
Disordini genomici - uniba.it · DNA segments provides a chance for ... Il fenotipo clinico e’...
46
By NA Disordini genomici Lezione 21
Transcript of Disordini genomici - uniba.it · DNA segments provides a chance for ... Il fenotipo clinico e’...

By NA
Disordini genomici
Lezione 21

By N.A.
Genome Research 2000Yonggang J, Evan E. Eichler, Stuart Schwartz, and Robert D. NichollsStructure of Chromosomal Duplicons and their Role in Mediating Human Genomic DisordersGenome Research Vol. 10, Issue 5, 597 -610, May 2000
Analysis of long stretches of available sequence from human chromosomes 22 and X suggest that 5% -10% of the genome may be duplicated (Mazzarella and Schlessinger 1998; Dunham et al. 1999). Sequence homology between duplicated DNA segments provides a chance for misalignment during meiosis, leading to unequal exchange and chromosome rearrangement, by either inter - or intrachromosomal or sister chromatid homologous recombination.This process with divergence between duplicated segments is essential to the generation of diversity and new genes over evolutionary time, although the more typical, short-term effect is genetic disease.

By N.A.
Trends in Genetics 2002P.Stankiewicz and J.R.Lupski
Genome , rearrangements and genomic disordersTRENDS in Genetics Vol. 18: 74 -82, 2002
architecture
Human Molecular Genetics 2004

By N.A.
Disordini Genomici
Il fenotipo clinico e’ conseguenza di alterato dosaggio genico di un gene o piu’ localizzati all’interno di segmenti genomici riarrangiati
L instabilita di alcune regioni genomiche e dovuta alla presenza di low copyrepeats: duplicazioni segmentali/dupliconi
All’origine ci possono essere :la Non-Allelic Homologous Recombination: NAHR
’ ’ ’

By N.A.
Disordini Genomici NAHR
cromosoma duplicato
cromosoma deleto
Ricombinazione omologa non allelica
Dupliconi altamente omologhiRegione contenentealmeno un gene dosage sensitive
All’origine ci possono essere :la Non Homologous End Joining: NHEJ
By N.A.
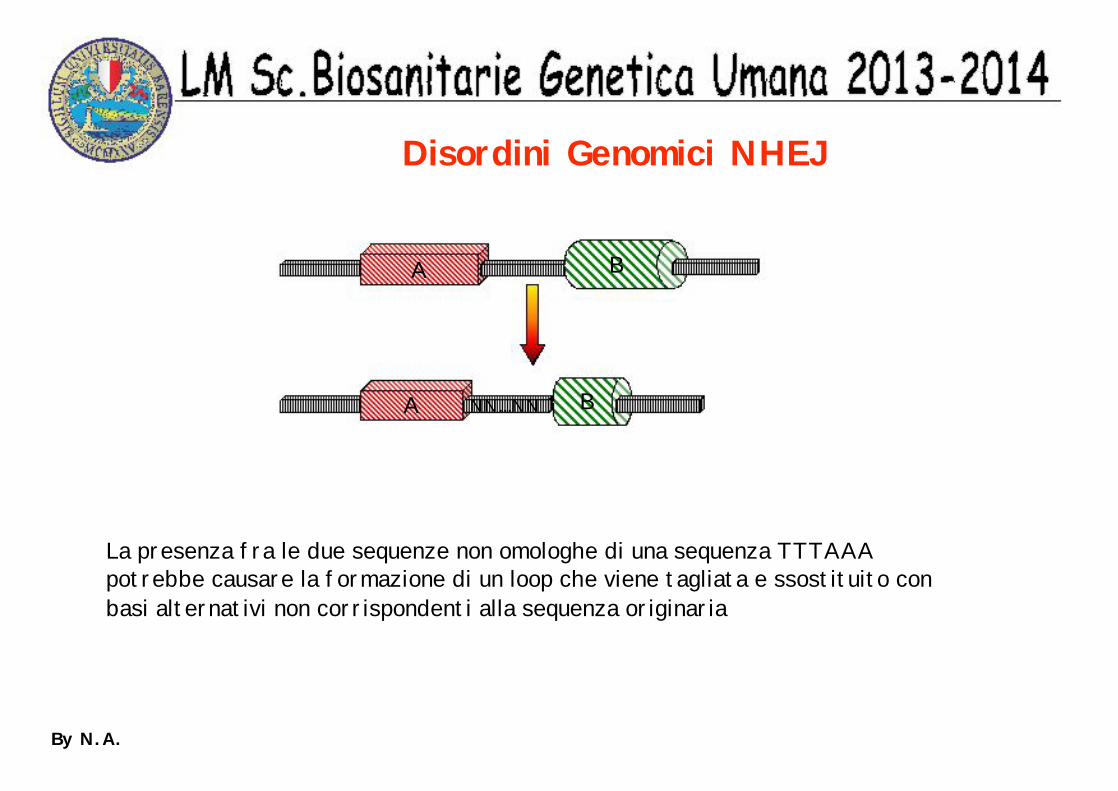
Disordini Genomici NHEJ
A
A NN...NN
B
B
La presenza fra le due sequenze non omologhe di una sequenza TTTAAApotrebbe causare la formazione di un loop che viene tagliata e ssostituito conbasi alternativi non corrispondenti alla sequenza originaria

By N.A.
Disordini GenomiciPossiamo classificarli sulla base del contenuto di geni sensibili alla dose presenti nel segmento compreso fra i dupliconi che vanno incontro alla NAHR
Disordini mendeliani: Un solo gene (piccoli riarrangiamenti)
Sindromi cromosomiche : molti geni sensibili (riarrangiamenti molto grandi)
Sindromi da geni contigui: piu’ geni sensibili alla dose (riarrangiamenti piu’ ampi)

By N.A.
Caratteri mendeliani IBanter syndrome type III AD 1p36 del 11kbGaucher disease AR 1q21 del 16kbFam. juv. nephronophthisis AR 2q13 del 290kbSpinal muscular atrophy AR 5q13.2 inv/dup Cong adrenal hyperplasia (21 hydroxylase def.) AR 6p21.3 del 30kbGlucocorticoid -remediable aldosteronism (GRA) AD 8q21 dup 45kbBeta -thalassemia AR 11p15.5 delalfa -thalassemia AR 16p13.3Polycystic kidney disease 1 AD 16p13.3Charcot -Marie -Tooth (CMT1A) AD 17p12 dup 1400kbHer. neur. with liability to pressure palsies (HNPP) AD 17p12 del idemNeurofibromatosis type 1 (NFl) AD 17q11.2 del 1500kbPituitary dwarfism AR 17q23.3 del 6.7kbCYP2D6 pharmacogenetic trait AR 22q13.1 del/dup 9.3kbIchthyosis XL Xp22.32 del 1900kbRed -green color blindness XL Xq28 delIncontinentia pigmenti XL Xq28 del 10kbHemophilia A XL Xq28 inv 300 -500kbEmery -Dreifuss musc. dystr. XL Xq28 del/dup/inv 48kbHunter syndrome XL Xq28 inv/del 20kb
By N.A.
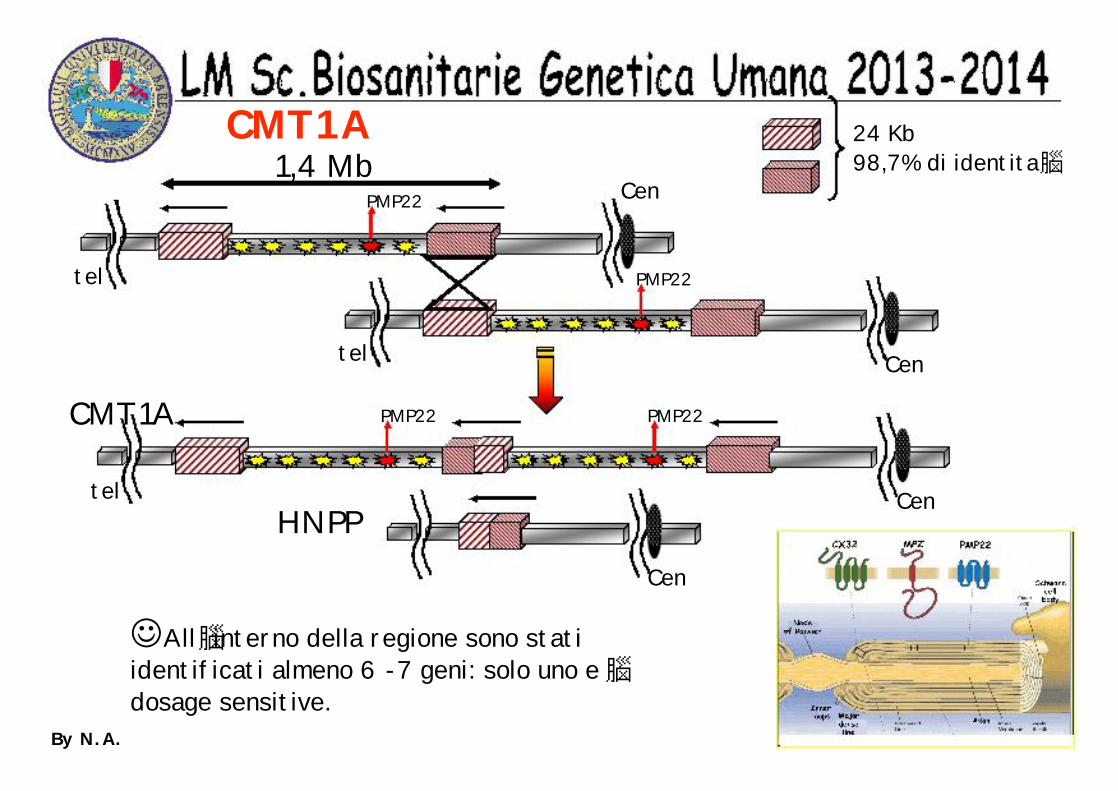
CMT1A 24 Kb98,7% di identita1,4 Mb
tel Cen
tel
Cen
Cen
tel Cen
PMP22
PMP22
PMP22PMP22
All interno della regione sono stati identificati almeno 6 -7 geni: solo uno edosage sensitive.
CMT1A
HNPP
’
’
’
☺

By N.A.
Caratteri mendeliani IBanter syndrome type III 1p36Gaucher disease 1q21Fam. juv. nephronophthisis 2q13Fascioscapulohumeral muscular dystrophy 4q35Spinal muscular atrophy 5q13.2Cong adrenal hyperplasia (21 hydroxylase def.) 6p21.3Glucocorticoid -remediable aldosteronism (GRA) 8q21Beta -thalassemia 11p15.5alfa -thalassemia 16p13.3Polycystic kidney disease 1 16p13.3Charcot -Marie -Tooth (CMT1A) 17p12Her. neur. with liability to pressure palsies (HNPP) 17p12Neurofibromatosis type 1 (NFl) 17q11.2Pituitary dwarfism 17q23.3CYP2D6 pharmacogenetic trait 22q13.1Ichthyosis Xp22.32Red -green color blindness Xq28Incontinentia pigmenti Xq28Hemophilia A Xq28Emery -Dreifuss musc. dystr. Xq28Hunter syndrome Xq28

By N.A.
Haemophilia A (X-linked): Factor VIII
Sostituzioni nucleotidiche (missense / nonsense) 515Sostituzioni nucleotidiche (splicing) 49Sostituzioni nucleotidiche (regulatory) 0Piccole delezioni 116Piccole inserzioni 36Piccole inserzioni/delezioni 5Grandi delezioni 86Grandi delezioni & duplicazioni 6Riarrangiamenti complessi (incluse inversioni) 9
TOTAL 822
By N.A.
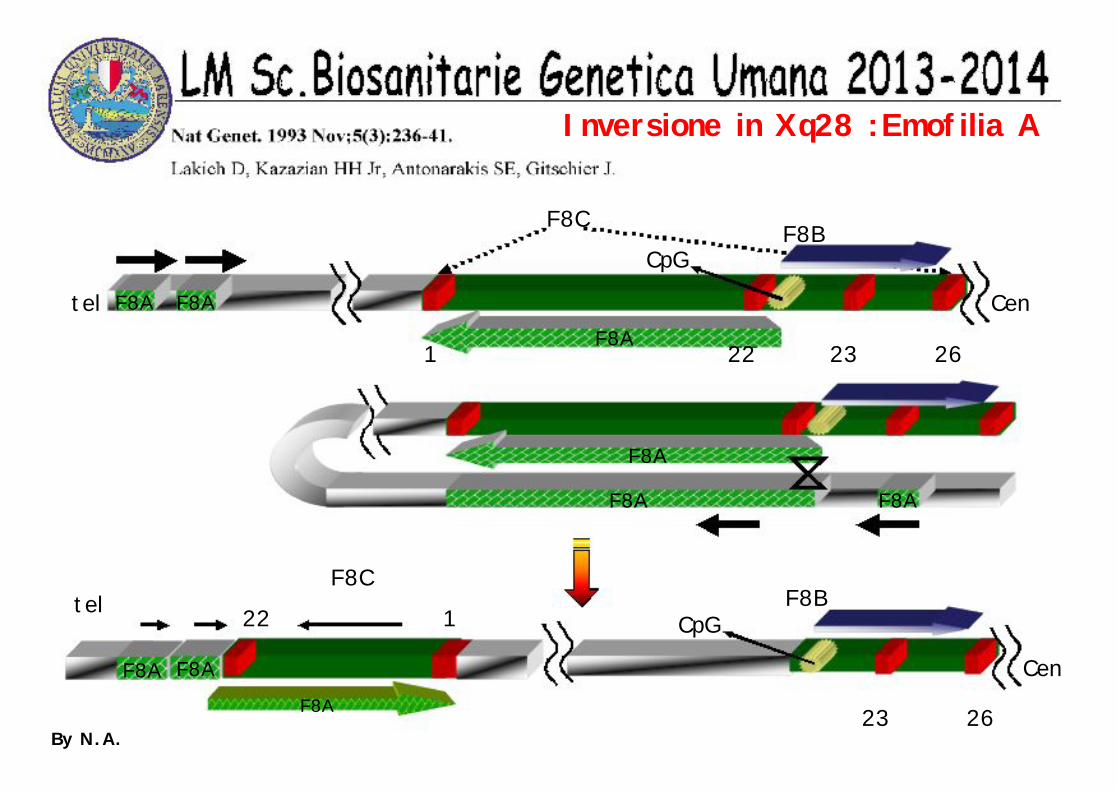
Inversione in Xq28 :Emofilia A
Cen
F8B
1 22 23 26
F8C
F8A
CpG
tel F8A F8A
F8A
F8AF8A
F8BF8C
tel
F8A
122
23 26
CpG
CenF8AF8A
By N.A.
Inversione in Xq28 :Emofilia A
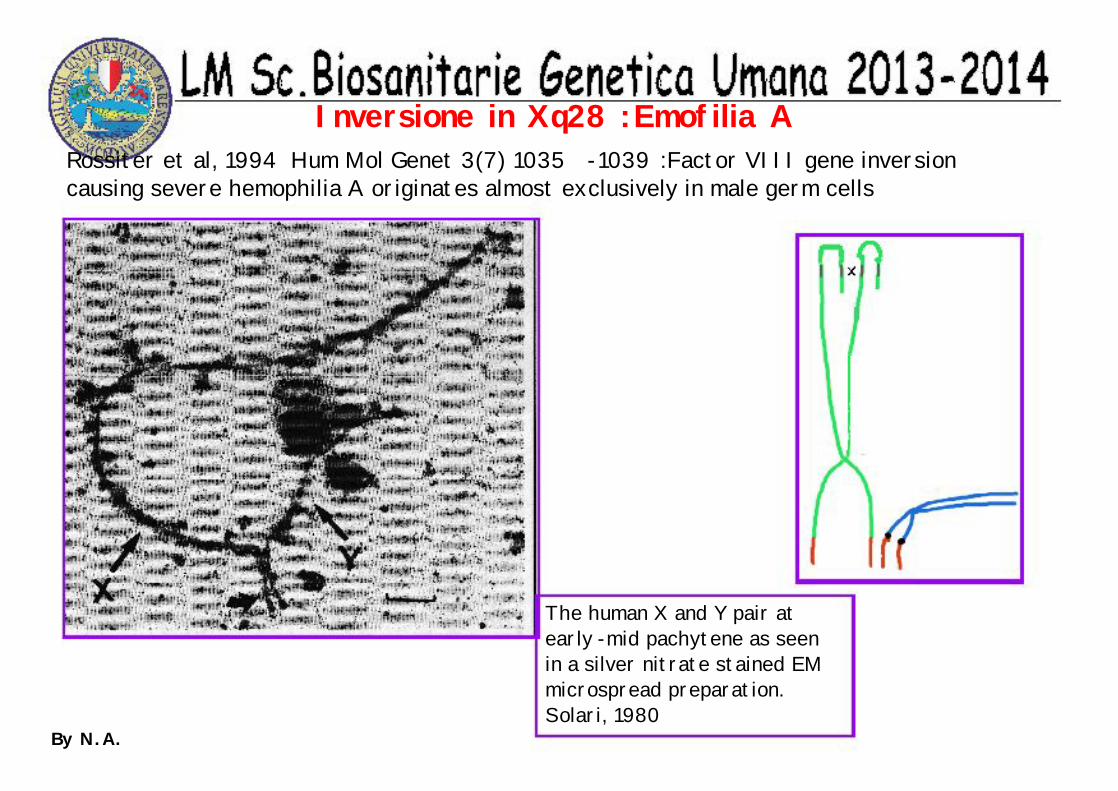
The human X and Y pair at early -mid pachytene as seen in a silver nitrate stained EM microspread preparation. Solari, 1980
Rossiter et al, 1994 Hum Mol Genet 3(7) 1035 -1039 :Factor VIII gene inversion causing severe hemophilia A originates almost exclusively in male germ cells

Banter syndrome type III AD 1p36 del 11kbGaucher disease AR 1q21 del 16kbFam. juv. nephronophthisis AR 2q13 del 290kbSpinal muscular atrophy AR 5q13.2 inv/dup Cong adrenal hyperplasia (21 hydroxylase def.) AR 6p21.3 del 30kbGlucocorticoid -remediable aldosteronism (GRA) AD 8q21 dup 45kbBeta -thalassemia AR 11p15.5 delalfa -thalassemia AR 16p13.3Polycystic kidney disease 1 AD 16p13.3Charcot -Marie -Tooth (CMT1A) AD 17p12 dup 1400kbHer. neur. with liability to pressure palsies (HNPP) AD 17p12 del idemNeurofibromatosis type 1 (NFl) AD 17q11.2 del 1500kbPituitary dwarfism AR 17q23.3 del 6.7kbCYP2D6 pharmacogenetic trait AR 22q13.1 del/dup 9.3kbIchthyosis XL Xp22.32 del 1900kbRed -green color blindness XL Xq28 delIncontinentia pigmenti XL Xq28 del 10kbHemophilia A XL Xq28 inv 300 -500kbEmery -Dreifuss musc. dystr. XL Xq28 del/dup/inv 48kbHunter syndrome XL Xq28 inv/del 20kb
Caratteri mendeliani I

Spinal muscular atrophy(SMA)Fenotipo autosomico recessivo letale,1/10.000 nati vivi. In circa il 18% dei casi e associato ad un riarrangiamento di una regione di ~500kb in 5q13
Sindrome di HunterFenotipo X -linked recessivo, dovuto a mutazioni del gene IDS (Xq28).Nel 20% dei casi la mutazione e dovuta a ricombinazione fra IDS e un suo pseudogene localizzato 90Kb a valle.
’
’
Caratteri mendeliani I

Banter syndrome type III AD 1p36 del 11kbGaucher disease AR 1q21 del 16kbFam. juv. nephronophthisis AR 2q13 del 290kbSpinal muscular atrophy AR 5q13.2 inv/dup Cong adrenal hyperplasia (21 hydroxylase def.) AR 6p21.3 del 30kbGlucocorticoid -remediable aldosteronism (GRA) AD 8q21 dup 45kbBeta -thalassemia AR 11p15.5 delalfa -thalassemia AR 16p13.3Polycystic kidney disease 1 AD 16p13.3Charcot -Marie -Tooth (CMT1A) AD 17p12 dup 1400kbHer. neur. with liability to pressure palsies (HNPP) AD 17p12 del idemNeurofibromatosis type 1 (NFl) AD 17q11.2 del 1500kbPituitary dwarfism AR 17q23.3 del 6.7kbCYP2D6 pharmacogenetic trait AR 22q13.1 del/dup 9.3kbIchthyosis XL Xp22.32 del 1900kbRed -green color blindness XL Xq28 delIncontinentia pigmenti XL Xq28 del 10kbHemophilia A XL Xq28 inv 300 -500kbEmery -Dreifuss musc. dystr. XL Xq28 del/dup/inv 48kbHunter syndrome XL Xq28 inv/del 20kb
Caratteri mendeliani I
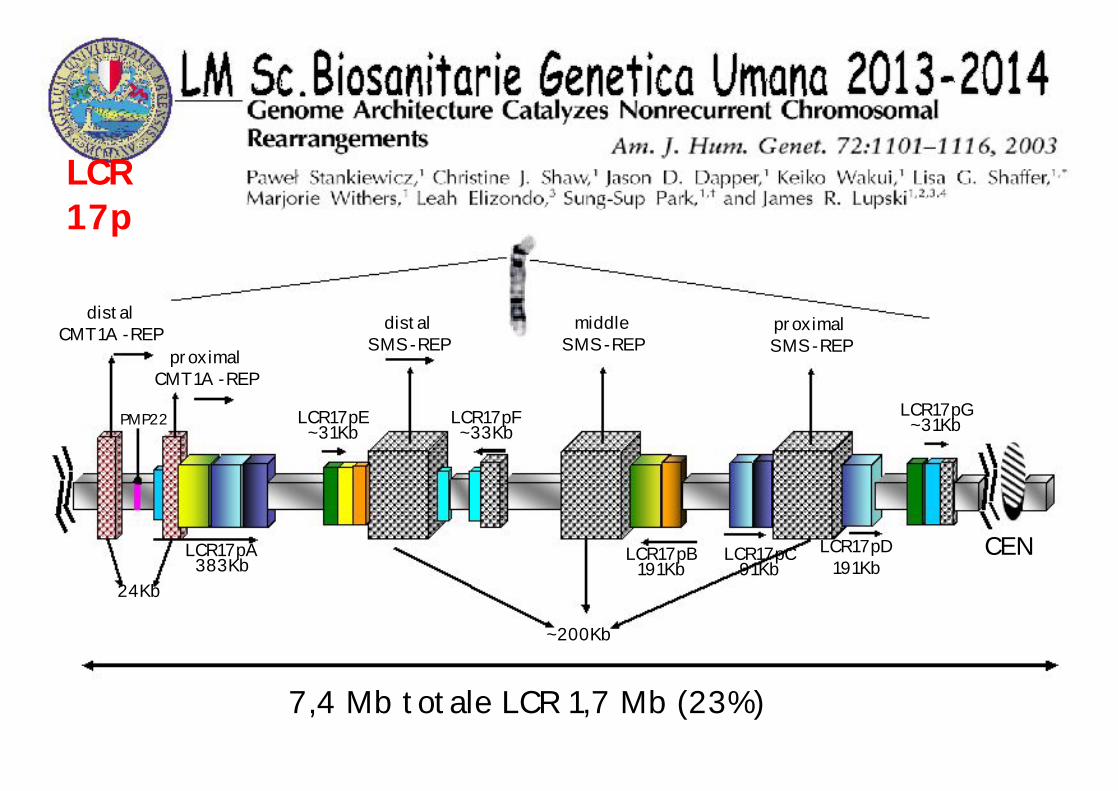
LCR 17p
PMP22
distalCMT1A -REP
proximalCMT1A -REP
24Kb
LCR17pA383Kb
LCR17pE~31Kb
distalSMS-REP
LCR17pF~33Kb
middleSMS-REP
proximalSMS-REP
LCR17pB191Kb
~200Kb
LCR17pC91Kb
LCR17pD191Kb
LCR17pG~31Kb
CEN
7,4 Mb totale LCR 1,7 Mb (23%)

Williams-Beuren syndrome 7q11.23Prader-Willi syndrome 15q11.2q13Angelman syndrome 15q11.2q13dup(15)(q11.2q13) 15q11.2q13triplication 15q11.2q13 15q11.2q13Smith-Magenis syndrome 17p11.2dup(17)(p11.2p11.2) 17p11.2DiGeorge/VCFS 22q11.2Male infertilityAZFa microdeletion Yq11.2Male infertility AZFc microdel. Yq11.2
Sindromi da geni contigui I

Williams-Beuren syndrome 7q11.23Prader-Willi syndrome 15q11.2q13Angelman syndrome 15q11.2q13dup(15)(q11.2q13) 15q11.2q13triplication 15q11.2q13 15q11.2q13Smith-Magenis syndrome 17p11.2dup(17)(p11.2p11.2) 17p11.2DiGeorge/VCFS 22q11.2Male infertilityAZFa microdeletion Yq11.2Male infertility AZFc microdel. Yq11.2
Sindromi da geni contigui I
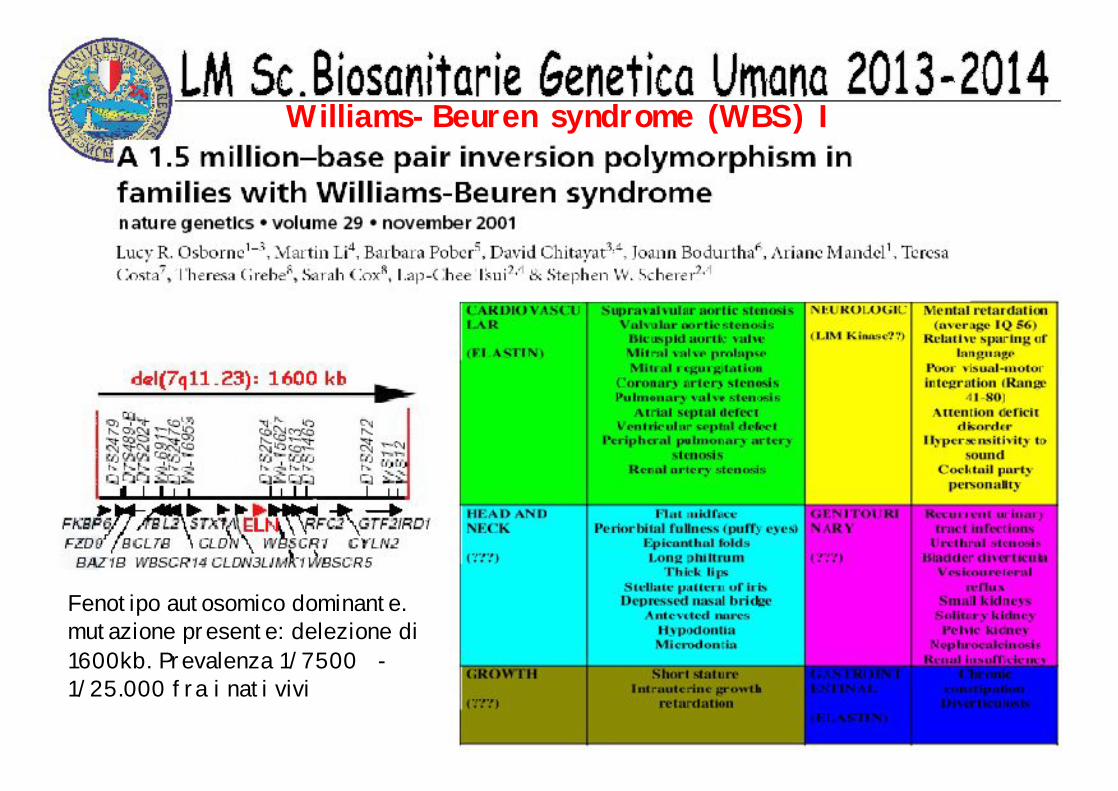
Fenotipo autosomico dominante. mutazione presente: delezione di 1600kb. Prevalenza 1/7500 -1/25.000 fra i nati vivi
Williams-Beuren syndrome (WBS) I
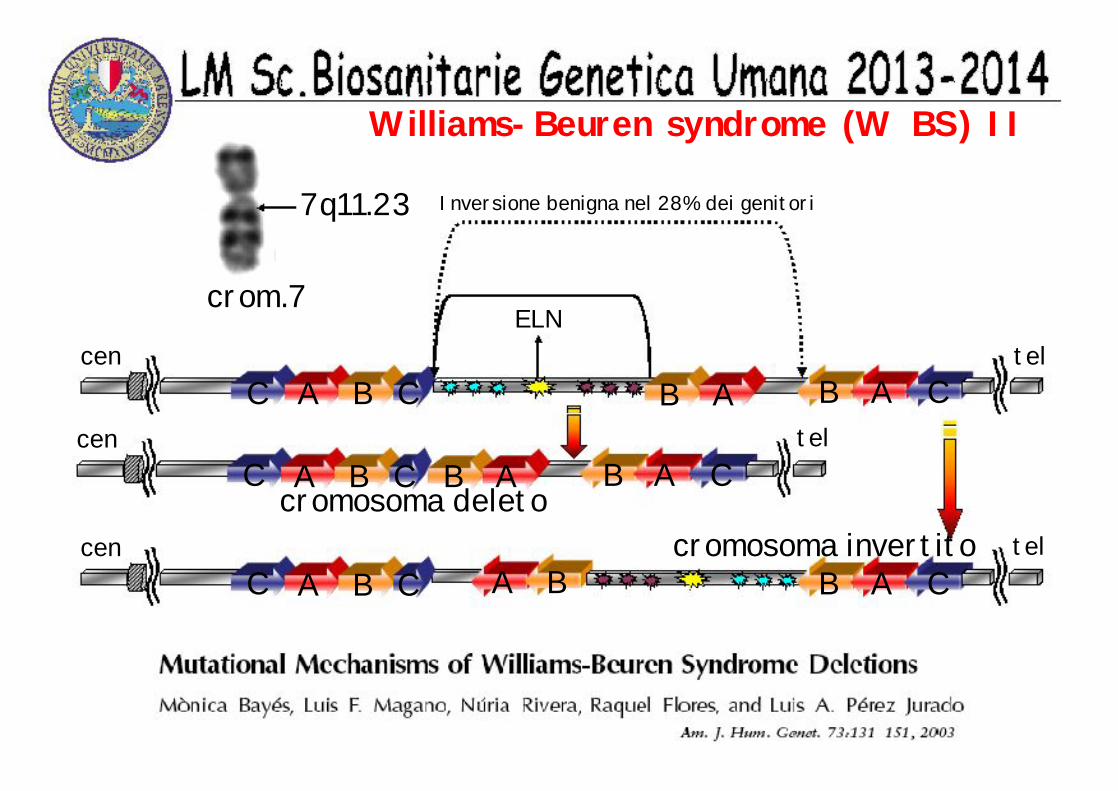
Williams-Beuren syndrome (W BS) II
C A B C B A B A Ccen tel
ELN
A BC A B C B A Ccen telcromosoma invertito
cromosoma deletoC A B C B A B A C
cen tel
Inversione benigna nel 28% dei genitori
crom.7
7q11.23

Williams-Beuren syndrome 7q11.23Prader-Willi syndrome 15q11.2q13Angelman syndrome 15q11.2q13dup(15)(q11.2q13) 15q11.2q13triplication 15q11.2q13 15q11.2q13Smith-Magenis syndrome 17p11.2dup(17)(p11.2p11.2) 17p11.2DiGeorge/VCFS 22q11.2Male infertilityAZFa microdeletion Yq11.2Male infertility AZFc microdel. Yq11.2
Sindromi da geni contigui I
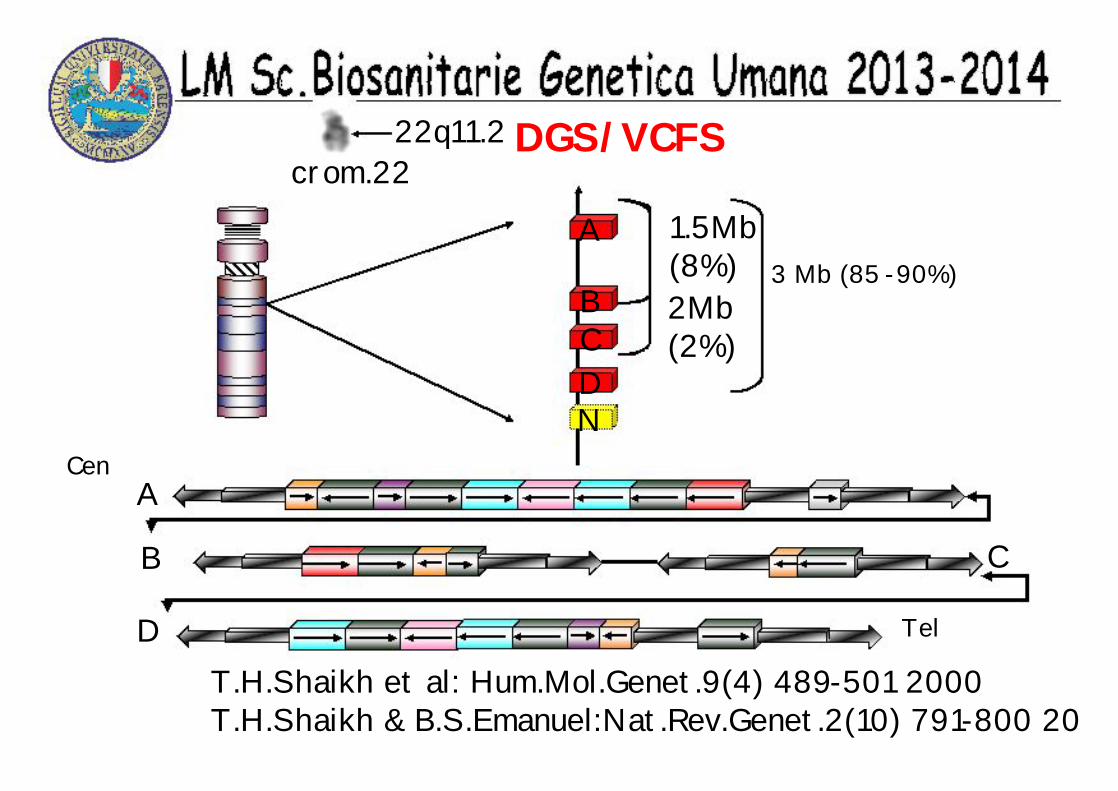
DGS/VCFS22q11.2
A
BCDN
3 Mb (85 -90%)1.5Mb(8%)2Mb(2%)
A
B C
Cen
D Tel
T.H.Shaikh et al: Hum.Mol.Genet.9(4) 489-501 2000T.H.Shaikh & B.S.Emanuel:Nat.Rev.Genet.2(10) 791-800 20
crom.22

Williams-Beuren syndrome 7q11.23Prader-Willi syndrome 15q11.2q13Angelman syndrome 15q11.2q13dup(15)(q11.2q13) 15q11.2q13triplication 15q11.2q13 15q11.2q13Smith-Magenis syndrome 17p11.2dup(17)(p11.2p11.2) 17p11.2DiGeorge/VCFS 22q11.2Male infertilityAZFa microdeletion Yq11.2Male infertility AZFc microdel. Yq11.2
Sindromi da geni contigui I
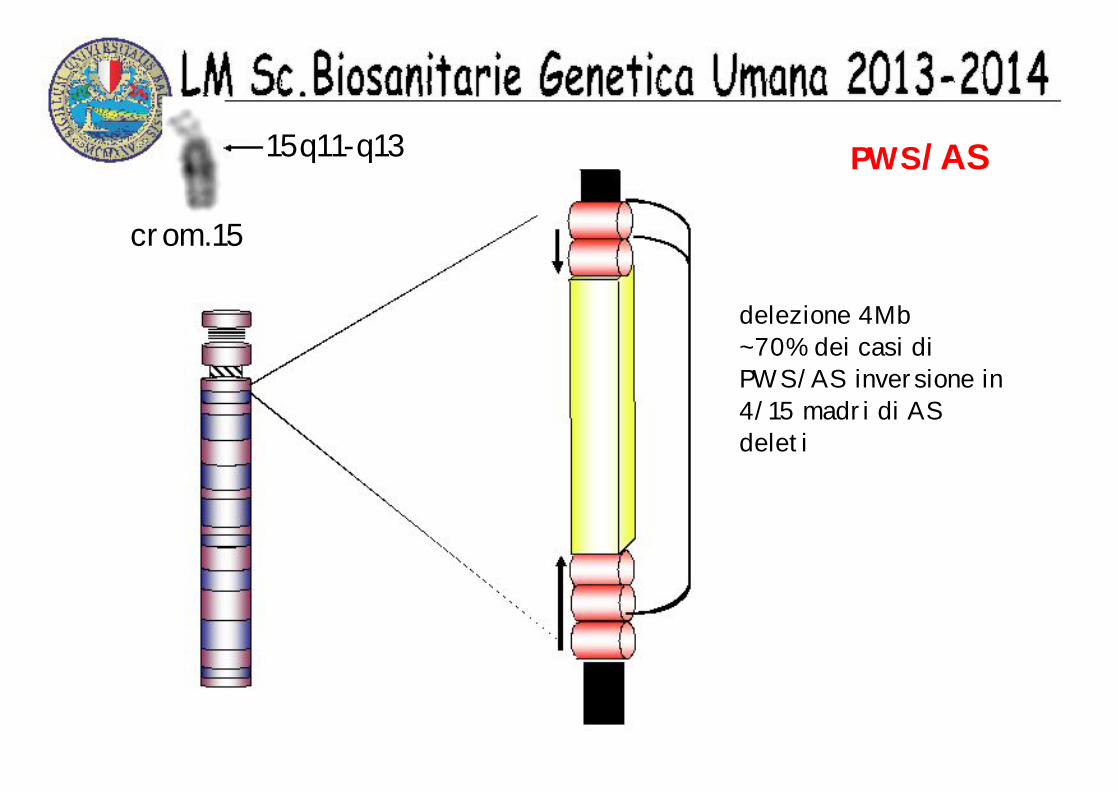
PWS/AS15q11-q13
crom.15
delezione 4Mb ~70% dei casi di PWS/AS inversione in 4/15 madri di AS deleti
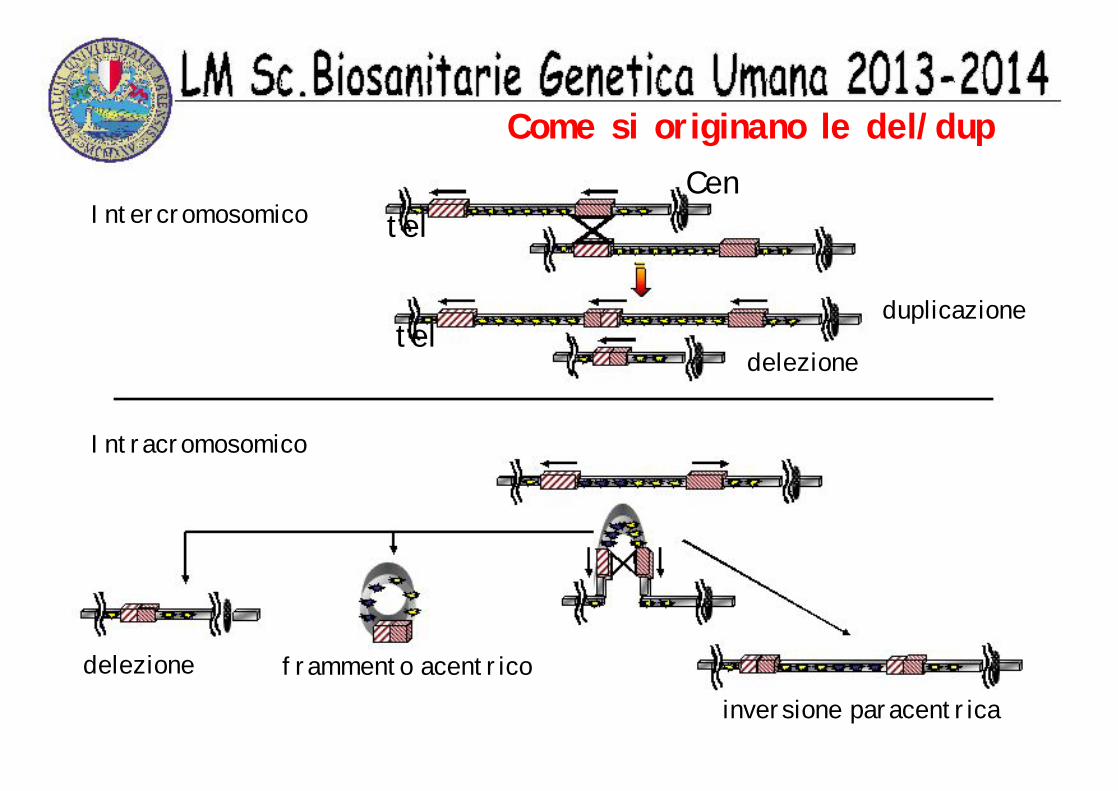
Come si originano le del/dup
Intercromosomico
duplicazionetel
delezione
telCen
Intracromosomico
delezione frammento acentricoinversione paracentrica

inv dup(15)(q11q13) Inverted dupinv dup(22)(q11.2) Inverted dupdic(X)(p11.2) Isodicentricinv dup(8p) inv/dup/delder(8)(pterp23.1::p23.2pter); inv/dup/deldel(8)(p23.1p23.2)dup(15)(q24q26) dupt(11;22)(11q23;22q11)
Riarragiamenti costituzionali
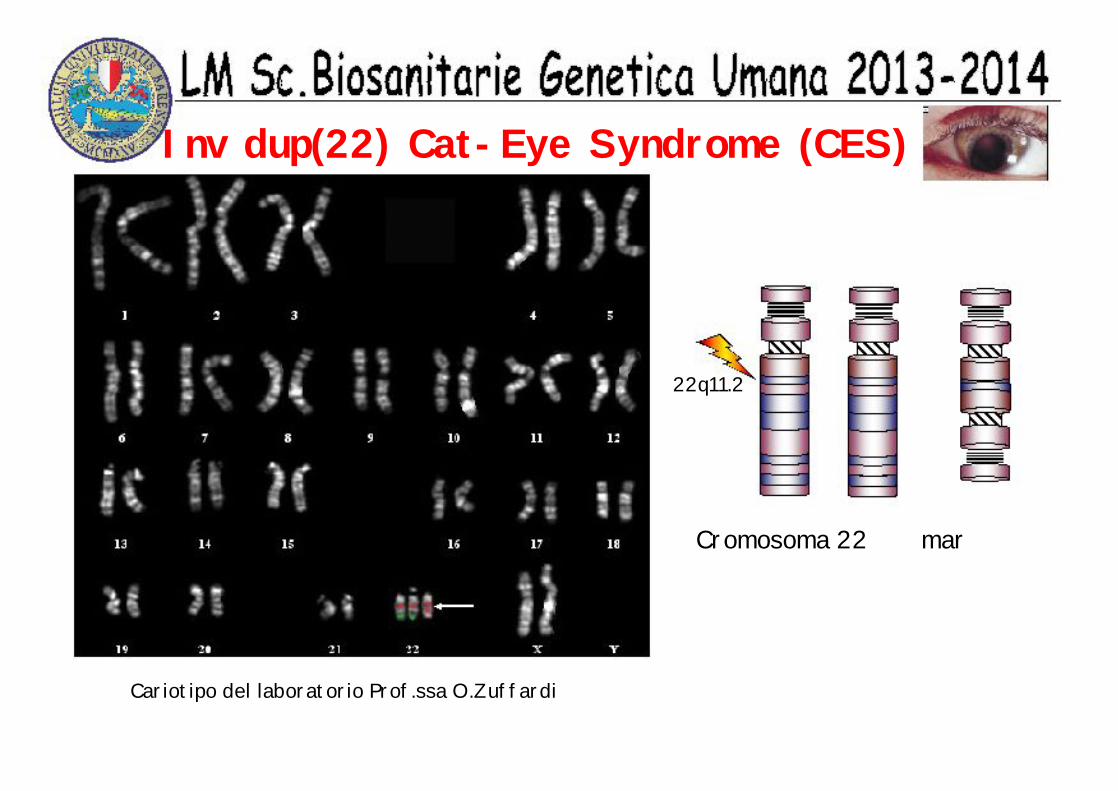
Inv dup(22) Cat-Eye Syndrome (CES)
Cariotipo del laboratorio Prof.ssa O.Zuffardi
22q11.2
Cromosoma 22 mar
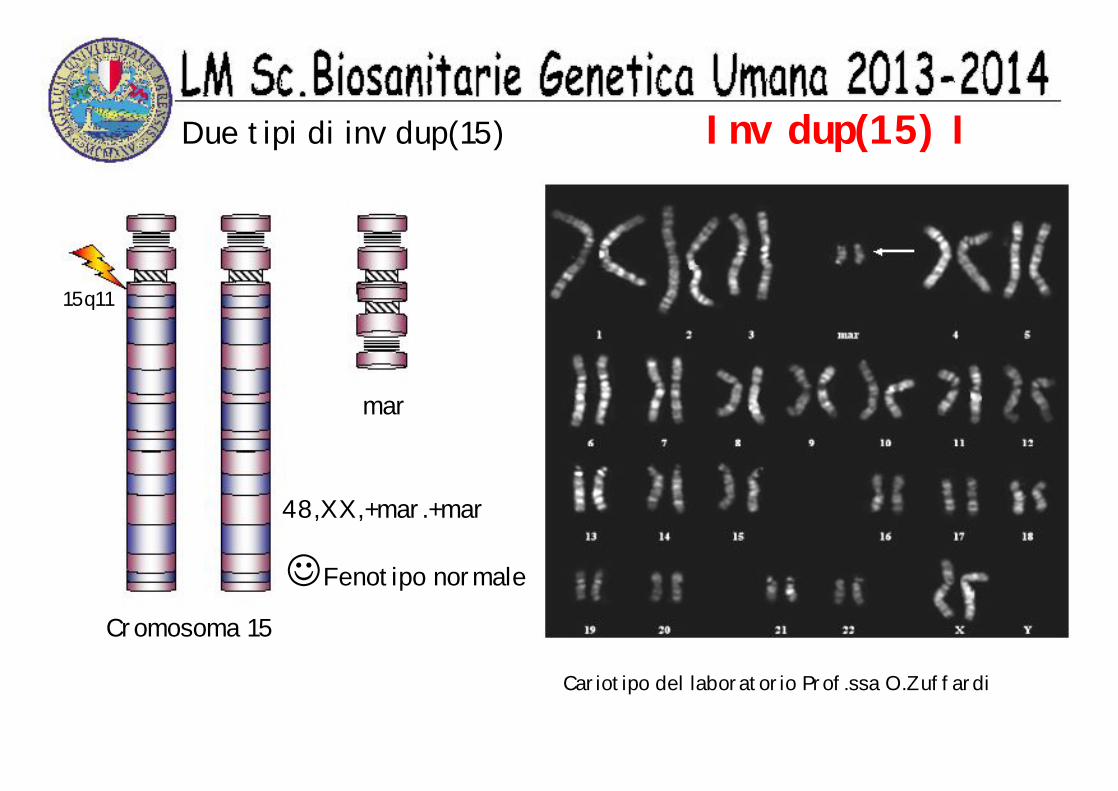
Inv dup(15) IDue tipi di inv dup(15)
Fenotipo normale
15q11
Cariotipo del laboratorio Prof.ssa O.Zuffardi
48,XX,+mar.+mar
Cromosoma 15
mar
☺
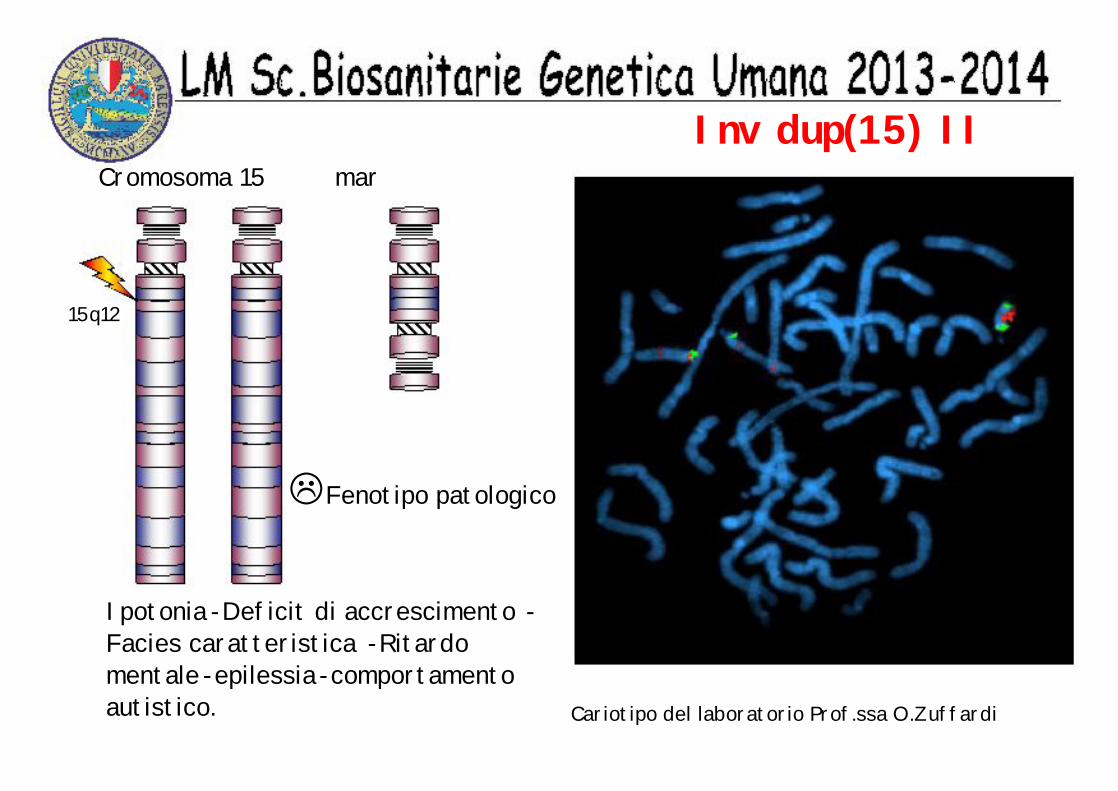
Inv dup(15) II
LFenotipo patologico
15q12
Cariotipo del laboratorio Prof.ssa O.Zuffardi
Ipotonia -Deficit di accrescimento -Facies caratteristica -Ritardo mentale -epilessia -comportamento autistico.
Cromosoma 15 mar
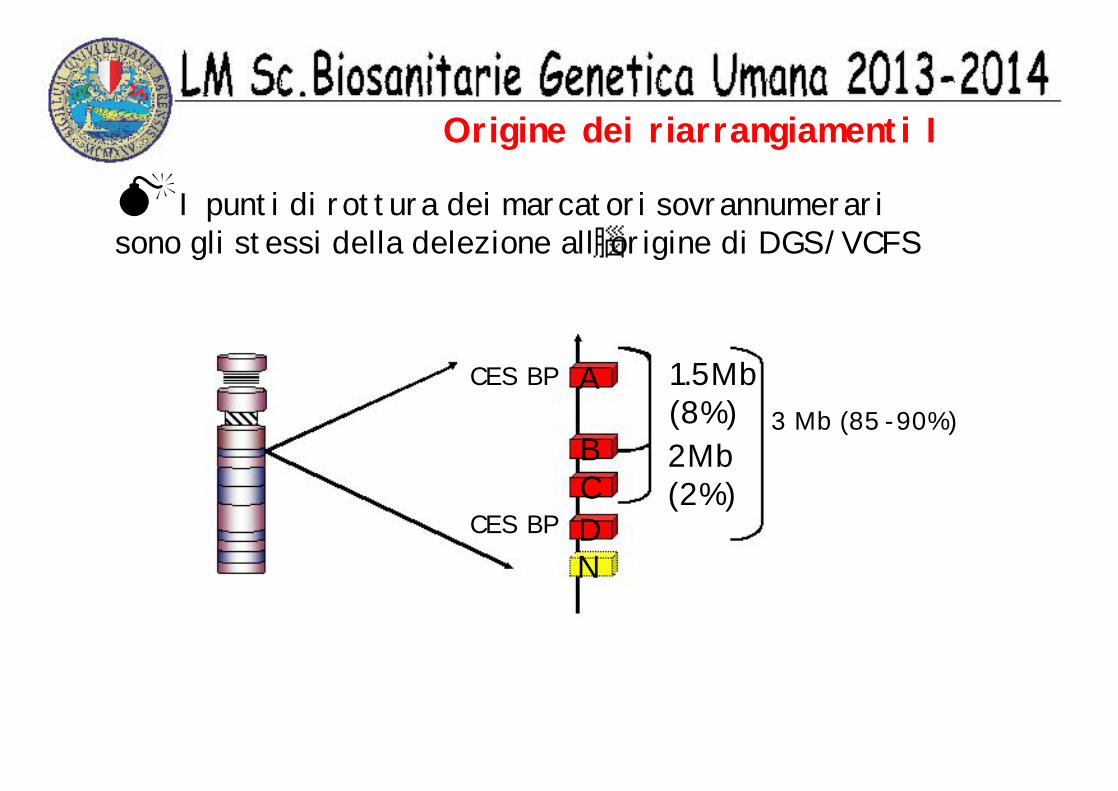
Origine dei riarrangiamenti I
MI punti di rottura dei marcatori sovrannumerari sono gli stessi della delezione all origine di DGS/VCFS
A
BCDN
3 Mb (85 -90%)1.5Mb(8%)2Mb(2%)
CES BP
CES BP
’
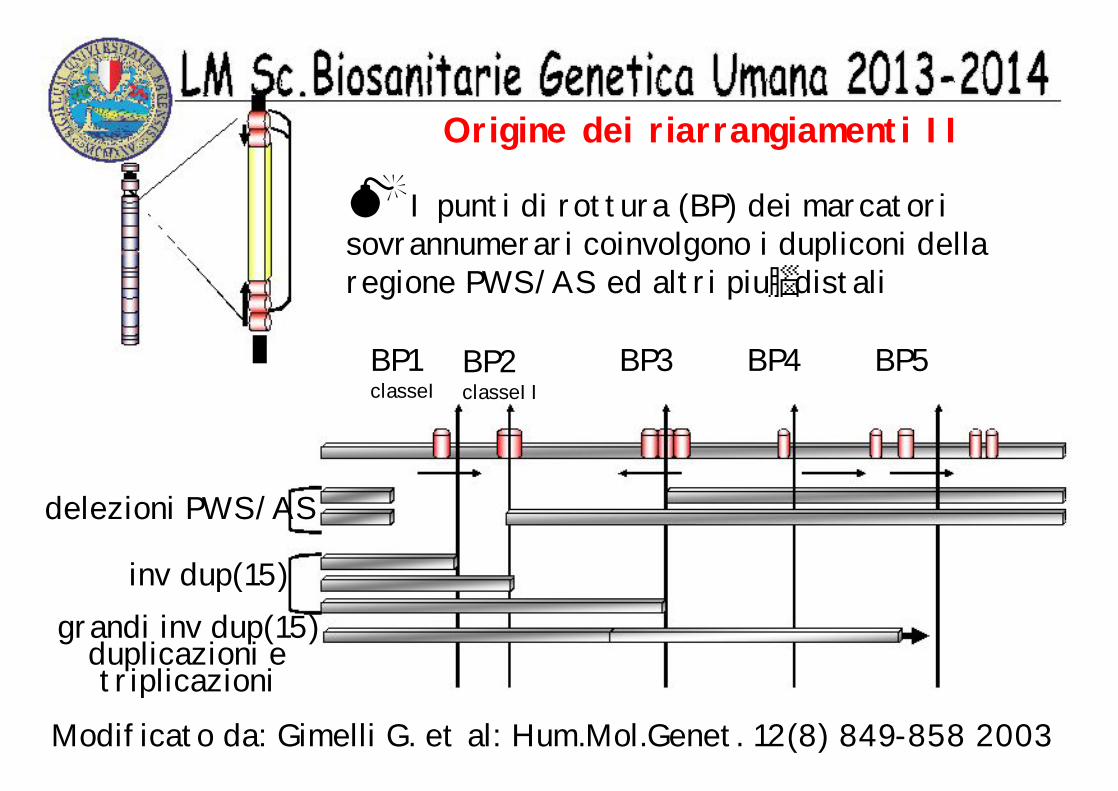
Origine dei riarrangiamenti II
MI punti di rottura (BP) dei marcatori sovrannumerari coinvolgono i dupliconi della regione PWS/AS ed altri piu distali
BP1classeI
BP2classeII
BP3 BP4 BP5
delezioni PWS/AS
inv dup(15)grandi inv dup(15)
duplicazioni etriplicazioni
Modificato da: Gimelli G. et al: Hum.Mol.Genet. 12(8) 849-858 2003
’
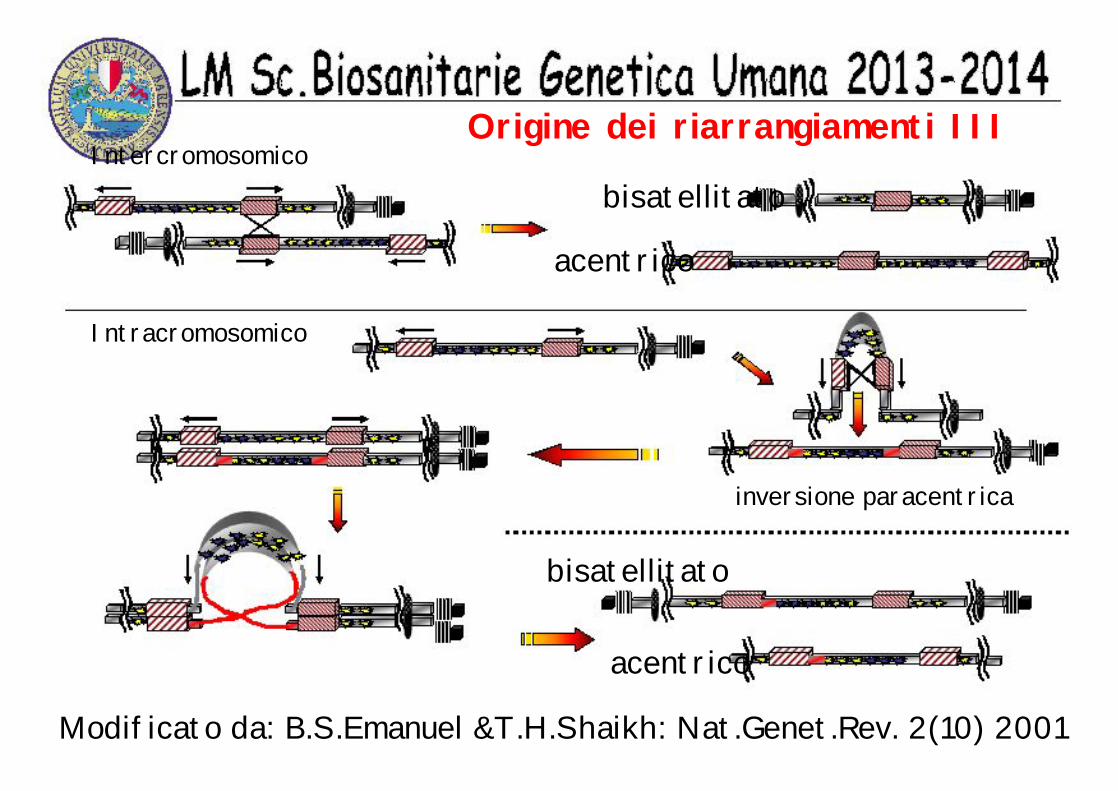
Origine dei riarrangiamenti III Intercromosomico
bisatellitato
acentrico
Intracromosomico
inversione paracentrica
bisatellitato
acentrico
Modificato da: B.S.Emanuel &T.H.Shaikh: Nat.Genet.Rev. 2(10) 2001

inv dup(15)(q11q13) Inverted dupinv dup(22)(q11.2) Inverted dupdic(X)(p11.2) Isodicentricinv dup(8p) inv/dup/delder(8)(pterp23.1::p23.2pter); inv/dup/deldel(8)(p23.1p23.2)dup(15)(q24q26) dupt(11;22)(11q23;22q11)
Riarragiamenti costituzionali
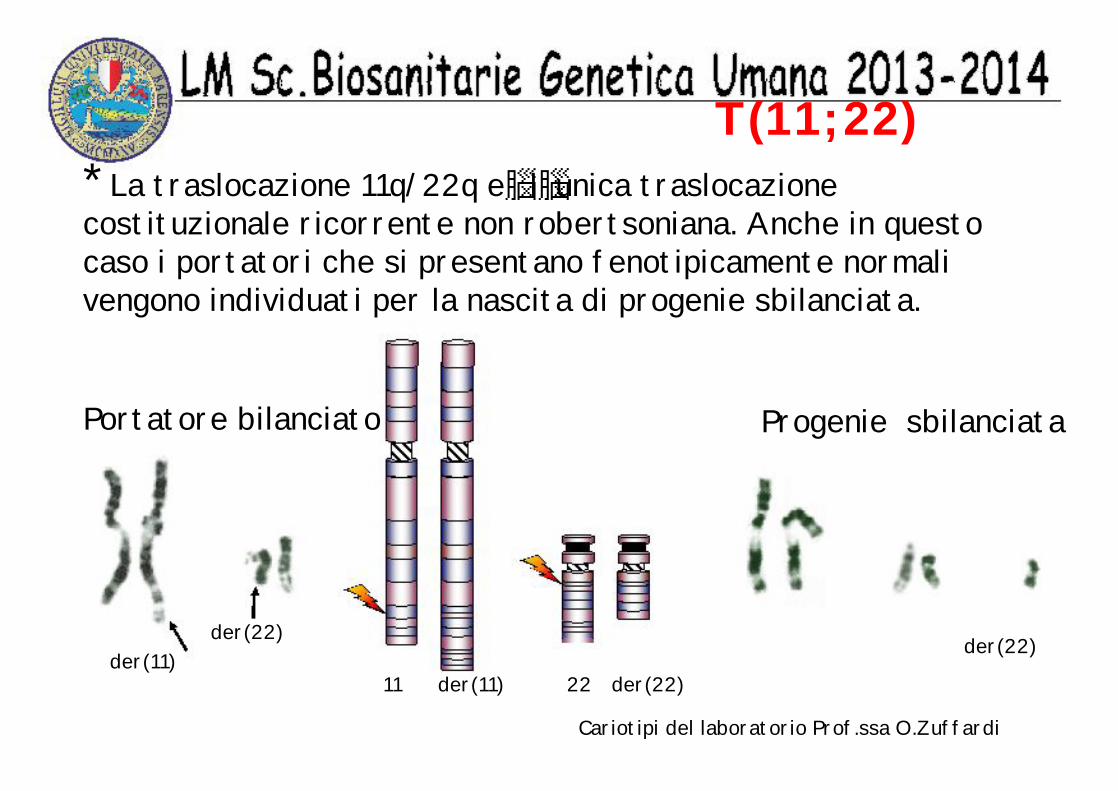
T(11;22)* La traslocazione 11q/22q e l unica traslocazione costituzionale ricorrente non robertsoniana. Anche in questo caso i portatori che si presentano fenotipicamente normali vengono individuati per la nascita di progenie sbilanciata.
11 der(11) 22 der(22)
Portatore bilanciato Progenie sbilanciata
der(22)der(11)
der(22)
Cariotipi del laboratorio Prof.ssa O.Zuffardi
’’
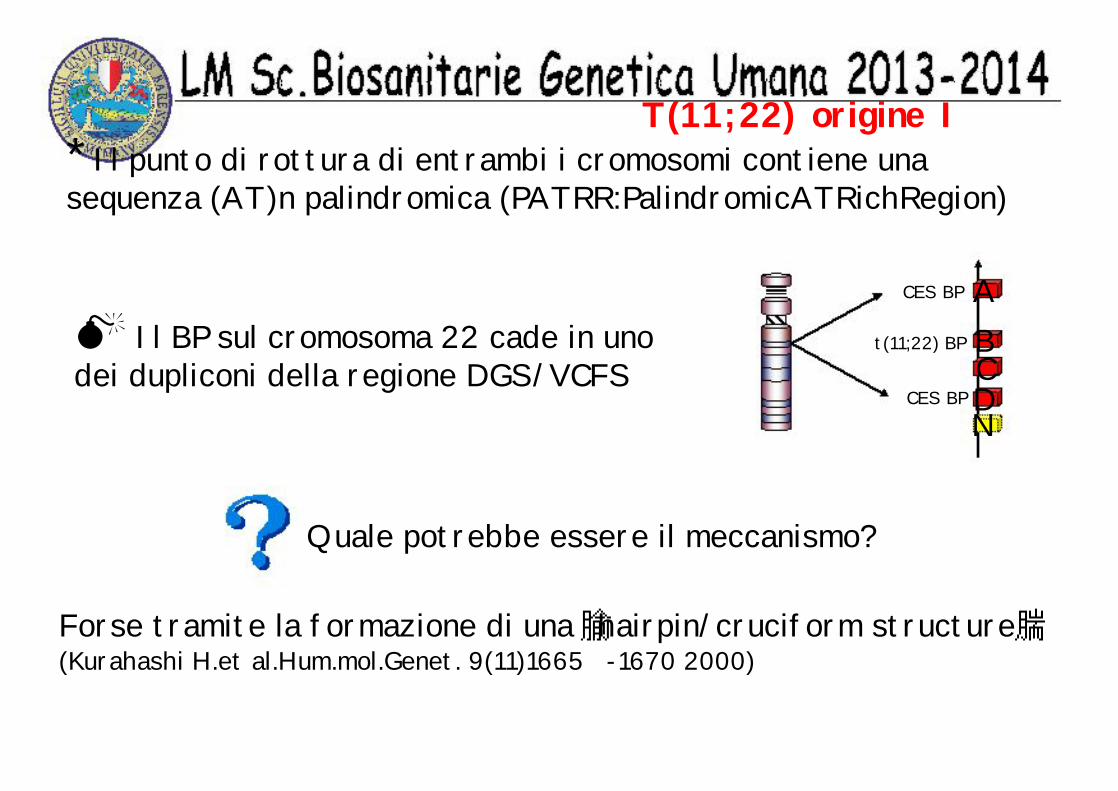
T(11;22) origine I* Il punto di rottura di entrambi i cromosomi contiene unasequenza (AT)n palindromica (PATRR:PalindromicATRichRegion)
Il BP sul cromosoma 22 cade in uno dei dupliconi della regione DGS/VCFS
ABCDN
CES BP
CES BP
t(11;22) BP
Forse tramite la formazione di una hairpin/cruciform structure(Kurahashi H.et al.Hum.mol.Genet. 9(11)1665 -1670 2000)
Quale potrebbe essere il meccanismo?
M
“ ”
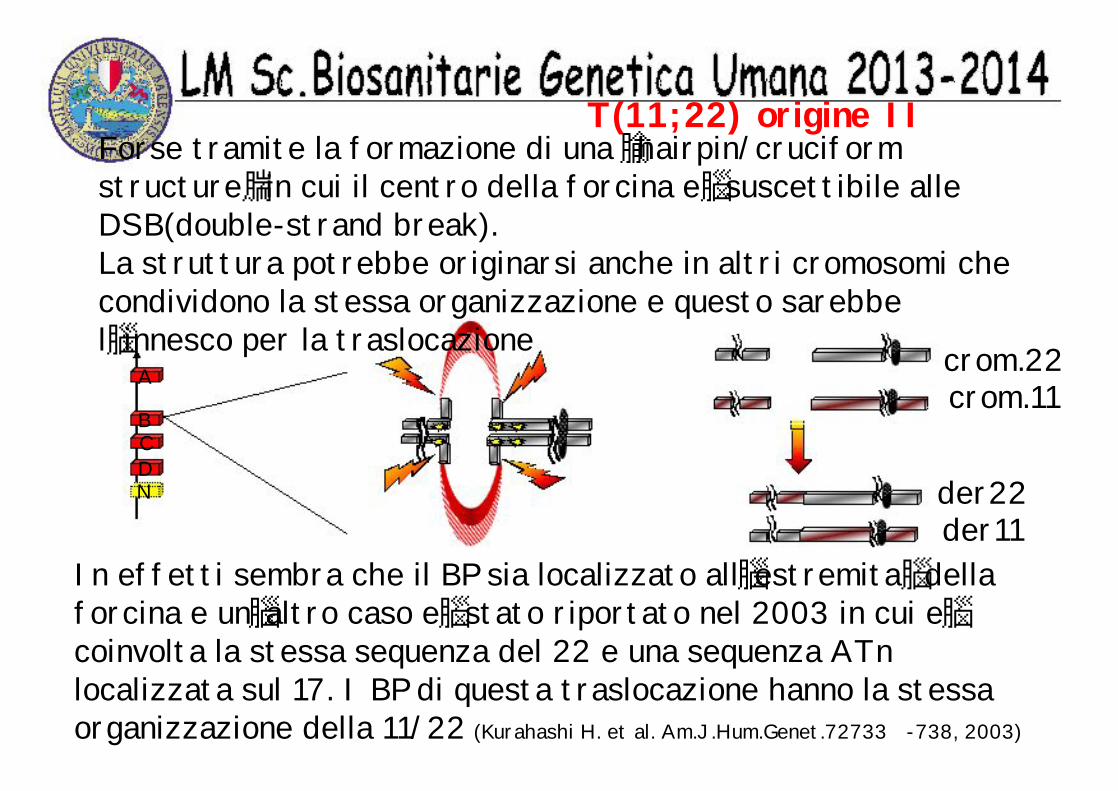
T(11;22) origine IIForse tramite la formazione di una hairpin/cruciform structure in cui il centro della forcina e suscettibile alle DSB(double-strand break).La struttura potrebbe originarsi anche in altri cromosomi che condividono la stessa organizzazione e questo sarebbe l innesco per la traslocazione
A
BCDN
In effetti sembra che il BP sia localizzato all estremita della forcina e un altro caso e stato riportato nel 2003 in cui ecoinvolta la stessa sequenza del 22 e una sequenza ATn localizzata sul 17. I BP di questa traslocazione hanno la stessa organizzazione della 11/22 (Kurahashi H. et al. Am.J.Hum.Genet.72733 -738, 2003)
crom.22crom.11
der22der11
“” ’
’
’ ’’ ’ ’

inv dup(15)(q11q13) Inverted dupinv dup(22)(q11.2) Inverted dupdic(X)(p11.2) Isodicentricinv dup(8p) inv/dup/delder(8)(pterp23.1::p23.2pter); inv/dup/deldel(8)(p23.1p23.2)dup(15)(q24q26) dupt(11;22)(11q23;22q11)
Riarragiamenti costituzionali
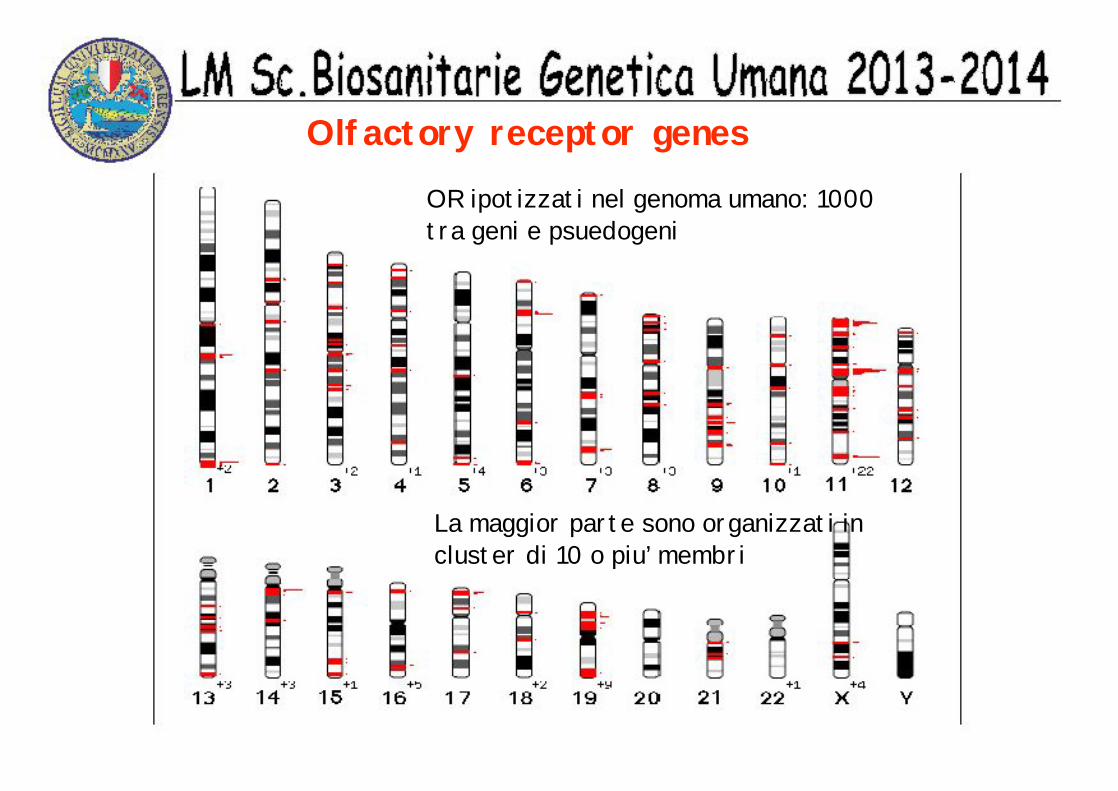
Olfactory receptor genesOR ipotizzati nel genoma umano: 1000 tra geni e psuedogeni
La maggior parte sono organizzati in cluster di 10 o piu’ membri
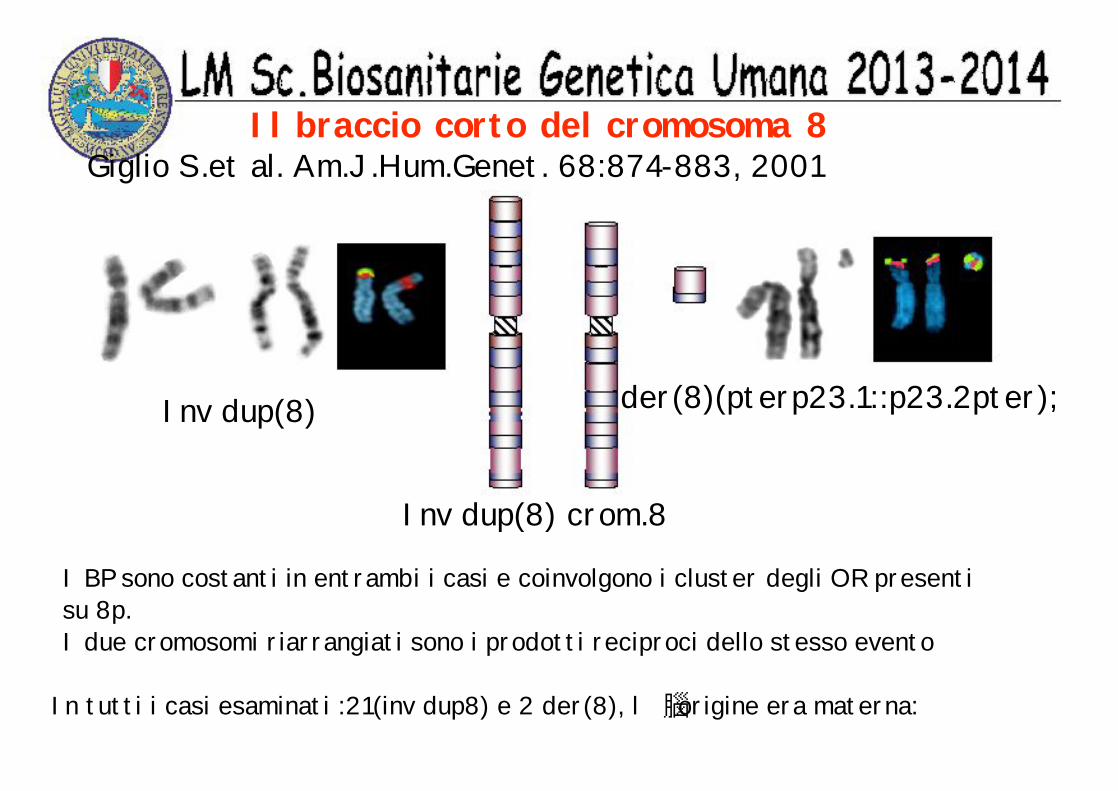
Il braccio corto del cromosoma 8
I BP sono costanti in entrambi i casi e coinvolgono i cluster degli OR presenti su 8p.I due cromosomi riarrangiati sono i prodotti reciproci dello stesso evento
Inv dup(8) der(8)(pterp23.1::p23.2pter);
Inv dup(8) crom.8
In tutti i casi esaminati :21(inv dup8) e 2 der(8), l origine era materna:
Giglio S.et al. Am.J.Hum.Genet. 68:874-883, 2001
’
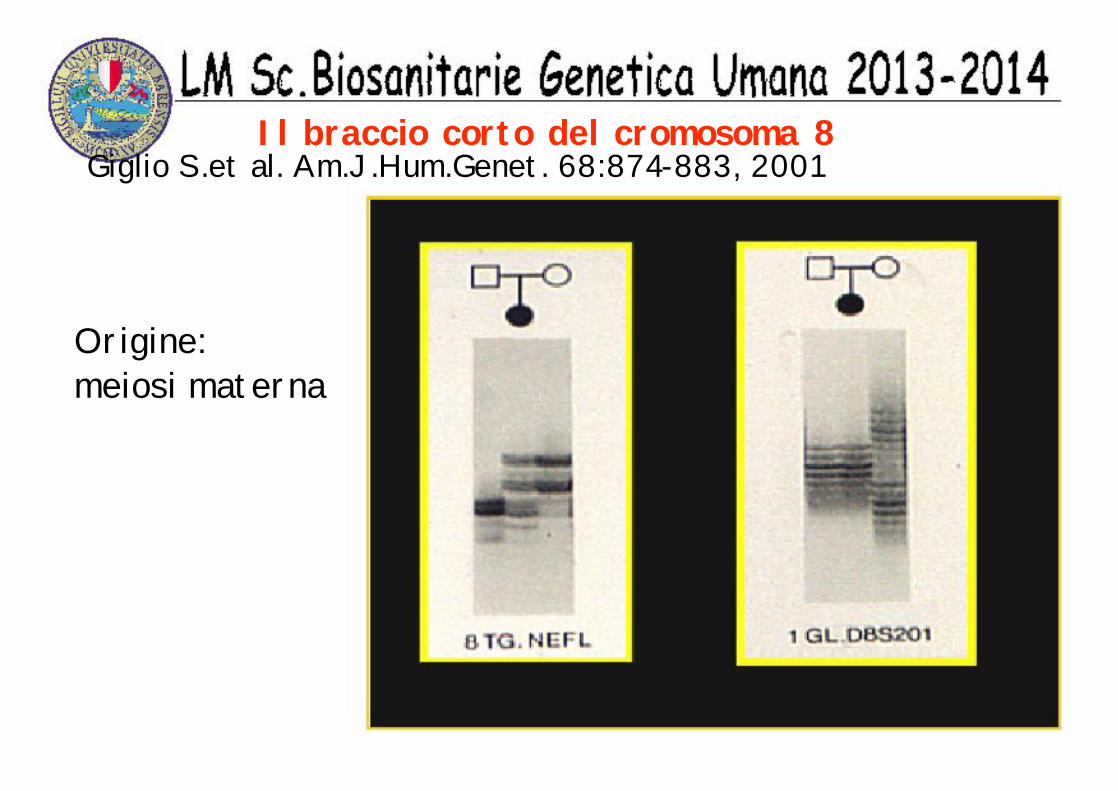
Il braccio corto del cromosoma 8Giglio S.et al. Am.J.Hum.Genet. 68:874-883, 2001
Origine:meiosi materna
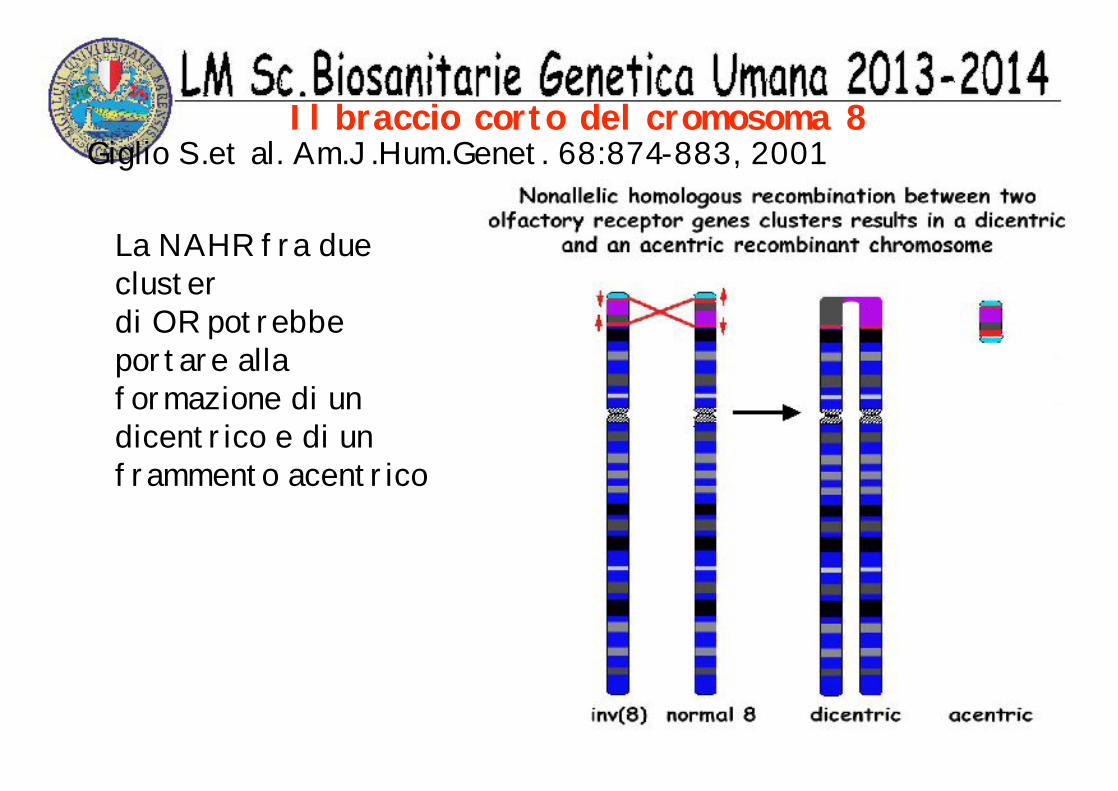
Il braccio corto del cromosoma 8Giglio S.et al. Am.J.Hum.Genet. 68:874-883, 2001
La NAHR fra due cluster di OR potrebbe portare alla formazione di un dicentrico e di un frammento acentrico
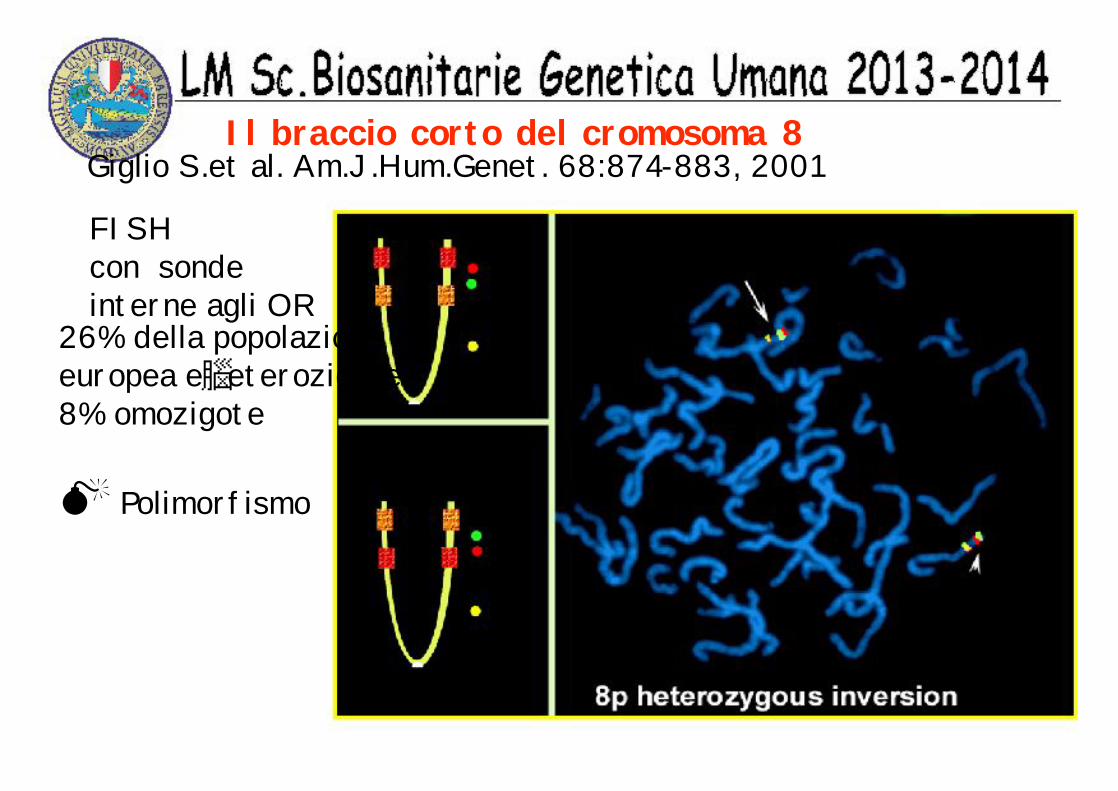
Il braccio corto del cromosoma 8Giglio S.et al. Am.J.Hum.Genet. 68:874-883, 2001
FISHcon sondeinterne agli OR
26% della popolazioneeuropea e eterozigote8% omozigote
Polimorfismo
’
M

Heterozygous submicroscopic inversions involving olfactory receptor -gene clusters mediate the recurrent t(4;8)(p16;p23) translocation . Giglio S.et al. Am. J. Hum. Genet. 71:276 -285 (2002)
Anche in questo caso l origine era la meiosi materna. Anche in questo caso le madri erano eterozigoti per la regione compresa fra i cluster degli OR del cromosoma 4 e del cromosoma 8
T(4;8)(p16;p23)
12,5% eterozigoti per il crom.4
26% eterozigoti per il crom.8
~2.5% doppie eterozigoti
’

CONCLUSIONIParticolari strutture genomiche predispongono all’insorgenza di riarrangiamenti genomici e cromosomici, in meiosi e/o in mitosi
La variabilita’ individuale potrebbe essere il principale fattore di suscettibilita’ alla loro insorgenza

By NA
NON sono dispense, ma un ausilio allo studio sullibro
















![1 –cosa: worldview(s) · (Parmenide, Poema sulla Natura, frammento 2) SeedQuestions (b) ... Subjectarea of design: pattern [schemi di relazioni] Relazionismo SeedQuestions](https://static.fdocuments.us/doc/165x107/5c6925e109d3f242168c91fa/1-cosa-worldviews-parmenide-poema-sulla-natura-frammento-2-seedquestions.jpg)

