
Evaluation of the Information Content of Sedimentation Equilibrium Data in Self-Interacting Systems
10
Evaluation of the Information Content of Sedimentation Equilibrium Data in Self-Interacting Systems Shirley Ang, Arthur J Rowe* Introduction The analytical ultracentrifuge (AUC) is increasingly used as a sensitive and theoretically sound approach to the definition of macromolecular solution properties and interactions. [1,2] Among its particular virtues are the absence of unwanted interaction of the solute components with instrumental surfaces and structures, as can happen with column-based instrumentation, and the potentially high sensitivity of the method when employing recently developed software. [3] AUC analysis can detect protein– protein interaction at a level (K d > 1 10 3 M) undetectable using alternative approaches. It is the identification and quantitation of interaction terms which form the primary focus of most contemporary AUC work. With protein solutes at least, the emphasis from earlier decades on ‘measuring molecular weights’ no longer obtains: in most cases the molecular weight of the (recombinant) solute(s) is given, and indeed is routinely fixed in non-linear fitting of data sets. Two experimental modes are available to the AUC user: sedimentation velocity (SV) analysis and sedimentation equilibrium (SE) analysis. In the former mode very large numbers of scans of the (continuously varying) signal vs. radial distance within the experimental cells are logged. This data-rich environment enables analysis using widely employed software such as SEDFIT [4,5] to largely eliminate both radially independent (RI) and time-independent (TI) noise. The sheer amount of data then fitted, using numerical fitting to multiple Lamm equation solution sets, effects a major reduction in the contribution of residual stochastic noise to the final profiles generated of the diffusion- deconvoluted function c(s) vs. s. In contrast, SE analysis must be of necessity carried out on a single data set, since when the equilibrium state is attained that state does not, by definition, change with elapse of further time. Hence the issue of the elimination of systematic (RI þ TI) and stochastic error takes on much greater significance in SE as compared to SV. Nonetheless the use of the SE mode of analysis remains of interest, Full Paper S. Ang, A. J Rowe NCMH, University of Nottingham, School of Biosciences, Sutton Bonington, Leicestershire, LE12 5RD UK Fitting r ¼ f(c) as opposed to the usual c ¼ f(r) to the inverted form of the sedimentation equilibrium equation for interacting solute (INVEQ algorithm), it is shown by detailed simulation and by experimentation that stable, simultaneous estimates can be retrieved for both virial (2nd BM/3rd CM) and specific inter- action (K a ) terms. In suitable systems estimates for two distinct second virial (BM) and single K a terms can equally be defined. Whilst cell loading level is critical, noise level in the interference fringe data is shown to have surprisingly little influence on these outcomes. 798 Macromol. Biosci. 2010, 10, 798–807 ß 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim DOI: 10.1002/mabi.201000065
-
Upload
shirley-ang -
Category
Documents
-
view
212 -
download
0
Transcript of Evaluation of the Information Content of Sedimentation Equilibrium Data in Self-Interacting Systems

Full Paper
798
Evaluation of the Information Contentof Sedimentation Equilibrium Data inSelf-Interacting Systems
Shirley Ang, Arthur J Rowe*
Fitting r¼ f(c) as opposed to the usual c¼ f(r) to the inverted form of the sedimentationequilibrium equation for interacting solute (INVEQ algorithm), it is shown by detailedsimulation and by experimentation that stable, simultaneous estimates can be retrievedfor both virial (2nd BM/3rd CM) and specific inter-action (Ka) terms. In suitable systems estimatesfor two distinct second virial (BM) and single Ka
terms can equally be defined. Whilst cell loadinglevel is critical, noise level in the interferencefringe data is shown to have surprisingly littleinfluence on these outcomes.
Introduction
The analytical ultracentrifuge (AUC) is increasingly used as
a sensitive and theoretically sound approach to the
definition of macromolecular solution properties and
interactions.[1,2] Among its particular virtues are the
absence of unwanted interaction of the solute components
with instrumental surfaces and structures, as can happen
with column-based instrumentation, and the potentially
high sensitivity of the method when employing recently
developed software.[3] AUC analysis can detect protein–
protein interaction at a level (Kd> 1� 10�3M) undetectable
using alternative approaches. It is the identification and
quantitation of interaction terms which form the primary
focus of most contemporary AUC work. With protein
solutes at least, the emphasis from earlier decades on
‘measuring molecular weights’ no longer obtains: in most
cases themolecularweight of the (recombinant) solute(s) is
S. Ang, A. J RoweNCMH, University of Nottingham, School of Biosciences, SuttonBonington, Leicestershire, LE12 5RD UK
Macromol. Biosci. 2010, 10, 798–807
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
given, and indeed is routinely fixed in non-linear fitting of
data sets.
Two experimental modes are available to the AUC user:
sedimentation velocity (SV) analysis and sedimentation
equilibrium (SE) analysis. In the former mode very large
numbers of scans of the (continuously varying) signal vs.
radial distance within the experimental cells are logged.
This data-rich environment enables analysis using widely
employed software such as SEDFIT[4,5] to largely eliminate
both radially independent (RI) and time-independent (TI)
noise.Thesheeramountofdata thenfitted,usingnumerical
fitting to multiple Lamm equation solution sets, effects a
major reduction in the contribution of residual stochastic
noise to the final profiles generated of the diffusion-
deconvoluted function c(s) vs. s.
In contrast, SE analysis must be of necessity carried out
on a single data set, since when the equilibrium state is
attained that state does not, by definition, change with
elapse of further time. Hence the issue of the elimination of
systematic (RIþ TI) and stochastic error takes on much
greater significance in SE as compared to SV. Nonetheless
the use of the SE mode of analysis remains of interest,
DOI: 10.1002/mabi.201000065

Evaluation of the Information Content of Sedimentation . . .
inasmuch as the time-invariance of the final state of the
system satisfies the requirements for true thermodynamic
analysis of that state, in which the ‘virial’ terms tradition-
ally used in the parameterization of the expansion with
respect to concentration of the chemical activity of the
solute(s) can be directly employed,[6] without a need for
consideration of ‘hydrodynamic non-ideality’ terms, as is
necessary in SV mode. An additional – but not necessarily
trivial – advantage of the SE mode is that both the total
experimental time takenand the amountof solutematerial
required for a seriesof experimentsareappreciably lower in
SE than in SV mode: especially since the use of multi-
channel cells enables three times more experiments to
be conducted in a given run in the formermode than in the
latter. To quantify this, let us briefly consider the resource
cost (i.e., 10mg�ml�1 solutematerial plus instrument time)
of estimatingasingleKavalue. For SVthedefinitionofans-c
isotherm over a set of seven concentrations (0.5–
10mg �ml�1, load volume 400 ml) would require � 13mg
in total over seven cells. Only 1Ka value can be evaluated
per run. In contrast, by SE, with only 80ml load volume, just
0.8mg is required, as a single SE experiments suffices to
establishvalues for the interaction term, the SEdistribution
itself sampling over a range of solute concentration. The
resource advantage of SE over SV become even more
markedwhenwe note that in a single SE run no fewer than
21Kavalues canbeevaluated (7� 3multi-sector cells). True,
an SE run takes longer than an SV run: but the difference in
resource requirement totally outweighs this factor.
However, the issue of the effects of error in the data sets
acquired in SE mode needs to be addressed, before we
confirm with confidence the above assertion, that ‘one
experiment suffices’. We find that in any attempt to fit SE
data, all of the following conditions need to be satisfied:
(i) t
Macro
� 20
he errors (systematic and stochastic) in the raw signal
values, and also in the radial values should have been
pre-determined for the instrument (the latter value is
trivial, it may be set at �0.5 pixel for Rayleigh
interference optics)
(ii) i
n every fit, the algorithm employed (Marquardt–Levenberg or other) should incorporate the appro-
priate use of these provided error estimates in the
minimization (of the SSR or whatever) procedure
(iii) e
specially where the error surface is complex, theMarquardt–Levenberg (ML) algorithm may not be
stable in use, even when initial parameter guesses
which are not unreasonable are provided.[7] Alter-
native algorithms need to be available for initial or
even final fitting, algorithms which may be slower
than ML but which can give stable initial estimates –
which should themselves be multiple[7] – for the
‘floated’ parameters. Our approach to obtaining a
stable fit involves three progressive steps: (i) use of
mol. Biosci. 2010, 10, 798–807
10 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
real-time interactive manual fitting to obtain a good
approximation to optimal parameter values; (ii)
improvement of these estimates by re-fitting (without
statistical analysis but with input of data error
estimates) using a Robust algorithm; (iii) final re-
fitting using theML algorithmwith 500 iterations and
a confidence interval of 68.4%.
Wehavebeenable to satisfyall of theaboveconditionsby
performing our fits within the curve fitting and data
analysis packagepro Fit (QuantumSoft, Zurich) onanApple
Macintosh computer running under OS-X. This software is
capable, without user programming input, of meeting in a
user-friendly mode all of the requirements defined above.
Boot-strapping of parameter estimates and errors, for
example, is effected by simply entering in a dialog box
the estimated errors in the raw data, the number of iter-
ations required and the associated confidence interval (we
use the default of 68.4%). Completion of the fit then gen-
erates, in addition to the results, a full set of 500 estimates
for each floated parameter together with estimates for the
appropriate confidence intervals. Via a ‘Binning’ option
histogramsof theseparameterestimateare thengenerated.
As will be seen, the distribution of these estimates can be
very ‘non-Gaussian’ indeed.
Thevalueswhichweuse for our estimates of errors in the
raw data values are obtained experimentally, by a simple
subtractive procedure which eliminates – to a first
approximation – TI noise (RI noise is not relevant, since a
‘baseline offset’, which is in essence an RI noise value, is
floated in all our fits). Details of our approach, including our
handling of the issue of baseline distortion, are given below
(Experimental Part).
All our analyses are carried out using the INVEQ
functions, in which instead of fitting signal values as a
function of radial values, an inverse fit is performed, of
radial values as a function of signal value. The details are
given below. It needs to be emphasized that no ‘new
equation’ is being employed, merely a conventional
equation for SE, fitted after inversion as a means for
avoiding problems with the transcendental nature of the
equation as conventionally written. The INVEQ approach
hasbeenemployedsuccessfullybyourselvesandothers ina
range of applications.[6,8–12] In this present studywe define
more fully the extent to which a range of parameters can
be defined, showing that it is possible to retrieve sensible
values for interaction termsup to at least the 3rd virial term
in a single (but potentially self-interacting) solute system,
and we define the solute concentration values which are
called for. We find, perhaps surprisingly, that (all other
factors being equal) the level of the cell-loading concentra-
tion is considerably more crucial to success than is the
level of error in the raw signal data. We confirm via the
distribution of boot-strapped parameter estimates that our
www.mbs-journal.de 799

S. Ang, A. J. Rowe
800
earlier finding[6] concerning the complexnature of the error
surface in SE fitting is correct. Finally, we present
experimental data on a well-defined protein (RNAse A)
which show consistency with the results from our
simulations, and illustrate that results obtained by varying
the ionic strength of the solvent are consistent with
expectation in respect of the values of the interaction terms
yielded.
Experimental Part
Simulation
Simulated sedimentation equilibrium data were produced by
using the ‘Tabulate’ optionwithin pro Fit after appropriate values
for the selected parameters had been entered into the parameter
window, within the selected INVEQ function (inveq5aBC). Values
for the reference radius (rref) and for the reduced flotational
molecular weight (s) were fixed: other parameters, namely
reference solute concentration (cref), baseline offset (E), second
virial term(s) BMi, the third virial term CM, and the interaction
constant Ka were fixed for simulation, but floated for data
analysis. Note, the second and third virial ‘terms’ are the algebraic
product of the monomer molecular mass and the appropriate
‘virial coefficient’.
The simulations presented (selected from a more extensive set)
are for cell loading concentrations of ten fringe (�3mg �ml�1
protein) and 30 fringe (�10mg �ml�1 protein) with a reduced
floatational weight (see below) of s¼1.5, Ka¼0.0015 fringe�1,
BM¼0.0015ml � fringe�1 and CM¼ � 2e–6 fringe�2. These would
correspond to a Kd�14� 10�3M (at least one order of magnitude
weaker than a value measurable by other technologies), and
2BM�10 g �ml�1, not untypical for a globular proteinwith a small
net charge. The value for CM is at the lowest end of themagnitude
expected to be found for such a protein. It is not readily compared
with literature values, these being close to non-existent. All these
values are ‘conservative’, in the sense that they present a ‘case of
maximum difficulty’ for analysis. The reduced floatational weight
of s¼1.5 is again chosen partly in the interests of being
conservative. However there is also a practical reason for not
using larger values: at 30 fringe cell loading, no higher level is
feasible in ‘real life’ as thegradientsbecometoo steep tobe resolved
by the camera system of the Beckman XL-I. If one is performing
single runs at lower concentrations then of course the s value
(based upon rotor speed, see equation (2) below) can be increased,
with consequent gain in precision in parameter estimates. But
often inpractice onewill be runningmultiple samples together in a
rotorovera rangeof concentrations, and in this case the speedmust
be restricted to the speed appropriate to the highest concentration
used.
Data Analysis
The INVEQfunctions, as initiallydefined [8], fit thedatasetof (r,c) to
the followingequation,which is the simple algebraic inverse of the
conventionally defined equation for sedimentation equilibrium in
a system where a single second virial term BM defines the
Macromol. Biosci. 2010, 10, 798–807
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
expansion in c of the chemical potential at any radial position (r)
within the cell:
r ¼n�
ln cr=cið Þ þ 0:5� sw= 1þ 2BMcrð Þð Þ � r2i
�.�0:5� sw= 1þ 2BMcrð Þð Þ
�o0:5
(1)
where r is any radial position at which the solute concentration c
has the value cr, and ri and ci are the values of these parameters at
a defined reference position. The choice of this latter radial
position is not critical: it can sensibly be taken as being close to the
‘hinge point’ at which initial, cell-loading concentration is
conserved. The parameter s is the reduced molecular weight of
the solute, defined as
s ¼ M 1� vrð Þv2�RT (2)
where M is the molecular weight of the solute, v (ml � g�1) partial
specific volume, r the density of the solvent, v (radians/sec)
the angular velocity of the rotor, R the gas constant and T the
temperature (K). sw is the weight averaged value over the
monomer and the dimer, computed during every iteration during
the non-linear fitting of the parameters associated with
Equation (1) by solving from current value of Ka within the
iteration for a, the weight fraction of the total mass concentration
of the solute in dimer form at defined radial locus,
It must be stressed that Equation (1) is in no sense a ‘new’
equation. It is simply an inversion of the basic equation describing
the distribution at sedimentation equilibrium for the i’th solute
component as:
cr ¼ ca exp 0:5si 1þ @ ln g ið Þ=@crð Þcrf g r2a � r2� �� �
(3)
where g i is the activity coefficient of that component. Clearly
for fitting purposes the activity term must be expressed in
terms of definite parameters, and a summation effected over all
species present. For present purposes we restrict ourselves to
two species (monomer and its dimer) being present, whose
weights are given via the law of mass action, characterized by
an equilibrium constant Ka. We follow conventional practice
and express the interaction in terms of the inverse of Ka, namely
Kd. In terms of a single second virial term, BM, (optionally)
assumed to be common to monomer and dimer, then
Equation (3) is written as
cr ¼ ca exp 0:5sw 1þ 2BMcrf g r2a � r2� �� �
Z (4)
and the inversion of Equation (4) is seen to yield Equation (1).
There is however no limit in principle to the number of interaction
terms which could be written into Equation (3) prior to inversion.
The inner bracketed term can be written as
sw 1þ 2BMcr þ 3CMc2r�
(5)
for the case of values common to monomer and to dimer of
the second (2BM) and third (3CM) virial terms. Equally, for
two non-identical second virial terms, (BM)1 and (BM)2
DOI: 10.1002/mabi.201000065

Evaluation of the Information Content of Sedimentation . . .
characterizing the monomer and dimer, respectively, the inner
term is given as
Macrom
� 2010
s 1þ 2 a� 1ð Þ BMð Þ1cr þ 2a BMð Þ2cr�
(6)
where a represents the fraction of the total concentration of
monomers in dimer form, again derived directly from the Ka value
(which within the fit will be the current estimate of that
parameter).
It is important to understand however that there is a practical
limit to thenumberof interaction termswhich canbeadded, not in
terms of theory but in terms of the stability of the numerical
solutionswhichcanbeextracted. There isnopoint inaddinga large
numberof terms inanattempttoachieve ‘rigour’ if theoutcomeisa
set of equations incapable of simultaneous solution in terms of
numerical estimates obtainable for all these parameters.
In this latter context we must briefly address a recent doubt
whichhas been expressed[13]with regard to the INVEQapproach. It
is alleged that our derivation of the inverted equation (Equation 1)
requires the use of the Fujita-Adams[14] approximation; and also
attempts the algebraically impossible in defining both thermo-
dynamic and mass interaction parameters. Leaving aside the
(irrelevant) issue as towhether the assumption that (BM)1¼ (BM)2is or is not correctly attributed to ‘Fujita-Adams’, this assumption is
indeed onewhichwe havemade formany purposes: but it is in no
way required. No new equation is being ‘derived’, merely a re-
arrangementby trivial algebraofanexisting (universally accepted)
equation (Equation (3) above), with assignation of discrete
parameters to the thermodynamic interaction terms. As regards
the second allegation, we accept of course that in the limit of
infinite dilution the resolution of thermodynamic and interaction
termscannotbepossible, sinceonlya ‘limitingslope’, single-valued
of necessity, can be defined for the c-dependence: but we are not
restricting ourselves to this limiting state. Our present work
establishes further that not only is the INVEQ approach algebrai-
cally sound, but that the precision in experimental data is more
than sufficient for its practical implementation. Finally, as wewill
discuss further, we note that other workers using other methods
have successfully implemented an algorithm and practical
methods for the separate resolution of the thermodynamic and
mass interaction terms.[15] In particular, Zorrilla et al[15] – not
cited[13] – have described a ‘general model for self-association and
non-ideal repulsive interaction in a solution containing a single
solute component’ and formulated ‘an algorithm for simulta-
neously calculating the composition of solute species and signal
average buoyant molar mass of the solute as a function of its total
concentration’ (loci cit). A particular model (the long-established
‘hard sphere’ model) is used, and a long-duration, long-column
experimental system is employed, but the outcome is similar to
that given by the use of the (model free) INVEQ algorithm.
At this juncture, we note that there is an infinite set of
combinations of parameters which could be assigned when
we parameterize Equation (3). After second and third, why could
we not add fourth, fifth or higher virial terms? Why not assign
multiple Ka values for complex interactions? It has been our
intention in the work now presented to define just how many
stable parameter estimates it is possible to retrieve froma set of SE
data, at a level of precisionwhichyields results ‘of interest’. Initially
ol. Biosci. 2010, 10, 798–807
WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
and in our work published to date we have confined our enquiry –
subjectively – to the evaluation of lower (i.e., second) order
‘thermodynamic interaction parameters’, defined by conventional
virial terms, and by a single specific ‘weak’ (i.e., Kd� 0.1�10�3M)
interaction term. Thesehavebeen entered into the programcodeof
the function used as given in Equation (5) and (6).
Finally, the analysis of the data set, whether for simulated or for
real data, is routinely carried out within pro Fit (although the
precise software used is not critical, provided that the conditions
earlier defined aremet in the implementation). A single data point
in the region of the ‘hinge point’ of the equilibrium is selected, and
the co-ordinates (rref, cref) entered into the parameter window
associated with the function being employed. Estimates are
entered for the s value of the solute monomer derived via
SEDNTERP software for real solutes, for all the interaction terms
which are to be floated, and for a baseline offset E. Errors in initial
estimates for the latter quantity are generally large. But the INVEQ
functionshavebeencoded insuchawaythat cref is expectedtobe in
the form (signal þE) rather than signal, which causes the function
curve displayed in the Preview Window to rotate around the
selected central point as E is varied. Hence a surprisingly good
estimate for E can be obtained by simply ‘dragging’ this curvewith
E as the variable.
If this manual fit is clearly very close to the curve, then a ML fit
with 500 iterations using input error estimates for raw data is
performed, followed by binning and plotting out of histograms for
the parameter estimates. Otherwise, and in most cases, further
fitting in which the estimates for the interaction parameters are
refined is carried out, followed by the use of a Robust algorithm to
generate near-optimal parameter estimates prior toMLfitting. The
limits given for each parameter by the boot-strapping procedure at
the end of the MANUAL/Robust/ML fitting procedure are taken as
defining the quality of the estimate.
AUC Analysis
Highly purified salt-free lyophilized Bovine Pancreatic Ribonu-
clease A was purchased from Teknova (Hollister, California, USA).
After dissolving in PBS (phosphate buffered saline) adjusted to pH
7.0, or in this solvent pre-diluted to varying ionic strengths, the
concentration was checked spectrophotometrically and adjusted
by dilution to the required concentration. The nominal ionic
strengthof PBSwas taken for practical purposes to be 150�10�3M;
the exact value would vary slightly with pH. Simple proportional
dilution with water was performed to give a range of (again,
nominal) ionic strengths. A nominal ‘zero ionic strength’ solution
was achieved by dissolving the protein in water without pH
adjustment. Ultracentrifugal analysis was performed at 20 8C in a
Beckman XL-I Analytical Ultracentrifuge, using Rayleigh Inter-
ference Optics. A 2.5mm solution column length for both solution
and reference solventwas employed,withmeniscus level carefully
matched. Initial scans at 3 000 rpm.were recorded, to check for the
presence of any large aggregates. None were found. After
acceleration of the rotor to final speed (20 000 rpm) scans were
recorded immediately, and then every 2h until the patterns
recorded became time-invariant. Our instrument gives very level
fringepatterns for the ‘no redistribution’ case, andsuchvariation in
level along the course of the data region (TI noise) is small and not
www.mbs-journal.de 801
S. Ang, A. J. Rowe
802
obviously reproducible from cell to cell. The use of a ‘blank’ cell is
thus pointless – a better removal of the TI noise is achieved by
subtracting the first scan at speed from the final selected scan, and
this we do. All scans were logged to disc using the Beckman
ProteomeLab software.
Results
Estimation of Error in Raw Data
AsimpleExcel routine (FringeNoise.xls)hasbeenemployedto
attempt to eliminate TI noise and to give an estimate for the
stochasticnoise.Thetabulatedvaluesforthefringeincrement
over a range of 200 radial values are subtracted from the
values for the increment at the same radial values in the
preceding scan, and the tabulated differences thus produced
are plotted against radial value. This should eliminate TI
noise. Andwhere the average difference is clearly close to an
integral, then thealgebraic valueof that integral is subtracted
from the initial difference value, whichmostly eliminates RI
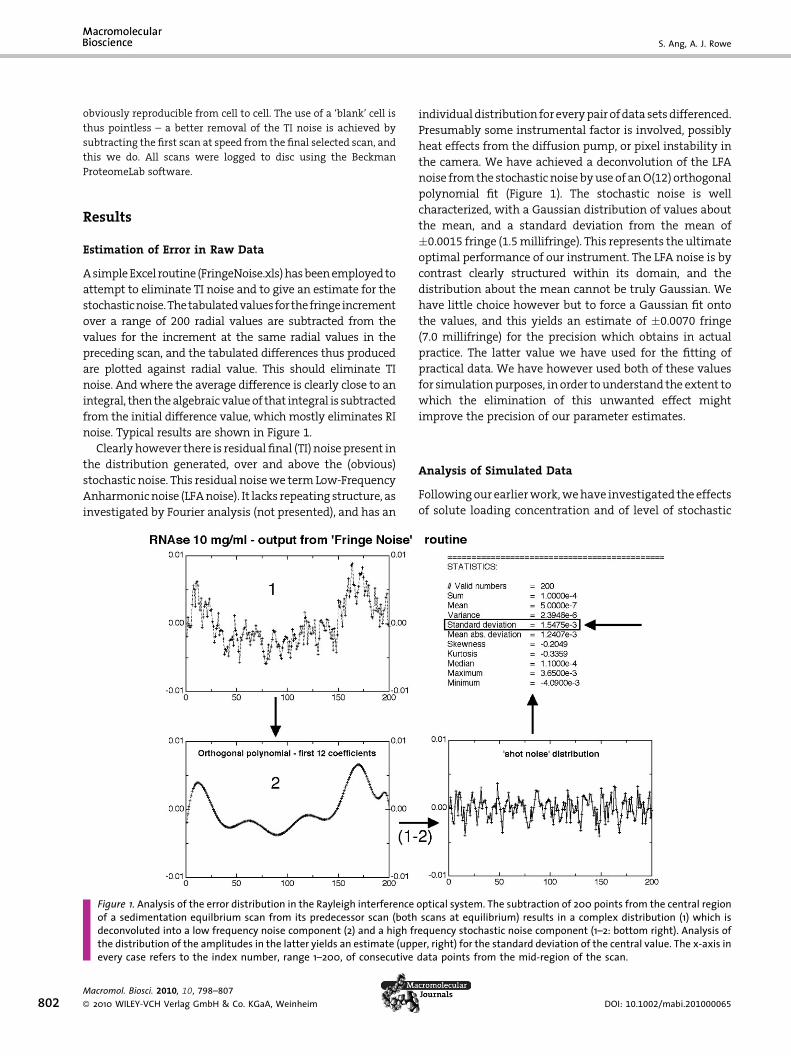
noise. Typical results are shown in Figure 1.
Clearlyhowever there is residual final (TI) noise present in
the distribution generated, over and above the (obvious)
stochastic noise. This residual noisewe termLow-Frequency
Anharmonicnoise (LFAnoise). It lacks repeating structure, as
investigated by Fourier analysis (not presented), and has an
Figure 1. Analysis of the error distribution in the Rayleigh interferenceof a sedimentation equilbrium scan from its predecessor scan (bothdeconvoluted into a low frequency noise component (2) and a high frthe distribution of the amplitudes in the latter yields an estimate (uppevery case refers to the index number, range 1–200, of consecutive
Macromol. Biosci. 2010, 10, 798–807
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
individualdistribution foreverypairofdata setsdifferenced.
Presumably some instrumental factor is involved, possibly
heat effects from the diffusion pump, or pixel instability in
the camera. We have achieved a deconvolution of the LFA
noise fromthe stochastic noise byuse of anO(12) orthogonal
polynomial fit (Figure 1). The stochastic noise is well
characterized, with a Gaussian distribution of values about
the mean, and a standard deviation from the mean of
�0.0015 fringe (1.5millifringe). This represents the ultimate
optimal performance of our instrument. The LFA noise is by
contrast clearly structured within its domain, and the
distribution about the mean cannot be truly Gaussian. We
have little choice however but to force a Gaussian fit onto
the values, and this yields an estimate of �0.0070 fringe
(7.0 millifringe) for the precision which obtains in actual
practice. The latter value we have used for the fitting of
practical data. We have however used both of these values
for simulationpurposes, in order tounderstand the extent to
which the elimination of this unwanted effect might
improve the precision of our parameter estimates.
Analysis of Simulated Data
Followingourearlierwork,wehave investigated theeffects
of solute loading concentration and of level of stochastic
optical system. The subtraction of 200 points from the central regionscans at equilibrium) results in a complex distribution (1) which is
equency stochastic noise component (1–2: bottom right). Analysis ofer, right) for the standard deviation of the central value. The x-axis indata points from the mid-region of the scan.
DOI: 10.1002/mabi.201000065

Evaluation of the Information Content of Sedimentation . . .
noise in the data on the precision of retrieval of input
parameters. Selected data from regions critical for valid
parameter retrieval are presented. Initiallywehave studied
the case of floating (Ka, BM) together with the baseline
offsetEandareferenceconcentrationvalue,uncorrected for
E, at a radial value located (butnot critically) in the regionof
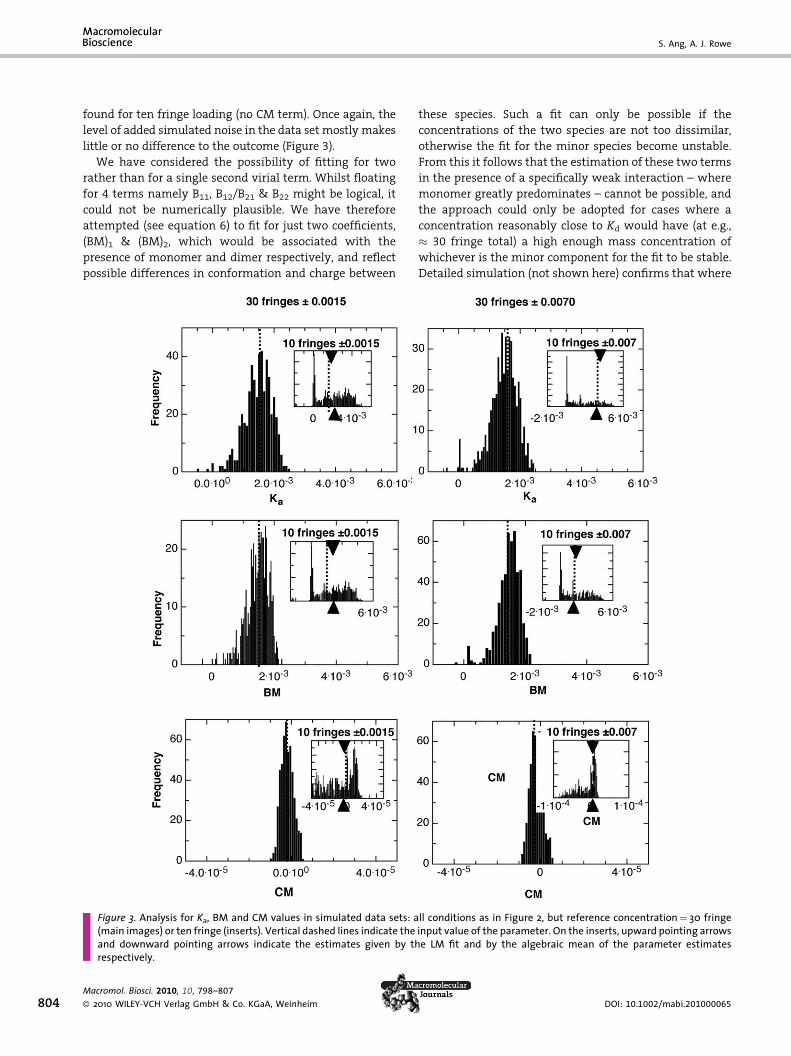
the ‘hingepoint’. Figure 2 (mainplots) shows the results of a
comparison of two cell loading concentrations (10 fringe or
30 fringe load) at two different levels of precision: namely
an ‘optimal’ data set with true stochastic noise only
(�0.0015 fringe) and a ‘realistic’ data set, in which the
LFA noise is present, and the overall distribution of the
total noise set is approximated by a gaussian distribution
(�0.0070 fringe). As is evident, very little difference
between the twoparameter sets is returned. Theparameter
distributions are all close to Gaussian, and on this basis a
standard deviation (SD) of the parameters Ka and BM
around the final estimate is calculated as � �1–3%. Thus
the level of error assumed to be present in the datamakes –
perhaps surprisingly– little ornodifference to theoutcome.
When the cell loading concentration is reduced to 3
fringe, however, the parameter distributions are very
Figure 2. Analysis for Ka and BM values in simulated data sets: s¼concentration¼ 10 fringe (main images) or 3 fringe (smaller inserted iParameter estimates are obtained from 500 iterations of the fit, errincrement) and �0.0002 (radial value) at 68.4% confidence interval. Vinserts, upward pointing arrows and downward pointing arrows indicaparameter estimates respectively
Macromol. Biosci. 2010, 10, 798–807
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
different (Figure 2, inset plots). Reflecting the known
complexity of the error surface for these fits, there is no
single peak present, but rather an approximately bi-modal
distribution, strongly skewed. The fit by ML is better than
might be expected, within �50% of input value, and the
weight-average of the parameters in the distribution is
even closer to input value (Figure 2, inset plots). However,
we are clearly on (or over) the borderline beyondwhich the
methodology is of practical use. Again, the level of error
added to the simulated data seems to have little relevance.
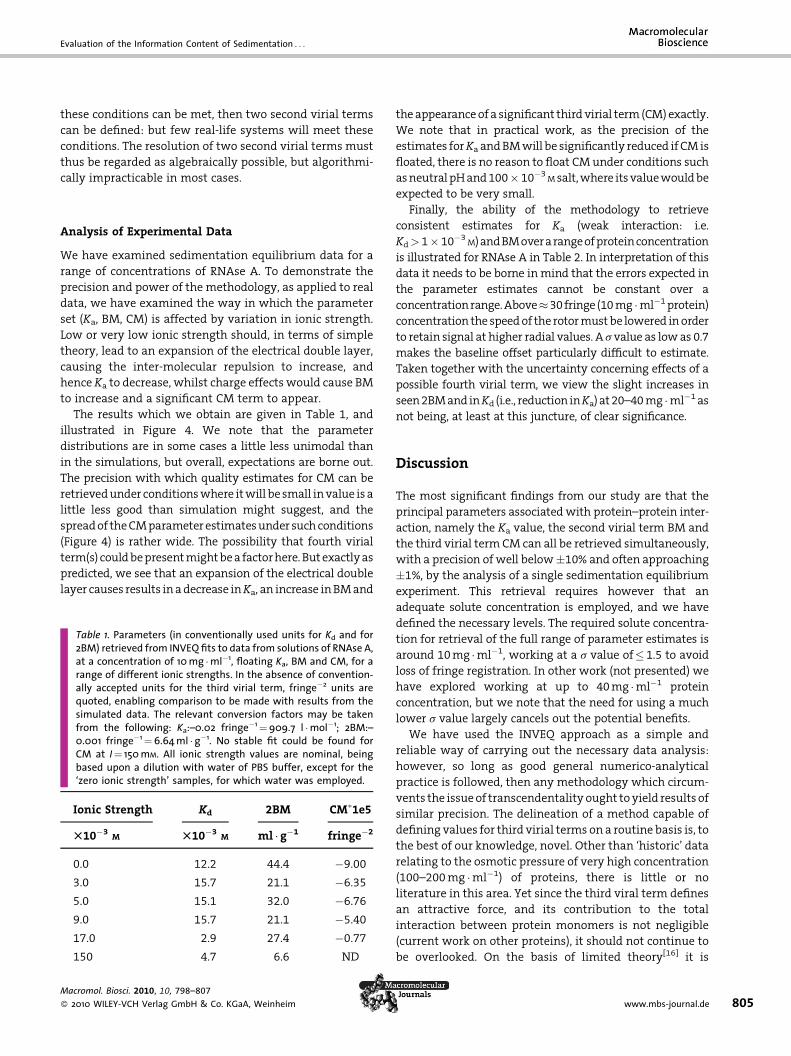
When an additional term (the third virial term, CM) is
added, then we would expect that higher cell loading
concentrations would need to be employed to obtain stable
fits. This is found to be the case. In Figures (main and inset
plots)we see the effect of theuse of concentrations of 30 and
10 fringe. At 30 fringe cell loading (main plots) it is clearly
possible to retrieve stable estimates for CM, SD� 5%, even at
the verynumerically low simulated value employed. For the
parameters Ka and BM the additional cell loading concen-
tration approximately compensates for the employment of
an additional parameter (CM) in the fit, and the precision
found for the parameters Ka and BM approximates to that
1.5; Ka¼BM¼0.0015 (fringe concentration units), with referencemages) and with normal random error added at the levels indicated.ors in the fitting routine fixed at �0.007 or þ0.0015 (for the fringeertical dashed lines indicate the input value of the parameter. On thete the estimates given by the LM fit and by the algebraic mean of the
www.mbs-journal.de 803
S. Ang, A. J. Rowe
804
found for ten fringe loading (no CM term). Once again, the
level of added simulated noise in the data set mostly makes
little or no difference to the outcome (Figure 3).
We have considered the possibility of fitting for two
rather than for a single second virial term. Whilst floating
for 4 terms namely B11, B12/B21 & B22 might be logical, it
could not be numerically plausible. We have therefore
attempted (see equation 6) to fit for just two coefficients,
(BM)1 & (BM)2, which would be associated with the
presence of monomer and dimer respectively, and reflect
possible differences in conformation and charge between
Figure 3. Analysis for Ka, BM and CM values in simulated data sets: a(main images) or ten fringe (inserts). Vertical dashed lines indicate theand downward pointing arrows indicate the estimates given by threspectively.
Macromol. Biosci. 2010, 10, 798–807
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
these species. Such a fit can only be possible if the
concentrations of the two species are not too dissimilar,
otherwise the fit for the minor species become unstable.
From this it follows that the estimation of these two terms
in the presence of a specifically weak interaction – where
monomer greatly predominates – cannot be possible, and
the approach could only be adopted for cases where a
concentration reasonably close to Kd would have (at e.g.,
� 30 fringe total) a high enough mass concentration of
whichever is the minor component for the fit to be stable.
Detailed simulation (not shown here) confirms that where
ll conditions as in Figure 2, but reference concentration¼ 30 fringeinput value of the parameter. On the inserts, upward pointing arrowse LM fit and by the algebraic mean of the parameter estimates
DOI: 10.1002/mabi.201000065
Evaluation of the Information Content of Sedimentation . . .
these conditions can be met, then two second virial terms
can be defined: but few real-life systems will meet these
conditions. The resolution of two second virial terms must
thus be regarded as algebraically possible, but algorithmi-
cally impracticable in most cases.
Analysis of Experimental Data
We have examined sedimentation equilibrium data for a
range of concentrations of RNAse A. To demonstrate the
precision and power of the methodology, as applied to real
data, we have examined the way in which the parameter
set (Ka, BM, CM) is affected by variation in ionic strength.
Low or very low ionic strength should, in terms of simple
theory, lead to an expansion of the electrical double layer,
causing the inter-molecular repulsion to increase, and
henceKa to decrease, whilst charge effects would cause BM
to increase and a significant CM term to appear.
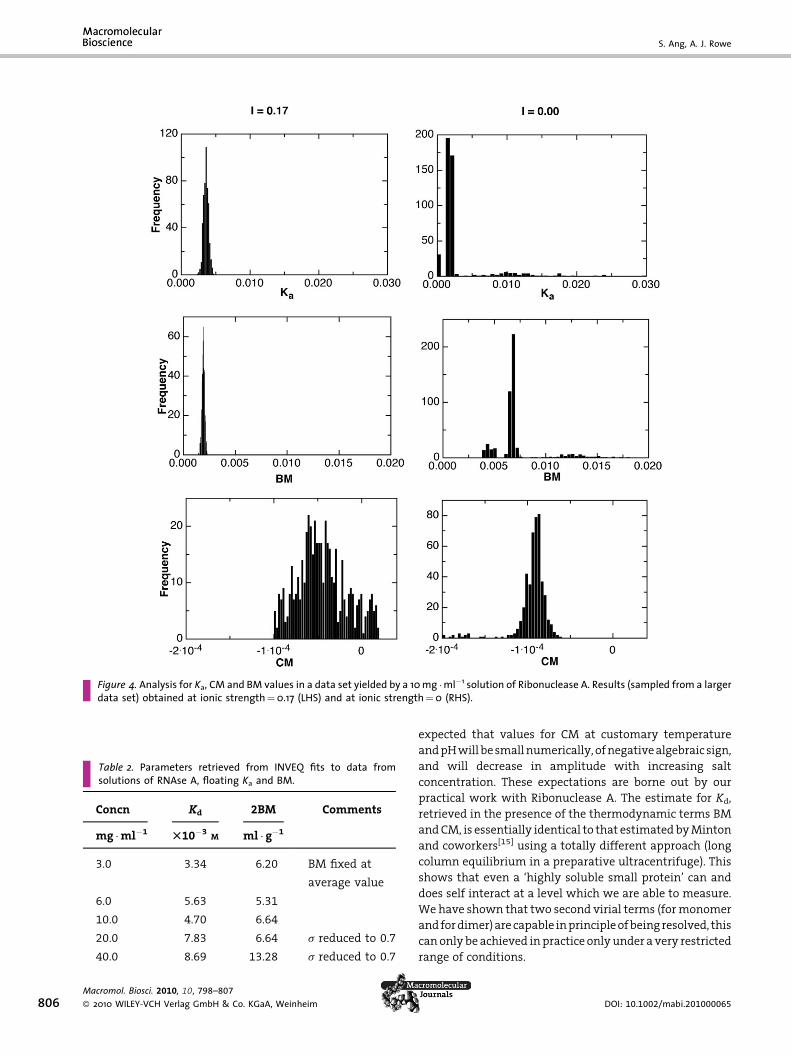
The results which we obtain are given in Table 1, and
illustrated in Figure 4. We note that the parameter
distributions are in some cases a little less unimodal than
in the simulations, but overall, expectations are borne out.
The precision with which quality estimates for CM can be
retrievedunder conditionswhere itwill besmall invalue isa
little less good than simulation might suggest, and the
spreadof theCMparameterestimatesunder suchconditions
(Figure 4) is rather wide. The possibility that fourth virial
term(s) couldbepresentmightbea factorhere.Butexactlyas
predicted, we see that an expansion of the electrical double
layer causes results inadecrease inKa, an increase inBMand
Table 1. Parameters (in conventionally used units for Kd and for2BM) retrieved from INVEQ fits to data from solutions of RNAse A,at a concentration of 10 mg �ml�1, floating Ka, BM and CM, for arange of different ionic strengths. In the absence of convention-ally accepted units for the third virial term, fringe�2 units arequoted, enabling comparison to be made with results from thesimulated data. The relevant conversion factors may be takenfrom the following: Ka:–0.02 fringe�1¼909.7 l �mol�1; 2BM:–0.001 fringe�1¼6.64 ml �g�1. No stable fit could be found forCM at I¼ 150 mM. All ionic strength values are nominal, beingbased upon a dilution with water of PBS buffer, except for the‘zero ionic strength’ samples, for which water was employed.
Ionic Strength Kd 2BM CM�1e5
T10�3M T10�3
M ml � g�1 fringe�2
0.0 12.2 44.4 �9.00
3.0 15.7 21.1 �6.35
5.0 15.1 32.0 �6.76
9.0 15.7 21.1 �5.40
17.0 2.9 27.4 �0.77
150 4.7 6.6 ND
Macromol. Biosci. 2010, 10, 798–807
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
theappearanceof a significant thirdvirial term (CM)exactly.
We note that in practical work, as the precision of the
estimates forKa andBMwill be significantly reduced if CMis
floated, there is no reason to float CMunder conditions such
asneutralpHand100� 10�3Msalt,where itsvaluewouldbe
expected to be very small.
Finally, the ability of the methodology to retrieve
consistent estimates for Ka (weak interaction: i.e.
Kd> 1� 10�3M)andBMoverarangeofproteinconcentration
is illustrated for RNAse A in Table 2. In interpretation of this
data it needs to be borne inmind that the errors expected in
the parameter estimates cannot be constant over a
concentrationrange.Above�30fringe (10mg �ml�1protein)
concentration the speedof the rotormustbe lowered inorder
to retain signal at higher radial values. A s value as lowas 0.7
makes the baseline offset particularly difficult to estimate.
Taken together with the uncertainty concerning effects of a
possible fourth virial term, we view the slight increases in
seen2BMand inKd (i.e., reduction inKa) at20–40mg �ml�1as
not being, at least at this juncture, of clear significance.
Discussion
The most significant findings from our study are that the
principal parameters associatedwith protein–protein inter-
action, namely the Ka value, the second virial term BM and
the third virial termCM can all be retrieved simultaneously,
with a precision ofwell below�10% and often approaching
�1%, by the analysis of a single sedimentation equilibrium
experiment. This retrieval requires however that an
adequate solute concentration is employed, and we have
defined the necessary levels. The required solute concentra-
tion for retrieval of the full range of parameter estimates is
around 10mg �ml�1, working at a s value of� 1.5 to avoid
loss of fringe registration. In other work (not presented) we
have explored working at up to 40mg �ml�1 protein
concentration, but we note that the need for using a much
lower s value largely cancels out the potential benefits.
We have used the INVEQ approach as a simple and
reliable way of carrying out the necessary data analysis:
however, so long as good general numerico-analytical
practice is followed, then any methodology which circum-
vents the issueof transcendentality ought toyield results of
similar precision. The delineation of a method capable of
defining values for third virial terms on a routine basis is, to
the best of our knowledge, novel. Other than ‘historic’ data
relating to the osmotic pressure of very high concentration
(100–200mg �ml�1) of proteins, there is little or no
literature in this area. Yet since the third viral term defines
an attractive force, and its contribution to the total
interaction between protein monomers is not negligible
(current work on other proteins), it should not continue to
be overlooked. On the basis of limited theory[16] it is
www.mbs-journal.de 805
S. Ang, A. J. Rowe
Figure 4. Analysis for Ka, CM and BM values in a data set yielded by a 10 mg �ml�1 solution of Ribonuclease A. Results (sampled from a largerdata set) obtained at ionic strength¼0.17 (LHS) and at ionic strength¼0 (RHS).
Table 2. Parameters retrieved from INVEQ fits to data fromsolutions of RNAse A, floating Ka and BM.
Concn Kd 2BM Comments
mg �ml�1 T10�3M ml � g�1
3.0 3.34 6.20 BM fixed at
average value
6.0 5.63 5.31
10.0 4.70 6.64
20.0 7.83 6.64 s reduced to 0.7
40.0 8.69 13.28 s reduced to 0.7
806Macromol. Biosci. 2010, 10, 798–807
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
expected that values for CM at customary temperature
andpHwillbesmallnumerically, ofnegativealgebraic sign,
and will decrease in amplitude with increasing salt
concentration. These expectations are borne out by our
practical work with Ribonuclease A. The estimate for Kd,
retrieved in the presence of the thermodynamic terms BM
andCM, is essentially identical to that estimatedbyMinton
and coworkers[15] using a totally different approach (long
column equilibrium in a preparative ultracentrifuge). This
shows that even a ‘highly soluble small protein’ can and
does self interact at a level which we are able to measure.
We have shown that two second virial terms (formonomer
andfordimer)arecapable inprincipleofbeingresolved, this
canonlybe achieved inpractice onlyunder a very restricted
range of conditions.
DOI: 10.1002/mabi.201000065

Evaluation of the Information Content of Sedimentation . . .
Asurprisingfindingwhichcomes fromoursimulations is
that the level of noise in the basic SE data is of relatively
little importance in the quality of the interaction para-
meters retrieved. This may simply reflect the fact that the
(realistic) error range of �0.0015 to �0.0070 fringe is
populated by valueswhich are very small in comparison to
the total signal of 10–30 fringe. As noted above, we must
usually restrict ourselves in simulation and in practice to
rotor speeds yielding s values of� 1.5, to avoid loss of
registration of fringe patterns.
The MANUAL/Robust/ML composite fitting procedure
whichweemploy is rooted in several decades of practice in
the optimization of non-linear fitting routines, where it
has long been apparent than initial choices of parameters
are crucial. It may be compared with the MANUAL/
Simplex/ML procedure suggested[17] as optimal for the
application of SEDPHAT software to SE data. The account
given therein by Balbo[17] of the limitations of a search-
linked Monte Carlo routine as the second stage or the
procedure is mirrored by our own experience in the
development of INVEQ fitting.
Conclusion
Analysis of both computer-simulated and real data (for
RNAse A) yields parameter distributions which show that
for solute concentrations of at least 30 fringe, a single
sedimentation equilibrium experiment can be analysed to
yield stable estimates for the third virial term (CM), in
addition to estimates for the specificmass interaction term
(Ka) and the second virial term (BM): although the third
virial term is expected to be very small and belowdetection
range at a physiological pH and ionic strength. A solute
concentration of only ten fringe is too low for this to be
achievable, but in this case fitting for (Ka, BM) returns
stable and precise estimates for these parameters, as has
been shown in a range of published studies.[6,8–12] The
procedure presented for the routine determination of
values for the third virial interaction term in protein
solutionsappears tobenovel, andknowledge thusobtained
for this protein–protein interaction term could be of
importance. Concerning the limits to which the use of
the AUC in this area might be pressed, the errors in
the estimation of fringe increment values in the
Rayleigh interference optical system have been defined.
In addition to simple, high frequency stochastic noise a
large but non-reproducible level of ‘Low Frequency
Anharmonic’ noise (LFA noise) is invariably present.
However detailed simulation shows that the elimination
Macromol. Biosci. 2010, 10, 798–807
� 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
of this LFAnoisewouldachievesurprisingly little in theway
of improvement of precision in retrieved interaction
parameters.
Acknowledgements: This work was supported by the NCMHBusiness Centre.
Received: February 12, 2010; Revised: April 5, 2010; Publishedonline: June 30, 2010; DOI: 10.1002/mabi.201000065
Keywords: interaction; protein; RNAse; sedimentation equili-brium ;simulation; virial coefficients
[1] Ultracentrifugation in Biochemistry & Polymer Science, S. E.Harding, J. C. Horton, A. J. Rowe, Eds., The Royal Society ofChemistry, London 1992.
[2] Analytical Ultracentrifugation: Techniques and Methods, D. J.Scott, S. E. Harding, A. J. Rowe, Eds., The Royal Society ofChemistry, London 1992.
[3] J. S. Philo, AAPS J . 2005, 8, Article 65.[4] P. Schuck, Biophys. J. 2000, 78, 1606.[5] P. H. Brown, A. Balbo, P. Schuck, AAPS J. 2008, 10, 481.[6] A. J. Rowe, ‘‘Weak Interactions: Optimal Algorithms for Their
Study in the AUC,’’ in: The Analytical Ultracentrifuge: Tech-niques&Methods,D. J. Scott, S. E. Harding, A. J. Rowe, Eds., TheRoyal Society of Chemistry, London 2005.
[7] T. S. Ahearn, R. T. Staff, T. W. Redpath, I. K. Semple, Phys. Med.Biol. 2005, 50, N85–N92.
[8] R. Luo, B. Mann, W. S. Lewis, A. Rowe, R. Heath, M. L. Stewart,A. E. Hamburger, S. Sivakolundu, E. Lacy, P. J. Bjorkman,E. Tuomanen, R. W. Kriwacki, EMBO J. 2005, 24, 1.
[9] C. L. Gee, A. Nourse, A.-Y. Hsin, Q. Wu, J. D. Tyndall, G. G.Grunwald, M. J. McLeish, J. L. Martin, Biochim. Biophys. Acta2005, 1750, 82.
[10] W. Zeng, H. E. Seward, A. Malnasi-Csizmadia, S. Wakelin, R. J.Woolley, G. S. Cheema, J. Basran, T. R. Patel, A. J. Rowe, C. R.Bagshaw, Biochemistry 2006, 45, 10482.
[11] A. Nyarko, K. Moshabi, A. J. Rowe, A. Leech, M. Boter,K. Shirasu, C. Kleanthous, Biochemistry 2007, 46, 11331.
[12] N. P. Mullin, P. Nicholas, A. Yates, A. J. Rowe, B. Mijmeijer,D. Colby, P. N. Barlow, M. Walkinshaw, I. Chambers, Biochem.J. 2008, 4, 227.
[13] D. R. Winzor, P. R. Wells, Anal. Biochem. 2007, 368, 168.[14] E. T. Adams, Jr, H. Fujita, Ultracentrifugal Analysis, J. W.
Williams, Ed., Academic Press, New York 1963, p. 119.[15] S. Zorilla, M. Jimenez, P. Lillo, G. Rivas, A. P. Minton, Biophys.
Chem. 2004, 108, 89.[16] D. X. Liu, H. W. Xiang, Int. J. Thermophys. 2003, 24, 1667.[17] Andrea Balbo http://www.analyticalultracentrifugation.
com/sedphat/MixTutorial.htm#Step%2019:%20On%20the%20statistical%20error%20analysis.
www.mbs-journal.de 807


















