
FLUORINATED FULLERENE DERIVATIVES: SYNTHESIS, STRUCTURE, AND ELECTRONIC ... files/papers/2-26... ·...
12
2-26 FLUORINATED FULLERENE DERIVATIVES: SYNTHESIS, STRUCTURE, AND ELECTRONIC PROPERTIES Alexey A. Goryunkov, Marina G. Apenova, Eugenia V. Borkovskaya, Victor A. Brotsman, Nikita M. Belov, Ilya N. Ioffe, Vitaliy A. Ioutsi, Natalia S. Lukonina, Vitaliy Yu. Markov, Alexey V. Rybalchenko, Olesya O. Semivrazhskaya Chemistry Department, Lomonosov Moscow State University, Leninskie Gory, 1, 119991, Moscow (Russia) Abstract The structural features and synthetic procedures for three main classes of the fluorinated fullerene derivatives, C 60 R n , R=F, CF 2 , and CF 3 are discussed. The electronic properties of these compounds are largely determined by the number of addends and their attachment pattern. New approach with low-computational cost for predictions of the isomeric composition of fullerene polyadducts was developed and one was verified on the example of perfluoroalkylated fullerenes. Introduction Fullerenes and their derivatives have been proposed to be employed in the design of materials for a considerable variety of applications including nano- and molecular electronics (photovoltaic cells, optical limiters, organic light-emission diodes and field-effect transistors)(1- 3) and medicine (antioxidants, neuroprotective agents, antimicrobial agents, agents for photodynamic therapy and magnetic resonance imaging)(4). Their value in electronics stems from their spherically conjugated π-electron system that gives rise to n-type conductivity, low reduction potential and low reorganization energy in electron-transfer reactions. In addition, there is a broad selection of fullerene derivatives suitable for solution-based technology. However, there is still a need for improvement and fine tuning of their electronic properties, which requires development of the regioselective techniques of exohedral functionalization as well as skeletal transformation. Contrary to the anticipated enormous compositional and isomeric complexity to arise from concurrence of a multitude of reaction sites, these fullerene derivatives can be prepared as relatively simple mixtures of products or even as individual isomers. According to our extensive quantum chemical DFT surveys, the observed selectivity can be explained by both thermodynamic and kinetics factors. We found that the addition motifs of the initially attached groups controls the regioselectivity of further derivatization. Efficient and reliable methods to predict the structure of the diversely functionalized fullerene compounds are presented below.
-
Upload
nguyendang -
Category
Documents
-
view
218 -
download
0
Transcript of FLUORINATED FULLERENE DERIVATIVES: SYNTHESIS, STRUCTURE, AND ELECTRONIC ... files/papers/2-26... ·...

2-26
FLUORINATED FULLERENE DERIVATIVES: SYNTHESIS,
STRUCTURE, AND ELECTRONIC PROPERTIES
Alexey A. Goryunkov, Marina G. Apenova, Eugenia V. Borkovskaya, Victor A. Brotsman,
Nikita M. Belov, Ilya N. Ioffe, Vitaliy A. Ioutsi, Natalia S. Lukonina, Vitaliy Yu. Markov,
Alexey V. Rybalchenko, Olesya O. Semivrazhskaya
Chemistry Department, Lomonosov Moscow State University, Leninskie Gory, 1, 119991,
Moscow (Russia)
Abstract
The structural features and synthetic procedures for three main classes of the fluorinated
fullerene derivatives, C60Rn, R=F, CF2, and CF3 are discussed. The electronic properties of these
compounds are largely determined by the number of addends and their attachment pattern. New
approach with low-computational cost for predictions of the isomeric composition of fullerene
polyadducts was developed and one was verified on the example of perfluoroalkylated
fullerenes.
Introduction
Fullerenes and their derivatives have been proposed to be employed in the design of
materials for a considerable variety of applications including nano- and molecular electronics
(photovoltaic cells, optical limiters, organic light-emission diodes and field-effect transistors)(1-
3) and medicine (antioxidants, neuroprotective agents, antimicrobial agents, agents for
photodynamic therapy and magnetic resonance imaging)(4). Their value in electronics stems
from their spherically conjugated π-electron system that gives rise to n-type conductivity, low
reduction potential and low reorganization energy in electron-transfer reactions. In addition,
there is a broad selection of fullerene derivatives suitable for solution-based technology.
However, there is still a need for improvement and fine tuning of their electronic properties,
which requires development of the regioselective techniques of exohedral functionalization as
well as skeletal transformation.
Contrary to the anticipated enormous compositional and isomeric complexity to arise from
concurrence of a multitude of reaction sites, these fullerene derivatives can be prepared as
relatively simple mixtures of products or even as individual isomers. According to our extensive
quantum chemical DFT surveys, the observed selectivity can be explained by both
thermodynamic and kinetics factors. We found that the addition motifs of the initially attached
groups controls the regioselectivity of further derivatization. Efficient and reliable methods to
predict the structure of the diversely functionalized fullerene compounds are presented below.

2-27
Synthetic pathways, structural features and electronic properties of fluorinated fullerenes
derivatives
Enhancement of electron withdrawing
properties of a fullerene cage is readily and
efficiently achievable via derivatization with
fluorinated moieties. To date, three rather broad
and extensively explored classes of the fluorine-
containing fullerene derivatives are available.
These are fluorofullerenes (e.g. C60Fn, n=2–
48),(5-9) trifluoromethylfullerenes (C60(CF3)2n
and C70(CF3)2m,(10-12) n=1–9 and m=1–10),
and difluoromethylenofullerenes (C60(CF2)n,
C70(CF2)n, n=1–3).(13-15) Most of these
compounds exhibit enhancement the electron
withdrawing properties and processability due
to high solubilities in the common organic
solvents and capability to sublime without
decomposition (except for
difluoromethylenofullerenes).
The methods for the synthesis of polyfluorinated derivatives of fullerenes can be divided
into three groups: (i) fluorination of fullerene by molecular fluorine, (ii) fluorination by fluorides
of transitional metals in high oxidation states and (iii) fluorination of chloro- or bromofullerenes.
(16, 17) Fluorination of C60 with molecular fluorine yields two D3- and S6-symmetrical isomers
C60F48,(7) whereas heating of the C60 with MnF3 under the dynamic vacuum conditions leads to
mixture of the three T-, C3-, and C1-symmetrical isomers C60F36.(9) Low fluorinated fullerenes
C60Fn, n=2, 4, 6, 18, and 20 were prepared in the reaction of the C60 with complex fluorides like
K2PtF6 and KMnF4 during heating under the dynamic vacuum conditions.(5, 6, 8, 18, 19)
According to X-Ray single crystal and 19
F NMR data the addition of the fluorine atoms occurs at
C-C double bonds with formation of the continues patterns of ortho-attached atoms (see Fig 1).
High fluorination degree of the fullerenes increases the electron affinities (EA) of the
compounds. So, measured EA values for C60F2 and C60F48 are 2.74(7)(20) and 4.06(3)(21) eV,
whereas EA of C60 is 2.683(8) eV.(22) The same trend was demonstrated by electrochemical
studies of fluorofullerenes.(23-25) The cathodic peaks (Epc) of the first reduction potentials of
Fig. 1. Schlegel diagrams of fluorofullerenes
C60Fn, n=2, 4, 6, 18, 36, and 48 (a–f).

2-28
C60F2, C60F4, C60F18, C60F36, and C60F48 are anodic shifted relatively Epc of C600/–
couple for
values of 0.04, 0.08, 0.26, 0.63, and 1.38 V, respectively. The electron transfer occurs
electrochemically and chemically irreversible: defluorination of the compounds is observed
during reduction. Therefore applicability of the fluorofullerenes for doping of semiconducting
polymers is restricted by their degradability during electron transfer.
There are about 70 structurally characterized trifluoromethylfullernes, C60(CF3)2n and
C70(CF3)2m, n=1–9 and m=1–10.(10-12) The available synthetic techniques make it possible to
control the average number of addends. Thus, when reacting C60 or C70 with silver(I)
trifluoroacetate under the dynamic vacuum conditions, the number of the CF3 addends may be
varied from a minimum of two up to maximum of 20–22.(26, 27) Trifluoromethylation under the
atmosphere of CF3I allows formation of molecules with intermediate to high degrees of addition.
In particular, the reaction with CF3I in a sealed ampoule shifts the composition toward higher
functionalized derivatives C60/70(CF3)n, n = 12–20,(28, 29) while in a flow of CF3I, which
conditions enable rapid removal of the products from the reaction zone, one generally obtains
C60/70(CF3)n compounds within the range of n = 6–14.(30, 31) Further, lower functionalized
trifluoromethylated fullerenes can be prepared via thermal treatment of the higher functionalized
ones. For example, sublimation of products of the fullerene/CF3COOAg reaction (n = 2–20)(10,
27, 32) or heating the C60/70(CF3)n, n = 2–20, in a mixture with pristine fullerene(33-35)
facilitates conversion into lower CF3-derivatives.
Attachment moieties for several trifluoromethylfullerenes are shown on Fig 2. One can
see, attachment moieties form a ribbon of edge-sharing para-C6(CF3)2 and meta-C6(CF3)2
hexagons. Compact arrangement of the groups is sterically prohibited because of bulkiness of
CF3 groups, that leads to a greater isomeric variety of trifluoromethylfullerenes in comparison
with fluorofullerenes. It is important, that attachment patterns dictates the electronic withdraw
properties of the trifluoromethylfullerenes. For example, family of C70(CF3)n demonstrates
variability of the first reduction potentials within range from –0.11 to 0.34 V vs C700/–
.(36) At
contrary to fluorofullerenes, the reduction of vast majority of trifluoromethylfullerenes is
electrochemical and chemical reversible. However, variation of the reduction potentials is
counterintuitive. So, compound with 10 CF3 groups, C70(CF3)10 (Fig 2b, 1070-I), is weaker
oxidizing agent comparatively to pristine C70 (Epc –0.11 V vs C700/–
couple), whereas dramatic
anodic shift of the reduction potential (0.34 V vs C700/–
couple) is observed for C2-symmetrical
C70(CF3)8 (870-II). Isomeric Cs-symmetrical C70(CF3)8 (870-I) differs position of only one CF3
group. But it results in cathodic shift of the reduction potential for 0.3 V (0.04 V vs C700/–
couple). This behavior is correlated with DFT estimated LUMO energies.(36)

2-29
Fig. 2. Schlegel diagrams of trifluoromethylfullerenes (a) C60(CF3)n, n=2–8, and (b) C70(CF3)m,
m=2–10.
Thus, a broad family of trifluoromethylated fullerenes bearing from 2 to 20 CF3 groups
per fullerene cage and manifold addition patterns enlarges the library of the fullerene-based
compounds with “tuneable” electronic properties and attracts attention as prospective building
blocks for organic electronics useful as electron acceptors due to chemical stability during
electron transfer.
Third class of the fluorinated fullerenes
derivatives is difluoromethylene fullerenes. These
compounds are prepared in the reaction of the
fullerene with :CF2 carbene generated in situ by
thermolysis of alkaline difluorochloroacetates.(13-15)
Insertion of difluoromethylene fragment into a [6,6]-
bond leads to cleavage of the said bond with
associated redistribution of the π-electron bonding in
the cage (see Fig 3a). The resulting so-called “[6,6]-
open” adducts contain 1,6-
difluoromethano[10]annulene moeity where the sp2
hybridization of the bridgehead carbon atoms is
retained and, accordingly, all carbon atoms of the
parent fullerene remain within the spherical π-
system, though its connectivity slightly decreases.
Fig. 3. Molecular structures of C60(CF2) (a),
cis-2-C60(CF2)2 (b), and isomers C70(CF2)-I
and -II (c, d).
2-30
These compounds with open [6,6] C–C bond (homofullerenes) are quite unusual among fullerene
exohedral derivatives. Though still very little is known about the reactivity of homofullerenes, it
is clear that the preservation of the spherical π-system of fullerenes with all carbon atoms
remaining sp2 hybridized makes homofullerenes more sensitive than methanofullerenes to any
sort of further functionalization. Our recent electrochemical investigation of the [6,6]-open
C60(CF2) and C70(CF2) demonstrated that the electron affinity of this compound is well above the
values obtained for the other known C60 adducts with one bivalent or two monovalent
addends.(14, 15)
Genetic protocol for identification of favorable isomers of trifluoromethylfullerenes
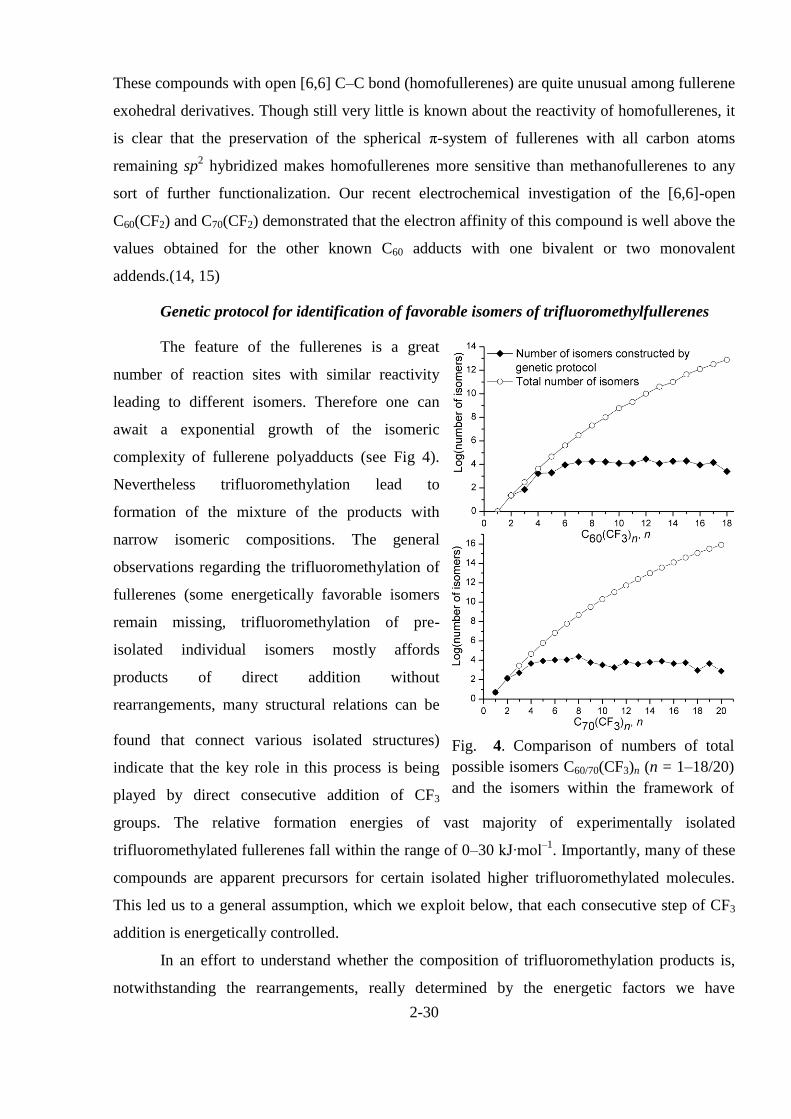
The feature of the fullerenes is a great
number of reaction sites with similar reactivity
leading to different isomers. Therefore one can
await a exponential growth of the isomeric
complexity of fullerene polyadducts (see Fig 4).
Nevertheless trifluoromethylation lead to
formation of the mixture of the products with
narrow isomeric compositions. The general
observations regarding the trifluoromethylation of
fullerenes (some energetically favorable isomers
remain missing, trifluoromethylation of pre-
isolated individual isomers mostly affords
products of direct addition without
rearrangements, many structural relations can be
found that connect various isolated structures)
indicate that the key role in this process is being
played by direct consecutive addition of CF3
groups. The relative formation energies of vast majority of experimentally isolated
trifluoromethylated fullerenes fall within the range of 0–30 kJ∙mol–1
. Importantly, many of these
compounds are apparent precursors for certain isolated higher trifluoromethylated molecules.
This led us to a general assumption, which we exploit below, that each consecutive step of CF3
addition is energetically controlled.
In an effort to understand whether the composition of trifluoromethylation products is,
notwithstanding the rearrangements, really determined by the energetic factors we have
Fig. 4. Comparison of numbers of total
possible isomers C60/70(CF3)n (n = 1–18/20)
and the isomers within the framework of
our kinetic model.

2-31
performed massive quantum chemical calculations on the formation energies of the possible
molecular and radical products of the consecutive CF3 addition to fullerene. In order to decrease
the number considered structures, the following genetic algorithm was used. On the first step, we
considered the complete sets of the isomeric C60(CF3)2 bisadducts and C70(CF3)• radicals at the
semi-empirical AM1 level. Then, the isomers that fell within a predetermined range of relative
AM1 energy 0–ΔE (50–80 kJ∙mol–1
to ensure completeness of our survey) were used as
precursors, and all possible closed-shell C60/70(CF3)2 products of second addition to any of them
were generated. The same procedure of energy-based selection and subsequent addition was then
repeated for each consecutive passage from C60/70(CF3)k to C60/70(CF3)k+1 up to 18 and 20 CF3
groups added per C60 and C70 cages, respectively. Finally, the molecular geometry and the
relative energies of the above obtained sets of C60/70(CF3)n isomers with relative energy within
20–90 kJ∙mol–1
AM1 energy gap were refined at the DFT level of theory. It turns out that the
number of structures under consideration rapidly becomes dramatically lower than the total
number of possible isomers, as demonstrated in Fig. 4. The number of structures considered at
the AM1 level did not exceed 3·104 for any particular degree of addition, which made it possible
to accomplish these preliminary semiempirical calculations with the use of several conventional
PCs.
The above approach appears to be highly predictive for both C60 and C70 as it captured all
the experimentally isolated C60/70(CF3)n isomers with n up to 18/20, even though it ignores the
possibility of isomerization. On the other hand, it does not provide any means to filter off those
stable isomers that were not found to form (or to accumulate). A useful hint comes from a
consideration that our procedure missed none of the synthesized compounds despite there might
exist some stable structures that have no sufficiently stable precursors. This suggests that some
energy-based criteria act on each consecutive stage and, accordingly, they must be carefully
considered stepwise rather than globally.
Kinetic model based on the BEP relations
The selectivity in formation of particular stable isomers of C60/70(CF3)n can be
rationalized using the Bell-Evans-Polanyi (BEP) principle. The BEP model states that the
activation barriers (and, consequently, the rates) for a given type of processes must correlate with
their enthalpies. When applied to our case of sequential trifluoromethylation, this means that (i)
CF3 addition to a given structure will follow the most exothermic routes and (ii) accumulation of
each particular isomer will depend on exothermicity of its formation and of its further
trifluoromethylation. Perhaps, more important should be the slower stages of addition to closed-

2-32
shell molecules yielding radicals with odd number of addends while their subsequent
recombination with yet another CF3 group will proceed much faster.
Fig. 5. Most energetically favorable pathways from C70 to C70(CF3)2. Hereinafter we indicate the
corresponding enthalpies of CF3 addition (kJ∙mol–1
, DFT), numberation is given in the order of
relative DFT energy (shown in parentheses, kJ·mol–1
). Roman numerals indicate the
experimentally obtained compounds. The dotted arrows indicate lower-rate processes.
As an introductory example, we consider consecutive addition of two CF3 groups to
fullerene C70 (see Fig. 5). There are five nonequivalent carbon atoms in the D5h-C70 cage, so one
can obtain five different radical species ·C70CF3. Four addition enthalpies that correspond to the
structures 1170
–1470
are almost indistinguishable while the fifth alternative turns out to be
significantly less favorable. Thus, according to the BEP principle, radical intermediate 1570
can
be discarded. The addition of further CF3 group to the intermediates 1170
–1470
gives 136 possible
structures C70(CF3)2; however, only those designated 2170
–2570
fall within a range of 30 kJ·mol–1
.
Furthermore, clearly preferable is formation of 2170
, 2270
, 2370
. Indeed, synthetically available are
only 2170
and 2270
also known in the literature as 270
-II and 270
-I, respectively. The third one,
2370
, shows higher reactivity towards further addition and its motif can be found in many higher
trifluoromethylated derivatives of C70. Such considerations make it possible to track the whole
sequence of structures up to C70(CF3)10 consisting of isomers with relative energy less then 30
kJ·mol–1
.
Consecutive trifluoromethylation of C60 can be rationalized on the same basis as above.
In fig. 6 we exemplify this with the most likely kinetic pathways between C60 and C60(CF3)4.
While addition of the first CF3 to C60 produces only one possible radical intermediate, C60(CF3)2
already has 23 possible isomers. However, the para- or 1,7-isomer finds no competition: the next

2-33
stable 1,9-C60(CF3)2 (with ortho-addition) is founds 33 kJ·mol–1
higher, and 1,7-C60(CF3)2 is the
only experimentally available product with two CF3 groups.
Fig. 6. Major pathways of the consecutive trifluoromethylation of fullerene C60. The
corresponding enthalpies of ·CF3 addition as well as relative energies of isomers (values in
parentheses) are indicated (kJ∙mol–1
, DFT). Roman numerals indicate the experimentally
obtained compounds. The dotted arrows indicate lower-rate processes.
The course of further addition is illustrated on Fig. 6. The most energetically preferable
addition of the third CF3 group occurs in the para position, resulting in C60(CF3)3· 31 radical.
Formation of the next by energy isomers 32–36 are less preferable for 13–20 kJ·mol–1
.
Nevertheless attachment of the forth CF3 group to these isomers lead to experimental found
isomers of C60(CF3)4 4-II, 4-III, and 4-I (latter one was observed as an oxide, C60(CF3)4O,
because of high reactivity of fulvene moiety). According to BEP principle, the rate limiting stage
is formation of C60(CF3)3· radical species again, whereas addition of the fourth CF3 group will be
easily occurs.
In discussing the highest degrees of addition, 18 CF3 groups, the key role direct
consecutive addition of CF3 groups should be used with caution and the isomerization processes
cannot be neglected at these stages. First of all there are the direct experimental data of the
rearrangement of C70(CF3)12 isomers at trifluoromethylation.(37) Possible pathways of the
formation of C3v-C60(CF3)18 provided direct addition CF3 groups are given on fig. 7. In the
presented cases the isomers with high relative energy (up to 52 kJ·mol–1
) can not be excluded

2-34
from the sequence of structures up to abovementioned compounds. Such isomers never were
found in experiments and their absents give additional proof in favor of rearrangements.
Theoretical consideration states that the activation energy of the CF3 rearrangements became less
when the energy difference of isomers increases and can be considered as a reason of elimination
isomers with high relative energies.(38, 39)
Fig. 7. Possible pathways of the formation of C3v-C60(CF3)18 not involving rearrangements of
CF3 groups. The reaction enthalpies and the relative energies (kJ·mol-1
, DFT) are shown.
Conclusions
The richly branched process of fullerene trifluoromethylation requires careful analysis in
order to achieve good quantitative predictability. However, the energetic correlations in kinetic
behavior simplify the task. We found that the purely thermodynamic model affords at least
satisfactory prediction of the results even for evidently non-equilibrium syntheses. Still, such
simplified description is imperfect, and the most problematic aspect is hindered formation of
certain kinetically inaccessible isomers or, on the contrary, fast conversion of certain structures
that are kinetically labile towards further trifluoromethylation. These special cases can be
identified by means of detailed kinetic modeling based on the BEP approach that includes
stepwise energetic analysis of the possible trifluoromethylation sequences. The BEP survey
makes it possible to explain the discrepancies of thermodynamic modeling and the experimental
results. To eliminate such discrepancies in practice, one can employ additional post-annealing of
the trifluoromethylated products, which brings the trifluoromethylated fullerene mixtures closer
to equilibrium and promotes formation of otherwise inaccessible stable isomers, possibly
through rearrangements in the shells of CF3 addends.

2-35
The modeling approaches developed in this work are quite general and may be thus
applied to other processes of stepwise functionalization of fullerenes. However, necessary
alterations are to be introduced in accordance with the reaction mechanisms (e.g. nucleophilic
rather than radical) and the nature of the intermediates.
Experimental
Initially, molecular geometry of the isomers was optimized by TINKER 4.2 molecular
mechanic package with MM2 parameter sets.(40) Further, preliminary geometry optimization for
these molecules was carried out at the AM1 level of theoryPreliminary geometry optimizations
were carried out sequentelly by molecular mechanics with MM2 force field with at the AM1
level of theory with the use of Firefly QC 8.0 package(41) partially based on the GAMESS (US)
software.(42) Single point DFT (spDFT) calculations of the isomers within the AM1 energy
range of 20–90 kJ∙mol–1
, and final optimization of those of them that fell within the spDFT
energy range of 20–50 kJ∙mol–1
were performed with the use of PRIRODA package.(43) The
PBE exchange-correlation functional(44) and the built-in TZ2P-quality basis set were used.
References
1. J. L. Delgado, P. Bouit, S. Filippone, M. A. Herranz, N. Martin, "Organic photovoltaics: a chemical
approach", Chem Commun 2010, 46, 4853–4865.
2. A. J. Ferguson, J. L. Blackburn, N. Kopidakis, "Fullerenes and carbon nanotubes as acceptor materials in
organic photovoltaics", Materials Lett 2013, 90, 115–125.
3. R. Meerheim, S. Olthof, M. Hermenau, S. Scholz, A. Petrich, N. Tessler, O. Solomeshch, B. Lüssem, M.
Riede, K. Leo, "Investigation of C60F36 as low-volatility p-dopant in organic optoelectronic devices", J. Appl. Phys.
2011, 109, 103102.
4. P. Anilkumar, F. Lu, L. Cao, P. G. Luo, J. Liu, S. Sahu, K. N. Tackett II, Y. Wang, Y. Sun, "Fullerenes for
applications in biology and medicine", Current Medicinal Chemistry 2011, 18, 2045–2059.
5. O. V. Boltalina, A. Y. Lukonin, J. M. Street, R. Taylor, "C60F2 exists!", Chem. Commun. 2000, 17, 1601-
1602.
6. O. V. Boltalina, A. D. Darwish, J. M. Street, R. Taylor, X. W. Wei, "Isolation and characterisation of
C60F4, C60F6, C60F8, C60F7CF3 and C60F2O, the smallest oxahomofullerene; the mechanism of fluorine addition to
fullerenes", J. Chem. Soc., Perkin Trans. 2 2002, 251-256.
7. A. A. Gakh, A. A. Tuinman, J. L. Adcock, R. A. Sachleben, R. N. Compton, "Selective synthesis and
structure determination of C60F48", J. Am. Chem. Soc. 1994, 116, 819-820.
8. O. V. Boltalina, V. Y. Markov, R. Taylor, M. P. Waugh, "Preparation and characterisation of C60F18",
Chem. Commun. 1996, 22, 2549-2550.
9. O. V. Boltalina, A. Y. Borschevskiy, L. N. Sidorov, J. M. Street, R. Taylor, "Preparation of C60F36 and
C70F36/38/40", Chem. Commun. 1996, 4, 529-530.
10. E. I. Dorozhkin, A. A. Goryunkov, I. N. Ioffe, S. M. Avdoshenko, V. Y. Markov, N. B. Tamm, D. V.
Ignat'eva, L. N. Sidorov, S. I. Troyanov, "Synthesis, structure, and theoretical study of lower trifluoromethyl
derivatives of [60]fullerene", Eur. J. Org. Chem. 2007, 5082–5094.
11. E. I. Dorozhkin, D. V. Ignat'eva, N. B. Tamm, A. A. Goryunkov, P. A. Khavrel, I. N. Ioffe, A. A. Popov, I.
V. Kuvychko, A. V. Streletskiy, V. Y. Markov, J. Spandal, S. H. Strauss, O. V. Boltalina, "Synthesis,
characterization, and theoretical study of stable isomers of C70(CF3)n (n = 2, 4, 6, 8, 10)", Chem. Eur. J. 2006, 12,
3876–3889.
12. D. V. Ignat’eva, A. A. Goryunkov, I. N. Ioffe, L. N. Sidorov, "Trifluoromethylation of fullerenes: kinetic
and thermodynamic control", J. Phys. Chem. A 2013, 117, 13009–13017.
13. A. S. Pimenova, A. A. Kozlov, A. A. Goryunkov, V. Y. Markov, P. A. Khavrel, S. M. Avdoshenko, I. N.
Ioffe, S. G. Sakharov, S. I. Troyanov, L. N. Sidorov, "Synthesis and characterization of difluoromethylene-
homo[60]fullerene, C60(CF2)", Chem. Commun. 2007, 374 - 376.

2-36
14. A. A. Goryunkov, E. S. Kornienko, T. V. Magdesieva, A. A. Kozlov, V. A. Vorobiev, S. M. Avdoshenko,
I. N. Ioffe, O. M. Nikitin, V. Y. Markov, P. A. Khavrel, A. K. Vorobiev, L. N. Sidorov, "Electrochemical, esr and
theoretical studies of [6,6]-opened C60(CF2), cis-2-C60(CF2)2 and their anions", Dalton Trans 2008, 6886-6893.
15. N. A. Samoylova, N. M. Belov, V. A. Brotsman, I. N. Ioffe, N. S. Lukonina, V. Y. Markov, A. Ruff, A. V.
Rybalchenko, P. Schuler, O. O. Semivrazhskaya, B. Speiser, S. I. Troyanov, T. V. Magdesieva, A. A. Goryunkov,
"[6,6]-open and [6,6]-closed isomers of C70(CF2): synthesis, electrochemical and quantum chemical investigation",
Chem. Eur. J. 2013, 19, 17969–17979.
16. A. A. Goryunkov, N. S. Ovchinnikova, I. V. Trushkov, M. A. Yurovskaya, "Synthesis, structures and
reactivity of polyhalo[60]fullerenes", Russ. Chem. Rev. 2007, 76, 289-312.
17. R. Taylor, "Why fluorinate fullerenes?", J. Fluorine Chem. 2004, 125, 359-368.
18. A. A. Goryunkov, I. N. Ioffe, P. A. Khavrel, S. M. Avdoshenko, V. Y. Markov, Z. Mazej, L. N. Sidorov, S.
I. Troyanov, "The former “C60F16” is actually a double-caged adduct: (C60F16)(C60)", Chem. Commun. 2007, 704–
706.
19. N. B. Shustova, Z. Mazej, Y. Chen, A. A. Popov, S. H. Strauss, O. V. Boltalina, "Saturnene revealed: x-ray
crystal structure of D5d-C60F20 formed in reactions of C60 with axmfy fluorinating agents (a=alkali metal; m=3d
metal)", Angew. Chem. Int. J. 2010, 49, 812–815.
20. O. V. Boltalina, L. N. Sidorov, E. V. Sukhanova, I. D. Sorokin, "Observation of difluorinated higher
fullerene anions by Knudsen cell mass spectrometry and determination of electron affinities of C60F2 and C70F2",
Chem. Phys. Lett. 1994, 230, 567-570.
21. C. Jin, R. Hettich, R. Compton, A. Tuinman, A. Derecskei-Kovacs, D. Marynick, B. Dunlap, "Attachment
of two electrons to C60F48: coulomb barriers in doubly charged anions", Phys. Rev. Lett. 1994, 73, 2821-2824.
22. X. Wang, H. Woo, L. Wang, "Vibrational cooling in a cold ion trap: vibrationally resolved photoelectron
spectroscopy of cold C60− anions", J. Phys. Chem. 2005, 123, 051106.
23. F. Zhou, G. J. Van Berkel, B. T. Donovan, "Electron-transfer reaction of C60F48", J. Am. Chem. Soc. 1994,
116, 5485-5486.
24. A. A. Popov, J. Tarábek, I. E. Kareev, S. F. Lebedkin, S. H. Strauss, O. V. Boltalina, L. Dunsch,
"Poly(trifluoromethyl)fullerene radical anions. an ESR/Vis-NIR spectroelectrochemical study of C60F2,4 and
C60(CF3)2,10", J. Phys. Chem. A 2005, 109, 9709 - 9711.
25. D. Paolucci, F. Paolucci, M. Marcaccio, M. Carano, R. Taylor, "Electrochemistry of perfluorinated
fullerenes: the case of three isomers of C60F36", Chem. Phys. Lett. 2004, 400, 389-393.
26. A. D. Darwish, A. G. Avent, A. K. Abdul-Sada, R. Taylor, "[60]- and [70]fullerenes are
trifluoromethylated across 5:6-bonds", Chem. Commun. 2003, 1374-1375.
27. A. A. Goryunkov, I. V. Kuvychko, I. N. Ioffe, D. L. Dick, L. N. Sidorov, S. H. Strauss, O. V. Boltalina,
"Isolation of C60(CF3)n (n = 2, 4, 6, 8, 10) with high compositional purity", J. Fluorine Chem. 2003, 124, 61–64.
28. N. A. Samokhvalova, P. A. Khavrel, V. Y. Markov, P. S. Samokhvalov, A. A. Goruynkov, E. Kemnitz, L.
N. Sidorov, S. I. Troyanov, "Isolation and structural characterization of the most stable, highly symmetric isomer of
C60(CF3)18", Eur. J. Org. Chem. 2009, 2935–2938.
29. D. V. Ignat'eva, A. A. Goryunkov, N. B. Tamm, I. N. Ioffe, L. N. Sidorov, S. I. Troyanov, "Isolation and
structural characterization of the most highly trifluoromethylated C70 fullerenes: C70(CF3)18 and C70(CF3)20", New J.
Chem. 2013, 299-302.
30. I. E. Kareev, N. B. Shustova, I. V. Kuvychko, S. F. Lebedkin, S. M. Miller, O. P. Anderson, A. A. Popov,
S. H. Strauss, O. V. Boltalina, "Thermally stable perfluoroalkylfullerenes with the skew-pentagonal-pyramid
pattern: C60(C2F5)4O, C60(CF3)4O, and C60(CF3)6", J. Am. Chem. Soc. 2006, 128, 12268-12280.
31. I. E. Kareev, I. V. Kuvychko, A. A. Popov, S. F. Lebedkin, S. M. Miller, O. P. Anderson, S. H. Strauss, O.
V. Boltalina, "High-temperature synthesis of the surprisingly stable C1-C70(CF3)10 isomer with a para7–meta–para
ribbon of nine C6(CF3)2 edge-sharing hexagons", Angew. Chem. Int. Ed. 2005, 44, 7984–7987.
32. A. A. Goryunkov, I. N. Ioffe, I. V. Kuvychko, T. S. Yankova, V. Y. Markov, A. V. Streletskii, D. L. Dick,
L. N. Sidorov, O. V. Boltalina, "Trifluoromethylated [60]fullerenes: synthesis and characterization", Fullerenes,
Nanotubes, Carbon Nanostr. 2004, 12, 181-185.
33. T. Mutig, I. N. Ioffe, E. Kemnitz, S. I. Troyanov, "Crystal and molecular structures of C2-C70(CF3)8·1.5
PhMe", Mendeleev Commun. 2008, 18, 73–75.
34. T. Mutig, E. Kemnitz, S. I. Troyanov, "Trifluoromethyl derivatives of fullerene C70, C70(CF3)2, C70(CF3)8
and C70(CF3)14", Mendeleev Commun. 2009, 19, 30–31.
35. N. M. Belov, M. G. Apenova, A. V. Rybalchenko, E. V. Borkovskaya, N. S. Lukoniona, A. A. Goryunkov,
I. N. Ioffe, S. I. Troyanov, L. N. Sidorov, "Transalkylation of higher trifluoromethylated fullerenes with C70: a
pathway to new addition patterns of C70(CF3)8", Chem. Eur. J. 2014, 20, 1126–113.
36. A. A. Popov, I. E. Kareev, N. B. Shustova, S. F. Lebedkin, S. H. Strauss, O. V. Boltalina, L. Dunsch,
"Synthesis, spectroscopic and electrochemical characterization, and DFT study of seventeen C70(CF3)n derivatives
(n=2, 4, 6, 8, 10, 12)", Chem. Eur. J. 2008, 14, 107–121.

2-37
37. D. V. Ignat'eva, T. Mutig, A. A. Goryunkov, N. B. Tamm, E. Kemnitz, S. I. Troyanov, L. N. Sidorov, "New
C70(CF3)n isomers (n = 12, 14, 16). realkylation and addend rearrangements", Russ. Chem. Bull. 2009, 58, 1146–
1154.
38. N. A. Romanova, T. S. Papina, V. A. Luk'yanova, A. G. Buyanovskaya, R. M. Varuschenko, A. I.
Druzhinina, A. A. Goryunkov, V. Y. Markov, R. A. Panin, L. N. Sidorov, "S6 isomer of C60(CF3)12: synthesis,
properties and thermodynamic functions", J. Chem. Thermodyn. 2013, 66, 59–64.
39. S. M. Avdoshenko, I. N. Ioffe, L. N. Sidorov, "Theoretical study of isomerization mechanisms in
fluorinated fullerene derivatives", J. Phys. Chem. A 2009, 113 (40), 10833-10838.
40. J. W. Pownder, F. M. Richards, "An efficient newton-like method for molecular mechanics energy
minimization of large molecules", J. Comput. Chem. 1987, 8, 1016-1024.
41. Firefly (formerly pc gamess). http://classic.chem.msu.su/gran/gamess/index.html.
42. M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. J. Jensen, S. Koseki, N.
Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis, J. A. Montgomery, "General atomic and molecular
electronic structure system", J. Comput. Chem. 1993, 14, 1347–1363.
43. D. N. Laikov, "Fast evaluation of density functional exchange-correlation terms using the expansion of the
electron density in auxiliary basis sets", Chem. Phys. Lett. 1997, 281, 151–156.
44. J. P. Perdew, K. Burke, M. Ernzerhof, "Generalized gradient approximation made simple", Phys. Rev. Lett.
1996, 77, 3865–3868.



![Research Article The Activity of [60]Fullerene Derivatives …downloads.hindawi.com/journals/jnm/2014/907435.pdf · · 2015-11-22We report a comparative investigation of the antibacterial](https://static.fdocuments.us/doc/165x107/5aee20247f8b9a662590f06b/research-article-the-activity-of-60fullerene-derivatives-report-a-comparative.jpg)














