
EpCAM/CD3-Bispecific T-cell Engaging Antibody...
11
Cancer Therapy: Preclinical EpCAM/CD3-Bispecific T-cell Engaging Antibody MT110 Eliminates Primary Human Pancreatic Cancer Stem Cells Michele Cioffi 1 , Jorge Dorado 1 , Patrick A. Baeuerle 2 , and Christopher Heeschen 1 Abstract Purpose: Tumor-initiating cells with stem-like properties, also termed cancer stem cells (CSC), have been shown to sustain tumor growth as well as metastasis and are highly resistant to chemotherapy. Because pancreatic CSCs have been isolated on the basis of EpCAM expression, we investigated whether a targeted immunotherapy to EpCAM using the bispecific T-cell–engaging antibody MT110 is capable of eradicating CSCs. Experimental Design: We studied in vitro and in vivo the effects of MT110 on CSCs using both established cell lines as well as primary cells of human pancreatic cancer. Results: Although established cell lines were more responsive to MT110-engaged T cells, also primary cells showed a time- and dose-dependent response to treatment with the bispecific antibody. In addition, the population of highly tumorigenic CSCs was efficiently targeted by the EpCAM/CD3-bispecific anti- body MT110 in vitro and in vivo using a mouse model of established primary pancreatic cancer. Pan- creatic cancer cells derived from metastases were slightly more resistant to MT110 treatment on the basis of in vivo tumorigenicity studies. This appeared to be related to a higher frequency of an EpCAM-negative subpopulation of CSCs. Conclusions: Cytotoxic T cells can be effectively redirected against primary human pancreatic cancer cells by T-cell–engaging BiTE antibody MT110 including a subpopulation of highly tumorigenic CSCs. Clin Cancer Res; 18(2); 465–74. Ó2011 AACR. Introduction Pancreatic cancer is the fourth most frequent cause for cancer-related death (1) and is characterized by early metas- tasis and pronounced resistance to chemotherapy and radi- ation. Despite extensive research efforts, hardly any sub- stantial progress with regard to improvement in clinical endpoints has occurred over the past decades. The only currently available effective treatment modality for pancre- atic cancer requires a very invasive and complex surgical procedure, also known as Whipple procedure. Although these patients show an extended median survival of 20 months, only a minority of patients with local disease are suitable for surgical intervention (20% of patients; ref. 2). For patients with advanced disease, the introduction of the antimetabolite gemcitabine more than a decade ago improved clinical response by reducing pain and weight loss (3). However, the prognosis of patients with metastatic pancreatic cancer has remained extremely poor with a 5- year survival rate of 3% to 4% and a median survival period of 4 to 6 months (4). Therefore, new approaches for targeting the complex biology of pancreatic cancer are desperately needed to pave the way for the development of more effective treatments for these patients. Increasing evidence indicates that stem cells may play a decisive role not only in the generation of complex multi- cellular organisms but also in the development and pro- gression of tumors (5, 6). Cells bearing stem cell properties may have an integral part in the development and perpet- uation of different human cancers (7–11). The current consensus definition describes a cancer stem cell (CSC) as a cell within a tumor that is able to self-renew and to produce the heterogeneous lineages of cancer cells that comprise the majority of the tumor mass (6). We and others have recently provided supportive evidence for a hierarchi- cal organization of human pancreatic cancer (12, 13). Importantly, pancreatic CSCs are heterogenic, and a sub- population of the CD133 þ cells identified by additional expression of the chemokine receptor CXCR4 bear exclusive metastatic activity (12). As these tumorigenic CSCs are highly resistant to standard chemotherapy, such cells may be the source of the virtually inevitable relapse of pancreatic Authors' Affiliations: 1 Stem Cells & Cancer Group, Clinical Research Programme, Spanish National Cancer Research Centre (CNIO), Madrid, Spain; and 2 Micromet, Inc., Rockville, Maryland Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/). Corresponding Author: Christopher Heeschen, Stem Cells & Cancer Group, Clinical Research Programme, Spanish National Cancer Research Centre (CNIO), Madrid 28034, Spain. Phone: +3491-732-8000; Fax: +3491- 732-8000; E-mail: [email protected] doi: 10.1158/1078-0432.CCR-11-1270 Ó2011 American Association for Cancer Research. Clinical Cancer Research www.aacrjournals.org 465 on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270
Transcript of EpCAM/CD3-Bispecific T-cell Engaging Antibody...

Cancer Therapy: Preclinical
EpCAM/CD3-Bispecific T-cell Engaging Antibody MT110Eliminates Primary Human Pancreatic Cancer Stem Cells
Michele Cioffi1, Jorge Dorado1, Patrick A. Baeuerle2, and Christopher Heeschen1
AbstractPurpose: Tumor-initiating cells with stem-like properties, also termed cancer stem cells (CSC), have been
shown to sustain tumor growth as well as metastasis and are highly resistant to chemotherapy. Because
pancreatic CSCs have been isolated on the basis of EpCAM expression, we investigated whether a targeted
immunotherapy to EpCAM using the bispecific T-cell–engaging antibody MT110 is capable of eradicating
CSCs.
ExperimentalDesign:Westudied in vitro and in vivo the effects ofMT110onCSCs using both established
cell lines as well as primary cells of human pancreatic cancer.
Results: Although established cell lines were more responsive to MT110-engaged T cells, also primary
cells showed a time- and dose-dependent response to treatment with the bispecific antibody. In addition,
the population of highly tumorigenic CSCs was efficiently targeted by the EpCAM/CD3-bispecific anti-
body MT110 in vitro and in vivo using a mouse model of established primary pancreatic cancer. Pan-
creatic cancer cells derived from metastases were slightly more resistant to MT110 treatment on the basis
of in vivo tumorigenicity studies. This appeared to be related to a higher frequency of an EpCAM-negative
subpopulation of CSCs.
Conclusions: Cytotoxic T cells can be effectively redirected against primary human pancreatic cancer
cells by T-cell–engaging BiTE antibody MT110 including a subpopulation of highly tumorigenic CSCs.
Clin Cancer Res; 18(2); 465–74. �2011 AACR.
Introduction
Pancreatic cancer is the fourth most frequent cause forcancer-related death (1) and is characterized by earlymetas-tasis and pronounced resistance to chemotherapy and radi-ation. Despite extensive research efforts, hardly any sub-stantial progress with regard to improvement in clinicalendpoints has occurred over the past decades. The onlycurrently available effective treatment modality for pancre-atic cancer requires a very invasive and complex surgicalprocedure, also known as Whipple procedure. Althoughthese patients show an extended median survival of 20months, only a minority of patients with local disease aresuitable for surgical intervention (�20% of patients; ref. 2).For patients with advanced disease, the introduction of the
antimetabolite gemcitabine more than a decade agoimproved clinical response by reducing pain and weightloss (3). However, the prognosis of patients withmetastaticpancreatic cancer has remained extremely poor with a 5-year survival rate of 3% to 4% and amedian survival periodof 4 to 6 months (4). Therefore, new approaches fortargeting the complex biology of pancreatic cancer aredesperately needed to pave the way for the developmentof more effective treatments for these patients.
Increasing evidence indicates that stem cells may play adecisive role not only in the generation of complex multi-cellular organisms but also in the development and pro-gression of tumors (5, 6). Cells bearing stem cell propertiesmay have an integral part in the development and perpet-uation of different human cancers (7–11). The currentconsensus definition describes a cancer stem cell (CSC) asa cell within a tumor that is able to self-renew and toproduce the heterogeneous lineages of cancer cells thatcomprise themajority of the tumormass (6).We and othershave recently provided supportive evidence for a hierarchi-cal organization of human pancreatic cancer (12, 13).Importantly, pancreatic CSCs are heterogenic, and a sub-population of the CD133þ cells identified by additionalexpression of the chemokine receptor CXCR4 bear exclusivemetastatic activity (12). As these tumorigenic CSCs arehighly resistant to standard chemotherapy, such cells maybe the source of the virtually inevitable relapse of pancreatic
Authors' Affiliations: 1Stem Cells & Cancer Group, Clinical ResearchProgramme, Spanish National Cancer Research Centre (CNIO), Madrid,Spain; and 2Micromet, Inc., Rockville, Maryland
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
Corresponding Author: Christopher Heeschen, Stem Cells & CancerGroup, Clinical Research Programme, Spanish National Cancer ResearchCentre (CNIO),Madrid 28034, Spain. Phone: +3491-732-8000; Fax: +3491-732-8000; E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-11-1270
�2011 American Association for Cancer Research.
ClinicalCancer
Research
www.aacrjournals.org 465
on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270

cancer. Indeed, in cases in which bulk disease is eradicatedby chemotherapy, only to be followed by a relapse, aplausible explanation is that the CSCs have not beendestroyed completely.
Epithelial cell adhesion molecule (EpCAM; CD326) isfrequently overexpressed and functionally altered in malig-nant cells (14, 15), including CSCs (16–18). Certain nor-mal epithelial tissues and embryonic stem cells also expressEpCAM (19–21), but emerging evidence indicates thatEpCAM on normal epithelial tissues is largely sequesteredwithin intercellular boundaries while becoming accessibleon the surface of disintegrated cancer cells (22). Therefore,EpCAMmay represent a promising target for immunother-apy of EpCAM-expressing cancer cells including tumorigen-ic CSCs. As MT110 is a bispecific T-cell–engaging (BiTE)antibody construct, which simultaneously targets EpCAMon tumor cells and the T-cell receptor–CD3 complex on Tcells, we hypothesized that MT110 may also target anderadicate EpCAM-expressing pancreatic cancer cells includ-ing CSCs by redirecting cytotoxic effector T cells.
MT110 and related EpCAM-specific BiTE antibodies havealready shown high antitumor activity in diverse xenograftmodels (23–26). Importantly, studies in syngeneic mousemodels using a BiTE binding tomurine EpCAMandmurineCD3 showed antitumor activity without affecting normalepithelia, which expressed EpCAM at similar level anddistribution as seen in human tissues (27, 28). MT110 iscurrently tested in a dose-escalating phase I clinical trialenrolling patients with diverse epithelial cancers for safetyand initial signs of antitumor activity. Because EpCAM isalso expressed in pancreaticCSCs (12, 13), EpCAM-directedimmunotherapy may provide a new treatment opportunityfor the effective eradication of pancreatic cancer includingCSCs as the putative root of pancreatic cancer.
Methods
Primary human pancreatic cancer cellsHuman pancreatic tumors were obtained with written
informed consent from all patients. For in vitro studies,tissue fragments were minced, enzymatically digested withcollagenase (Stem Cell Technologies) for 90 minutes at
37�C (29) and, after centrifugation for 5 minutes at1,200 r.p.m., the pellets were resuspended and cultured inRPMI, 10% FBS, and 50 units/mL penicillin/streptomycin.Pancreatic cancer spheres were generated by placingpancreatic cancer cells in suspension (20,000 cells/mL) inserum-free Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 medium, supplemented with B27 (1:50; Invitrogen),20 ng/mL basic fibroblast growth factor (bFGF), and50 units/mL penicillin/streptomycin. For serial passaging,7-day-old sphereswere harvested using 40-mmcell strainers,dissociated to single cells with trypsin, and then re-grownfor 7 days. Cultures were kept no longer than 4 weeks afterrecovery from frozen stocks.
CytometryTo identify pancreatic CSCs, cells were trypsinized,
washedwith PBS, and stainedwith the following antibodiesfor 30minutes at 4�C: anti-CD133/1-APC or phycoerythrin(PE; Miltenyi), EpCAM-FITC or APC, CD44-PE, SSEA-1APC, or appropriate isotype-matched control antibodies(Becton Dickinson). To estimate the percentage of apopto-tic cells, samples were labeled for 20 minutes at 4�C withfluorescein isothiocyanate (FITC)-conjugated Annexin Vdiluted in calcium-containing binding buffer and 40,6-dia-midino-2-phenylindole (DAPI), or 7-AAD was used forexclusion of dead cells (eBiosciences). Samples were ana-lyzed by flow cytometry using a FACS Canto II (BectonDickinson) and data were analyzed with FlowJo 9.2software.
Cytotoxicity assayRedirected cellular cytotoxicity was assayed using human
peripheral blood mononuclear cells (PBMC) as effectorcells and various EpCAM-positive human pancreatic cancercells. PBMC were isolated from healthy donors by Ficolldensity gradient centrifugation using standard procedures,washedwith PBS, and resuspended in RPMI-1640 completemedium. The cells were seeded in 96-well plates (NalgenNunc International) at a concentration of 1 � 104 cells perwell in 100 mL of complete medium. The cells were incu-bated for 24 hours to allow sufficient cell adhesion, and allthe microplates were incubated for a total of 72 hours afteradministration of compounds. The cytotoxic activity wasmeasured by sulforhodamine B (SRB)-based cytotoxicityassay as described previously (30). The protein absorbanceof the viable cells at each concentrationwas expressed as therelative percentage of absorbance compared with the con-trol well without drug exposure. Each experiment wascarried out using 3 replicated wells at same drug concentra-tions, and all the experiments were repeated 3 times.
Western blot analysisFor the analysis of protein expression, human primary
pancreatic cancer cells and spheres were homogenized inlysis buffer (40 mmol/L HEPES pH 7.5, 120 mmol/LNaCl, 5 mmol/L MgCl2, 1 mmol/L EGTA, 0.5 mmol/LEDTA, 1% Triton X-100) containing protease and phos-phatase (20 mmol/L a-glycerolphosphate, 2.5 mmol/L
Translational Relevance
On the basis of our previous work showing the exis-tence of cancer stem cells (CSC) in pancreatic cancer andtheir strong resistance to standard chemotherapy, wenowprovidemultiple lines of evidence for the efficacy ofa novel treatment regimen using the EpCAM/CD3-bis-pecific T-cell–engaging antibody MT110. BecauseMT110 has already been tested in clinical trials in othertumor entities, translation of our findings into the clin-ical setting using CSCs as an important biomarker fortherapeutic response could be rapidly achieved.
Cioffi et al.
Clin Cancer Res; 18(2) January 15, 2012 Clinical Cancer Research466
on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270

Na-pyrophosphate) inhibitors. Twenty-five microgramsof cell lysates were analyzed on 12% to 10% SDS-PAGE,and the following primary antibodies were used: anti-Oct4a (2890 Cell Signalling), anti-Nanog (ab21624;Abcam, Inc.), anti-GAPDH (ab8245-100; Abcam, Inc.).Anti-mouse and anti-rabbit immunoglobulin G (IgG)coupled to peroxidase were used as secondary antibodies(Santa Cruz Biotechnology) and the signal was revealedthrough a chemiluminescent detection kit (ECL DetectionKit; Amersham Biosciences).
RNA preparation and real-time PCRTotal RNAs from human primary pancreatic cancer cells
and spheres were extracted with the TRIzol Kit (Life Tech-nologies Inc.) according to the manufacturer’s instructions.A 1 mg total RNA sample was used for cDNA synthesis withSuperScript II reverse transcriptase (Life Technologies Inc.)and random hexamers. Quantitative real-time PCR (qRT-PCR) was carried out using SYBR Green PCR master mix(Qiagen), according to themanufacturer’s instructions. Thefollowing primers were used: Nanog: sense-TGAACCT-CAGCTACAAACAGGT, antisense-AACTGCATGCAGGAC-TGCAGAG; Oct3/4: sense-CTTGCTGCAGAAGTGGGTG-GAGGAA, antisense-CTGCAGTGTGGGTTTCGGGCA;Klf4:sense-ACCCACACAGGTGAGAAACC, antisense-ATGTG-TAAGGCGAGGTGGTC; Sox2: sense-AGAACCCCAAGATG-CACAAC, antisense-CGGGGCCGGTATTTATAATC; andglyceraldehyde-3-phosphate dehydrogenase (GAPDH):sense-CAGGAGCGAGATCCCT, antisense-GGTGCTAAG-CAGTTGGT.
In vivo model for primary human pancreatic cancerSingle-cell suspensions of primary pancreatic cancer cells
together with freshly isolated donor-derived PBMCs at aratio of 1:2 (Cells:PBMC)were injected subcutaneously intothe flanks of 6- to 8-week-old female nude mice (HarlanEurope). The mice were housed and maintained underspecific pathogen-free conditions in accordance with cur-rent regulations and standards of the Instituto de Salud
Carlos III, Madrid, Spain. The tumors weremeasured with acaliper and tumor volumes were calculated according to theformula: tumor volume ¼ [(width2 � length)/2].
Histologic analysisFor histologic evaluation of the tumors, 1 part of the
tumor tissue is fixed in formalin and embedded in paraffin.Another part of the tumor is embedded in O.C.T. com-pound (Sakura Finetek) and stored at �80�C. For thedetection of CSCs, sections were double-stained with PE-labeled CD133 monoclonal antibodies (Miltenyi orAbcam) and antibodies against cytokeratin. Nuclei wereidentified by DAPI (Vector Labs). The CSC frequency wasdefined as the number of CD133þ cells per high-powerfield. All images were generated on a Leica SP5 ConfocalLaser Scanning Microscope.
Statistical analysisResults for continuous variables are expressed asmeans�
SD. Treatment groupswere comparedwith the independentsamples t-test. In case of non-normal distribution, theMann–Whitney U test was used. Pair-wise multiple com-parisons are carried out with the 1-way ANOVA (2-sided)with Bonferroni adjustment. P values < 0.05 were consid-ered statistically significant. All analyses were carried outwith SPSS 19.0 (SPSS Inc.).
Results
Redirected lysis of EpCAM expressing primary humanpancreatic cancer cells by MT110-engaged T cells
The efficacy of T-cell–mediated redirected lysis byMT110of EpCAM-expressing cells was investigated for several pri-mary human pancreatic cancer cells. A total of 3 humanpancreatic adenocarcinoma xenografts (A6L, 185, and198), some of which have been described previously(31), as well as 2 established pancreatic cancer cell lines(L3.6pl and MiaPaCa2) were investigated (12). Primarypancreatic cancer tissues were expanded as xenografts.
Figure 1. Activity of MT110-engaged T cells against humanpancreatic cancer cell lines. AnMT110 dose response for redirectedlysis of human pancreatic cancerlines L3.6pl (A) and MiaPaCa2 (B)pancreatic cancer cells is shownusing an SRB colorimetric assaymeasuring the loss of cell protein.Unstimulated human PBMCs fromhealthy donors were used at aneffector-to-target (E:T) ratio of 5:1.Bar graphs are mean of percentageof viable cells after treatment for 24,48, and 72 hours in the presence ofPBMC alone, or in combination withthe indicated concentrations ofcontrol BiTE antibody or MT110.
0%
20%
40%
60%
80%
100%
120%
24h 48h 72h
0%
20%
40%
60%
80%
100%
120%
24h 48h 72h
*P < 0.05 vs. PBMC + control BiTE
Control BiTEng/mL
MT110 BiTEng/mL
100
––100– 100101–
– ––100– – –
+PBMC
Control BiTEng/mL
MT110 BiTEng/mL
100
––100– 100101–
– ––100– – –
+ PBMC
BA
Cell
via
bili
ty
Cell
via
bili
ty
L3.6pl MiaPaCa2
24 h 48 h 72 h24 h 48 h 72 h
*
*
*
*
*
*
Immunotherapy against Pancreatic Cancer Stem Cells
www.aacrjournals.org Clin Cancer Res; 18(2) January 15, 2012 467
on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270

Isolated cells from these xenografts were cultured at lowpassage number as adherent cells or spheres for functionalenrichment of CSCs (Supplementary Fig. S1). All cellsexpressed EpCAM, CD133, and CD44, as determined byflow cytometry. Redirected lysis of pancreatic cancer cells byMT110-engaged T cells was monitored by loss of cellularprotein using a SRB-based cytotoxicity assay. Unstimulatedallogeneic human PBMCs from healthy donors were thesource of effector cells that were used at an effector-to-target(E:T) ratio of 5:1, which typically contain between 10% and20% cytotoxic CD8þ T cells (32). A Control BiTE antibodyshared the CD3-binding arm with MT110, but otherwiserecognized an herbicide as an irrelevant antigen.
Established pancreatic cancer cell lines L3.6pl and Mia-PaCa2 showed lysis at 1 ng/mL MT110, which was slightlyincreased at 10 and 100 ng/mL MT110 (Fig. 1A and B).Statistically significant redirected lysis of target cells byMT110 required at least 48 hours of coculture. In addition,an insignificant reduction of cell viability was noted forL3.6p1 cells in the presence of healthy donor PBMCs alone.Otherwise, incubation of pancreatic cancer cell lines withMT110, a CD3 only-binding control BiTE, or with PBMCalone did not significantly affect cell viability. Protein loss incell cultures treated with MT110 and PBMC reached up to80% for L3.6pl cells and 70% for MiaPaCa2 cells, whichoverall were less sensitive for redirected lysis by MT110than L3.6pl cells (compare Fig. 1A and B). The remainingfraction of viable cells (i.e., of cell protein) of �20% maycorrespond to the PBMC fraction, which had been presentin the coculture at a 1:5 ratio to target cells.
Primary human pancreatic cancer cell lines A6L, 185, and198 were more resistant to lysis by cytotoxic T cells as
compared with established cell lines (compare Fig. 1A andB with Fig. 2A and B). First, and in contrast to establishedcell lines, primary cells cocultured with allogeneic PBMCand control BiTE maintained a viability of 80% to 90% ascompared with 50% to 70% for established cell lines.Secondly, a statistically significant decrease in cell viabilitywas only observed at a concentration of 100 ng/mLMT110after, minimally, 48 or 72 hours of coculture with compa-rable responsiveness for the 3 different primary cell lines.MT110-induced lysis of target cells was dose- and time-dependent. Treatment with 1 ng/mL MT110 for 48 hoursresulted in 21% cell lysis (P ¼ 0.10) whereas 100 ng/mLMT110 resulted in 41% cell lysis (P¼ 0.03). After 72 hoursat 1 ng/mLMT110, we observed specific lysis in 31%of cells(P ¼ 0.10), and at 100 ng/mL MT110 in 53% of cells (P ¼0.01). Representative images show themicroscopic changesseen in coculture experiments with primary cell line A6L inthe presence and absence of 100 ng/mL each of MT110 andCD3-only binding Control antibody after 72 hours oftreatment (Fig. 2C). Clearance of the cell culture plate fromadhered cells and compilation of effector and target cells inclumps was evident after treatment with MT110.
Activation of T cells in cocultures with primarypancreatic cancer cells by MT110
Next, we explored whether MT110 is capable of inducingT-cell activation in the presence of pancreatic cancer cells.CD8þ T cells from 7-day cocultures of PBMCs with primarypancreatic cancer cell lines A6L and 185 were analyzed byflow cytometry for the de novo expression of early T-cellactivation marker CD69 and late activation marker CD25.CD8þ T cells in PBMCs were initially resting as they
0%
20%
40%
60%
80%
100%
120%
A6L 185 198
0%
20%
40%
60%
80%
100%
120%
A6L 185 198
*P < 0.05 vs. PBMC + control BiTE
48 h treatment
Ce
ll via
bili
ty
PBMC + control BiTE Control PBMC + MT110
A
C
72 h treatment
Ce
ll via
bili
ty
Control BiTEng/mL
MT110 BiTEng/mL
100
––100– 100101–
– ––100– – –
+ PBMC
Control BiTEng/mL
MT110 BiTEng/mL
100
––100– 100101–
– ––100– – –
+ PBMC
B
**
*
**
*Figure 2. Activity of MT110-engaged T cells against humanprimary pancreatic cancer cells. AnMT110 dose response forredirected lysis of primary humanpancreatic cancer cells A6L, 185,and 198 is shown using an SRBcolorimetric assay measuring theloss of cell protein. Bar graphs aremean values of the percentage oflive cells observed after (A) 48 hoursand (B) 72 hours of treatment withdifferent concentrations of MT110at control BiTE. UnstimulatedPBMCs were used as effectors atan E:T ratio of 5:1. C, representativeimages of A6L cells after 72-hourtreatment in the presence ofPBMCs with MT110 (100 ng/mL) orcontrol BiTE (100 ng/mL)antibodies.
Cioffi et al.
Clin Cancer Res; 18(2) January 15, 2012 Clinical Cancer Research468
on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270

expressed only very low levels of CD69 or CD25 (data notshown). Only a slight increase of the fraction of CD69þ/CD8þ T cells was observed after 7 days in the presence ofMT110, which was similar to that seen with the CD3-onlybinding control BiTE in cocultures with A6L and 185 cells(Fig. 3A). In contrast, after 7 days, a robust increase of theCD8þ/CD25þ T cell population to 15.9% in A6L and to21% in 185 cocultures was seen by MT110 treatment vis-�a-vis treatment with the control BiTE (Fig. 3B). In addition,the fraction of non-CD8þ T cells within PBMCs showed acomprehensive upregulation of CD25 surface expressioncomparable with that seen for the CD8þ T-cell population.It is likely that, after 7 days in coculture, only the late T-cellactivationmarker CD25 but not the immediate-early mark-er CD69 was still expressed on CD8þ T cells.
Induction of apoptosis in primary human pancreaticcancer cells by MT110Redirected lysis of A6L and 185 primary pancreas cancer
cells by MT110-engaged T cells was further studied bysurface expression of the proapoptotic marker Annexin Vusing fluorescence-activated cell-sorting (FACS) analysis.Treatment of cells with MT110 at 100 ng/mL for 7 days inthe presence of PBMCs resulted in a significant increase ofthe population of the Annexin Vþ cell population (Fig. 4A).Comparedwith the control BiTE, treatmentwith 100ng/mLMT110 increased the percentage of DAPIþAnnexin Vþ cellsby 10-fold to 29% � 4.2% in A6L cells and by 3-fold to51%� 8.9% in 185 cells. In addition, apoptosis was slightlyincreased by treatment with 100 ng/mL control BiTE com-pared with no addition, that is, from 1.5%� 1% to 3.5%�1.9% in A6L cells and from 9.5%� 2.2% to 17%� 4.9% in
185 cells. In the Annexin V assay, A6L cells appeared to bemore resistant to T-cell–mediated apoptosis byMT110 than185 cells, which was less apparent in the SRB test that alsoused shorter assay periods (see Figs. 1 and 2). Consistently,EpCAMþ Annexin Vþ cells also increased in MT110-treatedcells, with a subsequent decrease in EpCAMþ cells confirm-ing effective targeting of the EpCAMþ cell population (Fig.4B). Importantly, a significant decrease was also detectablenot only for CD133þ cells, in general but also forEpCAMþCD133þ CSC, in particular.
MT110 treatment reduces capacity of pancreatic cancercells to form spheres
We next examined the impact of MT110 treatment onsubsequent sphere formation by pancreatic cancer cells as asurrogate assay for the self-renewal capacity of CSCs. Adher-ent A6L and 185 primary pancreatic cancer cell lines weretreated for 7 days with PBMCs and either control BiTE orMT110 at a concentration of 100 ng/mL. The remainder ofviable cells was plated under sphere-promoting conditionsto assess their remaining sphere-forming capacity. Sphereswere quantified after 7 days by a cell counter. A significantdecrease in sphere formation of 3- and4.2-fold for both A6Land 185 cells, respectively, was observed for cells treatedwithMT110 comparedwith control BiTE (Fig. 5A and B). Inaddition, treatment with control BiTE significantly reducedsphere formation by 185 cells, but not by A6L cells. Itappeared that larger spheres were formed in the presenceof the control BiTE, eventually explaining a reduction innumber (Fig. 5A, middle). Consistent with the data fromAnnexin V staining, the sphere-formation capacity wasmore strongly reduced in 185 cells comparedwith A6L cells.
0 102
103
104
105
<PE-A>
0
102
103
104
105
<F
ITC
-A>
17.8 6.88
20.754.6
0 102
103
104
105
<PE-A>
0
102
103
104
105
<F
ITC
-A>
8.27 16.9
38.935.9
0 102
103
104
105
<PE-A>
0
102
103
104
105
<F
ITC
-A>
21.1 3.81
16.159
0 102
103
104
105
<PE-A>
0
102
103
104
105
<F
ITC
-A>
7.7 21
57.314
CD
8
CD25
185A6L
5.5%
15.9%
3.8%
21%
0 102
103
104
105
<APC-A>
0
102
103
104
105
<F
ITC
-A>
19.5 3.07
10.666.8
0 102
103
104
105
<APC-A>
0
102
103
104
105
<F
ITC
-A>
18 3.28
20.358.5
0 102
103
104
105
<APC-A>
0
102
103
104
105
<F
ITC
-A>
20.2 3.2
7.2869.4
0 102
103
104
105
<APC-A>
0
102
103
104
105
<F
ITC
-A>
20.1 5.79
20.253.9
CD
8
CD69
185A6L
3.3%
3.1%
3.2%
5.8%
PBMC + control BiTE
PBMC + MT110
BA
Figure 3. Activation of CD8þ T lymphocytes in coculture with MT110 and pancreas cancer cells. Stimulation of CD8þ T-cell populations was determined inprimary humanpancreatic cancer cells A6L and 185 treatedwith 100 ng/mL control BiTE orMT110 for 7 days. The expression of the (A) early activationmarkerCD69 and (B) the late activation marker CD25 were used to monitor MT110-dependent T-cell activation. The low fraction of CD25- and CD69-expressingT cells in the control BiTE samples indicates that the cells were unstimulated before MT110 treatment.
Immunotherapy against Pancreatic Cancer Stem Cells
www.aacrjournals.org Clin Cancer Res; 18(2) January 15, 2012 469
on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270
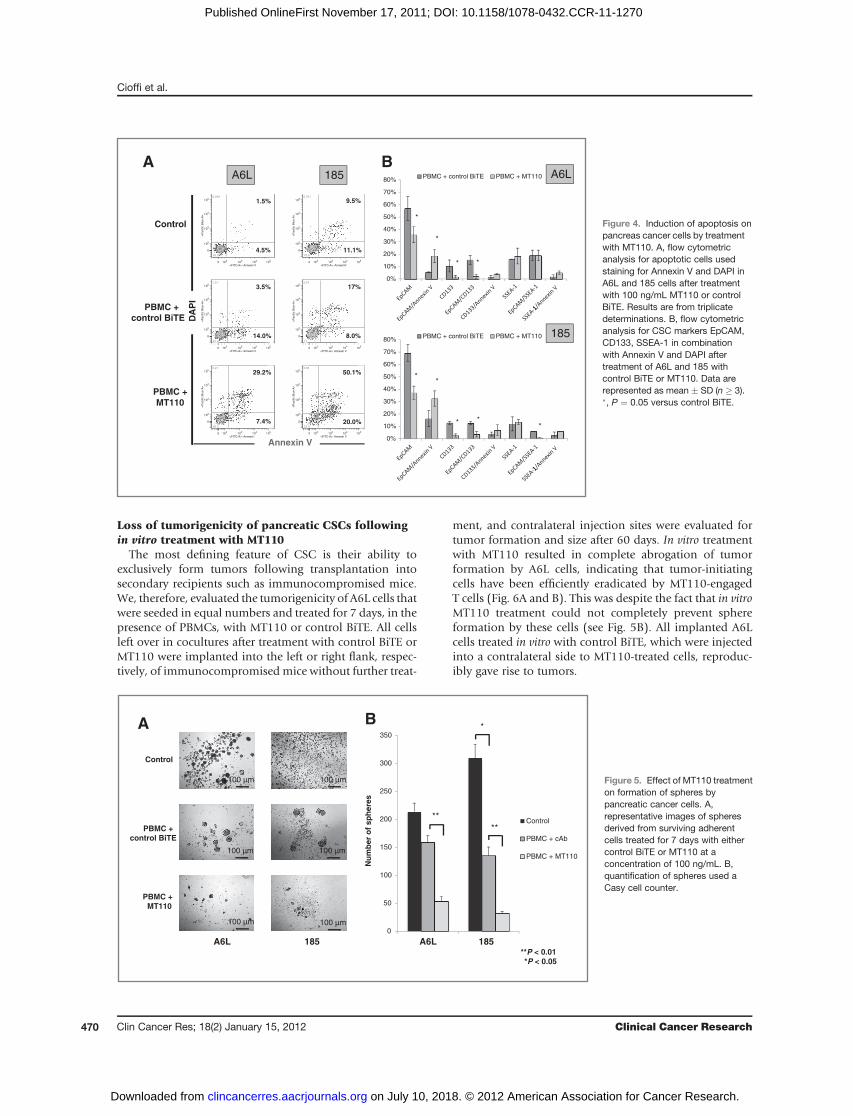
Loss of tumorigenicity of pancreatic CSCs followingin vitro treatment with MT110
The most defining feature of CSC is their ability toexclusively form tumors following transplantation intosecondary recipients such as immunocompromised mice.We, therefore, evaluated the tumorigenicity of A6L cells thatwere seeded in equal numbers and treated for 7 days, in thepresence of PBMCs, with MT110 or control BiTE. All cellsleft over in cocultures after treatment with control BiTE orMT110 were implanted into the left or right flank, respec-tively, of immunocompromised mice without further treat-
ment, and contralateral injection sites were evaluated fortumor formation and size after 60 days. In vitro treatmentwith MT110 resulted in complete abrogation of tumorformation by A6L cells, indicating that tumor-initiatingcells have been efficiently eradicated by MT110-engagedT cells (Fig. 6A and B). This was despite the fact that in vitroMT110 treatment could not completely prevent sphereformation by these cells (see Fig. 5B). All implanted A6Lcells treated in vitro with control BiTE, which were injectedinto a contralateral side to MT110-treated cells, reproduc-ibly gave rise to tumors.
0%
10%
20%
30%
40%
50%
60%
70%
80%PBMC + control BiTE PBMC + MT110
1002
103
104
105
<FITC-A>: Annexin V
0
102
103
104
105
<P
acific
Blu
e-A
>
0.239 1.47
4.5193.8
1002
103
104
105
<FITC-A>: Annexin V
0
102
103
104
105
<P
acific
Blu
e-A
>
1.21 3.45
1481.4
1002
103
104
105
<FITC-A>: Annexin V
0
102
103
104
105
<P
acific
Blu
e-A
>
3.21 29.2
7.4360.2
1.5%
3.5%
29.2%
1002
103
104
105
<FITC-A>: AnnexinV
0
102
103
104
105
<P
acific
Blu
e-A
>
0.761 9.54
11.178.6
1002
103
104
105
<FITC-A>: Annexin V
0
102
103
104
105
<P
acific
Blu
e-A
>
3.91 17.1
8.0471
1002
103
104
105
<FITC-A>: Annexin V
0
102
103
104
105
<P
acific
Blu
e-A
>
6.65 50.1
2023.3
9.5%
17%
50.1%
Annexin V
DA
PI
Control
PBMC + control BiTE
PBMC +MT110
4.5%
14.0%
7.4%
11.1%
8.0%
20.0%
A6L
*
*
* *
0%
10%
20%
30%
40%
50%
60%
70%
80%PBMC + control BiTE PBMC + MT110
**
* **
185
A6L 185BA
-1 -1-1
-1 -1-1
Figure 4. Induction of apoptosis onpancreas cancer cells by treatmentwith MT110. A, flow cytometricanalysis for apoptotic cells usedstaining for Annexin V and DAPI inA6L and 185 cells after treatmentwith 100 ng/mL MT110 or controlBiTE. Results are from triplicatedeterminations. B, flow cytometricanalysis for CSC markers EpCAM,CD133, SSEA-1 in combinationwith Annexin V and DAPI aftertreatment of A6L and 185 withcontrol BiTE or MT110. Data arerepresented as mean � SD (n � 3).�, P ¼ 0.05 versus control BiTE.
0
50
100
150
200
250
300
350
185A6L
Control
PBMC + cAb
PBMC + MT110
**
Nu
mb
er o
f sp
her
es
**P < 0.01*P < 0.05
Control
PBMC +control BiTE
PBMC + MT110
A6L 185
100 µm 100 µm
100 µm 100 µm
100 µm 100 µm
*BA
A6L 185
**
Figure 5. Effect of MT110 treatmenton formation of spheres bypancreatic cancer cells. A,representative images of spheresderived from surviving adherentcells treated for 7 days with eithercontrol BiTE or MT110 at aconcentration of 100 ng/mL. B,quantification of spheres used aCasy cell counter.
Cioffi et al.
Clin Cancer Res; 18(2) January 15, 2012 Clinical Cancer Research470
on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270

In the next experiment, we further enriched for putativelysurviving CSCs by sphere formation during 7 days after apreceding in vitro treatment withMT110–PBMCs or controlBiTE–PBMCs for 7 days and before implantation of the cellsinto immunocompromised mice. After 60 days, tumor takeoccurred in only one third of themice in the group receivingspheres obtained after MT110 treatment, whereas all micedeveloped tumors in the group receiving spheres aftertreatment in the control BiTE group (Fig. 6C). Consistentwith formation of larger but fewer spheres after treatmentwith control BiTE (see Fig. 5A), tumors induced by suchspheres were larger than under control conditions. Thesedata indicate that CSCs enriched within spheres and beingresistant to a 1-week treatment with MT110 have a much-reduced tumorigenicity, if any.To directly compare metastasis-derived A6L cells with
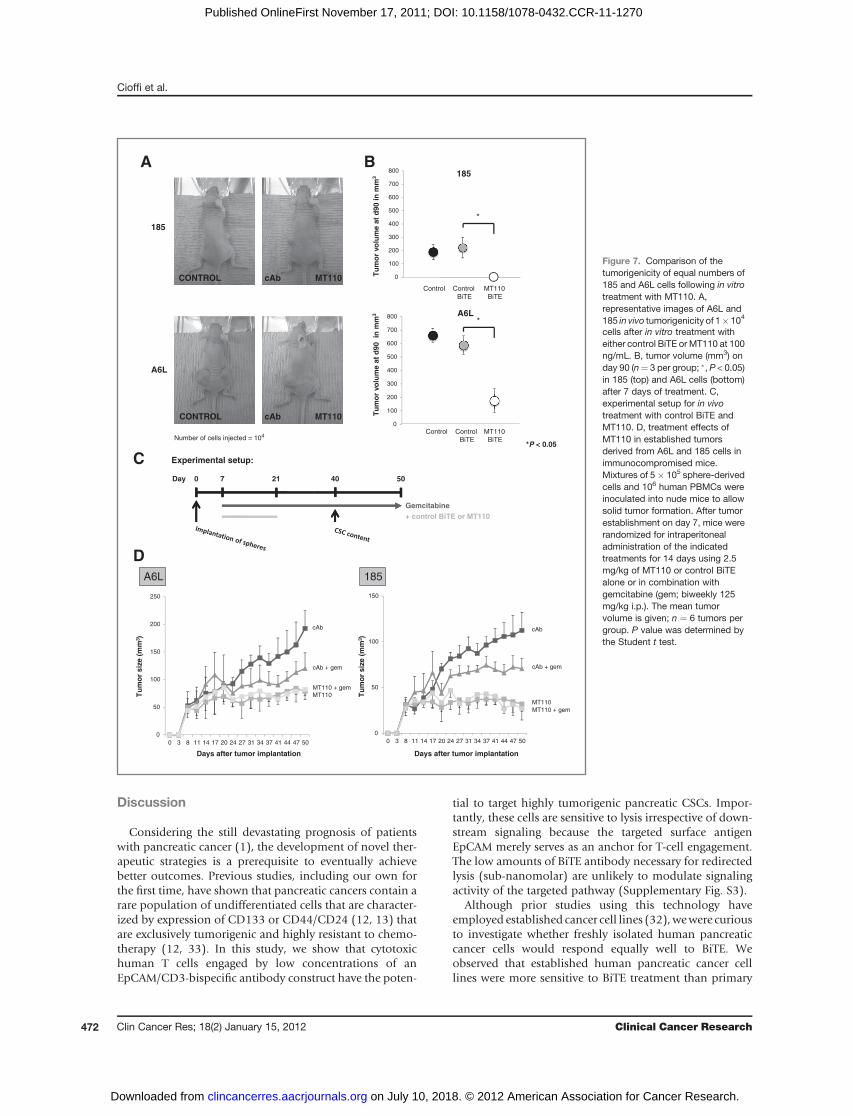
primary tumor-derived 185 cells for their susceptibilitytoward redirected lysis by MT110 a further in vivo experi-ment used equal numbers of cells (104) pretreated witheither control BiTE–PBMC or MT110–PBMC for 7 days.Equal cell numberswere implanted into contralateral flanksof immunocompromised mice and tumorigenicity, andtumor volumewasdeterminedonday 90 following implan-tation. The less aggressively growing and in vitro less resis-tant 185 cells had entirely lost their in vivo tumorigenicityafter MT110 pretreatment as compared with control BiTE–treated cells (Fig. 7A and B). In contrast, tumorigenicity andtumor growth by the same number of metastasis-derivedA6L cells was significantly reduced, but not abrogated.Flow cytometric analysis revealed that A6L cells harbored
a much more prominent EpCAM-negative subpopulationof 22% of cells compared with 185 cells with only 2%(Supplementary Fig. S5). This subpopulation of A6L cells
becomes slightly enriched during sphere formation toapproximately 32% of cells, which was also evident in thesubpopulation of CD133þ CSCs. In addition, 185 cellsenriched EpCAM-negative cells on sphere formation toapproximately 15%. EpCAM-low or -negative CSCs arelikely to evade redirected lysis by T cells during MT110treatment in vitro. Nonetheless, they appear to be compro-mised in their in vivo tumor formation given the signifi-cantly reduced tumorigenicity seen after implantation ofcells obtained after anti-EpCAM treatment.
Finally, we studied the effects of MT110 in vivo using amodel of established primary human pancreatic cancercoimplanted with healthy donor-derived PBMCs. Treat-ment was initiated on day 7 after cell implantation(Fig. 7C). Control BiTE and MT110, respectively, wereadministered by intraperitoneal (IP) injection for 14 dayswith orwithout coadministration of gemcitabine. Althoughgemcitabine (in combinationwith control BiTE) resulted ina modest slowdown of tumor growth, we observed a com-plete stall in tumor growth for MT110-treated mice(Fig. 7D). As the treatment effectsweremaintained through-out the follow-up period of several weeks after discontin-uation of treatment, these data are consistent with ourin vitro findings that the tumors were depleted for tumor-igenic/tumor-promoting CSCs. Indeed, on day 40 and,therefore, 19 days after termination of MT110 treatment,investigation of the CSC content in representative tumorsusing flow cytometric analysis revealed depletion for cellsexpressing CD133, SSEA-1, and CXCR4 as well as signifi-cantly reduced sphere-formation capacity in the MT110group as compared with control BiTE (Supplementary Fig.S2). The combination of MT110 with gemcitabine did notresult in a further reduction in tumor size.
Figure 6. Loss of tumorigenicity ofpancreatic cancer stem cells andspheres following in vitro treatmentwith MT110. A, representativepictures of in vivo tumorigenicity ofsurviving cells after in vitro treatmentwith either control BiTE or MT110 at100 ng/mL. B, tumorigenicity andtumor volume (mm3) on day 60 (n¼ 3per group; �, P < 0.05) in cells after 7days treatment and (C) in spheresformed from pretreated cells.
0
100
200
300
400
500
600
700
800
43210
0
100
200
300
400
500
600
700
800
0 1 2 3 4
Number of cells injected = surviving cells after treatment
MT110cAbCONTROL
MT110cAbMT110cAb
A
Tu
mo
r vo
lum
e at
d60
in m
m3
A6L cellsB
Tu
mo
r vo
lum
e at
d60
in m
m3
A6L spheres derived from pretreated cells
Control ControlBiTE
MT110BiTE
Control ControlBiTE
MT110BiTE
C
0
20
40
60
80
100
120
ControlControl
BiTE
MT110
BiTE
0
20
40
60
80
100
120
ControlControl
BiTE
MT110
BiTE
*P < 0.05T
um
or
take
rat
e (%
)T
um
or
take
rat
e (%
)
Control ControlBiTE
* *
**
MT110BiTE
Control ControlBiTE
MT110BiTE
Immunotherapy against Pancreatic Cancer Stem Cells
www.aacrjournals.org Clin Cancer Res; 18(2) January 15, 2012 471
on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270
Discussion
Considering the still devastating prognosis of patientswith pancreatic cancer (1), the development of novel ther-apeutic strategies is a prerequisite to eventually achievebetter outcomes. Previous studies, including our own forthe first time, have shown that pancreatic cancers contain arare population of undifferentiated cells that are character-ized by expression of CD133 or CD44/CD24 (12, 13) thatare exclusively tumorigenic and highly resistant to chemo-therapy (12, 33). In this study, we show that cytotoxichuman T cells engaged by low concentrations of anEpCAM/CD3-bispecific antibody construct have the poten-
tial to target highly tumorigenic pancreatic CSCs. Impor-tantly, these cells are sensitive to lysis irrespective of down-stream signaling because the targeted surface antigenEpCAM merely serves as an anchor for T-cell engagement.The low amounts of BiTE antibody necessary for redirectedlysis (sub-nanomolar) are unlikely to modulate signalingactivity of the targeted pathway (Supplementary Fig. S3).
Although prior studies using this technology haveemployed established cancer cell lines (32),wewere curiousto investigate whether freshly isolated human pancreaticcancer cells would respond equally well to BiTE. Weobserved that established human pancreatic cancer celllines were more sensitive to BiTE treatment than primary
MT110cAbCONTROL
A6L
Number of cells injected = 104
MT110cAbCONTROL
185
Tu
mo
r vo
lum
e at
d90
in
mm
3
0
100
200
300
400
500
600
700
800
*P < 0.05
Tu
mo
r vo
lum
e at
d90
in m
m3
Control Control
BiTE
MT110
BiTE
BA
0
100
200
300
400
500
600
700
800
1 2 3 4
A6L
Control Control
BiTE
MT110
BiTE
185
*
*
Days after tumor implantation
0
50
100
150
200
250
50474441373431272420171411830
A6L 185
5021Day 0 7
Gemcitabine
Experimental setup:C
D
0
50
100
150
50474441373431272420171411830
Tu
mo
r si
ze (
mm
3 )
Tu
mo
r si
ze (
mm
3 )
+ control BiTE or MT110
Days after tumor implantation
40
cAb
cAb + gem
MT110 + gem MT110
cAb
cAb + gem
MT110 + gem
MT110
Figure 7. Comparison of thetumorigenicity of equal numbers of185 and A6L cells following in vitrotreatment with MT110. A,representative images of A6L and185 in vivo tumorigenicity of 1� 104
cells after in vitro treatment witheither control BiTE or MT110 at 100ng/mL. B, tumor volume (mm3) onday 90 (n¼ 3 per group; �, P < 0.05)in 185 (top) and A6L cells (bottom)after 7 days of treatment. C,experimental setup for in vivotreatment with control BiTE andMT110. D, treatment effects ofMT110 in established tumorsderived from A6L and 185 cells inimmunocompromised mice.Mixtures of 5� 105 sphere-derivedcells and 106 human PBMCs wereinoculated into nude mice to allowsolid tumor formation. After tumorestablishment on day 7, mice wererandomized for intraperitonealadministration of the indicatedtreatments for 14 days using 2.5mg/kg of MT110 or control BiTEalone or in combination withgemcitabine (gem; biweekly 125mg/kg i.p.). The mean tumorvolume is given; n ¼ 6 tumors pergroup. P value was determined bythe Student t test.
Cioffi et al.
Clin Cancer Res; 18(2) January 15, 2012 Clinical Cancer Research472
on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270

humanpancreatic cancer cells. This observation emphasizesthe rather limited predictive value of established cancer celllines for treatment studies. Moreover, the used primarypancreatic cancer cells showed a much more robust in vivotumorigenicity because usually less than 1,000 isolated cellsare capable of tumor formation in immunocompromisedmice, whereas up to 106 cells derived from establishedpancreatic cancer cell lines are required for reproducible invivo tumor formation. Therefore, the model system used inthe present study represents themost challenging setting fortesting new treatment modalities against pancreatic cancer.In this respect, our study adds important data to the
growing evidence that treatment with EpCAM/CD3-bispe-cific antibody constructs can target highly tumorigenicCSCs. A recent study has already shown the highly efficientelimination of colorectal cancer–derived tumor-initiatingcells using BiTE technology (31). In the present study, wehave now extended these findings to pancreatic cancer asone of the most metastatic and highly therapy-resistantcancers (2). The population of tumor-initiating cells wasvirtually erased from in vitro treated primary human pan-creatic cancer cells as the remaining cells could no longerform tumors after transfer into immunocompromisedmice. More importantly, in vivo studies in established pri-mary pancreatic cancers revealed disease stabilization inresponse to MT110 treatment whereas tumors treated withcontrol BiTE or gemcitabine continued to grow and even-tually required sacrifice of the animals. The addition ofgemcitabinedidnot lead to a further reductionof tumor sizenor did the chemotherapy apparently interfere with theactivity of MT110. Harvesting of the tumors revealed thatthe small remaining tumors were depleted for CSCs andcontainedmostly differentiated cancer cells as evidenced byflow cytometric and sphere-formation assays. Therefore, thefailure of gemcitabine to further reduce the size of thesesmall reminiscent tumors can be rationalized by the lack ofresponse of nonproliferating tumor cells to cytotoxic agents.For A6L cells, which were derived from a metastatic
pancreatic cancer lesion in the liver (29), responsivenesstoMT110-engagedT cellswasmore diverse than for primarytumor-derived 185 cells. If A6L cells were pretreated and allsurviving cells implanted, in vivo tumorigenicity wascompletely abrogated. However, if putatively survivingCSCs were enriched before implantation by sphere forma-tion or the number of injected viable cells was adjusted tothe number of viable cells harvested from control culturesand brought to 104 cells, we still observed some tumor takefor BiTE-treated cells. Importantly, however, the arisingtumors grew extremely slowly andwere significantly smallercomparedwith those arising fromcontrol BiTE-treated cells.Flow cytometric analysis revealed that A6L cells contained alarger proportion of EpCAM-negative cells than 185 cells,which was also found to increase upon sphere formation
(Supplementary Fig. S4). These data are consistent with thenotion that sphere-derived EpCAM-negative cells are inva-sive CSC that have undergone epithelial-to-mesenchymaltransition (EMT; ref. 34) and have likely escaped MT110treatment. Their reduced tumorigenicity, however, indi-cates that loss of EpCAM is of disadvantage for their CSCproperties. Indeed, EpCAM knockdown experiments usingsiRNA in CSC derived from colorectal cancer tissue supporta functional role of EpCAM for tumor initiation, and CSCproliferation and migration (P. Baeuerle, unpublisheddata). Future studies will have to address the importanceof EpCAM signaling for pancreatic CSCs.
An important characteristic of pancreatic cancer tissue isits poor vascularization and strong desmoplastic response(35). Although the in vivo response of BiTE antibody admin-istrationonhumanpancreatic cancer cells embedded in thisstromal fortress will still need to be determined, the elim-ination of circulating cancer (stem) cells by antibody-engaged T cells can already be predicted from the currentset of data. Indeed, it is important to note that, based on thesystemic nature of pancreatic cancer, elimination of circu-lating and/or isolatedmetastatic CSCsmay be an importanttherapeutic approach. Although these cells are still highlyresistant to standard therapy, they aremore easily accessibleto immunotherapy, and their elimination already bearsgreat potential for improving the outcome of patients withpancreatic cancer. Indeed, a recent study in breast cancerpleural effusates showed efficient lysis of these potentiallymetastatic cancer cells (36).
Disclosure of Potential Conflicts of Interests
C.Heeschenholds a commercial grant fromMicromet Inc. P.A. Baeuerle isan employee of Micromet and has equity. No potential conflicts of interestwere disclosed by other authors.
Acknowledgments
The authors thank Mercedes Alonso and Sonia Alcala (both CNIO) forexcellent technical assistance and Matthias Munz (Micromet) for technicalsupport, supply of materials, and helpful discussions.
Grant Support
This work was financially supported by a research grant from MicrometInc., an ERC Advanced Investigator Grant (Pa-CSC 233460), the Sub-direcci�on General de Evaluaci�on y Fomento de la Investigaci�on, Fondo deInvestigaci�on Sanitaria (PS09/02129), and the Programa Nacional de Inter-nacionalizaci�on de la IþD, Subprograma: FCCI 2009 (PLE2009-0105;both Ministerio de Ciencia e Innovaci�on, Spain). M. Cioffi is financiallysupported by the La Caixa Predoctoral Fellowship Program.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received May 21, 2011; revised October 7, 2011; accepted October 31,2011; published OnlineFirst November 17, 2011.
References1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J
Clin 2010;60:277–300.2. Philip PA, Mooney M, Jaffe D, Eckhardt G, Moore M, Meropol N, et al.
Consensus report of the National Cancer Institute Clinical Trials
Immunotherapy against Pancreatic Cancer Stem Cells
www.aacrjournals.org Clin Cancer Res; 18(2) January 15, 2012 473
on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270

Planning Meeting on pancreas cancer treatment. J Clin Oncol2009;27:5660–9.
3. MatanoE, Tagliaferri P, LibroiaA,DamianoV, Fabbrocini A,DeLorenzoS, et al. Gemcitabine combinedwith continuous infusion 5-fluorouracilin advanced and symptomatic pancreatic cancer: a clinical benefit-oriented phase II study. Br J Cancer 2000;82:1772–5.
4. Rothenberg ML, Moore MJ, Cripps MC, Andersen JS, Portenoy RK,BurrisHA3rd, et al. A phase II trial of gemcitabine in patientswith 5-FU-refractory pancreas cancer. Ann Oncol 1996;7:347–53.
5. Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med2006;355:1253–61.
6. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al.Cancer stem cells–perspectives on current status and future directions:AACR Workshop on cancer stem cells. Cancer Res 2006;66:9339–44.
7. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison S, Clarke MF.Prospective identification of tumorigenic breast cancer cells. ProcNatlAcad Sci U S A 2003;100:3983–8.
8. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cellcapable of initiating tumour growth in immunodeficient mice. Nature2007;445:106–10.
9. Ricci-Vitiani L, Lombardi D, Pilozzi E, Biffoni M, Todaro M, Peschle C,et al. Identification and expansion of human colon-cancer-initiatingcells. Nature 2007;445:111–5.
10. Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S,et al. Identification of bronchioalveolar stem cells in normal lung andlung cancer. Cell 2005;121:823–35.
11. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al.Identification of human brain tumour initiating cells. Nature 2004;432:396–401.
12. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al.Distinct populations of cancer stem cells determine tumor growth andmetastatic activity in human pancreatic cancer. Cell Stem Cell2007;1:313–23.
13. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identi-fication of pancreatic cancer stem cells. Cancer Res 2007;67:1030–7.
14. Baeuerle PA, Gires O. EpCAM (CD326) finding its role in cancer. Br JCancer 2007;96:417–23.
15. Went P, Vasei M, Bubendorf L, Terracciano L, Tornillo L, Riede U, et al.Frequent high-level expression of the immunotherapeutic target Ep-CAM in colon, stomach, prostate and lung cancers. Br J Cancer2006;94:128–35.
16. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accu-mulating evidence and unresolved questions. Nat Rev Cancer2008;8:755–68.
17. Gires O, Klein CA, Baeuerle PA. On the abundance of EpCAM oncancer stem cells. Nat Rev Cancer 2009;9:143; author reply 143.
18. MunzM, Baeuerle PA, Gires O. The emerging role of EpCAM in cancerand stem cell signaling. Cancer Res 2009;69:5627–9.
19. Schmelzer E, Reid LM.EpCAMexpression in normal, non-pathologicaltissues. Front Biosci 2008;13:3096–100.
20. Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al.An embryonic stem cell-like gene expression signature in poorlydifferentiated aggressive human tumors. Nat Genet 2008;40:499–507.
21. Gonzalez B, Denzel S, Mack B, ConradM, Gires O. EpCAM is involvedin maintenance of the murine embryonic stem cell phenotype. StemCells 2009;27:1782–91.
22. McLaughlin PM,HarmsenMC, DokterWH, Kroesen BJ, van derMolenH, Brinker MG, et al. The epithelial glycoprotein 2 (EGP-2) promoter-
driven epithelial-specific expression of EGP-2 in transgenic mice: anew model to study carcinoma-directed immunotherapy. Cancer Res2001;61:4105–11.
23. Brischwein K, Schlereth B, Guller B, Steiger C, Wolf A, Lutterbuese R,et al. MT110: a novel bispecific single-chain antibody construct withhigh efficacy in eradicating established tumors.Mol Immunol 2006;43:1129–43.
24. Amann M, Brischwein K, Lutterbuese P, Parr L, Petersen L, Lorenc-zewski G, et al. Therapeutic window of MuS110, a single-chain anti-body construct bispecific for murine EpCAM and murine CD3. CancerRes 2008;68:143–51.
25. Schlereth B, Fichtner I, Lorenczewski G, Kleindienst P, Brischwein K,da Silva A, et al. Eradication of tumors from a human colon cancer cellline and from ovarian cancer metastases in immunodeficient mice by asingle-chain Ep-CAM-/CD3-bispecific antibody construct. CancerRes 2005;65:2882–9.
26. Schlereth B, Kleindienst P, Fichtner I, Lorenczewski G, Brischwein K,Lippold S, et al. Potent inhibition of local and disseminated tumorgrowth in immunocompetent mouse models by a bispecific antibodyconstruct specific for murine CD3. Cancer Immunol Immunother2006;55:785–96.
27. Amann M, D'Argouges S, Lorenczewski G, Brischwein K, Kischel R,Lutterbuese R, et al. Antitumor activity of an EpCAM/CD3-bispecificBiTE antibody during long-term treatment of mice in the absence of T-cell anergy and sustained cytokine release. J Immunother 2009;32:452–64.
28. Amann M, Friedrich M, Lutterbuese P, Vieser E, Lorenczewski G,Petersen L, et al. Therapeutic window of an EpCAM/CD3-specificBiTE antibody in mice is determined by a subpopulation of EpCAM-expressing lymphocytes that is absent in humans. Cancer ImmunolImmunother 2009;58:95–109.
29. MuellerM-T,HermannPC,Huber S, Leicht SF,Witthauer J,MustafaM,et al. Multimodal treatment to eliminate CSCs in human pancreaticcancer. Gastroenterology 2009;137:1102–13.
30. Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cyto-toxicity screening. Nat Protoc 2006;1:1112–6.
31. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al.Core signaling pathways in human pancreatic cancers revealed byglobal genomic analyses. Science 2008;321:1801–6.
32. Herrmann I, Baeuerle PA, Friedrich M, Murr A, Filusch S, Ruttinger D,et al. Highly efficient elimination of colorectal tumor-initiating cells byan EpCAM/CD3-bispecific antibody engaging human T cells. PLoSOne 2010;5:e13474.
33. Jimeno A, Feldmann G, Suarez-Gauthier A, Rasheed Z, Solomon A,ZouGM,et al. A direct pancreatic cancer xenograftmodel as aplatformfor cancer stem cell therapeutic development. Mol Cancer Ther2009;8:310–4.
34. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. Theepithelial–mesenchymal transition generates cells with properties ofstem cells. Cell 2008;133:704–15.
35. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D,Honess D, et al. Inhibition of Hedgehog signaling enhances deliveryof chemotherapy in a mouse model of pancreatic cancer. Science2009;324:1457–61.
36. Witthauer J, Schlereth B, Brischwein K, Winter H, Funke I, Jauch KW,et al. Lysis of cancer cells by autologous T cells in breast cancer pleuraleffusates treated with anti-EpCAM BiTE antibody MT110. BreastCancer Res Treat 2009;117:471–81.
Cioffi et al.
Clin Cancer Res; 18(2) January 15, 2012 Clinical Cancer Research474
on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270

2012;18:465-474. Published OnlineFirst November 17, 2011.Clin Cancer Res Michele Cioffi, Jorge Dorado, Patrick A. Baeuerle, et al. Primary Human Pancreatic Cancer Stem CellsEpCAM/CD3-Bispecific T-cell Engaging Antibody MT110 Eliminates
Updated version
10.1158/1078-0432.CCR-11-1270doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2011/11/17/1078-0432.CCR-11-1270.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/18/2/465.full#ref-list-1
This article cites 36 articles, 11 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/18/2/465.full#related-urls
This article has been cited by 7 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/18/2/465To request permission to re-use all or part of this article, use this link
on July 10, 2018. © 2012 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 17, 2011; DOI: 10.1158/1078-0432.CCR-11-1270


















