4f), confirming that the effect wasnot cell type specific. As such, the endogenous levels
Fig. 3. Transient siRNA knockdown of talin-1 expressionincreases cell susceptibility to pseudotyped HIV-1. HeLacells were transfected with either a control GFP duplex ortwo independent siRNAs (labeled TLN-B and TLN-C)targeting human talin-1 on two consecutive dayswith equalamounts of RNA duplexes and were subsequently seededand infected with two different dilutions (1:2 and 1:5) ofHIV-1-VSV-luc (a) or various amounts of HIV-1-VSV-puro(b) viruses. Noninfected (N.I.) cells were included asnegative control. Susceptibility to infection was measuredas described in legend to Fig. 1. Similar results wereobtained in at least three independent experiments. (c)qPCR was used to show endogenous levels of talin-1transcript in HeLa cells transfected with siRNA duplexes.Cytoplasmic RNAwas reverse transcribed andwas used asa template for qPCR. Data are median copy numbersnormalized to cypA obtained in triplicates from twoindependent RNA preparations. (d) Western blottingshowing the levels of talin-1 knockdown in HeLa cells.Talin-1 protein levels measured with anti-talin-1 antibodies(top panel). Loading of equal amounts of lysates wasconfirmed with anti-GAPDH antibodies (lower panel).

Fig. 5. Talin-1 and vinculin block pseudotyped HIV-1 early in the viral life cycle. Quantitative PCR indicating thelocalization of the block induced by talin-1 and vinculin in HeLa cells. HeLa cells were transiently transfected with siRNAduplexes targetingGFP, talin-1 (TLN-B), or vinculin (VCL-C) as described in (c), then infectedwithHIV-1-VSV-puro. Low-molecular-weight DNA was extracted 24 h postinfection and was measured by real-time PCR. Using primers specific forMSSDNAor puro sequences, we determined the amounts ofMSSDNA (a and b) and total viral DNA (c and d) normalizedto input DNA, respectively. Similar results were obtained in at least three independent experiments.
767Talin-1 and Vinculin Limit Retroviral Infection
of talin-1 and vinculin in human cells were sufficientto limit virus infection and suggested that theseproteins were important natural determinants ofcellular sensitivity to HIV-1 infection.
Talin-1 and vinculin block early steps of theHIV-1 life cycle
Infection assays using pseudotyped retrovirusesonly assess the early stages of viral infection as they
Fig. 4. Transient siRNA knockdown of vinculin expressioncells were transfected with either a control GFP duplex or two ihuman vinculin on two consecutive days with equal amountinfected with two different dilutions (1:2 and 1:5) of HIV-1viruses. Noninfected (N.I.) cells were included as negative conin legend to Fig. 1. Similar results were obtained in at least thendogenous levels of vinculin transcript in HeLa cells transCytoplasmic RNA was reverse transcribed and was used anormalized to cypA obtained in triplicates from two indepenlevels of vinculin knockdown in HeLa cells. Vinculin proteinLoading of equal amounts of lysates was confirmed with anti-transfected with siRNA duplexes targeting vinculin or GFPabove. (f) Vinculin levels in 293A cells after siRNA knockdow
are not replication competent, and a successfulinfection is therefore completed after integration ofthe viral genome and expression of the marker gene.To investigate more precisely at which stage talin-1and vinculin blocked HIV-1 infection, we examinedviral DNA synthesis in siRNA-transfected cells afterinfection with HIV-1-VSV-puro. The siRNA du-plexes TLN-B and VCL-C were chosen to knockdown talin-1 and vinculin, respectively, inHeLa cellsas they were found previously to elicit the greatest
increases cell susceptibility to pseudotyped HIV-1. HeLandependent siRNAs (labeled VCL-B and VCL-C) targetings of siRNA duplexes and were subsequently seeded and-VSV-luc (a) or various amounts of HIV-1-VSV-puro (b)trol. Susceptibility to infection was measured as describedree independent experiments. (c) qPCR was used to showfected with the same siRNA duplexes as in (a) and (b).s a template for qPCR. Data are median copy numbersdent RNA preparations. (d) Western blotting showing thelevels measured with anti-vinculin antibodies (top panel).GAPDH antibodies (lower panel). (e) 293A cells were alsoand were subsequently infected with HIV-1-VSV-luc asn were measured by immunoblotting as above.

Fig. 6. Talin-1 and vinculin block infection by a numberof retroviruses independently of route of viral entry. HeLacells were transiently transfected with siRNA duplexestargeting GFP, talin-1 (TLN-B), or vinculin (VCL-C) asdescribed in (c), then infectedwithHIV-1-luc pseudotypedwith an amphotropic envelope (a) or SIV-VSV-luc (b), andluciferase expression was measured 48 h postinfection todetermine susceptibility to infection. (c) CHME3 cells weretransiently transfected with either empty vector control orplasmids expressing talin-1 or vinculin, infected with M-MuLV-VSV-puro 48 h later. Susceptibility to infection wasmeasured by the addition of puromycin 48 h postinfectionand staining and counting puromycin-resistant coloniesafter a further 10 days. Similar results were obtained in atleast three independent experiments.
768 Talin-1 and Vinculin Limit Retroviral Infection
effect (Figs. 3d and 4d). After transfection of HeLacells with siRNAs targeting talin-1, vinculin, or GFPas a control, cultureswere then infectedwith VSV-G-pseudotyped HIV-1-puro, and HIRT DNA wasprepared for PCR analysis of viral DNA. Whenexamined, minus-strand strong stop (MSS) DNAand total viral DNA were found to be increased incells treated with siRNAs targeting talin-1 (Fig. 5aand c, 4.33-fold and 4.10-fold, respectively) orvinculin (Fig. 5b and d, 1.83-fold and 6.13-fold,respectively) compared to the GFP knockdowncontrol cells. As MSS DNA is the first step in thereverse transcription process, its accumulation intalin-1-depleted or vinculin-depleted cells demon-strated that the block to infection in both casesoccurred at or before the initiation of reversetranscription.
Talin-1 and vinculin block infection by a numberof retroviruses independently of the route ofviral entry
VSV-G-pseudotyped viruses enter the cell byreceptor-mediated endocytosis. Retroviral entrycan occur through specific receptor-mediated fusionor endocytic processes.34,35 As such, to ensure thatthe effects of talin-1 and vinculin were not solely dueto effects on VSV-G-mediated entry processes, wenext used luciferase-expressing HIV-1 pseudotypedwith an amphotropic envelope to infect HeLa cellsthat had been transfected with siRNAs targetingtalin-1 (TLN-B), vinculin (VCL-C), or GFP as acontrol. Similar to findings with VSV-G-pseudo-typed virus, siRNAs targeting either talin-1 orvinculin also resulted in an increase in infection(2.05-fold and 3.60-fold, respectively) by amphotro-pic virus (Fig. 6a). This demonstrated that bothfactors also blocked HIV-1 infection mediated byamphotropic envelope-mediated entry processesand that their inhibitory effects were independentof the route of viral entry. Finally, HeLa cellstransfected with siRNAs targeting talin-1 (TLN-B),vinculin (VCL-C), or GFP control were infected withVSV-G-pseudotyped simian immunodeficiencyvirus (SIV), while cells overexpressing each factorwere infected with VSV-G-pseudotyped Moloneymurine leukemia virus (M-MuLV), in order toexamine whether the block to infection was uniqueto HIV-1 or whether these factors also inhibitedother retroviruses. In the case of SIV infection,knockdown of talin-1 or vinculin increased infection(3.36-fold and 5.36-fold, respectively) relative to thecontrol cells, demonstrating that endogenous talin-1and vinculin also blocked SIV (Fig. 6b). Notably, thedegree to which siRNAs targeting each factoraffected infection with either amphotropic HIV-1or VSV-G-SIV was very similar (Fig. 6a and b),suggesting that this occurs through a similarmechanism for these two closely related lentivi-

769Talin-1 and Vinculin Limit Retroviral Infection
ruses. M-MuLV, however, is a gammaretrovirus andis therefore more distantly related to HIV-1. To testtheir effects on M-MuLV, we transiently transfectedCHME3 cells with talin-1, vinculin, or empty vectorcontrol plasmids then infected with M-MuLV-VSV-puro, which confers puromycin resistance ininfected cells. Results demonstrated that overex-pression of either talin-1 or vinculin significantlyreduced infection (2.34-fold and 2.82-fold, respec-tively) as measured by the number of drug-resistantcolonies in each case (Fig. 6c). This demonstratedthat both talin-1 and vinculin not only blockedlentiviruses such as HIV-1 and SIV but also wereable to reduce infection by a more distantly relatedgammaretrovirus, M-MuLV. Taken together, thesefindings suggest that talin-1 and vinculin areheterologous inhibitors of infection by a number ofretroviruses.
The effects of talin-1 and vinculin are notdependent on actin or MT networks
The main function of these two interacting cellularproteins is to attach actin filaments (F-actin) tointegrin receptors at the cell periphery, which isimportant for the transmission of force from theECM through the cell and for cell signaling. It isknown that F-actin plays a role in the early stages ofretroviral infection.13 As such, to investigate wheth-er the inhibition of retroviral infection by eitherfactor was dependent on F-actin, we first examinedthe effects of the actin depolymerizing agentcytochalasin D in distinct cell types either under-expressing or overexpressing talin-1 or vinculin.HeLa cells were transfected with siRNAs targetingtalin-1, vinculin, or GFP control then treated withdimethyl sulfoxide (DMSO) solvent control(untreated) or cytochalasin D for 5 h. Cultureswere then infected with HIV-1-VSV-luc in thecontinued presence of inhibitors and were assayedfor luciferase activity. Results demonstrated that, incontrol lines, infection was reduced by cytochalasinD (Fig. 7a), in-line with the established role forF-actin in retroviral infection.13 In untreated cul-tures, siRNAs targeting either talin-1 or vinculinincreased infection (2.14-fold and 3.02-fold, respec-tively), but infection was also increased (3.27-foldand 4.14-fold for talin-1 and vinculin, respectively)by siRNAs targeting either factor relative to controlsiRNA-treated cells in the presence of cytochalasinD, suggesting that both factors could alter cellularsensitivity to infection independently of actin. Todetermine whether this was limited to conditionsunder which factor expression was reduced andwhether this was a cell-type-specific effect, wetransfected CHME3 cells with an empty vector, thetalin-1 expression plasmid, or the vinculin expres-sion plasmid, then treated with DMSO control(untreated) or cytochalasin D for 5 h. Cells were
then infected with HIV-1-VSV-luc in the continuedpresence of cytochalasin D. When luciferase activitywas measured, it was found to be reduced bycytochalasin D in control lines, as expected given theimportance of F-actin for infection (Fig. 7b). Inaddition, both talin-1 and vinculin overexpressionsreduced the levels of infection (1.80-fold and1.95-fold, respectively) in CHME3 cells treated withDMSO control (untreated). However, in the presenceof cytochalasin D, talin-1 and vinculin continued tonegatively affect retroviral infection to extents(1.76-fold and 2.84-fold for talin-1 and vinculin,respectively) similar to those observed in untreatedcontrol cells. These findings were in agreementwith results from siRNA experiments above andsuggested that both talin-1 and vinculin werecapable of regulating retroviral infection indepen-dently of actin.Another cytoskeletal network, MTs, consists of
stable and dynamic filaments and has also beensuggested to be important for efficient retroviralinfection at both early and late stages.13 We thereforeinvestigated whether the inhibitory effects of talin-1or vinculin were dependent on these filaments byusing nocodazole to disrupt MT dynamics. In HeLacells, it was found that treatment with 1 μMnocodazole for 1 h efficiently disrupted MTscompared to the DMSO-treated control cells(untreated), as determined by staining of these cellsusing an antibody against dynamic tyrosinated MTs(Fig. 7c). To examine the potential contribution ofMTs to the block to infection imposed by endoge-nous talin-1 or vinculin, we transfected cells withsiRNAs targeting talin-1, vinculin, or GFP controlduplexes, then infected with HIV-1-VSV-luc in thepresence of DMSO (untreated) or nocodazole.Infection levels were reduced about 2-fold uponnocodazole treatment of control lines, in agreementwith previous reports,8,12 yet cells in which eithertalin-1 or vinculin had been knocked down exhibitedthe same degree of increased infection compared tothe control GFP knockdown cells either in thepresence (2.72-fold and 4.88-fold, respectively) or inthe absence (1.92-fold and 3.42-fold, respectively) ofnocodazole. This demonstrated that endogenoustalin-1 and vinculin were able to inhibit HIV-1infection independently of MTs (Fig. 7d). Finally,we examined whether either factor affected theabundance of stable MTs, a small subset of MTsthat are detyrosinated (lacking the carboxy-terminaltyrosine) and that are more stable than the majority(90%) of MTs. This stable detyrosinated subset (alsotermed Glu-MTs) of MTs has been shown to berelatively long lasting and resistant to noco-dazole.36,37 We previously showed that the antire-troviral activity of the cytoskeletal regulatoryproteins moesin and ezrin was accompanied bytheir ability to disrupt Glu-MT formation13,15 Wetherefore measured the levels of Glu-MTs in cells in
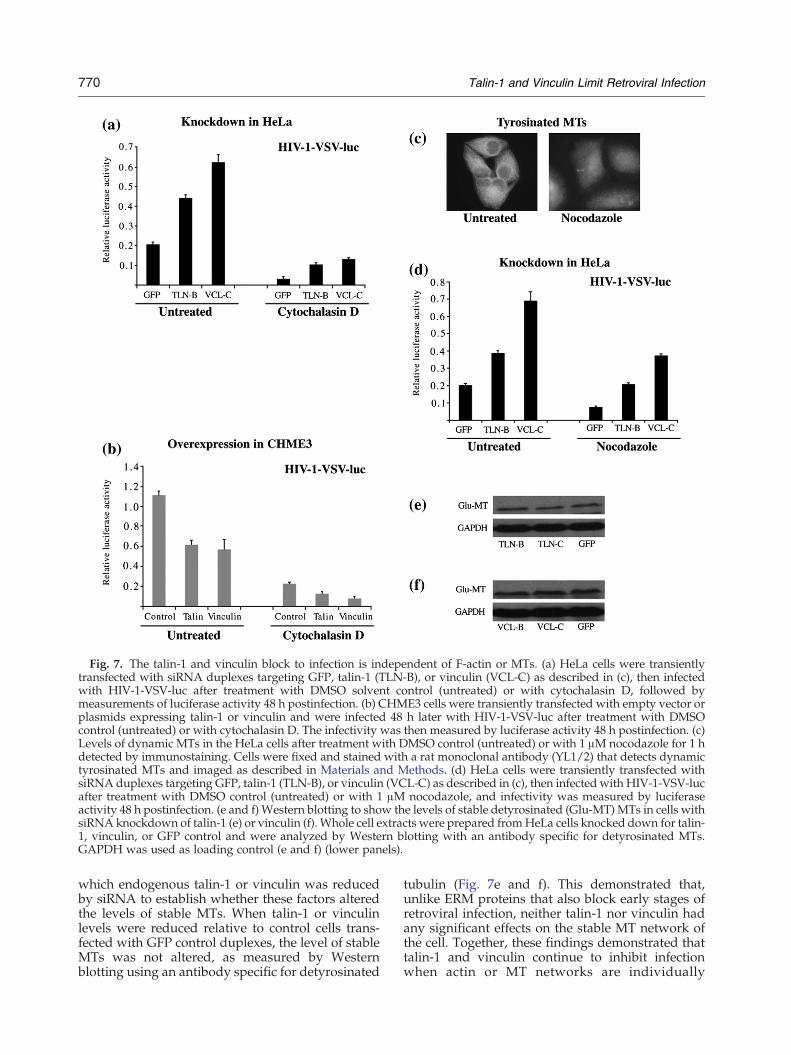
Fig. 7. The talin-1 and vinculin block to infection is independent of F-actin or MTs. (a) HeLa cells were transientlytransfected with siRNA duplexes targeting GFP, talin-1 (TLN-B), or vinculin (VCL-C) as described in (c), then infectedwith HIV-1-VSV-luc after treatment with DMSO solvent control (untreated) or with cytochalasin D, followed bymeasurements of luciferase activity 48 h postinfection. (b) CHME3 cells were transiently transfected with empty vector orplasmids expressing talin-1 or vinculin and were infected 48 h later with HIV-1-VSV-luc after treatment with DMSOcontrol (untreated) or with cytochalasin D. The infectivity was then measured by luciferase activity 48 h postinfection. (c)Levels of dynamic MTs in the HeLa cells after treatment with DMSO control (untreated) or with 1 μM nocodazole for 1 hdetected by immunostaining. Cells were fixed and stained with a rat monoclonal antibody (YL1/2) that detects dynamictyrosinated MTs and imaged as described in Materials and Methods. (d) HeLa cells were transiently transfected withsiRNA duplexes targeting GFP, talin-1 (TLN-B), or vinculin (VCL-C) as described in (c), then infected with HIV-1-VSV-lucafter treatment with DMSO control (untreated) or with 1 μM nocodazole, and infectivity was measured by luciferaseactivity 48 h postinfection. (e and f) Western blotting to show the levels of stable detyrosinated (Glu-MT) MTs in cells withsiRNA knockdown of talin-1 (e) or vinculin (f). Whole cell extracts were prepared fromHeLa cells knocked down for talin-1, vinculin, or GFP control and were analyzed by Western blotting with an antibody specific for detyrosinated MTs.GAPDH was used as loading control (e and f) (lower panels).
770 Talin-1 and Vinculin Limit Retroviral Infection
which endogenous talin-1 or vinculin was reducedby siRNA to establish whether these factors alteredthe levels of stable MTs. When talin-1 or vinculinlevels were reduced relative to control cells trans-fected with GFP control duplexes, the level of stableMTs was not altered, as measured by Westernblotting using an antibody specific for detyrosinated
tubulin (Fig. 7e and f). This demonstrated that,unlike ERM proteins that also block early stages ofretroviral infection, neither talin-1 nor vinculin hadany significant effects on the stable MT network ofthe cell. Together, these findings demonstrated thattalin-1 and vinculin continue to inhibit infectionwhen actin or MT networks are individually
771Talin-1 and Vinculin Limit Retroviral Infection
disrupted, suggesting that theymight block infectionby exerting effects on other processes.
Talin-1 and vinculin negatively regulate tyrosinephosphorylation of paxillin, a positive regulatorof HIV-1 infection
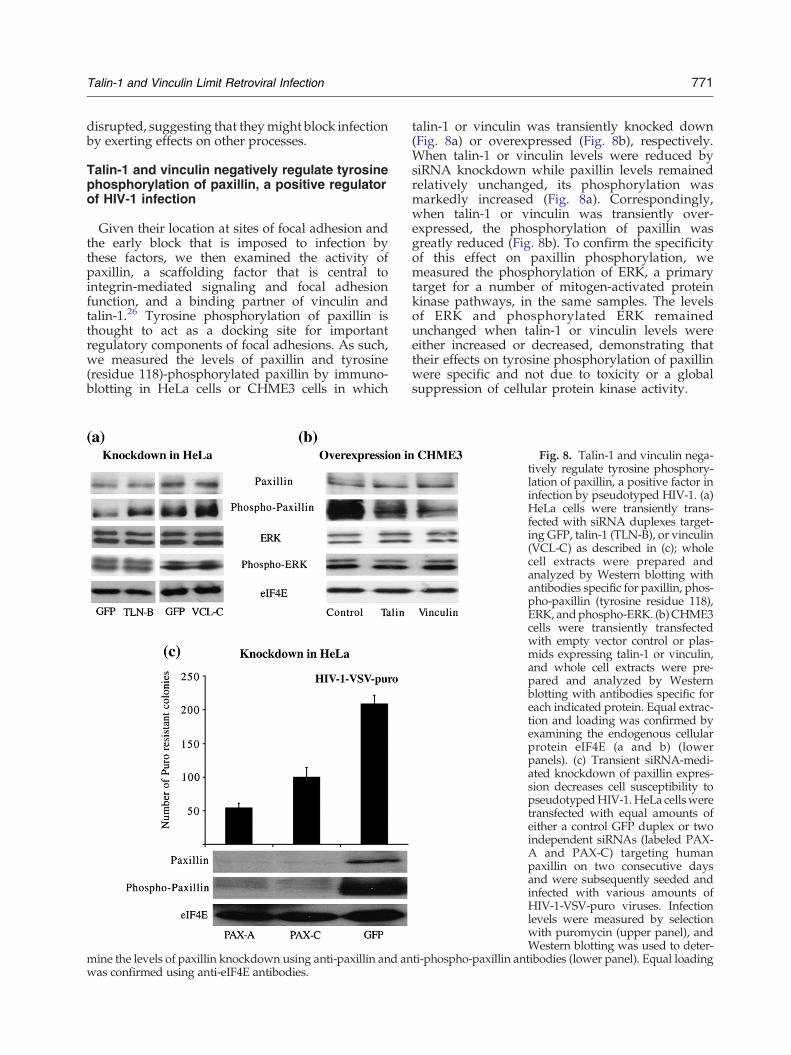
Given their location at sites of focal adhesion andthe early block that is imposed to infection bythese factors, we then examined the activity ofpaxillin, a scaffolding factor that is central tointegrin-mediated signaling and focal adhesionfunction, and a binding partner of vinculin andtalin-1.26 Tyrosine phosphorylation of paxillin isthought to act as a docking site for importantregulatory components of focal adhesions. As such,we measured the levels of paxillin and tyrosine(residue 118)-phosphorylated paxillin by immuno-blotting in HeLa cells or CHME3 cells in which
mine the levels of paxillin knockdown using anti-paxillin and anwas confirmed using anti-eIF4E antibodies.
talin-1 or vinculin was transiently knocked down(Fig. 8a) or overexpressed (Fig. 8b), respectively.When talin-1 or vinculin levels were reduced bysiRNA knockdown while paxillin levels remainedrelatively unchanged, its phosphorylation wasmarkedly increased (Fig. 8a). Correspondingly,when talin-1 or vinculin was transiently over-expressed, the phosphorylation of paxillin wasgreatly reduced (Fig. 8b). To confirm the specificityof this effect on paxillin phosphorylation, wemeasured the phosphorylation of ERK, a primarytarget for a number of mitogen-activated proteinkinase pathways, in the same samples. The levelsof ERK and phosphorylated ERK remainedunchanged when talin-1 or vinculin levels wereeither increased or decreased, demonstrating thattheir effects on tyrosine phosphorylation of paxillinwere specific and not due to toxicity or a globalsuppression of cellular protein kinase activity.
Fig. 8. Talin-1 and vinculin nega-tively regulate tyrosine phosphory-lation of paxillin, a positive factor ininfection by pseudotyped HIV-1. (a)HeLa cells were transiently trans-fected with siRNA duplexes target-ing GFP, talin-1 (TLN-B), or vinculin(VCL-C) as described in (c); wholecell extracts were prepared andanalyzed by Western blotting withantibodies specific for paxillin, phos-pho-paxillin (tyrosine residue 118),ERK, andphospho-ERK. (b)CHME3cells were transiently transfectedwith empty vector control or plas-mids expressing talin-1 or vinculin,and whole cell extracts were pre-pared and analyzed by Westernblotting with antibodies specific foreach indicated protein. Equal extrac-tion and loading was confirmed byexamining the endogenous cellularprotein eIF4E (a and b) (lowerpanels). (c) Transient siRNA-medi-ated knockdown of paxillin expres-sion decreases cell susceptibility topseudotypedHIV-1.HeLa cellsweretransfected with equal amounts ofeither a control GFP duplex or twoindependent siRNAs (labeled PAX-A and PAX-C) targeting humanpaxillin on two consecutive daysand were subsequently seeded andinfected with various amounts ofHIV-1-VSV-puro viruses. Infectionlevels were measured by selectionwith puromycin (upper panel), andWestern blotting was used to deter-
ti-phospho-paxillin antibodies (lower panel). Equal loading

772 Talin-1 and Vinculin Limit Retroviral Infection
These findings suggested that talin-1 and vinculinmight act through negative regulation of paxillinand that paxillin, a major scaffolding protein thatplays a central role in focal adhesions, mightfunction to facilitate infection. To examine this, wetransiently reduced the levels of endogenous pax-illin by transfection of HeLa cells with siRNAduplexes followed by infection with pseudotypedHIV-1, as described above. As shown in Fig. 8c(lower panel), the abundance of paxillin protein wasefficiently reduced in cells treated with either of thepaxillin-specific duplexes, PAX-A and PAX-C, whencompared to the GFP control duplex, and bothpaxillin siRNA duplexes decreased infection (2.08-to 3.82-fold) with pseudotyped HIV-1-VSV-purocompared to the GFP control duplex (upper panel),indicating that endogenous paxillin promotes HIV-1infection. Taken together, these findings demon-strated that the antiviral proteins vinculin and talin-1 negatively affect the phosphorylation of paxillin, acentral scaffold protein in focal adhesions thatfacilitates HIV-1 infection.
Discussion
Although factors and functions involved in avariety of events that occur during retroviralinfection have been identified, many early processesand the cellular factors involved remain poorlydefined. Notably, the host cytoskeletal networkscomposed of actin and MTs along with severalsignal transduction pathways are broadly known toplay important roles in early stages of retroviralinfection.13,24 Filamin-A, an actin binding protein,was identified as an adaptor that links CD4 andHIV-1 coreceptors to the actin cytoskeleton toregulate receptor clustering on the cell surface andpromote gp120 activation of RhoA, which mayfacilitate virus infection.22 HIV-1 envelope-mediatedsignaling was also recently shown to activate cofilinto overcome a cortical actin restriction to an earlypost-entry step of viral infection in resting T cells.23Given the functions of focal adhesion complexes assites of actin and MT organization, as well as in theregulation of cellular signaling pathways from thecell surface, their components are likely to playimportant roles in the retroviral life cycle.Talin-1 is a 270-kDa elongated antiparallel dimer
with a 4.1/ezrin/radixin/moesin (FERM) domaincontaining a globular head. The talin-1 FERMdomain contains its actin binding site, as well asbinding sites for β-integrin, FAK, and PIP kinase.38
The talin-1 rod domain has a second integrin bindingsite and three vinculin binding sites. Vinculin is a117-kDa protein with a globular head domain thatcontains its actin binding site and a tail regioncontaining binding sites for F-actin and paxillin,which is a multidomain protein that functions as a
focal adhesion scaffolding protein to recruit diversecytoskeletal and signaling factors.26 Vinculin associ-ates with talin-1 to bind F-actin and regulates thebinding of talin-1 to the β-subunit of the integrinreceptor.39,40 As such, vinculin is important forconnecting actin filaments to integrin receptors andis therefore a key player in focal adhesion assembly.The present study examines for the first time the
effect that the focal adhesion proteins talin-1 andvinculin have on retroviral infection. Our datademonstrate that talin-1 and vinculin are negativeregulators of HIV-1 infection. Transient overexpres-sion of talin-1 or vinculin efficiently reducedinfection with pseudotyped HIV-1 in a dose-depen-dent manner in several human cell lines, includinghuman microglia CHME3, a natural target cell typefor HIV-1 infection. In agreement with these over-expression studies, human cells become moresusceptible to retroviral infection upon transientknockdown of endogenous talin-1 or vinculin, alsoin a dose-dependent manner, indicating that thesefactors are natural determinants of cellular sensitiv-ity to retroviral infection. In addition to HIV-1, talin-1 and vinculin also block infection by SIV and themore distantly related gammaretrovirus, M-MuLV,suggesting that these factors are heterologous in-hibitors of retroviral infection.As these proteins function in the assembly of focal
adhesions and cell–cell contact,38–41 they are likely toalter several properties of the host cell that areimportant for viral entry and/or early post-entrystages of the viral life cycle. This appears not to bedue to generalized changes in cell viability, as cellsappeared healthy and ERK activity remained unaf-fected by transient increases or decreases in eithertalin-1 or vinculin expression. These proteinsblocked infection by pseudotyped retroviruses thatenter the cell by either fusion at the plasmamembrane or endocytosis, indicating that the blockto viral infection imposed by talin-1 and vinculinwas independent of the route of viral entry. As weare unable to determine whether these differentvirions actually entered into cells, it is possible thatfocal adhesions are critical sites for the entry ofviruses that employ distinct mechanisms. Indeed,focal adhesion signaling and assembly have beenreported to facilitate entry of several viruses,including HIV-1.27,33,42–47 This would make talin-1and vinculin broad-spectrum regulators of virionentry. On the other hand, analyses of HIV-1 DNAsynthesis localized the block imposed by talin-1 andvinculin to a stage at or prior to the initiation ofreverse transcription, suggesting that, if the blockoccurs post-entry, it occurs at a very early stage. Asimilar block to early post-entry events of retroviralinfection was recently described for the two closelyrelated ERM family member proteins, moesin andezrin, which attach the cell membrane to the actincytoskeleton.14,15 Focal adhesions have been

773Talin-1 and Vinculin Limit Retroviral Infection
suggested to be major sites of cross-talk betweenactin and MT cytoskeletons.48 Given that talin-1 andvinculin bind to actin and that functional actindynamics are required for efficient retroviral infec-tion, we investigated whether their effects weredependent on actin filaments.13,38–40 However, whencells that overexpressed talin-1 or vinculin wereinfected in the presence of cytochalasin D, bothfactors continued to negatively influence retroviralinfection, while siRNA-mediated reduction in en-dogenous levels increased the levels of infection evenin the presence of inhibitor. This suggested that theireffects were independent of actin-mediated process-es that regulate retroviral infection. In addition, theMT depolymerizing agent nocodazole did not alterthe ability of either factor to negatively regulateinfection, suggesting that they functioned indepen-dently of the MT network of the host cell. Indeed,changes in talin-1 or vinculin expression had noeffect on stableMT levels, distinguishing their effectsfrom other FERM domain proteins, moesin andezrin, both of which are actin cross-linking factorsthat regulate stable MT formation and also blockinfection at an early stage.14,15 These findingssuggest that the antiviral activity of talin-1 andvinculin is not uniquely dependent on either theactin cytoskeleton or the MT cytoskeleton.It is possible that these proteins, as well as focal
adhesions in general, may influence both viraltrafficking on cortical actin and the transition toMTs, which might explain their continued antiviralactivity in the individual presence of inhibitors ofone network or the other networks. Alternatively, itmay be that the focal adhesions themselves arecritical sites for the passage of virus into the cell, andalterations in their structure or function directlyaffect the ability of virions to penetrate into the cell.Paxillin is a large scaffolding protein that plays acentral role in focal adhesion function. Paxillin isphosphorylated on multiple (tyrosine, serine, andthreonine) residues in response to cell adhesionand/or exposure to a variety of growth factors andcytokines, as well as viral or bacterial infection.26 Inparticular, tyrosine phosphorylation on residue 118is thought to act as a docking site for severalimportant regulatory factors.26 Paxillin is known tointeract with both vinculin and talin-1. With this inmind, we examined the effects of talin-1 andvinculin expression on paxillin phosphorylationand found that these two proteins negatively affecttyrosine 118 phosphorylation without altering ERKphosphorylation, suggesting specificity in theireffects on host signal pathways and phosphoryla-tion of cellular proteins. In agreement with ourfindings, an increase in paxillin phosphorylationupon vinculin knockout has been reportedpreviously.49 By influencing the phosphorylationof this critical scaffold protein, talin-1 and vinculinmight negatively impact on focal adhesion activity
or assembly and impair the ability of these viruses toexploit these sites for viral entry or early post-entryprocesses. In support of this, we also found thatreducing paxillin expression impaired infection ofHeLa cells with pseudotyped HIV-1. Indeed, pax-illin might be an important mediator of infectivityby several viruses, as a direct association of the E6oncoproteins with human papilloma virus 16 andbovine papillomavirus with paxillin has also beensuggested to regulate paxillin phosphorylation andits function.50,51 Paxillin phosphorylation in re-sponse to Kaposi's sarcoma (KS)-associated herpes-virus infection has also been reported.52 In addition,paxillin was shown to be modulated during KS cellfunctions related to migration and adhesion,53
suggesting that it might have roles in cellulartransformation by this virus. HIV-1 Tat has alsobeen reported to enhance the phosphorylation andassociation of focal adhesion components such aspaxillin in KS cells, and this activity was recentlyshown to be the target for a novel anti-AIDS drugcandidate in these cells.54,55 As such, the antiviralactivity of vinculin and talin-1 may not be mediatedby their effects on actin but, instead, may bemediated through their effects on paxillin andthrough direct effects on the properties of focaladhesions. Notably, paxillin knockout cells can stillgenerate Glu-MTs upon integrin signaling,56 whichmay explain why we failed to observe the effects onGlu-MT networks in response to changes in vinculinor talin-1 expression and why actin or MT depoly-merizing agents did not affect the antiviral proper-ties of either of these proteins.It has also been demonstrated that a number of
focal adhesion proteins including talin-1 are specif-ically cleaved by HIV-1 protease.29 This is particu-larly intriguing given our findings that both talin-1and vinculin are negative regulators of viralinfection. Interestingly, elevated vinculin levelsand vinculin-peptide-specific CTL (cytotoxic Tlymphocytes) activity were previously reported inlymphoid cells from HIV-1-infected individuals.57
In addition, increased protein levels of vinculin,filamin-A, and talin-1 were recently reported inperipheral blood mononuclear cells from HIV-positive patients.58 As such, focal adhesion compo-nents such as talin-1 and vinculin, which negativelyregulate infection, may be induced as part ofimmune responses to infection. Our findings dem-onstrate that both exogenous and endogenouslyexpressed forms of the focal adhesion proteins talin-1 and vinculin negatively regulate tyrosine phos-phorylation of paxillin, a novel positive regulator ofHIV-1 infection, and impose an early block toinfection by a number of distinct retroviruses.Given their induction in immune cells in responseto HIV-1, developing our understanding of howthese factors function will provide important in-sights into virus–host interacts that are functionally

774 Talin-1 and Vinculin Limit Retroviral Infection
important determinants of the outcome of viralinfection.
Materials and Methods
Cells and viruses
HeLa, 293A, 293T, and CHME3 cells were cultured inDulbecco's modified Eagle's medium supplemented withL-glutamine, penicillin/streptomycin, sodium pyruvate,and 5% FBS, as described previously.18,59 HIV-1-luc andHIV-1-puro (HIV-based viral vectors carrying a luciferasereporter or a puromycin resistance marker, respectively)and M-MuLV-puro (M-MuLV-based viral vector carryinga puromycin resistance marker) pseudotyped with VSV-G envelope were produced by transfection of 293T cellsusing a combination of three expression vectors, asdescribed previously.18 VSV-G-pseudotyped SIV-luc(SIVmac-based viral vector carrying a luciferase reporter)virus was generated by cotransfection of 293T cells withpSIVmac239-luc-E
−60 (a kind gift from Dr. NathanielLandau) and the VSV-G envelope-expressing vectorpMDG.61 HIV-1-puro carrying an amphotropic envelopewas generated by replacing pMDG61 with the pHit45662
vector in previously described protocols.14
Infection of cultured cells and transduction assays
Target cells (1×105) were infectedwithHIV-1-VSV-puroor M-MuLV-VSV-puro as previously described.18 Poly-brene was added to infections at a final concentration of8 μg/ml. Levels of infection were measured by colonycounting after selection in puromycin: 1 μg/ml for HeLacells and 0.35 μg/ml for CHME3 cells.59 For HIV-1-lucinfections, 2×104 target cells were seeded 24 h beforeinfection and were lysed 48 h postinfection, then assayedfor luciferase activity as per manufacturer's protocol(Promega).
Drug treatment of cultured cells
For cytochalasin D treatment, target cells were pre-treated with 0.4 μg/ml cytochalasin D for 5 h then infectedin the continuous presence of cytochalasin D for 16 h. Thecells were then cultured in normal medium and wereassayed as above. For nocodazole treatment, HeLa cellswere treated with 1 μMnocodazole for 1 h before infectionand were infected in the presence of nocodazole for 5 h.The treated cells were then returned to normal medium fora further 43 h before being assayed as above.
Transient overexpression
Target cells (1×105) were transfected with increasingamounts of vector up to 500 ng of plasmid in OPTI-MEMusing FuGENE HD (Roche) and were seeded fortransfection or Western blotting 24 h later. For full-lengthvinculin expression, pcDNA-vinculin was made byexcising the NotI fragment from pDNR-Dual vinculin(PlasmID repository, Harvard) and ligating it into the
mammalian expression vector pcDNA 3.1+ (Invitrogen).pcDNA-HA-talin-1 was used for full-length talin-1 over-expression (kindly provided by Dr. Mark Ginsberg).pcDNA 3.1+ (Invitrogen) was used for the empty vectorcontrol.
Quantitative real-time PCR
Cytoplasmic RNAwas reverse transcribed and was usedas a template for PCR using SYBR Green JumpStart TaqReadyMix (Sigma).18 Talin-1 transcript levels were deter-mined using forward primer of TLN1-S1, 5′GTGCCATTC-CAGCCAATGCA3′, and reverse primer of TLN1-A1, 5′CAGAAGGCTTTGGTAGTGGCA3′. Primers for detectionof vinculin transcript levels were VCL-S, 5′CCAGAACCT-CATGCAGTCTGT3′, and VCL-A, 5′GGTATGGTTGG-CAGCAACATG3′. The number of target copies in eachsample was interpolated from its detection cycle threshold(CT) value using a cyclophilin A15 standard curve. Theprimers used for detection of cyclophilin A transcript levelswere hCypA-up: 5′TTGAGCTGTTTGCAGACA3′ andhCypA-down: 5′ACCCGTATGCTTTAGGAT3′. Ampliconsize and reaction specificity were confirmed by agarose gelelectrophoresis.
Immunoblotting
Protein levels were measured by Western blotting ofwhole cell extracts prepared in Laemmli buffer [62.5 mMTris–HCl, 10% glycerol, 2% SDS, and 0.0025% bromophe-nol blue (pH 6.8)] fractionated by SDS-PAGE and weretransferred onto nitrocellulose membrane (Whatman).Membranes were blocked and then probed with theindicated antibodies diluted in 3% bovine serum albuminin Tris-buffered saline and 0.1% Tween (TBS-T). Vinculinantibody (Ab18058; Abcam) and paxillin antibody(610619; BD Biosciences) were used at 1:500 dilution.Talin-1 (Ab57758; Abcam), phospho-paxillin (44-722G;BioSource), ERK (9102; Cell Signaling), and phospho-ERK(9101; Cell Signaling) antibodies were used at 1:1000dilution. The levels of stable detyrosinated tubulin (Glu-MTs) were detected using a 1:1000 dilution of a rabbitpolyclonal antibody (AB3201; Chemicon International).As a loading control, GAPDH antibody (SC-25778; SantaCruz Biotechnology) and eIF4E antibody (610269; BDBiosciences) were used at 1:1000 dilution.
RNA interference
HeLa cells (2×105) were transfected with two indepen-dent pre-designed siRNAs specific to human talin-1,vinculin, and paxillin (Ambion) or with a control siRNAduplex targeted to GFP (Sigma) using LipofectamineRNAiMax transfection reagent (Invitrogen) as describedpreviously.18 Cells were transfected with 300 pmol RNAduplex and subsequently seeded 48 h post-transfection,infectedwith various amounts of HIV-1-VSV-puro orHIV-1-VSV-luc and either selected in puromycin for colonycounting or lysed for luciferase activity assays, respective-ly, as described above. Endogenous talin-1, vinculin, andpaxillin levels were measured by quantitative real-timePCR (qPCR) and/or immunoblotting as described above.

775Talin-1 and Vinculin Limit Retroviral Infection
siRNA knockdown and quantification of viral DNA
HeLa cells were transfected with pre-designed siRNAs, asdescribed above. Cells (post-transfected 48 h) were infectedwith DNase-I-digested supernatant containing HIV-1-VSV-puro.63 HIRT DNA was isolated at 24 h after infection, and1 μl was used as template for 40 cycles (15 s at 94 °C, 60 s at60 °C, and 60 s at 72 °C) of three-step PCR of MSS and totalviral DNA using SYBR Green JumpStart Taq ReadyMix(Sigma) including 200 nM each primer: HIV-MSS-S5′-GGCTAACTAGGGAACCCACTG and HIV-MSS-A5′-CTGCTAGAGATTTTCCACACTGAC or puro-S2 5′CTCGACATCGGCAAGGTGTG3′ and puro-A2 5′GCCTTCCATCTGTTGCTGCG3′, respectively, as describedpreviously.63 The number of target copies in each samplewasinterpolated from its detection threshold (CT) value using afull-length HIV-1, pNL4-3,64 or a puromycin pCSPW (a giftfrom Dr. Greg Towers) plasmid standard curve.63
Immunofluorescence microscopy
For staining of dynamic MTs, cells grown on coverslipswere washed once in serum-free medium then fixed in ice-coldmethanol for 10min and rehydrated in TBS-T. Sampleswere pre-blocked in 10%normal goat serum in TBS-T for 1 hthen probed with a rat monoclonal antibody YL1/2 (1:10; akind gift from Gregg Gundersen, Columbia University,New York) against dynamic tyrosinatedMTs (Tyr-MTs) for2 h at room temperature. After washing, primary antibodywas detected using cross-absorbed FITC-conjugated anti-rat secondary antibody (Jackson ImmunoResearch Labora-tories, West Grove, PA) at a concentration of 1:200 for 1 h atroom temperature. Afterwashing,Hoechst stainwas addedat room temperature for 5 min to stain nuclei, followed bythree additional washes with TBS-T. All staining wasperformed in TBS-T containing 1% normal goat serum.For fluorescence microscopy, cells were observed with anOlympus BX50microscope, and imageswere capturedwithan Olympus camera DP70 (Olympus) using DP Controllersoftware (Olympus) as previously described.15
Acknowledgements
We thankMarkGinsberg (University of California,San Diego, California), Gregg Gundersen (ColumbiaUniversity, New York), Nathaniel Landau (NewYork University, New York), Greg Towers (Univer-sity College London, London), and Alex Eustace(Dublin City University, Dublin) for reagents. Thiswork was supported by grants from the ScienceFoundation Ireland under grant nos. (06/IN.1/B78)and (09-RFP-BMT2130) and by the Health ResearchBoard under grant no. (RP/2008/21).
References
1. Iyengar, S., Hildreth, J. E. & Schwartz, D. H. (1998).Actin-dependent receptor colocalization required for
human immunodeficiency virus entry into host cells. J.Virol. 72, 5251–5255.
2. Lehmann, M. J., Sherer, N. M., Marks, C. B., Pypaert,M. & Mothes, W. (2005). Actin- and myosin-drivenmovement of viruses along filopodia precedes theirentry into cells. J. Cell Biol. 170, 317–325.
3. Campbell, E. M., Nunez, R. & Hope, T. J. (2004).Disruption of the actin cytoskeleton can complementthe ability of Nef to enhance human immunodeficiencyvirus type 1 infectivity. J. Virol. 78, 5745–5755.
4. Radtke, K., Dohner, K. & Sodeik, B. (2006). Viralinteractions with the cytoskeleton: a hitchhiker's guideto the cell. Cell. Microbiol. 8, 387–400.
5. Greber, U. F. & Way, M. (2006). A superhighway tovirus infection. Cell, 124, 741–754.
6. Zennou, V., Petit, C., Guetard, D., Nerhbass, U.,Montagnier, L. & Charneau, P. (2000). HIV-1 genomenuclear import is mediated by a central DNA flap.Cell, 101, 173–185.
7. Arhel, N., Souquere-Besse, S., Munier, S., Souque, P.,Guadagnini, S., Rutherford, S. et al. (2007). HIV-1DNA flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J. 26,3025–3037.
8. Bukrinskaya, A., Brichacek, B., Mann, A. & Stevenson,M. (1998). Establishment of a functional humanimmunodeficiency virus type 1 (HIV-1) reversetranscription complex involves the cytoskeleton. J.Exp. Med. 188, 2113–2125.
9. Hottiger, M., Gramatikoff, K., Georgiev, O., Chapon-nier, C., Schaffner, W. & Hubscher, U. (1995). Thelarge subunit of HIV-1 reverse transcriptase interactswith beta-actin. Nucleic Acids Res. 23, 736–741.
10. Wilk, T., Gowen, B. & Fuller, S. D. (1999). Actinassociates with the nucleocapsid domain of thehuman immunodeficiency virus Gag polyprotein. J.Virol. 73, 1931–1940.
11. Turlure, F., Devroe, E., Silver, P. A. & Engelman, A.(2004). Human cell proteins and human immunode-ficiency virus DNA integration. Front. Biosci. 9,3187–3208.
12. McDonald, D., Vodicka, M. A., Lucero, G., Svitkina, T.M., Borisy, G. G., Emerman, M. & Hope, T. J. (2002).Visualization of the intracellular behavior of HIV inliving cells. J. Cell Biol. 159, 441–452.
13. Naghavi, M. H. & Goff, S. P. (2007). Retroviralproteins that interact with the host cell cytoskeleton.Curr. Opin. Immunol. 19, 402–407.
14. Naghavi, M. H., Valente, S., Hatziioannou, T., de LosSantos, K., Wen, Y., Mott, C. et al. (2007). Moesinregulates stable microtubule formation and limitsretroviral infection in cultured cells. EMBO J. 26,41–52.
15. Haedicke, J., de Los Santos, K., Goff, S. P. & Naghavi,M. H. (2008). The Ezrin-radixin-moesin family mem-ber ezrin regulates stable microtubule formation andretroviral infection. J. Virol. 82, 4665–4670.
16. Valenzuela-Fernandez, A., Alvarez, S., Gordon-Alonso, M., Barrero, M., Ursa, A., Cabrero, J. R. et al.(2005). Histone deacetylase 6 regulates human immu-nodeficiency virus type 1 infection. Mol. Biol. Cell, 16,5445–5454.
17. Leung, J., Yueh, A., Appah, F. S., Jr., Yuan, B., de losSantos, K. & Goff, S. P. (2006). Interaction of Moloney

776 Talin-1 and Vinculin Limit Retroviral Infection
murine leukemia virus matrix protein with IQGAP.EMBO J. 25, 2155–2166.
18. Naghavi, M.H., Hatziioannou, T., Gao, G. &Goff, S. P.(2005). Overexpression of fasciculation and elongationprotein ζ-1 (FEZ1) induces a post-entry block toretroviruses in cultured cells.Genes Dev. 19, 1105–1115.
19. Kim, W., Tang, Y., Okada, Y., Torrey, T. A.,Chattopadhyay, S. K., Pfleiderer, M. et al. (1998).Binding of murine leukemia virus Gag polyproteinsto KIF4, a microtubule-based motor protein. J. Virol.72, 6898–6901.
20. Tang, Y., Winkler, U., Freed, E. O., Torrey, T. A., Kim,W., Li, H. et al. (1999). Cellular motor protein KIF-4associates with retroviral Gag. J. Virol. 73, 10508–10513.
21. Martinez, N. W., Xue, X., Berro, R. G., Kreitzer, G. &Resh, M. D. (2008). Kinesin KIF4 regulates intracellu-lar trafficking and stability of the human immunode-ficiency virus type 1 Gag polyprotein. J. Virol. 82,9937–9950.
22. Jimenez-Baranda, S., Gomez-Mouton, C., Rojas, A.,Martinez-Prats, L., Mira, E., Ana Lacalle, R. et al.(2007). Filamin-A regulates actin-dependent cluster-ing of HIV receptors. Nat. Cell Biol. 9, 838–846.
23. Yoder, A., Yu, D., Dong, L., Iyer, S. R., Xu, X., Kelly, J.et al. (2008). HIV envelope-CXCR4 signaling activatescofilin to overcome cortical actin restriction in restingCD4 T cells. Cell, 134, 782–792.
24. Wu, Y. & Yoder, A. (2009). Chemokine coreceptorsignaling in HIV-1 infection and pathogenesis. PLoSPathog. 5, e1000520.
25. Mitra, S. K., Hanson, D. A. & Schlaepfer, D. D. (2005).Focal adhesion kinase: in command and control of cellmotility. Nat. Rev., Mol. Cell Biol. 6, 56–68.
26. Brown, M. C. & Turner, C. E. (2004). Paxillin: adaptingto change. Physiol. Rev. 84, 1315–1339.
27. Cicala, C., Arthos, J., Ruiz, M., Vaccarezza, M.,Rubbert, A., Riva, A. et al. (1999). Induction ofphosphorylation and intracellular association of CCchemokine receptor 5 and focal adhesion kinase inprimary human CD4+ T cells by macrophage-tropicHIV envelope. J. Immunol. 163, 420–426.
28. Cicala, C., Arthos, J., Rubbert, A., Selig, S., Wildt, K.,Cohen, O. J. & Fauci, A. S. (2000). HIV-1 envelopeinduces activation of caspase-3 and cleavage of focaladhesion kinase in primary human CD4+ T cells. Proc.Natl Acad. Sci. USA, 97, 1178–1183.
29. Shoeman, R. L., Hartig, R., Hauses, C. & Traub, P.(2002). Organization of focal adhesion plaques isdisrupted by action of the HIV-1 protease. Cell Biol.Int. 26, 529–539.
30. Avraham, H. K., Jiang, S., Lee, T. H., Prakash, O. &Avraham, S. (2004). HIV-1 Tat-mediated effects onfocal adhesion assembly and permeability in brainmicrovascular endothelial cells. J. Immunol. 173,6228–6233.
31. Urbinati, C., Bugatti, A., Giacca, M., Schlaepfer, D.,Presta, M. & Rusnati, M. (2005). αvβ3-integrin-dependent activation of focal adhesion kinase medi-ates NF-κB activation and motogenic activity by HIV-1 Tat in endothelial cells. J. Cell Sci. 118, 3949–3958.
32. Hoefen, R. J. & Berk, B. C. (2006). The multifunctionalGIT family of proteins. J. Cell Sci. 119, 1469–1475.
33. Garron, M. L., Arthos, J., Guichou, J. F., McNally, J.,Cicala, C. & Arold, S. T. (2008). Structural basis for the
interaction between focal adhesion kinase and CD4. J.Mol. Biol. 375, 1320–1328.
34. Anderson, J. L. & Hope, T. J. (2005). Intracellulartrafficking of retroviral vectors: obstacles and ad-vances. Gene Ther. 12, 1667–1678.
35. Miyauchi, K., Kim, Y., Latinovic, O., Morozov, V. &Melikyan, G. (2009). HIV enters cells via endocytosisand dynamin-dependent fusion with endosomes. Cell,137, 433–444.
36. Westermann, S. & Weber, K. (2003). Post-translationalmodifications regulate microtubule function. Nat.Rev., Mol. Cell Biol. 4, 938–947.
37. Bartolini, F. & Gundersen, G. G. (2009). Formins andmicrotubules. Biochim. Biophys. Acta, 1803, 164–173.
38. Critchley, D. R. (2004). Cytoskeletal proteins talin andvinculin in integrin-mediated adhesion. Biochem. Soc.Trans. 32, 831–836.
39. Humphries, J. D., Wang, P., Streuli, C., Geiger, B.,Humphries, M. J. & Ballestrem, C. (2007). Vinculincontrols focal adhesion formation by direct interac-tions with talin and actin. J. Cell Biol. 179, 1043–1057.
40. Ziegler, W. H., Gingras, A. R., Critchley, D. R. &Emsley, J. (2008). Integrin connections to the cytoskel-eton through talin and vinculin. Biochem. Soc. Trans.36, 235–239.
41. Maddugoda, M. P., Crampton, M. S., Shewan, A. M. &Yap, A. S. (2007). Myosin VI and vinculin cooperateduring the morphogenesis of cadherin cell–cell con-tacts in mammalian epithelial cells. J. Cell Biol. 178,529–540.
42. Li, E., Stupack, D. G., Brown, S. L., Klemke, R.,Schlaepfer, D. D. & Nemerow, G. R. (2000). Associa-tion of p130CAS with phosphatidylinositol-3-OHkinase mediates adenovirus cell entry. J. Biol. Chem.275, 14729–14735.
43. Lee, C., Liu, Q. H., Tomkowicz, B., Yi, Y., Freedman,B. D. & Collman, R. G. (2003). Macrophage activationthrough CCR5- and CXCR4-mediated gp120-elicitedsignaling pathways. J. Leukoc. Biol. 74, 676–682.
44. Chu, J. J. & Ng, M. L. (2004). Interaction of West Nilevirus with αvβ3 integrin mediates virus entry intocells. J. Biol. Chem. 279, 54533–54541.
45. Veettil, M. V., Sharma-Walia, N., Sadagopan, S.,Raghu, H., Sivakumar, R., Naranatt, P. P. & Chan-dran, B. (2006). RhoA-GTPase facilitates entry ofKaposi's sarcoma-associated herpesvirus into adher-ent target cells in a Src-dependent manner. J. Virol. 80,11432–11446.
46. Krishnan, H. H., Sharma-Walia, N., Streblow, D. N.,Naranatt, P. P. & Chandran, B. (2006). Focal adhesionkinase is critical for entry of Kaposi's sarcoma-associated herpesvirus into target cells. J. Virol. 80,1167–1180.
47. Abban, C. Y. &Meneses, P. I. (2010). Usage of heparansulfate, integrins, and FAK in HPV16 infection.Virology, 403, 1–16.
48. Palazzo, A. F. & Gundersen, G. G. (2002). Microtu-bule-actin cross-talk at focal adhesions. Sci STKE,2002, pe31.
49. Subauste, M. C., Pertz, O., Adamson, E. D., Turner, C.E., Junger, S. & Hahn, K. M. (2004). Vinculinmodulation of paxillin–FAK interactions regulatesERK to control survival and motility. J. Cell Biol. 165,371–381.

777Talin-1 and Vinculin Limit Retroviral Infection
50. Vande Pol, S. B., Brown, M. C. & Turner, C. E. (1998).Association of Bovine Papillomavirus Type 1 E6oncoprotein with the focal adhesion protein paxillinthrough a conserved protein interaction motif. Onco-gene, 16, 43–52.
51. Wade, R., Brimer, N. & Vande Pol, S. (2008).Transformation by bovine papillomavirus type 1 E6requires paxillin. J. Virol. 82, 5962–5966.
52. Wang, F. Z., Akula, S. M., Sharma-Walia, N., Zeng, L.&Chandran, B. (2003). Human herpesvirus 8 envelopeglycoprotein B mediates cell adhesion via its RGDsequence. J. Virol. 77, 3131–3147.
53. Liu, Z. Y., Ganju, R. K., Wang, J. F., Ona, M. A., Hatch,W. C., Zheng, T. et al. (1997). Cytokine signalingthrough the novel tyrosine kinase RAFTK in Kaposi'ssarcoma cells. J. Clin. Invest. 99, 1798–1804.
54. Ganju, R. K., Munshi, N., Nair, B. C., Liu, Z. Y., Gill, P.& Groopman, J. E. (1998). Human immunodeficiencyvirus tat modulates the Flk-1/KDR receptor, mitogen-activated protein kinases, and components of focaladhesion in Kaposi's sarcoma cells. J. Virol. 72,6131–6137.
55. Lu, C. X., Li, J., Sun, Y. X., Qi, X., Wang, Q. J., Xin, X. L.& Geng, M. Y. (2007). Sulfated polymannurogulur-onate, a novel anti-AIDS drug candidate, inhibitsHIV-1 Tat-induced angiogenesis in Kaposi's sarcomacells. Biochem. Pharmacol. 74, 1330–1339.
56. Palazzo, A. F., Eng, C. H., Schlaepfer, D. D.,Marcantonio, E. E. & Gundersen, G. G. (2004).Localized stabilization of microtubules by integrin-and FAK-facilitated Rho signaling. Science, 303,836–839.
57. di Marzo Veronese, F., Arnott, D., Barnaba, V., Loftus,D. J., Sakaguchi, K., Thompson, C. B. et al. (1996).
Autoreactive cytotoxic T lymphocytes in humanimmunodeficiency virus type 1-infected subjects. J.Exp. Med. 183, 2509–2516.
58. Zhang, L., Jia, X., Zhang, X., Sun, J., Peng, X., Qi, T.et al. (2010). Proteomic analysis of PBMCs: character-ization of potential HIV-associated proteins. ProteomeSci. 8, 12.
59. Janabi, N., Peudenier, S., Heron, B., Ng, K. H. &Tardieu, M. (1995). Establishment of human micro-glial cell lines after transfection of primary cultures ofembryonic microglial cells with the SV40 large Tantigen. Neurosci. Lett. 195, 105–108.
60. Yu, Q., Chen, D., Konig, R., Mariani, R., Unutmaz, D.& Landau, N. R. (2004). APOBEC3B and APOBEC3Care potent inhibitors of simian immunodeficiencyvirus replication. J. Biol. Chem. 279, 53379–53386.
61. Yee, J. K., Miyanohara, A., LaPorte, P., Bouic, K.,Burns, J. C. & Friedmann, T. (1994). A general methodfor the generation of high-titer, pantropic retroviralvectors: highly efficient infection of primary hepato-cytes. Proc. Natl Acad. Sci. USA, 91, 9564–9568.
62. Yap, M.W., Kingsman, S. M. & Kingsman, A. J. (2000).Effects of stoichiometry of retroviral components onvirus production. J. Gen. Virol. 81, 2195–2202.
63. Haedicke, J., Brown, C. & Naghavi, M. H. (2009). Thebrain-specific factor FEZ1 is a determinant of neuronalsusceptibility to HIV-1 infection. Proc. Natl Acad. Sci.USA, 106, 14040–14045.
64. Adachi, A., Gendelman, H. E., Koenig, S., Folks, T.,Willey, R., Rabson, A. & Martin, M. A. (1986).Production of acquired immunodeficiency syn-drome-associated retrovirus in human and nonhu-man cells transfected with an infectious molecularclone. J. Virol. 59, 284–291.


















