
DETERMINAÇÃO DE BENZO(A)PIRENO EM CHÁ … · SPE Solid Phase Extration ... HPLC High Performance...
78
RAQUEL GARCIA PEREIRA BRATHWAITE DETERMINAÇÃO DE BENZO(A)PIRENO EM CHÁ VERDE CANOAS, 2010.
Transcript of DETERMINAÇÃO DE BENZO(A)PIRENO EM CHÁ … · SPE Solid Phase Extration ... HPLC High Performance...

RAQUEL GARCIA PEREIRA BRATHWAITE
DETERMINAÇÃO DE BENZO(A)PIRENO EM CHÁ VERDE
CANOAS, 2010.

RAQUEL GARCIA PEREIRA BRATHWAITE
DETERMINAÇÃO DE BENZO(A)PIRENO EM CHÁ VERDE
Trabalho de Conclusão do Curso realizado no Centro Universitário Unilasalle, como parte dos requisitos para obtenção do título de Bacharel em Química.
Orientação: Profa. Dr
a Marisa Tsao.
CANOAS, 2010

RAQUEL GARCIA PEREIRA BRATHWAITE
DETERMINAÇÀO DE BENZO(A)PIRENO EM CHÁ VERDE
Trabalho de conclusão aprovado como requisito parcial para obtenção do grau de Bacharel no Curso de Química do Centro Universitário La Salle – Unilasalle.
Aprovado pela avaliadora em 07 de julho de 2010.
__________________________________________ Prof
a. Dr
a Marisa Tsao
Unilasalle

À minha mãe, irmãos e ao Marcelo, pelo amor, amizade, compreensão e
confiança, o tempo todo.

AGRADECIMENTOS
Primeiramente a Deus, que me deu uma família maravilhosa, saúde e força,
tornando possível a realização deste trabalho.
À minha mãe, Daunila, agradeço a todo seu carinho, por sua atenção e
compreensão, por sua dedicação e amor incondicional. Obrigada pelo exemplo de
vida e luta pelo papel de mãe e pai. A minha vitória é tua também, te amo.
Aos meus queridos irmãos, Felipe, Rafael, Kely e Gabriela (famosa e
complicada família Brath), e ao meu amado, Marcelo, família a qual amo muito e é
minha base para tudo nessa vida.
À minha orientadora, Dra. Marisa Tsao, pelo exemplo de profissional, pela
atenção, ajuda, compreensão e importantes ensinamentos e incentivos em todos os
momentos e também a todos os professores que puderam me ajudar nesta
caminhada.
A Souza Cruz, empresa da qual faço parte atualmente, pela grande
oportunidade que tive de crescer profissionalmente, pela disponibilidade, que
possibilitaram a realização deste trabalho.
A minha grande amiga Lilian, fiel companheira de trabalho e de faculdade
pelos grandes e ótimos momentos que vivemos, espero continuar a compartilhar
bons momentos da minha e da tua vida.
Agradeço, enfim, a todos que direta ou indiretamente contribuíram para que
este trabalho se tornasse realidade.

“Você ganha forças, coragem e confiança a cada experiência em que você enfrenta
o medo. Você tem que fazer exatamente aquilo que acha que não consegue”.
Eleanor Roosevelt

RESUMO
Este trabalho descreve o estudo de determinação de Benzo(a)pireno, em amostras
de chá verde, utilizando Cromatografia Gasosa acoplada a Espectrometria de
Massas utilizando Impacto de Elétrons .
Palavras-chaves: Chá Verde. HPAs. Benzo(a)pireno. Cromatografia Gasosa.
Espectrometria de massas.
ABSTRACT
This work describes Benzo(a)pyrene determination, in green tea samples, using Gas
Chromatography Mass Spectrometry using electron impact.
Keywords: Green Tea. HPAs. Benzo(a)pyrene. gaseous Cromatografia.
Espectrometry of masses.

LISTA DE FIGURAS
Figura 1 - O chá. ..................................................................................................16
Figura 2 - Campos de chá verde..........................................................................18
Figura 3 - A colheita de chá. ................................................................................19
Figura 4 - Estrutura molecular de alguns hidrocarbonetos policíclicos aromáticos. .
.............................................................................................................22
Figura 5 - Estrutura molecular do B(a)P...............................................................23
Figura 5 - Esquema de um cromatógrafo a gás. ..................................................35
Figura 6 - Cromatograma ideal para uma amostra contendo três substâncias. ...36
Figura 7 - Esquema de componentes de um sistema de cromatografia gasosa
com espectrometria de massas (GC/MS) ............................................37
Figura 8 - Esquema representativo de um analisador tipo quadrupolo ................38
Figura 10 - Amostras pesadas e prontas para iniciar o procedimento ...................51
Figura 11 - Etapa de filtração .................................................................................52
Figura 12 - Secagem em evaporador rotatório.......................................................52
Figura 13 - Etapa de eluição da amostra ...............................................................53
Figura 14- Coleta do Benzo(a)pireno após a eluição ............................................54
Figura 15 - Transferência da amostra para frasco (vial) cromatográfico................54
Figura 16 - Injeção em CG/MS...............................................................................55
Figura 17 - Curva de calibração de B(a)P..............................................................62
Figura 18 - Cromatograma do ponto 7 da curva de calibração. .............................65
Figura 19 - Espectro de massas do B(a)P D12 (PI), íons 260 e 264. ....................66
Figura 20 - Espectro de massas do Padrão B(a)P, íons 250 e 252. ......................66
Figura 21 - Cromatograma da amostra 4, extração do chá com diclorometano.....67
Figura 22 - Cromatograma da amostra 4, extração da infusão com diclorometano...
.............................................................................................................68
Figura 23 - Espectro de massas da amostra 4, massa 260 e 264 (B(a)P D12.) ....68
Figura 24 - Espectro de massas da amostra 4, massa 250 e 252 (B(a)P).............69

LISTA DE QUADROS
Quadro 1 - Classificação em grupos de acordo com a evidência de
carcinogenicidade de HPAs em humanos e em animais. ....................27
Quadro 2 - Classificação de alguns HPAs de acordo com grupos estabelecidos
pela IARC, relacionada carcinogenicidade...........................................28
Quadro 3 - Níveis de (B(a)P) reportados na literatura para alguns alimentos........31
Quadro 4 - Níveis máximos de B(a)P em alguns tipos de alimentos de acordo com
o Regulamento (CE) nº 208, de 04 de fevereiro de 2005, da
Comunidade Européia..........................................................................32
Quadro 5 - Padrões analíticos utilizados. ..............................................................46
Quadro 6 - Soluções estoque primárias.................................................................47
Quadro 7 - Soluções estoque secundárias. ...........................................................47
Quadro 8 - Soluções estoque terciárias. ................................................................48
Quadro 9 - Soluções para estabelecer a curva de calibração................................49
Quadro 10 - Íons Selecionados e Tempo de Retenção ...........................................57
Quadro 11 - Condições de análise do extrato de chá verde por GC-MS .................58
Quadro 12 - Dados de área versus concentração dos padrões...............................61
Quadro 13 - Estudo de Limites de Detecção e Quantificação..................................64
Quadro 14 - Limites de Detecção e Quantificação do método CG/MS. ...................64
Quadro 15 - Percentagem de recuperação de B(a)P em 3 níveis de fortificação. ...64
Quadro 16 - Resultados de B(a)P obtidos pelo método CG/MS proposto. ..............69

LISTA DE ABREVIATURAS E SIGLAS
B(a)P Benzo(a)pireno
B(a)PD12 Benzo(a)pireno dodecadeuterado
CCD Cromatografia em Camada Delgada
CG Cromatografia Gasosa
CG/MS Cromatografia Gasosa com Detector de Massas
SIM Monitoramento de Íons Seletivos
DCM Diclorometano (CH2Cl2)
HPAs Hidrocarbonetos Policíclicos Aromáticos
ANVISA Agência Nacional de Vigilância Sanitária
IUPAC International Union of Pure and Applied Chemistry
MAPA Ministério da Agricultura, Pecuária e Abastecimento
IARC International Agency for Research on Câncer
EPA United States Environmental Protection Agency
LD Limite de Detecção
LQ Limite de Quantificação
PI Padrão Interno
ND Não Detectado
mg Miligrama
ng Nanograma
r2 Coeficiente de Correlação
SPE Solid Phase Extration (Extração em Fase Sólida – EFS)
Ai Área do analito na amostra
Api Área do padrão interno na amostra
I Coeficiente linear da curva analítica
Qpi Quantidade de padrão interno adicionado (ng)
S Coeficiente angular da curva analítica
m Massa da amostra
EUA Estados Unidos da América
CTC Cortar, Triturar, Concentrar
OMS Organização Mundial de Saúde
FAO Food and Agriculture Organization

JECFA Joint Expert Commite on Food Additive (Comitê Conjunto de
Peritos em Aditivos Alimentares - CCPAA)
IPCS International Programme on Chemical Safety (Programa
Internacional de Segurança Química – PISQ)
CE Comunidade Européia
ELL Extração Líquido-Líquido
HPLC High Performance Liquid Chromatography (Cromatografia
líquida de alta eficiência – CLAE)
FE Fase Estacionária
FM Fase Móvel
CH Cicloexil
IE Impacto de Elétrons

SUMÁRIO
1 INTRODUÇÃO .............................................................................................14
2 OBJETIVOS .................................................................................................15
2.1 Objetivo geral ..............................................................................................15
2.2 Objetivos específicos .................................................................................15
3 REVISÃO BIBLIOGRÁFICA ........................................................................16
3.1 A história do chá .........................................................................................16
3.2 Os chás ........................................................................................................17
3.2.1 Etapas de processamento do chá ................................................................19
3.2.1.1 Drenagem.....................................................................................................20
3.2.1.2 Vaporização..................................................................................................20
3.2.1.3 Rotação ........................................................................................................20
3.2.1.4 Secagem.......................................................................................................21
3.2.1.5 Triagem.........................................................................................................21
3.3 Hidrocarbonetos Policíclicos Aromáticos ................................................21
3.3.1 Benzo(a)pireno .............................................................................................23
3.3.1.1 Propriedades físico-químicas........................................................................23
3.3.1.2 Aspectos históricos.......................................................................................24
3.3.1.3 Fontes emissoras..........................................................................................25
3.3.1.4 Toxicidade ....................................................................................................26
3.3.1.5 Contaminação em alimentos ........................................................................29
3.4 Legislação vigente ......................................................................................30
3.5 Evolução da metodologia analítica para determ inação de B(a)P em
alimentos, bebidas e águas . ......................................................................32
3.6 Cromatografia .............................................................................................33
3.6.1 Cromatografia em fase gasosa .....................................................................34
3.7 Espectrometria de Massas .........................................................................36
3.7.1 Ionização por impacto de elétrons ................................................................37
3.7.2 Analisador.....................................................................................................38

3.8 Validação de resultados .............................................................................39
3.8.1 Linearidade ...................................................................................................39
3.8.2 Limite de Detecção (LD) ...............................................................................41
3.8.3 Recuperação ................................................................................................42
4 PROCEDIMENTO EXPERIMENTAL ............................................................44
4.1 Amostragem ................................................................................................44
4.2 Materiais ......................................................................................................44
4.3 Equipamentos .............................................................................................45
4.4 Reagentes ....................................................................................................45
4.5 Padrões ........................................................................................................46
4.6 Preparo de Padrões ....................................................................................46
4.6.2 Soluções estoques secundárias ...................................................................47
4.6.3 Soluções estoques terciárias ........................................................................48
4.6.4 Solução de trabalho de padrão interno - B(a)P D12 .....................................48
4.6.6 Soluções padrão para estabelecer a curva de calibração ............................48
4.7 Método de extração ....................................................................................50
4.7.1 Extração do chá seco ...................................................................................51
4.7.2 Extração em fase sólida (SPE- Solid Phase Extration).................................53
4.7.3 Extração da infusão ......................................................................................55
4.7.3.1 Método 1.......................................................................................................55
4.7.3.2 Método 2.......................................................................................................56
4.7.3.3 Extração em fase sólida................................................................................56
4.8 Método de Análise ......................................................................................57
4.8.1 Condições de CG/MS ...................................................................................57
4.9 Quantificação ..............................................................................................58
4.10 Linearidade ..................................................................................................59
4.11 Limite de detecção ......................................................................................59
4.12 Limite de quantificação ..............................................................................59
4.13 Recuperação ...............................................................................................60
5 RESULTADOS E DISCUSSÃO ...................................................................61

5.1 Parâmetros de validação de resultados ...................................................61
5.1.1 Linearidade ...................................................................................................61
5.1.2 Limite de detecção e limite de Quantificação................................................62
5.1.3 Recuperação ................................................................................................64
5.2 Cromatogramas ..........................................................................................65
5.2.1. Cromatograma de Padrão ............................................................................65
5.2.1.1 Extração dos íons monitorados do Padrão ...................................................66
5.2.2 Cromatograma de amostra ...........................................................................67
5.2.2.1 Extração dos íons monitorados de amostra contaminada. ...........................68
5.3 Avaliação de resultados obtidos por extração com solvente .................69
5.4 Avaliação de resultados obtidos da extração d a infusão .......................70
6 CONCLUSÕES ............................................................................................71
REFERÊNCIAS ............................................................................................72

14
1 INTRODUÇÃO
Nas últimas décadas, a contaminação de alimentos por substâncias tóxicas
tem sido objeto de intensas pesquisas (TFOUNE, 2005; BETTIN, FRANCO, 2005).
Dentre essas, destacam-se os estudos relacionados aos Hidrocarbonetos
Policíclicos Aromáticos (HPAs), compostos químicos que têm sido detectados em
alimentos e bebidas (CARUSO, 2007), formados principalmente em processos de
combustão incompleta de matéria orgânica. Encontram-se amplamente distribuídos
na natureza como contaminantes de solos, ar, água e alimentos e esses compostos
são considerados importantes contaminantes devido a seu elevado potencial
genotóxico, mutagênico e carcinogênico, sendo capazes de desencadear doenças
degenerativas no ser humano, dependendo da susceptibilidade genética de cada
indivíduo e freqüência de exposição a esses compostos (CAMARGO, TOLEDO,
2002b; PEREIRA NETO et al, 2000).
Dentre os HPAs carcinogênicos, destaca-se o Benzo(a)Pireno (B(a)P), potente
carcinógeno que tem sido utilizado por diversos pesquisadores como indicador da
presença desses compostos em alimentos. No Brasil, já foram desenvolvidas
metodologias direcionadas à determinação desta substância tóxica em produtos
cárneos, óleos, gorduras e derivados, além de pesquisas de avaliação dos efeitos
tóxicos no organismo humano (CAMARGO, TOLEDO, 2002a).
A contaminação de alimentos e bebidas por B(a)P pode ocorrer por meio de
duas fontes principais: fontes naturais (processos geoquímicos, atividades
vulcânicas e biossíntese por algas) e fontes antropogênicas (queimadas em
florestas; atividades industriais como defumação, secagem direta com madeira e
torrefação e poluição ambiental como tráfego, sistemas de aquecimento,
vazamentos de óleo) (BETTIN, FRANCO, 2005; SIMAL-GÁNDARA et al., 2005;
GODOI et al., 2004; CONDE et al., 2004; EC, 2002; IPCS, 1998).
Com base no exposto, este trabalho visa avaliar a possível contaminação do
ser humano com a ingestão do composto químico Benzo(a)Pireno, através do chá
verde, além de avaliar suas implicações como substância tóxica para o organismo
humano.

15
2 OBJETIVOS
Os objetivos deste trabalho dividem-se em objetivos gerais e específicos.
2.1 Objetivo geral
Determinar a presença de Benzo(a)pireno em amostras de chá verde.
2.2 Objetivos específicos
a) Determinar Benzo(a)pireno em amostras de chá verde, por Cromatografia
Gasosa acoplada a Espectrometria de massa com Impacto de Elétrons
por:
- Extração com diclorometano (solvente);
- Extração com água, por infusão.
b) Comparar os resultados obtidos pelos dois tipos de extração estudados;
c) Discutir o alto potencial toxicológico do benzo(a)pireno à saúde humana.

16
3 REVISÃO BIBLIOGRÁFICA
O levantamento bibliográfico foi realizado para elucidar questões polemicas a
respeito da toxicidade do benzo(a)pireno.
3.1 A história do chá
O chá é a segunda bebida mais consumida no planeta, logo depois da água.
Cada cultura tem seus próprios costumes envolvendo o chá. Os japoneses, por
exemplo, consideram o chá muito importante e criaram a Cerimônia do Chá, ou
Chanoyu, como uma celebração ritualista da bebida. Muitos norte-americanos
bebem chá gelado - e os que moram no sul dos EUA são famosos por consumir
grandes quantidades de chá doce. A "hora do chá" ou o "chá da tarde" é uma
tradição britânica (HOWSTUFFWORKS, 2008).
Figura 1 - O chá
Fonte: (HOWSTUFFWORKS, 2008).
Embora seja considerado, em grande parte, uma bebida social, os estudiosos
reconhecem o chá como uma substância medicinal há milhares de anos. Uma
pesquisa recente parece confirmar essas afirmações, sugerindo que o chá oferece

17
vários benefícios para a saúde, incluindo a redução do risco de câncer ou de doença
cardíaca (KANG, 2010).
Sabemos muito pouco sobre as origens do chá. Os humanos podem ter
consumido chá por dezenas de milhares de anos, mas os registros que fazem
referência ao chá têm apenas cerca de 2 mil anos. Um mito permaneceu, o do
lendário imperador chinês Shen Nung que, segundo dizem, governou há mais de 5
mil anos. Shen Nung era conhecido como um talentoso cientista e artista e, para
reforçar a higiene na China, pediu que todas as pessoas fervessem a água que seria
bebida. Durante uma visita a uma região distante da China, ele parou com sua corte
para descansar. Enquanto seus criados preparavam a água, uma rajada de vento
levou algumas folhas de um arbusto próximo para dentro da água, deixando o
líquido com uma coloração marrom-amarelada. Como era um cientista curioso, Shen
Nung decidiu experimentar a água. Ele bebeu a infusão e achou a bebida
revigorante. Assim, estava criado o chá (HOWSTUFFWORKS, 2008).
3.2 Os chás
Existem quatro tipos principais de chá: o verde, o preto, o de oolong (vermelho)
e o chá branco. A cor do chá verde é variável, sendo que os tons variam entre o
verde e o amarelo, além de o gosto e o aroma da bebida serem bastante
característicos.
O que muitas pessoas não sabem é que esses quatro tipos de chá vêm de uma
única planta, a Camellia sinensis e não de quatro espécies de plantas diferentes.é a
maneira como as folhas de chá são processadas que são originados os diversos
tipos de chás, com seus sabores, cores e aromas específicos.

18
Figura 2 - Campos de chá verde Fonte: (HOWSTUFFWORKS, 2008).
O chá, sob certos aspectos, é parecido com o vinho porque o ambiente em que
ele é cultivado determina a maior parte de seu sabor e qualidade. As plantas
utilizadas para a produção de chá costumam se adaptar melhor em solos ácidos e
em regiões com alto índice de chuvas (cerca de 1 m por ano) (HOWSTUFFWORKS,
2008).
Chás produzidos em grandes quantidades são cultivados em plantações em
mais de 30 países, mas os quatro maiores produtores são a China, a Índia, o Quênia
e o Sri Lanka. A maioria dos chás é colhida à mão para que se possa conseguir a
qualidade necessária, enquanto que o uso de máquinas tende a danificar muito as
folhas. Geralmente são feitas duas colheitas durante o ano, a primeira no início da
primavera e a segunda no verão. Os cultivadores mantêm a planta do chá no estágio
inicial do cultivo com podas constantes, e colhem apenas duas folhas e um botão
dos topos das plantas (HOWSTUFFWORKS, 2008).

19
Figura 3 - A colheita de chá
Fonte: (HOWSTUFFWORKS, 2008).
Assim que os trabalhadores recolhem quantidades suficientes de folhas de chá, o
estoque é levado rapidamente a uma fábrica que deve ficar localizada próxima da
origem das folhas, porque, a partir do momento em que a folha é colhida, a oxidação
já começa. O que distingue o chá verde dos demais é o seu processo de oxidação e
no caso do chá verde, este não é oxidado, o que faz dele o mais claro de todos os
chás – varia entre o verde e o amarelo claro – e aquele que mantém um dos maiores
e mais potentes níveis de antioxidantes, apenas superado pelo chá branco. Colhido
manualmente, são apanhadas sempre duas folhas e um rebento da planta Camellia
sinensis para confeccionar o chá verde. Em vez de serem oxidadas, as folhas são
imediatamente cozidas ao vapor, enroladas e deixadas para secar – só assim é que
se consegue preservar a sua cor verde.
3.2.1 Etapas de processamento do chá
Existem duas maneiras de produzir o chá verde: por meio do método ortodoxo
e pelo método "Cortar, Triturar, Concentrar" (CTC). Os dois possuem cinco etapas
comuns, sendo a principal diferença é que no método ortodoxo o processamento é
feito à mão, ao passo que pelo CTC, são feitos por máquinas.

20
A seguir serão detalhadas as etapas de processamento do chá.
3.2.1.1 Drenagem
Na etapa de drenagem, as folhas de chá são espalhadas em grandes grupos e
drenadas, isso é feito para que as folhas liberem um pouco da umidade retida nas
folhas.
3.2.1.2 Vaporização
Depois que as folhas foram drenadas elas são colocadas em bandejas
quentes, onde sofrem o processo denominado de vaporização. Neste, o processo de
oxidação é interrompido, fazendo com que a folha de chá permaneça em seu estado
atual, mantendo a coloração naturalmente verde.
3.2.1.3 Rotação
No processo de rotação pelo método ortodoxo, as folhas são giradas para que
a umidade restante seja liberada, revestindo a superfície das folhas. Esse método é
particularmente delicado, onde as folhas de chá costumam ficar inteiras. Já pelo
método CTC, ocorre o corte das folhas de chá em pedaços pequenos e o resultado
é um produto com aspecto de pó.

21
3.2.1.4 Secagem
No processo de secagem as folhas são secas com ar quente e as suas cores
podem mudar desde de o tom cobre até o marrom ou preto.
3.2.1.5 Triagem
O processo final de triagem envolve a seleção das folhas de chá de acordo
com o tamanho e a qualidade.
3.3 Hidrocarbonetos Policíclicos Aromáticos
Os Hidrocarbonetos Policíclicos Aromáticos (HPAs) são produzidos por
combustão incompleta ou pirólise da matéria orgânica. A formação pirolítica de
HPAs é bastante complexa e variável, dependendo das condições reacionais. O
esquema mecanístico aceito para esta reação envolve a polimerização via radicais
livres, em várias etapas, até a formação de núcleos aromáticos condensados
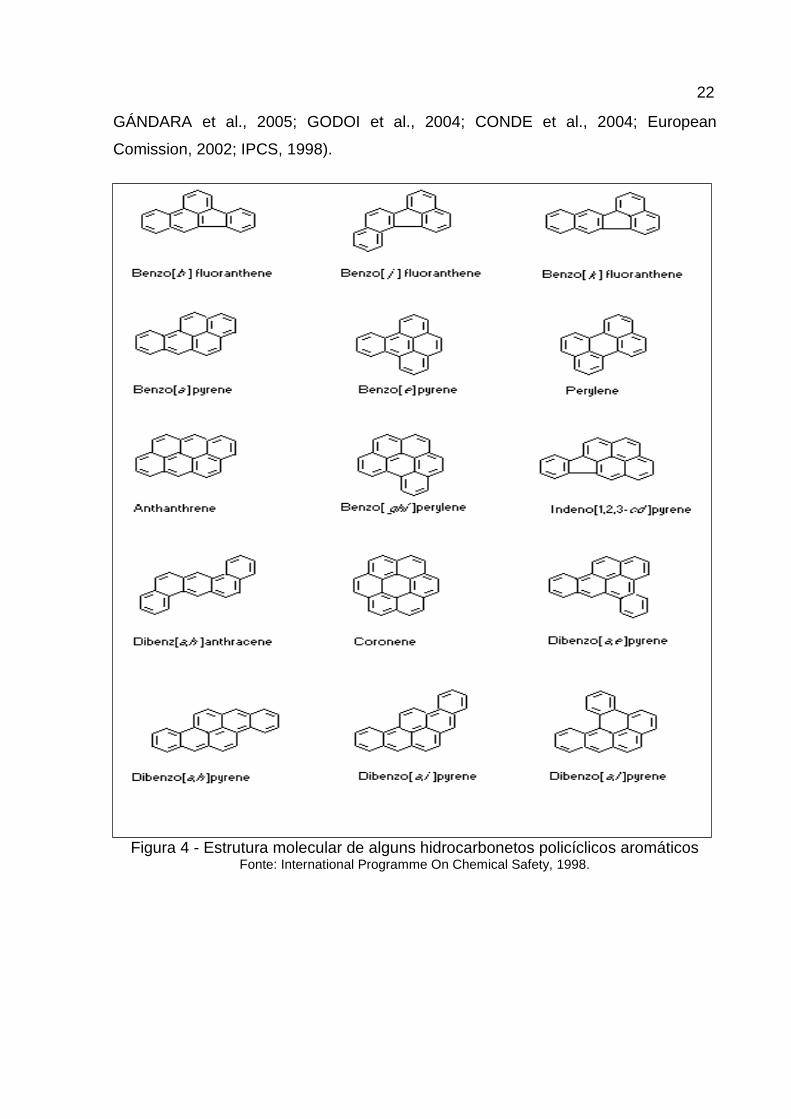
(LOPES, ANDRADE, 1996) (Figura 4). A formação destes compostos depende de
fatores como tipo da biomassa presente, quantidade de oxigênio disponível, pressão
e, principalmente, de calor, pois a concentração de HPAs aumenta linearmente na
faixa de temperatura de 400 a 1000 ºC (CONDE et al., 2004).
Estudos revelam que os HPAs podem ser provenientes de várias fontes
antropogênicas como: queima do carvão, escapamentos de veículos, óleos
lubrificantes usados em motores, fumaça de cigarro, dentre outras, bem como de
fontes ambientais como erupções vulcânicas e queimadas espontâneas. A
contribuição de fontes naturais é muito limitada, contribuindo com pequenas
quantidades de HPAs, enquanto que as fontes antropogênicas representam o
principal processo de emissão destes compostos (BETTIN, FRANCO, 2005; SIMAL-
22
GÁNDARA et al., 2005; GODOI et al., 2004; CONDE et al., 2004; European
Comission, 2002; IPCS, 1998).
Figura 4 - Estrutura molecular de alguns hidrocarbonetos policíclicos aromáticos Fonte: International Programme On Chemical Safety, 1998.

23
3.3.1 Benzo(a)pireno
Dentre os hidrocarbonetos policíclicos aromáticos, o benzo(a)pireno é um dos
mais conhecidos e estudados. Segundo a IUPAC, a grafia recomendada é
benzo[a]pireno, enquanto que o Chemical Abstract adota benzo(a)pireno. São
sinônimas as seguintes formas: benzo(def)criseno; 1,2-benzopireno; 3,4-
benzopireno; 6,7-benzopireno; alfabenzopireno; benzo(alfa)pireno; 3,4-benzpireno;
3,4-benz(a)pireno; B(a)P e B(a)P (IPCS, 1998; IARC, 1983).
Abaixo serão descritas algumas das principais características do composto
Benzo(a)pireno.
Figura 5 - Estrutura molecular do B(a)P Fonte: Chempider, 2010.
3.3.1.1 Propriedades físico-químicas
O Benzo(a)Pireno é um dos HPAs mais conhecidos e estudados. Sua fórmula
molecular é C20H12, apresentando massa molecular de 252,3 g/mol. Possui a
aparência de cristais em forma de agulhas de cor amarelo-pálido; apresenta baixa
volatilidade; pontos de fusão e ebulição de 178,1°C e 310 – 312°C (a 10 mmHg),

24
respectivamente; pressão de vapor a 25°C é de 2,13 x 10-5 Pa e a constante de
Henry a 20°C, 1,86x10 -5. Além disso, esse composto sofre foto-oxidação quando
exposto à luz solar ou radiação fluorescente (CARUSO, 2007).
Como os demais HPAs, o B(a)P é lipossolúvel, apresentando coeficiente de
partição octanol/água (log/Kow = logaritmo da concentração de equilíbrio (K) ou do
coeficiente de partição octanol (o) e água (w=water)) igual a 6,04 e solubilidade em
água a 25°C de 3,8 µg/L. Desta forma, em sistemas aquosos, o B(a)P tende a
concentrar-se em sedimentos ou permanecer associado à matéria orgânica em
suspensão (PEREIRA NETTO et al, 2000).
3.3.1.2 Aspectos históricos
As primeiras provas dos riscos ocupacionais e ambientais dos HPAs foram
obtidas em 1922 de estudos em que extratos orgânicos de fuligem tinham ação
tumoral em animais e, também pela ação neoplásica do extrato de material
particulado. De acordo com Cunha (2007), pode-se considerar como o início da
química dos HPAs o isolamento do B(a)P do carvão, em 1931 e, subseqüentemente,
a sua síntese no mesmo ano. A identificação do B(a)P como uma nova substância
química, em 1933, permitiu identificar como sendo esta o agente cancerígeno em
animais. Em 1970 o B(a)P e outros HPAs foram caracterizados como agentes
cancerígenos, de distribuição mundial, em ambientes respiráveis e como
constituintes de aerossóis urbanos. Neste mesmo ano, foi reconhecido o poder
carcinogênico dos extratos de partículas atmosféricas até então atribuído ao B(a)P,
demonstrando que a atividade cancerígena não era somente devida a esta
substância, mas, também, à presença de outras substâncias orgânicas ainda
desconhecidas e também presentes no material orgânico de fontes de emissão
primárias (COSTA, 2001).
Também nos anos 70 foi introduzido um método muito sensível e eficaz para a
determinação da mutagenicidade de substâncias químicas, por meio de ensaios
envolvendo bactérias do gênero Salmonella que ficariam conhecidos como ensaio
de mutagenicidade ou "Teste de Ames – Salmonella" em homenagem a seus

25
autores (Ames et al., 1975). A partir desta época, muita atenção tem sido dada à
avaliação de HPA em matrizes ambientais e biológicas (COSTA, 2001).
3.3.1.3 Fontes emissoras
Os HPAs são emitidos por fontes naturais e antropogênicas. A contribuição
das fontes naturais é muito limitada restringindo-se, praticamente, à queima
espontânea de florestas e emissões vulcânicas. As fontes antropogênicas
representam o principal processo de produção de HPAs (Lee et al. 1981). A
queima de combustíveis como petróleo e seus derivados, carvão, madeira, gás de
carvão etc; produz HPAs e muitos outros poluentes atmosféricos. A quantidade e
os tipos de HPAs formados dependem das condições específicas do processo e
do tipo de combustível, sendo que processos mais eficientes emitem menores
quantidades de HPAs. A fumaça de cigarro, queimadas e calefação
(especialmente em países de clima temperado) são importantes fontes de HPAs e
derivados (Lopes et al. 1996). Os HPAs são oriundos também de fontes
tecnológicas que podem ser móveis ou estacionárias. Entre as fontes móveis,
destaca-se o motor de combustão interna como o principal emissor destas
substâncias para o ambiente. Este tipo de motor é o mais comum em diversos
veículos de transporte de cargas e passageiros. As fontes estacionárias são
subdivididas entre as utilizadas na geração de energia elétrica e calor e aquelas
ligadas à atividade industrial (produção de alumínio) e de incineração
(principalmente de rejeitos químicos) e podem emitir uma grande variedade de
produtos de combustão incompleta (Netto et al. 1999). As fontes veiculares de
emissão têm uma grande importância devido à complexidade e quantidade, cada
vez maior, de material que é lançado na atmosfera. O material particulado emitido
por veículos a diesel, por exemplo, é constituído principalmente de carbono
elementar que atua como superfície de condensação de HPAs e de outros
compostos aromáticos.

26
3.3.1.4 Toxicidade
O interesse pelo estudo da contaminação por HPAs e seus derivados reside no
fato de que muitos deles são potencialmente carcinogênicos e mutagênicos (IARC,
1983). Os HPAs estão entre aqueles poluentes que apresentam atividade
cancerígena e mutagênica, podendo provocar tumoração em animais e mutação em
bactérias (EC, 2002). A exposição humana aos HPAs pode ocorrer por diferentes
vias, como inalação, pele ou por ingestão. A ação exercida pelos HPAs é ativada
durante o seu processo metabólico, visando à formação de compostos
hidrossolúveis para facilitar a sua excreção. O mecanismo de eliminação envolve a
formação de epóxidos, seguidos de compostos polihidroxilados, os quais são mais
solúveis em água, viabilizando a sua eliminação pela via urinária. Um destes
intermediários pode reagir com a guanina do DNA e formar um aduto dando origem
a processos de tumoração (LOPES, ANDRADE, 1996). Segundo a International
Agency for Research on Câncer (IARC), os HPAs são classificados de acordo com a
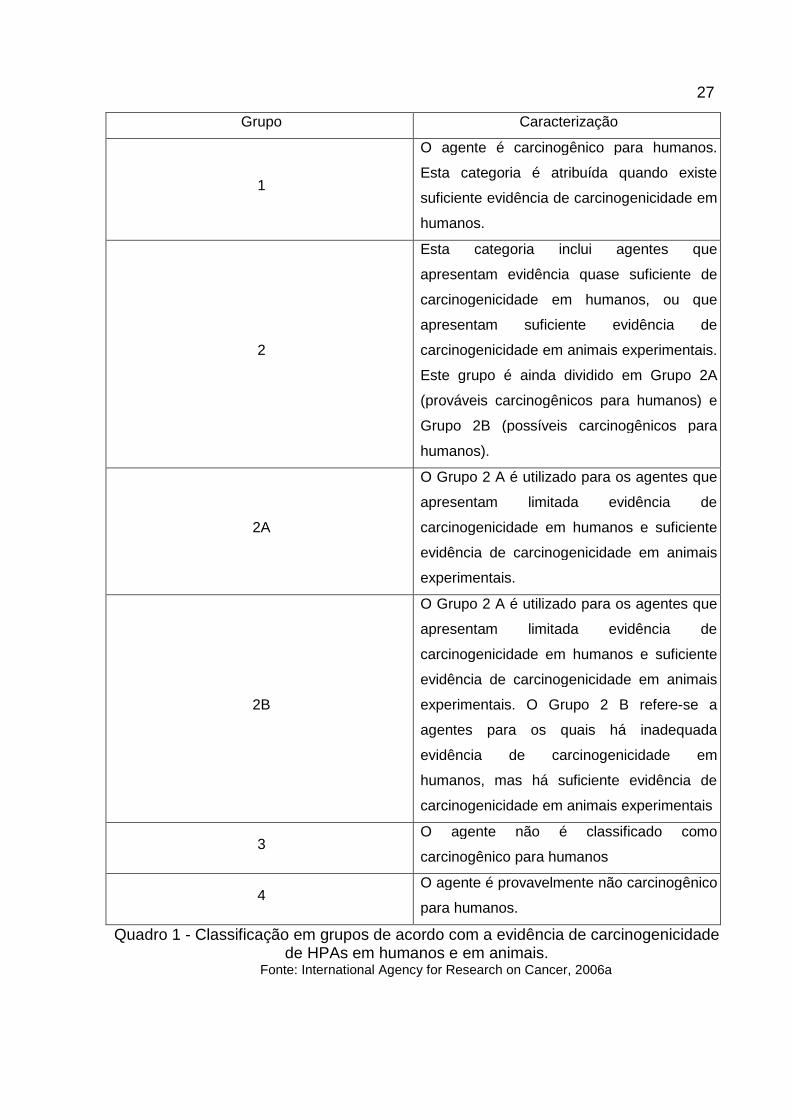
evidência de carcinogenicidade em humanos e em animais experimentais.
Os agentes são divididos nos grupos 1, 2A, 2B, 3 e 4 e estão descritos no
quadro 1.
27
Grupo Caracterização
1
O agente é carcinogênico para humanos.
Esta categoria é atribuída quando existe
suficiente evidência de carcinogenicidade em
humanos.
2
Esta categoria inclui agentes que
apresentam evidência quase suficiente de
carcinogenicidade em humanos, ou que
apresentam suficiente evidência de
carcinogenicidade em animais experimentais.
Este grupo é ainda dividido em Grupo 2A
(prováveis carcinogênicos para humanos) e
Grupo 2B (possíveis carcinogênicos para
humanos).
2A
O Grupo 2 A é utilizado para os agentes que
apresentam limitada evidência de
carcinogenicidade em humanos e suficiente
evidência de carcinogenicidade em animais
experimentais.
2B
O Grupo 2 A é utilizado para os agentes que
apresentam limitada evidência de
carcinogenicidade em humanos e suficiente
evidência de carcinogenicidade em animais
experimentais. O Grupo 2 B refere-se a
agentes para os quais há inadequada
evidência de carcinogenicidade em
humanos, mas há suficiente evidência de
carcinogenicidade em animais experimentais
3 O agente não é classificado como
carcinogênico para humanos
4 O agente é provavelmente não carcinogênico
para humanos.
Quadro 1 - Classificação em grupos de acordo com a evidência de carcinogenicidade de HPAs em humanos e em animais.
Fonte: International Agency for Research on Cancer, 2006a

28
O quadro 2 apresenta a classificação de alguns HPAs de acordo com a
evidência de carcinogenicidade
HPA Classificação
Antraceno Grupo 3
Benzo(a)antraceno Grupo 2B
Benzo(b)fluoranteno Grupo 2B
Benzo(j)fluoranteno Grupo 2B
Benzo(k)fluoranteno Grupo 2B
Benzo(g,h,i)fluoranteno Grupo 3
Benzo(c)fenantreno Grupo 2B
Benzo(a)pireno Grupo 1
Benzo(e)pireno Grupo 3
Criseno Grupo 2B
Coronono Grupo 3
Dibenzo(a,c)antraceno Grupo 3
Dibenzo(a,h)antraceno Grupo 2A
Dibenzo(a,j)antraceno Grupo 3
Fluoranteno Grupo 3
Fluoreno Grupo 3
Indeno 1,23-cd-pireno Grupo 2B
Naftaleno Grupo 3
Pireno Grupo 3 Quadro 2 - Classificação de alguns HPAs de acordo com grupos estabelecidos pela
IARC, relacionada carcinogenicidade Fonte: International Agency for Research on Cancer, 2006b.
De acordo com a proposta do trabalho, que é a contaminação de
benzo(a)pireno em chá verde, no próximo item será abordada a somente a
contaminação de benzo(a)pireno em alimentos.

29
3.3.1.5 Contaminação em alimentos
Existem muitos estudos sobre a ocorrência de B(a)P em diversos tipos de
alimentos, incluindo óleos vegetais, margarinas, maionese, produtos lácteos, frutas,
vegetais, produtos defumados, chás, café, cereais, água, alimentos de origem
marinha, alimentos grelhados entre outros (TFOUNI, 2005).
No caso dos óleos vegetais, a presença desses compostos é atribuída à etapa
de secagem dos grãos, sendo a secagem à lenha a técnica que mais contribui para
a formação dos compostos e sua presença no produto final. Embora a
desodorização reduza os teores totais de HPAs, seu efeito não é substancial, sendo
necessário o controle do óleo bruto ou a utilização de carvão ativo para remoção
dessas substâncias (TOLEDO, CAMARGO, 1998).
A ocorrência de B(a)P em frutas e vegetais se deve principalmente à poluição
ambiental. Neste caso, o material particulado se deposita na superfície destes, onde
são concentrados pela camada de cera através de adsorção superficial (CAMARGO,
TOLEDO, 2002c). O nível de contaminação depende da localização da plantação e
da área de superfície do alimento exposta à contaminação. As plantações situadas
próximas a rodovias dão origem a produtos com maiores teores de B(a)P
(CAMARGO, TOLEDO, 2002a).
A contaminação de cereais se dá de maneira análoga à de frutas e vegetais,
sendo a área de cultivo um fator muito importante. Outra via de contaminação dos
grãos é a secagem dos mesmos pela aplicação de gases de combustão, com os
teores de B(a)P variando em função do tipo de combustível utilizado e das condições
de sua queima (CAMARGO, TOLEDO, 2002b).
A contaminação do café ocorre no processo de torrefação dos grãos. Quando
se prepara a bebida no modo tradicional (café coado em processo direto), 20 a 30%
dos HPAs totais presentes no pó de café são extraídos para a bebida; entretanto,
quando o pó de café é fervido juntamente com a água antes da filtração, forma-se
um complexo HPA-cafeína que facilita a passagem dos hidrocarbonetos para a
bebida, resultando em um café com maiores níveis de HPAs (CAMARGO, TOLEDO,
2002b).
O chá geralmente apresenta níveis superiores de B(a)P em relação ao café
torrado. A contaminação do chá se dá devido à poluição ambiental aliada à secagem

30
direta das folhas de chá (CAMARGO, TOLEDO, 2002b; CAMARGO, TOLEDO,
2002c).
Em produtos defumados os B(a)P são formados pela queima da madeira
utilizada no processo. A maior concentração de B(a)P nesses produtos se dá
imediatamente após a defumação, quando a sua concentração na fumaça diminui
devido à decomposição na presença de luz e à interação com outros compostos.
Estes podem penetrar no alimento, onde ficam protegidos da luz e do oxigênio;
depois de algum tempo, a concentração se estabiliza em um valor constante
(SIMKO, 2002). Segundo Azeredo (2001), o controle de alguns parâmetros
importantes como temperatura, distância da fonte geradora e concentração de
fumaça pode contribuir para a diminuição da contaminação de produtos defumados
por B(a)P. Nos processos de defumação industriais mais modernos, a fumaça é
gerada numa câmara separada e tratada antes de passar à câmara de defumação.
Passagem por filtros eletrostáticos e “lavagem” da fumaça (formação de fumaça
líquida) são algumas das técnicas utilizadas para diminuir a contaminação da
fumaça por B(a)P.
Quando alimentos são preparados em contato com a chama, como acontece
com as carnes de churrasco, a gordura da carne em contato com o fogo, sofre
pirólise e então retorna à carne na forma de fumaça, carregando os HPAs que
contaminam a carne. Quanto maior o teor de gordura da carne, maior a quantidade
de HPAs presente no produto, conseqüentemente maior teor de B(a)P (PHILLIPS,
1999; CAMARGO, TOLEDO, 2002c).
3.4 Legislação vigente
De acordo com Caruso (2007), vários estudos vêm sendo realizados com o
objetivo de avaliar quais são os alimentos ou grupo de alimentos que mais
contribuem na ingestão diária de B(a)P. Estes estudos revelaram que a maioria dos
alimentos citados no item 3.3.1.5 apresentam partículas deste composto. Além
disso, tem sido verificado que as fontes de exposição variam de acordo com o país e
o respectivo hábito alimentar.

31
A Quadro 3 apresenta a faixa dos níveis de Benzo(a)Pireno encontrados em alguns
tipos de alimentos:
Alimentos B(a)P(µg/Kg) Alimentos defumados nd* – 66,90
Alface 0,1 – 31,7 Óleo de oliva nd – 164,4 Óleo de soja nd – 42,90
Óleo de girassol nd – 8,0 Óleo de milho 1,51 – 58,9
Peixes e frutos do mar <0,005 – 4,54 * nd – não detectado
Quadro 3 - Níveis de (B(a)P) reportados na literatura para alguns alimentos Fonte: Camargo, Toledo, 2002c.
A Portaria N° 518/2004 vigente estabelece limite má ximo de B(a)P apenas para
a potabilidade de água (0,7 µg/L) (BRASIL, 2004) e para aroma de fumaça (0,03
µg/kg de produto final) (BRASIL, 2007). Somente alguns países possuem limites
estabelecidos para um número restrito de alimentos (SIMKO, 2002): na Alemanha,
Áustria e Polônia, o teor máximo permitido de B(a)P em carnes defumadas é 1µg/Kg
e este valor tem sido usado como limite de referencia para avaliação do grau de
contaminação de outros alimentos. Para óleos e gorduras, as indústrias alemãs têm
suas próprias recomendações quanto aos limites. Por exemplo, a soma dos resíduos
dos HPAs leves não deve exceder a 25 µg/Kg, enquanto que a soma dos HPAs
pesados, que inclui o B(a)P, deve permanecer abaixo de 5 µg/Kg (CAMARGO,
TOLEDO, 1998).
De acordo com decisão do Comitê Científico da Alimentação Humana, da
Comunidade Européia (CE), os níveis de HPAs e de B(a)P nos gêneros alimentícios
devem ser reduzidos a concentrações tão baixas quanto possível. Desta forma,
através do Regulamento (CE) nº 208, de 04 de fevereiro de 2005, este Comitê
determinou que se utilizasse o Benzo(a)Pireno como marcador relativo à ocorrência
de outros HPAs cancerígenos e determinou limites máximos para este contaminante
para alguns tipos de alimentos. Tais valores estão apresentados no Quadro 3.

32
Produto alimentício B(a)P (µg/Kg de peso fresco)
Óleos e gorduras 2,0
Alimentos para lactentes e crianças 1,0 Carnes defumadas e produtos defumados à base
de carnes 5,0
Partes comestíveis de peixes defumados 5,0
Partes comestíveis de peixes 2,0
Moluscos bivalves 10,0
Crustáceos e cefalópodes 5,0 Quadro 4 - Níveis máximos de B(a)P em alguns tipos de alimentos de acordo com o
Regulamento (CE) nº 208, de 04 de fevereiro de 2005, da Comunidade Européia Fonte: Caruso, 2007.
3.5 Evolução da metodologia analítica para determin ação de B(a)P em
alimentos, bebidas e águas.
Na década de 60, a metodologia utilizada para análise de HPAs em alimentos
envolvia Extração Líquido-Líquido (ELL), purificação em coluna cromatográfica
clássica, separação em cromatografia em papel e em camada delgada e
quantificação por espectrofotometria na região do ultravioleta. Eram empregados
grandes volumes de solventes (em torno de 3000 mL), como etanol, dimetilsulfóxido,
isooctano, e benzeno, sendo este último considerado cancerígeno (HOWARD et al.,
1965, 1966 abc; 1968; MANOSKI et al., 1968; THE MERCK INDEX, 1989). A partir
de 1975, Grimmer e Böhnke, propuseram uma metodologia que vem sendo bastante
utilizada por diversos pesquisadores. Envolve ELL com cicloexano, com
dimetilformamida/água e novamente com cicloexano. A etapa de purificação é feita
em coluna clássica. Neste método, ainda eram gastos grandes volumes de
solventes, acima de 2500 mL, aproximadamente. Esta metodologia, no entanto, vem
sofrendo várias modificações desde a sua publicação, sendo que atualmente o
volume de solvente empregado é quase 1/6 desse total (TFOUNI et al., 2007;
FALCÓ et al., 2003; CAMARGO, TOLEDO, 2002ab; CAMARGO, TOLEDO, 2001,
2000, 1998; CAMARGO, TOLEDO, 1998). Por outro lado, alguns pesquisadores têm
adotado o aparelho de ultrassom para extração de HPAs, obtendo resultados
satisfatórios com simplicidade, menor tempo de análise e pequenos volumes de
solvente. De um modo geral, os solventes mais utilizados para este fim são

33
acetona/n-hexano (JÁNSKÁ et al., 2006; BANJOO, NELSON, 2005), diclorometano
(LIN et al., 2005), diclorometano/acetona (LIN, ZHU, 2004), éter etílico/cloreto de
metileno (NIEVA-CANO et al., 2001), clorofórmio (CEJPEK et al., 1995), e
cicloexano (JOE et al., 1982).
Nos últimos anos, observa-se um crescente número de trabalhos em que a
extração em fase sólida vem sendo utilizada para extração ou purificação dos
extratos de HPAs. Isto ocorre pela praticidade, custos relativamente pequenos,
volume de solventes e tempo reduzidos, bem como pelos excelentes resultados de
recuperação e repetitividade que este método proporciona (CARUSO, 2007).
A seguir é abordada a técnica de cromatografia, já que foi esta a técnica
utilizada para elaboração da pesquisa.
3.6 Cromatografia
A cromatografia faz parte de um importante grupo de métodos de separação,
que nos permite separar, isolar, identificar e quantificar substâncias, mesmo em
misturas muito complexas.
Existem várias técnicas cromatográficas, que vão das mais simples, como a
cromatografia em papel, até as mais sofisticadas, como a Cromatografia Líquida de
Alta Eficiência (HPLC). O ponto comum entre essas técnicas, e que caracteriza o
método, é o fato dos componentes da mistura que compõem uma amostra, serem
distribuídos entre duas fases. Uma delas permanece fixa, e por isso é chamada de
fase estacionária (F.E.), enquanto a outra percola através da F.E., sendo então
chamada de fase móvel (F.M.). Esta situação dinâmica resulta em uma migração
diferenciada, ou seja, os componentes da amostra têm diferentes velocidades ao
passarem pela fase estacionária. (SKOOG et al., 2001).
A fase estacionária, de forma geral, é acondicionada nas chamadas colunas
cromatográficas, que na sua maioria são tubos de vidro ou metal de dimensões
diversas. Quando essa fase é um sólido, basta que a coluna seja preenchida com o
mesmo, de acordo com técnicas especiais. Por outro lado, quando a fase
estacionária é um líquido, esse pode tanto revestir as paredes da coluna, no caso de

34
colunas capilares, quanto estar aderido a um suporte sólido com o qual se preenche
a coluna.
A amostra é introduzida na coluna e a fase móvel carreia os diversos
componentes dessa amostra através da coluna e, dependendo do tipo de
cromatografia, pode participar ou não da separação. Assim, a separação dos
diversos componentes será dada em função de uma maior ou menor afinidade de
cada um deles, por cada uma das fases. Logo, o componente com maior afinidade
pela fase estacionária fica mais tempo retido na coluna, enquanto aquele que tiver
mais afinidade pela fase móvel percorrerá a coluna com maior rapidez. Outras
características importantes na separação serão o tamanho da molécula, sua massa
e, no caso da cromatografia em fase gasosa, sua pressão de vapor (ou ponto de
ebulição) (SKOOG et al., 2001).
3.6.1 Cromatografia em fase gasosa
A cromatografia gasosa veio possibilitar, de maneira rápida e eficiente, uma
série de separações extremamente difíceis, ou mesmo impossíveis pelos métodos
convencionais, como por exemplo, a separação da mistura benzeno-cicloexano,
impossível de ser feita por destilação fracionada, ou ainda, a análise de gases
provenientes de automóveis, contendo mais de cem componentes, que podem ser
separados por essa técnica. Os princípios do método cromatográfico são os mesmos
para todas as formas de cromatografia, ou seja, é um processo no qual uma mistura
de substâncias pode ser separada nos seus constituintes graças à passagem de
uma fase móvel gasosa, que carreia a mistura, sobre uma fase estacionária.
Como o nome denota, a Cromatografia Gasosa (CG) é aquela em que a fase
móvel é um gás , podendo a fase estacionária ser um sólido ou um líquido.
A função do gás usado como fase móvel é apenas a de carrear os
componentes da amostra através da coluna, sem participar dos processos de
interação. Por este motivo é chamado gás de arraste. Exemplos de gases mais
utilizados em CG são o hélio (He), hidrogênio (H2) e o nitrogênio (N2 ).
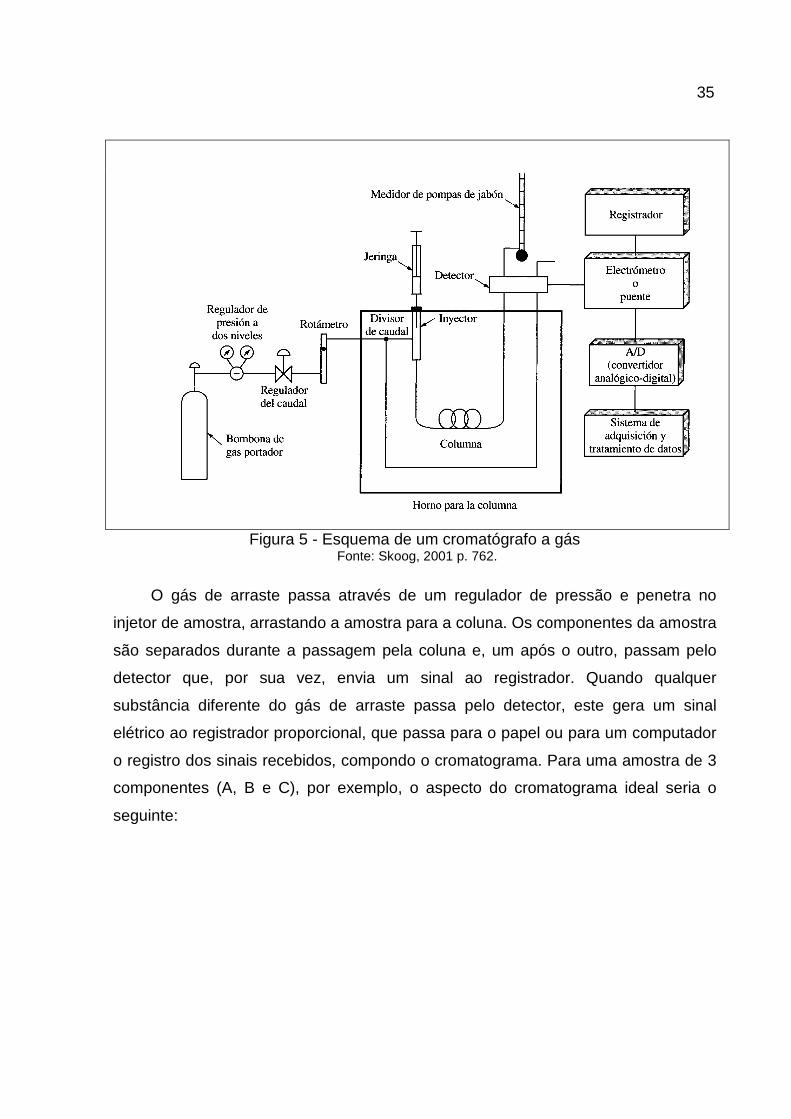
Na figura abaixo se observam os principais componentes de um Cromatógrafo
a Gás.
35
Figura 5 - Esquema de um cromatógrafo a gás Fonte: Skoog, 2001 p. 762.
O gás de arraste passa através de um regulador de pressão e penetra no
injetor de amostra, arrastando a amostra para a coluna. Os componentes da amostra
são separados durante a passagem pela coluna e, um após o outro, passam pelo
detector que, por sua vez, envia um sinal ao registrador. Quando qualquer
substância diferente do gás de arraste passa pelo detector, este gera um sinal
elétrico ao registrador proporcional, que passa para o papel ou para um computador
o registro dos sinais recebidos, compondo o cromatograma. Para uma amostra de 3
componentes (A, B e C), por exemplo, o aspecto do cromatograma ideal seria o
seguinte:

36
Figura 6 - Cromatograma ideal para uma amostra contendo três substâncias Fonte: Adaptado de Skoog, 2006
Atualmente, existem muitos detectores que podem ser acoplados tanto a
cromatografia gasosa, quanto à cromatografia líquida. No item a seguir, será
abordado o detector Espectrometria de Massa (EM), o qual foi utilizado para
elaboração deste trabalho.
3.7 Espectrometria de Massas
A espectrometria de massas é uma técnica de detecção que utiliza o
movimento de íons em campo elétrico e magnético para classificá-los de acordo com
sua relação massa/carga. Desta maneira, a espectrometria de massas é uma
técnica analítica por meio da qual as substancias químicas são identificadas e/ou
quantificadas, separando os íons gasosos através do uso de campos elétricos e
magnéticos. O dispositivo que realiza esta operação e utiliza meios elétricos para
detectar os íons classificados é conhecido como espectrômetro de massas.
Um espectrômetro de massa é composto por sistema de vácuo, fonte de íons,
analisador de massas, detector de íons e sistema de processamento de dados,
assim como é demonstrado na figura 7.

37
Figura 7 - Esquema de componentes de um sistema de cromatografia gasosa com espectrometria de massas (GC/MS)
Fonte: Adaptado de Skoog, 2001, p. 272.
Na fonte de íons os componentes de uma amostra são convertidos em íons
positivos ou negativos por bombardeamento das moléculas vaporizadas da amostra
e são imediatamente acelerados em direção ao analisador de massa. Existem várias
técnicas de ionização, sendo a de ionização química e ionização por impacto de
elétrons (IE) as mais conhecidas. A seguir será detalhada de uma melhor forma a
técnica de ionização por impacto de elétrons, sendo que esta foi à utilizada para a
construção deste trabalho.
3.7.1 Ionização por impacto de elétrons
Na técnica de impacto de elétrons, comumente a mais utilizada, por ter maior
sensibilidade e maior abrangência, um espectrômetro de massas bombardeia

38
moléculas na fase vapor com feixe de elétrons de alta energia e registra o resultado
do impacto dos elétrons como um espectro de íons separados na base da razão
massa/carga (m/z).
Um analisador separa tais íons de acordo com a sua relação massa/carga
(m/z), que geralmente é selecionada previamente de acordo com as massas de
interesse para o analito em questão e então ocorre o registro dos sinais gerados
num software, no qual é possível verificar o cromatograma gerado e após investigar
as massas e áreas geradas, podendo ser calculado através de uma curva de
calibração a concentração obtida na amostra.
3.7.2 Analisador
Um analisador bastante utilizado é o quadrupolo, porque este instrumento é
considerado o mais barato, compacto e robusto espectrômetro de massas do
mercado sendo considerado um filtro de massas de alta velocidade de varredura
(SKOOG et al., 2001).
A Figura 8 mostra um esquema do analisador de massas quadrupolar e abaixo
o sistema de seleção de massas, onde no eixo x são transmitidas razões massa-
carga inferior a um determinado valor e no eixo y superior. Isto ocorre de forma que
apenas uma unidade de massa seja estável em sua trajetória até o detector.
Figura 8 - Esquema representativo de um analisador tipo quadrupolo Fonte: Adaptado de Skoog, 2006, p. 825.
Rolos do quadrupolo Detector
Íons que serão detectados
Íons descarragados

39
Um campo elétrico é formado por quatro barras paralelas as quais é aplicada
uma corrente contínua que afeta o percurso dos íons. Para determinadas voltagens,
somente os íons de uma relação massa/carga específica podem passar através do
filtro do quadrupolo, enquanto os outros são varridos como moléculas
descarregadas.
Finalmente um detector recebe os íons que foram separados pelo analisador,
transformando a corrente de íons em sinais elétricos que são processados.
A seguir são descritos alguns parâmetros utilizados na validação de
resultados.
3.8 Validação de resultados
A necessidade de se mostrar a qualidade de medições químicas está sendo
cada vez mais reconhecida e exigida. Dados analíticos não confiáveis podem
conduzir a decisões desastrosas e a prejuízos financeiros irreparáveis. Para garantir
que um novo método analítico gere informações confiáveis ele deve sofrer uma
avaliação denominada validação (RIBANI et al., 2004).
É fundamental que os laboratórios disponham de meios e critérios objetivos
para demonstrar, através da validação, que os métodos de ensaio que executam
conduzem para resultados confiáveis e adequados à qualidade pretendida
(INMETRO, 2003).
3.8.1 Linearidade
A linearidade é a habilidade do método em gerar resultados diretamente
proporcionais à concentração do analito dentro da faixa de trabalho a qual
corresponde às concentrações máximas e mínimas do analito que podem ser
quantificadas com exatidão e precisão (RIBANI et al., 2004; SHABIR, 2003; LEITE,
2000). Na maior parte dos casos, a relação matemática entre o sinal e a

40
concentração da espécie de interesse deve ser determinada empiricamente, a partir
de sinais medidos para diversas concentrações conhecidas dessa espécie (LEITE,
2000)
Essa relação matemática, muitas vezes, pode ser expressa através de uma
equação de reta chamada de curva analítica. A equação da reta que relaciona as
variáveis dependentes (y) e independentes (x) é:
Onde:
y: variável dependente;
x: variável independente;
a: coeficiente angular (expressa a inclinação da curva de calibração);
b: coeficiente linear (expressa a interseção da curva com o eixo y).
Matematicamente, a estimativa dos coeficientes de uma curva analítica a
partir de um conjunto de medições experimentais pode ser efetuada usando o
método matemático. Um dos mais conhecidos é o de regressão linear, também
chamado de método dos mínimos quadrados, utilizado para estimar qual a melhor
reta que representa os pontos da curva analítica. Além dos coeficientes de
regressão a e b, também é possível calcular, a partir dos pontos experimentais, o
coeficiente de correlação r, que expressa à relação de x e y na curva. Este
parâmetro permite uma estimativa da qualidade da curva obtida, pois quanto mais
próximo de 1,0, menor a dispersão do conjunto de pontos experimentais e menor a
incerteza dos coeficientes de regressão estimados. Para verificar se a equação de
regressão é estatisticamente significativa podem ser efetuados os testes de ajuste
do modelo linear, validade da regressão. Um coeficiente de correlação maior que
0,990 é considerado como evidência de um ajuste ideal dos dados para a linha de
regressão.
y = ax + b
... (1)

41
3.8.2 Limite de Detecção (LD)
O limite de detecção (LD) de uma metodologia analítica é definido como a
menor concentração de um analito que pode ser detectada em uma dada amostra,
porém, não quantificada dentro dos limites de precisão e exatidão estabelecidos
(SHABIR, 2003).É geralmente determinado através de soluções padrões muito
diluídas, como uma comparação da relação sinal/ruído, ou seja, compara-se o sinal
obtido das soluções com aqueles obtidos de brancos ou ruídos do sistema,
estabelecendo assim o nível do ruído e o nível mínimo de sinal realmente detectável,
sendo geralmente aceitas razões sinal/ruído de 2:1 a 3:1.
Dentre os métodos utilizados para calcular o limite de detecção, estão os
procedimentos recomendados pela Eurachem, que é uma rede de organizações
nacionais européias que juntamente com a Comissão Europeia tem por objetivo
estabelecer um sistema para a rastreabilidade internacional dos resultados de
medições químicas e promover as boas práticas laboratoriais. Os guias da
Eurachem são traduzidos no Brasil pela Anvisa recomendam que o LD seja
estabelecido efetuando-se a leitura ou injeção de amostras em branco, ou brancos
fortificados na menor concentração aceitável, isto é, com um grau de incerteza
aceitável. Em geral o limite de detecção é calculado através da média dos valores
encontrados mais três desvios padrões, sendo também necessário estabelecer a
média e o desvio padrão, conforme fórmulas abaixo:
Média: X = ∑ x i
n
n
Desvio Padrão: S = ∑ (xi - x)
n - 1
i
n
2
... (2)
... (3)

42
Onde:
X = média;
xi = resultado individual;
S = desvio padrão;
n = número de dado
3.8.3 Limite de Quantificação (LQ)
O limite de quantificação (LQ) é o menor valor determinado, em confiabilidade
de precisão e exatidão aceitáveis, para uma determinada condição analítica.
Determina-se reduzindo a concentração do analito numa amostra até que seja
encontrado um valor mínimo a partir do qual a precisão e exatidão do método já não
são mais aceitáveis.
É normalmente expresso na mesma unidade das amostras, e pode também
ser relatado como a concentração cuja relação sinal/ruído=10, através da análise do
ruído do sistema. Para calcular o limite de quantificação a seguinte fórmula deve ser
utilizada.
3.8.3 Recuperação
A recuperação é definida com uma proporção da quantidade da substância de
interesse, presente ou adicionada na porção analítica do material teste, que é
extraída e passível de ser quantificada. A informação de recuperação pode ser
estimada através da fortificação de uma quantidade conhecida de um padrão de
LD = 3 X S ... (4)
LQ = 10 X S ... (5)

43
referência, ou de um composto substituto (RIBANI et al., 2004).
Conforme a ANVISA, pode ser calculada através da fortificação de uma
amostra em três níveis diferentes, como por exemplo:
1º nível - ponto médio da faixa de trabalho menos 50% (concentração baixa)
2º nível - ponto médio da faixa de trabalho (concentração média)
3º nível - ponto médio da faixa de trabalho mais 50% (concentração alta)
As determinações devem ser feitas usando o mesmo procedimento de
quantificação especificado no método final, mas normalmente emprega-se um
mínimo de seis replicatas de cada concentração. Os valores médios aceitos, em
geral, dependem da faixa de concentração estudada, utilizando-se em muitos casos
valores de recuperações aceitáveis de 80 – 120%.
A recuperação pode ser calculada através das fórmulas:
Onde:
Cfort. = concentração do analito na amostra fortificada;
Cref = concentração do analito na amostra não fortificada;
Cadic = concentração do analito adicionada à amostra;
A seguir é descrito o procedimento experimental, no qual é relatado como o
processo de análise foi realizado.
% Rec = Cfort – C ref
C adic X 100
... (6)
% Erro = 100 - %Rec ... (7)

44
4 PROCEDIMENTO EXPERIMENTAL
O procedimento experimental utilizado para a análise esta descrito abaixo.
4.1 Amostragem
As amostras de chá verde foram adquiridas no período de abril de 2009, no
comércio de Porto Alegre/RS. Foram selecionadas sete amostras de chá verde de
sache, de marcas diferentes, de forma aleatória, que posteriormente foram
submetidas à análise para a identificação do contaminante Benzo(a)pireno.
As amostras foram codificadas como 1, 2, 3, 4, 5, 6 e 7.
4.2 Materiais
a) Frasco de 50 mL com tampa;
b) Balões volumétricos calibrados de 10, 50 e 250 mL;
c) Micropipetadores, Transferpette Dig, Brand – volume fixo: 250, 500 e 1000
µL e volume variável de 10 a 100 µL e 1,0 a 10,0 mL
d) Funil de separação de vidro, volume de 250 mL, com tampa e torneira de
Teflon;
e) Balão de fundo redondo 50 mL, Pyrex;
f) Microseringas de 10 µL, Hamilton;
g) Papel de filtro qualitativo;
h) Pipeta Pasteur descartável;
i) Funil;
j) Tubo de ensaio;
k) Dispensador para frasco Dispensette de 50 mL;
l) Frasco (vial) com tampa para o amostrador automático.

45
4.3 Equipamentos
a) Sistema cromatográfico, Agilent, composto de cromatógrafo a gás (GC
6890), injetor automático modelo 7683, detector seletivo de espectrometria
de massas (5973N), com acessório para ionização por impacto de elétrons,
software (MSD Chemstation G 1701CA version D.03.00SP1);
b) Balança analítica com precisão de 0,1mg, Metler AG204;
c) Banho de ultra-som Chubby Thornton;
d) Chapa aquecedora Eletrolab;
e) Agitador orbital New Brunswick Scientific;
f) Misturador (mixer) Termolyne;
g) Sistema para extração em fase sólida Merck Lichrolut;
h) Coluna capilar de sílica fundida 5% fenil metilpolisiloxano (DB-5), 30 m,
0,32 mm, 0,25 µm (J&W Scientific 12-5032);
i) Rota evaporador Yamato RE540.
4.4 Reagentes
a) Diclorometano (CH2Cl2 ) - grau HPLC, marca TÉDIA;
b) Cicloexano (C6H12) - grau HPLC, marca TÉDIA;
c) Pentano (C5 H12) - grau HPLC, marca TÉDIA;
d) Metanol (CH3OH) - grau HPLC, marca TÉDIA;
e) Água deionizada qualidade American Society for Testing and Materials
(ASTM - Sociedade Americana para Ensaios e Materiais) grau 1;
f) Hydromatrix® (terra diatomácea) Chem Elut Varian 198004;
g) Cartucho contendo 1,0g de sílica gel e capacidade de 6,0mL, Applied
Separations SPE 2107;
h) Cartucho contendo 1,0g de ciclohexil e capacidade de 6,0mL, Applied
Separations SPE 2077.

46
4.5 Padrões
No quadro 5, estão descritas as especificações dos padrões utilizados.
Padrão Fórmula Molecular Pureza (%) Marca
Benzo(a)pireno -B(a)P C20H12 98 Dr. Enrenstorfer
Benzo(a)pireno
dodecadeuterado -
B(a)P D12
C20D12
98
Dr. Enrenstorfer
Quadro 5 – Padrões analíticos utilizados. Fonte: Autoria própria, 2010
4.6 Preparo de Padrões
A seguir estão descritas as etapas necessárias para a realização do preparo
das soluções padrão e curva de calibração. Foi utilizado padrão analítico com pureza
mínima de 98% de pureza, de benzo(a)pireno (B(a)P) e também de Benzo(a)pireno
dodecadeuterado (B(a)P D12), que é utilizado como padrão interno do processo.
4.6.1 Soluções estoques primárias
a) Benzo(a)pireno (1,0 mg/mL)
Foi pesado aproximadamente 50 mg de benzo(a)pireno em balança
analítica, transferido para balão volumétrico de 50 mL e elevado ao volume
final com cicloexano.
47
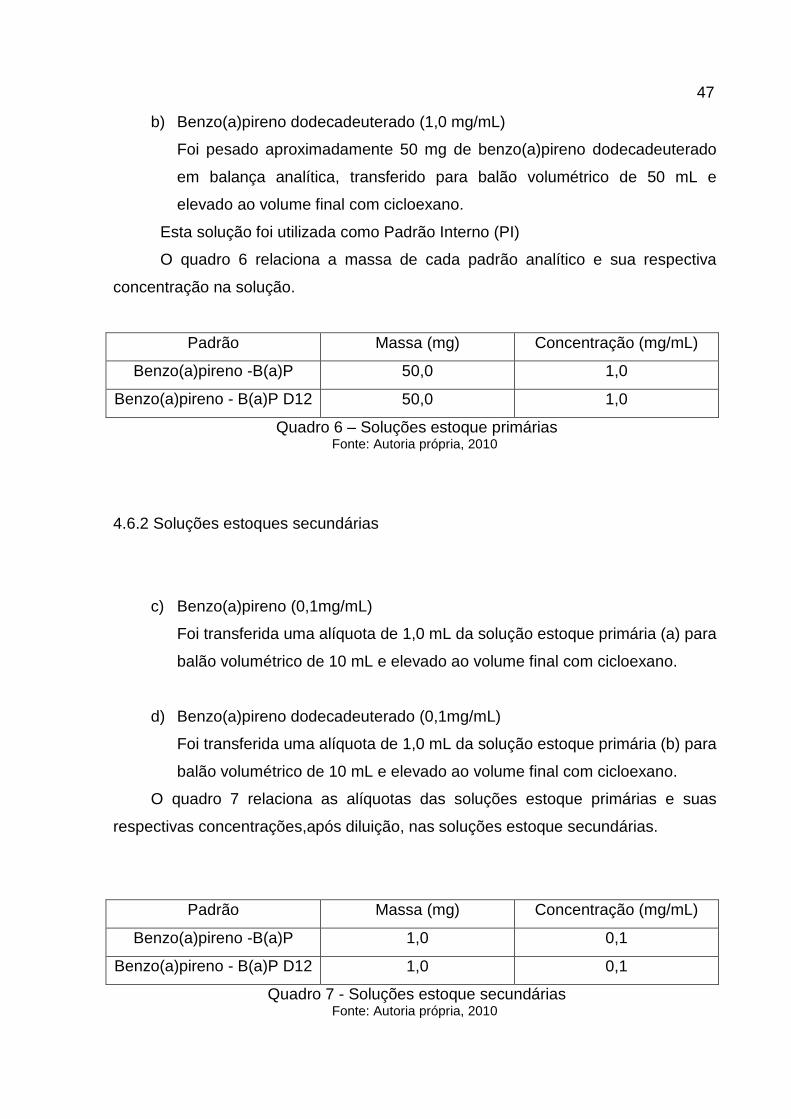
b) Benzo(a)pireno dodecadeuterado (1,0 mg/mL)
Foi pesado aproximadamente 50 mg de benzo(a)pireno dodecadeuterado
em balança analítica, transferido para balão volumétrico de 50 mL e
elevado ao volume final com cicloexano.
Esta solução foi utilizada como Padrão Interno (PI)
O quadro 6 relaciona a massa de cada padrão analítico e sua respectiva
concentração na solução.
Padrão Massa (mg) Concentração (mg/mL)
Benzo(a)pireno -B(a)P 50,0 1,0
Benzo(a)pireno - B(a)P D12 50,0 1,0
Quadro 6 – Soluções estoque primárias Fonte: Autoria própria, 2010
4.6.2 Soluções estoques secundárias
c) Benzo(a)pireno (0,1mg/mL)
Foi transferida uma alíquota de 1,0 mL da solução estoque primária (a) para
balão volumétrico de 10 mL e elevado ao volume final com cicloexano.
d) Benzo(a)pireno dodecadeuterado (0,1mg/mL)
Foi transferida uma alíquota de 1,0 mL da solução estoque primária (b) para
balão volumétrico de 10 mL e elevado ao volume final com cicloexano.
O quadro 7 relaciona as alíquotas das soluções estoque primárias e suas
respectivas concentrações,após diluição, nas soluções estoque secundárias.
Padrão Massa (mg) Concentração (mg/mL)
Benzo(a)pireno -B(a)P 1,0 0,1
Benzo(a)pireno - B(a)P D12 1,0 0,1
Quadro 7 - Soluções estoque secundárias Fonte: Autoria própria, 2010

48
4.6.3 Soluções estoques terciárias
e) Benzo(a)pireno (2000ng/mL)
Foi transferida uma alíquota de 1,0 mL da solução estoque secundária (c)
para balão volumétrico de 50 mL e elevado ao volume final com cicloexano.
f) Benzo(a)pireno dodecadeuterado (2000ng/mL)
Foi transferida uma alíquota de 1,0 mL da solução estoque secundária (d)
para balão volumétrico de 50 mL e elevado ao volume final com cicloexano.
O quadro 8 relaciona as alíquotas das soluções estoque secundárias e suas
respectivas concentrações,após diluição, nas soluções estoque terciárias.
Padrão Massa (mg) Concentração (mg/mL)
Benzo(a)pireno -B(a)P 1,0 2000,0
Benzo(a)pireno - B(a)P D12 1,0 2000,0
Quadro 8 - Soluções estoque terciárias Fonte: Autoria própria, 2010
4.6.4 Solução de trabalho de padrão interno - B(a)P D12
Foi transferida uma alíquota 500µL da solução estoque secundária de padrão
de Benzo(a)pireno dodecadeuterado (d) para balão volumétrico de 250 mL e
elevado ao volume final com cicloexano.
4.6.6 Soluções padrão para estabelecer a curva de calibração
As soluções padrão utilizadas para o preparo da curva de calibração são
preparadas a partir das soluções estoques terciárias dos padrões analíticos.

49
Foram utilizadas alíquotas de 31,25 µL a 2000 µL da solução estoque terciária
se benzo(a)pireno (e) e um volume fixo de 1,0mL de solução estoque terciária do
padrão interno de benzo(a)pireno dodecadeuterado (f) para o preparo de 7 soluções
para a curva de calibração da B(a)P, com concentrações de 1,25 a 80,0 ng/mL em
balões volumétricos de 50mL, cujo volume foi completado com cicloexano.
A quadro a seguir reúne as diversas concentrações que foram utilizadas para a
construção da curva de calibração de benzo(a)pireno e suas concentrações finais.
Solução
Padrão
Alíquota de solução
estoque terciária
B(a)P
(µL)
Concentração B(a)P
(ng/mL)
1 31,25 1,25
2 75,00 3,00
3 250,00 10,00
4 500,00 20,00
5 1000,00 40,00
6 1500,00 60,00
7 2000,00 80,00
Quadro 9 - Soluções para estabelecer a curva de calibração Fonte: Autoria própria, 2010.
50
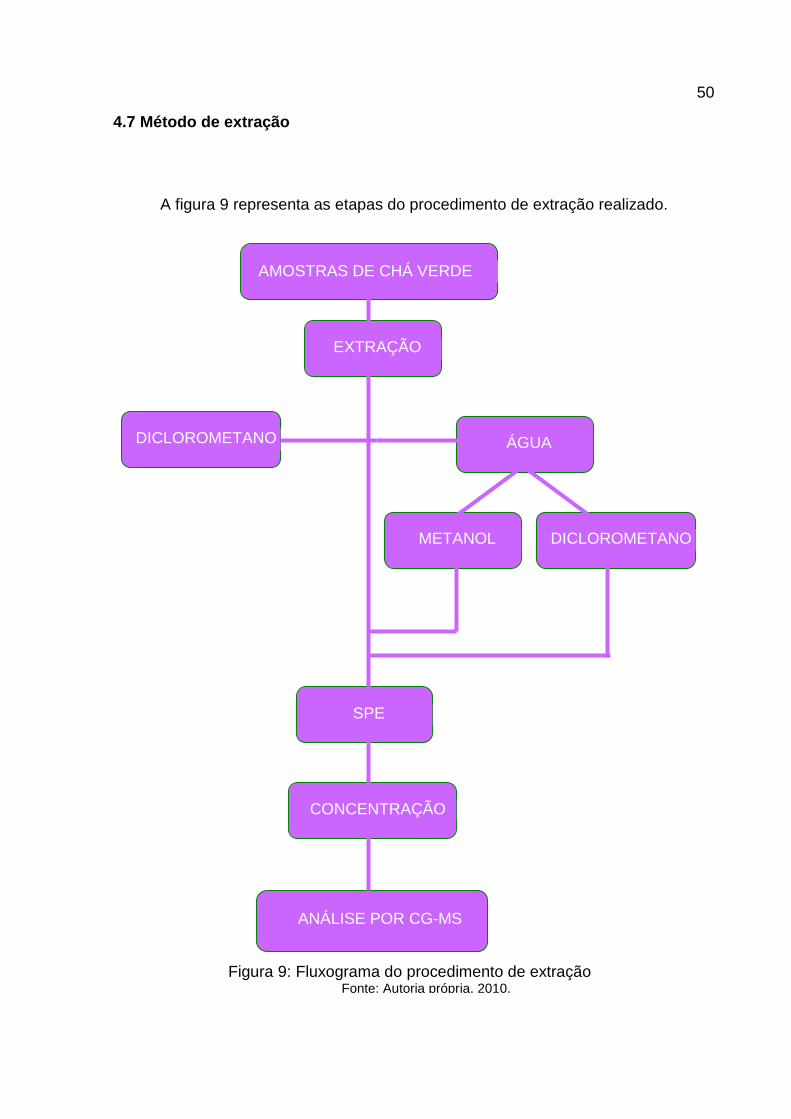
4.7 Método de extração
A figura 9 representa as etapas do procedimento de extração realizado.
AMOSTRAS DE CHÁ VERDE
EXTRAÇÃO
ÁGUA
ANÁLISE POR CG-MS
SPE
CONCENTRAÇÃO
METANOL DICLOROMETANO
Figura 9: Fluxograma do procedimento de extração Fonte: Autoria própria, 2010.
DICLOROMETANO

51
4.7.1 Extração do chá seco
Foi pesado 1,000 ± 0,005 g de chá verde em frasco de 50 mL, adicionar 0,5 mL
de solução de trabalho de padrão interno de Benzo(a)pireno dodecadeuterado. Logo
após adicionar 2g de Hydromatrix® (Chem Elut Variam 198004) que tem a função de
absorver qualquer umidade presente na solução e 25 mL de diclorometano.
Na figura 10, estão as amostras pesadas e prontas para iniciar o processo.
Figura 10 - Amostras pesadas e prontas para iniciar o procedimento
Fonte: Autoria própria, 2010.
A etapa de extração foi realizada em ultra-som por 20 minutos e após filtrada
em papel de filtro qualitativo para balão de fundo redondo de 50 mL, lavando o
frasco com mais 25 mL de diclorometano, obtendo um volume final de 50 mL.
A figura 11 demonstra a etapa de filtração.

52
Figura 11 - Etapa de filtração
Fonte: Autoria própria, 2010.
O extrato foi evaporado até a secura em rota evaporador, sob vácuo, à
temperatura de aproximadamente 50 °C. Foi reconstit uído com 3mL de cicloexano
sob agitação constante em misturador.
Figura 12 - Secagem em evaporador rotatório
Fonte: Autoria própria, 2010.

53
4.7.2 Extração em fase sólida (SPE- Solid Phase Extration)
A extração do possível contaminante B(a)P presente no extrato de chá verde
obtido foi realizada através do processo denominado extração em fase solida (SPE).
Na sequência de figuras 13, 14, 15 e 16 esta apresentado o processo final
onde foi transferido todo o extrato reconstituído para um cartucho contendo 1,0 g de
sílica gel e previamente condicionado com, aproximadamente, 3 mL de cicloexano
(figura 13). O extrato foi lavado com 2,5 mL de pentano, sendo descartando o
eluente. O benzo(a)pireno foi eluído com, aproximadamente, 4,0 mL de uma mistura
composta por duas partes de cicloexano para uma parte de diclorometano (figura
14). O eluato foi homogeneizado e transferiu-se uma alíquota de, aproximadamente,
1 mL para um “vial” de cromatografia e analisado por GC/MS,como mostram a figura
15 e 16.
Figura 13 - Etapa de eluição da amostra
Fonte: Autoria própria, 2010.

54
Figura 14- Coleta do Benzo(a)pireno após a eluição
Fonte: Autoria própria, 2010.
Figura 15 - Transferência da amostra para frasco (vial) cromatográfico
Fonte: Autoria própria, 2010.

55
Figura 16 - Injeção em CG/MS Fonte: Autoria própria, 2010.
4.7.3 Extração da infusão
Foram utilizadas duas metodologias diferentes para a extração da infusão de
chá verde, denominadas Método1 e Método 2.
A infusão do chá foi obtida conforme descrevem os fabricantes: um sache de
chá verde em uma xícara de água fervente, aproximadamente 200 mL.
Abaixo estão descritas as duas formas de extração.
4.7.3.1 Método 1
Após o resfriamento, foi transferida uma alíquota de 60 mL desta infusão
aquosa para um funil de separação e adicionou-se 25 ml de diclorometano,
procedendo a extração e filtrando em papel qualitativo contendo 2g de

56
Hydromatrix®. A fração contendo diclorometano foi transferida para um balão de
fundo redondo de 50 ml, após, foi feita uma segunda extração, com mais 25 ml de
diclorometano totalizando 50 ml, filtrando para o mesmo balão. Adicionou-se 0,5 ml
de solução de trabalho de padrão interno de Benzo(a)pireno dodecadeuterado.
Evaporou-se o extrato até a secura em rota evaporador, sob vácuo, à
temperatura de aproximadamente 50 °C. Reconstituiu- se o extrato com 3 mL de
cicloexano sob agitação constante em “mixer”, após esse procedimento a amostra
foi submetida ao procedimento descrito no item 4.7.2.
4.7.3.2 Método 2
Após o resfriamento, transferiu-se uma alíquota de 60 mL desta infusão aquosa
para um funil de separação e adicionou-se 40 mL de metanol, 0,5 mL de solução de
trabalho de padrão interno de Benzo(a)pireno dodecadeuterado e agitou-se.
4.7.3.3 Extração em fase sólida
Condicionou-se um cartucho de CH com 10 ml de metanol e após com 10 ml
de uma mistura composta por seis partes de água para quatro partes de metanol.
Transferiu-se todo o extrato para o cartucho, lavaou-se o funil de separação com 10
ml de uma mistura composta por seis partes de água para quatro partes de metanol
e passou-se pelo cartucho. Aguardou-se 30 minutos com o sistema de vácuo ligado
para que houvesse a passagem de ar pelo cartucho para sua secagem. Após este
tempo, eluiu-se o benzo(a)pireno do cartucho de CH com, aproximadamente 15 mL
de cicloexano, reduziu-se o volume em rota evaporador , até obter um volume
próximo a 1 mL, transferiu-se para um frasco (vial) de cromatografia e analisou-se
por GC/MS.

57
4.8 Método de análise
A análise do possível contaminante B(a)P foi realizada por Cromatografia
Gasosa acoplada à Espectrometria de Massas, utilizando o modo de Impacto de
Elétrons (EI) e a técnica de Monitoramento Seletivo de Íons (SIM)
O quadro 10 mostra os íons selecionados para a quantificação dos
Benzo(a)pireno juntamente com o tempo de retenção,
Substância Íons Selecionados (m/z) Tempo de Retenção (min.)
Benzo(a)pireno -B(a)P 250 e 252 11,806
Benzo(a)pireno - B(a)P D12 260 e 264 11,760
Quadro 10 - Íons Selecionados e Tempo de Retenção Fonte: Autoria própria, 2010.
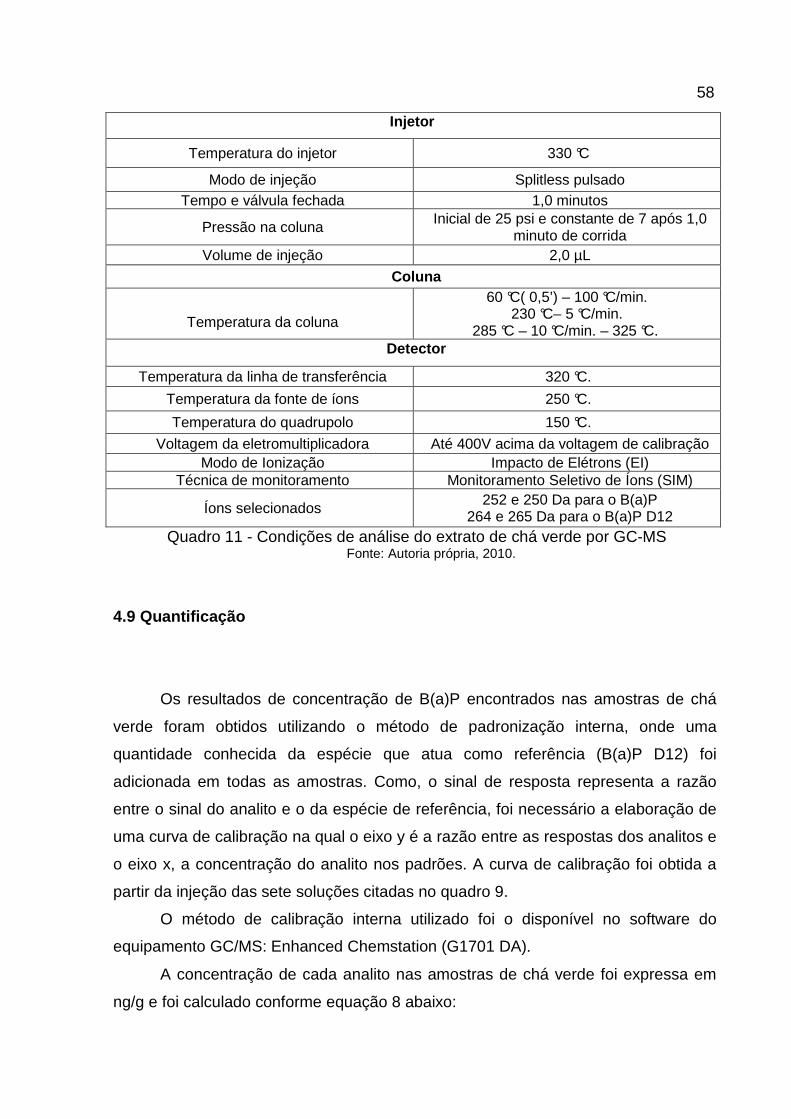
4.8.1 Condições de CG/MS
No quadro 11 estão descritas as condições cromatográficas utilizadas na
analise do extrato de chá verde.
58
Injetor
Temperatura do injetor 330 °C
Modo de injeção Splitless pulsado Tempo e válvula fechada 1,0 minutos
Pressão na coluna Inicial de 25 psi e constante de 7 após 1,0 minuto de corrida
Volume de injeção 2,0 µL
Coluna
Temperatura da coluna
60 °C( 0,5’) – 100 °C/min. 230 °C– 5 °C/min.
285 °C – 10 °C/min. – 325 °C. Detector
Temperatura da linha de transferência 320 °C.
Temperatura da fonte de íons 250 °C.
Temperatura do quadrupolo 150 °C.
Voltagem da eletromultiplicadora Até 400V acima da voltagem de calibração Modo de Ionização Impacto de Elétrons (EI)
Técnica de monitoramento Monitoramento Seletivo de Íons (SIM)
Íons selecionados 252 e 250 Da para o B(a)P 264 e 265 Da para o B(a)P D12
Quadro 11 - Condições de análise do extrato de chá verde por GC-MS Fonte: Autoria própria, 2010.
4.9 Quantificação
Os resultados de concentração de B(a)P encontrados nas amostras de chá
verde foram obtidos utilizando o método de padronização interna, onde uma
quantidade conhecida da espécie que atua como referência (B(a)P D12) foi
adicionada em todas as amostras. Como, o sinal de resposta representa a razão
entre o sinal do analito e o da espécie de referência, foi necessário a elaboração de
uma curva de calibração na qual o eixo y é a razão entre as respostas dos analitos e
o eixo x, a concentração do analito nos padrões. A curva de calibração foi obtida a
partir da injeção das sete soluções citadas no quadro 9.
O método de calibração interna utilizado foi o disponível no software do
equipamento GC/MS: Enhanced Chemstation (G1701 DA).
A concentração de cada analito nas amostras de chá verde foi expressa em
ng/g e foi calculado conforme equação 8 abaixo:

59
Onde:
Ai: área do analito na amostra (u.a -unidades de área)
API: área do padrão interno na amostra
I: coeficiente linear da curva de calibração
S: coeficiente angular da curva de calibração
m: massa da amostra (ng)
QPI: quantidade de padrão interno adicionada na amostra (ng)
4.10 Linearidade
A linearidade foi estudada através da construção das curvas analíticas obtidas
utilizando soluções padrões de B(a)P em diclorometano descritos no quadro 9, em 7
níveis de concentração diferentes, em uma faixa que variou de 1,25 a 80,0 ng/mL. O
estudo da linearidade foi realizado através de dez injeções da curva de calibração.
4.11 Limite de detecção
O Limite de detecção foi estudado a partir da análise de injeções de extrato
diluído de chá verde. Com os resultados dessas injeções, foi calculado o desvio
padrão e logo após o LD, conforme equação abaixo:
Canalito (ng/g) = Ai
Api - I
x Qpi
S x m
... (8)

60
A amostra de chá verde utilizada para determinar os LD foi a amostra 5, que
obteve um valor médio de Benzo(a)pireno de 10,02 ng/g.Este extrato foi diluído em
uma proporção de 1:4 (200 µL de extrato e 800 µL de cicloexano/diclorometano 2:1).
4.12 Limite de quantificação
O Limite de quantificação foi estudado a partir da análise de dez injeções de
extrato diluído de erva mate. Com os resultados dessas injeções, foi calculado o
desvio padrão e logo após o LQ, conforme equação abaixo:
4.13 Recuperação
A recuperação de Benzo(a)pireno foi avaliada em três níveis de fortificação. O
estudo foi realizado a partir da adição de concentrações conhecidas de solução
padrão de B(a)P em amostra de chá verde, de concentração: baixa (50,32 ng/g),
média (145,15 ng/g)e alta (251,60 ng/g) adicionados em amostra isenta do
contaminante B(a)P. O ensaio foi realizado por três dias e as amostras injetadas em
duplicatas no cromatógrafo.
LD = 3 X S ... (9)
LQ = 10 X S
... (10)
61
5 RESULTADOS E DISCUSSÃO
Neste capitulo serão discutidos os resultados obtidos com os dados
experimentais frente aos dados pesquisados.
5.1 Parâmetros de validação de resultados
Foram avaliados alguns parâmetros considerados importantes para a
validação dos resultados obtidos.
5.1.1 Linearidade
O quadro 12 apresenta os resultados das médias das áreas dos sinais
cromatográficos correspondentes as concentrações dos pontos de curvas avaliados.
Concentração (ng/g) Média de áreas (unidades de área - ua) Pontos de
Curva B(a)P B(a)P D12 B(a)P B(a)P D12
1 5,100 102,93 70.816 1.993.675
2 11,60 102,93 177.786 1.845.060
3 38,80 102,93 538.912 1.841.279
4 77,60 102,93 1084.405 1.795.269
5 155,6 102,93 2466.656 2.060.817
6 232,4 102,93 3394.177 1.847.155
7 310,5 102,93 4352.056 1.797.944
Quadro 12 - Dados de área versus concentração dos padrões Fonte: Autoria própria, 2010.
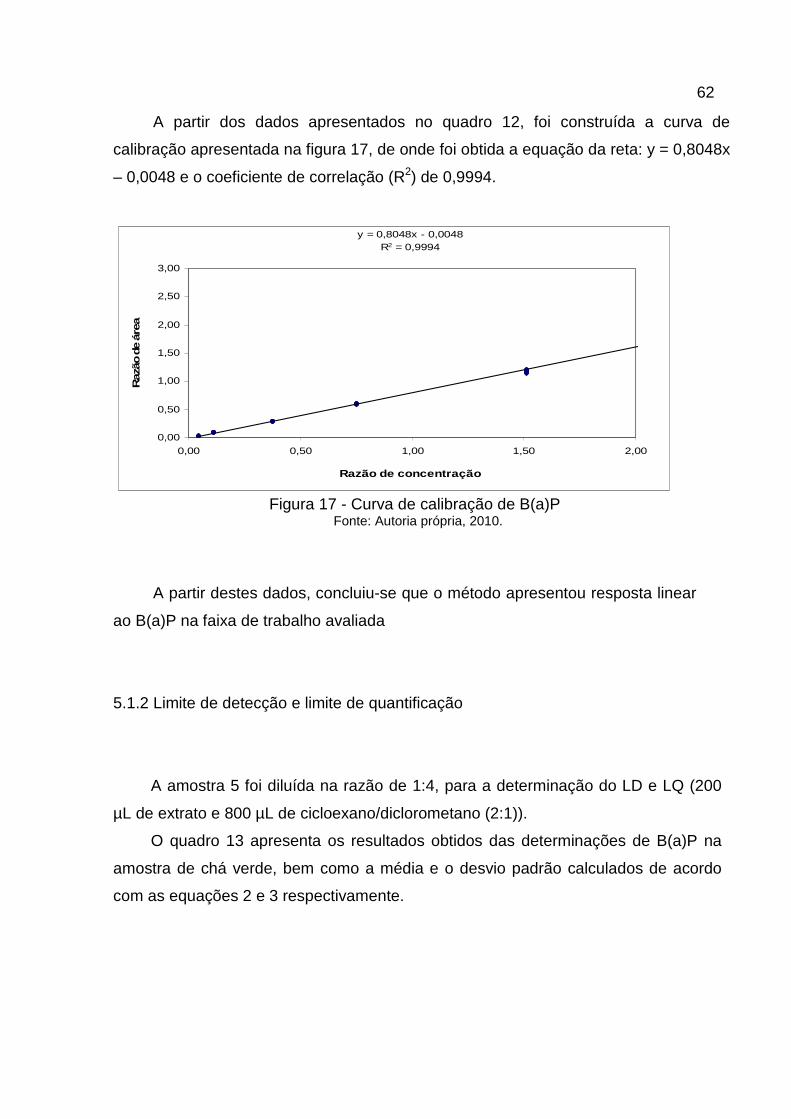
62
A partir dos dados apresentados no quadro 12, foi construída a curva de
calibração apresentada na figura 17, de onde foi obtida a equação da reta: y = 0,8048x
– 0,0048 e o coeficiente de correlação (R2) de 0,9994.
y = 0,8048x - 0,0048R2 = 0,9994
0,00
0,50
1,00
1,50
2,00
2,50
3,00
0,00 0,50 1,00 1,50 2,00
Razão de concentração
Raz
ão d
e ár
ea
Figura 17 - Curva de calibração de B(a)P
Fonte: Autoria própria, 2010.
A partir destes dados, concluiu-se que o método apresentou resposta linear
ao B(a)P na faixa de trabalho avaliada
5.1.2 Limite de detecção e limite de quantificação
A amostra 5 foi diluída na razão de 1:4, para a determinação do LD e LQ (200
µL de extrato e 800 µL de cicloexano/diclorometano (2:1)).
O quadro 13 apresenta os resultados obtidos das determinações de B(a)P na
amostra de chá verde, bem como a média e o desvio padrão calculados de acordo
com as equações 2 e 3 respectivamente.
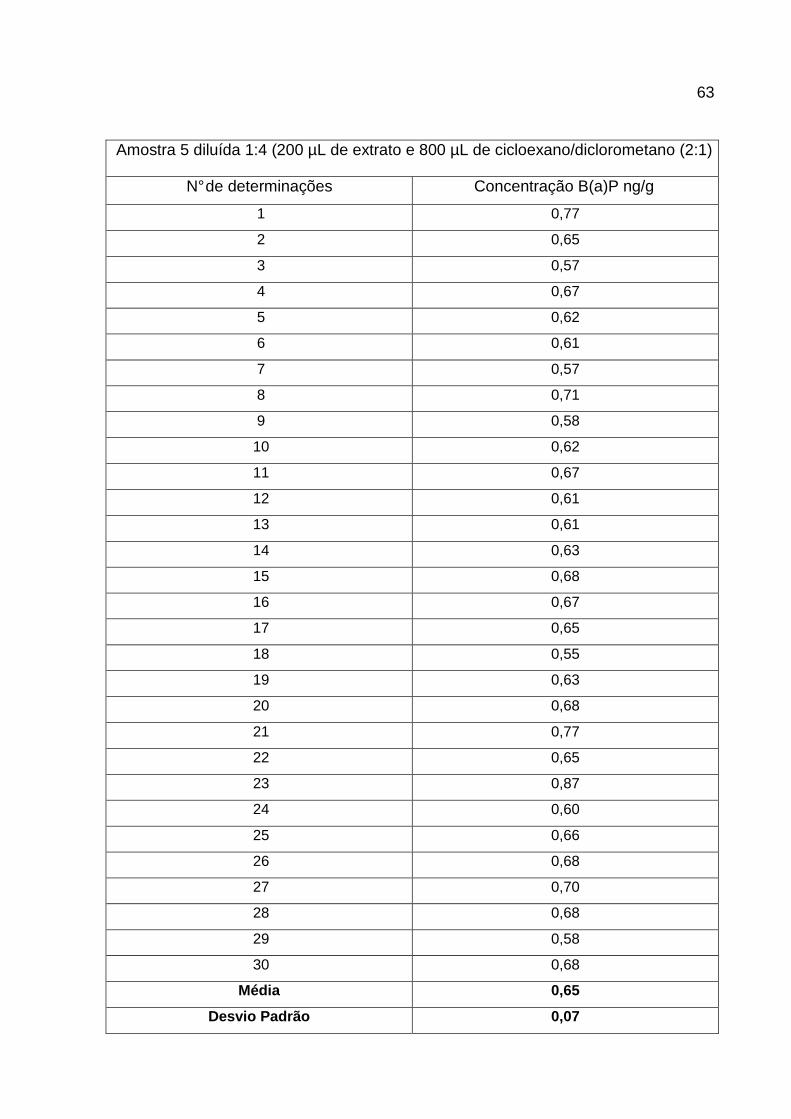
63
Amostra 5 diluída 1:4 (200 µL de extrato e 800 µL de cicloexano/diclorometano (2:1)
N° de determinações Concentração B(a)P ng/g
1 0,77
2 0,65
3 0,57
4 0,67
5 0,62
6 0,61
7 0,57
8 0,71
9 0,58
10 0,62
11 0,67
12 0,61
13 0,61
14 0,63
15 0,68
16 0,67
17 0,65
18 0,55
19 0,63
20 0,68
21 0,77
22 0,65
23 0,87
24 0,60
25 0,66
26 0,68
27 0,70
28 0,68
29 0,58
30 0,68
Média 0,65
Desvio Padrão 0,07
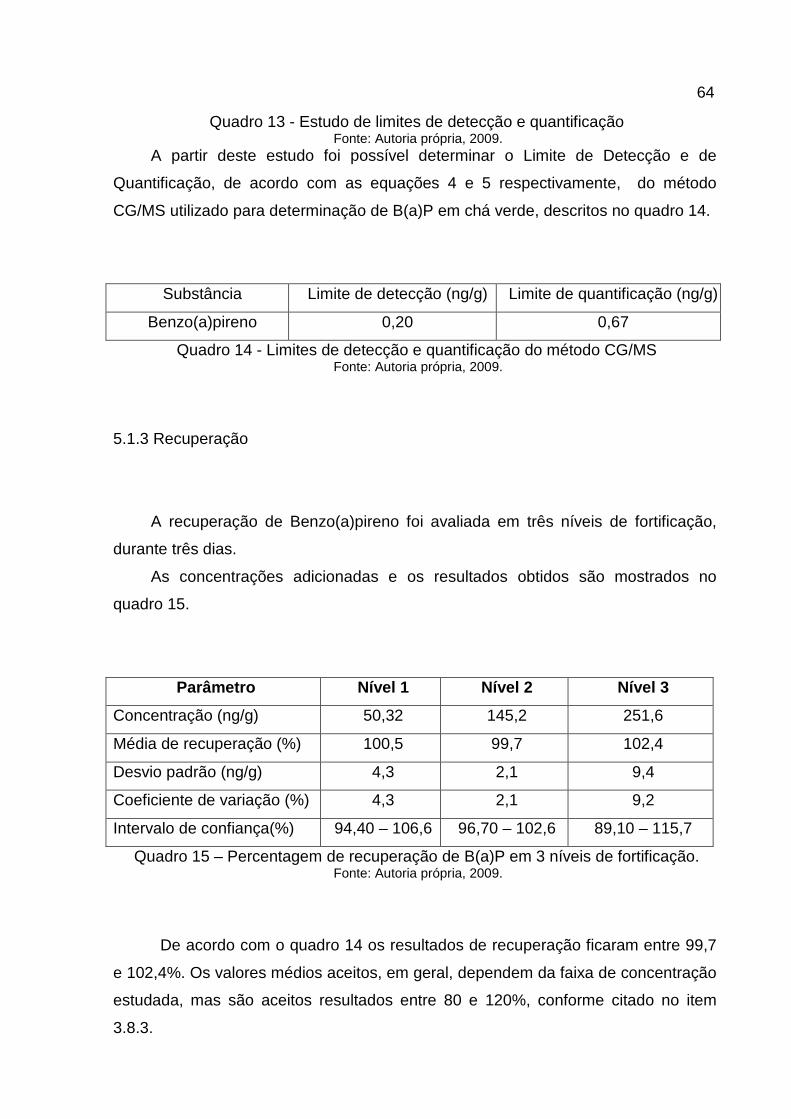
64
Quadro 13 - Estudo de limites de detecção e quantificação Fonte: Autoria própria, 2009.
A partir deste estudo foi possível determinar o Limite de Detecção e de
Quantificação, de acordo com as equações 4 e 5 respectivamente, do método
CG/MS utilizado para determinação de B(a)P em chá verde, descritos no quadro 14.
Substância Limite de detecção (ng/g) Limite de quantificação (ng/g)
Benzo(a)pireno 0,20 0,67
Quadro 14 - Limites de detecção e quantificação do método CG/MS Fonte: Autoria própria, 2009.
5.1.3 Recuperação
A recuperação de Benzo(a)pireno foi avaliada em três níveis de fortificação,
durante três dias.
As concentrações adicionadas e os resultados obtidos são mostrados no
quadro 15.
Parâmetro Nível 1 Nível 2 Nível 3
Concentração (ng/g) 50,32 145,2 251,6
Média de recuperação (%) 100,5 99,7 102,4
Desvio padrão (ng/g) 4,3 2,1 9,4
Coeficiente de variação (%) 4,3 2,1 9,2
Intervalo de confiança(%) 94,40 – 106,6 96,70 – 102,6 89,10 – 115,7
Quadro 15 – Percentagem de recuperação de B(a)P em 3 níveis de fortificação. Fonte: Autoria própria, 2009.
De acordo com o quadro 14 os resultados de recuperação ficaram entre 99,7
e 102,4%. Os valores médios aceitos, em geral, dependem da faixa de concentração
estudada, mas são aceitos resultados entre 80 e 120%, conforme citado no item
3.8.3.

65
Como pode se concluir a partir dos resultados obtidos, o método utilizado
tem uma boa recuperação nas concentrações avaliadas.
5.2 Cromatogramas
Estão apresentados a seguir os cromatogramas de padrões e de amostras para
que se obtenha um comparativo entre os resultados.
5.2.1. Cromatograma de padrão
Na figura 18 esta representado um cromatograma típico, este se refere ao
ponto 7 da curva de calibração, onde no tempo de retenção de 11,760min. é
caracterizado o B(a)P D12 e no tempo de retenção de 11,806 min. o B(a)P.
Figura 18 - Cromatograma do ponto 7 da curva de calibração
Fonte: Autoria própria, 2010.

66
5.2.1.1 Extração dos íons monitorados do padrão
No espectro de massas apresentado na figura 19, obtido através do
cromatograma ilustrado na figura 18, foi realizada a extração dos íons (m/z)
característicos do B(a)P D12, que são respectivamente 260 e 264, que foi utilizado
como padrão interno para a determinação realizada.
Figura 19 - Espectro de massas do B(a)P D12 (PI), íons 260 e 264
Fonte: Autoria própria, 2010.
No espectro de massas, representado pela figura 20, também obtido através do
cromatograma ilustrado na figura 18, foi realizada a extração dos íons (m/z)
característicos do B(a)P, que são respectivamente 250 e 252.
Figura 20 - Espectro de massas do Padrão B(a)P, íons 250 e 252
Fonte: Autoria própria, 2010.
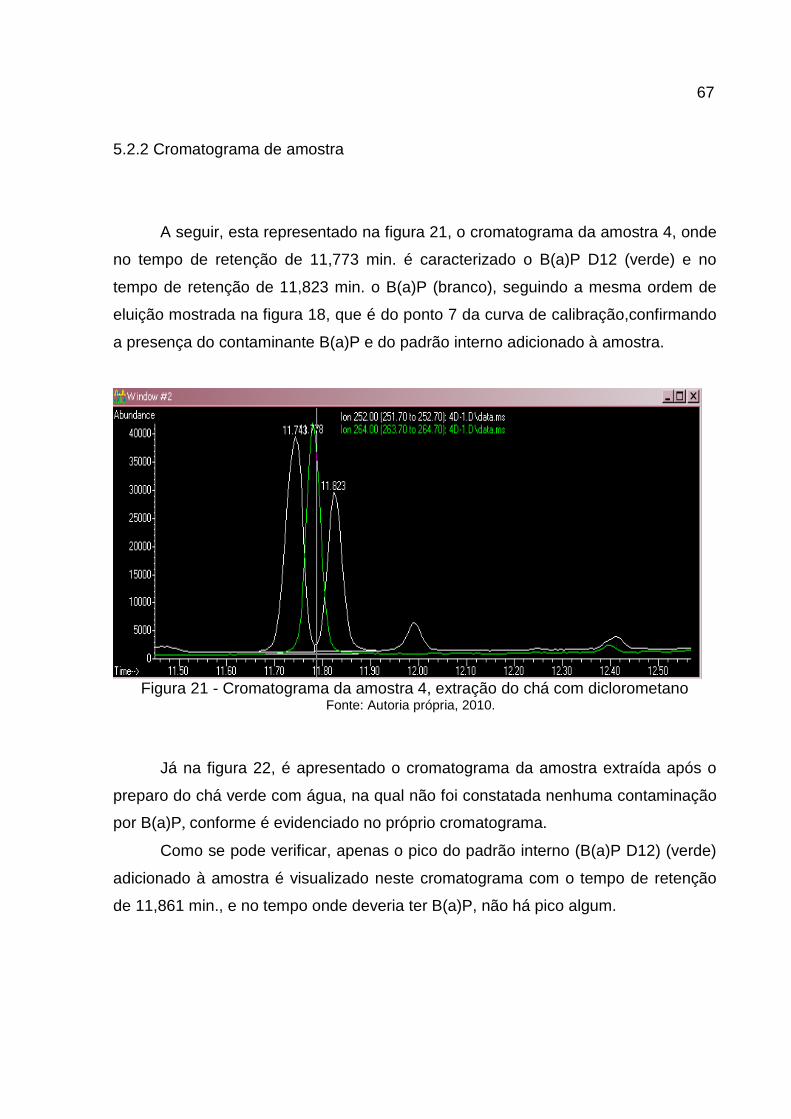
67
5.2.2 Cromatograma de amostra
A seguir, esta representado na figura 21, o cromatograma da amostra 4, onde
no tempo de retenção de 11,773 min. é caracterizado o B(a)P D12 (verde) e no
tempo de retenção de 11,823 min. o B(a)P (branco), seguindo a mesma ordem de
eluição mostrada na figura 18, que é do ponto 7 da curva de calibração,confirmando
a presença do contaminante B(a)P e do padrão interno adicionado à amostra.
Figura 21 - Cromatograma da amostra 4, extração do chá com diclorometano
Fonte: Autoria própria, 2010.
Já na figura 22, é apresentado o cromatograma da amostra extraída após o
preparo do chá verde com água, na qual não foi constatada nenhuma contaminação
por B(a)P, conforme é evidenciado no próprio cromatograma.
Como se pode verificar, apenas o pico do padrão interno (B(a)P D12) (verde)
adicionado à amostra é visualizado neste cromatograma com o tempo de retenção
de 11,861 min., e no tempo onde deveria ter B(a)P, não há pico algum.

68
Figura 22 - Cromatograma da amostra 4, extração da infusão com diclorometano
Fonte: Autoria própria, 2010.
5.2.2.1 Extração dos íons monitorados de amostra contaminada.
A figura 23 mostra o espectro de massas obtido da analise ilustrada pelo
cromatograma da figura 21. A extração dos íons (m/z) característicos do B(a)P D12,
que são respectivamente 260 e 264, que foi utilizado como padrão interno para a
determinação de B(A)P realizada, da mesma forma que foi utilizado na curva de
calibração.
Figura 23 - Espectro de massas da amostra 4, massa 260 e 264 (B(a)P D12.)
Fonte: Autoria própria, 2010.
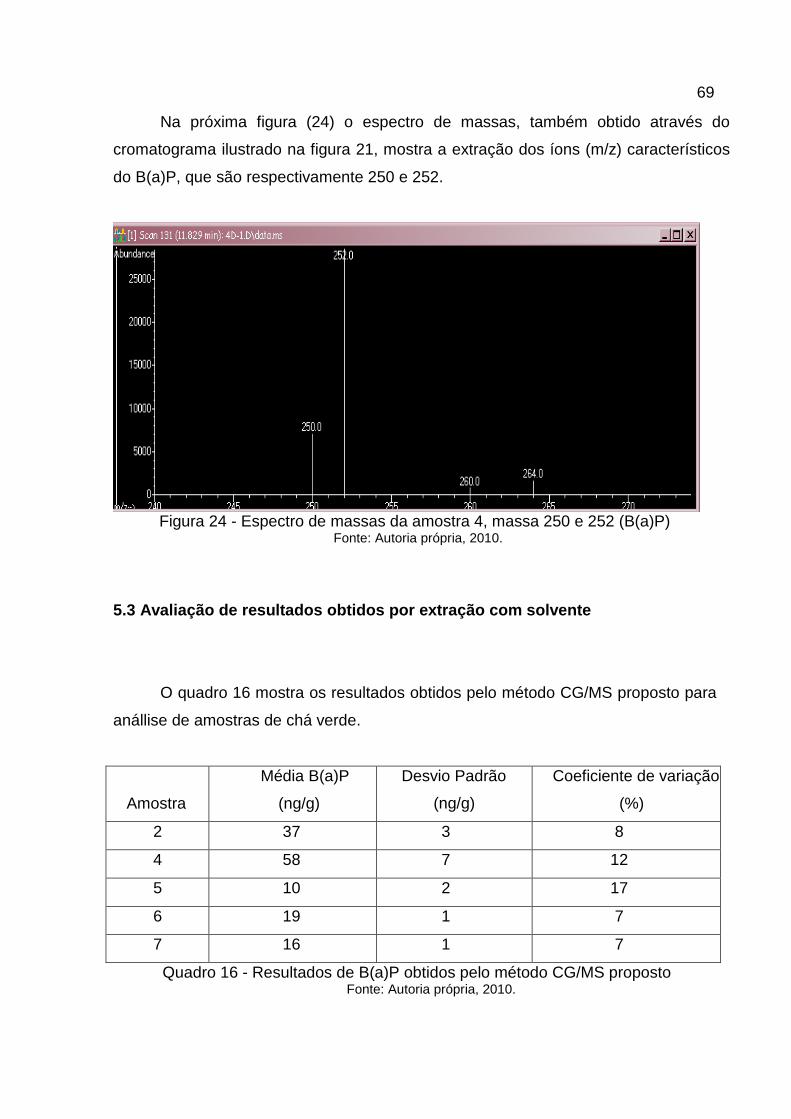
69
Na próxima figura (24) o espectro de massas, também obtido através do
cromatograma ilustrado na figura 21, mostra a extração dos íons (m/z) característicos
do B(a)P, que são respectivamente 250 e 252.
Figura 24 - Espectro de massas da amostra 4, massa 250 e 252 (B(a)P)
Fonte: Autoria própria, 2010.
5.3 Avaliação de resultados obtidos por extração co m solvente
O quadro 16 mostra os resultados obtidos pelo método CG/MS proposto para
anállise de amostras de chá verde.
Amostra
Média B(a)P
(ng/g)
Desvio Padrão
(ng/g)
Coeficiente de variação
(%)
2 37 3 8
4 58 7 12
5 10 2 17
6 19 1 7
7 16 1 7
Quadro 16 - Resultados de B(a)P obtidos pelo método CG/MS proposto Fonte: Autoria própria, 2010.

70
As análises foram realizada nas sete amostras adquiridas, dentre as quais,
duas não apresentaram contaminação por Benzo(a)pireno (amostras 1 e 3). As
demais, 2, 4, 5, 6, e 7 apresentaram resultados positivos para a presença de B(a)P,
como apresentado no quadro 16.
De acordo com os resultados apresentados neste estudo, podemos confirmar a
contaminação de amostras comercializadas de chá verde pelo hidrocarboneto
policíclico aromático Benzo(a)pireno, determinadas a partir da extração com solvente
orgânico, no caso deste estudo, com diclorometano, indicando a necessidade da
continuidade de pesquisas nesta área.
5.4 Avaliação de resultados obtidos da extração da infusão
De acordo com as análises realizadas, não foi detectado o B(a)P em nenhuma
das amostras de chá verde que foram analisadas, ao contrário do resultado
encontrado no extrato seco, ou seja, no método de extração que utiliza
diclorometano diretamente com o chá, sem utilização de água, como neste caso, da
infusão.
Com os métodos utilizados para extração de B(a)P em meio aquoso, que são,
extrair a infusão com diclorometano e o outro é extraí-la com metanol, foi permitido
observar, através deste trabalho, que o chá que bebemos, ou seja, a infusão não é
contaminada pelo B(a)P.
Espera-se com isso, que outros estudos possam aprofundar as pesquisas e
abrir uma nova discussão: até que ponto ou limites podemos ingerir este chá
contaminado? Ou as folhas podem ser utilizadas na composição de outros
alimentos?

71
6 CONCLUSÕES
Em vista dos resultados obtidos por este estudo, as indústrias produtoras de
chá devem ser alertadas quanto à necessidade e importância de se mapear
possíveis fontes de contaminação dos chás, identificando-se as etapas do
processamento da erva de chá que podem estar envolvidas na contaminação.
Constatou-se também com este estudo a necessidade de uma atenção por
parte dos órgãos brasileiros fiscalizadores, como a ANVISA, em relação à presença
desse composto em chás e em outros alimentos, de uma forma geral que sejam
processadas por alguma das fontes/formas citadas que possam produzir
potencialmente B(a)P ou outros HPAs.
A presença do contaminante Benzo(a)pireno identificado em amostras de chá
verde seco, extraído com solvente, mostra à sociedade e órgãos responsáveis pela
saúde pública a necessidade de se estabelecer um monitoramento da presença de
B(a)P em chá verde, outros chás e, caso a contaminação, medidas devem ser
indicadas para sua redução e controle. O estabelecimento de limites para a presença
deste contaminante em chás e outros alimentos poderia também ser outra iniciativa
importante de órgãos reguladores protegendo a saúde da população.
É evidente que o chá não é utilizado pela sociedade somente desta forma, chá
em infusão, e foi uma grata conclusão deste trabalho constatar que a infusão do
sache de chá verde em água, não consegue extrair o B(a)P, pelo menos em
concentrações acima do LD estabelecido no método utilizado neste Trabalho de
Conclusão de Curso (TCC). A partir desses resultados podemos ficar mais tranqüilos
quanto ao baixo risco de ingestão deste contaminante carcinogênico, pelo menos nas
amostras de chá verde analisadas, pelo habito milenar de beber chá. Mas há de se
ter cautela e devemos nos manter atentos quanto aos contaminantes que possam
estar presentes nos nossos alimentos.

72
REFERÊNCIAS AMES, et. al., Method for detecting carcinogens and mutagens with Salmonella/mammalian microsome teste. Mutatation Research , 1975. V. 31, p. 347-64. Disponível em: < http://ntp.niehs.nih.gov/?objectid=16D6D969-A9F4-0B6F-2B81F2FB53BBFD3E> Acesso em: 02 abr. 2010. AZEREDO, A. Determinação de Benzo(a)pireno em pescados comercia lizados em campinas-SP .2001, 76f. Dissertação (Mestrado em Ciências dos Alimentos) – Universidade Estadual de Campinas, Campinas, 2001. Disponível em: <http://www.unicamp.br>. Acesso em: 02 abr. 2010. BANJOO DR, Nelson PK. Improved ultrasonic extraction procedure for the determination of polycyclic aromatic hydrocarbons in sediments. J Chromatogr A , 2005; n.1066, p.9-18. BETTIN, S. M.; FRANCO, D. W. Hidrocarbonetos policíclicos aromáticos (HPAs) em aguardentes. Ciênc. Tecnol. Aliment. Campinas, v. 25, n.2, abr./jun. 2005. Disponível em: em: <http://www.ifpi.edu.br/eventos/iiencipro/arquivos/ALIMENTOS/ 2a095ac9453c3a2f02c42074449c0b7b.pdf>. Acesso em: 02 abr.2010. BRASIL. Agência Nacional de Vigilância Sanitária. Resolução RDC nº 02, de 15 de janeiro de 2007. Aprova o Regulamento Técnico sobre Aditivos Aromatizantes. Disponível em: <http://www.anvisa.gov.br>. Acesso em: 02 abr. 2010. BRASIL. Ministério da Saúde. Portaria nº 518, de 25 de março de 2004 . Estabelece os procedimentos e responsabilidades relativos ao controle e vigilância da qualidade da água para consumo humano e seu padrão de potabilidade, e dá outras providências. Disponível em:<http://www.anvisa.gov.br>. Acesso em: 02 abr. 2010. BRASIL, Portaria n. 36, de 19 de janeiro de 1990. Aprova normas e o padrão de portabilidade de água destinada ao consumo humano. Diário Oficial da União , Brasília, DF, 23 de jan. 1990. Disponível em: <http://www.anvisa.gov.br>. Acesso em: 15 mar. 2010. BRASIL. Resolução n° 277 de 22 de setembro de 2005. Regulamento técnico para café, cevada, chá, erva-mate e produtos solúveis. Diário Oficial da República Federativa do Brasil , Brasília, DF, 23 set. 2005. Disponível em: <http://e-legis.anvi sa.gov.br/leisref/public/showAct.php>. Acesso em: 12 ago. 2009

73
CAMARGO, M. C. R.; TOLEDO, M. C. F. Avaliação da contaminação de diferentes grupos de alimentos por hidrocarbonetos policíclicos aromáticos. Braz. J. Food. Technol , v. 5, 2002a. Disponível em: <http://www.ital.sp.gov.br>. Acesso em: 01 abr. 2010. CAMARGO, M. C. R.; TOLEDO, M. C. F. Chá-mate e café como fontes de hidrocarbonetos policíclicos aromáticos (HPAs) na dieta da população de Campinas. Ciênc. Tecnol. Aliment. Campinas, v. 22, n. 1, jan./abr., 2002b. Disponível em: <http://www.scielo.br>. Acesso em: 01 abr. 2010. CAMARGO, M. C. R.; TOLEDO, M. C. F. Hidrocarbonetos policíclicos aromáticos – uma revisão. Boletim da Sociedade Brasileira de Ciência e Tecnol ogia de Alimentos , v. 36, n.1, p. 69-78, 2002c. Disponível em: < http://www.ifpi.edu.br /eventos/iiencipro/arquivos/ALIMENTOS/2a095ac9453c3a2f02c42074449c0b7b.pdf >. Acesso em: 01 abr. 2010. CAMARGO, MSFO, Toledo MCF. Avaliação da ingestão de hidrocarbonetos policíclicos aromáticos (HPAs) através da dieta, em diferentes regiões do Brasil. Revista Brasileira de Toxicologia . v. 14,n. 2, p. :23-30, 2001. CAMARGO, MSF, Toledo MCF. Hidrocarbonetos aromáticos policíclicos em margarina, creme vegetal e maionese. Ciênc Tecnol Aliment ..v. 20, n. 1, p. 51-55, 2000.Disponível em:<ftp://ftp.cip.saude.sp.gov.br/teses/Tese_CCD_Caruso, %20MSF_2007.pdf >. Acesso em: 01 fev. 2010. CAMARGO, Mônica Cristiane Rojo et al . Determinação de hidrocarbonetos policíclicos aromáticos (HPAS) em guaraná em pó (Paullinia cupana). Ciênc. Tecnol. Aliment. , Campinas, v. 26, p. 230-234, 2006. Disponível em: <http://www. scielo.br/scielo.php?script=sci_arttext&pid=S010120612006000100036&lng=en&nrm=iso>. Acesso em: 03 jan. 2010 CARUSO, M. S. F. Otimização de metodologia para determinação de benzo(a)pireno em cachaças por CLAE com detecção de fluorescência e avaliação de sua ocorrência . 2007. 119p. Dissertação (Mestre em Ciências) – Fundação de Amparo à Pesquisa do Estado de São Paulo, São Paulo, 2007. Disponível em: <http://www.ial.sp.gov.br>. Acesso em: 01 fev. 2010. CAVALCANTI, ASS; ROSA, JAB; LIMA, MSCS ; SILVA, AG. O uso do chá verde, Camellia sinensis L. (Theaceae) em produtos tópicos – uma revisão. Natureza on line , v.5, n.2, p.76-84, 2007. Disponível em:<http://www.naturezaonline.com.br> Acesso em: 10 mai. 2010.

74
CEC – The Commission of the European Communities. Commission Regulation (EC) No 208/2005 of 4 February 2005 . Official Journal of European Union, 8.2.2005. Disponível em: <http://ec.europa.eu/index_en.htm>. Acesso em: 01 mar. 2010. CHEMSPIDER, Building community for chemists. Disponível em: <http://www. chemspider.com/RecordView.aspx?rid=f55fd9c5-8488-44a9-89fc-3a0431a90cf1>. Acesso em: 01 maio 2010. COSTA, A. F. Avaliação da contaminação humana por hidrocarboneto s policíclicos aromáticos: determinação de 1-hidroxip ereno urinário . 2001. 80p. Dissertação (Mestre em Saúde Pública) – Fundação Oswaldo Cruz, Escola Nacional de Saúde Pública. Rio de Janeiro, 2001. Disponível em: <http://portalteses.cict. fiocruz.br>. Acesso em: 01 mar. 2010. CUNHA, A. L. M. C. Desenvolvimento e validação de método fosforimétric o em substrato de celulose para determinação de criseno e pireno . 2007. 187p. Tese (Doutorado em Química) – Pontifícia Universidade Católica do Rio de Janeiro, Rio de janeiro, 2007. Disponível em: <http://www.puc-rio.br>. Acesso em: 11 dez. 2009. EUROPEAN COMMISSION. Health and Consumer Protection Directorate-General.. Polycyclic aromatic hydrocarbons – Occurrence in fo ods, dietary exposure and health effects. SCF/CS/CNTM/PAH/29 ADD1 Final. Brussels, 2002. Disponível em: <http://ec.europa.eu/food/fs/sc/scf/out154_en.pdf>. Acesso em: 15 mar. 2010. EUROPEAN COMMISSION, SCF/CS/CNTM/PAH/29 Final Opinion of the Scientific. Committee on Food on the risks to human health of polycyclic aromatic hydrocarbons infood, 2002. Disponível em: <http://europa.mm/food/food/ chemicalsafety/contaminants/out153 em.pdf>. Acesso em: 15 mar. 2010. EVANGELISTA, J. Tecnologia de alimentos . São Paulo: Atheneu, 2000. MASTRANDEA, C. et. al., Hidrocarburos aromáticos policíclicos. Riesgos para la salud y marcadores biológicos. Acta Bioquím. Clín. Lationam ., La Plata,v. 39, n. 1, jan./mar., 2005. Disponível em: <http://www.scielo.br>. Acesso em: 11 dez. 2009. FREI, B.; HIGDON, J. V. Antioxidant activity of tea polyphenols in vivo: evidence from animal studies. Journal of. Nutrition . 2003, 133, 3275S–3284S. HowStuffWorks.com.,Tea. 15 October 2008. Disponível em: http://science. howstuffworks.com/flowering-plants/tea-info.htm. Acesso em: 08 Jul 2010.

75
INTERNATIONAL AGENCY FOR RESEARCH OF CANCER. Polynuclear aromatic compounds. IARC, Monographs on the evaluation of the carcinogenic risk of chemical to humans. Part 1, Chemical, Environmental and Experimental Data , v. 32, Lyon, France, 1983.Disponível em:< http://monographs.iarc.fr/ENG/ recentpub/mono89.pdf>. Acesso em: 08 Jul 2010. INTERNATIONAL AGENCY FOR RESEARCH ON CANCER. Monographs on the evaluation of carcinogenic risk of chemicals to humans. Preamble , 2006a. Disponível em: <http://monographs.iarc.fr/ENG/Preamble/CurrentPreamble.pdf>. Acesso em: 10 jan. 2010. INTERNATIONAL AGENCY FOR RESEARCH ON CANCER. International Agency for Research on Cancer. Monographs on the evaluation of carcinogenic risk of chemicals to humans. Polycyclic Aromatic Hydrocarbons , 2006b. Disponível em: <http:// monographs.iarc.fr/ ENG/Meetings/92_pahs.pdf>. Acesso em: 10 mar. 2010. INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY. World Health Organization. Environmental Helth Criteia 2002. Selected non-heterocyclic. Polycyclic aromatic hydrocarbons . Geneva, 1998. JÁNSKÁ M, et.al... Optimization of the procedure for determination of polycyclic aromatic hydrocarbons and their derivatives in fish tiss Estimation of measurements uncertainty, 2006. Food Addit Contam . v.23, n.3, p. 309-25. JOE, FL Jr, et. al.. Liquid chromatography determination of trace of residues of polynuclear aromatic hydrocarbons in food, 1984. J AOAC . v. 67, n.6, p.1072-82. KANG M. K., et. al.. Metabolomics Analysis Reveals the Compositional Differences of Shade Grown Tea (Camellia sinensis L.). Journal of. Agriculture and. Food Chemistry ., v. 58, n. 1, 2010. Disponível em:< http://pubs.acs.org/doi/pdfplus/ 10.1021/jf902929h>. Acesso em: 19 jun 2010 LEE RB, et. al.. Cervical carcinoma in pregnancy. Obstet Gynecol ,1981. v.58(5), p. 584-9. PubMed PMID: 7301234. Disponível em: < http://pubs.acs.org/doi/pdfplus/10.1021/jf902929h>. Acesso em: 19 jun 2010 LEITE F. Validação em análise química . 4 ed. Campinas: Editora Átomo; 2002. LIN D, Tu Y, ZHU L. Concentrations and health risk of polycyclic aromatic hydrocarbons in tea, 2005. Food Chem Toxicol . v. 43 p.41-8.

76
LOPES W.A., ANDRADE J.B. Fontes, formação, reatividade e quantificação de hidrocarbonetos policíclicos aromáticos (HPA) na atmosfera,1996. Química Nova , v.9, n.5, p.497-516. Disponível em: < Disponível em: <http://www.scielo.br>. Acesso em: 11 dez. 2009.>. Acesso em: 11 abr. 2010. MEIRE, R. O.; AZEREDO, A.; TORRES, J. P. M. Aspectos ecotoxicológicos de hidrocarbonetos policíclicos aromáticos. Oecol. Brás. , v. 11, n. 2, 2007. Disponível em: <http://www.scielo.br>. Acesso em: 11 dez. 2009. MUKHTAR H; Ahmad N. Green tea in chemoprevention of cancer. Toxicological Sciences . v. 52, p. 111-117, 1999. Disponível em: <http://jn.nutrition.org/cgi/ content/full/135/12/2978S>. Acesso em: 08 abr. 2010. M. Inoue, K. Robien, R. Wang, DJ Van Den Berg, W.-P. Koh, and MC Yu E MC Koh Yu. Green tea intake, MTHFR/TYMS genotype and breast cancer risk: the Singapore Chinese Health Study. Carcinogenesis , v. 29, n. 10, p.1967 – 1972, oct. 2008. Disponível em: <http://carcin.oxfordjournals.org/cgi/content/short/27/7/1310>. Acesso em: 08 abr. 2010. NIEVA-CANO MJ, et. al.. Determination of polycyclic aromatic hydrocarbons with fluorimetric detection following sonication without sample clean-up.Comparison of two spectrofluorimetric methods apllied to water samples, 2001. Analyst . v.126, p. 1326-31. PEREIRA NETO, A. D.,et.al.. Avaliação da contaminação humana por hidrocarbonetos policíclicos aromáticos (HPAs) e seus derivados (NHPAs): uma revisão metodológica. Química Nova , 2000. Disponível em: <http://www.scielo.br>. Acesso em: 13 dez. 2009. PHILLIPS, D. H. Polycyclic aromatic hydrocarbons in the diet. Mutation Research , v. 443, p. 139- 147, 1999. Disponível em: <http://www.sciencedirect.com> Acesso em: 11 dez. 2009. SILVA, B. A. K.; et. al.. Usefulness of argyrophilic nucleolar organizer regions in detection of lung cells alterations after benzo(a)pyrene instillation. Acta Cirúrgica Brasileira , v. 21, n. 4, 2006. Disponível em: <http://www.scielo.br>. Acesso em: 30 mar. 2010. SIMKO, P. Determination of polycyclic aromatic hydrocarbons in smoked meat products andsmoking flavoring food additives. Journal of Cromatography B , v. 770, 2002. Disponível em: <http://www.sciencedirect.com>. Acesso em: 30 mar. 2010.

77
SKOOG, Douglas A. et al. Fundamentos de química analítica. São Paulo: Pioneira Thomson Learning, 2006. SKOOG, Douglas A.;et.al.. Princípios de análise instrumental. 5. ed. Porto Alegre: Artmed, 2001. TFOUNI, S. A. V. Estudo do efeito do processamento na contaminação d e cana-de-açúcar e derivados por hidrocarbonetos policícli cos aromáticos . 2005. 113f. Tese (Doutorado em Ciência dos Alimentos) – Universidade Estadual de Campinas, Campinas, 2005. Disponível em: <http://www.unicamp.br>. Acesso em: 30 dez. 2009. TOLEDO, M. Cecilia de F.; CAMARGO, M. Sílvia F.O.. Benzo(a)pireno em óleos de milho produzidos e comercializados no Brasil. Ciênc. Tecnol. Aliment. , Campinas, v. 18, n. 1, Apr. 1998. Disponível em: <http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0101-20611998000100016&lng=en&nrm=iso>. Acesso em: 01 abr. 2010. ZHAO JF. Green tea protects against psoralen plus ultraviolet A-induced photochemical damage to skin. Journal of Investigative Dermatology , v. 113, p. 1070-1075, 1999. Disponível em: <http://www.naturezaonline.com.br/natureza/conteudo/pdf/04_CavalcantiASSetal_7684.pdf>. Acesso em: 08 abr. 2010.


















