

Disorders of sex development (DSD) · DSD: definition Congenital conditions in which development of...
27
Transcript of Disorders of sex development (DSD) · DSD: definition Congenital conditions in which development of...

Clinical features
▪Ambiguous genitalia (in females: enlarged clitoris, labial fusion, in males: hypospadia, undescended testes)
▪ primary amennorhea
▪ streak gonads, inguinal hernia
▪ delayed development of secondary sex characteristics
▪ signs of virilization in female – enlarged clitoris, acne, hirsutism, deepening of the voice, increased muscle strength, menstrualdisruption due to anovulation

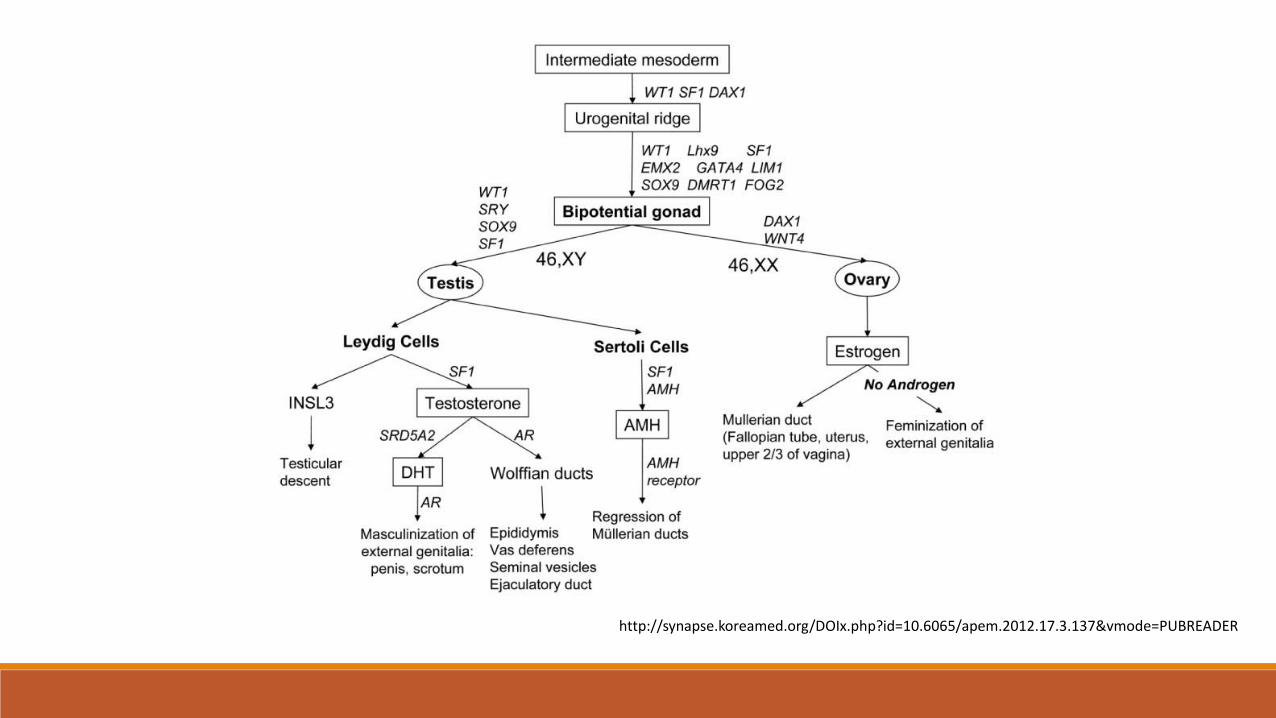
DSD: definition
▪ Congenital conditions in which development of chromosomal, gonadal or anatomical sex is atypical
Level 1 – Genetic / chromosomal sex 46 XX vs 46, XY
Level 2 - Gonadal sex – testes / ovariesUndifferentiated gonad – testicular development dependent on presence of TDF
Level 3 - Phenotypic sex1. Internal reproductive structures (dependent on Mullerian inhibiting factor) 2. External reproductive structures (dependent on testosterone exposure)3. Secondary sex characteristics

Embryology of the human genital tract
http://www.glowm.com/section_view/heading/Genetics%20of%20Sexual%20Differentiation/item/346

Important issues
▪ Gender assignment
▪ Risk of gonadal cancer
▪ Infertility
http://synapse.koreamed.org/DOIx.php?id=10.6065/apem.2012.17.3.137&vmode=PUBREADER

https://www.scieeopen.com/document?vid=be86306e-3e81-44d4-8fb5-448b21bdef60nc

Management of disorders of sex development
Hiort O et al., Nature Reviews Endocrinology 10, 520–529 (2014)

http://clinicalgate.com/normal-puberty-and-pubertal-disorders/
Tanner staging
https://en.wikipedia.org/wiki/Tanner_scale

Diagnostic procedures
❑Cytogenetic tests (kariotype)
❑ Visualisation of internal sex organs throughultrasonography or ascending urethrography (ovaries, uterus)
❑ Hormonal analyses (17-OH progesterone, GC/MS steroid profile, LH, FSH, testosterone, AMH)
❑ DSD gene chip – around 200 genomic regions

Turner syndrome
Responsible genes: X genes that escape inactivation, SHOX
Proteins: SHOX: Short stature homeobox protein
Cytogenetic locus: SHOX: Xpter-p22.32
Inheritance: sporadic
Clinical Features and Diagnostic Criteria: congenital lymphedema, growth failure, normal intelligence (10%
significant delays), coarctation of the aorta, bicuspid aortic valve, HLHS, hyperlipidemia, gonadal dysgenesis
(10% go into puberty), hypothyroidism, diabetes, strabismus, renal malformation, osteoporosis.
Molecular Tests: Karyotype, FISH SRY
Disease Mechanism: SHOX: thought to act as a transcription regulator with many down-stream targets that
modify growth and stature. SHOX protein has been identified in the growth plate from 12 weeks to late
childhood.
Treatment/Prognosis: GH, HRT, gonadectomy if Y chromosome mosaicism (risk for gonadoblastoma).
Lifelong cardiac follow-up, at risk for aortic dilation and dissection with bicuspid aortic valve.

Klinefelter syndromeClinical Features and Diagnostic Criteria: tall stature, slightly delayed motor and languageskills, learning problems, testosterone plateaus age 14, small fibrosed testes, azoospermia and infertility, gynecomastia, increased cholesterol, slightly increased risk of autoimmunedisorders and mediastinal germ cell tumors (1% risk)
Molecular Tests: karyotype, at least one extra chromosome to a 46,XY
Disease Mechanism: 1st or 2nd meiotic division nondisjunction of either parent. Maternal>paternal origin. + advanced maternal effect
Treatment/Prognosis: Testosterone in mid-late adolescence for bone density, development of secondary sex characteristics, muscle mass, cholesterol, improved energy.

Groupwork▪ Discuss briefly: etiology, diagnosis, clinical symptoms, treatment / management
Group A: Congenital adrenal hyperplasia (CAH)
Group B: Complete / partial gonadal dysgenesis
Group C: Complete / partial androgen insensitivity syndrome (CAIS / PAIS)
Ovotesticular DSD
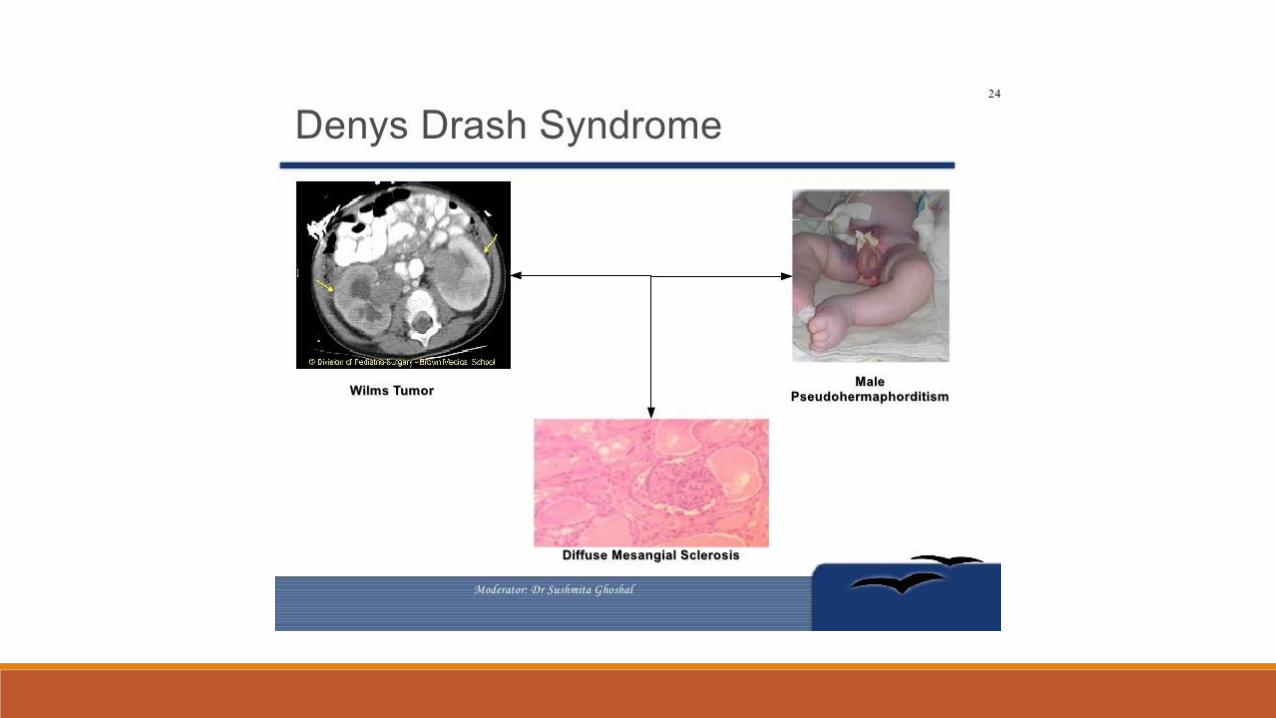
Denys-Drash syndrome

Congenital adrenal hyperplasia (CAH)
https://labtestsonline.org/understanding/analytes/6-17hydroxy/tab/sample/

Congenital adrenal hyperplasia❑ Autosomal recessive, prevalence of 1/12000-1/15000
❑ Deficiency of one of the 5 enzymes required for cortisol synthesis
❑ 90% of cases: 21-hydroxylase deficiency. CYP21 gene on 6p. Carrier frequency 1/50.
❑ Girls with Classic CAH: virilization
❑ Boys with classic CAH: poor feeding, dehydration, collapse (low plasma Na, high K, acidosis)
❑Non-classic CAH: asymptomatic or signs of postnatal androgen excess
❑ Treatment if recognized at birth: steroid therapy (hydrocortisone +/- fludrocortisone and salt

Complete gonadal dysgenesis
❑Kariotype 46,XY; 46,XX
❑ Female phenotype
❑ Vagina, uterus, fallopian tubes present
❑ Streak gonads in the place of ovaries
❑ Absence of secondary sex characteristics
❑ High FSH, low testosterone, low estradiol


Partial gonadal dysgenesis
❑Kariotype 45,X/46,XY or 46,XY
❑ Ambiguous genitalia
❑ Vagina, uterus, vas deferens present
❑ Bilateral dysgenetic testes
❑ Low testosterone, low AMH, low estradiol, high FSH


https://courses.washington.edu/conj/bess/differentiation/differentiation.htm



Ovotesticular DSD❑Kariotype 46,XX or 46,XY or mosaics
❑ Ambiguous genitalia
❑ Vagina, uterus, vas deferens present
❑ Presence of ovarian tissue or testicular tissue
❑ Hormonal tests depending on gonadal function

Complex management of DSD❑ CAH with salt-wasting – life-threatening
❑ Optimal gender assignment/choice, reconstruction of sex organs
❑ Therapy
❑ Risk of gonadal cancer
❑ Hormone secretion
❑ Fertility, sexual issues, psychosocial implications
❑ Genetic counselling

Child with DSD - Consensus of 2006❑ interdisciplinary diagnosis
❑ Each child should be given a gender.
❑ Parents should take part in decision-making on the child’s gender.
❑ In doubtful cases, not to do reconstruction operations prematurely
❑ Gonadectomy in high-risk groups

Gonadectomy in DSD
5th I-DSD Symposium Programme. 11th-13th June 2015, Ghent, Belgium


















