
electrophoresis
43
Electrophoresis • Submitted by • Saubhagya • Sri venkateswara college • University of Delhi
-
Upload
saubhagya1994 -
Category
Science
-
view
140 -
download
1
Transcript of electrophoresis

Electrophoresis
• Submitted by
• Saubhagya
• Sri venkateswara college
• University of Delhi

Electrophoresis Principle & Application

• It is the process of moving charged biomolecules in
solution by applying an electrical field across the mixture.
• This technique was first developed by Arne Tiselius in 1930
for the study of serum proteins.
• Their rate of migration (electrophoretic mobility) through
the electrical field, depends on following factors
• strength of the field,
• net charge, size, and shape of the molecules,
• concentration of the molecule in solution
• ionic strength of buffer, viscosity, and temperature of
the medium in which the molecules are moving.
• Applied voltage
• Electrophoresis is used for:
• For analysis and purification of very large molecules
(proteins, nucleic acids)
• For analysis of simpler charged molecules (sugars,
amino acids, peptides, nucleotides, and simpler ions).
Introduction

• Electrophoresis can be one dimensional (i.e. one plane of
separation) or two dimensional.
• One dimensional electrophoresis is used for most routine
protein and nucleic acid separations. Two dimensional
separation of proteins is used for finger printing , and when
properly constructed can be extremely accurate in resolving
all of the proteins present within a cell (greater than 1,500).
• Most common stabilizing media are polyacrylamide or
agarose gels.

• Molecules can have distinct charges
• Positive or Negative
• Net charge will cause different movement through gel
• Molecules can have different shapes
• Linear
• Globular
• Alpha helix
When charged molecules are placed in an electric field, they
migrate toward either the positive (anode) or negative (cathode)
pole according to their charge.
Basic Principle of Electrophoresis

Electrophoretic mobility
• The principle involved in both slab and capillary electrophoresis are same which involves electrophoreticmobility of the ions.
• Electrophoretic mobility μ = v / E = q / f
– V=Velocity, E=Electric field, q=Charge f=Frictional coefficient
• Depends on
– Particle property :Surface, charge, density & size
– Solution properties: Ionic strength, pH, Conductivity,viscosity
– Temperature & Voltage

General operations performed in conventional
electrophoresis include:
(1) separation
(2) staining
(3) detection
(4) Quantification
General Procedure

The Basic Components
• Support Medium (Powdered of agarose or polyacrylamise
• The electric power supply provides the electricity that carries the molecules through the gel
• The electrophoresis chamber is where it all takes place.
• Boiling Buffer solution• Gel stain

• The “gel” in gel electrophoresis is the component that
physically separates the molecules.
• Use the proper gel concentration for sample size
range.
0.5–5% agarose (for DNA & RNA)
3.5–20% polyacrylamide (for Protein)
• Agarose and polyacrylamide gels are across-linked,
spongelike structure
• It is important that the support media is electrically
neutral. Presence of charge group may cause:
-Migration retardation
-The flow of water toward one or the other electrode so
called ‘Electroendosmosis (EEO)’, which decrease
resolution of the separation
Support media

Agarose – highly purified polysaccharide derived from
agar (extracted from seeweed), long sugar polymers held
together by hydrogen and hydrophobic bonds.
Acrylamide (CH2=CH-CO-NH2) Polyacrylamide gel
structure held together by covalent cross-links

Buffer additives modify sample molecules.
• Commonly used buffer
• Tris-acetate-EDTA , Tris Phosphate EDTA & Tris-borate-
EDTA (50mmol/L; pH 7.5-7.8) used most often for DNA.
• 10 mM sodium phosphate or MOPS buffer used for RNA.
• Barbital buffer & Tris-EDTA for protein
• Function of buffer
1. carries the applied current
2. established the pH
3. determine the electric charge on the solute
• High ionic strength of buffer
– produce sharper band
– produce more heat
Buffers

Combs are used to put wells in the cast gel for sample loading.
– Regular comb: wells separated by an “ear” of gel
– Hound stooth comb: wells immediately adjacent
• Use the proper comb (well) and gel size.
Combs

Applied voltage
• Increase:
-charged molecule migrate faster
-Increases current
- temperature
Temperature
• Better at low temp(40C) : Otherwise density difference convection current Disturbed diffusion
• Distortion of Zones: Column gel migrating Center (warmer) faster than outer (cooler)
• Evaporation: increase in ionic strength of the buffer
• Viscosity: agarose gels becomes softer

Classification
1. Gel electrophoresis
1(a) Electrophoreis of nucleic acids
Agarose gel electrophoresis of DNA
Pulsed- field gel electrophoresis
1(b) Electrophoesis of proteins
Sodium dodecyl sulphate (SDS)- polyacrylamide gel
electrophoresis
– Native (buffer) gels
– Gradient gels
– Isoelectric focusing gel
Two-dimentional polyacrylamide gel electrophoresis
– Cellulose acetate electrophoresis
– Detection estimation and recovery of proteins in gels
– Protein (western blotting) blotting
2. Capillary electrophoresis
3. Microchip electrophoresis

Electrophoresis
• Horizontal Agarose Gels
• Agarose forms a gel or molecular sieve that
supports the movement of small materials in solution
used for DNA
• Vertical Polyacrylamide Gels
• Made of Polyacrylamide
• Used for Protein molecular size, shape, charge
• IEF electrophoresis
• Western Blot technique

Horizontal Gels Vertical gel

• Gel is a colloid in a solid form (99% is water).
• The separation here is brought about through molecular
sieving technique, based on molecular size of substances.
• During electrophoresis, macromolecules are forced to move
through the pores when the electrical current is applied.
Support media
• Agarose for nucleic acid and polyacrylamide gels for protein
are a cross-linked, sponge like structure.
• It is important that the support media is electrically neutral.Presence of charge group may cause:
-Migration retardation
-The flow of water toward one or the other electrode so
called ‘Electroendosmosis (EEO)’, which decreaseresolution of the separation
Gel electrophoresis

Agrose gel electrophoresis
• Agarose gel electrophoresis is a method to separate DNA, or RNA molecules by size. This is achieved by moving negatively charged nucleic acid molecules through an agarose matrix with an electricfield (electrophoresis).
• The pore size is determined by adjusting the concentration of
agarose in a gel (normally in the rank of 0.4-4%
• Shorter molecules move faster and migrate farther than longer ones .


PFGE allows investigators to separate much larger pieces of DNA than conventional agarose gel electrophoresis. In conventional gels, the current is applied in a single direction (from top to bottom). But in PFGE, the direction of the current is altered at a regular interval
Pulsed Field Gel Electrophoresis (PFGE)



• Detect bands by staining during or after electrophoresis
• Ethidium bromide: for double-stranded DNA
• SyBr green or SyBr gold: for single- or double-stranded DNA or for RNA
• Silver stain: more sensitive for single- or double-stranded DNA or for RNA and proteins
Nucleic acid stain

An ethidium-stained gel photographed under UV light

1(a). Electrophoresis of proteins Sodiumdodecylsulphate(SDS)-Polyacrylamide Gel Electrophoresis• Polyacrylamide gel electrophoresis (PAGE), describes a technique widely
used in biochemistry, forensics, genetics, molecularbiology and biotechnology to separate biological macromolecules, usuallyproteins or nucleic acids, according to their electrophoretic mobility.
• Mobility is a function of the length, conformation and charge of themolecule
• As with all forms of gel electrophoresis, molecules may be run intheir native state, preserving the molecules' higher-order structure, or achemical denaturant may be added to remove this structure and turn themolecule into an unstructured linear chain whose mobility depends onlyon its length and mass-to-charge ratio.
• For nucleic acids, urea is the most commonly used denaturant. For
proteins, sodium dodecyl sulfate (SDS) also called lauryl sulfate is ananionic detergent applied to protein sample to linearize proteins and toimpart a negative charge to linearized proteins. This procedure iscalled SDS-PAGE.

• SDS is an anionic detergent that denatures secondary and non–disulfide–linked tertiary structures, and additionally applies a negative charge toeach protein in proportion to its mass.
• Urea breaks the hydrogen bonds between the base pairs of the nucleicacid, causing the constituent strands to separate. Heating the samples toat least 60°C further promotes denaturation.
• In most proteins, the binding of SDS to the polypeptide chain imparts aneven distribution of charge per unit mass, thereby resulting in afractionation by approximate size during electrophoresis.
• Proteins that have a greater hydrophobic content, for instance manymembrane proteins, and those that interact with surfactants in theirnative environment, are intrinsically harder to treat accurately using thismethod, due to the greater variability in the ratio of bound SDS.
• In addition to SDS, proteins may optionally be briefly heated to nearboiling in the presence of a reducing agent, such as dithiothreitol(DTT) or 2-mercaptoethanol (beta-mercaptoethanol/BME), which furtherdenatures the proteins by reducing disulfide linkages, thus overcomingsome forms of tertiary protein folding, and breaking up quaternaryprotein structure (oligomeric subunits). This is known as reducing SDS-PAGE.

SDS -PAGE

Electrophoretic method that separates proteins according
to the iso-electric points
Is ideal for seperation of amphoteric substances
Each protein has own pI = pH at which the protein has
equal amount of positive and negative charges (the net
charge is zero)
PI of proteins can be theoretically predicted. Therefore, IEF
can also be used for protein identification.
Seperation is achieved by applying a potential difference
across a gel that contain a pH gradient
Mixtures of ampholytes, small amphoteric molecules with
high buffering capacity near their pI, are used to generate
the pH gradient.
Isoelectric focusing requires solid support such as
polyacrylamide gel
Isoelectric Focusing


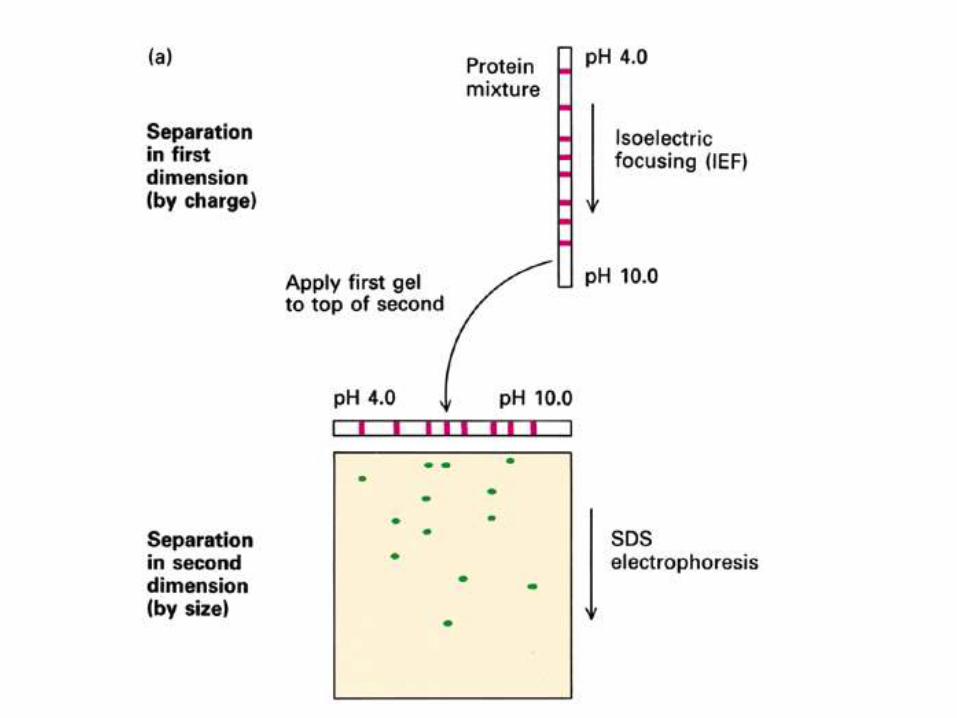
In the first dimension, proteins are resolved in according to their isoelectric points (pIs) using immobilized pH gradient electrophoresis (IPGE), isoelectric focusing (IEF), or non-equilibrium pH gradient electrophoresis.
In the second dimension, proteins are separated according to their approximate molecular weight using sodium dodecyl sulfate poly-acrylamide-electrophoresis (SDS-PAGE).
The combination of these two technique to give two-dimension (2-D)PAGE provides a highly sophisticated analytical method for analysingprotein mixtures.
2D-PAGE

Stain Detection limit
Ponceau S 1-2 mg
Amido Black 1-2 mg
Coomassie Blue 1.5 mg
India Ink 100 ng
Silver stain 10 ng
Colloidal gold 3 ng
Commonly used protein stains
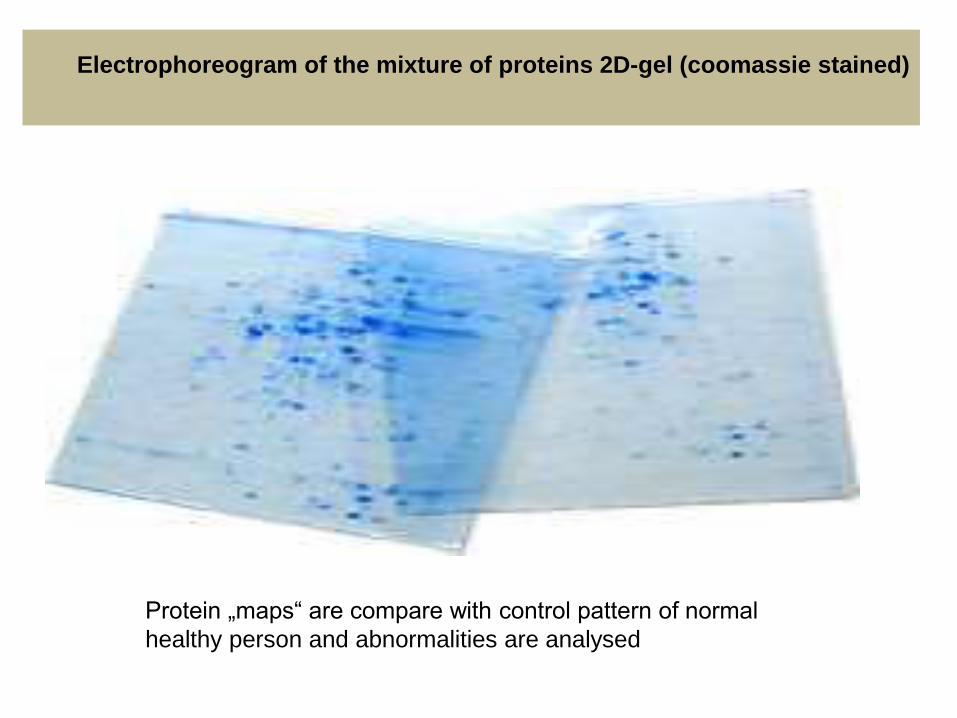
Protein „maps“ are compare with control pattern of normal
healthy person and abnormalities are analysed
Electrophoreogram of the mixture of proteins 2D-gel (coomassie stained)

Example of silver stained gel
Silver staining is usually 10-100 times more sensitive than Coomassie Blue staining, but it is more complicated.
Faint but still visible bands on this gel contain less than 0.5 ng of protein!


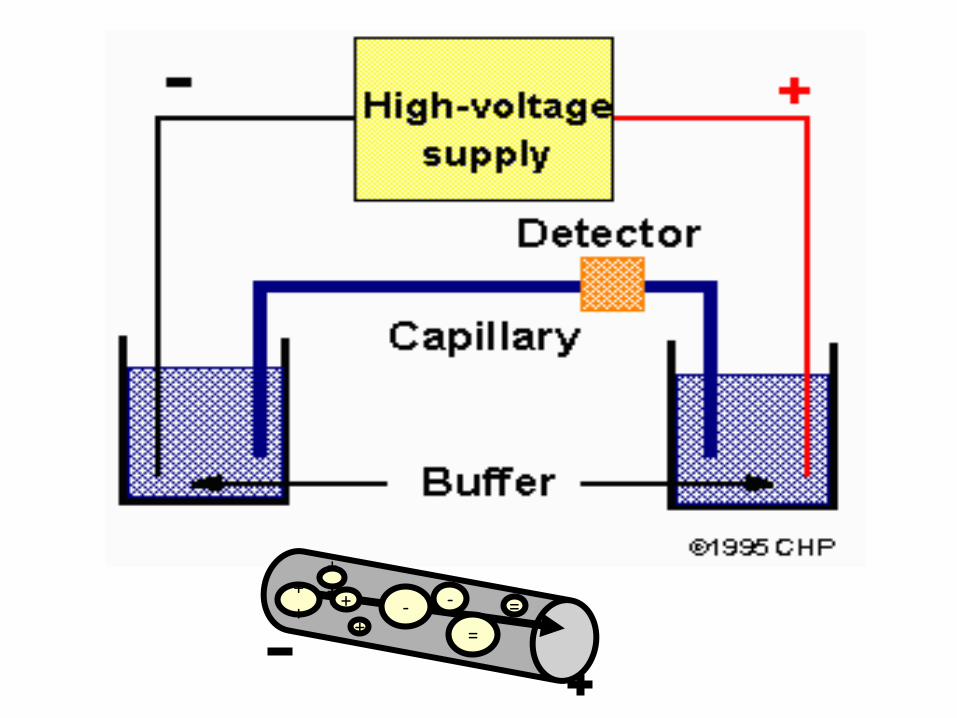
Capillary electrophoresis
Capillary gel electrophoresis is used for separation of biological
molecule including amino acid, peptides, proteins, DNA fragments, and
nucleic acids well as any number of small organic molecules such as drugs or even metal ions. to separate nucleic acids.
It is classic techniques of electrophoresis are carried out in a small-
bore, fused silica capillary tube, the outer diameter of such tubes
typically varies from 180 to 375 micrometer, the inner diameter from 20
to 180 micrometer, and the total length from 20 cm up to several
meters.
The capillary can also be filled with a gel, which eliminates the
electroosmotic flow. This capillary tube serves as a capillary
electrophoretic chamber that is connected to a detector at its terminal
end and, via buffer reservoirs, to a high-voltage power supply
Separation is accomplished as in conventional gel electrophoresis but
the capillary allows higher resolution, greater sensitivity, and on-line
detection.

One advantage of using capillary is that they reduce problem resulting from
heating effects. Because of small diameter of the tubing there is a larger
surface-to-volume ratio, which give enhanced heat dissipation. Improved
heat dissipation permits the application of voltages in the range of 20 to 30
kV, which enhances separation efficiency and reduces separation time in
some cases to less than 1 minute
•During a separation, uncharged molecules move at the same velocity as
the electroosmotic flow (with very little separation). Positively-charged
ions move faster and negatively-charged ions move slower.
•The surface of the silicate glass capillary contains negatively-charged
functional groups that attract positively-charged counterions. The
positively-charged ions migrate towards the negative electrode and carry
solvent molecules in the same direction. This overall solvent movement is
called electroosmotic flow.
=+ +
+
+
++
- -
=

3. Microchip electrophoresis
• Microchip analysis completed in tens of seconds where as capillaryelectrophoresis can take 20 min and conventional gel electrophoresis atleast2h.
• Using new detection system, such as laser induced fluorescence,picomole to attomole (10-8 moles) sensitivity can be achieved, which is atleast two order of magnitude greater than for conventional capillaryelectrophoresis.
• The microchip provide an electrophoretic system similar to CE but withmore flexibility.
• Current developments of this technology are based integrating functionsother than just separation in to the chip.
• For example: sample extraction, pre concentration of sample prior toseparation, PCR amplification of DNA sample using infrared mediatedthermo cycling for rapid on-chip amplification and the extraction ofseparated molecules using micro chamber –bound solid phases are allexamples of where further functions have been built into a microchipelectrophoresis system.
• An interface has also been developed for microchip electrophoresis-massspectrometry where drug have been separated by MCE and thenidentified by MS.


Applications
The vast application of electrophoresis include:
• Vaccine analysis such as influenza vaccine, hepatitis vaccine and polio vaccine.
• Protein and DNA analysis
• To see the map and the differences in the genetic code of species on the earth.
• Electrophoretic DNA analysis also provides a reliable tool in forensic investigations.
• Determination of impurities
• Chiral analysis
• Analysis of carbohydrates and other macromolecules.
• Analysis of inorganic anions/metal ions

Protein analysis• Electrophoresis has advanced our understanding on the structure and
function of proteins. These molecules are needed by our body cells andmay be analyzed, for instance by getting blood and urine samples. Thenthrough electrophoresis, the amount of protein in your blood or in yoururine is measure d and compare to established normal value lower orhigher than the normal levels usually indicates a disease.
DNA analysis• Electrophoresis is one way of analyzing DNA, which is the unique code of
every individual. Through electrophoresis, specific DNA sequences can beanalyzed isolated and cloned. The analyzed DNA may be used in forensicinvestigation and paternity tests.
• For this well are formed at one end of an agarose gel for the loading ofthe DNA sample. The slab is then placed horizontally in to theelectrophoresis buffer chamber. The DNA migrates in bands towards thepositive electrode. The smaller molecule migrate through the matrix morerapidly than the larger ones, which are restricted. The DNA bands are thenstained gel is then viewed directly under ultraviolet light andphotographed.


















