
DNA Microarrays Comp602 Bioinformatics Algorithms -m werner 2011.
65
DNA Microarrays Comp602 Bioinformatics Algorithms -m werner 2011
-
date post
22-Dec-2015 -
Category
Documents
-
view
215 -
download
0
Transcript of DNA Microarrays Comp602 Bioinformatics Algorithms -m werner 2011.

DNA Microarrays
Comp602 Bioinformatics Algorithms-m werner 2011

Cell Snapshot
• What if you could capture what a cell is doing at any given instant?
• You could compare diseased cells to normal ones
• You could follow changes over the cell’s lifetime
04/19/23 m werner WIT Page 2Comp602 Bioinformatics Algorithms

Expressed Proteins Provide a Snapshot
• In humans every cell (almost) contains genetic coding for all human functions
• i.e. It can express any human protein• But specialized cells only express proteins
appropriate to their function• Further – they only express proteins as needed• Knowing which proteins are expressed by a cell at
any instant reveals information on what kind of cell it is and what it is currently doing
04/19/23 m werner WIT Page 3Comp602 Bioinformatics Algorithms

DNA Microarrays to Measure Protein Expression
• Protein expression can be measured efficiently• Given a sample (cytoplasm, bloodstream, etc.)• Can measure mRNA present• A gene is:
– On if the cell is currently transcribing its mRNA– Off otherwise
04/19/23 m werner WIT Page 4Comp602 Bioinformatics Algorithms

Probes for mRNA
• To see if a gene is on use a probe• The probe is the DNA complement of the mRNA we
are checking• i.e. if the mRNA has the sequence AGGTATC the
probe would have TCCATAG• So the mRNA would stick to the probe• You may not need the entire gene for the probe• A short oligonucleotide of 25 – 40 base pairs may
suffice
04/19/23 m werner WIT Page 5Comp602 Bioinformatics Algorithms

Probing• To test a sample for the expression of a particular
protein• Paint a glass slide with lots of its probes
(complements of its mRNA)• Add a dye to the sample• Bathe the slide in the sample and rinse off• If the protein is being expressed the slide glows
with the dye color • The intensity of the glow indicates the level of the
protein expression
04/19/23 m werner WIT Page 6Comp602 Bioinformatics Algorithms

Parallel Probes
• A DNA Microarray allows for simultaneous probes for multiple proteins
• The slide is divided into small squares• The squares are printed (sometimes with
inkjet technology) with the probes• A single slide can test for 1000 or more
proteins
04/19/23 m werner WIT Page 7Comp602 Bioinformatics Algorithms

Ink Jet Printing
• Four cartridges are loaded with the four nucleotides: A, G, C,T
• As the printer head moves across the array, the nucleotides are deposited in pixels where they are needed.
• This way (many copies of) a 20-60 base long oligonucleotide are deposited in each pixel.
By Steve Hookway lecture and Sorin Draghici’s book “Data Analysis Tools for DNA Microarrays”
04/19/23 m werner WIT Page 8Comp602 Bioinformatics Algorithms

Inkjet Printed Microarrays
Inkjet head, squirting phosphor-ammodites
From Zohar Yakhini04/19/23 m werner WIT Page 9Comp602 Bioinformatics Algorithms

cDNA array,Inkjet deposition
In-Situ synthesized oligonucleotide array. 25-60 mers.
Thermal Ink Jet Arrays, by Agilent Technologies
04/19/23 m werner WIT Page 10
Comp602 Bioinformatics Algorithms

Wash the slide with two samples labeled with green and red dye
Results:•Green – Sample 1 expressed•Red – Sample 2 expressed•Yellow – Samples 1 and 2 co-expressed
Flash Demo
04/19/23 11Comp602 Bioinformatics Algorithms

Two Channel Detection
• Two-channel microarrays are hybridized with cDNA prepared from two samples to be compared (e.g. diseased tissue versus healthy tissue) and that are labeled with two different fluorophores, red and green.
@mw04/19/23 m werner WIT Page 12Comp602 Bioinformatics Algorithms

Log Ratios
• For a particular protein (gene) we are interested in the ratio of expression between the malignant sample m and the benign sample b.
• The ratio m/b is not symetric. i.e if malignant is more expressed the ratios go from 1 to ∞, but if benign is more expressed the ratios are squeezed between 0 and 1.
• To equalize, take log2(b/n). Now, ratios >1 have logs ranging up from 0 and negative ones ranging down from 0.
04/19/23 m werner WIT Page 13Comp602 Bioinformatics Algorithms

Using Microarrays (cont’d)
• Green: expressed only from control
• Red: expressed only from experimental cell
• Yellow: equally expressed in both samples
• Black: NOT expressed in either control or experimental cells
04/19/23 14Comp602 Bioinformatics Algorithms

Microarray Data • Microarray data are usually transformed into an intensity
matrix (below)• The intensity matrix allows biologists to make correlations
between different genes (even if they are dissimilar) and to understand how genes functions might be
related
Time: Time X Time Y Time Z
Gene 1 10 8 10
Gene 2 10 0 9
Gene 3 4 8.6 3
Gene 4 7 8 3
Gene 5 1 2 3
Intensity (expression level) of gene at measured time
04/19/23 15Comp602 Bioinformatics Algorithms

Clustering of Microarray Data • Plot each datum as a point in N-dimensional
space• Make a distance matrix for the distance
between every two gene points in the N-dimensional space
• Genes with a small distance share the same expression characteristics and might be functionally related or similar.
• Clustering reveals groups of functionally related genes
04/19/23 16Comp602 Bioinformatics Algorithms

Clustering of Microarray Data (cont’d)
Clusters
04/19/23 17Comp602 Bioinformatics Algorithms

Homogeneity and Separation Principles
• Homogeneity: Elements within a cluster are close to each other
• Separation: Elements in different clusters are further apart from each other
• …clustering is not an easy task!
Given these points a clustering algorithm might make two distinct clusters as follows
04/19/23 18Comp602 Bioinformatics Algorithms

Bad ClusteringThis clustering violates both Homogeneity and Separation principles
Close distances from points in separate clusters
Far distances from points in the same cluster
04/19/23 19Comp602 Bioinformatics Algorithms

Good ClusteringThis clustering satisfies both Homogeneity and Separation principles
04/19/23 20Comp602 Bioinformatics Algorithms

Clustering Techniques• Agglomerative: Start with every element in
its own cluster, and iteratively join clusters together
• Divisive: Start with one cluster and iteratively divide it into smaller clusters
• Hierarchical: Organize elements into a tree, leaves represent genes and the length of the pathes between leaves represents the distances between genes. Similar genes lie within the same subtrees
04/19/23 21Comp602 Bioinformatics Algorithms

Hierarchical Clustering
04/19/23 22Comp602 Bioinformatics Algorithms

Hierarchical Clustering: Example
04/19/23 m werner WIT Page 23Comp602 Bioinformatics Algorithms

Hierarchical Clustering: Example
04/19/23 m werner WIT Page 24Comp602 Bioinformatics Algorithms

Hierarchical Clustering: Example
04/19/23 m werner WIT Page 25Comp602 Bioinformatics Algorithms

Hierarchical Clustering: Example
04/19/23 m werner WIT Page 26Comp602 Bioinformatics Algorithms

Hierarchical Clustering: Example
04/19/23 m werner WIT Page 27Comp602 Bioinformatics Algorithms
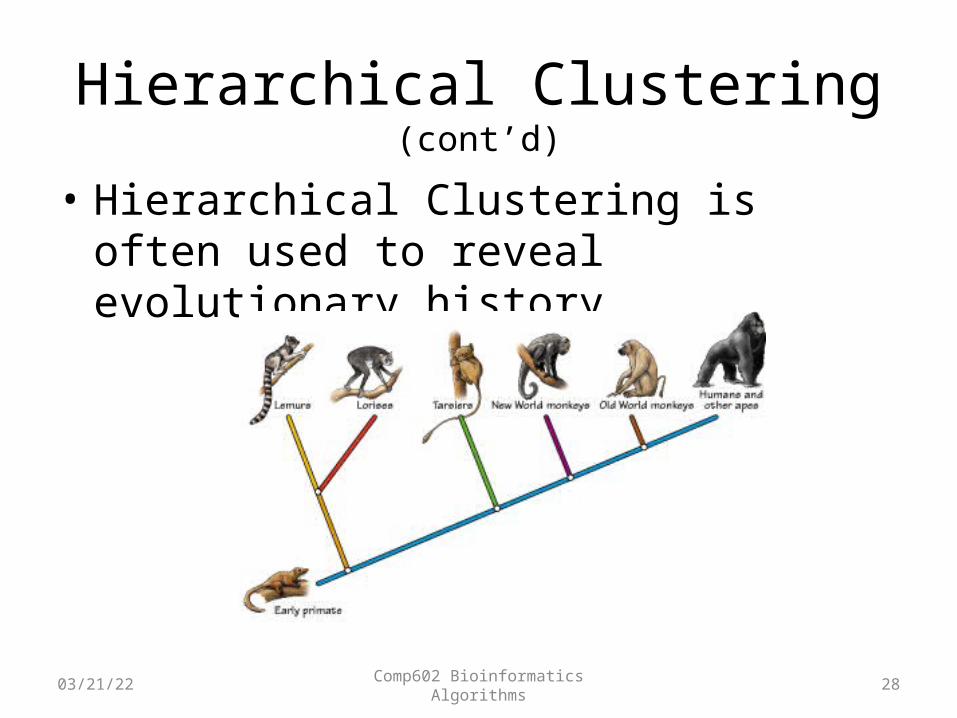
Hierarchical Clustering (cont’d)
• Hierarchical Clustering is often used to reveal evolutionary history
04/19/23 28Comp602 Bioinformatics Algorithms

Hierarchical Clustering Algorithm
1. Hierarchical Clustering (d , n)2. Form n clusters each with one element3. Construct a graph T by assigning one vertex to each cluster4. while there is more than one cluster5. Find the two closest clusters C1 and C2
6. Merge C1 and C2 into new cluster C with |C1| +|C2| elements7. Compute distance from C to all other clusters8. Add a new vertex C to T and connect to vertices C1 and C2
9. Remove rows and columns of d corresponding to C1 and C2
10. Add a row and column to d corrsponding to the new cluster C
11. return T
The algorithm takes a nxn distance matrix d of pairwise distances between points as an input.
04/19/23 m werner WIT Page 29Comp602 Bioinformatics Algorithms

Hierarchical Clustering Algorithm
1. Hierarchical Clustering (d , n)2. Form n clusters each with one element3. Construct a graph T by assigning one vertex to each cluster4. while there is more than one cluster5. Find the two closest clusters C1 and C2
6. Merge C1 and C2 into new cluster C with |C1| +|C2| elements7. Compute distance from C to all other clusters8. Add a new vertex C to T and connect to vertices C1 and C2
9. Remove rows and columns of d corresponding to C1 and C2
10. Add a row and column to d corrsponding to the new cluster C11. return T
Different ways to define distances between clusters may lead to different clusterings
04/19/23 m werner WIT Page 30Comp602 Bioinformatics Algorithms

Hierarchical Clustering: Recomputing Distances
• dmin(C, C*) = min d(x,y) for all elements x in C and y in C*
– Distance between two clusters is the smallest distance between any pair of their elements
• davg(C, C*) = (1 / |C*||C|) ∑ d(x,y) for all elements x in C and y in C*
– Distance between two clusters is the average distance between all pairs of their elements
04/19/23 m werner WIT Page 31Comp602 Bioinformatics Algorithms

Squared Error Distortion• Given a data point v and a set of points X, define the distance from v to X
d(v, X)
as the (Euclidian) distance from v to the closest point from X.
• Given a set of n data points V={v1…vn} and a set of k points X, define the Squared Error Distortion
d(V,X) = ∑d(vi, X)2 / n 1 < i < n
04/19/23 m werner WIT Page 32Comp602 Bioinformatics Algorithms

04/19/23 m werner WIT Page 33Comp602 Bioinformatics Algorithms

Clustering a Heat Map
• Each sample (column) is a point in n-space where n is the number of proteins tested (rows)
• On the top samples are clustered hierarchically into a dendrogram according to some distance measure. Columns have been swapped to show close samples next to each other.
• On the right proteins are clustered to show how closely they are co-expressed in the various samples. Rows have been swapped.
04/19/23 m werner WIT Page 34Comp602 Bioinformatics Algorithms

A: Hierarchical cluster analysis of gene expression patterns of normal and cancerous prostate specimens. The dendrogram indicates relationships among samples based on gene expression profiles. Cluster analysis is seen to distinguish malignant from normal prostate (pink branches). Cluster analysis also defines three subtypes of prostate cancer (numbered above) not distinguishable histologically.
A Perspective on DNA Microarrays in Pathology Research and Practice, Jonathan R. Pollack
04/19/23 m werner WIT Page 35Comp602 Bioinformatics Algorithms

A Data Mining Problem• On a given microarray, we test on the order of 10k
elements in one time• Number of microarrays used in typical experiment is no more than 100.• Insufficient sampling.• Data is obtained faster than it can be processed.• High noise.• Algorithmic approaches to work through this large
data set and make sense of the data are desired.
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 m werner WIT Page 36Comp602 Bioinformatics Algorithms

A Data Mining Problem
• On a given Microarray we test on the order of 10k elements at a time
• Data is obtained faster than it can be processed
• We need some ways to work through this large data set and make sense of the data
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 m werner WIT Page 37Comp602 Bioinformatics Algorithms

Grouping and Reduction
• Grouping: discovers patterns in the data from a microarray
• Reduction: reduces the complexity of data by removing redundant probes (genes) that will be used in subsequent assays
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 m werner WIT Page 38Comp602 Bioinformatics Algorithms

Unsupervised Grouping: Clustering
• Pattern discovery via grouping similarly expressed genes together
• Three techniques most often used
k-Means ClusteringHierarchical ClusteringKohonen Self Organizing Feature Maps
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 m werner WIT Page 39Comp602 Bioinformatics Algorithms

Clustering Limitations
• Any data can be clustered, therefore we must be careful what conclusions we draw from our results
• Clustering is non-deterministic and can and will produce different results on different runs
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 m werner WIT Page 40Comp602 Bioinformatics Algorithms

K-means Clustering
• Given a set of n data points in d-dimensional space and an integer k
• We want to find the set of k points in d-dimensional space that minimizes the mean squared distance from each data point to its nearest center
• No exact polynomial-time algorithms are known for this problem
“A Local Search Approximation Algorithm for k-Means Clustering” by Kanungo et. al
04/19/23 m werner WIT Page 41Comp602 Bioinformatics Algorithms

K-means Algorithm (Lloyd’s Algorithm)
• Has been shown to converge to a locally optimal solution
• But can converge to a solution arbitrarily bad compared to the optimal solution
•“K-means-type algorithms: A generalized convergence theorem and characterization of local optimality” by Selim and Ismail
•“A Local Search Approximation Algorithm for k-Means Clustering” by Kanungo et al.
K=3
Data Points
Optimal Centers
Heuristic Centers
04/19/23 m werner WIT Page 42Comp602 Bioinformatics Algorithms

Distance Measures Between Two Datapoints in n-Space
• Euclidean Distance• Cosine Coefficient – Compare 2 vectors for direction using the dot
product:• x∙y = ∑xi∙yi = |x||y|cos(Ѳ )
• Manhattan Distance• Pearson Correlation Coefficient
04/19/23 m werner WIT Page 43Comp602 Bioinformatics Algorithms

Pearson Correlation Coefficient, r. values in [-1,1] interval
• Gene expression over d experiments is a vector in Rd, e.g. for gene C: (0, 3, 3.58, 4, 3.58, 3)
• Given two vectors X and Y that contain N elements, we calculate r as follows:
Cho & Won, 2003
04/19/23 m werner WIT Page 44Comp602 Bioinformatics Algorithms

Euclidean Distance
n
iiiE yxyxd
1
2)(),(
543),( 22 AOd E
Now to find the distance between two points, say the origin and the point (3,4):
Simple and Fast! Remember this when we consider the complexity!
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 m werner WIT Page 45Comp602 Bioinformatics Algorithms

Finding a Centroid
We use the following equation to find the n dimensional centroid point amid k n dimensional points:
),...,2
,1
(),...,,( 11121 k
xnth
k
ndx
k
stxxxxCP
k
ii
k
ii
k
ii
k
Let’s find the midpoint between 3 2D points, say: (2,4) (5,2) (8,9)
)5,5()3
924,
3
852(
CP
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 m werner WIT Page 46Comp602 Bioinformatics Algorithms

K-means Algorithm1. Choose k initial center points randomly2. Cluster data using Euclidean distance (or other distance
metric)3. Calculate new center points for each cluster using only
points within the cluster4. Re-Cluster all data using the new center points
1. This step could cause data points to be placed in a different cluster
5. Repeat steps 3 & 4 until the center points have moved such that in step 4 no data points are moved from one cluster to another or some other convergence criteria is met
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici04/19/23 m werner WIT Page 47Comp602 Bioinformatics Algorithms

An example with k=2
1. We Pick k=2 centers at random
2. We cluster our data around these center points
Figure Reproduced From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 48Comp602 Bioinformatics Algorithms

K-means example with k=2
3. We recalculate centers based on our current clusters
Figure Reproduced From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 49Comp602 Bioinformatics Algorithms

K-means example with k=2
4. We re-cluster our data around our new center points
Figure Reproduced From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 50Comp602 Bioinformatics Algorithms

K-means example with k=2
5. We repeat the last two steps until no more data points are moved into a different cluster
Figure Reproduced From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 51Comp602 Bioinformatics Algorithms

Choosing k
• Use another clustering method• Run algorithm on data with several different
values of k• Use advance knowledge about the
characteristics of your test– Cancerous vs Non-Cancerous
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 m werner WIT Page 52Comp602 Bioinformatics Algorithms

Cluster Quality
• Since any data can be clustered, how do we know our clusters are meaningful?– The size (diameter) of the cluster vs. The inter-cluster
distance– Distance between the members of a cluster and the
cluster’s center– Diameter of the smallest sphere
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 m werner WIT Page 53Comp602 Bioinformatics Algorithms

Cluster Quality Continued
size=5
size=5distance=20
distance=5
Quality of cluster assessed by ratio of distance to nearest cluster and cluster diameter
Figure Reproduced From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 54Comp602 Bioinformatics Algorithms

Cluster Quality Continued
Quality can be assessed simply by looking at the diameter of a cluster
A cluster can be formed even when there is no similarity between clustered patterns. This occurs because the algorithm forces k clusters to be created.
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici04/19/23 55Comp602 Bioinformatics Algorithms

Characteristics of k-means Clustering
• The random selection of initial center points creates the following properties– Non-Determinism– May produce clusters without patterns
• One solution is to choose the centers randomly from existing patterns
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 m werner WIT Page 56Comp602 Bioinformatics Algorithms

Squared Error Distortion• Given a data point v and a set of points X, define the distance from v to X
d(v, X)
as the (Euclidian) distance from v to the closest point from X.
• Evaluate the error induced by mapping each data point to one of the k-means centroids
• Given a set of n data points V={v1…vn} and a set of k centroids X={c1…ck}, define the Squared Error Distortion
d(V,X) = ∑d(vi, X)2 / n 1 < i < n

Algorithm Complexity
• Linear in the number of data points, N• Can be shown to have time of cN
– c does not depend on N, but rather the number of clusters, k
• Low computational complexity• High speed
From “Data Analysis Tools for DNA Microarrays” by Sorin Draghici
04/19/23 m werner WIT Page 58Comp602 Bioinformatics Algorithms

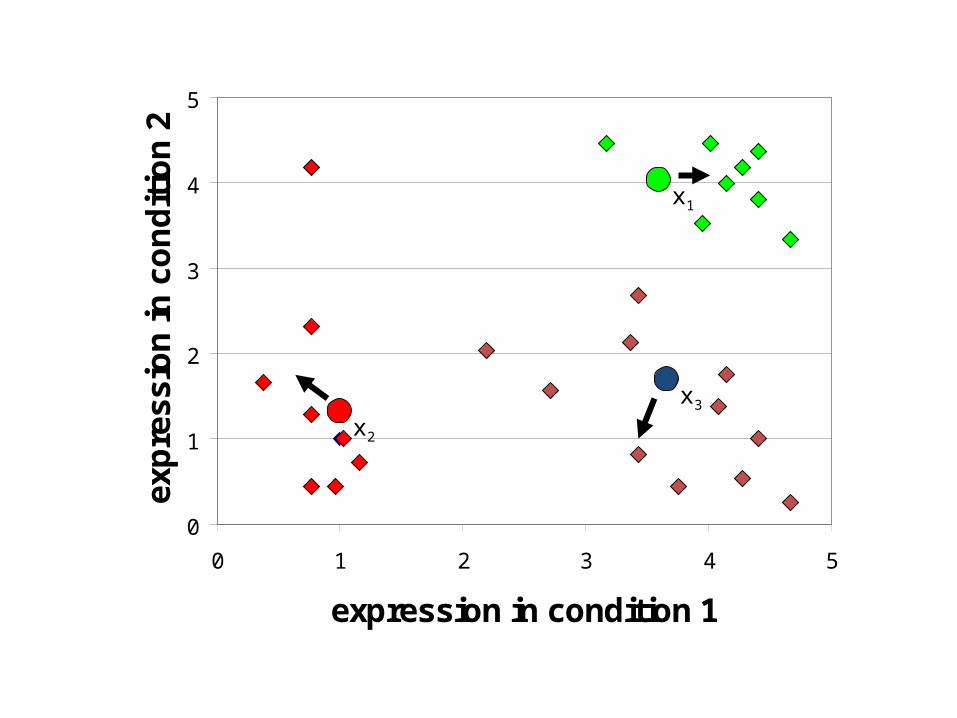
K-Means Clustering: Lloyd Algorithm1. Lloyd Algorithm2. Arbitrarily assign the k cluster centers3. while the cluster centers keep changing4. Assign each data point to the cluster Ci
corresponding to the closest cluster representative (center) (1 ≤ i ≤ k)
5. After the assignment of all data points, compute new cluster representatives according to the center of gravity of each
cluster, that is, the new cluster representative is ∑v / |C| for all v in C for every cluster C
*This may lead to merely a locally optimal clustering.

0
1
2
3
4
5
0 1 2 3 4 5
expression in condition 1
exp
ressio
n in
co
nd
itio
n 2
x1
x2
x3

0
1
2
3
4
5
0 1 2 3 4 5
expression in condition 1
exp
ress
ion
in c
on
dit
ion
2
x1
x2
x3
0
1
2
3
4
5
0 1 2 3 4 5
expression in condition 1
exp
ress
ion
in c
on
dit
ion
2x1
x2
x3

0
1
2
3
4
5
0 1 2 3 4 5
expression in condition 1
exp
ress
ion
in c
on
dit
ion
2x1
x2x3

Conservative K-Means Algorithm• Lloyd algorithm is fast but in each iteration it moves
many data points, not necessarily causing better convergence.
• A more conservative method would be to move one point at a time only if it improves the overall clustering cost
– The smaller the clustering cost of a partition of data points is the better that clustering is
– Different methods (e.g., the squared error distortion) can be used to measure this clustering cost

K-Means “Greedy” Algorithm1. ProgressiveGreedyK-Means(k)2. Select an arbitrary partition P into k clusters3. while forever4. bestChange 05. for every cluster C6. for every element i not in C7. if moving i to cluster C reduces its clustering cost8. if (cost(P) – cost(Pi C) > bestChange9. bestChange cost(P) – cost(Pi C) 10. i* I11. C* C12. if bestChange > 013. Change partition P by moving i* to C*
14. else15. return P


















