
Evidence Project Final Report -...
13
EVID4 Evidence Project Final Report (Rev. 06/11) Page 1 of 13 General Enquiries on the form should be made to: Defra, Procurements and Commercial Function (Evidence Procurement Team) E-mail: [email protected] Evidence Project Final Report Note In line with the Freedom of Information Act 2000, Defra aims to place the results of its completed research projects in the public domain wherever possible. The Evidence Project Final Report is designed to capture the information on the results and outputs of Defra-funded research in a format that is easily publishable through the Defra website An Evidence Project Final Report must be completed for all projects. This form is in Word format and the boxes may be expanded, as appropriate. ACCESS TO INFORMATION The information collected on this form will be stored electronically and may be sent to any part of Defra, or to individual researchers or organisations outside Defra for the purposes of reviewing the project. Defra may also disclose the information to any outside organisation acting as an agent authorised by Defra to process final research reports on its behalf. Defra intends to publish this form on its website, unless there are strong reasons not to, which fully comply with exemptions under the Environmental Information Regulations or the Freedom of Information Act 2000. Defra may be required to release information, including personal data and commercial information, on request under the Environmental Information Regulations or the Freedom of Information Act 2000. However, Defra will not permit any unwarranted breach of confidentiality or act in contravention of its obligations under the Data Protection Act 1998. Defra or its appointed agents may use the name, address or other details on your form to contact you in connection with occasional customer research aimed at improving the processes through which Defra works with its contractors. Project identification 1. Defra Project code SE0786 2. Project title Investigate virulence correlates of NDV with respect to refining current in vitro tests 3. Contractor organisation(s) Animal Health and Veterinary Laboratories Agency Woodham Lane New Haw Addlestone Surrey KT15 3NB 4. Total Defra project costs £657,864 (agreed fixed price) 5. Project: start date ................ 01/04/2007 end date ................. 30/09/2013
-
Upload
truongthuan -
Category
Documents
-
view
218 -
download
0
Transcript of Evidence Project Final Report -...

EVID4 Evidence Project Final Report (Rev. 06/11) Page 1 of 13
General Enquiries on the form should be made to:
Defra, Procurements and Commercial Function (Evidence Procurement Team) E-mail: [email protected]
Evidence Project Final Report
Note
In line with the Freedom of Information Act 2000, Defra aims to place the results of its completed research projects in the public domain wherever possible. The Evidence Project Final Report is designed to capture the information on the results and outputs of Defra-funded research in a format that is easily publishable through the Defra website An Evidence Project Final Report must be completed for all projects.
This form is in Word format and the boxes may be expanded, as appropriate.
ACCESS TO INFORMATION
The information collected on this form will be stored electronically and may be sent to any part of Defra, or to individual researchers or organisations outside Defra for the purposes of reviewing the project. Defra may also disclose the information to any outside organisation acting as an agent authorised by Defra to process final research reports on its behalf. Defra intends to publish this form on its website, unless there are strong reasons not to, which fully comply with exemptions under the Environmental Information Regulations or the Freedom of Information Act 2000.
Defra may be required to release information, including personal data and commercial information, on request under the Environmental Information Regulations or the Freedom of Information Act 2000. However, Defra will not permit any unwarranted breach of confidentiality or act in contravention of its obligations under the Data Protection Act 1998. Defra or its appointed agents may use the name, address or other details on your form to contact you in connection with occasional customer research aimed at improving the processes through which Defra works with its contractors.
Project identification
1. Defra Project code SE0786
2. Project title
Investigate virulence correlates of NDV with respect to refining current in vitro tests
3. Contractor organisation(s)
Animal Health and Veterinary Laboratories Agency
Woodham Lane
New Haw
Addlestone
Surrey KT15 3NB
54. Total Defra project costs £657,864
(agreed fixed price)
5. Project: start date ................ 01/04/2007
end date ................. 30/09/2013

EVID4 Evidence Project Final Report (Rev. 06/11) Page 2 of 13
6. It is Defra’s intention to publish this form.
Please confirm your agreement to do so. ................................................................................... YES NO
(a) When preparing Evidence Project Final Reports contractors should bear in mind that Defra intends that they be made public. They should be written in a clear and concise manner and represent a full account of the research project which someone not closely associated with the project can follow.
Defra recognises that in a small minority of cases there may be information, such as intellectual property or commercially confidential data, used in or generated by the research project, which should not be disclosed. In these cases, such information should be detailed in a separate annex (not to be published) so that the Evidence Project Final Report can be placed in the public domain. Where it is impossible to complete the Final Report without including references to any sensitive or confidential data, the information should be included and section (b) completed. NB: only in exceptional circumstances will Defra expect contractors to give a "No" answer.
In all cases, reasons for withholding information must be fully in line with exemptions under the Environmental Information Regulations or the Freedom of Information Act 2000.
(b) If you have answered NO, please explain why the Final report should not be released into public domain
Executive Summary
7. The executive summary must not exceed 2 sides in total of A4 and should be understandable to the intelligent non-scientist. It should cover the main objectives, methods and findings of the research, together with any other significant events and options for new work.
Newcastle disease (ND), caused by virulent avian paramyxovirus type 1 (APMV-1) strains is regarded throughout the world as one of the most important diseases of poultry. This is not only due to the serious animal health and welfare impacts, including high flock mortality that may result from some ND virus (NDV) infections, but also due to the economic impacts due to trading restrictions and embargoes placed on areas and countries where ND outbreaks have occurred. NDV remains enzootic in many areas of the world and continues to cause sporadic outbreaks of disease in countries where it is considered to be exotic, including the UK (Aldous et al 2007; Irvine et al 2009). Disease presentation in infected birds varies widely, ranging from asymptomatic infections to devastating disease with very high mortality. The variation observed in clinical signs is influenced by a number of factors including the strain of the virus, the species, condition and health status of the host and environmental conditions. The NDV fusion (F) protein cleavage site amino acid motif has been demonstrated to be the principle determinant of virus virulence. However, considerable variation in disease signs may be seen where this motif is similar or identical and other viral properties (genotype) may also influence the observed clinical signs (phenotype). The aims and key outcomes of this project were as follows:
1. Validation of APMV-1 F gene real time RT-PCR (RRT-PCR) pathotyping assay for the simultaneous detection and rapid differentiation of avirulent APMV-1 and virulent ND viruses.
2. Develop and utilise Reverse Genetics (RG) and full genome sequencing capacity and capability at AHVLA Weybridge to investigate and identify specific viral properties of APMV-1 strains that demonstrate a discordant genotype (based on predicted virulence characteristics from the F gene cleavage site amino acid motif) and phenotype (observed pathogenicity in vivo).
The AMPV-1 F gene RRT-PCR pathotyping assay is based on two probes, both designed to hybridise specifically with the fusion gene cleavage site of either virulent or avirulent APMV-1 viruses (Fuller et al 2009). As a result of this project the F gene pathotyping assay is routinely used as part of the UKAS-accredited avian notifiable disease (AND) diagnostic test portfolio at AHVLA Weybridge. This provides a molecular capability to determine the pathotype of APMV-1 viruses more rapidly than the conventional method of virus isolation (VI) and intra-cerebral pathogenicity index (ICPI) test. Hence, the F gene assay supports statutory disease control decision-making and risk management, enabling faster and proportionate implementation of restriction zones in the event of a confirmed ND outbreak in Great Britain. The F gene RRT-PCR can be used on clinical samples collected as part of statutory and report case investigations and directly complements the initial AMPV-1 L gene screening RRT-PCR assay and

EVID4 Evidence Project Final Report (Rev. 06/11) Page 3 of 13
conventional ‘gold standard’ VI and ICPI tests, as described in the relevant internationally-recognised standards laid down by the EU and OIE (CEU, 1992; OIE, 2012). The utility of the RG system has been demonstrated successfully, including the generation, rescue and in vivo evaluation of RG APMV-1 viruses with identified putative virulence markers (Dortmans et al 2010). More specifically, the variability in pathogenicity of two pigeon paramyxoviruses was studied by generating clones of altered pathogenicity through the passage of the parental viruses in SPF embryonated fowls’ eggs or in 6-week-old chickens. Several mutations were identified within these clones. These viruses were generated by RG technologies to study the effect of these individual mutations on pathogenicity. Interestingly, a change in a proline residue within the fusion protein for these viruses was responsible for the increase in pathogenicity. One of these mutations in the Fusion protein - S475P - was shown to affect the replication rate of these viruses in multicycle growth curves (i.e. those viruses with lower pathogenicity scores carrying this mutation would have a lower replication rate). Through the development and routine implementation of the RG and full genome sequencing systems we have been able to identify specific viral factors that may influence the variation in disease presentation observed in poultry infected with virulent NDV strains. This is of direct relevance to informing surveillance programmes for the detection of NDV in poultry in GB. In turn, knowledge of genetic markers of virulence in APMV-1 strains provides evidence to support hazard identification and risk management for specific NDV lineages and strains that pose a threat to animal health and welfare, international trade and food security. The outputs of this project provide robust evidence and an improved understanding of the determinants and characteristics of the evolution and emergence of virulent ND viruses and tools that enable rapid detection of emerging threats posed by NDV and directly supports Defra’s objective of the proportionate, timely implementation of risk management and statutory disease control measures through early warning surveillance mechanisms. Outputs from this project have been of direct relevance to the development of further work in project SE0797 (“Investigation of the molecular basis for ND virulence to allow improved assessment of risk factors for infection”). This project aims to build on the scientific outputs from project SE0786 and investigate further the mechanisms that drive the events that may lead to outbreaks of virulent NDV in domestic poultry. Therefore, it is envisaged that evidence will be forthcoming to inform threat detection and risk management, In addition, insights are anticipated into the persistence and maintenance mechanisms of virulent ND viruses, including the identification of host species, and indeed possible host/virus combinations that may represent a higher risk for the covert introduction and ‘silent spread’ of NDV to the UK.
Project Report to Defra
8. As a guide this report should be no longer than 20 sides of A4. This report is to provide Defra with details of the outputs of the research project for internal purposes; to meet the terms of the contract; and to allow Defra to publish details of the outputs to meet Environmental Information Regulation or Freedom of Information obligations. This short report to Defra does not preclude contractors from also seeking to publish a full, formal scientific report/paper in an appropriate scientific or other journal/publication. Indeed, Defra actively encourages such publications as part of the contract terms. The report to Defra should include:
the objectives as set out in the contract;
the extent to which the objectives set out in the contract have been met;
details of methods used and the results obtained, including statistical analysis (if appropriate);
a discussion of the results and their reliability;
the main implications of the findings;
possible future work; and
any action resulting from the research (e.g. IP, Knowledge Exchange).

EVID4 Evidence Project Final Report (Rev. 06/11) Page 4 of 13
Objective 1: Validate real-time assay, using novel probe with two innovations, in tests with clinical samples. Objective 1 of this project was the validation of a Newcastle Disease virus (NDV) pathotyping real-time reverse transcription polymerase chain reaction (RRT-PCR) assay with the ultimate goal of implementing this as a new test in the AHVLA’s diagnostic test portfolio. This NDV pathotyping assay, to discriminate between virulent ND and avirulent APMV-1 viruses (and includes the two innovations: inosines and locked nucleic acids), was initially developed in a previous ROAME (SE0774). The current ‘gold standard’ method for diagnosis of NDV is virus isolation (VI) in 9-11 day old SPF embryonated fowl’s eggs (EFEs) followed by either nucleotide sequencing of the F-gene cleavage site or the intracerebral pathogenicity index (ICPI) test. Development of an in vitro molecular method to determine pathogenicity of any isolated ND virus will therefore contribute to a reduction of EFEs (3Rs) and provide a more rapid test turnaround time. Ultimately, the application of this assay complements the current avian notifiable disease (AND) test portfolio at AHVLA Weybridge by providing rapid molecular determination of the F gene cleavage site for samples that have tested positive by a APMV-1 L-gene RRT-PCR screening assay. The two real time assays, APMV-1 screening and pathotyping, are not anticipated to replace VI as a diagnostic tool, but provide the possibility for a more rapid confirmation of AMPV-1. It is important to note that virus isolation detects viable virus whereas RRT-PCR detects viral RNA so, results from these two tests are not analogous. During this study a total of 623 clinical samples (574 swabs and 49 tissues) were sourced from chickens, turkeys, ducks, partridges and pheasants that had been previously submitted to the VLA. These were tested by virus isolation and analysed by RRT-PCR pathotyping assay (Table1). For further details of methods and results, please refer to the following published manuscript Fuller, C. M., M. S. Collins, et al. (2009). "Development of a real-time reverse-transcription PCR for the detection and simultaneous pathotyping of Newcastle disease virus isolates using a novel probe." Arch Virol 154(6): 929-937. Table 1: 2x2 box analysis comparing NDV pathotyping assay to VI
Virus isolation in EFEs (gold standard)
+ - Total
NDV pathotyping RRT-PCR
+ 25 0 25
- 12 586 598
Total 37 586 623
From these results we have determined that the sensitivity of the RRT-PCR assay was only 67.57% when compared to VI; twelve false negative results were reported by the RRT-PCR assay. Specificity of the RRT-PCR assay was 100% with no false positives recorded. It is important to note that the F gene is a confirmatory assay to compliment the L gene screening RRT-PCR (Fuller et al., 2010) and is therefore used on positive samples. Therefore this assay requires high specificity (i.e. no false positives) as opposed to a screening assay (L gene) that requires high sensitivity (no false negatives). The end point of detection for the NDV pathotyping RRT-PCR assay was determined using one virus from each of the six lineages (1-6) of NDV (Aldous et al., 2003). A 10-fold dilution series of Ulster, Hitchner B1, Herts 33, pi/CY/819/07 3256/1, ck/ET/1028/07 762 and dk/GB/7800/06 was prepared and used to determine the 50% egg infectious dose per 0.1ml (EID50/0.1ml) of each neat virus stock. The EID50 of each virus was determined as 10
8.1, 10
8.5, 10
6.9, 10
6.7, 10
7.1, and 10
8.3 respectively. The same dilution
series was then tested in the NDV pathotyping RRT-PCR assay. The results of this data are presented in Table 2, where the titre (EID50/0.1ml) of the last positive dilution detected by the relevant probe is given. Above this value all dilutions were RRT-PCR positive.
EVID4 Evidence Project Final Report (Rev. 06/11) Page 5 of 13
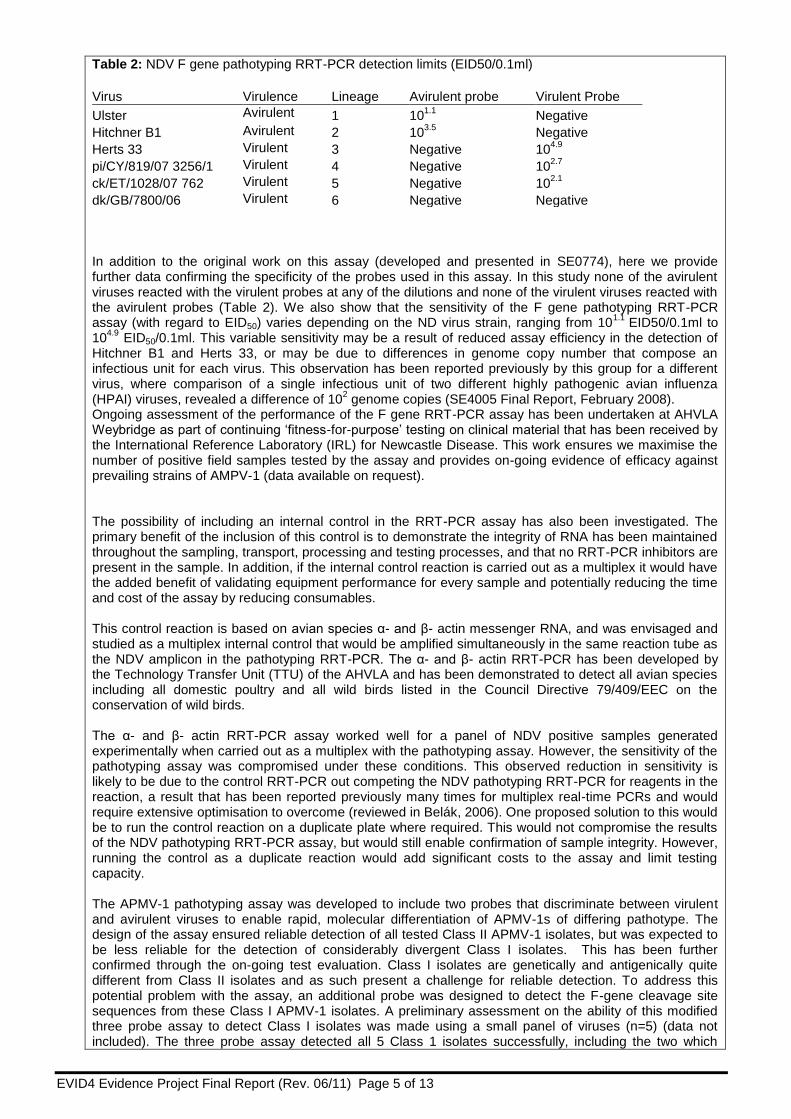
Table 2: NDV F gene pathotyping RRT-PCR detection limits (EID50/0.1ml)
Virus Virulence Lineage Avirulent probe Virulent Probe
Ulster Avirulent 1 101.1
Negative
Hitchner B1 Avirulent 2 103.5
Negative
Herts 33 Virulent 3 Negative 104.9
pi/CY/819/07 3256/1 Virulent 4 Negative 102.7
ck/ET/1028/07 762 Virulent 5 Negative 102.1
dk/GB/7800/06 Virulent 6 Negative Negative In addition to the original work on this assay (developed and presented in SE0774), here we provide further data confirming the specificity of the probes used in this assay. In this study none of the avirulent viruses reacted with the virulent probes at any of the dilutions and none of the virulent viruses reacted with the avirulent probes (Table 2). We also show that the sensitivity of the F gene pathotyping RRT-PCR assay (with regard to EID50) varies depending on the ND virus strain, ranging from 10
1.1 EID50/0.1ml to
104.9
EID50/0.1ml. This variable sensitivity may be a result of reduced assay efficiency in the detection of Hitchner B1 and Herts 33, or may be due to differences in genome copy number that compose an infectious unit for each virus. This observation has been reported previously by this group for a different virus, where comparison of a single infectious unit of two different highly pathogenic avian influenza (HPAI) viruses, revealed a difference of 10
2 genome copies (SE4005 Final Report, February 2008).
Ongoing assessment of the performance of the F gene RRT-PCR assay has been undertaken at AHVLA Weybridge as part of continuing ‘fitness-for-purpose’ testing on clinical material that has been received by the International Reference Laboratory (IRL) for Newcastle Disease. This work ensures we maximise the number of positive field samples tested by the assay and provides on-going evidence of efficacy against prevailing strains of AMPV-1 (data available on request). The possibility of including an internal control in the RRT-PCR assay has also been investigated. The primary benefit of the inclusion of this control is to demonstrate the integrity of RNA has been maintained throughout the sampling, transport, processing and testing processes, and that no RRT-PCR inhibitors are present in the sample. In addition, if the internal control reaction is carried out as a multiplex it would have the added benefit of validating equipment performance for every sample and potentially reducing the time and cost of the assay by reducing consumables. This control reaction is based on avian species α- and β- actin messenger RNA, and was envisaged and studied as a multiplex internal control that would be amplified simultaneously in the same reaction tube as the NDV amplicon in the pathotyping RRT-PCR. The α- and β- actin RRT-PCR has been developed by the Technology Transfer Unit (TTU) of the AHVLA and has been demonstrated to detect all avian species including all domestic poultry and all wild birds listed in the Council Directive 79/409/EEC on the conservation of wild birds. The α- and β- actin RRT-PCR assay worked well for a panel of NDV positive samples generated experimentally when carried out as a multiplex with the pathotyping assay. However, the sensitivity of the pathotyping assay was compromised under these conditions. This observed reduction in sensitivity is likely to be due to the control RRT-PCR out competing the NDV pathotyping RRT-PCR for reagents in the reaction, a result that has been reported previously many times for multiplex real-time PCRs and would require extensive optimisation to overcome (reviewed in Belák, 2006). One proposed solution to this would be to run the control reaction on a duplicate plate where required. This would not compromise the results of the NDV pathotyping RRT-PCR assay, but would still enable confirmation of sample integrity. However, running the control as a duplicate reaction would add significant costs to the assay and limit testing capacity. The APMV-1 pathotyping assay was developed to include two probes that discriminate between virulent and avirulent viruses to enable rapid, molecular differentiation of APMV-1s of differing pathotype. The design of the assay ensured reliable detection of all tested Class II APMV-1 isolates, but was expected to be less reliable for the detection of considerably divergent Class I isolates. This has been further confirmed through the on-going test evaluation. Class I isolates are genetically and antigenically quite different from Class II isolates and as such present a challenge for reliable detection. To address this potential problem with the assay, an additional probe was designed to detect the F-gene cleavage site sequences from these Class I APMV-1 isolates. A preliminary assessment on the ability of this modified three probe assay to detect Class I isolates was made using a small panel of viruses (n=5) (data not included). The three probe assay detected all 5 Class 1 isolates successfully, including the two which

EVID4 Evidence Project Final Report (Rev. 06/11) Page 6 of 13
were negative previously using the two probe approach, and seemed to have no impact on the results for the other viruses. This modified assay was used in the annual National reference laboratory molecular ring trial (2010) circulated by the NDV EU Reference Laboratory. Using the modified three probe protocol the assay successfully characterised all eight samples in the ring trial which included Class I and Class II isolates. Objective 2: Rescue of NDV from cloned cDNA In Objective 2 we developed and applied reverse genetics techniques to NDV. Our system utilises the transfection of a plasmid containing the entire ND viral genome, as well as expression plasmids for the nucleoprotein (N), phosphoprotein (P) and polymerase (L) genes to produce the proteins of the viral RNA polymerase complex. These plasmids are under the control of the T7 RNA polymerase promoter; the T7 RNA polymerase enzyme will bind to these plasmid DNA templates and make complementary RNA in vitro. The T7 RNA polymerase is provided inside the cells by co-infecting the cells with recombinant Fowl pox virus (FPV) which expresses T7 polymerase enzyme. This polymerase binds to the cDNA full genome plasmid and the N, P and L expression plasmids and uses them as a template to make both the negative sense viral RNA genome, and mRNA for the N, P and L proteins. The N, P and L mRNA enables the generation of the N, P and L proteins which then function in their normal way, using the viral genome to make progeny virus. Using this approach we are able to produce mutant viruses by manipulating the full genome plasmids by introducing selected mutations prior to recovery of the new, mutant virus. By comparing the phenotypes of mutant viruses produced we can investigate the effects that the individual mutations have on the pathogenicity of NDV. Due to the large size of the insert required (15.1KB), construction of a full genome infectious clone of NDV is both technically challenging and time consuming. Infectious clones are designed using full genome sequences with the addition of a T7 promoter and terminator as well as an autocatalytic ribozyme sequence. The position of the T7 promoter on the full genome plasmid ensures the transcription of the plasmid DNA into negative sense RNA genome by the T7 DNA dependent RNA polymerase is initiated at the correct location. The transcription termination sequence must be robust to ensure that an efficient termination of RNA transcription occurs. The position of the autocatalytic ribozyme sequence, in this case from hepatitis delta virus, is critical to ensure that the transcribed RNA is post-transcriptionally cleaved to the terminal end of the NDV genome producing viral RNA of the correct length for the RNP to then initiate replication and transcription to produce viral progeny. In a previous project SE0774, a PPMV-1 isolate (Belgium/1073/98 248 VB) that had a F gene cleavage site amino acid motif usually associated with virulent viruses (RRQKRF) and an avirulent intracerebral pathogenicity index (ICPI) of 0.39 was identified. From this parent strain a number of cloned viruses were isolated, all with the same virulent cleavage site motif but with different ICPI values. The cloned viruses with ICPI values of 0.025 (hereafter termed virus 0.025) and 1.3 (hereafter termed virus 1.3) were selected for further study. To generate an infectious clone by reverse genetics under the SE0786 project, the sequence of 5’ and 3’ non-coding sequence regions of viruses 1.3 and 0.025 were required. The 5’ and 3’ non-coding regions were determined using the ‘RACE’ (Rapid Amplification of cDNA Ends) technique. The completed sequence of the genome ends of virus 0.025 and virus 1.3 identified no additional mutations in the genome ends. Full genome sequencing of the 15,192kb genome of the 0.025 and 1.3 viruses identified only four nucleotide/amino acids differences (Table 3). Table 3. Heterogeneity observed between ND viruses 0.025 and 1.3
Location of mutation (gene and location in genome)
Virus Matrix aaa 100 Fusion aa 453 L polymerase aa
1378 L polymerase aa 1945
Virus 0.025 Aspartic acid (D) Proline (P) Valine (V) Asparagine (N)
Virus 1.3 Asparagine (N) Serine (S) Methionine (M) Serine (S)
a Amino acid measured from the start of the gene
Strong collaborative links have been maintained throughout this project with the Central Veterinary Institute (CVI) (formerly Central Institute for Animal Disease Control) Lelystad, The Netherlands. This collaboration has provided an essential technical resource for this project whereby technical problems and protocols can be discussed and investigated effectively, with rapid and often positive outcomes. Communication and interaction with the collaborating institutes has taken the form of residential training

EVID4 Evidence Project Final Report (Rev. 06/11) Page 7 of 13
for VLA technical staff at CVI, manuscript critique and review by AHVLA, and frequent email and phone conversations between the principle investigators in both institutes. Through this collaboration a number of technical issues have been resolved, a manuscript has been produced (Dortmans et al., 2010) and the relationship between the two institutes has been further consolidated. Our collaborators carried out an initial investigation of two of these mutations – Fusion P453S and L polymerase V1378M - using a reverse genetics system. The Fusion P453S and L polymerase V1378M mutations were introduced into an infectious cDNA clone of a virulent Newcastle disease virus. The mutations were introduced individually and combined, and the effects of the mutations on pathogenicity by ICPI, were examined. Only the S453P substitution in the F protein had a modest effect on pathogenicity (Dortmans et al. 2010). For further details of methods and results, please refer to the following published manuscript Dortmans JC, Fuller CM, Aldous EW, Rottier PJ, Peeters BP. (2010). Two genetically closely related pigeon paramyxovirus type 1 (PPMV-1) variants with identical velogenic fusion protein cleavage sites but with strongly contrasting virulence. Vet Microbiol. 2010 Jul 14; 143(2-4):139-44. To study the molecular basis for the phenotype difference between the 0.025 and 1.3 PPMV-1 viruses, two infectious clones were designed on the 0.025 and 1.3 virus full genome sequences with the addition of a T7 promoter and terminator as well as an autocatalytic ribozyme sequence. These were constructed into a low copy plasmid (pJ211). The plasmid construct for both 0.025 and 1.3 viruses were produced commercially, which reduces the staff resources and production costs significantly. Virus 0.025 and 1.3 were rescued from this infectious clone using the optimised protocol reported above. To produce the “intermediate” clones of the 0.025 and 1.3 viruses, the four mutations identified in the 1.3 virus (Table 3) were produced by PCR and cloned into the plasmid construct of the 0.025 virus. Unfortunately only the generation of the P453S mutation in the Fusion protein was successful using this approach. Three viruses were generated by reverse genetics – the 0.025 virus, the 1.3 virus and the virus containing the P453S mutation. After rescue the virus were isolated by limiting dilution in CEF tissue culture to remove any residual fowl pox virus which might interfere with the ICPI test and a stock was grown in EFEs. The rescued viruses were subjected to full genome sequencing and pathotyping by ICPI test. As expected, the full genome sequence of the 0.025 and 1.3 rescued viruses were identical to the original virus confirming the virulent cleavage site sequence of RRQKRF. The ICPI observed for the rescued 0.025 and 1.3 viruses was 0.025 and 1.075 respectively. Interestingly, the virus containing the P453S mutation in the Fusion protein produced an ICPI of 1.24. This mutation in the fusion protein is clearly important for the virulence of this virus and is likely to be due to a conformational change in the protein (a ‘twist’) introduced by the removal of a proline residue. The slight decrease in ICPI of the 1.3 virus to 1.075 is intriguing but this variation may be within the variability of the test, and is an acceptable result as an ICPI of 1.075 is still considered virulent. However, the reduction in the ICPI of the 1.3 virus does suggest that there are additional factors, possibly non-genetic in nature, affecting the pathogenicity of these viruses in the ICPI assay. These results confirm that the rescued virus displayed the unusual phenotypic properties of the original virus - an avirulent pathotype and a genotype normally associated with virulent viruses. Through further collaborative work with our colleagues at Lelystad, The Netherlands, when the P453S mutation was introduced into a Herts 33 backbone by reverse genetics, this mutation only reduced the ICPI from 1.6 to 1.3. The only difference between the 0.025 and 1.3 viruses is the P453S mutation in the F protein. Since the interaction of F and HN is important functionally, the entire F and HN genes from the 0.025 and the 1.3 viruses were inserted into the Herts 33 backbone to determine if this would reduce the ICPI dramatically. There was no difference in the ICPI obtained for these two viruses - 0.9 and 1.0. This is in contrast to the results obtained from our studies where the 0.025 and 1.3 viruses produced by reverse genetics resulted in a dramatic difference in ICPI – 0.025 and 1.075 respectively. I our work the introduction of the P453S mutation also resulted in a modest change in the ICPI. This must mean that the mechanism for reducing pathogenicity through the Fusion mutation (P) must be through an interaction with some other part of the genome other than HN. This is a significant finding. F interacts with HN to initiate Fusion but it is not known to interact with any other part of the genome. Objective 3 and 4: Obtain cloned virus from single PPMV-1 isolate with an IVPI of 0.00 and an IVPI that is significantly raised following passage. Sequence entire genome of the cloned virus with IVPI 0.00 and the cloned virus with increased IVPI following passage from Objective 3. Understanding the factors that influence the virulence of APMV-1s is a complex task. Throughout this project we investigated naturally occurring, unusual viruses that may enable further insights into virulence. Analysis of the phenotypic properties of virus submissions received by the International Reference Laboratory (IRL) has led to the identification of further, unusual isolates that display contrasting pathotyping results, where the ICPI is high, indicating a virulent virus, but the intravenous pathogenicity

EVID4 Evidence Project Final Report (Rev. 06/11) Page 8 of 13
index (IVPI) is low, suggesting an avirulent virus. Further analysis of these unusual isolates provided an opportunity to investigate factors that may influence these phenotypic properties. The aim for this objective was to investigate these properties through cloning, passage and selection of viruses. This work was carried out according to the experiences and lessons learnt from the previous project, SE0774. This work was presented as a poster at the 2010 Negative Strand Virus conference in Bruges, Belgium. Fuller C, Seekings J, Irvine R, Aldous E, and Brown I. (2010). Two genetically similar viruses, differing in pathogenicity, obtained from a single isolate of pigeon paramyxovirus type 1 (PPMV-1). Conference proceedings p. 172: XIV International Conference on Negative Strand Viruses, June 20-25, 2010. Bruges, Belgium. We selected APMV1/UK/pigeon/617/83 as an original virus stock [virus A]; it has an ICPI of 1.46 (virulent) and an IVPI of 0.7 (low virulence). This original stock of virus had a mean death time (MDT) in embryonated fowls’ eggs (EFEs) of 72 hours (mesogenic/intermediate virulence). From this initial virus stock [A], three viruses [B-D] have been produced (Figure 1). The complete genome sequence for all four isolates was also determined.
[A]APMV-1/pigeon/England/617/83
Original
ICPI 1.47 IVPI 0.7
MDT 72 hrs EID50 106.83
Passaged 1x in EFE
to make working stock
Plaque purified in CEFs 3x
[C] Brain from one 6-week-old chicken
Passaged 1x in EFE
IVPI 2.03
Plaque purified in CEFs 3x
[B] Purified stock grown in EFE
ICPI 1.55 IVPI 0.0
[D] Purified stock grown in EFE
ICPI 1.67 IVPI 1.19
Figure1. Summary of selection procedure
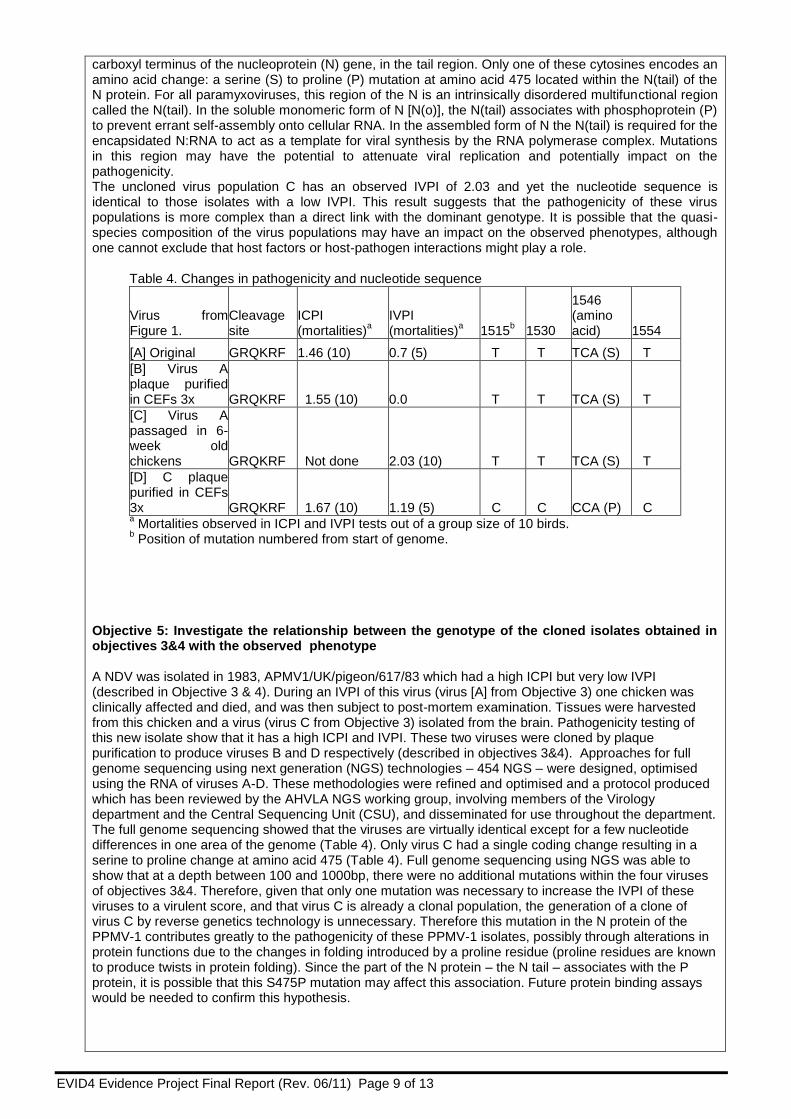
Briefly, the original stock of APMV1/UK/pigeon/617/83 virus [A] was used in an IVPI and resulted in score of 0.7 (Table 4). Virus was isolated using EFEs from the brain material of one of the chickens that died on day 7 [Virus C]. When tested in an IVPI this new isolate [C], was shown to have an elevated IVPI of 2.03. Cloned virus populations of viruses A and C were then produced by three rounds of plaque purification in chick embryo fibroblast cells (CEFs), generating new virus samples B and D. The complete genomes of the four virus preparations - the original stock virus [A], a plaque purified virus from this stock [B], a 6-week old chicken passaged virus [C] and a plaque purified virus from this chicken passaged isolate [D] - were sequenced to a minimum of two fold coverage (Table 4) using methods adapted from work in previous ROAMEs (SE0774, SE0779). All primers were screened against a panel of PPMV-1 full genome sequences and selected based upon primer location within conserved sequence. Briefly, viral RNA was extracted from allantoic-amniotic fluid containing each of the viruses A, B, C and D using the Qiagen Viral RNA extraction kit. First-strand DNA synthesis was carried out using the Superscript™ III Reverse Transcriptase kit (Invitrogen) with random hexamers. Four over-lapping PCR fragments were generated for each virus using the Elongase® Amplification kit (Invitrogen); each amplification step was carried out five times and the resulting amplicons pooled to ensure adequate template volumes. PCR fragments of the expected size were separated by electrophoresis in a 1% agarose gel and purified using the QiaQuick (Qiagen) gel extraction kit. Nucleotide sequencing of the purified PCR amplicon was carried out using the ‘Big-Dye’ DNA sequencing kit V3.1 in a plate format and capillary sequenced on an Applied Biosystems 3130 genetic analyser. Sequence analysis and alignment was carried out using the Lasergene DNASTAR software version 8. Viruses A, B and C have identical sequence across the entire genome despite the considerable difference in IVPIs observed. Virus D differs from the others in four thymidine to cytosine point mutations near the
EVID4 Evidence Project Final Report (Rev. 06/11) Page 9 of 13
carboxyl terminus of the nucleoprotein (N) gene, in the tail region. Only one of these cytosines encodes an amino acid change: a serine (S) to proline (P) mutation at amino acid 475 located within the N(tail) of the N protein. For all paramyxoviruses, this region of the N is an intrinsically disordered multifunctional region called the N(tail). In the soluble monomeric form of N [N(o)], the N(tail) associates with phosphoprotein (P) to prevent errant self-assembly onto cellular RNA. In the assembled form of N the N(tail) is required for the encapsidated N:RNA to act as a template for viral synthesis by the RNA polymerase complex. Mutations in this region may have the potential to attenuate viral replication and potentially impact on the pathogenicity. The uncloned virus population C has an observed IVPI of 2.03 and yet the nucleotide sequence is identical to those isolates with a low IVPI. This result suggests that the pathogenicity of these virus populations is more complex than a direct link with the dominant genotype. It is possible that the quasi-species composition of the virus populations may have an impact on the observed phenotypes, although one cannot exclude that host factors or host-pathogen interactions might play a role.
Table 4. Changes in pathogenicity and nucleotide sequence
Virus from Figure 1.
Cleavage site
ICPI (mortalities)
a IVPI (mortalities)
a 1515
b 1530
1546 (amino acid) 1554
[A] Original GRQKRF 1.46 (10) 0.7 (5) T T TCA (S) T
[B] Virus A plaque purified in CEFs 3x GRQKRF 1.55 (10) 0.0 T T TCA (S) T
[C] Virus A passaged in 6-week old chickens GRQKRF Not done 2.03 (10) T T TCA (S) T
[D] C plaque purified in CEFs 3x GRQKRF 1.67 (10) 1.19 (5) C C CCA (P) C a Mortalities observed in ICPI and IVPI tests out of a group size of 10 birds.
b Position of mutation numbered from start of genome.
Objective 5: Investigate the relationship between the genotype of the cloned isolates obtained in objectives 3&4 with the observed phenotype A NDV was isolated in 1983, APMV1/UK/pigeon/617/83 which had a high ICPI but very low IVPI (described in Objective 3 & 4). During an IVPI of this virus (virus [A] from Objective 3) one chicken was clinically affected and died, and was then subject to post-mortem examination. Tissues were harvested from this chicken and a virus (virus C from Objective 3) isolated from the brain. Pathogenicity testing of this new isolate show that it has a high ICPI and IVPI. These two viruses were cloned by plaque purification to produce viruses B and D respectively (described in objectives 3&4). Approaches for full genome sequencing using next generation (NGS) technologies – 454 NGS – were designed, optimised using the RNA of viruses A-D. These methodologies were refined and optimised and a protocol produced which has been reviewed by the AHVLA NGS working group, involving members of the Virology department and the Central Sequencing Unit (CSU), and disseminated for use throughout the department. The full genome sequencing showed that the viruses are virtually identical except for a few nucleotide differences in one area of the genome (Table 4). Only virus C had a single coding change resulting in a serine to proline change at amino acid 475 (Table 4). Full genome sequencing using NGS was able to show that at a depth between 100 and 1000bp, there were no additional mutations within the four viruses of objectives 3&4. Therefore, given that only one mutation was necessary to increase the IVPI of these viruses to a virulent score, and that virus C is already a clonal population, the generation of a clone of virus C by reverse genetics technology is unnecessary. Therefore this mutation in the N protein of the PPMV-1 contributes greatly to the pathogenicity of these PPMV-1 isolates, possibly through alterations in protein functions due to the changes in folding introduced by a proline residue (proline residues are known to produce twists in protein folding). Since the part of the N protein – the N tail – associates with the P protein, it is possible that this S475P mutation may affect this association. Future protein binding assays would be needed to confirm this hypothesis.

EVID4 Evidence Project Final Report (Rev. 06/11) Page 10 of 13
Objective 6: Evaluation of the growth characteristics and population composition of the isolates obtained in Objective 3 and 4 Growth curves were performed to determine whether the differences in pathogenicity for the PPMV-1 isolates produced in Objectives 3&4 - viruses A to D (Table 4) were due to altered growth kinetics. To establish the best protocol for assessing the viral titre in growth curves a number of pilot studies were carried out. Plaque assays (Avicell and agarose) were compared to TCID50 and the timing of measurements by cytopathic effect (CPE), haemagglutination (HA) and monoclonal antibody (MAb) staining over a time course was assessed. The titres of both viruses were determined by plaque assay. Growth curves were performed in CEFs using both high and low multiplicity of infection (MOI). High MOI is used to assess the replication rate of viruses as the cell monolayer is infected with enough virus to infect every cell. Low MOI is used to study multiple virus growth cycles as not all cells are infected. Therefore the efficiency of the virus spread to new cells is assessed. Unfortunately the low titre of virus C and the small volume produced prevented the use of this virus in the growth curves. Samples were taken in triplicate at timed intervals and stored at -70°C. The amount of virus in each sample was quantified by TCID50. Growth curve comparisons of viruses A (wild type), B (cloned virus with low IVPI) and D (cloned virus with high IVPI) infected in CEFs at a high MOI showed that the replication rate (slope) of these viruses were not significantly different (Fig 2). Therefore the difference in IVPI observed with these viruses cannot be attributed to their replication rates within a single round of replication. Virus replication (slope) of the viruses A (wild type), B (cloned virus with low IVPI) and D (cloned virus with high IVPI) infected in CEFs at a low MOI, showed that the replication of virus B (designated as V2, Figure 3) was lower than the wild type virus A and the cloned virus (D) with a high IVPI. This would suggest that the phenotype of low virulence by IVPI may be attributed to a reduced replication and spread of these viruses during multicycle replication. Figure 2. Growth curve comparisons of viruses A (wild type), B (cloned virus with low IVPI) and D (cloned virus with high IVPI) infected in CEFs at a high MOI.
H ig h M O I
0 1 0 2 0 3 0 4 0 5 0
0
2
4
6
8
1 0
W ild t y p e ( IV P I 0 .7 )
L o w IV P I v ir u s ( IV P I 0 .0 )
h ig h IV P I v ir u s ( IV P I 1 .1 9 )
h p i
lo
g 1
0 T
CID
50
/m
l
EVID4 Evidence Project Final Report (Rev. 06/11) Page 11 of 13
Figure 3. Growth curve comparisons of viruses A (wild type), B (cloned virus with low IVPI) and D (cloned virus with high IVPI) infected in CEFs at a low MOI.
V1 = Original E1 passage of parent virus (Virus A); V2 = Plaque purified low IVPI virus (Virus B); V4 = Plaque purified High IVPI virus (Virus D) Summary An APMV-1 F gene real time RT-PCR pathotyping assay for the simultaneous detection and rapid differentiation of avirulent APMV-1 and virulent ND viruses has been validated. The application of this assay complements the current avian notifiable disease (AND) test portfolio at AHVLA Weybridge by providing rapid molecular determination of the F gene cleavage site for samples that have tested positive by a APMV-1 L-gene RRT-PCR screening assay. This provides a molecular capability to determine the pathotype of APMV-1 viruses more rapidly than the conventional method of virus isolation and intra-cerebral pathogenicity index test. Hence, the F gene assay supports statutory disease control decision-making and risk management, enabling faster and proportionate implementation of restriction zones in the event of a confirmed ND outbreak in Great Britain Substantial progress has been made in the reverse genetics component of the project through rescuing a virus that confirms the unusual phenotypic properties of the original virus, specifically an avirulent ICPI pathotype and a genotype (F gene cleavage site sequence) normally associated with virulent ND viruses. The viruses produced in Objective 3 have displayed the expected discrepancy between ICPI and IVPI measures of pathogenicity and yet the observed genetic difference between these viruses is minimal. This provides us with further, fully characterised viruses to enable targeted analysis of the mutations identified and their possible role in APMV-1 virus virulence. In addition, the viruses generated that are genetically identical, but have varying virulence, enable us to begin investigating what other factors may be at work. The options for application of various in vivo approaches and molecular tools will be investigated to establish any potential links between these factors and the virulence of the viruses produced in Objective 3. We have propagated viruses in which the discrepancy between the ICPI and the IVPI is conserved and transmitted through the genetic sequence alone, thereby confirming that there are factors in addition to the cleavage site that are affecting virus virulence, and in this case attenuating what would be expected to be a virulent virus. In this example we are provided with confirmation of the importance of using genetic sequence analysis in the identification of notifiable ND. The identification of the factors that are attenuating this virus will contribute to understanding the potential of this, and other similar viruses that may be circulating the UK pigeon population, to cause outbreaks of notifiable disease in UK poultry. Interestingly, for the viruses with altered pathogenicity studied within this project, it was a change in a proline residue within the fusion protein that was responsible for the increase in pathogenicity. Further protein studies would be required to determine the biochemical basis for this altered pathogenicity. However, these mutations should be key during surveillance programs to identify virulent viruses. Future work The outputs of this project provide robust evidence and an improved understanding of the determinants and characteristics of the evolution and emergence of virulent ND viruses and tools that enable rapid detection of emerging threats posed by NDV and directly supports Defra’s objective of the proportionate, timely implementation of risk management and statutory disease control measures through early warning surveillance mechanisms.

EVID4 Evidence Project Final Report (Rev. 06/11) Page 12 of 13
Outputs from this project have been of direct relevance to the development of further work in project SE0797 (“Investigation of the molecular basis for ND virulence to allow improved assessment of risk factors for infection”). This project aims to build on the scientific outputs from project SE0786 and investigate further the mechanisms that drive the events that may lead to outbreaks of virulent NDV in domestic poultry. Therefore, it is envisaged that evidence will be forthcoming to inform threat detection and risk management, In addition, insights are anticipated into the persistence and maintenance mechanisms of virulent ND viruses, including the identification of host species, and indeed possible host/virus combinations that may represent a higher risk for the covert introduction and ‘silent spread’ of NDV to the UK.
References Aldous E. W., Manvell R. J., Cox W. J., Ceeraz V., Harwood D. G., Shell W., Alexander D. J. & Brown I. H.(2007) Outbreak of Newcastle disease in pheasants (Phasianus colchicus) in south-east England in July 2005. Veterinary Record 160, 482–484 Irvine R. M., Aldous E. W., Manvell R. J., Cox W. J., Ceeraz V., Fuller C. M. and others (2009) Outbreak of Newcastle disease due to pigeon paramyxovirus type 1 infection in grey partridges (Perdix perdix) in Scotland in October 2006. Veterinary Record 165, 531–535 COUNCIL OF THE EUROPEAN UNION (1992) Council Directive 92/66/EEC of 14 July 1992 introducing Community measures for the control of Newcastle disease. Official Journal of the European Communities L 260, 1–20. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:1992:260:0001:0020:EN:PDF. Accessed September 20, 2013 OIE (2012) Manual Of Diagnostic Tests And Vaccines For Terrestrial Animals. Chapter 2.3.14. Newcastle disease (version adopted in May 2012; contributors Dr C.L. Afonso, Dr P.J. Miller, Dr Ch. Grund, Dr G. Koch, Dr B. Peeters, Dr P.W. Selleck, Dr G.B. Srinivas) http://www.oie.int/fileadmin/Home/fr/Health_standards/tahm/2.03.14_NEWCASTLE_DIS.pdf. Accessed September 20, 2013

EVID4 Evidence Project Final Report (Rev. 06/11) Page 13 of 13
References to published material
9. This section should be used to record links (hypertext links where possible) or references to other published material generated by, or relating to this project.
Peer-review publication outputs from the project Dortmans JC, Fuller CM, Aldous EW, Rottier PJ, Peeters BP. (2010). Two genetically closely related pigeon paramyxovirus type 1 (PPMV-1) variants with identical velogenic fusion protein cleavage sites but with strongly contrasting virulence. Vet Microbiol. 2010 Jul 14; 143(2-4):139-44. Fuller, C. M., M. S. Collins, et al. (2009). "Development of a real-time reverse-transcription PCR for the detection and simultaneous pathotyping of Newcastle disease virus isolates using a novel probe." Arch Virol 154(6): 929-937. Conference Proceedings Abstract & Presentation Fuller C, Seekings J, Irvine R, Aldous E, and Brown I. (2010). Two genetically similar viruses, differing in pathogenicity, obtained from a single isolate of pigeon paramyxovirus type 1 (PPMV-1). Conference proceedings p. 172: XIV International Conference on Negative Strand Viruses, June 20-25, 2010. Bruges, Belgium.


















