
Dangling Bond Defects: The Critical Roadblock to E ...zhiwu/research/my_papers/JPhysChemC_11… ·...
23
Dangling Bond Defects: The Critical Roadblock to Efficient Photoconversion in Hybrid Quantum Dot Solar Cells Huashan Li,* Zhigang Wu,* and Mark T. Lusk* Department of Physics, Colorado School of Mines, Golden, Colorado 80401, United States * S Supporting Information ABSTRACT: Inorganic−organic hybrid materials based on silicon quantum dots (SiQDs) have been utilized for photovoltaic applications but suffer from rapid charge recombination and low carrier mobility. We present an ab initio investigation of charge dynamics to pinpoint the source of this severe problem, and our results indicate that such devices show great promise provided that dangling bond (DB) defects can be sufficiently removed. Without DBs, the predicted charge transfer (CT) rate is much higher than that of photoluminescence (PL), while the electron hopping (EH) proceeds more quickly than interfacial charge recombination (CR). In contrast, one DB in a SiQD leads to a dramatic enhancement, by 10 orders of magnitude, in the CR rate and a reduction of the EH rate by 4 orders of magnitude, so that the diffusion of carriers to electrodes becomes extremely difficult. Although other factors, such as dot size distribution and oxidation, also play a deleterious role in device performance, their effects are deemed much less important than the critical role played by dangling bonds. ■ INTRODUCTION SiQD-based inorganic−organic hybrid solar cells are an attractive candidate for photovoltaic applications as they combine the advantages of their constituents. While the conjugated polymers have strong absorption in the visible range, 1 SiQDs offer tunable energy levels, high stability 2,3 and the potential for multiple exciton generation and hot carrier collection. 4 Unfortunately, the solar conversion efficiency of the current SiQD/P3HT blend devices is only 1.1%, 3 much lower than the typical value of 5% for their purely organic counterparts. 5 To understand the origin of such limited performance, a novel broadband (UVVisNIR) transient absorption spectros- copy was recently employed to probe the relevant charge dynamics. The results indicate that in regioregular P3HT/SiQD films, the inherent photovoltaic conversion process is photo- excitation on P3HT followed by an ultrafast electron transfer from P3HT to the SiQD. Such exciton dissociation would primarily result in free charge carriers, but in reality only a small proportion of these carriers are able to diffuse to the electrode because of overwhelming nonradiative recombination at the interface. 6 Although the charge transfer (CT) rate of 7 × 10 12 s −1 is extremely high, the ultrafast charge recombination (CR), with a rate of 1× 10 12 s −1 , along with the low electron mobility (10 −6 ∼ 10 −5 cm 2 V −1 s −1 ) 7 causes the poor performance of the SiQD/P3HT solar cells. 6 It is widely believed that a more precise control of the dot size distribution and a lower density of trap states are required, but their influences on the charge dynamics are still unclear, 2,3,8 in particular, which factor is most critical to the deteriorated photoconversion? This has motivated our ab initio investigation to identify the main source responsible for the observed rapid recombination and low electron mobility, which would allow attention to be focused on the key roadblock to device performance. Four possible reasons for the low photoconversion efficiency have been postulated: (1) vibronically induced transitions to the ground state due to conical intersection at the degenerate points of the potential energy surfaces; 6 (2) dot size distribution; (3) oxidation; (4) surface dangling bonds. However, quantitative support for these possibilities is lacking, motivating the current analysis. As detailed in the subsequent section, the associated computational methodology calls for phonon-assisted transport theory beyond the standard Marcus and MJL (Marcus, Levich, and Jortner) theories, and excited- state electronic structure beyond those estimated from density functional theory (DFT) are required. The development of a successful computational approach is itself an important aspect of our investigation. Four rate estimates, depicted in Figure 1, are carried out using first-principles analysis: (Figure 1a) CT of singlet exciton; (Figure 1b) photoluminescence (PL) from P3HT; (Figure 1c) CR at the interface; (Figure 1d) electron hopping (EH) between two dots. Since phonon-assisted hopping is the Received: July 19, 2013 Revised: December 9, 2013 Published: December 9, 2013 Article pubs.acs.org/JPCC © 2013 American Chemical Society 46 dx.doi.org/10.1021/jp407190p | J. Phys. Chem. C 2014, 118, 46−53
-
Upload
phungkhanh -
Category
Documents
-
view
214 -
download
0
Transcript of Dangling Bond Defects: The Critical Roadblock to E ...zhiwu/research/my_papers/JPhysChemC_11… ·...

Dangling Bond Defects: The Critical Roadblock to EfficientPhotoconversion in Hybrid Quantum Dot Solar CellsHuashan Li,* Zhigang Wu,* and Mark T. Lusk*
Department of Physics, Colorado School of Mines, Golden, Colorado 80401, United States
*S Supporting Information
ABSTRACT: Inorganic−organic hybrid materials based onsilicon quantum dots (SiQDs) have been utilized forphotovoltaic applications but suffer from rapid chargerecombination and low carrier mobility. We present an abinitio investigation of charge dynamics to pinpoint the sourceof this severe problem, and our results indicate that suchdevices show great promise provided that dangling bond (DB)defects can be sufficiently removed. Without DBs, thepredicted charge transfer (CT) rate is much higher than thatof photoluminescence (PL), while the electron hopping (EH)proceeds more quickly than interfacial charge recombination(CR). In contrast, one DB in a SiQD leads to a dramaticenhancement, by 10 orders of magnitude, in the CR rate and a reduction of the EH rate by 4 orders of magnitude, so that thediffusion of carriers to electrodes becomes extremely difficult. Although other factors, such as dot size distribution and oxidation,also play a deleterious role in device performance, their effects are deemed much less important than the critical role played bydangling bonds.
INTRODUCTION
SiQD-based inorganic−organic hybrid solar cells are anattractive candidate for photovoltaic applications as theycombine the advantages of their constituents. While theconjugated polymers have strong absorption in the visiblerange,1 SiQDs offer tunable energy levels, high stability2,3 andthe potential for multiple exciton generation and hot carriercollection.4 Unfortunately, the solar conversion efficiency of thecurrent SiQD/P3HT blend devices is only 1.1%,3 much lowerthan the typical value of 5% for their purely organiccounterparts.5
To understand the origin of such limited performance, anovel broadband (UVVisNIR) transient absorption spectros-copy was recently employed to probe the relevant chargedynamics. The results indicate that in regioregular P3HT/SiQDfilms, the inherent photovoltaic conversion process is photo-excitation on P3HT followed by an ultrafast electron transferfrom P3HT to the SiQD. Such exciton dissociation wouldprimarily result in free charge carriers, but in reality only a smallproportion of these carriers are able to diffuse to the electrodebecause of overwhelming nonradiative recombination at theinterface.6 Although the charge transfer (CT) rate of 7 × 1012
s−1 is extremely high, the ultrafast charge recombination (CR),with a rate of 1× 1012 s−1, along with the low electron mobility(10−6 ∼ 10−5 cm2 V−1 s−1)7 causes the poor performance of theSiQD/P3HT solar cells.6 It is widely believed that a moreprecise control of the dot size distribution and a lower densityof trap states are required, but their influences on the charge
dynamics are still unclear,2,3,8 in particular, which factor is mostcritical to the deteriorated photoconversion?This has motivated our ab initio investigation to identify the
main source responsible for the observed rapid recombinationand low electron mobility, which would allow attention to befocused on the key roadblock to device performance. Fourpossible reasons for the low photoconversion efficiency havebeen postulated: (1) vibronically induced transitions to theground state due to conical intersection at the degeneratepoints of the potential energy surfaces;6 (2) dot sizedistribution; (3) oxidation; (4) surface dangling bonds.However, quantitative support for these possibilities is lacking,motivating the current analysis. As detailed in the subsequentsection, the associated computational methodology calls forphonon-assisted transport theory beyond the standard Marcusand MJL (Marcus, Levich, and Jortner) theories, and excited-state electronic structure beyond those estimated from densityfunctional theory (DFT) are required. The development of asuccessful computational approach is itself an important aspectof our investigation.Four rate estimates, depicted in Figure 1, are carried out
using first-principles analysis: (Figure 1a) CT of singlet exciton;(Figure 1b) photoluminescence (PL) from P3HT; (Figure 1c)CR at the interface; (Figure 1d) electron hopping (EH)between two dots. Since phonon-assisted hopping is the
Received: July 19, 2013Revised: December 9, 2013Published: December 9, 2013
Article
pubs.acs.org/JPCC
© 2013 American Chemical Society 46 dx.doi.org/10.1021/jp407190p | J. Phys. Chem. C 2014, 118, 46−53

dominant carrier transport mechanism,9 the calculations arecarried out within a nonadiabatic quantum tunneling setting.9,10
In particular, we employ the full quantum treatment thatexplicitly accounts for the effects of all phonon modes.9 Thisstate-of-the-art approach is computationally very demanding,and has rarely been applied to realistic systems.
COMPUTATIONAL APPROACHThe Marcus11 and MJL10,12 equations have been widely used inthe organic community to successfully calculate the chargetransfer rates. However, they are based on assumptions that arenot satisfied by the Si147H100/P3HT system. In particular, thefrequencies of most phonon modes far exceed the energy ofthermal fluctuations at room temperature so the high-temperature approximation on which Marcus theory is basedis not valid. In addition, the reorganization energy spectrum istoo broad to be represented by a single effective mode, so theMJL equation cannot be used either.At the SiQD/P3HT interfaces, coupling to the environment
is strong while coupling between donor and acceptor iscomparatively weak. The assumption of carrier transport viaincoherent nonadiabatic quantum tunneling scenario is there-fore reasonable. The full quantum treatment, based onperturbation theory, gives the charge transport rate as9,10
∫
∑
=ℏ
| |
Δℏ
− + − − +ω
ω
−∞
+∞
−⎪
⎪
⎪
⎪
⎧⎨⎩⎫⎬⎭
k V t
iG
t S n n n
1d
exp [(2 1) e ( 1)
e ]
jj j j
i tj
i t
fullQM 2 DA2
j
j
(1)
Here −ΔG is the driving force, VDA is the electron couplingbetween donor and acceptor, and the phonon occupationnumber is
ω=
ℏ −n
k T1
exp( / ) 1jj B (2)
The Huang−Rhys factor Sj is defined as
λ ω= ℏS /j j j (3)
where λj is the reorganization energy for vibrational mode j.
In the high-temperature limit, i.e., when kBT≫ ℏωj holds forall vibrational modes j, the full quantum treatment reduces tothat of Marcus theory11
πλ
λλ
= | |ℏ
− Δ +⎧⎨⎩⎫⎬⎭k V
k TG
k Texp
( )4DAMarcus
22
B
2
B (4)
with λ the total reorganization energy.For lower temperatures, CT may be coupled to both high-
frequency intramolecular modes and low-frequency solventmodes: ℏωj ≫ kBT ≫ ℏωs. In this regime it is appropriate touse the MJL equation10,12 provided that the phonon spectrumin the high frequency range is narrow enough to be representedby one effective mode. Under these conditions, the transportrate is given by
∑ππ λ ν
λ ν ωλ
=ℏ
| |!
−Δ + + ℏ
ν
ν
=
∞−
⎧⎨⎩⎫⎬⎭
k Vk T
S
Gk T
2 14
e
exp( )
4
s
S
s
s
MJL DA2
B 0
eff2
B (5)
Here λs is the reorganization energy related to solvent orsurrounding matrix, and ωeff is the frequency of the effectivemolecular mode. For our system, the calculated molecularmodes with energies lower than thermal fluctuations (24 meV)were merged into the λs term, and ωeff was taken to be theweighted average over molecular modes with energies higherthan 24 meV.In all of these methods, there exists an implicit assumption
that the phonon-assisted charge transport is slower thanvibrational relaxation, making it reasonable to employ anequilibrium phonon occupation. Although the frequencies ofboth molecular and solvent10 phonon modes exceed 10 cm−1,some of our calculated rates turned out to violate thisassumption. For those cases, we employed a self-consistentcorrection procedure wherein a cutoff phonon frequency is setto the original transfer rate. By excluding the contribution ofthe phonon modes with frequencies lower than this cutoff, anew rate and a corresponding new cutoff frequency wereobtained. Such iterations were continued until self-consistencywas reached.The driving forces were computed as the energy changes of
isolated donor and acceptor, plus the Coulomb interactionbetween pairwise charges localized on each atom obtained byMulliken population analysis.13 The reorganization energy λwas divided into an inner part λin and an external (solvent) partλs, corresponding to donor/acceptor and P3HT matrix,respectively. Within the harmonic approximation, the reorgan-ization energy spectrum was obtained by modal decomposition.The driving force and inner reorganization energy were
calculated from density functional theory (DFT) as imple-mented in the DMOL package.14 The conductor-like screeningmodel (COSMO)15 was used to account for the dielectric effectof the solvent. An all-electron approach was used with exchangeand correlation effects accounted for by the generalizedgradient approximation (GGA) parametrized by Perdew,Burke, and Ernzerhof (PBE).16 A real-space, double numericplus polarization (DNP) basis was used along with an octupoleexpansion to specify the maximum angular momentumfunction.14 One exception was the optical gap, which was setto be 2.0 eV as observed in experiments because of theincapability of our calculation to capture the π−π stacking
Figure 1. Initial states and final states: (a) charge transfer (CT); (b)photoluminescence (PL); (c) nonradiative charge recombination(CR); (d) electron hopping (EH); (e) polaron dissociation (PD).Tan spheres depict SiQDs and red wavy lines are P3HT. Yellowellipses represent exciton binding in part a and Coulomb trapping inparts b, c, and e.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp407190p | J. Phys. Chem. C 2014, 118, 46−5347

effect.13 Besides, an empirical formula was used for the externalreorganization energy,13 and we verified that the transition rateswere quite insensitive to both the frequencies and thereorganization energies of the solvent modes.The electronic coupling VDA for CT and CR between the dot
and molecule were calculated using a two-state model17 at theHartree−Fock (HF) level18 as implemented in the NWChemPackage.19 The anticrossing methodology9,20 was employed tocompute VDA for EH by taking advantage of the similar energylevels of initial and final states. Once the charge hopping rate isobtained, the drift mobility can be evaluated as21
μ = ek T N
r k1
2B
2EH
(6)
where N = 3 is the dimensionality and r is the distance betweenthe center of the neighboring SiQDs.This computationally intense approach restricts our explicit
calculations to dots with diameters less than 2 nm, while CTand CR rates for larger dots might be estimated viaextrapolation. In particular, the lowest unoccupied molecularorbital (LUMO) shifts of large dots are estimated to be half ofthe optical gap changes,22 and the solvent reorganization energycan be computed by fixing the face-to-face distance between thedot and the P3HT. In addition, the spectrally resolvedreorganization energy can be taken to be that of a computa-tionally tractable dot but rescaled so as to match the totalreorganization energy of each dot.9 A similar approach wasapplied to estimate the electron hopping rates between twodots with different diameters along with an additionalassumption that only the wave functions within a prescribeddistance of the dot surfaces contribute to the electroniccoupling.In addition to the CT, CR, and CH rates associated with
nonradiative processes, the photoluminescence (PL) rate atroom temperature was calculated via Fermi’s golden rule,4,23
∑ε
=ℏ
| |k p if E
cM( )
4e ( )
3i
l iiPL
2 2mol
3
3 32
(7)
Here p(i) is the room-temperature Boltzmann occupation, Ei isthe excitonic energy of excited state i, f l is the local field factorderived from the effective dielectric constant, and Mi is thedipole matrix element between excitonic state i and the groundstate. The excitonic state is described as a linear combination ofpairwise electron−hole wave functions. Both the coefficientsand the excitonic energies were obtained by solving the Bethe−Salpeter equation (BSE) using the RGWBS suite,24 whiledipole moments were computed using the MX code.25
It is also worth noting that the energy-level alignment andthe charge transfer rates are determined by the quasi-Fermilevel and thus depend on the device working conditions.26
However, it is very difficult to obtain the interaction betweenthe electrolyte and the SiQD/P3HT blend via quantumchemistry calculations, which is beyond the scope of ouranalysis. In this work, we focus on the rates in the limit ofisolated dots without connection to the electrodes.More details on the computational method are available in
the Supporting Information.
RESULTS AND DISCUSSIONFour Essential Rates to Determine Charge Dynamics.
Four rates, CT, CR, PL, and EH, were quantified to elucidatethe complex charge dynamics carried out in SiQD/P3HT films.
Three related processes were not considered because: (1) notriplet exciton is detected during the ultrafast charge transfer,ruling out intersystem crossing as a source of efficiency loss;6
(2) hole mobility through P3HT is measured to be muchhigher than electron mobility among the dots;27 and, (3) thedissociation rate of the interfacial charge transfer state(polarons) bound by Coulomb attraction is known to beextremely fast6 even though the relevant binding energies canbe as large as 0.1−0.5 eV.28
To elucidate the efficiency of polaron dissociation (Figure1e)i.e., why holes can easily escape from deep Coulomb trapsand create free carriers in the filmwe consider hole hoppingfrom a nearest neighbor (NN) P3HT to a next NN P3HT inthe vicinity of a 4 nm SiQD. Regioregular P3HT (rrP3HT)exhibits lamellar aggregation,29 so it is reasonable to assume aπ-stacking separation distance of 3.8 Å. In addition, our DFTcalculations indicate that the separation distance between thedot surface and a NN P3HT is 4 Å. Adopting an effectivedielectric constant of ε = 3 results in an estimated Coulombwell of 27 meV. This implies that the Coulomb well does notblock polaron from dissociation at room temperature, and it isactually an upper bound since the NN P3HT may not wraptightly around the dot.Such Coulomb attraction is expected to be stronger for
smaller dots, however. In particular, dots of the optimal size forproviding the largest open circuit voltage are predicted by ouranalysis to be 17.4 Å in diameter, and the associated Coulombwell depth for a hole to hop from a NN P3HT to a next NNP3HT increases to 87 meV. We argue that this should belargely offset by a larger energy gradient related to the stongerquantum confinement in more disordered P3HT closer to theinterface of such small dots. For instance, previous computa-tional analysis has shown that the highest occupied molecularorbital (HOMO) of the P3HT around the PCBM (withdiameters of 1−2 nm) is 150 meV lower than that of the P3HTfar away.30 As a result of this mitigating influence of the energygradient, we expect that holes can efficiently leave the interfaceat room temperature.In addition, wave function delocalization associated with the
crystalline environment should also mitigate the influence ofany Coulomb trap. Specifically, delocalization of the hole wavefunction in crystallized P3HT favors polaron dissociation dueto the significantly enhanced effective distance between thehole and electron. This is illustrated in Figure 2 where theHOMO in the P3HT close to the SiQD has a nearlyhomogeneous distribution over all of the three layers includedin our calculation.
Figure 2. Isosurfaces of HOMO (blue) and LUMO (red) at a Si35H36/P3HT interface. The isosurfaces are plotted at electron densities of0.02 Å−3/2.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp407190p | J. Phys. Chem. C 2014, 118, 46−5348

Charge Transfer, Recombination, and Photolumines-cence on Defect-free Interface. The HOMO and LUMOlevels of a SiQD depend on its size, and the construction of atype-II junction, in which not only HOMOs and LUMOs forma staggered level alignment but also the local excited-stateenergy is higher than the CT state energy, requires that the dotsbe sufficiently large. This is summarized in Figure 3a, obtained
by idealizing the P3HT to a 6-unit oligomer as in previousworks13 as shown in Figure 3b,c. However, photoconversionefficiency decreases for sizes beyond this threshold value.2,3 Ahydrogen-passivated Si147 dot with a diameter of 1.7 nm wasfound to be optimal with minimum magnitude of driving forceand was utilized here for the following rate analyses.Our calculated phonon-assisted CT/CR rates for defect-free
SiQD/P3HT interface and the EH rate between two dotsemploying three approaches are summarized in Table 1, withthe phonon spectrum shown in Figure 4. For the CT at theSi147H100/P3HT interface and EH between two dots at the
Marcus normal region, the three approaches gave similar rates.In contrast, within the Marcus inverted region, the CR ratespredicted by the Marcus and MJL formulas were many ordersof magnitude lower than the more accurate prediction of thefull quantum mechanical treatment.This observation could be explained within the scenario of
phonon-assisted quantum tunneling. The major contributionsto the transport rates come from the resonance channelsbetween the vibronic levels of initial and final states. In theMarcus normal range, the magnitude of driving force |ΔG| isclose to the reorganization energy λ, and the resonancechannels correspond to the molecular phonon modes. The lowfrequency modes in P3HT solvent are dominant so that bothMarcus and MJL theories give reasonable estimates of the rates.However, in the Marcus inverted range, the high-frequencymodes that are ignored in Marcus theory play a critical role,while using one effective mode in the MJL equation reduces thepossibility to achieve resonance. A full quantum treatment istherefore necessary in this range, which requires a substantialcomputational investment.31
In our analysis of the influence of dot size distribution andsurface defect on charge dynamics, CT, CR, and CH may all bein the Marcus inverted region as the driving forces far exceedthe reorganization energies, so the full quantum treatment isnecessary and has been carried out to obtain all associated rates.As illustrated in Figure 5, our calculated CT (1.5 × 1012 s−1)
and PL (7.9 × 108 s−1) rates are in good agreement with
experiments,6,32 demonstrating that our model accuratelycaptures the key dynamics. Crucially, though, the predictedCR rate (1.5 × 102 s−1) is lower by 10 orders while the electronmobility (2.9 × 10−2 cm2 V−1 s−1) is higher by 3−4 orders thanthe reported experimental values of 1012 s−16 and 10−6 ∼ 10−5
Figure 3. (a) Energy level alignment between P3HT and SiQDs ofdiffering size. The singlet exciton state (S1) of P3HT and the chargetransfer state (CT) of the optimized interface are illustratedschematically in red and green dashed lines. (b, c) Side and topviews of the Si147H100/P3HT interface with HOMO and LUMOplotted in blue and red, respectively. A small organic molecule, with 6units in the backbone, was used to represent the P3HT. Atom colors:Si (tan); H (white); C (gray); S (yellow). The isosurfaces are plottedfor an electron density of 0.015 Å−3/2.
Table 1. Calculated CT and CR Rates at the Si147H100/P3HTInterface and the EH Rate between Two Si147H100 DotsUsing Full Quantum Mechanical Treatment, Marcus Theory,and MJL Theorya
full QM Marcus MJL
kCT 4.0 × 1012 4.0 × 1012 4.4 × 1012
kCR 1.5 × 102 4.6 × 10−31 1.9 × 10−3
kEH 1.3 × 1011 1.2 × 1011 1.2 × 1011
aAll rates are in unit of s−1.
Figure 4. Spectrally resolved reorganization energy for excitondissociation on P3HT. The blue and purple lines correspond to thephonon modes of Si147H100 and P3HT respectively, while the greendashed line denotes the thermal energy at room temperature.
Figure 5. CT and CR rates at Si147H100/P3HT interface, PL rate onP3HT, and EH rate between dots from our ab initio calculations andpreviously reported experiments.6,7,32
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp407190p | J. Phys. Chem. C 2014, 118, 46−5349

cm2 V−1 s−1,7 respectively; therefore, the poor photoconversionefficiency is the result of something, not yet in the ideal model,that is causing the CR rate to be too high and the EH rate to betoo low. In the following, we quantify the impact of eachpreviously postulated source on CR and EH rates to pinpointthe discrepancy between theory and experiment.Influence of Conical Intersection, Dot Size Distribu-
tion, and Oxidation. The first possible reason for thediscrepancy between our calculated CR/EH rates and theexperimental data is that transitions to the ground state canrapidly occur due to conical intersections of charge transfer andground state energy surfaces at the degenerate points. In thevicinity of those points, the Born−Oppenheimer approximationbreaks down, and a large surface-hopping rate within thenonadiabatic transition channel rises due to strong vibroniccoupling. This has been shown to be responsible for theextremely fast charge recombination of many organicmolecules.33
For our interface, the scenario of conical intersection isphotoexcitation from the ground state (GS) to the local exciton(LE) manifold followed by a vertical transition to the CTmanifold at the point B in Figure 6. Subsequent relaxation
brings the system to the conical intersection from which anefficient, nonradiative surface hop to the GS manifold ispossible. The conical intersection cannot be reached by thermalexcitation from point C though. Suppose that this mechanism isresponsible to the ultrafast charge recombination at ourSi147H100/P3HT interface, then, during the relaxation of theCT state from the LE structure, the energy surface of the GShappens to intersect that of the CT state. So the prerequisite ofthis process is that the energy of the GS in the lowest energyCT geometry (point D) should be higher than the energy ofthe CT in lowest energy CT geometry (point C). However, ourcomputation gives ED − EC = −1.66 eV, indicating that there isno conical intersection between points B and C. Therefore, thisnonradiative process cannot be the reason for the observedultrafast CR.A second postulated source for rapid CR and slow EH is that
assemblies of SiQDs have a substantial size distribution. Toexamine this, we used the computed CT and CR rates for theSi147H100/P3HT interface to estimate analogous rates for dotsof differing sizes. Driving forces were estimated using opticalgaps obtained from previous theoretical studies,22 and thereorganization energies and electron couplings were scaled withrespect to dot size.9 As shown in Figure 7, the change in drivingforce is about 0.8 eV when the dot diameter increases from 1.7to 8 nm. This leads to a decrease of the CT rate and an increase
in the CR rate (Figure 7a). However, even for 8 nm dots, thecalculated CR rate is still lower by 7 orders than theexperimental value,6 suggesting that there must exist otherultrafast CR channels.An accompanying analysis was carried out for EH between
two SiQDs over a range of size mismatches (Figure 7b). Therates between two identical dots with different sizes are similar,varying within only two orders when diameter increases from1.7 to 8 nm, and electrons can hop efficiently from smaller dotsto larger dots provided that the size difference is not too large.However, hopping from larger dots to smaller dots isenergetically unfavorable so electrons should tend to gettrapped in larger dots. Thus, QD size fluctuation would be aplausible explanation for low electron mobilities, but it alonecannot explain the ultrafast CR, and our subsequent analysisindicates that the dangling bond defects offer even strongertrapping to electrons.The third possible source is surface oxidation, which was
investigated by inserting 12 OH ligands (Figure 8b) and 12backbond O atoms (Figure 8c) into the dot surface (Figure 8a).Our computations suggest that the OH ligand has a negligibleimpact on either the HOMO/LUMO levels (Figure 9) or thewave function localization. In contrast, while the shape of theLUMO remains similar to that of the nonoxidized dot, a highdensity of backbond O atoms (12 O/per dot) reduces the
Figure 6. Sketch of the conical intersection mechanism for chargerecombination. The parabolas are potential energy surfaces of ground(GS), local exciton (LE), and charge transfer (CT) states.
Figure 7. (a) Charge transfer and recombination rates and drivingforces at SiQD/P3HT interface as functions of dot diameter. (b)Electron hopping rate from one SiQD to another dot with equal orlarger size.
Figure 8. Isosurfaces of LUMO for (a) Si147H100, (b) Si147H100 with 12OH ligands, and (c) with 12 backbond O atoms. Here all isosurfacesare plotted at electron densities of 0.015 Å−3/2.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp407190p | J. Phys. Chem. C 2014, 118, 46−5350

LUMO energy by 0.4 eV. However, such high backbond Odensity is highly unlikely;34 moreover, this is still much smallerthan the change of 1.2 eV caused by the same density of DBs,and even smaller than the 0.9 eV caused by only one DB on aSiQD, indicating that although the O backbonds could serve asshallow electron trap to reduce the device performance, theireffect is much less important than the DB.Dangling Bonds: The Primary Source of Fast Charge
Recombination and Slow Electron Hopping. After rulingout the above possibilities, we now demonstrate that DBs arethe most likely source of high CR rates and low EH rates. Asingle DB defect was therefore created on the surface of theSi147H100 dot (Figure 10, parts c and d) and both CR and EH
hopping rates were then calculated. In the presence of the DB,the LUMO energy level of the dot decreases while the HOMOlevel increases significantly due to the generation of surfacestates inside the gap, leading to a much larger driving force(from 0.06 to 1.32 eV) for CT and a reduced driving force(from 1.84 to 0.58 eV) for CR (Figure 10a). In addition, amuch larger distortion of the SiQD with a DB present thanwithout a DB is induced by excitation, consequently thereorganization energy (λ) for the dot increases by a factor of 6(Figure 10b). The LUMO is localized around the DB (Figure
10, parts c and d) and now becomes closer to the HOMOwhich itself tends to be localized on the P3HT. As a result, theelectronic coupling increases for both CT and CR (Table S4 inthe Supporting Information).Combining all these together, our calculations suggest that
the introduction of only one DB on the Si147H100 dot onlyslightly decreases the CT rate, but increases the CR rate by 10orders of magnitude and decreases the EH rate by 4 orders ofmagnitude. With the DB accounted for, both CR and EH ratesare in reasonable agreement with experiment as shown in Table2. Moreover, the ratio of these rates asserts that electrons arevery unlikely to diffuse to the electrode prior to recombining.
As has been pointed out previously,26 the charge transfer ratecould be sensitive to the driving force in the inverted Marcusrange. In our case, introducing an 0.15 eV uncertainty in ΔG,which is the estimated upper bound for errors in our computeddriving forces, leads to a fluctuation of 1−2 orders of magnitudeon transition rates (Figure 11). For instance, the inaccuracy of
ΔG in EH mainly comes from the single-electron approx-imation. We have tested the case of an electron hopping fromthe dot Si147H100 with diameter d = 1.7 nm to the dot Si211H140with d = 2.0 nm, and the error in ΔG is only 0.03 eV comparingwith the delta SCF method. Even with an 0.15 eV uncertaintyin ΔG, the qualitative comparisons of rates among differentrelated processes do not change significantly, and thus ourconclusion is robust.
CONCLUSIONOur analysis suggests that the charge dynamics plays out in thethin film as follows. After photoexcitation of P3HT, chargetransfer occurs at the dot/polymer interface. The resultingpolarons are quickly dissociated into free charge carriers, andthe electrons subsequently diffuse efficiently through the film
Figure 9. Energy level alignments for HOMO and LUMO of P3HT,Si147H100, and Si147H100 with 12 dangling bonds, 12 OH ligands, and12 backbonded O atoms.
Figure 10. (a) Energy level alignment between P3HT and Si147H100without and with one dangling bond (DB). (b) Reorganization energyspectrum of Si147H100 neutral dot relaxed from anion structure withoutand with one DB. The inset shows the large excitation-induceddistortion around a DB. (c, d) Side and top views of localized HOMO(blue) and LUMO (red) of the interface between P3HT and Si147H100with one DB. The Si atom with a DB is highlighted in green. Theisosurfaces are plotted for an electron density of 0.015 Å−3/2.
Table 2. Charge Transfer (CT) and Charge Recombination(CR) Rates at Si147H100/P3HT Interface along with ElectronHopping (EH) Rate between Two Dots with and withoutOne Dangling Bond (DB) on Each Dota−1
no DBs DBs experiment
kCT 4.0 × 1012 2.8 × 1011 7 × 1012 [ref 6]kCR 1.5 × 102 2.7 × 1012 1 × 1012 [ref 6]kEH 1.3 × 1011 1.3 × 107 2 × 107 [ref 7]
aAll rates are in unit of s.
Figure 11. Changes in CT, CR, and EH rates for the interfaceswithout and with DB as functions of uncertainty in ΔG.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp407190p | J. Phys. Chem. C 2014, 118, 46−5351

before getting trapped at the DBs. As a result, electrons andholes eventually recombine on DBs.In order to extract free carriers to the external circuit, the
carriers must drift to the electrodes before CR occurs. Theefficiency of this process is determined by a comparisonbetween the carrier sweep out time and the CT state lifetime,where the carrier sweep out time is taken to be the periodrequired by the carrier to drift from the center of the activelayer to the electrode.35 Elimination of DB defects wouldimprove the EH hopping rate and reduce the CR rate, makingit possible for the CT state lifetime to be much longer than thecharacteristic sweep-out time of the carriers drifting in theinternal field.In summary, the charge dynamics of SiQD/P3HT solar cells
have been investigated from first-principles using a phonon-assisted full quantum treatment, which accurately captures theCT and PL rates and, with dangling bonds accounted for,recovers the CR and EH rates observed experimentally.6,7 Weconclude that the dangling bond defects are the critical reasonfor the deleterious ultrafast CR and low electron mobilitymeasured in these hybrid materials. Thus, the mitigation ofsuch defects is expected to dramatically improve the perform-ance of hybrid solar cells.
ASSOCIATED CONTENT
*S Supporting InformationDetailed computational methods, accuracy and sensitivityanalysis, and electron coupling with dangling bonds. Thismaterial is available free of charge via the Internet at http://pubs.acs.org.
AUTHOR INFORMATION
Corresponding Authors*E-mail: (H.L.) [email protected].*E-mail: (Z.W.) [email protected].*E-mail: (M.T.L.) [email protected].
NotesThe authors declare no competing financial interest.
ACKNOWLEDGMENTS
This research is supported by the Renewable Energy MaterialsResearch Science and Engineering Center (REMRSEC) and bystartup funds from Colorado School of Mines (CSM) and theU.S. DOE early career research award (No. DE-SC0006433).We also acknowledge the Golden Energy ComputingOrganization at the Colorado School of Mines for the use ofresources acquired with financial assistance from the NationalScience Foundation and the National Renewable EnergyLaboratories.
REFERENCES(1) Svrcek, V.; Yamanari, T.; Shibata, Y.; Kondo, M. Tailoring ofHybrid Silicon Nanocrystalbased Bulk Heterojunction PhotovoltaicProperties Upon Nanocrystal Laser Processing in Liquid Medium.Acta Mater. 2011, 59, 764−773.(2) Niesar, S.; Fabian, W.; Petermann, N.; Herrmann, D.; Riedle, E.;Wiggers, H.; Brandt Martin, S.; Stutzmann, M. Efficiency Enhance-ment in Hybrid P3HT/Silicon Nanocrystal Solar Cells. Green 2011, 1,339−350.(3) Liu, C.-Y.; Holman, Z. C.; Kortshagen, U. R. Hybrid Solar Cellsfrom P3HT and Silicon Nanocrystals. Nano Lett. 2009, 9, 449−452.
(4) Lin, Z.; Li, H.; Franceschetti, A.; Lusk, M. T. Efficient ExcitonTransport between Strongly Quantum-Confined Silicon QuantumDots. ACS Nano 2012, 6, 4029−4038.(5) Reyes-Reyes, M.; Kim, K.; Carroll, D. L. High-efficiencyPhotovoltaic Devices Based on Annealed Poly(3-hexylthiophene)and 1-(3-methoxycarbonyl)-propyl-1- phenyl-(6,6)C61 Blends. Appl.Phys. Lett. 2005, 87, 083506−083506−3.(6) Herrmann, D.; Niesar, S.; Scharsich, C.; Kohler, A.; Stutzmann,M.; Riedle, E. Role of Structural Order and Excess Energy on UltrafastFree Charge Generation in Hybrid Polythiophene/Si PhotovoltaicsProbed in Real Time by Near-Infrared Broadband TransientAbsorption. J. Am. Chem. Soc. 2011, 133, 18220−18233.(7) Holman, Z. C.; Liu, C.-Y.; Kortshagen, U. R. Germanium andSilicon Nanocrystal Thin-Film Field-Effect Transistors from Solution.Nano Lett. 2010, 10, 2661−2666.(8) Borchert, H. Elementary Processes and Limiting Factors inHybrid Polymer/Nanoparticle Solar Cells. Energy Environ. Sci. 2010, 3,1682−1694.(9) Chu, I.-H.; Radulaski, M.; Vukmirovic, N.; Cheng, H.-P.; Wang,L.-W. Charge Transport in a Quantum Dot Supercrystal. J. Phys. Chem.C 2011, 115, 21409−21415.(10) Jortner, J. Temperature Dependent Activation Energy forElectron Transfer between Biological Molecules. J. Chem. Phys. 1976,64, 4860−4867.(11) Marcus, R. A. Electron, Proton and Related Transfers. Farad.Discuss. 1982, 74, 7−15.(12) Barbara, P. F.; Meyer, T. J.; Ratner, M. A. Contemporary Issuesin Electron Transfer Research. J. Phys. Chem. 1996, 100, 13148−13168.(13) Liu, T.; Troisi, A. Absolute Rate of Charge Separation andRecombination in a Molecular Model of the P3HT/PCBM Interface. J.Phys. Chem. C 2011, 115, 2406−2415.(14) Delley, B. An All-electron Numerical-method for Solving theLocal Density Functional for Polyatomic-molecules. J. Chem. Phys.1990, 92, 508−517.(15) Klamt, A.; Jonas, V.; Burger, T.; Lohrenz, J. C. W. Refinementand Parametrization of COSMO-RS. J. Phys. Chem. A 1998, 102,5074−5085.(16) Perdew, J. P.; Wang, Y. Accurate and Simple AnalyticRepresentation of the Electron-Gas Correlation-Energy. Phys. Rev. B1992, 45, 13244−13249.(17) Farazdel, A.; Dupuis, M.; Clementi, E.; Aviram, A. Electric-fieldInduced Intramolecular Electron-transfer in Spiro Pi-electron Systemsand Their Suitability as Molecular Electronic Devices - A Theoretical-study. J. Am. Chem. Soc. 1990, 112, 4206−4214.(18) Wong, A. T.; Harrison, R. J. Approaches to Large-scale ParallelSelf-consistent-field Calculations. J. Comput. Chem. 1995, 16, 1291−1300.(19) Valiev, M.; Bylaska, E. J.; Govind, N.; Kowalski, K.; Straatsma,T. P.; Van Dam, H. J. J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A Comprehensive and Scalable Open-sourceSolution for Large Scale Molecular Simulations. Comput. Phys.Commun. 2010, 181, 1477−1489.(20) Shan, W.; Walukiewicz, W.; Ager, J. W.; Haller, E. E.; Geisz, J.F.; Friedman, D. J.; Olson, J. M.; Kurtz, S. R. Band Anticrossing inGaInNAs Alloys. Phys. Rev. Lett. 1999, 82, 1221−1224.(21) Deng, W. Q.; Goddard, W. A. Predictions of HoleMobilities inOligoacene Organic Semiconductors from Quantum MechanicalCalculations. J. Phys. Chem. B 2004, 108, 8614−8621.(22) Luo, J.-W.; Stradins, P.; Zunger, A. Matrix-embedded SiliconQuantum Dots for Photovoltaic Applications: a Theoretical Study ofCritical Factors. Energy Environ. Sci. 2011, 4, 2546−2557.(23) Califano, M.; Franceschetti, A.; Zunger, A. TemperatureDependence of Excitonic Radiative Decay in CdSe Quantum Dots:The Role of Surface Hole Traps. Nano Lett. 2005, 5, 2360−2364.(24) Tiago, M. L.; Chelikowsky, J. R. Optical Excitations inOrganicMolecules, Clusters, and Defects Studied by First-principlesGreen’s Function Methods. Phys. Rev. B 2006, 73, 205334.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp407190p | J. Phys. Chem. C 2014, 118, 46−5352

(25) Franceschetti, A.; Fu, H.; Wang, L. W.; Zunger, A. Many-bodyPseudopotential Theory of Excitons in InP and CdSe Quantum Dots.Phys. Rev. B 1999, 60, 1819−1829.(26) Maggio, E.; Martsinovich, N.; Troisi, A. Evaluating ChargeRecombination Rate in Dye-Sensitized Solar Cells from ElectronicStructure Calculations. J. Phys. Chem. C 2012, 116, 7638−7649.(27) Gunes, S.; Neugebauer, H.; Sariciftci, N. S. ConjugatedPolymer-Based Organic Solar Cells. Chem. Rev. 2007, 107, 1324−1338.(28) Clarke, T. M.; Durrant, J. R. Charge Photogeneration in OrganicSolar Cells. Chem. Rev. 2010, 110, 6736−6767.(29) Spano, F. C. Modeling Disorder in Polymer Aggregates: TheOptical Spectroscopy of Regioregular Poly(3-hexylthiophene) ThinFilms. J. Chem. Phys. 2005, 122, 234701.(30) McMahon, D. P.; Cheung, D. L.; Troisi, A. Why Holes andElectrons Separate So Well in Polymer/Fullerene Photovoltaic Cells. J.Phys. Chem. Lett. 2011, 2, 2737−2741.(31) Walker, G. C.; Akesson, E.; Johnson, A. E.; Levinger, N. E.;Barbara, P. F. Interplay of Solvent Motion and Vibrational-excitation inElectron-transfer Kinetics - Experiment and Theory. J. Phys. Chem.1992, 96, 3728−3736.(32) Piris, J.; Dykstra, T. E.; Bakulin, A. A.; Loosdrecht, P. H. v.;Knulst, W.; Trinh, M. T.; Schins, J. M.; Siebbeles, L. D. Photo-generation and Ultrafast Dynamics of Excitons and Charges in P3HT/PCBM Blends. J. Phys. Chem. C 2009, 113, 14500−14506.(33) Polli, D.; Altoe, P.; Weingart, O.; Spillane, K. M.; Manzoni, C.;Brida, D.; Tomasello, G.; Orlandi, G.; Kukura, P.; Mathies, R. A.; et al.Conical Intersection Dynamics of the Primary PhotoisomerizationEvent in Vision. Nature 2010, 467, 440−U88.(34) Li, H.; Lusk, M. T.; Collins, R. T.; Wu, Z. Optimal Size Regimefor Oxidation-Resistant Silicon Quantum Dots. ACS Nano 2012, 6,9690−9699.(35) Cowan, S. R.; Roy, A.; Heeger, A. J. Recombination in Polymer-Fullerene Bulk Heterojunction Solar Cells. Phys. Rev. B 2010, 82,245207.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp407190p | J. Phys. Chem. C 2014, 118, 46−5353

Supporting information: Dangling Bond Defects:
The Critical Roadblock to Efficient Photoconversion
in Hybrid Quantum Dot Solar Cells
Huashan Li,∗ Zhigang Wu,∗ and Mark T. Lusk∗
Department of Physics, Colorado School of Mines, Golden, CO 80401, USA
E-mail: [email protected]; [email protected]; [email protected]
∗To whom correspondence should be addressed
1

Computational Methods
Non-Radiative Charge Transfer Rate
The full quantum treatment, based on perturbation theory, gives the charge transport rate as1,2
k f ullQM =1h2 |VDA|2
∫ +∞
−∞
dt exp
i∆Gh
t−∑j
S j[(2n j +1)−n je−iω jt− (n j +1)eiω jt
], (1)
Here−∆G is the driving force, |VDA| is the electron coupling between donor and acceptor, n j is the
phonon occupation number and S j is the Huang-Rhys factor.
Energies released by the system following charge transfer (CT), charge recombination (CR),
or electron hopping (EH) are written as3
∆GCT = E+D +E−A +Ec−E0
D−E0A−Eopt
g (2)
∆GCR = E0D +E0
A−E+D −E−A −Ec (3)
∆GEH = ELUMOA −ELUMO
D (4)
E+/0D ,E−/0
A are ionic/ground state energy of donor and acceptor, Ec is the Coulomb stabilization
energy, Eoptg is the optical gap of the donor, and ELUMO
D/A are the LUMO levels of donor/acceptor.
The reorganization energy, λ , is divided into an inner part, λin, and an external part, λs, corre-
sponding to the donor/acceptor and the P3HT matrix respectively so that
λCTin = Eca/le
D +Ean/gsA −Eca/ca
D −Ean/anA (5)
λCRin = Egs/ca
D +Egs/anA −Egs/gs
D −Egs/gsA (6)
λEHin = Ean/gs
D +Egs/anA −Egs/gs
D −Ean/anA (7)
Here subscripts ca, an, le and gs represent cation, anion, local exiton and ground states, respec-
2

tively, while Ex/y denotes the energy of state x in y configuration, obtained by relaxation of state
y followed by energy calculation of state x occupation. Within the harmonic approximation, the
reorganization energy spectrum is obtained via a modal decomposition routine in which
λEHj =
12
ω2j ∆Q2
j (8)
The amplitude of the jth vibrational mode, ∆Q j, is calculated as
∆Q j = ∆~Q · e j (9)
where ∆~Q is the total displacement of the structure during the charge transfer. The vector, e j, is the
eigenvector of the jth vibrational mode.
Nearly all the above quantities were calculated from the density functional theory (DFT) as
implemented in the DMOL package.4 The Conductor-like Screening Model (COSMO)5 was used
to account for the dielectric screening of the solvent. An all-electron approach (no pseudopoten-
tial) was used with exchange and correlation effects accounted for by the generalized gradient
approximation (GGA) of Perdew, Burke, and Ernzerhof (PBE).6 A real-space, double numeric
plus polarization (DNP) basis was used along with an octupole expansion to specify the maximum
angular momentum function.4
Optical gaps cannot be reliably calculated using DFT, so we used the many-body perturbation
theory with the GW approximation to account for quasi-particle effects. This was followed by
solving the Bethe Salpeter Equation (BSE) to correctly obtain both energies and wave functions
for excitonic effects.7 Both quasi-particle and excitonic corrections were carried out using the
package including the RGWBS7 and Parsec8 code sets.
As a previously employed computational expedient,3 P3HT was idealized as a P3(C3H7)T
oligomer. Since it has comparable size to the dot, it is generally thought to preserve the essential
character of dot/polymer interaction. However, our calculated optical gap of P3HT is 2.4 eV, much
higher than the 2.0 eV observed in experiments due to the neglect of the π−π stacking effect.3 In
3

order to obtain accurate energy level alignments, the experimental value of the optical gap is used
along with the computational wave functions.
The following empirical formula is used for the external reorganization energy:3
λs =(∆q)2
2
(1
3√
(RxRyRz)D+
13√(RxRyRz)A
− 2lDA
)(1
εopt− 1
εs
)(10)
Here ∆q is the transferred charge, Ri are the semi-axes of the ellipses that separately encompass
the donor and acceptor: (9.7,8.8,9.4) Å for Si147H100 and (12.6,2.8,6.6) Å for P3(C3H7)T). The
parameter, lDA =10.7 Å is the center-to-center distance between donor and acceptor, and εopt/s
are the optical/static dielectric constants of the surrounding P3HT matrix.9 Because the evaluation
of these dielectric constants via ab initio methods is daunting, requiring an account of a linear
response to the electric field for an the ensemble average of molecule arrangements, we chose to
use experimental the experimental values of εopt =1.96 and εs =3.3
The electronic coupling, VDA, for CT and CR between the dot and molecule are calculated using
a two-state model10 at the Hartree Fock (HF) level11 as implemented in the NWChem Package:12
VDA =
∣∣H f i−S f i(Hii +H f f )/2∣∣
1−S2f i
(11)
Here S f i is the overlap between initial and final wavefunctions,
S f i = 〈Ψ f |Ψi〉 (12)
and the matrix elements of the total electron Hamiltonian are defined as
H f i = 〈Ψ f |H|Ψi〉, H f f = 〈Ψ f |H|Ψ f 〉, Hii = 〈Ψi|H|Ψi〉 (13)
The electronic coupling associated with EH between two dots must also be calculated, but the HF
method is a computationally expensive proposition in this case. By taking advantage of the similar
4

energy levels of initial and final states, an anticrossing methodology is employed instead. This
method forces the eigenvalue of one SiQD to cross the associated eigenvalues of the other dot by
applying a very small external electric field on the system. Minimizing the difference between
these two eigenvalues provides good estimation of 2VDA.2,13
Once the charge hopping rate is obtained, the drift mobility can be evaluated as14
µ =e
kBT1
2Nr2kEH (14)
where N = 3 is the dimensionality and r is the distance between the center of the neighboring
SiQDs.
Photoluminescence Rate
Starting from Fermi’s golden rule, the photoluminescence (PL) rate at room temperature is15,16
kPL = ∑i
p(i)4e2 f 2
l (εmol)E3i
3c3h3
∣∣∣~Mi
∣∣∣2 (15)
Here p(i) is the room-temperature Boltzmann occupation, Ei is the excitonic energy of excited
state i, fl is the local field factor derived from the effective dielectric constant, and Mi is the dipole
matrix element between excitonic state i and the ground state.
For the P3HT polymer film, the dielectric constant inside the molecule is similar to that of the
environment, so the local field factor for all multi-pole expansions reduces to17
fl(x) =2l +1
(εmol/εs +1)l +1≈ 1 (16)
The dipole matrix elements are calculated as the sum over pairwise electron-hole states,
~Mi = ∑h,e
Cih,e〈ψh|~r|ψe〉 (17)
5

where ψh/e are single-particle wave functions and Cih,e are coefficients that used to describe the
ith excitonic state as a linear combination of these wave functions. Both Cih,e and the associated
energies, Ei , are obtained from a combined GW-BSE analysis with the RGWBS suite,7 while
dipole moments are computed using the MX code.18
Sensitivity of Charge Transfer Rates to Solvent Modes
The main difficulty associated with the full quantum mechanical treatment lies on the computa-
tional challenge of using ab initio methods to quantitatively characterize the solvent modes. For-
tunately, our computational results demonstrate that when the frequency of the solvent mode is
much less than that of thermal fluctuations, the calculated rate is extremely insensitive to the sol-
vent phonon frequency. To verify this, a number of effective solvent frequencies (ωs) between 10
and 100 cm−1, a range typically associated with organic polymers,1 were tested and the corre-
sponding change of CT, CR and EH rates are less than 4% (Table S1). For convenience, the values
corresponding to the 10 cm−1 frequency are used for discussions in the manuscript.
Table S1: CS and CR rates at the Si147H100/P3HT interface and the EH rate between twoSi147H100 dots calculated using the full quantum mechanical treatment with varying effectivefrequencies for the solvent modes.
ωs(cm−1) 10 20 30 40 50kCT (s−1) 1.52E12 1.53E12 1.53E12 1.53E12 1.53E12kCR (s−1) 1.49E2 1.49E2 1.49E2 1.50E2 1.50E2kEH (s−1) 1.32E11 1.32E11 1.32E11 1.33E11 1.33E11ωs(cm−1) 60 70 80 90 100kCT (s−1) 1.53E12 1.53E12 1.53E12 1.53E12 1.54E12kCR (s−1) 1.51E2 1.51E2 1.52E2 1.53E2 1.54E2kEH (s−1) 1.33E11 1.33E11 1.33E11 1.33E11 1.34E11
Another potential arises from the possible inaccuracy of the empirical formula for solvent re-
organization energy (λs). We did observe a non-negligible variation of the CT, CR and EH rates
with varying values of λs, and this implies that a much more delicate solvent model should be used
in the future in order to quantitatively predict the rates. However, even if the error of λs is as large
6

as 30%, the rate inequalities that we obtained, CT PL and EHCR, do not change (Table S2).
Table S2: CS and CR rates at the Si147H100/P3HT interface and the EH rate between twoSi147H100 dots calculated by full mechanical quantum treatment with varying reorganizationenergies for the solvent modes.
Change in λs (%) -30 -20 -10 0 10 20 30kCT (s−1) 2.20E12 1.94E12 1.72E12 1.52E12 1.35E12 1.20E12 1.06E12kCR (s−1) 6.33E1 8.43E1 1.12E2 1.49E2 1.97E2 2.62E2 3.46E2kEH (s−1) 2.15E11 1.82E11 1.55E11 1.32E11 1.13E11 9.67E10 8.28E10
Use of an Oligomer to Represent the P3HT Polymer
In our calculations, the large P3HT polymer is idealized as a small molecule containing 6 thiophene
rings in the main chain and 3 carbon units in the side chains. This is computationally motivated
and is based on the measured effective conjugation length of polythiophene as being between 6
and 12 thiophene rings.19 In addition, the effect of the side alkyl chains on the electronic structure
and optical properties of the polymer are expected to be negligible.20,21 A similar approach was
used to represent P3HT in previous ab initio calculations that successfully reproduced the energy
level alignment, CT and CR rates at P3HT-PCBM interfaces when compared with experiment
measurements.3 A longer side chain in our molecule, as compared with that study, was adopted
in order to accommodate the larger sizes of the SiQDs as compared with PCBM. This idealization
may slightly change the energy level alignment at the interface, but this is not deemed to be too
important because the data is only used to identify an optimal dot size to match the experimental
energy levels of the P3HT, the goal being to find a dot size for which the driving force for CT is
positive but nearly zero. Because of this, our conclusions that the energy loss is small, the exciton
dissociation is fast, and the interfacial CR are not deemed to rest on the idealization of the P3HT.
7

Figure S1: LUMO (blue) and LUMO+1 (red) of an Si147H100 dimer. The values of the isosurfacesare 0.015 Å−3/2.
Anti-crossing Method
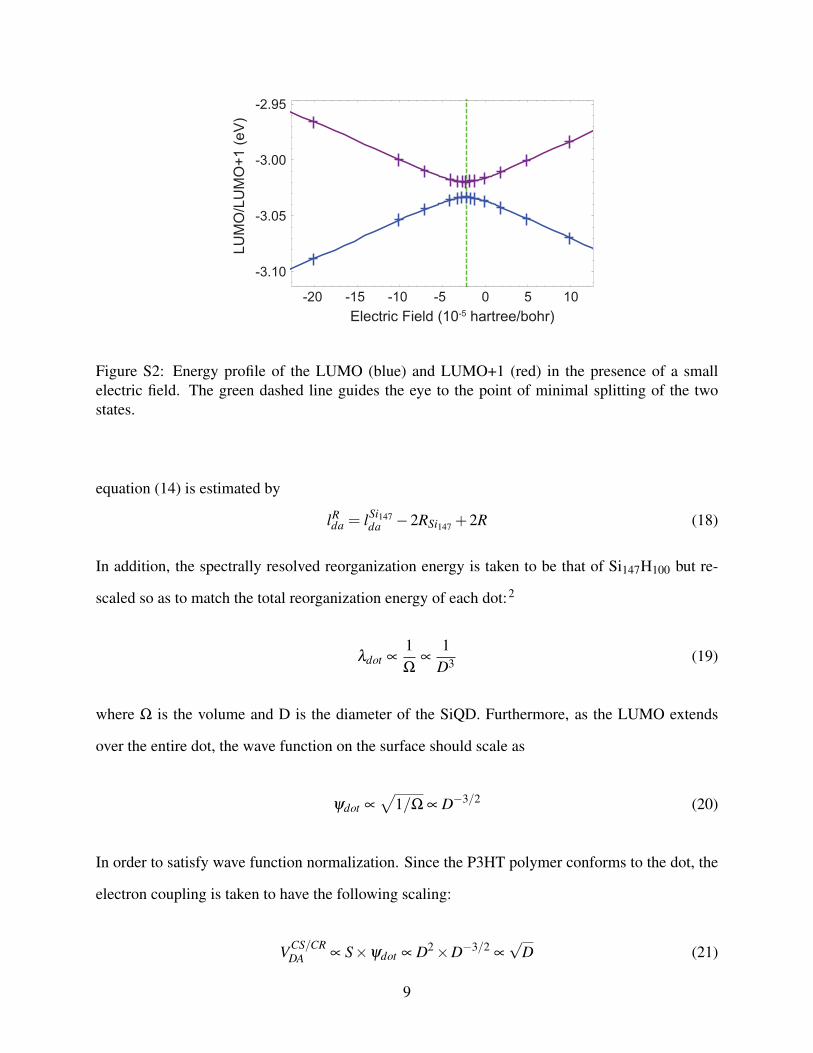
The anti-crossing method is a computationally efficient means of calculating the electronic cou-
pling between two nearly degenerate states.2 For the Si147H100 dimer, relaxed in a face-to-face
configuration, the LUMO and LUMO+1 orbitals (Figure S1) are localized on different dots. From
the energy profile of the LUMO and LUMO+1 in the presence of a small electric field (Figure S2),
we can determine the minimal splitting of these two states that can be shown to be a reasonable
estimate of twice of the electron coupling for electron hopping.2
Extrapolation of Charge Transfer Rates to Larger dots
To extrapolate the CT and CR rates from the Si147H100/P3HT interface with dot diameter of 1.7
nm to that associated with larger dot sizes (2-8 nm), the LUMO shifts of the dots are estimated as
being half of the optical gap change obtained from previous computational studies.22 The solvent
reorganization energy, as a function of dot size, is computed by preserving the face-to-face dis-
tance between the dot and the P3HT, namely for the dot with radius R, donor-acceptor distance in
8
-20 -15 -10 -5 0 5 10
-3.10
-3.05
-3.00
-2.95
LU
MO
/LU
MO
+1
(e
V)
Electric Field (10-5 hartree/bohr)
Figure S2: Energy profile of the LUMO (blue) and LUMO+1 (red) in the presence of a smallelectric field. The green dashed line guides the eye to the point of minimal splitting of the twostates.
equation (14) is estimated by
lRda = lSi147
da −2RSi147 +2R (18)
In addition, the spectrally resolved reorganization energy is taken to be that of Si147H100 but re-
scaled so as to match the total reorganization energy of each dot:2
λdot ∝1Ω
∝1
D3 (19)
where Ω is the volume and D is the diameter of the SiQD. Furthermore, as the LUMO extends
over the entire dot, the wave function on the surface should scale as
ψdot ∝√
1/Ω ∝ D−3/2 (20)
In order to satisfy wave function normalization. Since the P3HT polymer conforms to the dot, the
electron coupling is taken to have the following scaling:
VCS/CRDA ∝ S×ψdot ∝ D2×D−3/2
∝√
D (21)
9

D1 D2
h h
r1r2
Figure S3: Schematic of electronic coupling associated with electron hopping between dots ofdifferent size.
where S is the surface area of the dot. We used a similar approach to estimate the electron hopping
rates between two dots with diameters D1 and D2. As illustrated in Figure S3, we assume that the
two dots are circles, and only the wave functions within distance, h, of the dot surfaces contribute
to the electronic coupling so that the scaling of VDA can be approximated as
V EHDA ∝ min(S1,S2)×ψdot1×ψdot2 ∝ min(D1,D2)×D−3/2
1 ×D−3/22 (22)
Here
S1 = πr21 ≈ πD1h , S2 = πr2
2 ≈ πD2h (23)
provided that h D .
Influence of Dangling Bonds on Electronic Coupling
In contrast to the delocalized wave function of defect-free dots, the LUMO becomes localized on
the surface of the dot in the presence of a dangling bond (DB). The result is that it becomes closer
10

(a) (b) (c)
Figure S4: Three configurations of the Si147H100/P3HT interfaces with one dangling bond (green).
to the HOMO of the surrounding P3HT and this significantly enhances the electronic coupling for
both CT and CR (Table S3). Since the wave function of the dot with DB is spatially inhomoge-
neous, the electronic coupling depends strongly on the interfacial configuration. Three representa-
tive configurations (Figure S4) of Si147H100/P3HT interfaces are considered in our calculation, and
the average value is used in the rate expression of CT and CR. For the electron hopping between
two dots, we assumed that the electron coupling for all three DB configurations is the same as that
without DBs.
Table S3: Electronic coupling of charge transfer and recombination at the Si147H100/P3HTinterface without any dangling bonds and with one dangling bond. Three different configu-rations are considered.
No DB DB: config1 DB: config2 DB: config3CT (meV) 9.8 29.6 60.6 46.2CR (meV) 4.0 4.6 18.1 13.0
Performance of the Device without Dangling Bonds
The current density in an electric field E is
J = nevd = neµE ∝ µ (24)
11

where n is the carrier concentration and µ is the carrier mobility. Enhancement of the mobility
therefore leads to the same order of improvement in the current provided the internal voltage drop
does not change. In order to extract free carriers to the external circuit, the carriers must drift to the
electrodes before CR occurs. The efficiency of this process is determined by a comparison between
the carrier sweep out time and the CT state lifetime, where the carrier sweep out time is taken to
be the period required by the carrier to drift from the center of the active layer to the electrode:23
τs =d2
2µVin(25)
Here d is the distance between the electrodes and Vin is the internal potential drop.
As a example, we consider a set of typical solar cell parameters, with Vin = 0.2 V and d = 120
nm.24 For the Si147/P3HT blend with a DB on the dot surface,
τDBs =
d2
2µDBVin=
(120×10−9)2
2× (2.8×10−10)×0.2= 1.2×10−4s τ
DBCT =
1kDB
CR=
12.5×1012 = 4.0×10−13s
(26)
implying a noticeable proportion of carriers would be lost to CR. In contrast, for the assemblies
without a DB,
τPer f ects =
d2
2µPer f ectVin=
(120×10−9)2
2× (2.9×10−6)×0.2= 1.2×10−8s τ
Per f ectCT =
1
kPer f ectCR
=1
1.5×102 = 6.7×10−3s
(27)
indicating that most of the carriers will contribute to the photocurrent.
12

References
[1] Jortner, J. Temperature Dependent Activation Energy for Electron Transfer between Biolog-
ical Molecules. J. Chem. Phys. 1976, 64, 4860–4867.
[2] Chu, I.-H.; Radulaski, M.; Vukmirovic, N.; Cheng, H.-P.; Wang, L.-W. Charge Transport in
a Quantum Dot Supercrystal. J. Phys. Chem. C 2011, 115, 21409–21415.
[3] Liu, T.; Troisi, A. Absolute Rate of Charge Separation and Recombination in a Molecular
Model of the P3HT/PCBM Interface. J. Phys. Chem. C 2011, 115, 2406–2415.
[4] Delley, B. An All-electron Numerical-method for Solving the Local Density Functional for
Polyatomic-molecules. J. Chem. Phys. 1990, 92, 508–517.
[5] Klamt, A.; Jonas, V.; Burger, T.; Lohrenz, J. C. W. Refinement and Parametrization of
COSMO-RS. J. Phys. Chem. A 1998, 102, 5074–5085.
[6] Perdew, J. P.; Wang, Y. Accurate and Simple Analytic Representation of the Electron-Gas
Correlation-Energy. Phys. Rev. B 1992, 45, 13244–13249.
[7] Tiago, M. L.; Chelikowsky, J. R. Optical Excitations in Organic Molecules, Clusters, and
Defects Studied by First-principles Green’s Function Methods. Phys. Rev. B 2006, 73.
[8] Chelikowsky, J. R.; Troullier, N.; Saad, Y. Finite-Difference-Pseudopotential Method -
Electronic-structure Calculations without a Basis. Phys. Rev. Lett. 1994, 72, 1240–1243.
[9] Marcus, R. A. On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer.
I. J. Chem. Phys. 1956, 24, 966–978.
[10] Farazdel, A.; Dupuis, M.; Clementi, E.; Aviram, A. Electric-field Induced Intramolecular
Electron-transfer in Spiro Pi-electron Systems and Their Suitability as Molecular Electronic
Devices - A Theoretical-study. J. Am. Chem. Soc. 1990, 112, 4206–4214.
13

[11] Wong, A. T.; Harrison, R. J. Approaches to Large-scale Parallel Self-consistent-field Calcu-
lations. J. Comput. Chem. 1995, 16, 1291–1300.
[12] Valiev, M.; Bylaska, E. J.; Govind, N.; Kowalski, K.; Straatsma, T. P.; Van Dam, H. J. J.;
Wang, D.; Nieplocha, J.; Apra, E.; Windus, T. L. et al. NWChem: A Comprehensive and Scal-
able Open-source Solution for Large Scale Molecular Simulations. Comput. Phys. Commun.
2010, 181, 1477–1489.
[13] Shan, W.; Walukiewicz, W.; Ager, J. W.; Haller, E. E.; Geisz, J. F.; Friedman, D. J.; Ol-
son, J. M.; Kurtz, S. R. Band Anticrossing in GaInNAs Alloys. Phys. Rev. Lett. 1999, 82,
1221–1224.
[14] Deng, W. Q.; Goddard, W. A. Predictions of Hole Mobilities in Oligoacene Organic Semicon-
ductors from Quantum Mechanical Calculations. J. Phys. Chem. B 2004, 108, 8614–8621.
[15] Lin, Z.; Li, H.; Franceschetti, A.; Lusk, M. T. Efficient Exciton Transport between Strongly
Quantum-Confined Silicon Quantum Dots. ACS Nano 2012, 6, 4029–4038.
[16] Califano, M.; Franceschetti, A.; Zunger, A. Temperature Dependence of Excitonic Radiative
Decay in CdSe Quantum Dots: The Role of Surface Hole Traps. Nano Lett. 2005, 5, 2360–
2364.
[17] Baer, R.; Rabani, E. Theory of Resonance Energy Transfer Involving Nanocrystals: The Role
of High Multipoles. J. Chem. Phys 2008, 128.
[18] Franceschetti, A.; Fu, H.; Wang, L. W.; Zunger, A. Many-body Pseudopotential Theory of
Excitons in InP and CdSe Quantum Dots. Phys. Rev. B 1999, 60, 1819–1829.
[19] Roncali, J. Conjugated Poly(thiophenes) - Synthesis, Functionalization, and Applications.
Chem. Rev. 1992, 92, 711–738.
14

[20] Liu, T.; Gao, J. S.; Xia, B. H.; Zhou, X.; Zhang, H. X. Theoretical Studies on the Elec-
tronic Structures and Optical Properties of the Oligomers Involving Bipyridyl, Thiophenyl
and Ethynyl Groups. Polym. 2007, 48, 502–511.
[21] Goeb, S.; De Nicola, A.; Ziessel, R. Oligomeric Ligands Incorporating Multiple 5,5 ’-
diethynyl-2,2 ’-bipyridine Moieties Bridged and End-capped by 3,4-dibutylthiophene Units.
J. Org. Chem. 2005, 70, 1518–1529.
[22] Luo, J.-W.; Stradins, P.; Zunger, A. Matrix-embedded Silicon Quantum Dots for Photovoltaic
Applications: a Theoretical Study of Critical Factors. Energy Environ. Sci. 2011, 4, 2546–
2557.
[23] Cowan, S. R.; Roy, A.; Heeger, A. J. Recombination in Polymer-fullerene Bulk Heterojunc-
tion Solar Cells. Phys. Rev. B 2010, 82, 245207.
[24] Mihailetchi, V. D.; Koster, L. J. A.; Hummelen, J. C.; Blom, P. W. M. Photocurrent Genera-
tion in Polymer-Fullerene Bulk Heterojunctions. Phys. Rev. Lett. 2004, 93, 216601, PRL.
15


















