
Determination of protein structure and dynamics combining immune algorithms and pattern search
18
Abstract Natural proteins quickly fold into a complicated three-dimensional structure. Evolutionary algorithms have been used to predict the native structure with the lowest energy conformation of the primary sequence of a given protein. Successful structure prediction requires a free energy function sufficiently close to the true potential for the native state, as well as a method for exploring the conformational space. Protein structure prediction is a challenging problem be- cause current potential functions have limited accuracy and the conformational space is vast. In this work, we show an innovative approach to the protein folding (PF) problem based on an hybrid Immune Algorithm (IMMALG) and a quasi-Newton method starting from a population of promising protein confor- mations created by the global optimizer DIRECT. The new method has been tested on Met-Enkephelin peptide, which is a paradigmatic example of multiple– minima problem, 1POLY, 1ROP and the three helix protein 1BDC. DIRECT produces an initial population of promising candidate solutions within a poten- tially optimal rectangle for the funnel landscape of the PF problem. Hence, IMMALG starts from a population of promising protein conformations created by the global optimizer DIRECT. The experimental results show that such a multistage approach is a competitive and effective search method in the con- formational search space of real proteins, in terms of solution quality and computational cost comparing the results of the current state-of-art algorithms. Keywords Clonal Selection Algorithms DIRECT Immune Algorithms pattern search methods protein folding protein structure prediction structural bioinformatics A. M. Anile V. Cutello G. Narzisi G. Nicosia (&) Department of Mathematics and Computer Science, University of Catania, V.le A. Doria 6, 95125, Catania, Italy E-mail: [email protected] S. Spinella Department of Linguistics, University of Calabria, Ponte P. Bucci 17/B, 87036, Arcavata di Rende (CS), Italy 123 Nat Comput (2007) 6:55–72 DOI 10.1007/s11047-006-9027-3 ORIGINAL PAPER Determination of protein structure and dynamics combining immune algorithms and pattern search methods A. M. Anile V. Cutello G. Narzisi G. Nicosia S. Spinella Published online: 9 December 2006 Ó Springer Science+Business Media B.V. 2007
Transcript of Determination of protein structure and dynamics combining immune algorithms and pattern search

Abstract Natural proteins quickly fold into a complicated three-dimensionalstructure. Evolutionary algorithms have been used to predict the native structurewith the lowest energy conformation of the primary sequence of a given protein.Successful structure prediction requires a free energy function sufficiently closeto the true potential for the native state, as well as a method for exploring theconformational space. Protein structure prediction is a challenging problem be-cause current potential functions have limited accuracy and the conformationalspace is vast. In this work, we show an innovative approach to the proteinfolding (PF) problem based on an hybrid Immune Algorithm (IMMALG) and aquasi-Newton method starting from a population of promising protein confor-mations created by the global optimizer DIRECT. The new method has beentested on Met-Enkephelin peptide, which is a paradigmatic example of multiple–minima problem, 1POLY, 1ROP and the three helix protein 1BDC. DIRECTproduces an initial population of promising candidate solutions within a poten-tially optimal rectangle for the funnel landscape of the PF problem. Hence,IMMALG starts from a population of promising protein conformations createdby the global optimizer DIRECT. The experimental results show that such amultistage approach is a competitive and effective search method in the con-formational search space of real proteins, in terms of solution quality andcomputational cost comparing the results of the current state-of-art algorithms.
Keywords Clonal Selection Algorithms Æ DIRECT Æ Immune Algorithms Æpattern search methods Æ protein folding Æ protein structure prediction Æstructural bioinformatics
A. M. Anile Æ V. Cutello Æ G. Narzisi Æ G. Nicosia (&)Department of Mathematics and Computer Science, University of Catania, V.le A. Doria 6,95125, Catania, ItalyE-mail: [email protected]
S. SpinellaDepartment of Linguistics, University of Calabria, PonteP. Bucci 17/B, 87036, Arcavata di Rende (CS), Italy
123
Nat Comput (2007) 6:55–72DOI 10.1007/s11047-006-9027-3
ORI GI N A L P A PE R
Determination of protein structure and dynamicscombining immune algorithms and patternsearch methods
A. M. Anile Æ V. Cutello Æ G. Narzisi Æ G. Nicosia ÆS. Spinella
Published online: 9 December 2006� Springer Science+Business Media B.V. 2007

Introduction
Proteins are known to have many important functions in the cell, such as enzymaticactivity, storage and transport of material, signal transduction, antibodies and more(Eisenberg et al., 2000). The amino acids composition of a protein will usually uniquelydetermine its 3D structure, to which the protein’s functionality is directly related. Aprotein has a multiple level structure: Primary structure, chains of mono-dimensionalamino acid; Secondary structure, chains of structural regular elements (a-helices,b-sheets); Tertiary structure, 3D structure of protein in the native state; Quaternarystructure, contact of separate protein molecules forming very complex 3D assembling ofmultiple protein chains.
Common methods for finding protein 3D structures (such as X-ray crystallo-graphic and NMR – Nuclear Magnetic Resonance) are slow and costly, and may takeup several weeks (or months) of laboratory work. As a consequence, there has beena continuously growing interest in the design of ad hoc folding algorithms.
Successful structure prediction requires a free energy function sufficiently close tothe true potential for the native state, as well as a method for exploring the con-formational space. Protein structure prediction is a challenging problem becausecurrent potential functions have limited accuracy and the conformational space isvast. Several algorithmic approaches have been applied to the protein foldingproblem (PF) in the last 50 years: molecular dynamics (Levitt, 1983), Monte Carlomethods (Simons et al., 1997), simulated tempering (Hansmann and Okamoto,1997), evolutionary algorithms (Bowie and Eisemberg, 1994). In spite of all theseefforts, protein folding remains a challenging and computationally open problem(Tramontano, 2006).
The CHARMM potential energy function
We use energy functions in order to evaluate the structure of a molecule. It would be‘‘nice’’ to use quantum mechanics, but it is too computationally complex to be practicalto model large systems (long proteins). So, we use classical physics to deal with energyfunctions. Sometimes called potential energy functions or force fields, these functionsreturn a value for the energy based on the conformation of the molecule. They provideinformation on which conformations of the molecule are better or worse. The better is theconformation of the molecule the lower is the energy value. The most typical energyfunctions have the form:
EðRÞ ¼X
bonds
BðRÞ þX
angles
AðRÞ þX
torsions
TðRÞ þX
non�bonded
NðRÞ ð1Þ ð1Þ
where R is the vector representing the conformation of the molecule, typically incartesian coordinates or in torsion angles. The summation is over all pairs of atomsbetween which is present a chemical bond or local interaction (bonds, angles, tor-sions) and atoms not connected by chemical bonds (non-bonded).
The literature on cost functions is enormous. In this work, in order to evaluatethe conformation of a protein, we use the CHARMM energy function (version 27)(Brooks et al., 1983; MacKerell et al., 1998). CHARMM (Chemistry at HarvardMacromolecular Mechanics) is a popular all-atom force field used mainly forstudying macromolecules. It is a composite sum of several molecular mechanics
56 Nat Comput (2007) 6:55–72
123

equations: stretching, bending, torsion, Urey-Bradley, impropers, van-der-Walls,electrostatics. The CHARMM (Foloppe and MacKerell, 2000) energy function hasthe form:
Echarmm ¼X
bonds
kbðb� b0Þ2 þX
UB
kUBðS� S0Þ2 þX
angles
khðh� h0Þ2
þX
torsions
kv½1þ cosðnv� dÞ� þX
impropers
kimpð/� /0Þ2
þX
nonbond
eijRminij
rij
� �12
� Rminij
rij
� �6" #
þ qiqj
erij
ð2Þ
where, in order, b is the bond length, b0 is the bond equilibrium distance, and kb isthe bond force constant; S is the distance between two atoms separated by twocovalent bonds, S0 is the equilibrium distance, and kUB is the Urey Bradley forceconstant; h is the valence angle, h0 is the equilibrium angle, and Kh is the valenceangle force constant; v is the dihedral or torsion angle, kv is the dihedral forceconstant, n is the multiplicity, and d is the phase angle; / is the improper angle, /0
is the equilibrium improper angle, and kimp is the improper force constant; eij is theLennard Jones (LJ) well depth, rij is the distance between atoms i, and j, Rminij isthe minimum interaction radius, qi is the partial atomic charge for the atom i, and eis the dielectric constant. Typically, ei, and Rmini are obtained for individual atomtypes, and then combined together to yield eij, and Rminij for the interacting atomsvia combining rules. In CHARMM, eij values are obtained via the geometric meaneij ¼
ffiffiffiffiffiffiffieiejp
, and Rminij via the arithmetic mean, Rminij = (Rmini + Rminj)/2.Finally, the energy function CHARMM (Eq. 2) represents our minimization
objective, the torsion angles of the protein are the decision variables of the optimi-zation problem, and the constraint regions are the variable bounds. To evaluate theCHARMM energy function we use the routines of the TINKER (http://www.dasher.wustl.edu/tinker/) Molecular Modeling Package (Huang et al., 1999). First, theprotein structure in internal coordinates (torsion angles) is transformed in cartesiancoordinates using the PROTEIN routine. Then the conformation is evaluated usingthe ANALYZE routine, that gives back the CHARMM energy potential of a givenprotein structure.
The metrics: DME and RMSD
To evaluate how similar is the predicted conformation to the native one, we employtwo frequently used metrics: Root Mean Square Deviation (RMSD) and DistanceMatrix Error (DME). RMSD is computed using the formula:
RMSDða; bÞ ¼
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiPni¼1 jrai � rbij2
n
s
ð3Þ
where rai and rbi are the positions of atom i in structure a and structure b, respec-tively, and where structures a and b have been optimally superimposed. Fitting is
Nat Comput (2007) 6:55–72 57
123

performed using the McLachlan algorithm (McLachlan, 1982) as implemented in theprogram ProFit (http://www.bioinf.org.uk/software/profit/).
DME is calculated using the formula:
DMEða; bÞ ¼
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiPni¼1
Pnj¼1ðjrai � rajj � jrbi � rbjjÞ2
q
nð4Þ
which does not require the superposition of coordinates. For a particular pair ofstructures, the RMSD, which measures the similarity of atomic positions, is usuallylarger than the DME, which measures the similarity of inter-atomic distances.
DIRECT pattern search method
The DIRECT method is a global search algorithm for bound constrained optimi-zation based on Lipschitz constant estimation (Jones et al., 1993). It is classified as apattern search method. The method is based on three operations:
1. Lipschitz constant estimation,2. choice for potential optimality of domain subregions, and3. domain subdivision.
The method focuses on the ratio between local exploration and global explora-tion. The estimation of Lipschitz constant is interpreted as a scheme to assign thesuitable priorities for a set of rectangles which are a partition of the search domain.This priority is used to plan stratified sampling in order to balance exploration andconvergence and to obtain fast convergence with a broad domain coverage. More-over, unlike some other methods, Lipschitz global optimization requires only fewparameters. This algorithm has been applied in a variety of real applications (Carteret al., 2001; Hong and Bogy, 2002; Bartholomew-Biggs et al., 2002).
The choice for potential optimality is made according to the following definition:
Definition 1 (Potentially optimal rectangle). Given an objective function f, let Sbe the set of hyperrectangles generated by the algorithm after k iterations, and letfmin be the current best function value. An hyperrectangle ~R 2S with center c~R
and measure að~RÞ is said to be potentially optimal if there exists a Lipschitz constantK > 0 such that
f ðc~RÞ �Kað~RÞ � f ðcRÞ �KaðRÞ 8R 2S ð5Þ
f ðc~RÞ �Kað~RÞ � fmin� ejfminj: ð6Þ
where e ~ 10–4 is a constant to control the clustering during the search (Jones et al.,1993).
The choice of e was also improved by a runtime heuristic in order to speed up theconvergence process (Gablonsky and Kelley, 2001).
58 Nat Comput (2007) 6:55–72
123

The above definition gives a heuristic rule for the choice of hyperrectangles whichare potentially optimal in terms of increasing efficiency of the objective function.Equation (6) controls the clustering nearby the optimal points.
The algorithm can be structured in a main procedure (Algorithm 1) with a mainloop containing: the selection of potentially optimal rectangles, the pattern ofsampling, the subdivision (Algorithm 2) of these rectangles and the updating vari-able statements.
If an hyperrectangle R is potentially optimal then it will be sampled in the pointscR±dei, i = 1... N, where cR is the center point of the hyperrectangle, d is one-thirdthe side length of the hyperrectangle, and ei is the ith unit vector.
Next DIRECT chooses to leave the best function values in the largest space,therefore we define
wj ¼ minðf ðcR þ dejÞ; f ðcR � dejÞÞ ð7Þ
and divide the measure of the side length with the smallest wj into thirds, so thatci±dej are the centers of the new hyperrectangles. This pattern is repeated for alldimensions on the ‘‘center hyperrectangle’’, choosing the next dimension by deter-mining the next smallest wj. Figure 1 shows the sampling scheme of an hyperrec-tangle. This strategy increases the attractiveness of searching near points with goodfunction values (since large hyperrectangles are preferred for sampling, everythingelse is equal) (Finkel, 2003).
The whole procedure can be simplified in its exponential complexity, which is dueto the recursive subdivision of rectangles, stating a limit in the maximum number ofrectangle subdivisions. But in this case global convergence cannot be guaranteed andonly approximated solutions will be reached.
Without the previous subdivision limit, the algorithm guarantees the convergenceto the global minimum although it is not possible to predict how many iterations arerequired. The hypothesis for this claim is the continuity of the objective functionwhich, after an infinite number of iterations, guarantees a dense sampling of thedomain.
Algorithm 1. DIRECT pseudocodeRequire: Set of rectangles S1: n ‹ 0 {number of function calls}2: while n < TotCalls do3: Choose P � S , set of potential optimal rectangles;4: Sample the rectangles r2P updating the counter n;5: Subdivide the rectangles of P. Let subdivision be DP = {R1, R2, ..., Rm}6: S = S P[ DP
7: end while8: return the best minimum;
Algorithm 2. DIRECT Subdivision pseudocodeREQUIRE: A potentially optimal rectangle R with center cR
1: Identify the set I of dimension with the maximum side length. Let d equal one-thirdof this maximum side length.
2: Sample the function at points cR±dei " i 2I, where ei is the ith unit vector.3: Divide the rectangle R into thirds starting with the lowest value of
wj = min(f(cR + dej), f(cR–dej)) and continuing to the dimension with the highest wj.
Nat Comput (2007) 6:55–72 59
123

The Immune Algorithm
The theory of clonal selection (Burnet, 1959), suggests that among all possible cells,B and T lymphocytes, with different receptors circulating in the host organism, i.e., theones who are actually able to recognize the antigen (the fittest cells), will start toproliferate by cloning. Hence, when a B cell is activated by binding with an antigen,it produces many clones, in a process called clonal expansion. The resulting cells canundergo somatic hypermutation, creating offspring B cells with mutated receptors.Antigens compete for recognition with these new B cells, with their parents and withother clones. The higher is the affinity of a B cell to the available antigens, the morelikely it will clone. This results in a Darwinian process of variation and selectionwhich is called affinity maturation. The increasing size of these populations and theproduction of cells with longer expected lifetimes assure the organism a higherspecific responsiveness to an antigenic attack, by establishing a defense over time(immune memory). This principle inspired a new class of artificial immune systems,the Clonal Selection Algorithms (CSAs) (Nicosia et al., 2004).
CSAs and other Evolutionary Algorithms such as Genetic Algorithms (GA),Evolution Strategies (ES), Evolutionary Programming (EP) and Genetic Program-ming (GP), are methods that use simulated evolution processes to solve or to findapproximate solutions to complicated computational problems and NP-completeproblems that are difficult to tackle with deterministic and conventional methods(Nicosia, 2004; Cutello et al., 2004).
The Immune Algorithm proposed in this work uses an evolutionary approach withdifferent evolutionary operators, cloning, hypermutation, hypermacromutation, andaging, to find low-energy conformations of peptides. We have also used a Quasi-Newton procedure as a post-processing local optimization method to refine the qualityof the obtained solutions.
The proposed Immune Algorithm uses an internal coordinate representation(torsion angles) for the B cell or candidate solutions: each residue type requires afixed number of torsion angles to fix the 3D coordinates of all atoms. Bond lengthsand angles are fixed at their ideal values. The degrees of freedom in this represen-tation are the backbone and sidechain torsion angles (/, w, x, and vh). The numberof v angles depends on the residue type. The angles, measured in degrees, arerepresented by real numbers approximated to the third decimal digit.
Each structure of a given protein is encoded as a set of dihedral angles (/, w, x,and vh). In the real-valued representation, each B cell at time step t is a vector of realvariables:
Fig. 1 Sampling strategy of the DIRECT Algorithm
60 Nat Comput (2007) 6:55–72
123

xt ¼ ðx1; x2; . . . ; xnÞ 2 <n:
Two operators, hypermutation and hypermacromutation (Nicosia et al., 2004;Cutello et al., 2004) are used together in the Immune Algorithm to face the proteinfolding problem.
Usually, variation operators mutate an individual by adding a Gaussian distrib-uted random vector of mean zero and predefined deviation (r) as follows:
x0ðtÞ ¼ xt þ u
here the mutation vector u is
u ¼ ðu1; u2; . . . ; unÞ; uj ¼ Njð0; rÞ:
where Nj(0,r) is a real random number generated by a gaussian distribution of mean0 and standard deviation r.
The new hypermutation operator performs a local search of the conformationalspace. It will perturb a torsion angle 2(/,w,x,vh) of a randomly chosen residue withthe law:
xt;0i ¼ xt
i þNð0; rÞ
where r = 1.0.The new hypermacromutation operator may change the conformation dramati-
cally. When this operator acts on a peptide chain, a torsion angle of a randomlychosen residue is reselected from its corresponding constrained region (i.e., corre-sponding domain of the selected torsion angle).
Algorithm 3. Immune Algorithm for protein foldingREQUIRE ProteinSequence, d, dup, sB, I1: Nc ‹ d · dup;2: t ‹ 0;3: if I then4: P(t) ‹ Random_Initial_Pop (ProteinSequence);5: else6: P(t) ‹ DIRECT (ProteinSequence);7: end if8: Evaluate(P(t));9: while : Termination_Condition() do10: P(clo) ‹ Cloning (P(t), Nc);11: P(hyp) ‹ Hypermutation (P(clo));12: Evaluate (P(hyp));13: P(macro) ‹ Hypermacromutation (Pclo);14: Evaluate (P(macro));15: (P(t)
a,P(hyp)a, P(macro)
a ) ‹ Aging(P(t),P(hyp),P(macro),sB);16: P(t+1) ‹ (l + k)-Selection (P(t)
a ,P(hyp)a , P(macro)
a );17: t ‹ t + 1;18: end while19: BFGS(P(t))
Nat Comput (2007) 6:55–72 61
123

Methods
Given a sequence of amino acids, and the corresponding secondary structureassignment (using SSpro (http://www.ics.uci.edu/~baldig/scratch/) to perform proteinsecondary structure prediction (Baldi and Pollastri, 2003), we represent the proteinusing a sequence of torsion angles. The backbone angles (/,w) are selected in thefavorable Ramachandran region appropriate for the given secondary structure type;we set the last backbone angle x to the standard value of 180�. Sidechain torsionangles are constrained in regions derived from the backbone-independent rotamerlibrary of Roland L. Dunbrack (http://www.fccc.edu/research/labs/dunbrack/)(Dunbrack and Cohen, 1997). Sidechain constraint regions are of the form: [m–r,m + r]; where m and r are the mean and the standard deviation for each sidechaintorsion angle computed from the rotamer library. Under these constraints the con-formation is still highly flexible and the structure can take on various shapes that arevastly different from the native shape.
The Immune Algorithm designed in this paper can use two different initializationprocedures for the first population of conformations: random initialization of pop-ulation, the standard approach, and an initial population of conformations given byDIRECT global search procedure.
In the first method, the protein sequences are conformations of torsion anglesrandomly selected from their corresponding constrained regions. In the latter, theinitial population is constituted by a set of conformations created by the DIRECTalgorithm. DIRECT produces an initial population of promising candidate solutionsinside potentially optimal rectangles of the funnel landscape of the protein foldingproblem. This scheme reduces the number of fitness function evaluations of theoverall search process to the lowest energy value. This is the first known applicationof the DIRECT Algorithm to the protein folding problem. Considering the twodifferent initialization procedures for the population at the initial generation (t = 0),we have the IMMALG starting from a random initial population, and the IM-MALG–DIRECT approach for the IMMALG starting from a population ofpromising protein conformations created by the global optimizer DIRECT. Fromthe current population, a number dup of clones will be generated, producing thepopulation Pop(clo), which will be mutated into Pop(hyp) by the new hypermutationoperator, and into Pop(macro) by the new hypermacromutation operator. Algorithm 3shows the pseudo-code of the Immune Algorithm for the structure prediction of realproteins.
It is important to note that the IMMALG output is also improved by a deterministiclocal search, the BFGS scheme. The BFGS (Broyden–Fletcher–Goldfarb–Shanno)rule is a classical formula of the quasi-Newton methods. It is widely adopted in manyunconstrained optimization algorithms which compute a suitable approximation of theinverse Hessian matrix A–1 at every step. In this work we use the ‘‘limited memory’’version as implementation in Dennis and Schnabel (1983) and Byrd et al. (1994) whichuses few correction vectors in order to reconstruct A–1.
Finally, it is worth to observe that the immunological operators, DIRECT andBFGS procedure act on the internal representation, torsion angles, whereas theCHARMM potential uses the external representation in Cartesian coordinates.
62 Nat Comput (2007) 6:55–72
123

Results
The Met-Enkephalin peptide
The Immune Algorithm has been applied to determine the three- dimensionalstructure of the pentapeptide Met-Enkephalin (TYR-GLY-GLY-PHE-MET aminoacids). It is a very short polypeptide, only five amino acids, 22 variable backbone andside-chain torsion (or dihedral) angles (n ¼ 22) and 75 atoms. From an optimizationpoint of view the Met-Enkephalin polypeptide is a paradigmatic example of multi-ple–minima problem. It is estimated to have more than 1011 locally optimal con-formations. Nevertheless, the Met-Enkephalin (1MET) has defined structure, and anapparent global minimum (with conformational energy of –12.90 kcal/mol based onECEPP/2 routine) first located by a Monte Carlo-minimization method in 4 h only,in 1987 by Scheraga and Purisima (1987). For all these reasons, this peptide is anobvious ‘‘test bed’’, for which a substantial amount of in silico experiments has beendone (Purisima and Scheraga, 1987; Kaiser et al., 1997; Li and Scheraga, 1998;Bindewald et al., 1998). For frequent convergence to the global minimum, in Li andScheraga (1998) the authors set a maximum number of energy function evaluationsto Tmax ¼ 106. Using the multistage approach, the initial population of 10 individ-uals was generated by the global optimizer DIRECT after T1
max ¼ 11171 energyfunction evaluations. The Immune Algorithm starting from this ad hoc initial con-formation was executed for 500 generations with a population size of d = 10 indi-viduals, duplication parameter dup = 2, expected life time parameter sB = 5, for amaximum number of energy function evaluations equal to T2
max ¼ 20000 . Hence,considering the computational costs of the combined approach, DIRECT + IM-MALG, we have an overall Tmax ¼ T1
max þ T2max ¼ 31171 maximum number of
energy function evaluations for frequent convergence to the best energy value closeto the global minimum. For the 1MET peptide we omit running the BFGS proce-dure in order to perform a further local search.
The best conformation with the lowest energy value of –20.56 kcal/mol obtainedby the Immune Algorithm is reported in the left plot of Figure 2. The middle plotshows the backbone conformations of the Met-Enkephalin peptide obtained byScheraga and Li (1998), and the right plot shows superimposition of the two struc-tures using a full atom resolution. Computing the RMSD and DME metrics withrespect all atoms and the Ca atoms of the two structures, we have obtained,respectively, the following results out of five independent runs: best
Fig. 2 In the left plot the backbone structure of the 1MET peptide obtained by IMMALG; in themiddle plot the optimal conformation obtained by Scheraga (Li and Scheraga, 1998); in the right plotthe superimposition of the two structures. The RMSD of the two structures is 2.835 A, the RMSDca
is 0.490 A. In terms of DME measure for the two structures we have DME = 2.211 A andDMEca ¼ 0:454 A. The figure was made using the program UCSF Chimera (Huang et al., 1996)
Nat Comput (2007) 6:55–72 63
123

RMSD = 2.835 A (mean = 2.95,r = 0.061), and best RMSDca= 0.501 A, while in
terms of DME measure we have obtained the following best results:DME = 2.142 A and DMEca ¼ 0:468 A.
In Figure 3 we compare the three different approaches we have used in terms ofEnergy values (Kcal/mol) and fitness function evaluations (FFE): DIRECT globaloptimization procedure, Immune Algorithm and the hybrid approach, ImmuneAlgorithm with DIRECT (DIRECT+IA). Both individual methods reach proteinconformations with similar energy values with approximately 30000 energy functionevaluations. The hybrid approach is the only one able to yield the protein confor-mation near the native state (E = –20.47 kcal/mol), starting from a population ofpromising protein conformations with energy values equal to –2.11 kcal/mol createdby the global optimizer DIRECT using 11171 energy function evaluations.
Table 1 shows the comparisons of the designed hybrid Immune Algorithm withother folding algorithms. For each algorithm, the table reports (when available fromliterature) the mean and standard deviation energy values, the energy function used,the best RMSD measure with respect the Scheraga’s conformation, the acceptedoptimal conformation, and the maximum number of FFE. From a structural simi-
0
50
100
150
200
250
300
1 10 100 1000 10000
Ene
rgy
(Kca
l/mol
e)
Fitness Function Evaluations
Immune Algorithm, DIRECT and DIRECT + Immune Algorithm
DIRECTIA
DIRECT-IA
-22-20-18-16-14-12-10-8-6-4-2
10000 15000 30000
Fig. 3 Energy values versus fitness function evaluations for the DIRECT global search procedure,the Immune Algorithm, and the hybrid approach, Immune Algorithm with DIRECT, DIRECT+IA(axis x in log scale)
Table 1 The hybrid Immune Algorithm versus Folding algorithms for 1MET
Algorithm Energy (kcal/mol) RMSD FFE
Scheraga’s MC (Li and Scheraga, 1998) –12.90 ECEPP/2 n.a. 106
IMMALG-DIRECT –20.47 ± 1.54 CHARMM 2.835 A 3 · 104
REGAL (real cod.) Tight constr.(Kaiser et al., 1997)
–23.55 ± 1.69 CHARMM 3.23 A 1.5 · 105
Lamarkian (binary cod.)(Kaiser et al., 1997)
–28.35 ± 1.29 CHARMM 3.33A 1.5 · 105
Baldwinian (binary cod.)(Kaiser et al., 1997)
–22.57 ± 1.62 CHARMM 3.96 A 1.5 · 105
REGAL (real cod.) Loose constr.(Kaiser et al., 1997)
–22.01 ± 2.69 CHARMM 4.25 A 1.5 · 105
SGA (binary cod.) (Kaiser et al., 1997) –22.58 ± 1.57 CHARMM 4.51 A 1.5 · 105
REGAL (real cod.) (Kaiser et al., 1997) –24.92 ± 2.99 CHARMM 4.57 A 1.5 · 105
64 Nat Comput (2007) 6:55–72
123

larity point of view it is more significant to consider the RMSD. In terms of RMSDvalue, IMMALG–DIRECT obtains the more similar structure to the acceptedoptimal conformation of Scheraga with a computational cost of Tmax ¼ 31171 en-ergy function evaluations while the algorithms designed in Kaiser et al. (1997) wereallowed to reach 150000 evaluations.
In Figure 4, we report the dynamics of backbone angles / and w (omitting x andthe sidechain angles vh) obtained by the two methods DIRECT (left plot) andImmune Algorithms (right plot). As described in Section 4, the Immune Algorithmstarts from a promising region, that is, a promising conformation (a vector of torsionangles) previously determined by the DIRECT procedure after 104 energy functionevaluations.
After the first 5000/1000 energy function evaluations, the angle dynamics of DI-RECT reach a plateau region, and there are no significant improvements. Starting bythis conformation (the center point of the hyper-rectangle) the Immune Algorithmruns a local search inside the hyper-rectangle. The sequence of smooth perturbationsperformed by the hypermutation operator and the large angle variation performedby the hypermacromutation operator (see Figure 4 bottom plot at 500 and 13000energy function evaluations) drives the Immune Algorithm to obtain a 3D confor-mation near to the native state for the 1MET peptide. Finally, the angle dynamicsreported show the folding process of the Met-Enkephelin peptide.
1POLY peptide
The 1POLY polypeptide is a 14 residue model of Polyalanine. Its native structure isan a-helix. Polyalanine is a homogeneous molecule made up of 14 residues of theamino acid alanine (ALA), with 56 dihedral angles and 143 atoms (ALA1,...ALA14
amino acids). Immune Algorithm using DIRECT definitely formed the expecteda-helix secondary structure (Figure 5).
For the multistage approach, the initial population was generated by the DIRECTglobal optimization procedure using Tmax
1 = 10183 energy function evaluations. TheIMMALG starting from the ad hoc initial conformation was executed for 500 gen-erations with a population size of d = 10 individuals, duplication parameter dup = 2,expected life time parameter sB = 5, for a maximum number of energy functionevaluations equal to Tmax
2 = 20000. Hence, considering the computational costs of theDIRECT procedure and the IMMALG we have an overallTmax ¼ T1
max þ T2max ¼ 30183 maximum number of energy function evaluations for
frequent convergence to the lowest energy value close to the global minimum(E = 279.4 kcal/mole).
The multistage approach needs approximately 3 · 104 average number of energyfunction evaluations to the lowest energy value of Polyalanine while other ap-proaches, such as the evolutionary algorithms in Kaiser et al. (1997), need at least1.5 · 105.
In order to describe the hypermutation versus hypermacromutation phenomena,Figure 6 shows the Immune Algorithm dynamics for the two mutation operators.Figure 6 (top plot) reports the average energy values of hypermutated candidatesolutions, average energy values of the current population and the best energy value;all curves decrease monotonically. The bottom plot shows the conformationsperturbed by hypermacromutation; spikes correspond to new conformation(s) with
Nat Comput (2007) 6:55–72 65
123

unfeasible 3D structure caused by too close atoms interactions. This is consequenceof the way the hypermacromutation operator acts on a given conformation: a torsionangle of a randomly chosen residue is reselected from its corresponding domain.
-300
-250
-200
-150
-100
-50
0
50
100
150
200
0 5000 10000 15000 20000
Ang
les
in d
egre
es
Numbers of Energy Function Evaluations
Angle dynamics
TYR: φTYR: ψGLY: φGLY: ψGLY: φGLY: ψPHE: φPHE: ψMET: φMET: ψ
-250
-200
-150
-100
-50
0
50
100
150
200
20000160001200080004000
Ang
les
in d
egre
es
Numbers of Energy Function Evaluations
Angle dynamics
TYR: φTYR: ψGLY: φGLY: ψGLY: φGLY: ψPHE: φPHE: ψMET: φMET: ψ
Fig. 4 Angle dynamics. Top plot: backbone angle (/,w) dynamics versus energy functionevaluations obtained by the DIRECT algorithm. Bottom plot: angle dynamics obtained by ImmuneAlgorithm starting from promising conformation previously determined by DIRECT
66 Nat Comput (2007) 6:55–72
123

1ROP protein
In this paragraph we show the prediction capabilities of the multistage approach forthe 1ROP protein, a polypeptide of 56 amino acids with a 3D structure formed by 2parallel a-helix (see Figure 7). Figure 8 shows the predicted and the native structureof 1ROP. The RMSD of the two structure is 3.589 A, and the DME is 1.89 A. Forthe 1ROP protein DIRECT runs for 105 energy function evaluations while IM-MALG runs for 2 · 104 energy functions calls. Hence we have an overall 1.2 · 105
maximum number of energy function evaluations for frequent convergence to thelowest energy value close to the global minimum (E = –1117.249 kcal/mole).
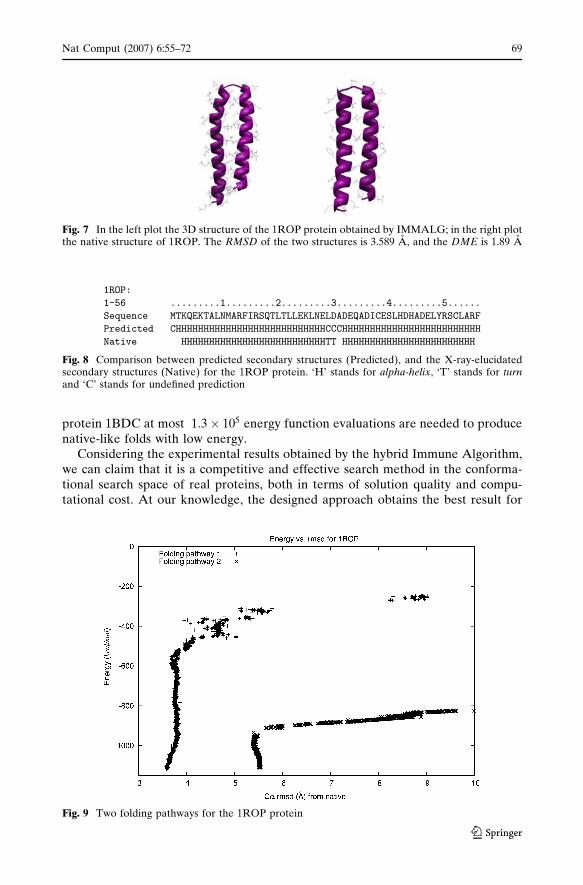
Figure 9 reports two possible folding pathways for the 1ROP protein in terms ofenergy value versus RMSD; Folding pathway 1 obtains the final structure with lowerRMSD value.
Finally, we present a possible folding process for the 1ROP protein (see Fig-ure 10). Starting from a promising conformation obtained by DIRECT. The figureshows the intermediate and native-like conformations.
1BDC protein
The last in silico experiment was performed on the 1BDC protein, a three-helixbundle protein (see Figure 11). 1BDC is formed by 60 residues. This hard proteininstance has been chosen to assess the final prediction capabilities of the designedmultistage approach. It is important to note that it is the number of helices and thepacking of the secondary structure elements, rather than the size of protein, thatseems to cause difficulties.
For the 1BDC protein DIRECT runs for 105 energy function evaluations whileIMMALG runs for 2 · 104 energy functions calls. Moreover, to improve the finalprotein conformation the quasi-Newton method BFGS procedure has been calledfor 104 energy function evaluations in order to perform a further local search. Hencefor 1BDC we have an overall 1:3� 105 maximum number of energy functionevaluations for frequent convergence to the lowest energy value close to the globalminimum ( E ¼ �1428:96 kcal/mole).
Figure 12 shows the predicted and native 3D structures for the 1BDC protein.The RMSD of the two structures is 8.780 A while the RMSDca is 7.368 A. In termsof DME measures we have DME = 6.15 A and DMEca ¼ 5:28 A. The RMSD forthe core region (amino acids [8–54]) is equal to 5.776 A.
Conclusions
We have developed an energy-based method to generate a variety of folds searchingthe conformational space with a multistage approach. The DIRECT procedure
Fig. 5 The 1POLY polypeptide structure obtained by the Immune Algorithm. Left plot: the fullatom resolution. Right plot: ribbon representation
Nat Comput (2007) 6:55–72 67
123

produces an initial population of promising candidate solutions inside potentiallyoptimal rectangles of the funnel landscape of the protein folding problem. Hence,the Immune Algorithm starts from a population of promising protein conformationscreated by the global optimizer DIRECT. This scheme reduces the number of fitnessfunction evaluations of the overall search process to reach low energy values. In fact,for the Met-Enkephelin and Polyalanine peptides an average number of energyfunction evaluations equal to 3� 104 are needed, and for 1ROP and the three-helix
278
280
282
284
286
288
290
292
50 100 150 200 250 300 350 400 450 500
Ene
rgy
(kca
l/m
ol)
Generations
Hypermutated clones average fitnessPopulation average fitness
Best fitness
100
1000
10000
100000
1e+06
1e+07
1e+08
1e+09
0 50 100 150 200 250 300 350 400 450 500
Ene
rgy
(kca
l/m
ol)
Generations
HyperMacromutated clones average fitness
Fig. 6 The Immune Algorithm dynamics, energy values versus generations, for the Polyalaninepeptide. Top plot: average energy value of hypermutated conformations, average energy value ofcurrent population and the best energy value. Bottom plot: energy values of hypermacromutatedconformations versus generations
68 Nat Comput (2007) 6:55–72
123
protein 1BDC at most 1:3� 105 energy function evaluations are needed to producenative-like folds with low energy.
Considering the experimental results obtained by the hybrid Immune Algorithm,we can claim that it is a competitive and effective search method in the conforma-tional search space of real proteins, both in terms of solution quality and compu-tational cost. At our knowledge, the designed approach obtains the best result for
Fig. 7 In the left plot the 3D structure of the 1ROP protein obtained by IMMALG; in the right plotthe native structure of 1ROP. The RMSD of the two structures is 3.589 A, and the DME is 1.89 A
1ROP:1-56 .........1.........2.........3.........4.........5......Sequence MTKQEKTALNMARFIRSQTLTLLEKLNELDADEQADICESLHDHADELYRSCLARFPredicted CHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCHHHHHHHHHHHHHHHHHHHHHHHHHNative HHHHHHHHHHHHHHHHHHHHHHHHHHTT HHHHHHHHHHHHHHHHHHHHHHHH
Fig. 8 Comparison between predicted secondary structures (Predicted), and the X-ray-elucidatedsecondary structures (Native) for the 1ROP protein. ‘H’ stands for alpha-helix, ‘T’ stands for turnand ‘C’ stands for undefined prediction
Fig. 9 Two folding pathways for the 1ROP protein
Nat Comput (2007) 6:55–72 69
123

the Met-Enkephelin peptide compared to the results obtained by the current state-of-art algorithms.
The analyzed protein instances have been guided by the DIRECT global searchprocedure and the Immune Algorithm to folds resembling their crystal structure.
Fig. 10 Folding process for the 1ROP protein. (a) Initial direct conformation. (b)-(e) Intermediateconformations at different time steps (generations). (f) Final native-like conformation
1BDC:1-60 .........1.........2.........3.........4.........5.........6Sequence TADNKFNKEQQNAFYEILHLPNLNEEQRNGFIQSLKDDPSQSANLLAEAKKLNDAQAPKANative S SS HHHHHHHTTTTTS HHHHHHHHHHHHH STHHHHHHHHHHHHHTTSPredicted CCCCCCCHHHHHHHHHHHCCCCCCHHHHHCHHHECCCCCHHHHHHHHHHHHHHHCCCCCC
Fig. 11 Comparison between predicted secondary structures (Predicted), and the X-ray-elucidatedsecondary structures (Native) for the 1BDC protein
Fig. 12 Predicted and native structures for the 1BDC protein. The RMSD of the two structures is8.780 A, the \raster="_"RMSDca is 7.368 A, and for core region [8–54] RMSD = 5.776 A. In termsof DME measure for the two structures we have DME = 6.15 A, and \raster="_"DMEca ¼ 5:28 A
70 Nat Comput (2007) 6:55–72
123

References
Baldi P, Pollastri G (2003) The principled design of large-scale recursive neural network architec-tures-DAG-RNNs and the protein structure prediction problem. Journal of Machine LearningResearch 4:575–603
Bartholomew-Biggs MC, Parkhurst SC, Wilson SP (2002) Using DIRECT to solve an aircraft routingproblem. Computational Optimization and Applications and International Journal 21(3):311–323
Bindewald E, Hesser J, Manner R (1998) Implementing genetic algorithms with sterical constrainsfor protein structure prediction. In: Proceedings of International Conference on Parallel Prob-lem Solving from Nature (PPSN V), pp. 959–967, Amsterdam, The Netherlands
Bowie JU, Eisemberg D (1994) An evolutionary approach to folding small alpha-helical proteinsthat uses sequence information and an empirical guiding fitness function. Proceedings of theNational Academy of Sciences of the United States of America 91: 4436–4440
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM:A program for macromolecular energy, minimization, and dynamics calculations. Journal ofComputational Chemistry 4:187–217
Burnet FM (1959) The Clonal Selection Theory of Acquired Immunity. Cambridge, UK: CambridgeUniversity Press
Byrd RH, Nocedal J, Schnabel RB (1994) Representation of quasi Newton matrices and their use inlimited memory methods. Mathematical Programming 63(4):129–156
Carter RG, Gablonsky JM, Patrick A, Kelley CT (2001) Algorithms for noisy problems in gastransmission pipeline optimization. Optimization and Engineering 2:139–157
Cutello V, Nicosia G, Pavone M (2004) Exploring the capability of Immune Algorithms: A char-acterization of hypermutation operators. In Proceedings of the Third International Conferenceon Artificial Immune Systems (ICARIS’04), vol. 3239, pp. 263–276, Catania, Italy
Dennis JE, Schnabel RB (1983) Numerical Methods for Unconstrained Optimization and NonlinearEquations. Prentice-Hall, Englewood Cliffs, NJ
Dunbrack RL Jr, Cohen FE (1997) Bayesian statistical analysis of protein sidechain rotamer pref-erences. Protein Science 6:1661–1681
Eisenberg D, Marcotte E, Xenarios I, Yeates TO (2000) Protein function in the post-genomic era.Nature 405(6788):823–826
Finkel DE (2003) DIRECT Optimization Algorithm User Guide. Technical Report, CRSC N.C.State University (ftp://ftp.ncsu.edu/pub/ncsu/crsc/pdf/crsc-tr03-11.pdf)
Foloppe N, MacKerell AD Jr (2000) All-atom empirical force field for nucleic acids: I. Parameteroptimization based on small molecule and condensed phase macromolecular target data. Journalof Computational Chemistry 21:86–104
Gablonsky JM, Kelley CT (2001) A locally-biased form of the DIRECT Algorithm. Journal ofGlobal Optimization 21:27–37
Hansmann UH, Okamoto Y (1997) Numerical comparisons of three recently proposed algorithms inthe protein folding problem. Journal of Computational Chemistry 18: 920–933
Hong Zhu, Bogy DavidB. (2002). DIRECT Algorithm and its application to slider Air-Bearingsurface optimization. IEEE Transactions on Magnetics 38(5): 2168–2170
Huang CC, Couch GS, Pettersen EF, Ferrin TE (1996) Chimera: An extensible molecular modelingapplication constructed using standard components. Pacific Symposium on Biocomputing 1, 724
Huang SE, Samudrala R, Ponder JW (1999) Ab initio folding prediction of small helical proteinsusing distance geometry and knowledge-based scoring functions. Journal of Molecular Biology290: 267–281
Jones DR, Perttunen CD, Stuckman BE (1993). Lipschitzian optimization without the lipschitzconstant. Journal of Optimization Theory and Application 79: 157–181
Kaiser CE Jr, Lamont GB, Merkle LD, Gates GH Jr, Patcher R (1997) Polypeptide structureprediction: Real-valued versus binary hybrid genetic algorithms. In: Proceedings of the ACMSymposium on Applied Computing (SAC), pp. 279–286, San Jose, CA
Levitt M (1983) Protein folding by restrained energy minimization and molecular dynamics. Journalof Molecular Biology 170:723–764
Li Z, Scheraga HA (1998) Structure and free energy of complex thermodynamics systems. Journal ofMolecular Structure 179:333–352
Nat Comput (2007) 6:55–72 71
123

MacKerell AD Jr, Brooks B, Brooks C L III, Nilsson L, Roux B, Won Y, Karplus M (1998)CHARMM: The energy function and its parameterization with an overview of the program. In:Schleyer PvR et al. (eds) The Encyclopedia of Computational Chemistry, vol. 1, pp. 271–277.John Wiley & Sons, Chichester
McLachlan AD (1982) Rapid comparison of protein structures. Acta cytologica A38: 871–873Nicosia G (2004) Immune Algorithms for Optimization and Protein Structure Prediction. Depart-
ment of Mathematics and Computer Science. University of Catania, ItalyNicosia G, Cutello V, Bentley P, Timmis J (2004) Proceedings of the Third International Conference
on Artificial Immune Systems. Springer-Verlag, Berlin, GermanyPurisima EO, Scheraga HA (1987) An approach to the multiple-minima problem in protein folding
by relaxing dimensionality. Test on enkephalin. Journal of Molecular Biology 196: 697–709Simons KT, Kooperberg C, Huang E, Baker D (1997) Assembly of protein tertiary structures from
fragments with similar local sequences using simulated annealing and bayesian scoring function.Journal of Molecular Biology 306:1191–1199
Tramontano A (2006) Protein Structure Prediction: Concepts and Applications. Wiley-VCH,Weinheim
72 Nat Comput (2007) 6:55–72
123


















