
DNA sequencing: methods I. Brief history of sequencing II. Sanger dideoxy method for sequencing III....
89
DNA sequencing: methods I. Brief history of sequencing II. Sanger dideoxy method for sequencing III. Sequencing large pieces of DNA VI. The “$1,000 dollar genome” On WebCT -- “The $1000 genome” -- review of new sequencing techniques by George Church
-
date post
20-Dec-2015 -
Category
Documents
-
view
238 -
download
1
Transcript of DNA sequencing: methods I. Brief history of sequencing II. Sanger dideoxy method for sequencing III....

DNA sequencing: methods
I. Brief history of sequencingII. Sanger dideoxy method for sequencingIII. Sequencing large pieces of DNAVI. The “$1,000 dollar genome”
On WebCT-- “The $1000 genome”-- review of new sequencing techniques by George Church

Why sequence DNA?
• All genes available for an organism to use -- a very important tool for biologists
• Not just sequence of genes, but also positioning of genes and sequences of regulatory regions
• New recombinant DNA constructs must be sequenced to verify construction or positions of mutations
• Etc.
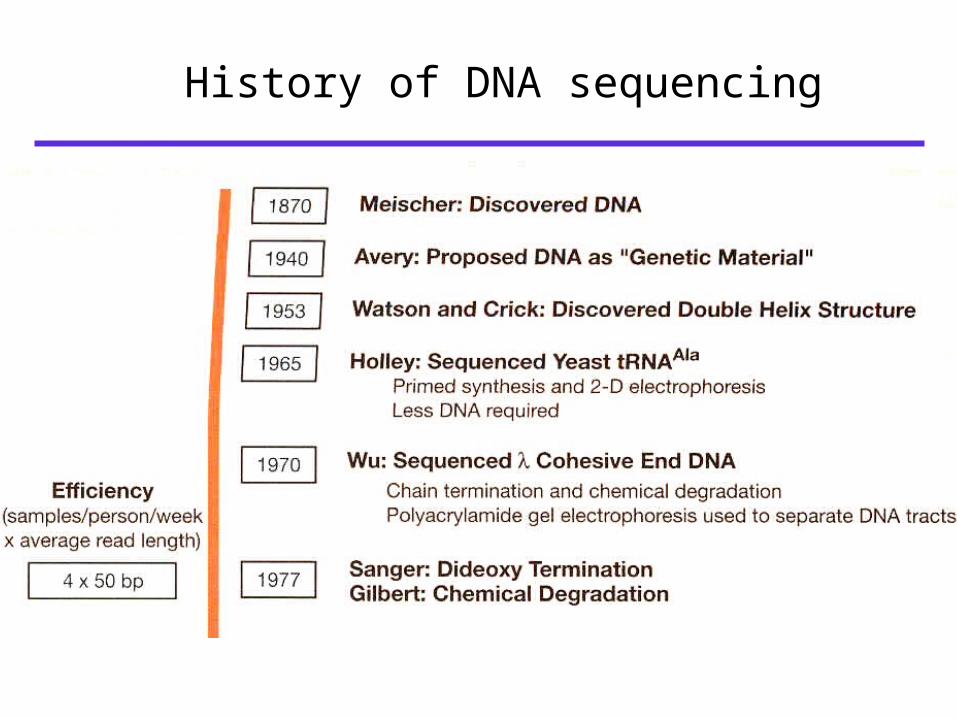
History of DNA sequencing
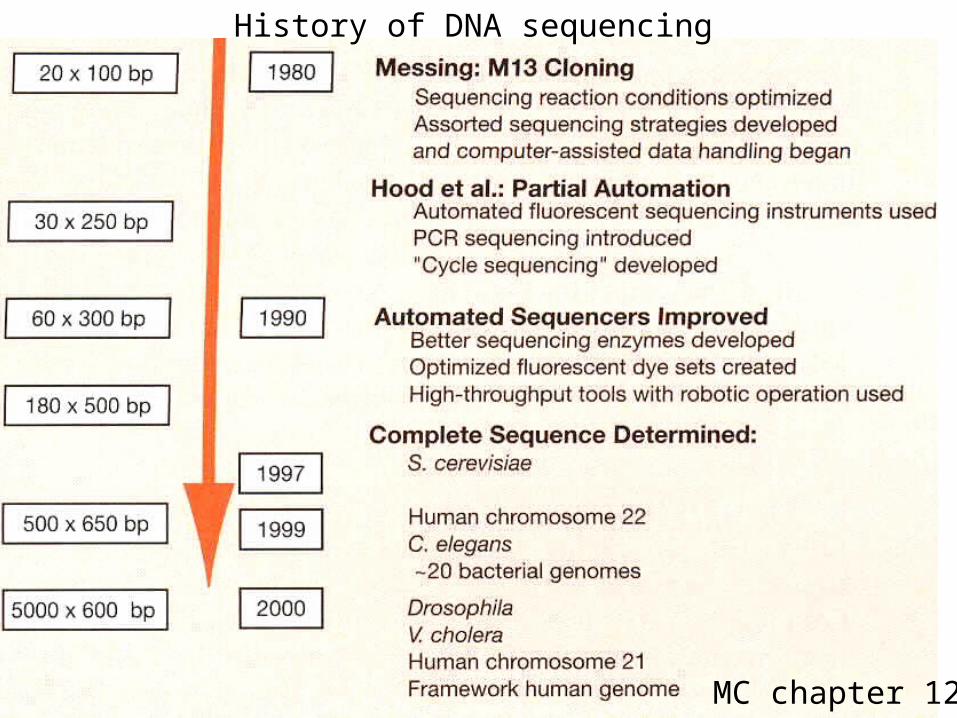
MC chapter 12
History of DNA sequencing

Methods of sequencing
A. Sanger dideoxy (primer extension/chain-termination) method: most popular protocol for sequencing, very adaptable, scalable to large sequencing projects
B. Maxam-Gilbert chemical cleavage method: DNA is labelled and then chemically cleaved in a sequence-dependent manner. This method is not easily scaled and is rather tedious
C. Pyrosequencing: measuring chain extension by pyrophosphate monitoring

for dideoxy sequencing you need:
1) Single stranded DNA template
2) A primer for DNA synthesis
3) DNA polymerase
4) Deoxynucleoside triphosphates and
dideoxynucleotide triphosphates

Primers for DNA sequencing
• Oligonucleotide primers can be synthesized by phosphoramidite chemistry--usually designed manually and then purchased
• Sequence of the oligo must be complimentary to DNA flanking sequenced region
• Oligos are usually 15-30 nucleotides in length

DNA templates for sequencing:
• Single stranded DNA isolated from recombinant M13 bacteriophage containing DNA of interest
• Double-stranded DNA that has been denatured
• Non-denatured double stranded DNA (cycle sequencing)
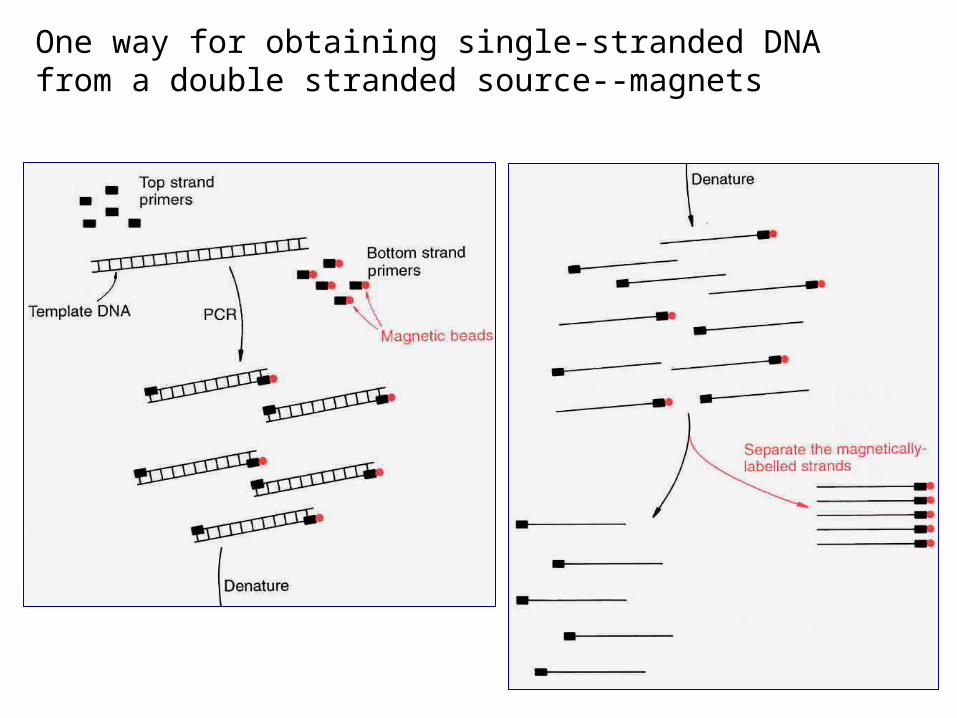
One way for obtaining single-stranded DNA from a double stranded source--magnets

Reagents for sequencing: DNA polymerases
• Should be highly processive, and incorporate ddNTPs efficiently
• Should lack exonuclease activity
• Thermostability required for “cycle sequencing”

Single stranded DNA 5’3’
5’ 3’
Sanger dideoxy sequencing--basic method
a) Anneal the primer
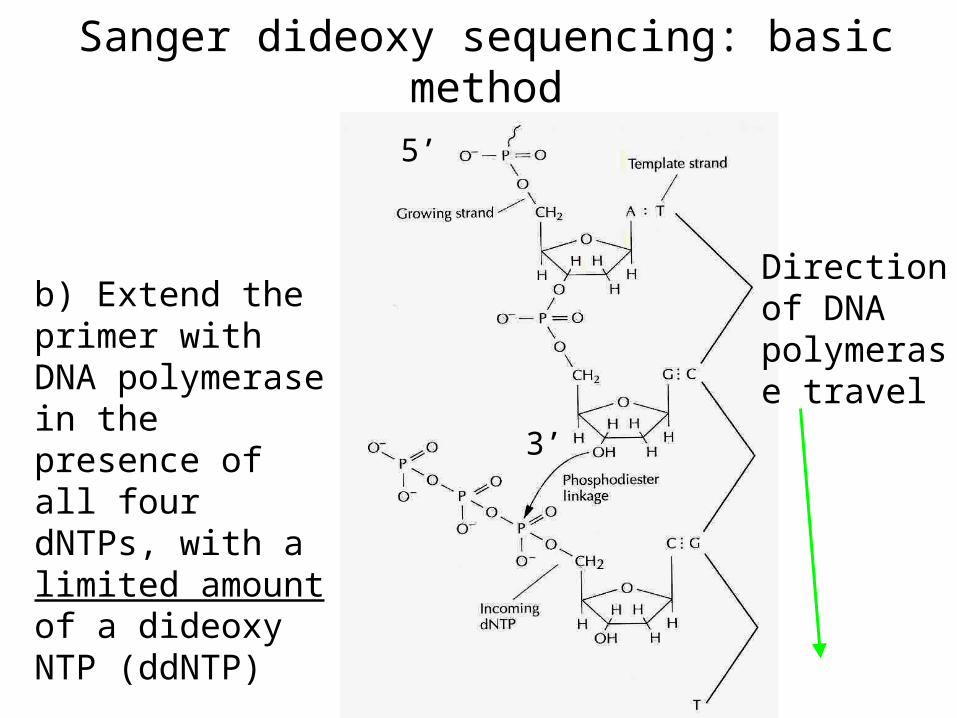
Sanger dideoxy sequencing: basic method
b) Extend the primer with DNA polymerase in the presence of all four dNTPs, with a limited amount of a dideoxy NTP (ddNTP)
5’
3’
Direction of DNA polymerase travel

DNA polymerase incorporates ddNTP in a template-dependent manner, but it works best if the DNA pol lacks 3’ to 5’ exonuclease (proofreading) activity
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
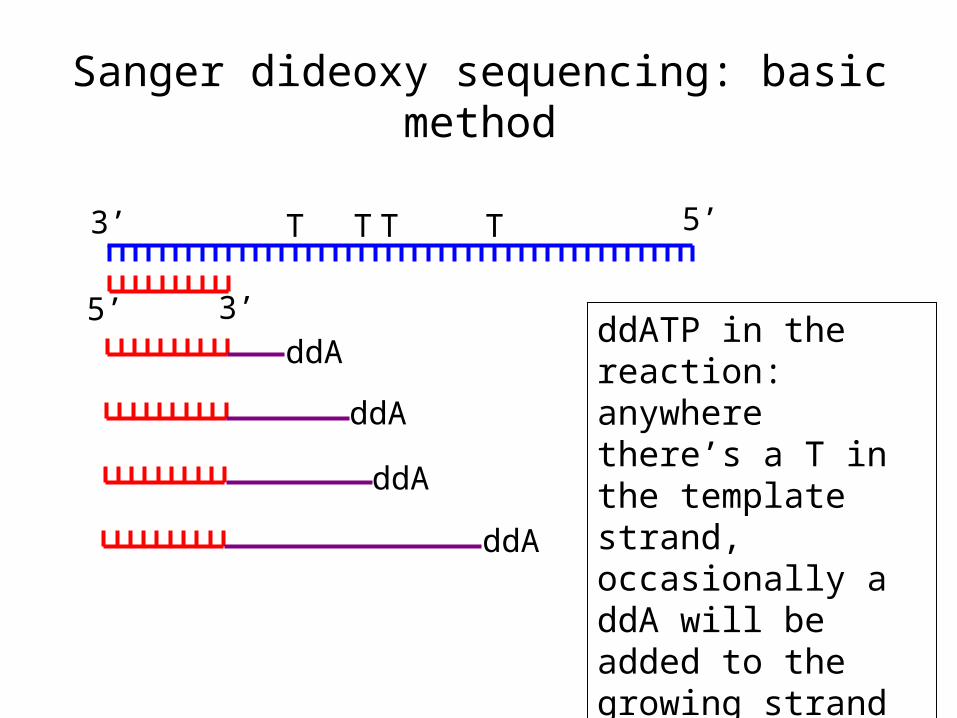
Sanger dideoxy sequencing: basic method
5’3’
5’ 3’
T T TT
ddA
ddA
ddA
ddA
ddATP in the reaction: anywhere there’s a T in the template strand, occasionally a ddA will be added to the growing strand

How to visualize DNA fragments?
• Radioactivity– Radiolabeled primers (kinase with 32P)– Radiolabelled dNTPs (gamma 35S or 32P)
• Fluorescence– ddNTPs chemically synthesized to contain
fluors– Each ddNTP fluoresces at a different
wavelength allowing identification

Analysis of sequencing products:
Polyacrylamide gel electrophoresis--good resolution of fragments differing by a single dNTP– Slab gels: as previously described– Capillary gels: require only a tiny
amount of sample to be loaded, run much faster than slab gels, best for high throughput sequencing

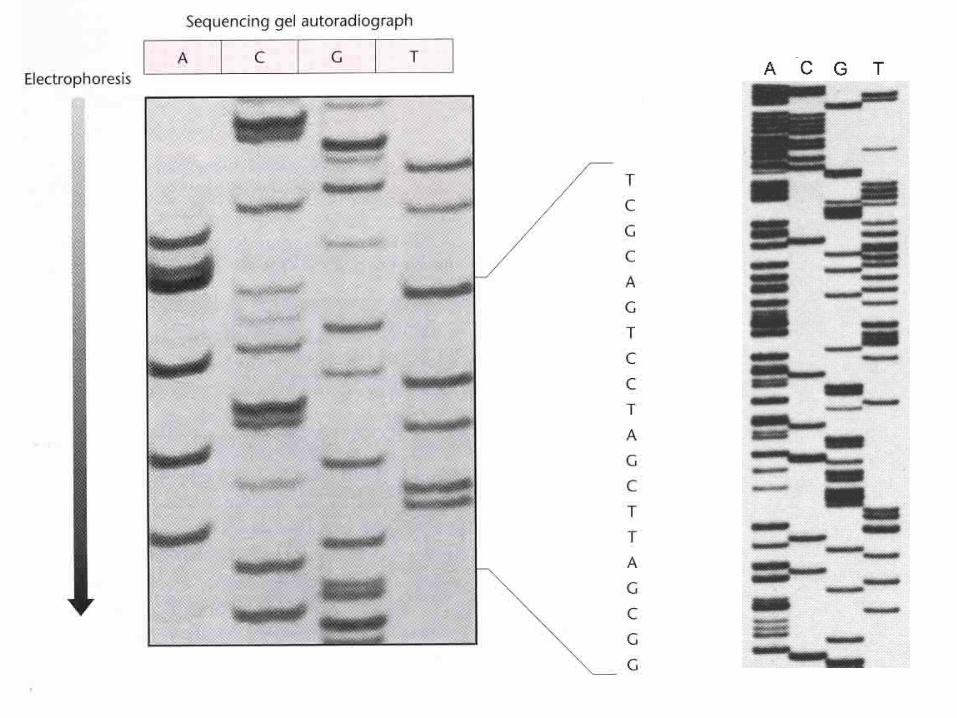
DNA sequencing gels: old school
Analyze sequencing products by gel electrophoresis, autoradiography
Different ddNTP used in separate reactions
Radioactively labelled primer or dNTP in sequencing reaction
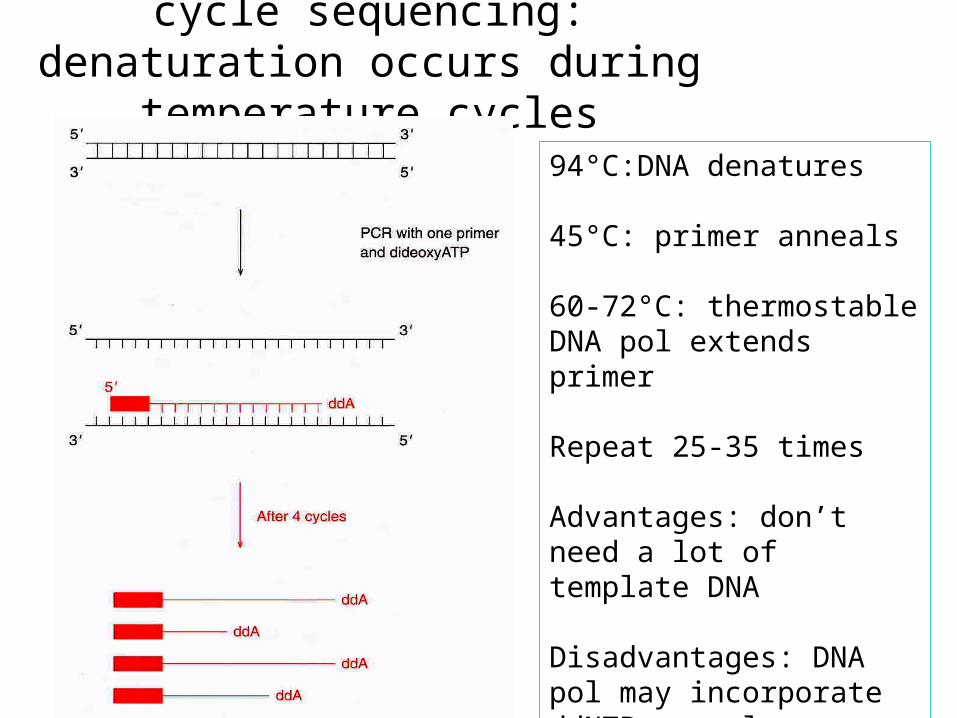
cycle sequencing: denaturation occurs during temperature cycles
94°C:DNA denatures
45°C: primer anneals
60-72°C: thermostable DNA pol extends primer
Repeat 25-35 times
Advantages: don’t need a lot of template DNA
Disadvantages: DNA pol may incorporate ddNTPs poorly

Animation of cycle sequencing: seehttp://www.dnai.org/
Click on:“manipulation”“techniques”“sorting and sequencing”
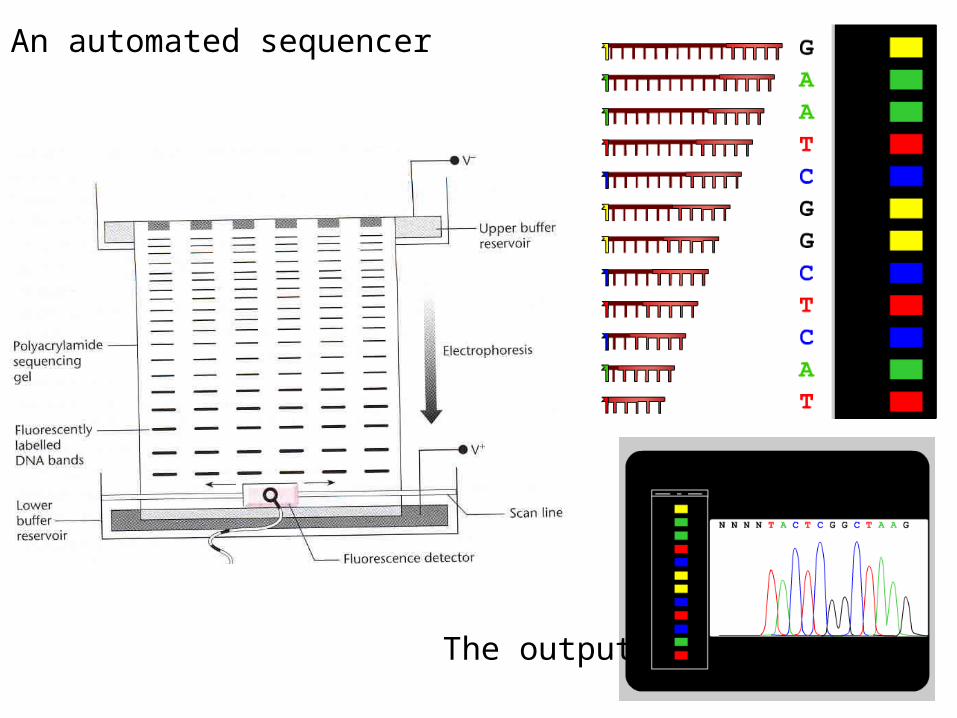
An automated sequencer
The output

Current trends in sequencing:
It is rare for labs to do their own sequencing:--costly, perishable reagents--time consuming--success rate varies
Instead most labs send out for sequencing:
--You prepare the DNA (usually plasmid, M13, or PCR product), supply the primer, company or university sequencing center does the rest
--The sequence is recorded by an automated sequencer as an “electropherogram”
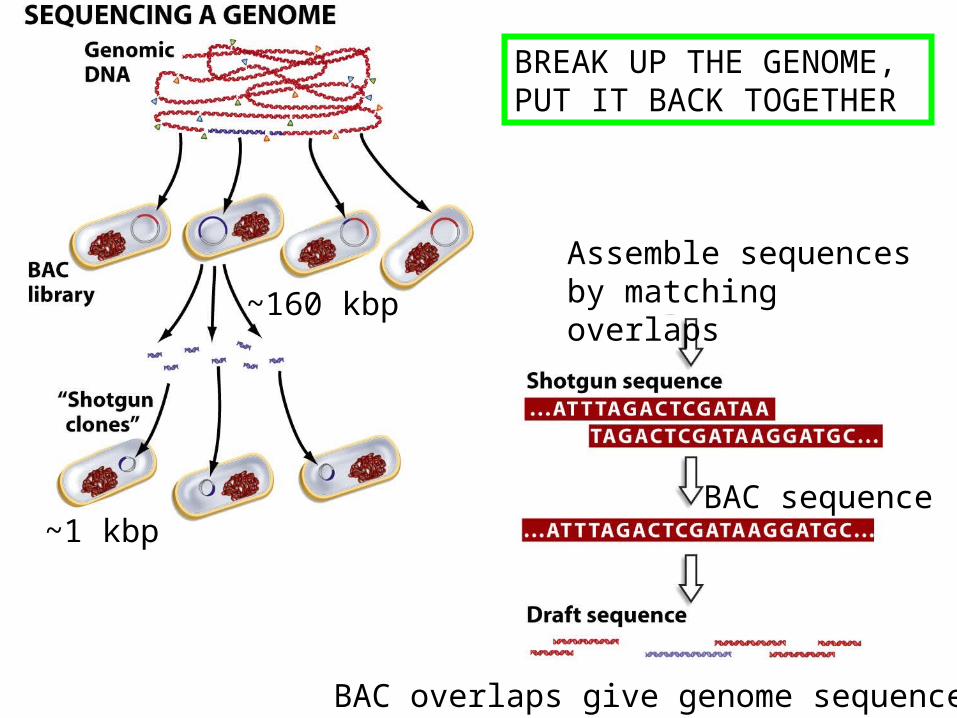
~160 kbp
~1 kbp
Assemble sequences by matching overlaps
BAC sequence
BAC overlaps give genome sequence
BREAK UP THE GENOME, PUT IT BACK TOGETHER

Sequencing large pieces of DNA:the “shotgun” method
• Break DNA into small pieces (typically sizes of around 1000 base pairs is preferable)
• Clone pieces of DNA into M13• Sequence enough M13 clones to ensure complete
coverage (eg. sequencing a 3 million base pair genome would require 5x to 10x 3 million base pairs to have a reliable representation of the genome)
• Assemble genome through overlap analysis using computer algorithms, also “polish” sequences using mapping information from individual clones, characterized genes, and genetic markers
• This process is assisted by robotics

Sequencing done by TIGR (Maryland) and The Sanger Institute (Cambridge, UK)
“Here we report an analysis of the genome sequence of P. falciparum clone 3D7, including descriptions of chromosome structure, gene content, functional classification of proteins, metabolism and transport, and other features of parasite biology.”

Sequencing strategyA whole chromosome shotgun sequencing
strategy was used to determine the genome sequence of P. falciparum clone 3D7. This approach was taken because a whole genome shotgun strategy was not feasible or cost-effective with the technology that was available at the beginning of the project. Also, high-quality large insert libraries of (A - T)-rich P. falciparum DNA have never been constructed in Escherichia coli, which ruled out a clone-by-clone sequencing strategy. The chromosomes were separated on pulsed field gels, and chromosomal DNA was extracted…

The shotgun sequences were assembled into contiguous DNA sequences (contigs), in some cases with low coverage shotgun sequences of yeast artificial chromosome (YAC) clones to assist in the ordering of contigs for closure. Sequence tagged sites (STSs)10, microsatellite markers11,12 and HAPPY mapping7 were also used to place and orient contigs during the gap closure process. The high (A /T) content of the genome made gap closure extremely difficult7–9.
Chromosomes 1–5, 9 and 12 were closed, whereas chromosomes 6–8, 10, 11, 13 and 14 contained 3–37 gaps (most less than 2.5 kb) per chromosome at the beginning of genome annotation. Efforts to close the remaining gaps are continuing.

Methods: Sequencing, gap closure and annotationThe techniques used at each of the three
participating centres for sequencing, closure and annotation are described in the accompanying Letters7–
9. To ensure that each centres’ annotation procedures produced roughly equivalent results, the Wellcome Trust Sanger Institute (‘Sanger’) and the Institute for Genomic Research (‘TIGR’) annotated the same100-kb segment of chromosome 14. The number of genes predicted in this sequence by the two centres was 22 and 23; the discrepancy being due to the merging of two single genes by one centre. Of the 74 exons predicted by the two centres, 50 (68%) were identical, 9 (2%) overlapped, 6 (8%) overlapped and shared one boundary, and the remainder were predicted by one centre but not the other. Thus 88% of the exons predicted by the two centres in the 100-kb fragment were identical or overlapped.
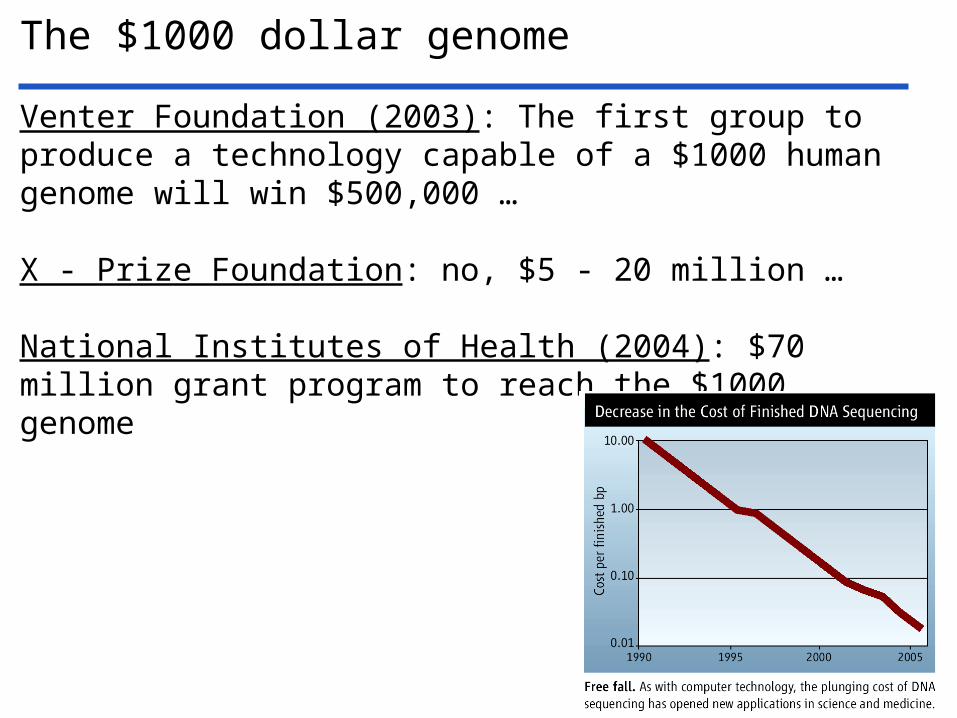
The $1000 dollar genome
Venter Foundation (2003): The first group to produce a technology capable of a $1000 human genome will win $500,000 …
X - Prize Foundation: no, $5 - 20 million …
National Institutes of Health (2004): $70 million grant program to reach the $1000 genome

Previous sequencing techniques: one DNA molecule at a timeNeeded: many DNA molecules at a time -- arrays
One of these: “pyrosequencing”
Cut a genome to DNA fragments 300 - 500 bases long
Immobilize single strands on a very small plastic bead (one piece of DNA per bead)
Amplify the DNA on each bead to cover each bead to boost the signal
Separate each bead on a plate with up to 1.6 million wells

Sequence by DNA polymerase -dependent chain extension, one base at a time in the presence of a reporter (luciferase)
Luciferase is an enzyme that will emit a photon of light in response to the pyrophosphate (PPi) released upon nucleotide addition by DNA polymerase
Flashes of light and their intensity are recorded
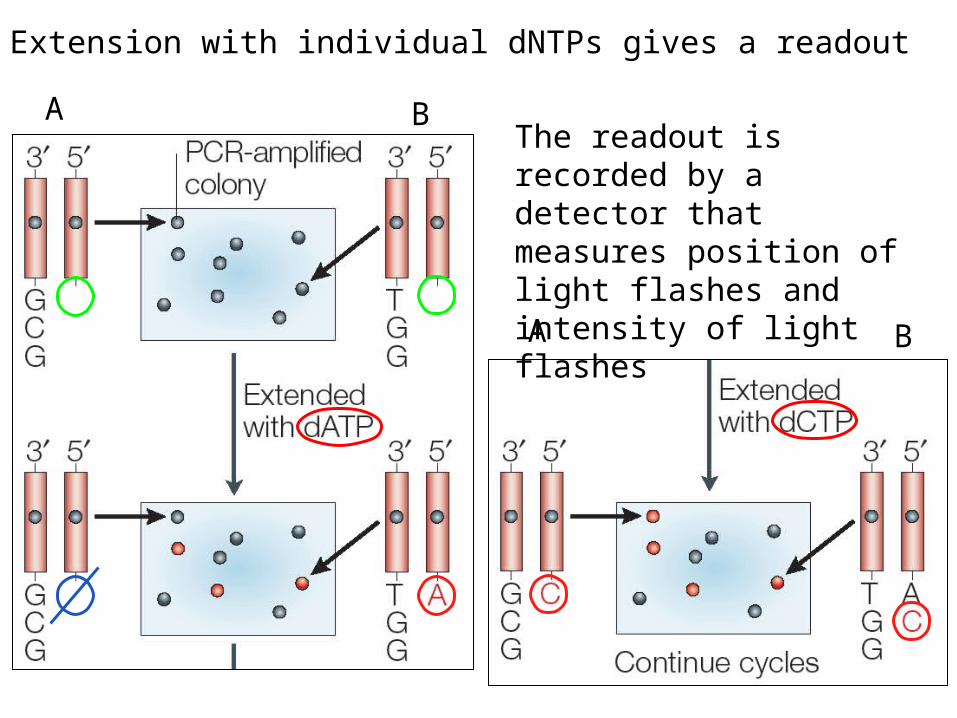
Extension with individual dNTPs gives a readout
A B
A B
The readout is recorded by a detector that measures position of light flashes and intensity of light flashes
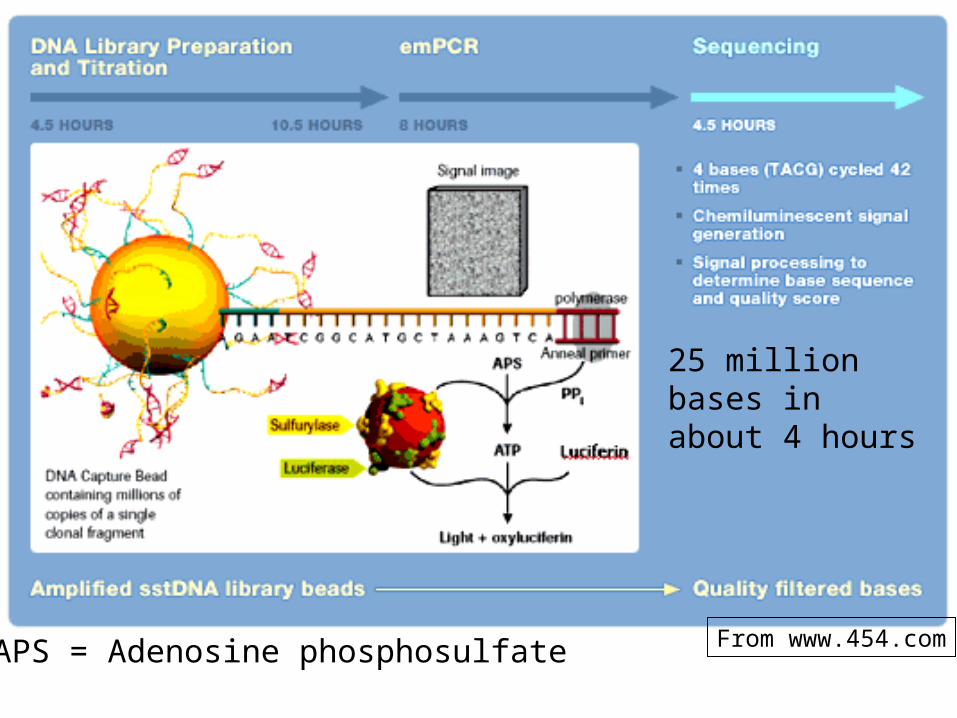
APS = Adenosine phosphosulfate From www.454.com
25 million bases in about 4 hours
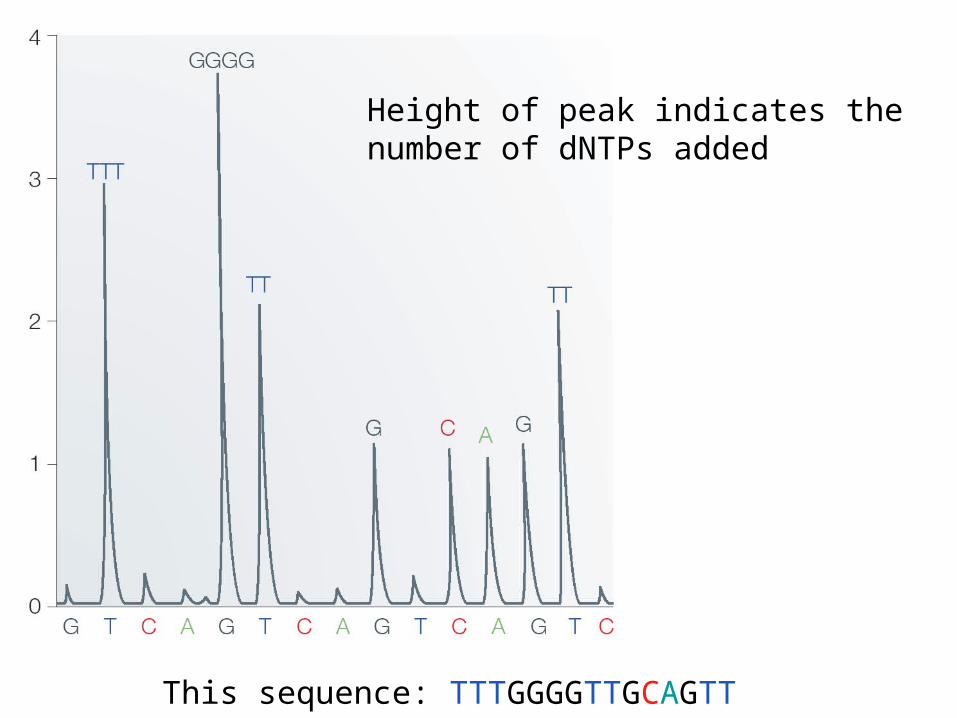
Height of peak indicates the number of dNTPs added
This sequence: TTTGGGGTTGCAGTT

DNA sequencing: methods
I. Brief history of sequencingII. Sanger dideoxy method for sequencingIII. Sequencing large pieces of DNAVI. The “$1,000 dollar genome”
On WebCT-- “The $1000 genome”-- review of new sequencing techniques by George Church

Introduction to bioinformatics
1) Making biological sense of DNA sequences
2) Online databases: a brief survey3) Database in depth: NCBI4) What is BLAST?5) Using BLAST for sequence
analysis6) “Biology workbench”, etc.
www.ncbi.nlm.nih.govwww.tigr.orghttp://workbench.sdsc.edu
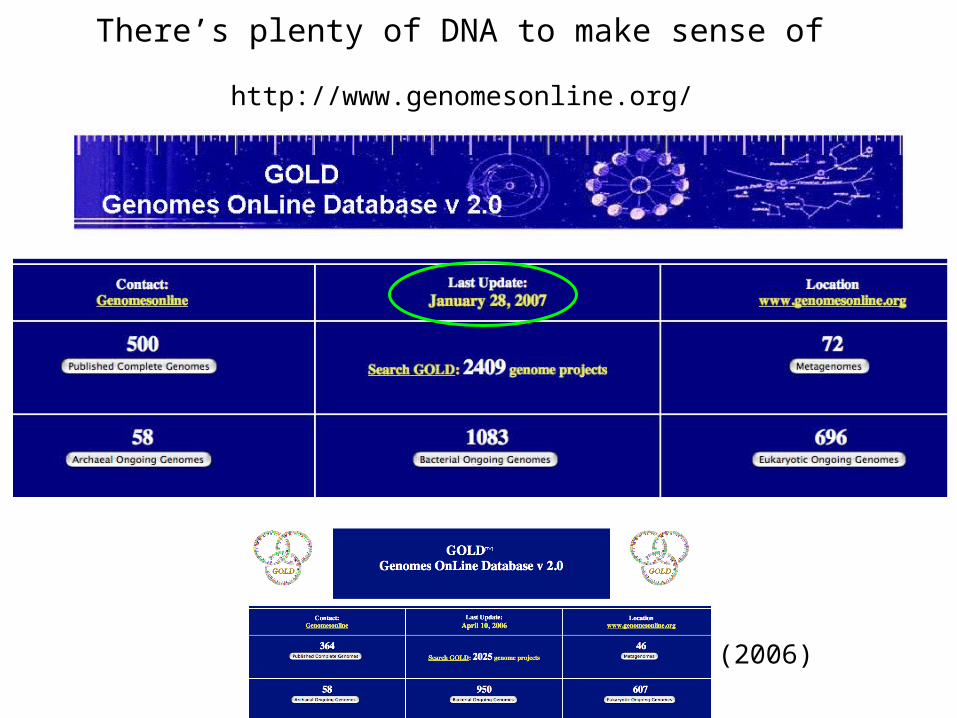
There’s plenty of DNA to make sense of
http://www.genomesonline.org/
(2006)

Making sense of genome sequences:
1) Genes
a) Protein-coding• Where are the open reading frames?• What are the ORFs most similar to?
(What is the function/structure/evolution history?)
b) RNA
2) Non-genes
• Regulation: promoters and factor-binding sites
• Transactions: replication, repair, and segregation, DNA packaging (nucleosomes)
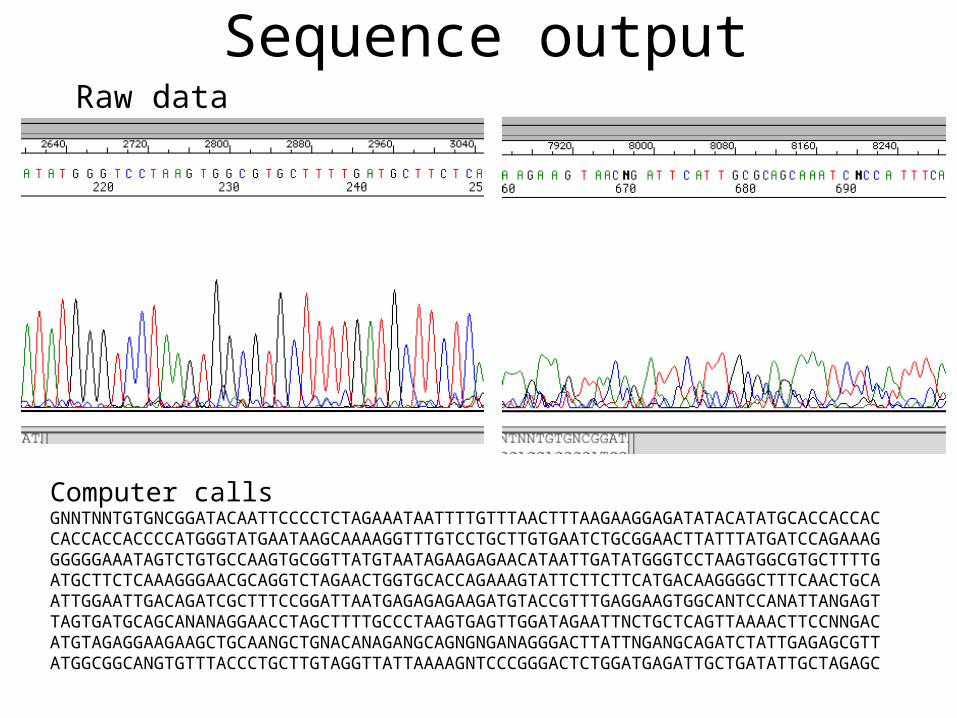
Sequence output
Computer calls
GNNTNNTGTGNCGGATACAATTCCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACATATGCACCACCACCACCACCACCCCATGGGTATGAATAAGCAAAAGGTTTGTCCTGCTTGTGAATCTGCGGAACTTATTTATGATCCAGAAAGGGGGGAAATAGTCTGTGCCAAGTGCGGTTATGTAATAGAAGAGAACATAATTGATATGGGTCCTAAGTGGCGTGCTTTTGATGCTTCTCAAAGGGAACGCAGGTCTAGAACTGGTGCACCAGAAAGTATTCTTCTTCATGACAAGGGGCTTTCAACTGCAATTGGAATTGACAGATCGCTTTCCGGATTAATGAGAGAGAAGATGTACCGTTTGAGGAAGTGGCANTCCANATTANGAGTTAGTGATGCAGCANANAGGAACCTAGCTTTTGCCCTAAGTGAGTTGGATAGAATTNCTGCTCAGTTAAAACTTCCNNGACATGTAGAGGAAGAAGCTGCAANGCTGNACANAGANGCAGNGNGANAGGGACTTATTNGANGCAGATCTATTGAGAGCGTTATGGCGGCANGTGTTTACCCTGCTTGTAGGTTATTAAAAGNTCCCGGGACTCTGGATGAGATTGCTGATATTGCTAGAGC
Raw data
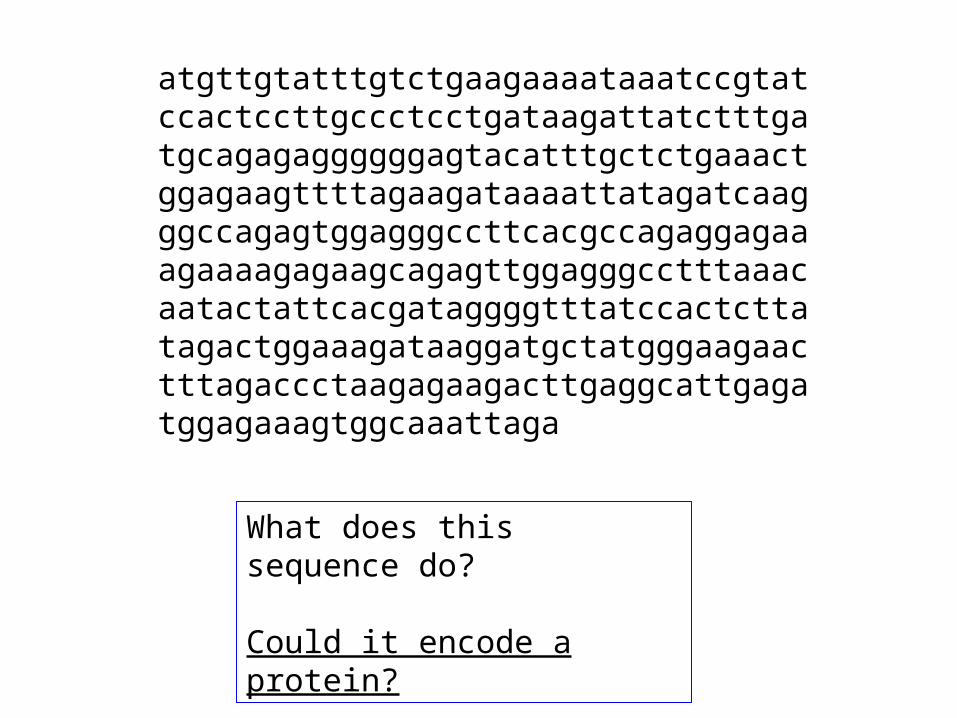
atgttgtatttgtctgaagaaaataaatccgtatccactccttgccctcctgataagattatctttgatgcagagaggggggagtacatttgctctgaaactggagaagttttagaagataaaattatagatcaagggccagagtggagggccttcacgccagaggagaaagaaaagagaagcagagttggagggcctttaaacaatactattcacgataggggtttatccactcttatagactggaaagataaggatgctatgggaagaactttagaccctaagagaagacttgaggcattgagatggagaaagtggcaaattaga
What does this sequence do?
Could it encode a protein?

Looking for ORFs (Open Reading Frames)
using “DNA Strider”
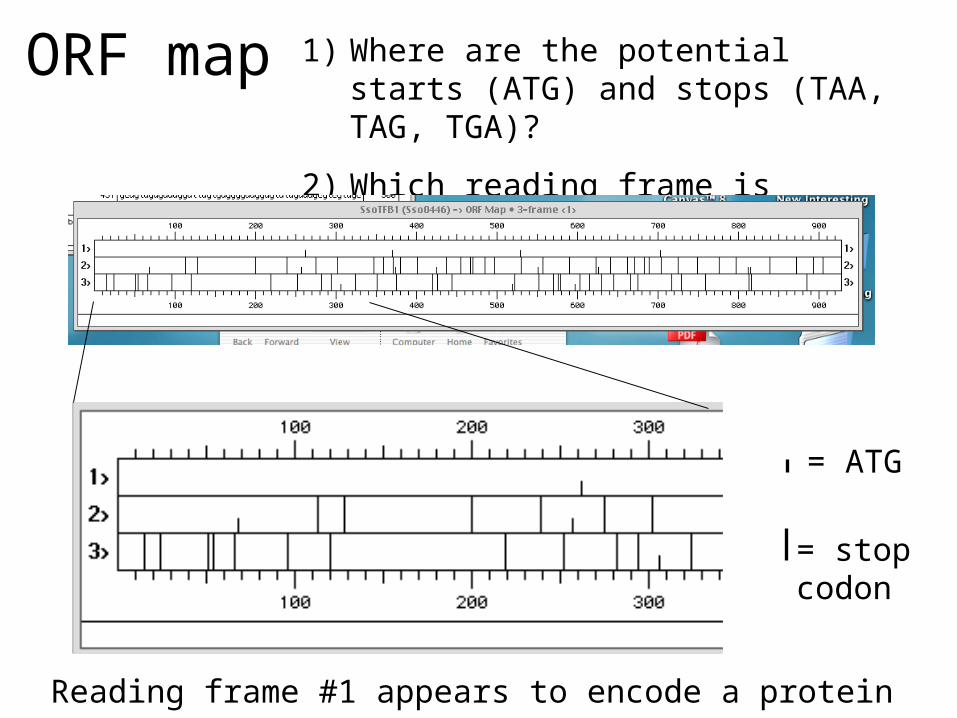
ORF map 1) Where are the potential starts (ATG) and stops (TAA, TAG, TGA)?
2) Which reading frame is correct?
= ATG
= stopcodon
Reading frame #1 appears to encode a protein

Cautions in ORF identification
• Not all genes initiate with ATG, particularly in certain microbes (archaea)
• What is the shortest possible length of a real ORF? 50 amino acids? 25 amino acids? Cut-off is somewhat arbitrary.
• In eukaryotes, ORFs can be difficult to identify because of introns
• Are there other sequences surrounding the ORF that indicate it might be functional?– promoter sequences for RNA polymerase binding– Shine-Dalgarno sequences for ribosome binding?

What is the function of the sequenced
gene?Classical methods:
-- mutate gene, characterize phenotype for clues to function (genetics)
-- purify protein product, characterize in vitro (biochemistry)
Comparison to previously characterized genes:
-- genes sequences that have high sequence similarity usually have similar functions
-- if your gene has been previously characterized (using classical methods) by someone else, you want to know right away! (avoid duplication of labor)

NCBINCBI home page --Go to www.ncbi.nlm.nih.gov for the following pages
Pubmed: search tool for literature--search by author, subject, title words, etc.
All databases: “a retrieval system for searching several linked databases”
BLAST: Basic Local Alignment Sequence Tool
OMIM: Online Mendelian Inheritance in Man
Books: many online textbooks available
Tax Browser: A taxonomic organization of organisms and their genomes
Structure: Clearinghouse for solved molecular structures

What does BLAST do?
1) Searches chosen sequence database and identifies sequences with similarity to test sequence
2) Ranks similar sequences by degree of homology (E value)
3) Illustrates alignment between test sequence and similar sequences

Alignment of sequences:
The principle: two homologous sequences derived from the same ancestral sequence will have at least some identical (similar) amino acid residues
Fraction of identical amino acids is called “percent identity”
Similar amino acids: some amino acids have similar physical/chemical properties, and more likely to substitute for each other--these give specific similarity scores in alignments
Gaps in similar/homologous sequences are rare, and are given penalty scores

Homology of proteins
Homology: similarity of biological structure, physiology, development, and evolution, based on genetic inheritance
Homologous proteins: statistically similar sequence, therefore similar functions (often, but not always…)
Alignment of TFB and TFIIB sequences
P h o T F B 1 1 - - - - - - - - - - - - - - - - - M T K Q K V C P V C G S T - - E F I Y D P E R G E I V C A R C G YP a b T F B 1 - - - - - - - - - - - - - - - - - M T K Q R V C P V C G S T - - E F I Y D P E R G E I V C A R C G YP f u T F B 1 1 - - - - - - - - - - - - - - - - - M N K Q K V C P A C E S A - - E L I Y D P E R G E I V C A K C G YT k o T F B 1 1 - - - - - - - - - - - - - - - - - M S G K R V C P V C G S T - - E F I Y D P S R G E I V C K V C G YT k o T F B 2 1 - - - - - - - - - - - - M R G - - I S P K R V C P I C G S T - - E F I Y D P R R G E I V C A K C G YP f u T F B 2 1 - - - - - - - M S S T E P G G G W L I Y P V K C P Y C K S R - - D L V Y D R Q H G E V F C K K C G SP h o T F B 2 _ d e d u c e d N T D i s f r o m B L A S T _ 1 - - - - - - - - - - - - Y G G - - - - S K I R C P V C G S S - - K I I Y D P E H G E Y Y C A E C G HS s o T F B 1 1 - - - - - - - - - - - - M L Y L S E E N K S V S T P C P P D - - K I I F D A E R G E Y I C S E T G ES s o T F B 2 1 - - - - - - - - - - - - - - - - - - - - - M K C P Y C K T D N - A I T Y D V E K G M Y V C T N C A SS c e T F I I B 1 M M T R E S I D K R A G R R G P N L N I V L T C P E C K V Y P P K I V E R F S E G D V V C A L C G Lc o n s e n s u s 1 m k v c p v C g s t e l i y d p e r G e i v C a r c g y
P h o T F B 1 3 2 V I E E N I I D M G P E W R A F D A S Q R - - E K R S R T G A P E S I L L H D K G L S T D I G I D RP a b T F B 3 2 V I E E N I V D M G P E W R A F D A S Q R - - E K R S R T G A P E S I L L H D K G L S T D I G I D RP f u T F B 1 3 2 V I E E N I I D M G P E W R A F D A S Q R - - E R R S R T G A P E S I L L H D K G L S T E I G I D RT k o T F B 1 3 2 V I E E N V V D E G P E W R A F D P G Q R - - E K R A R V G A P E S I L L H D K G L S T D I G I D RT k o T F B 2 3 5 V I E E N V V D E G P E W R A F E P G Q R - - E K R A R T G A P M T L M I H D K G L S T D I D W R DP f u T F B 2 4 2 I L A T N L V D S E L - - - - - - - - - - - - - - S R K T K T N D I P R Y - T K R I G - - - - - - -P h o T F B 2 _ d e d u c e d N T D i s f r o m B L A S T _ 3 3 V I K S - - F D T R V - - - - - - - - - - - - - - R T F S S P - - - P K F R S K G T S - - - - - - -S s o T F B 1 3 7 V L E D K I I D Q G P E W R A F T P E E K - - E K R S R V G G P L N N T I H D R G L S T L I D W K DS s o T F B 2 2 9 V I E D S A V D P G P D W R A Y N A K D R - - N E K E R V G S P S T P K V H D W G F H T I I G Y G RS c e T F I I B 5 1 V L S D K L V D T R S E W R T F S N D D H N G D D P S R V G E A S N P L L D G N N L S T R I G K G Ec o n s e n s u s 5 1 v i e e n i v D g p e w r a f d q r e k r s r t g a p e s i l l h d k g l s t d i g r
P h o T F B 1 8 0 - - - - - - S L T G L M R E K M Y R L R K W Q S R L R V S D A A E R N L A F A L S E L D R I T A Q LP a b T F B 8 0 - - - - - - S L T G L M R E K M Y R L R K W Q S R L R V S D A A E R N L A F A L S E L D R I T A Q LP f u T F B 1 8 0 - - - - - - S L S G L M R E K M Y R L R K W Q S R L R V S D A A E R N L A F A L S E L D R I T A Q LT k o T F B 1 8 0 - - - - - - S L T G L M R E K M Y R L R K W Q S R L R V S D A A E R N L A F A L S E L D R L A S N LT k o T F B 2 8 3 K D I H G N Q I T G M Y R N K L R R L R M W Q R R M R I N D A A E R N L A F A L S E L D R M A A Q LP f u T F B 2 7 0 - - - - - - - - - E F T R E K I Y R L R K W Q K K I - - - - S S E R N L V L A M S E L R R L S G M LP h o T F B 2 _ d e d u c e d N T D i s f r o m B L A S T _ 5 7 - - - - - - - - - D M V R E K I H R L K R L D S - - - - - - F G N K T E K L G V E E I S R I S S Q LS s o T F B 1 8 5 K D A M G R T L D P K R R L E A L R W R K W Q I R A R I Q S S I D R N L A Q A M N E L E R I G N L LS s o T F B 2 7 7 - - - - - - - - - A K D R L K T L K M Q R M Q N K I R V S - P K D K K L V T L L S I L N D E S S K LS c e T F I I B 1 0 1 - - - - - - - - - - T T D M R F T K E L N K A Q G K N V M D K K D N E V Q A A F A K I T M L C D A Ac o n s e n s u s 1 0 1 s l t g l m r e k m y r l r k w q s r l r v s d a a e r n l a f a l s e l d r i t a q l
P h o T F B 1 1 2 4 K L P K H V E E E A A R L Y R E A V R K G L I R G R S I E S V I A A C V Y A A C R L L K V P R T L DP a b T F B 1 2 4 K L P K H V E E E A A R L Y R E A V R K G L I R G R S I E S V I A A C V Y A A C R L L K V P R T L DP f u T F B 1 1 2 4 K L P R H V E E E A A R L Y R E A V R K G L I R G R S I E S V M A A C V Y A A C R L L K V P R T L DT k o T F B 1 1 2 4 S L P K H V E E E A A R L Y R E A V R K G L I R G R S I E A V I A A C V Y A A C R L L K V P R T L DT k o T F B 2 1 3 3 R L P R H L K E V A A S L Y R K A V M K K L I R G R S I E G M V S A A L Y A A C R M E G I P R T L DP f u T F B 2 1 0 7 K L P K Y V E E E A A Y L Y R E A A K R G L T R R I P I E T T V A A C I Y A T C R L F K V P R T L NP h o T F B 2 _ d e d u c e d N T D i s f r o m B L A S T _ 9 2 C L P K H V E R E A V R I Y R K L I K S G V T K G R S I E S V A A A C I Y I S C R L Y K V P R T L DS s o T F B 1 1 3 5 N L P K S V K D E A A L I Y R K A V E K G L V R G R S I E S V V A A A I Y A A C R R M K L A R T L DS s o T F B 2 1 1 7 E L P E H V K E T A S L I I R K M V E T G L T K R I D Q Y T L I V A A L Y Y S C Q V N N I P R H L QS c e T F I I B 1 4 1 E L P K I V K D C A K E A Y K L C H D E K T L K G K S M E S I M A A S I L I G C R R A E V A R T F Kc o n s e n s u s 1 5 1 k L P k h v e e e A a r l y r e a v r k g l i r g r s i e s v i a A c v y a a C r l l k v p R t l d
P h o T F B 1 1 7 4 E I S D I A R V E K K E I G R S Y R F I A R N L N - - - - - - - - - - L T P K K L F V K P T D Y V NP a b T F B 1 7 4 E I S D I A R V E K K E I G R S Y R F I A R N L N - - - - - - - - - - L T P K K L F V K P T D Y V NP f u T F B 1 1 7 4 E I A D I A R V D K K E I G R S Y R F I A R N L N - - - - - - - - - - L T P K K L F V K P T D Y V NT k o T F B 1 1 7 4 E I A D V S R V D K K E I G R S F R F I A R H L N - - - - - - - - - - L T P K K L F V K P T D Y V NT k o T F B 2 1 8 3 E I A S V S K V S K K E I G R S Y R F M A R G L G - - - - - - - - - - L N L R P - - T S P I E Y V DP f u T F B 2 1 5 7 E I A S Y S K T E K K E I M K A F R V I V R N L N - - - - - - - - - - L T P K M L L A R P T D Y V DP h o T F B 2 _ d e d u c e d N T D i s f r o m B L A S T _ 1 4 2 E I A K V A K E D K K V I A R V Y R L V V K K L G - - - - - - - - - - L S S K D M L I R P E Y Y I DS s o T F B 1 1 8 5 E I A Q Y T K A N R K E V A R C Y R L L L R E L D - - - - - - - - - - V S V P V S - - D P K D Y V TS s o T F B 2 1 6 7 E F K V R Y S I S S S E F W S A L K R V Q Y V A N S - - - - - - - - - I P G F R P K I K P A E Y I PS c e T F I I B 1 9 1 E I Q S L I H V K T K E F G K T L N I M K N I L R G K S E D G F L K I D T D N M S G A Q N L T Y I Pc o n s e n s u s 2 0 1 E i a i r v e k k e i g r s y r f i a r l n l t p k k l v k p t d Y v
P h o T F B 1 1 - - - - - - - - - - - - - - - - - M T K Q K V C P V C G S T - - E F I Y D P E R G E I V C A R C G YP a b T F B 1 - - - - - - - - - - - - - - - - - M T K Q R V C P V C G S T - - E F I Y D P E R G E I V C A R C G YP f u T F B 1 1 - - - - - - - - - - - - - - - - - M N K Q K V C P A C E S A - - E L I Y D P E R G E I V C A K C G YT k o T F B 1 1 - - - - - - - - - - - - - - - - - M S G K R V C P V C G S T - - E F I Y D P S R G E I V C K V C G YT k o T F B 2 1 - - - - - - - - - - - - M R G - - I S P K R V C P I C G S T - - E F I Y D P R R G E I V C A K C G YP f u T F B 2 1 - - - - - - - M S S T E P G G G W L I Y P V K C P Y C K S R - - D L V Y D R Q H G E V F C K K C G SP h o T F B 2 _ d e d u c e d N T D i s f r o m B L A S T _ 1 - - - - - - - - - - - - Y G G - - - - S K I R C P V C G S S - - K I I Y D P E H G E Y Y C A E C G HS s o T F B 1 1 - - - - - - - - - - - - M L Y L S E E N K S V S T P C P P D - - K I I F D A E R G E Y I C S E T G ES s o T F B 2 1 - - - - - - - - - - - - - - - - - - - - - M K C P Y C K T D N - A I T Y D V E K G M Y V C T N C A SS c e T F I I B 1 M M T R E S I D K R A G R R G P N L N I V L T C P E C K V Y P P K I V E R F S E G D V V C A L C G Lc o n s e n s u s 1 m k v c p v C g s t e l i y d p e r G e i v C a r c g y
P h o T F B 1 3 2 V I E E N I I D M G P E W R A F D A S Q R - - E K R S R T G A P E S I L L H D K G L S T D I G I D RP a b T F B 3 2 V I E E N I V D M G P E W R A F D A S Q R - - E K R S R T G A P E S I L L H D K G L S T D I G I D RP f u T F B 1 3 2 V I E E N I I D M G P E W R A F D A S Q R - - E R R S R T G A P E S I L L H D K G L S T E I G I D RT k o T F B 1 3 2 V I E E N V V D E G P E W R A F D P G Q R - - E K R A R V G A P E S I L L H D K G L S T D I G I D RT k o T F B 2 3 5 V I E E N V V D E G P E W R A F E P G Q R - - E K R A R T G A P M T L M I H D K G L S T D I D W R DP f u T F B 2 4 2 I L A T N L V D S E L - - - - - - - - - - - - - - S R K T K T N D I P R Y - T K R I G - - - - - - -P h o T F B 2 _ d e d u c e d N T D i s f r o m B L A S T _ 3 3 V I K S - - F D T R V - - - - - - - - - - - - - - R T F S S P - - - P K F R S K G T S - - - - - - -S s o T F B 1 3 7 V L E D K I I D Q G P E W R A F T P E E K - - E K R S R V G G P L N N T I H D R G L S T L I D W K DS s o T F B 2 2 9 V I E D S A V D P G P D W R A Y N A K D R - - N E K E R V G S P S T P K V H D W G F H T I I G Y G RS c e T F I I B 5 1 V L S D K L V D T R S E W R T F S N D D H N G D D P S R V G E A S N P L L D G N N L S T R I G K G Ec o n s e n s u s 5 1 v i e e n i v D g p e w r a f d q r e k r s r t g a p e s i l l h d k g l s t d i g r
P h o T F B 1 8 0 - - - - - - S L T G L M R E K M Y R L R K W Q S R L R V S D A A E R N L A F A L S E L D R I T A Q LP a b T F B 8 0 - - - - - - S L T G L M R E K M Y R L R K W Q S R L R V S D A A E R N L A F A L S E L D R I T A Q LP f u T F B 1 8 0 - - - - - - S L S G L M R E K M Y R L R K W Q S R L R V S D A A E R N L A F A L S E L D R I T A Q LT k o T F B 1 8 0 - - - - - - S L T G L M R E K M Y R L R K W Q S R L R V S D A A E R N L A F A L S E L D R L A S N LT k o T F B 2 8 3 K D I H G N Q I T G M Y R N K L R R L R M W Q R R M R I N D A A E R N L A F A L S E L D R M A A Q LP f u T F B 2 7 0 - - - - - - - - - E F T R E K I Y R L R K W Q K K I - - - - S S E R N L V L A M S E L R R L S G M LP h o T F B 2 _ d e d u c e d N T D i s f r o m B L A S T _ 5 7 - - - - - - - - - D M V R E K I H R L K R L D S - - - - - - F G N K T E K L G V E E I S R I S S Q LS s o T F B 1 8 5 K D A M G R T L D P K R R L E A L R W R K W Q I R A R I Q S S I D R N L A Q A M N E L E R I G N L LS s o T F B 2 7 7 - - - - - - - - - A K D R L K T L K M Q R M Q N K I R V S - P K D K K L V T L L S I L N D E S S K LS c e T F I I B 1 0 1 - - - - - - - - - - T T D M R F T K E L N K A Q G K N V M D K K D N E V Q A A F A K I T M L C D A Ac o n s e n s u s 1 0 1 s l t g l m r e k m y r l r k w q s r l r v s d a a e r n l a f a l s e l d r i t a q l
P h o T F B 1 1 2 4 K L P K H V E E E A A R L Y R E A V R K G L I R G R S I E S V I A A C V Y A A C R L L K V P R T L DP a b T F B 1 2 4 K L P K H V E E E A A R L Y R E A V R K G L I R G R S I E S V I A A C V Y A A C R L L K V P R T L DP f u T F B 1 1 2 4 K L P R H V E E E A A R L Y R E A V R K G L I R G R S I E S V M A A C V Y A A C R L L K V P R T L DT k o T F B 1 1 2 4 S L P K H V E E E A A R L Y R E A V R K G L I R G R S I E A V I A A C V Y A A C R L L K V P R T L DT k o T F B 2 1 3 3 R L P R H L K E V A A S L Y R K A V M K K L I R G R S I E G M V S A A L Y A A C R M E G I P R T L DP f u T F B 2 1 0 7 K L P K Y V E E E A A Y L Y R E A A K R G L T R R I P I E T T V A A C I Y A T C R L F K V P R T L NP h o T F B 2 _ d e d u c e d N T D i s f r o m B L A S T _ 9 2 C L P K H V E R E A V R I Y R K L I K S G V T K G R S I E S V A A A C I Y I S C R L Y K V P R T L DS s o T F B 1 1 3 5 N L P K S V K D E A A L I Y R K A V E K G L V R G R S I E S V V A A A I Y A A C R R M K L A R T L DS s o T F B 2 1 1 7 E L P E H V K E T A S L I I R K M V E T G L T K R I D Q Y T L I V A A L Y Y S C Q V N N I P R H L QS c e T F I I B 1 4 1 E L P K I V K D C A K E A Y K L C H D E K T L K G K S M E S I M A A S I L I G C R R A E V A R T F Kc o n s e n s u s 1 5 1 k L P k h v e e e A a r l y r e a v r k g l i r g r s i e s v i a A c v y a a C r l l k v p R t l d
P h o T F B 1 1 7 4 E I S D I A R V E K K E I G R S Y R F I A R N L N - - - - - - - - - - L T P K K L F V K P T D Y V NP a b T F B 1 7 4 E I S D I A R V E K K E I G R S Y R F I A R N L N - - - - - - - - - - L T P K K L F V K P T D Y V NP f u T F B 1 1 7 4 E I A D I A R V D K K E I G R S Y R F I A R N L N - - - - - - - - - - L T P K K L F V K P T D Y V NT k o T F B 1 1 7 4 E I A D V S R V D K K E I G R S F R F I A R H L N - - - - - - - - - - L T P K K L F V K P T D Y V NT k o T F B 2 1 8 3 E I A S V S K V S K K E I G R S Y R F M A R G L G - - - - - - - - - - L N L R P - - T S P I E Y V DP f u T F B 2 1 5 7 E I A S Y S K T E K K E I M K A F R V I V R N L N - - - - - - - - - - L T P K M L L A R P T D Y V DP h o T F B 2 _ d e d u c e d N T D i s f r o m B L A S T _ 1 4 2 E I A K V A K E D K K V I A R V Y R L V V K K L G - - - - - - - - - - L S S K D M L I R P E Y Y I DS s o T F B 1 1 8 5 E I A Q Y T K A N R K E V A R C Y R L L L R E L D - - - - - - - - - - V S V P V S - - D P K D Y V TS s o T F B 2 1 6 7 E F K V R Y S I S S S E F W S A L K R V Q Y V A N S - - - - - - - - - I P G F R P K I K P A E Y I PS c e T F I I B 1 9 1 E I Q S L I H V K T K E F G K T L N I M K N I L R G K S E D G F L K I D T D N M S G A Q N L T Y I Pc o n s e n s u s 2 0 1 E i a i r v e k k e i g r s y r f i a r l n l t p k k l v k p t d Y v
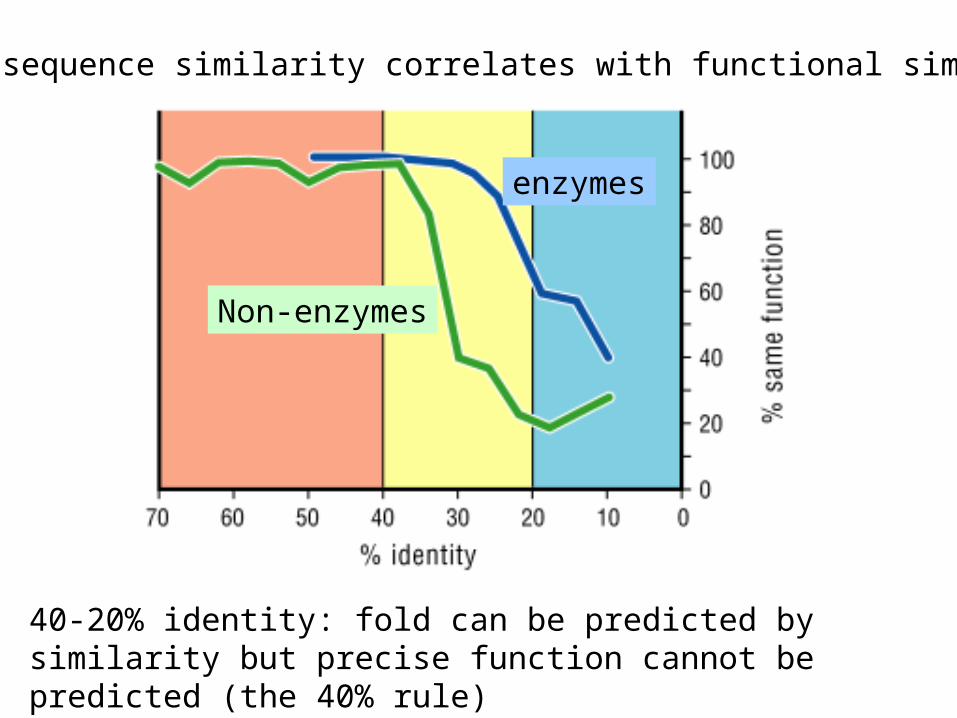
High sequence similarity correlates with functional similarity
40-20% identity: fold can be predicted by similarity but precise function cannot be predicted (the 40% rule)
enzymes
Non-enzymes

Programs available for BLAST searches
Protein sequence (this is the best option)blastp--compares an amino acid query sequence against a protein sequence database
tblastn--compares a protein query sequence against a nucleotide sequence database translated in all reading frames
DNA sequenceblastn--compares a nucleotide query sequence against a nucleotide sequence database
blastx--compares a nucleotide query sequence translated in all reading frames against a protein sequence database
tblastx--compares the six-frame translations of a nucleotide query sequence against the six-frame translations of a nucleotide sequence database.

BLAST considers all possible combinations of matchesmismatchesgaps
in any given alignment
Gives the “best” (highest scoring) alignment of sequences
Three scores1) percent identity2) similarity score3) E-value--probability that two sequences will
have the similarity they have by chance (lower number, higher probability of evolutionary homology, higher probability of similar function)

What is the E-value?
The E value represents the chance that the similarity is random and therefore insignificant. Essentially, the E value describes the random background noise that exists for matches between sequences. For example, an E value of 1 assigned to a hit can be interpreted as meaning that in a database of the current size one might expect to see 1 match with a similar score simply by chance.
You can change the Expect value threshold on most main BLAST search pages. When the Expect value is increased from the default value of 10, a larger list with more low-scoring hits can be reported.

E values (continued)
From the BLAST tutorial:
Although hits with E values much higher than 0.1 are unlikely to reflect true sequence relatives, it is useful to examine hits with lower significance (E values between 0.1 and 10) for short regions of similarity. In the absence of longer similarities, these short regions may allow the tentative assignment of biochemical activities to the ORF in question. The significance of any such regions must be assessed on a case by case basis.
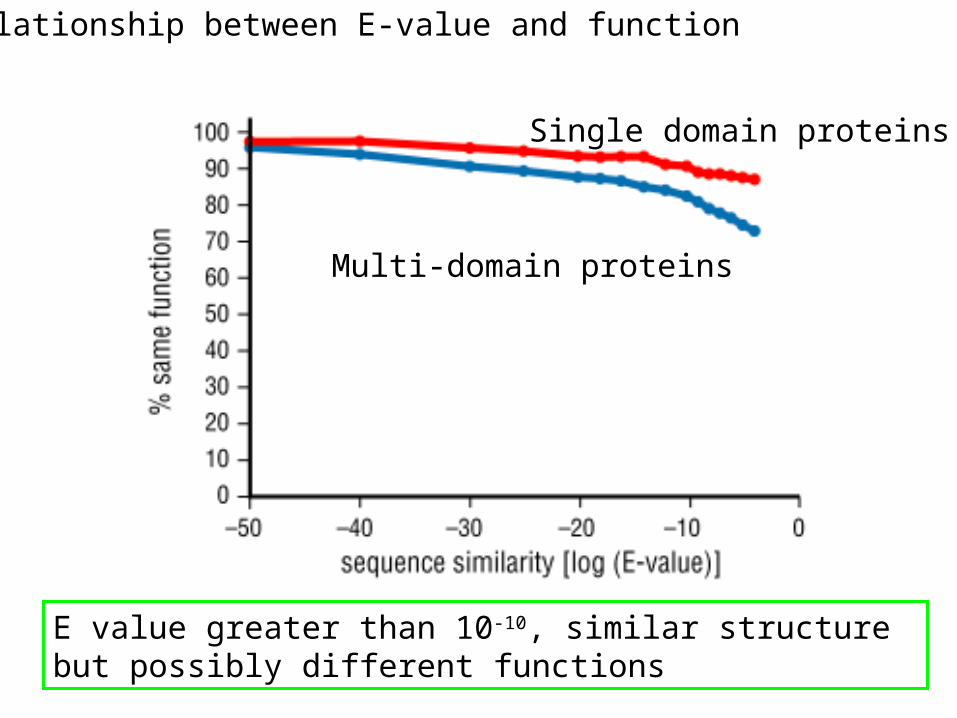
Relationship between E-value and function
Single domain proteins
Multi-domain proteins
E value greater than 10-10, similar structure but possibly different functions
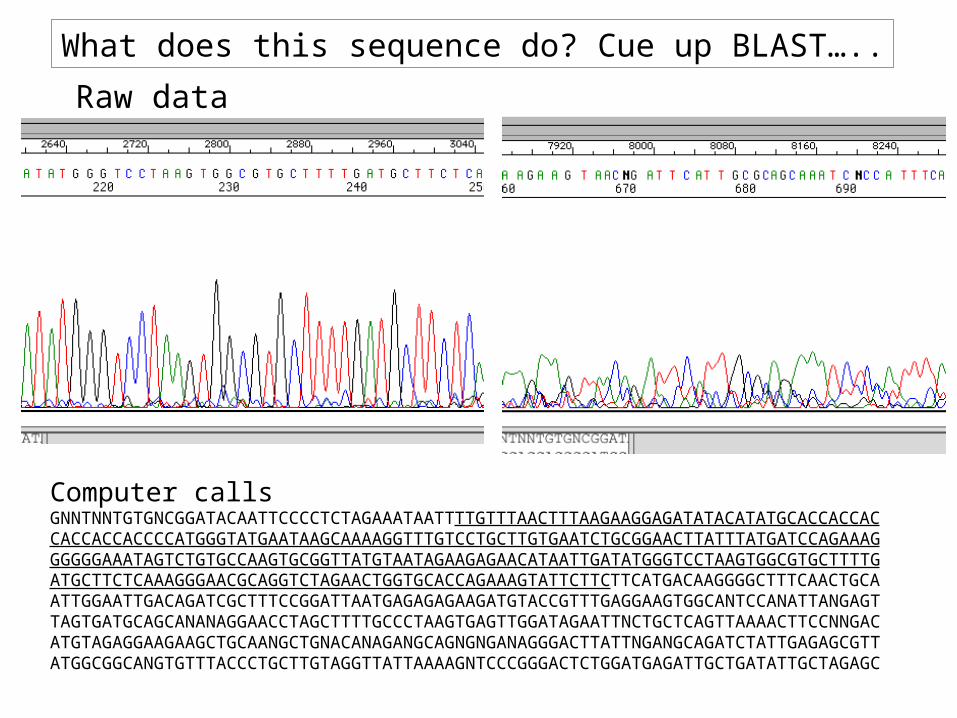
Computer calls
GNNTNNTGTGNCGGATACAATTCCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACATATGCACCACCACCACCACCACCCCATGGGTATGAATAAGCAAAAGGTTTGTCCTGCTTGTGAATCTGCGGAACTTATTTATGATCCAGAAAGGGGGGAAATAGTCTGTGCCAAGTGCGGTTATGTAATAGAAGAGAACATAATTGATATGGGTCCTAAGTGGCGTGCTTTTGATGCTTCTCAAAGGGAACGCAGGTCTAGAACTGGTGCACCAGAAAGTATTCTTCTTCATGACAAGGGGCTTTCAACTGCAATTGGAATTGACAGATCGCTTTCCGGATTAATGAGAGAGAAGATGTACCGTTTGAGGAAGTGGCANTCCANATTANGAGTTAGTGATGCAGCANANAGGAACCTAGCTTTTGCCCTAAGTGAGTTGGATAGAATTNCTGCTCAGTTAAAACTTCCNNGACATGTAGAGGAAGAAGCTGCAANGCTGNACANAGANGCAGNGNGANAGGGACTTATTNGANGCAGATCTATTGAGAGCGTTATGGCGGCANGTGTTTACCCTGCTTGTAGGTTATTAAAAGNTCCCGGGACTCTGGATGAGATTGCTGATATTGCTAGAGC
Raw data
What does this sequence do? Cue up BLAST…..
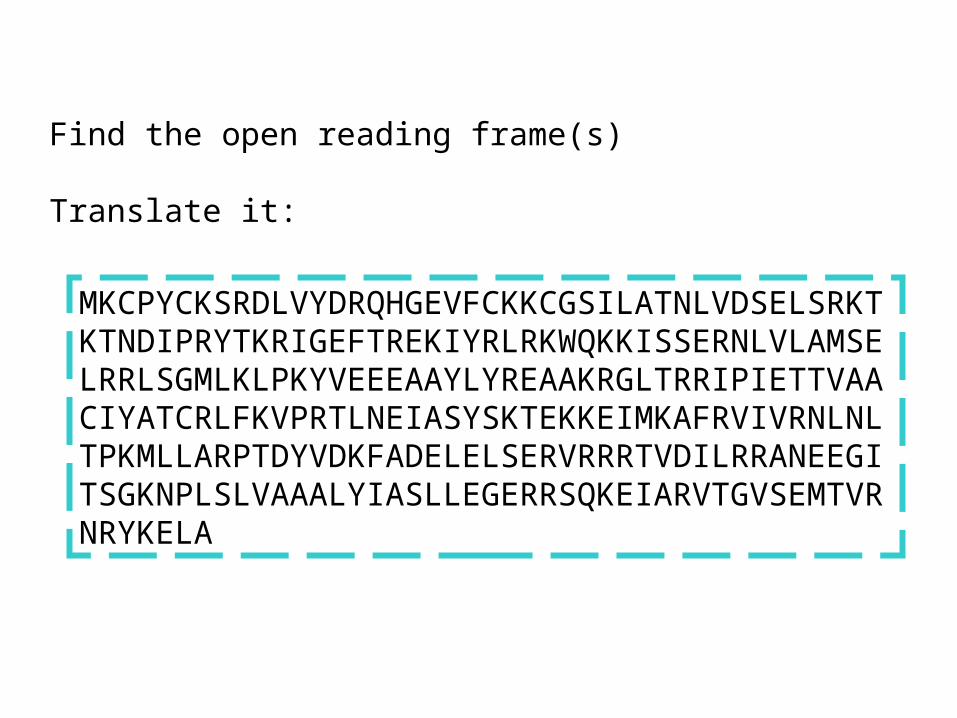
MKCPYCKSRDLVYDRQHGEVFCKKCGSILATNLVDSELSRKTKTNDIPRYTKRIGEFTREKIYRLRKWQKKISSERNLVLAMSELRRLSGMLKLPKYVEEEAAYLYREAAKRGLTRRIPIETTVAACIYATCRLFKVPRTLNEIASYSKTEKKEIMKAFRVIVRNLNLTPKMLLARPTDYVDKFADELELSERVRRRTVDILRRANEEGITSGKNPLSLVAAALYIASLLEGERRSQKEIARVTGVSEMTVRNRYKELA
Find the open reading frame(s)
Translate it:

BLAST against (go to genomes page):-- Microbial genomes-- environmental sequences (genomes)
Results:
1) Distribution of hits: query sequence and positions in sequence that gave alignments
2) Sequences producing significant alignments1) Accession number (this takes you to the sequence
that yielded the hit: gene or contig)2) Name of sequence (sometimes identifies the
gene)3) Similarity score4) E-value
3) Alignments arranged by E value, with links to gene reports

2) Large percentages of coding proteins cannot be assigned function based on homology
1) Homology? the function is only inferred (NOT known)
Two problems with BLAST
For a current list of databases and bioinformatics tools see: Nucleic Acids Research annual bioinformatics issue (comes out every January).
List of all the databases described, by category:
http://www.oxfordjournals.org/nar/database/cap/Guide to NCBI: see Webct

Bioinformatics:making sense of biological sequence
• New DNA sequences are analyzed for ORFs (Open Reading Frames: protein)
• Any DNA or protein sequence can then be compared to all other sequences in databases, and similar sequences identified
• There is much more -- a great diversity of programs and databases are available

Massively parallel measurements of gene expression: microarrays
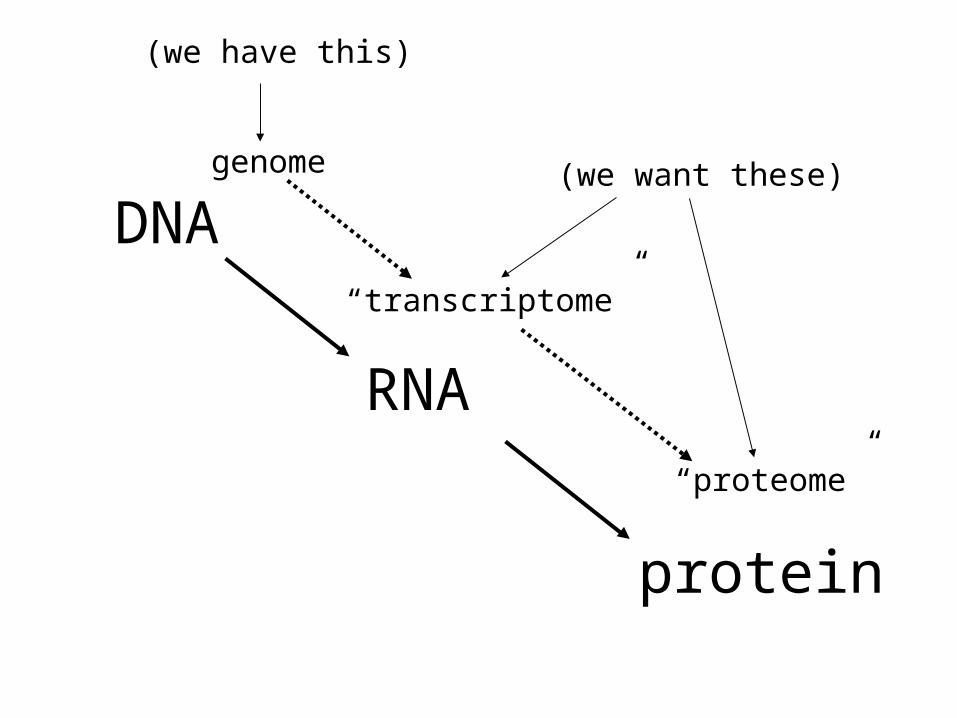
• Defining the “transcriptome”• The northern blot revisited• Detecting expression of many genes: arrays• A typical array experiment• What to do with all this data?
Brown and Botstein (1999) “Exploring the new world of the genome with DNA microarrays” Nature Genetics 21, p. 33-37.
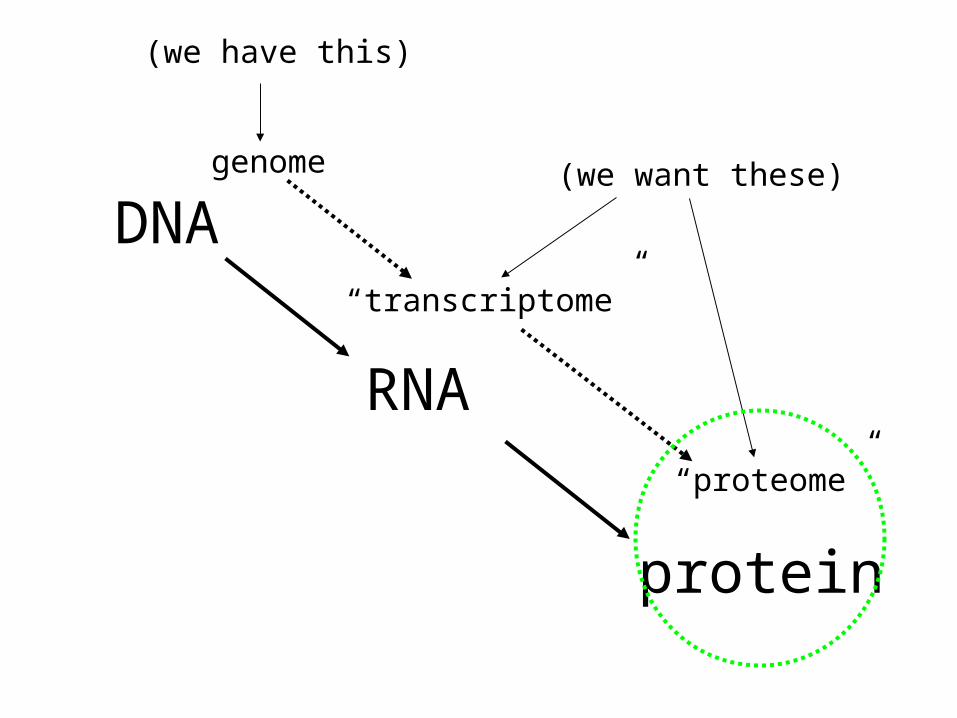
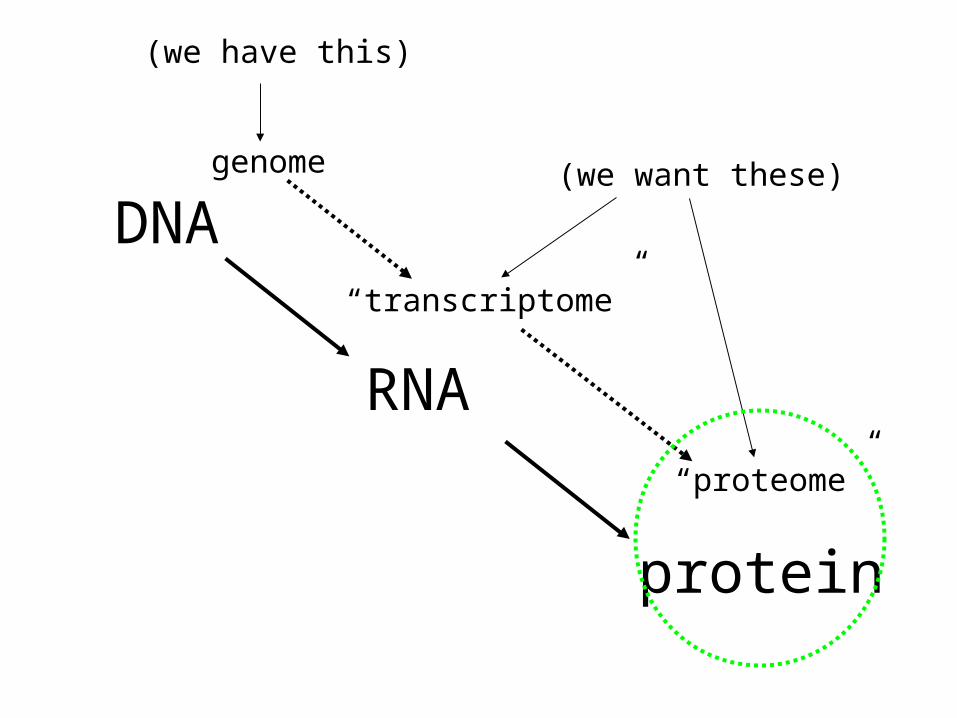
DNA
RNA
protein
genome
“transcriptome”
“proteome”
(we have this)
(we want these)

The value of DNA microarrays for studying gene expression
1) Study all transcripts at same time
2) Transcript abundance usually correlates with level of gene expression--much gene control is at level of transcription
3) Changes in transcription patterns often occur as a response to changing environment--this can be detected with a microarray

Detection of mRNA transcripts
• Northern Blot -- immobilize mRNA on membrane, detect specific sequence by hybridization with one labeled probe--requires a separate blotting for each probe
• DNA microarray -- immobilize many probes (thousands) in an ordered array, hybridize (base pair) with labelled mRNA or cDNA

Generating an array of probes
• Identify open reading frames (orfs)
1) PCR each orf (several for each orf), attach (spot) each PCR product to a solid support in a specific order (pioneered by Pat Brown’s lab, Stanford)
2) Chemically synthesize orf-specific oligonucleotide probes directly on microchip (Affymetrix)
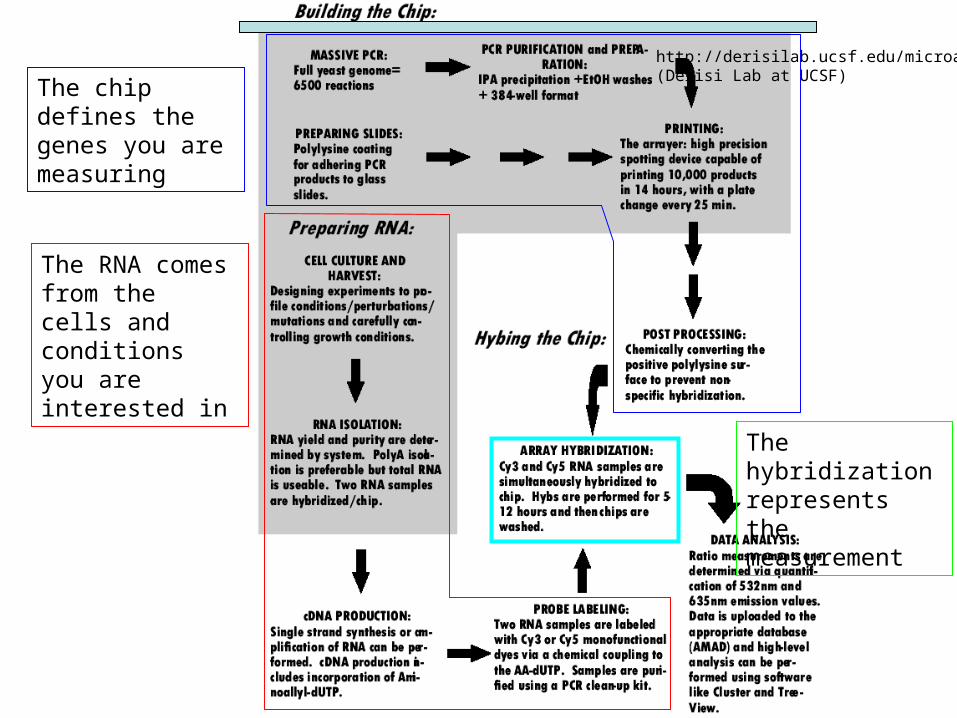
http://derisilab.ucsf.edu/microarray/(Derisi Lab at UCSF)
The chip defines the genes you are measuring
The hybridization represents the measurement
The RNA comes from the cells and conditions you are interested in
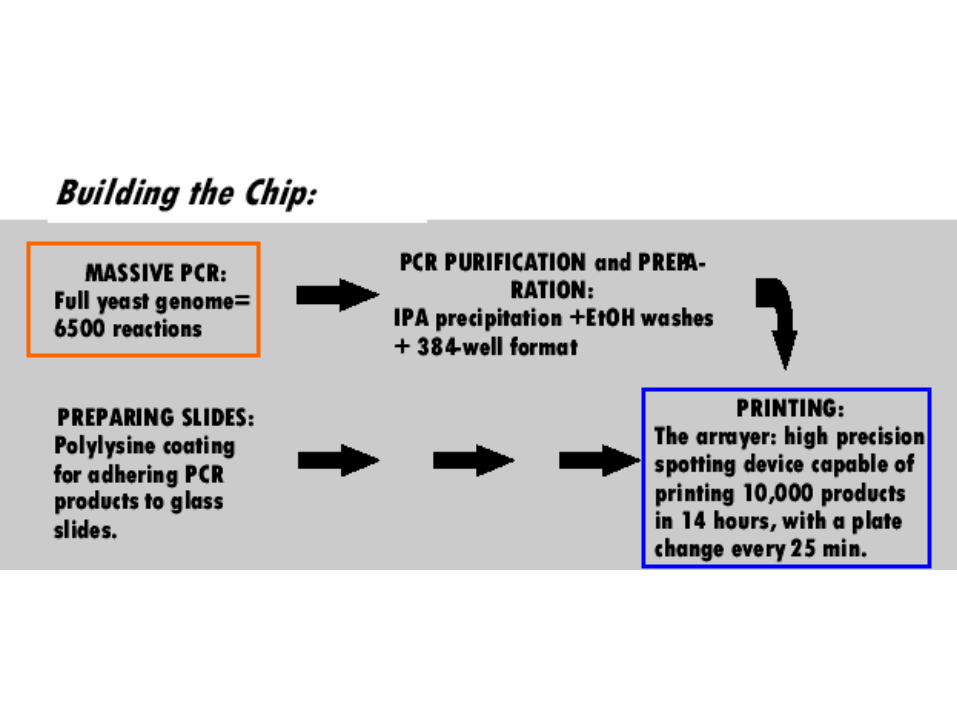

A print head for generating arrays of probes
Print head travels from DNA probe source (microtiter plate) to solid support (treated glass slide)
Small amount of DNA probe is put on a specific spot at a specific location
Each spot (DNA probe sequence) has a specific “address”
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.QuickTime™ and aTIFF (Uncompressed) decompressorare needed to see this picture.
Print head
Printing needles
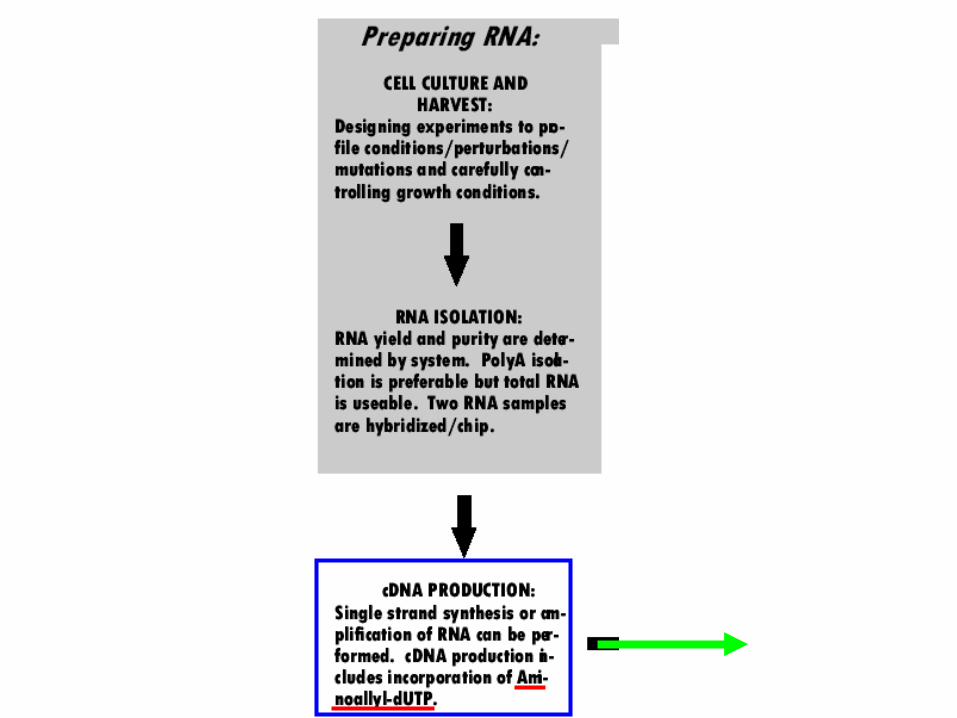
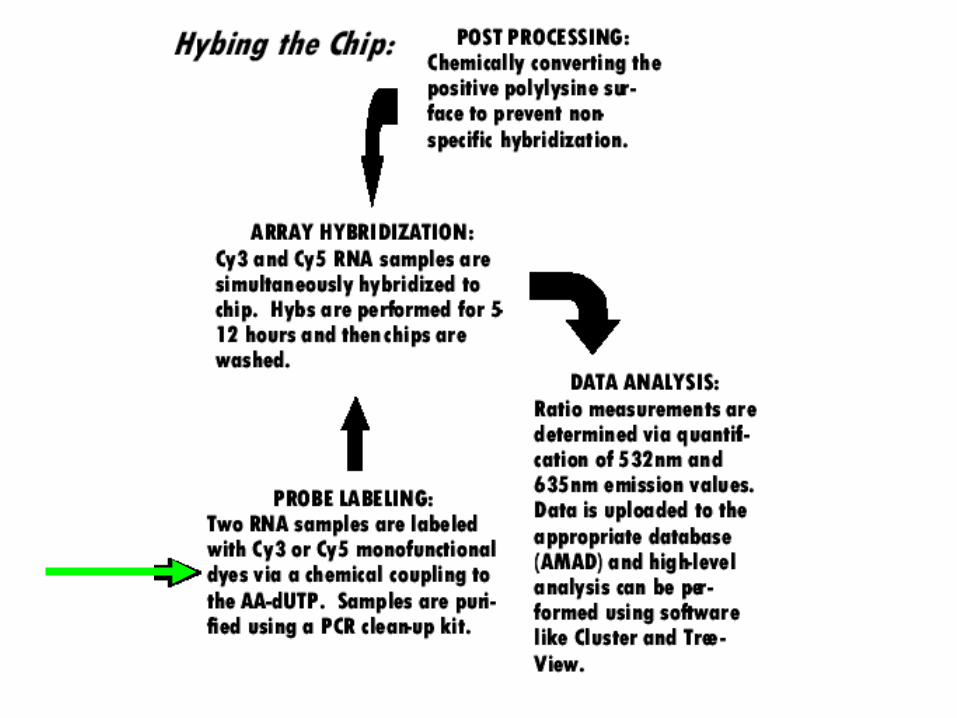
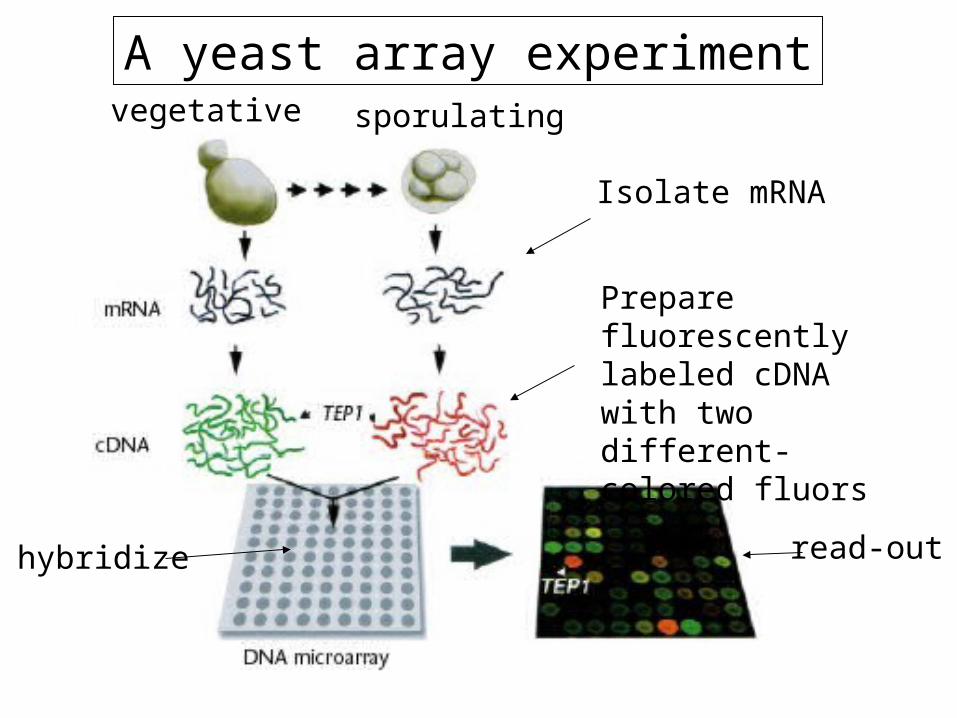
A yeast array experimentvegetative sporulating
Isolate mRNA
Prepare fluorescently labeled cDNA with two different-colored fluors
hybridize read-out
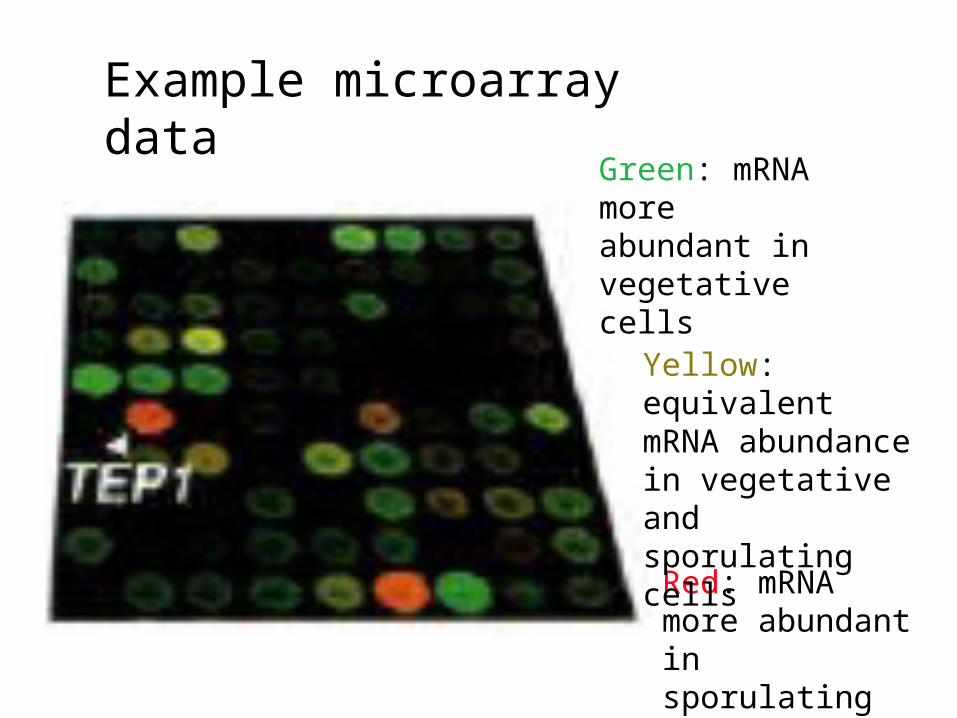
Example microarray data
Green: mRNA more abundant in vegetative cells
Red: mRNA more abundant in sporulating cells
Yellow: equivalent mRNA abundance in vegetative and sporulating cells

What to do with all that data?
Overarching patterns may become apparent
1) Organize data by hierarchical clustering, profiling to find patterns
2) Display data graphically to allow assimilation/comprehension
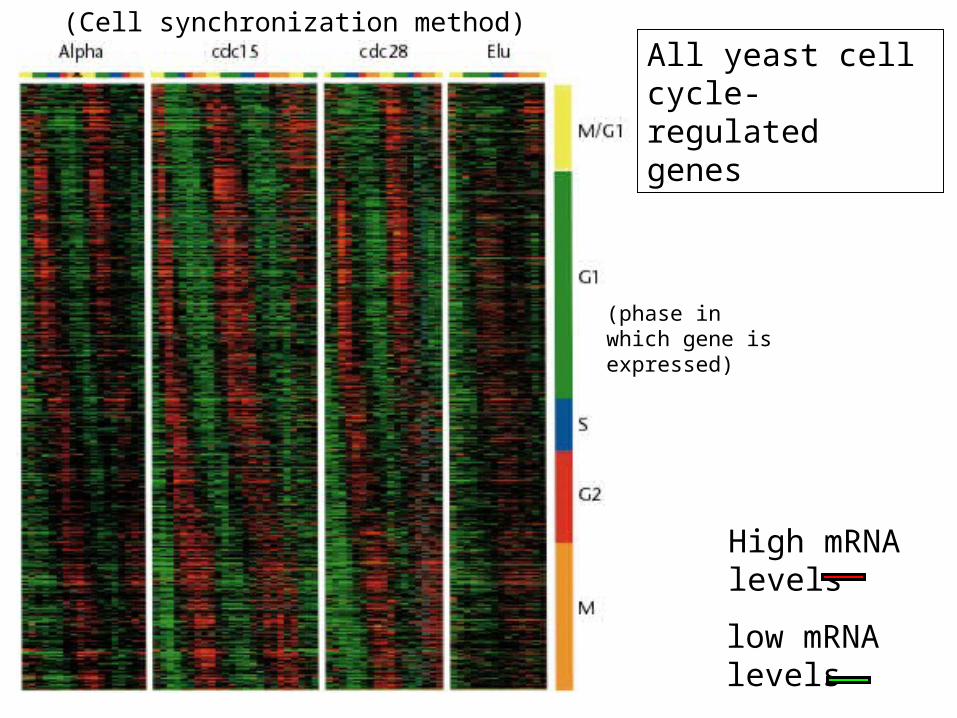
low mRNA levels
High mRNA levels
(Cell synchronization method)All yeast cell cycle-regulated genes
(phase in which gene is expressed)

MIAME:The Minimum Information About a Microarray Experiment
(#6 helps correct for variations in the quantity of starting RNA, and for variable labelling and detection efficiencies)
DNA
RNA
protein
genome
“transcriptome”
“proteome”
(we have this)
(we want these)

Analysis of the proteome: “proteomics”
• Which proteins are present and when?• What are the proteins doing?
– What interacts with what?•Protein-DNA interactions (chromatin
immunoprecipitation) •Protein-protein interactions
– Functions of proteins?
Phizicky et al. (2003) “Protein analysis on a proteomic scale” Nature 422, p. 208-215

Which proteins are expressed?
Classical method– Detect presence of a specific protein
•Using antibodies or specific assay•Measure changes in protein levels
with changing environment, in different tissues
– Very labor intensive, expensive to scale up to proteome

Massively parallel detection and identification of proteins
• 2D gel electrophoresis– Separate proteins in a given organism or tissue type by
migration in gel electrophoresis– Identify protein (cut out of gel, sequence or mass-spec) – Pattern of spots like a barcode for hi-throughput studies
• Mass spectrometry – Separate individual proteins from cell by charge and mass,
individual proteins can be identified (but need genome sequence information for this)
• Microarrays: isolate things that bind proteins
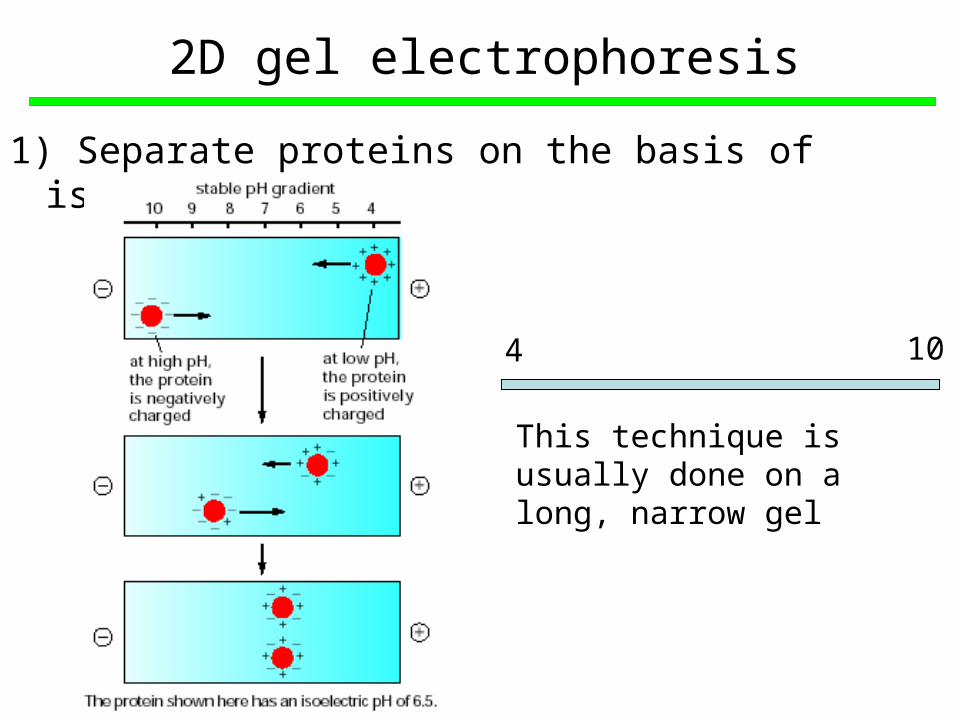
2D gel electrophoresis
1) Separate proteins on the basis of isoelectric point
This technique is usually done on a long, narrow gel
4 10
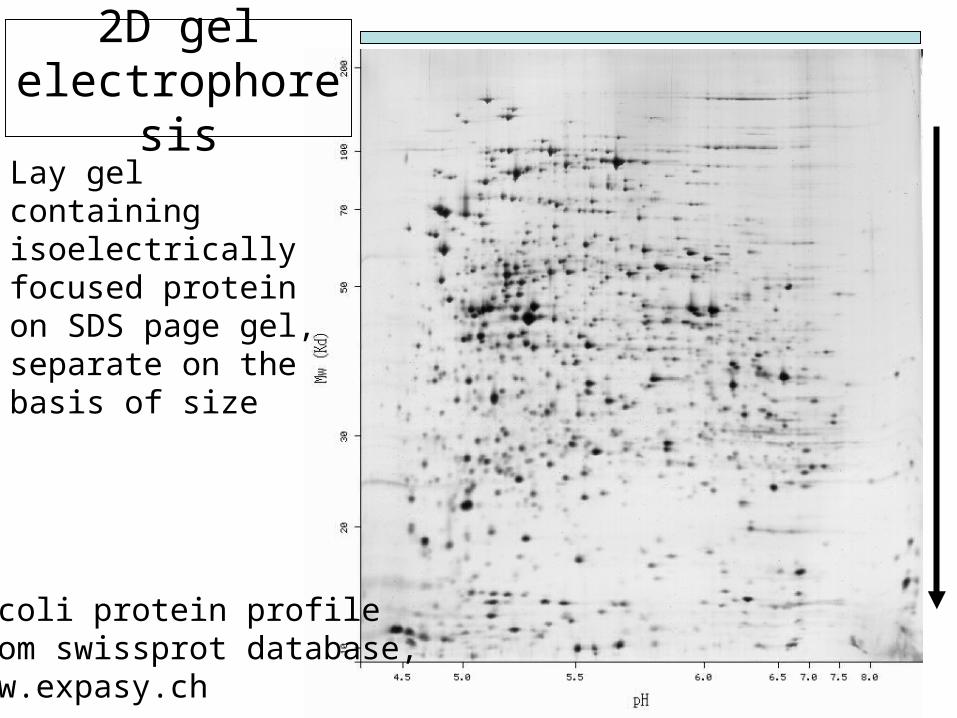
2D gel electrophoresi
sLay gel containing isoelectrically focused protein on SDS page gel, separate on the basis of size
E.coli protein profileFrom swissprot database, www.expasy.ch

Mass spectrometry for identifying proteins in a mixture
From J.R. Yates 1998 “Mass spectrometry and the age of the proteome” J Mass Spec. 33, p 1-19
Liquid chromatography and tandem mass spectrometry
Software for processing data

Defining protein function
• Classical methods:– Define activity of protein, develop an assay
for activity•Biochemistry: use assay to purify protein
from cell, characterize structure/function of protein in vitro
•Genetics: obtain mutants with change in activity, characterize phenotype of mutant, obtain suppressors to identify genes that interact with protein of interest
– Time intensive, expensive

Protein activity at the proteome level
• Protein-DNA interactions: identifying binding sites for DNA-binding proteins: regulation of gene expression
• Massively parallel screens for activity--protein arrays
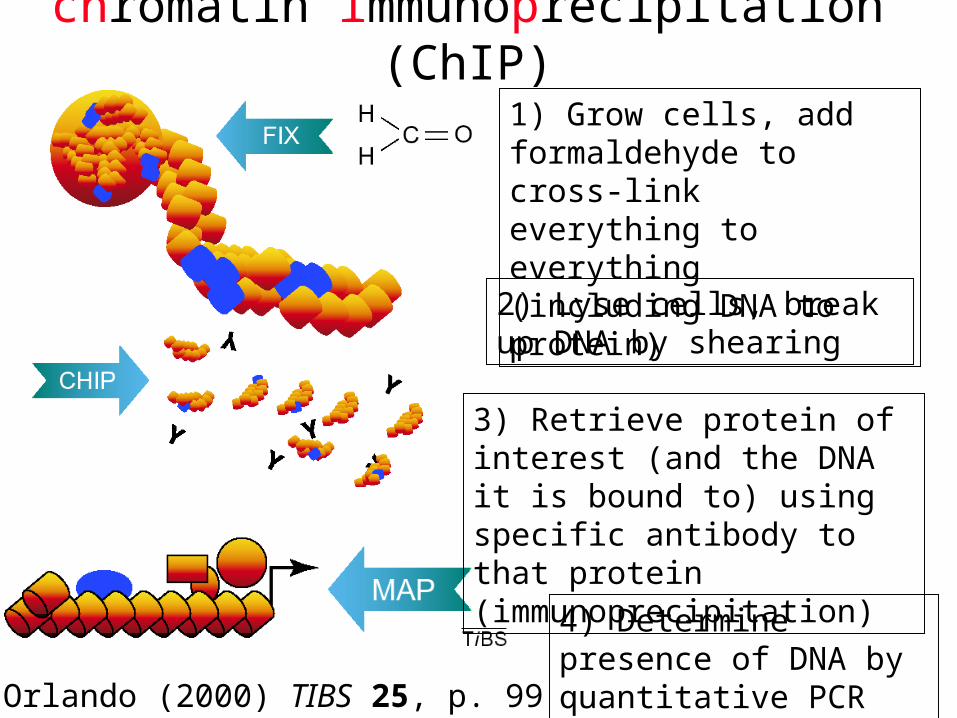
“chromatin immunoprecipitation” (ChIP)
1) Grow cells, add formaldehyde to cross-link everything to everything (including DNA to protein)
2) Lyse cells, break up DNA by shearing
3) Retrieve protein of interest (and the DNA it is bound to) using specific antibody to that protein (immunoprecipitation)
4) Determine presence of DNA by quantitative PCRV. Orlando (2000) TIBS 25, p. 99
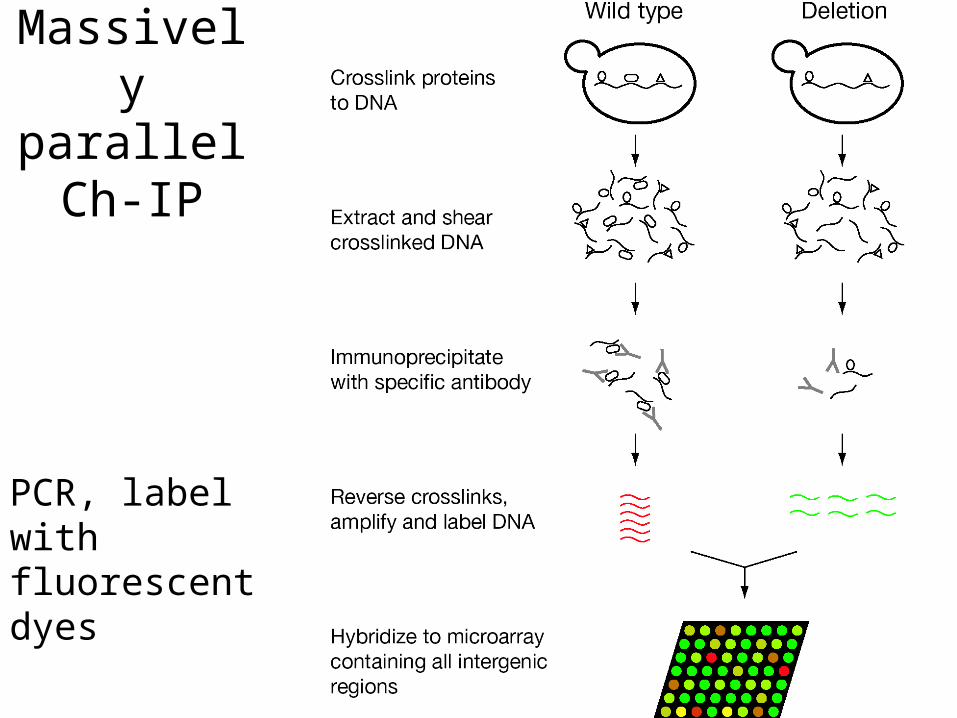
Massively parallel Ch-IP
PCR, label with fluorescent dyes
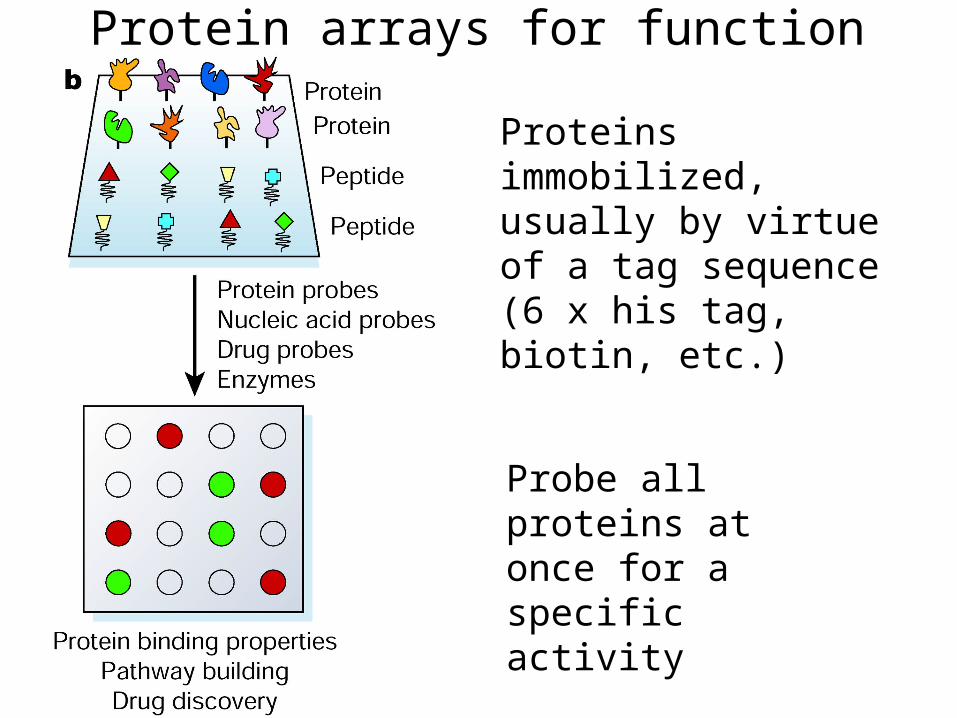
Protein arrays for function
Proteins immobilized, usually by virtue of a tag sequence (6 x his tag, biotin, etc.)
Probe all proteins at once for a specific activity

Example of a protein microarray
Proteins fused to GST with 6 x histidine tags, immobilized on Ni++ matrix
Anti-GST tells how much protein is immobilized on surface
Specific assays identify proteins with specific activities--calmodulin binding, phosphoinositide binding
DNA
RNA
protein
genome
“transcriptome”
“proteome”
(we have this)
(we want these)


















