

CSE182-L4: Scoring matrices, Dictionary Matching
60
Fa05 CSE 182 CSE182-L4: Scoring matrices, Dictionary Matching
-
Upload
rylee-rice -
Category
Documents
-
view
35 -
download
0
description
CSE182-L4: Scoring matrices, Dictionary Matching. Class Mailing List. [email protected] To subscribe, send email to [email protected] You can subscribe from the course web page Use the list for all course related queries, discussions,…. Protein Sequence Analysis. - PowerPoint PPT Presentation
Transcript of CSE182-L4: Scoring matrices, Dictionary Matching

Fa05 CSE 182
CSE182-L4: Scoring matrices, Dictionary Matching

Fa05 CSE 182
Class Mailing List
• To subscribe, send email to – [email protected]
• You can subscribe from the course web page• Use the list for all course related queries,
discussions,…

Fa05 CSE 182
Protein Sequence Analysis
• What can you do if BLAST does not return a hit?– Sometimes, homology (evolutionary similarity) exists at
very low levels of sequence similarity.
• A: Accept hits at higher P-value. – This increases the probability that the sequence similarity
is a chance event.– How can we get around this paradox?– Reformulated Q: suppose two sequences B,C have the
same level of sequence similarity to sequence A. If A& B are related in function, can we assume that A& C are? If not, how can we distinguish?

Fa05 CSE 182
Protein sequence motifs
• Premise: • The sequence of a protein sequence gives clues
about its structure and function.• Not all residues are equally important in
determining function.• How can we identify these key residues?

Fa05 CSE 182
Prosite
• In some cases the sequence of an unknown protein is too distantly related to any protein of known structure to detect its resemblance by overall sequence alignment. However, relationships can be revealed by the occurrence in its sequence of a particular cluster of residue types, which is variously known as a pattern, motif, signature or fingerprint. These motifs arise because specific region(s) of a protein which may be important, for example, for their binding properties or for their enzymatic activity are conserved in both structure and sequence. These structural requirements impose very tight constraints on the evolution of this small but important portion(s) of a protein sequence. The use of protein sequence patterns or profiles to determine the function of proteins is becoming very rapidly one of the essential tools of sequence analysis. Many authors ( 3,4) have recognized this reality. Based on these observations, we decided in 1988, to actively pursue the development of a database of regular expression-like patterns, which would be used to search against sequences of unknown function.
Kay Hofmann ,Philipp Bucher, Laurent Falquet and Amos Bairoch
The PROSITE database, its status in 1999

Fa05 CSE 182
Basic idea• It is a heuristic approach. Start with the following:
– A collection of sequences with the same function.– Region/residues known to be significant for maintaining
structure and function. • Develop a pattern of conserved residues around
the residues of interest• Iterate for appropriate sensitivity and specificity

Fa05 CSE 182
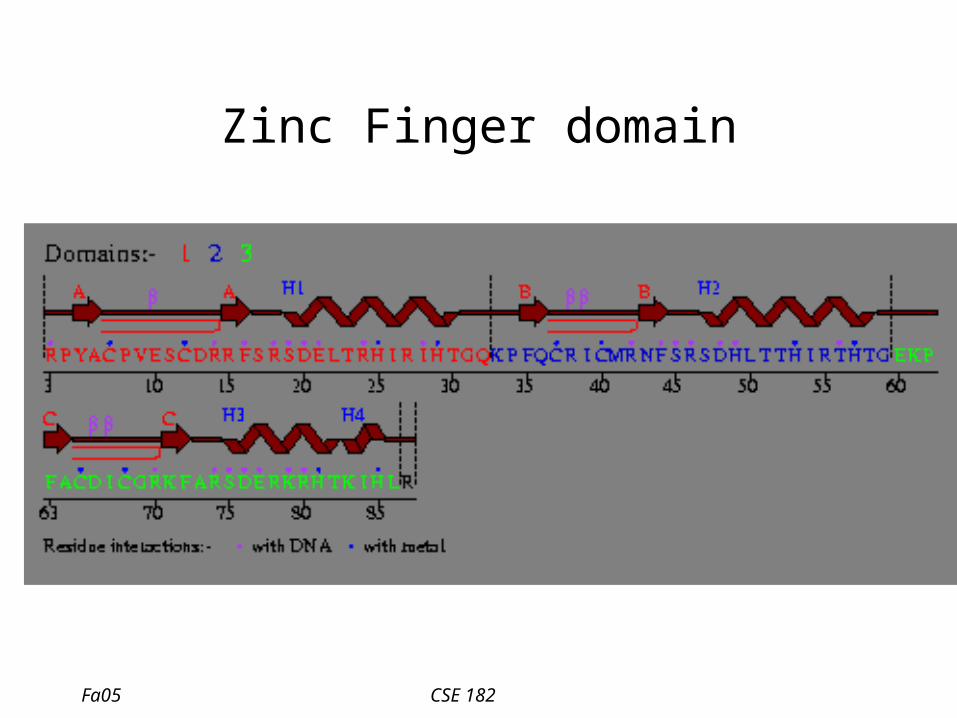
Zinc Finger domain

Fa05 CSE 182
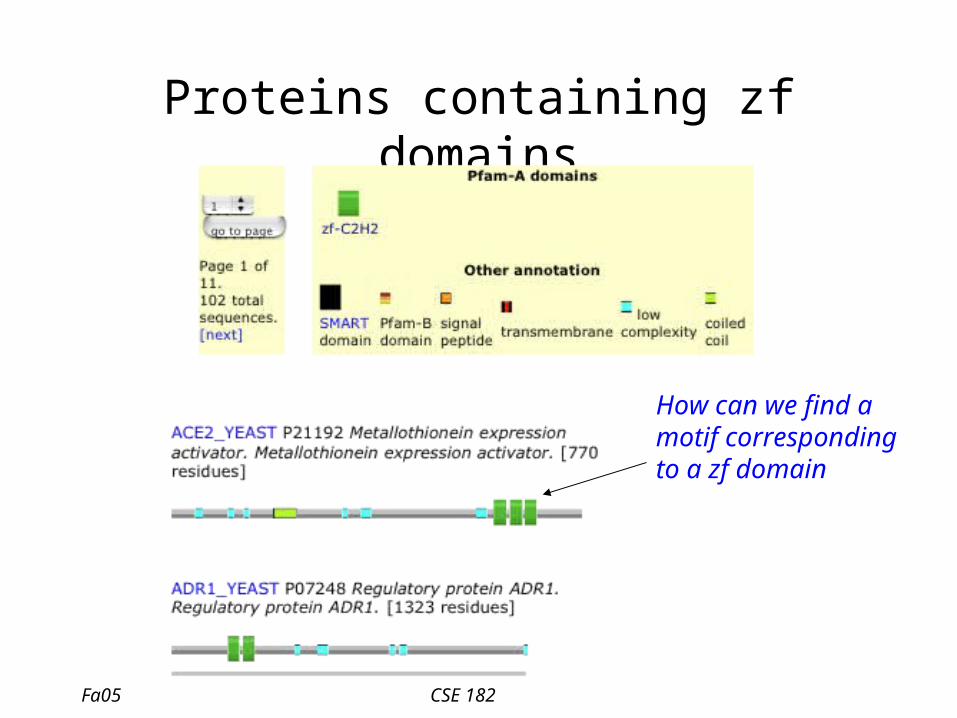
Proteins containing zf domains
How can we find a motif corresponding to a zf domain
Fa05 CSE 182
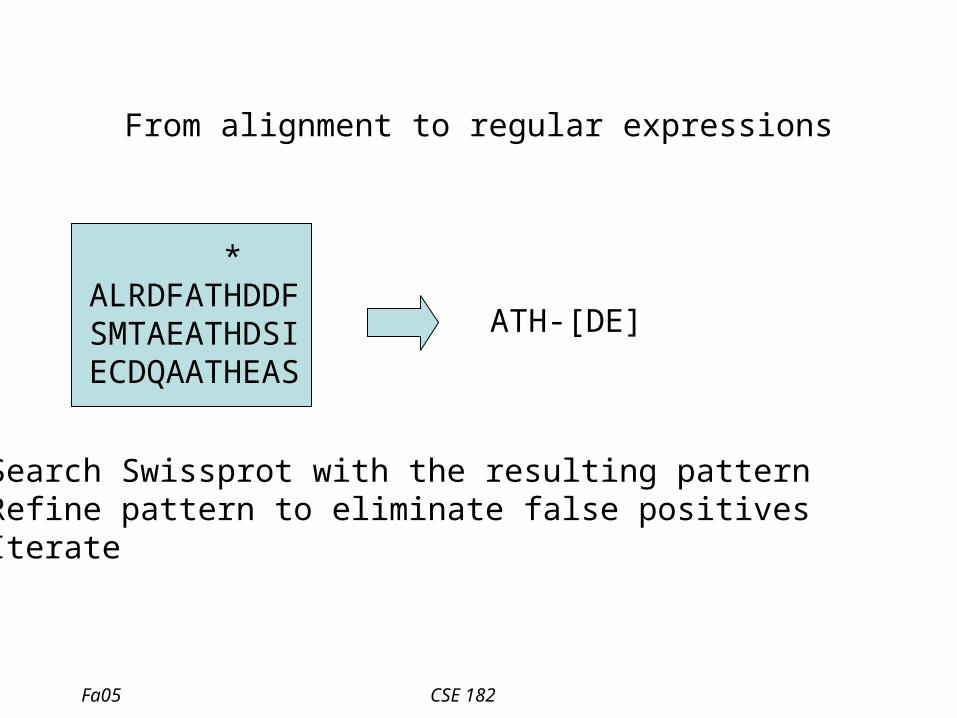
From alignment to regular expressions
* ALRDFATHDDF SMTAEATHDSI ECDQAATHEAS
ATH-[DE]
• Search Swissprot with the resulting pattern• Refine pattern to eliminate false positives• Iterate

Fa05 CSE 182
The sequence analysis perspective
• Zinc Finger motif
– C-x(2,4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3,5)-H – 2 conserved C, and 2 conserved H
• How can we search a database using these motifs?– The motif is described using a regular expression. What is
a regular expression?– How can we search for a match to a regular expression?
Not allowed to use Perl :-)• The ‘regular expression’ motif is weak. How can we make it
stronger

Fa05 CSE 182
Profiles
• Start with an alignment of strings of length m, over an alphabet A,
• Build an |A| X m matrix F=(fki)
• Each entry fki represents the frequency of symbol k in position i
0.71
0.14
0.71
0.28

Fa05 CSE 182
Scoring Profiles
€
S(i, j) = fki
k
∑ M rk,s j[ ]
k
i
s
fki
Scoring Matrix

Fa05 CSE 182
Psi-BLAST idea
• Multiple alignments are important for capturing remote homology.
• Profile based scores are a natural way to handle this.
• Q: What if the query is a single sequence.• A: Iterate:
– Find homologs using Blast on query– Discard very similar homologs– Align, make a profile, search with profile.

Fa05 CSE 182
Psi-BLAST speed
• Two time consuming steps.1. Multiple alignment of homologs2. Searching with Profiles.
1. Does the keyword search idea work?
• Pigeonhole principle again: – If profile of length m must
score >= T– Then, a sub-profile of length
l must score >= lT/m– Generate all l-mers that
score at least lT/M– Search using an automaton
• Multiple alignment:– Use ungapped
multiple alignments only

Fa05 CSE 182

Fa05 CSE 182
CSE182-L6
Regular Expression MatchingProtein structure basics
Fa05 CSE 182
Zinc Finger domain

Fa05 CSE 182
The sequence analysis perspective
• Zinc Finger motif
– C-x(2,4)-C-x(3)-[LIVMFYWC]-x(8)-H-x(3,5)-H – 2 conserved C, and 2 conserved H
• How can we search a database using these motifs?– The motif is described using a regular expression. What is
a regular expression?

Fa05 CSE 182
Regular Expressions
• Concise representation of a set of strings over alphabet .
• Described by a string over• R is a r.e. if and only if
€
Σ,⋅,∗,+{ }
€
R = {ε} Base caseR = {σ },σ ∈ ΣR = R1 + R2 Union of stringsR = R1 ⋅R2 ConcatenationR = R
1
* 0 or more repetitions

Fa05 CSE 182
Regular Expression
• Q: Let ={A,C,E}– Is (A+C)*EEC* a regular expression?– *(A+C)?– AC*..E?
• Q: When is a string s in a regular expression?– R =(A+C)*EEC*– Is CEEC in R?– AEC?– ACEE?

Fa05 CSE 182
Regular Expression & Automata
• Every R.E can be expressed by an automaton (a directed graph) with the following properties:– The automaton has a start and end node– Each edge is labeled with a symbol from , or
Suppose R is described by automaton ASuppose R is described by automaton AS S R if and only if there is a path from start to end R if and only if there is a path from start to end in A, labeled with s.in A, labeled with s.

Fa05 CSE 182
Examples: Regular Expression & Automata
• (A+C)*EEC*
CA
C
start endE E

Fa05 CSE 182
Constructing automata from R.E
• R = {}• R = {}, • R = R1 + R2
• R = R1 · R2
• R = R1*

Fa05 CSE 182
Regular Expression Matching
• Given a database D, and a regular expression R, is a substring of D in R?
• Is there a string D[l..c] that is accepted by the automaton of R?
• Simpler Q: Is D[1..c] accepted by the automaton of R?

Fa05 CSE 182
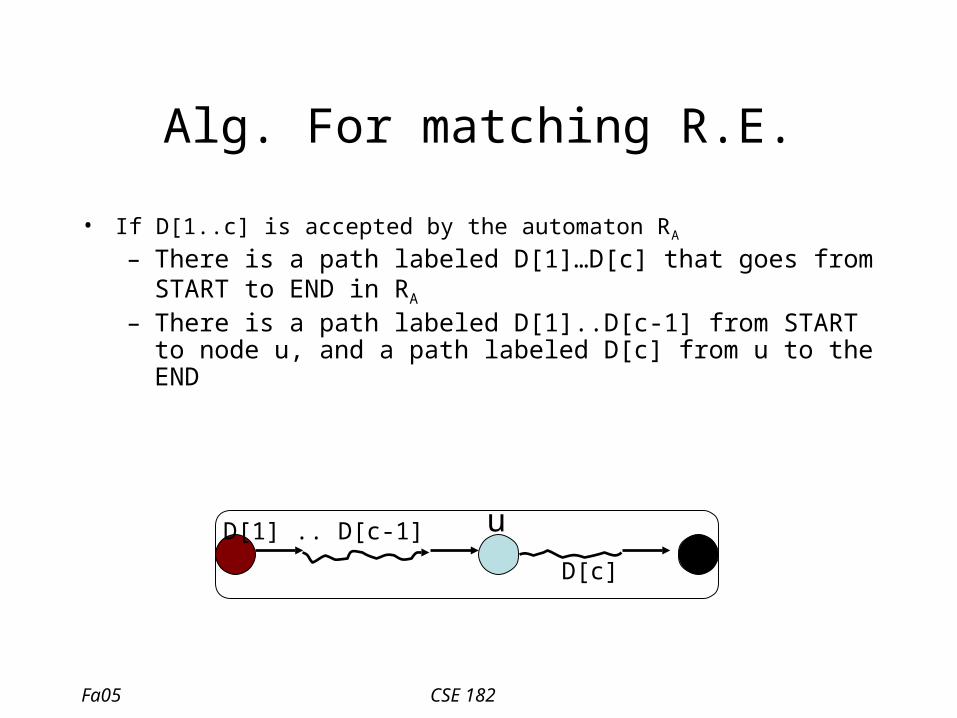
Alg. For matching R.E.
• If D[1..c] is accepted by the automaton RA
– There is a path labeled D[1]…D[c] that goes from START to END in RA
D[1] D[2] D[c]
Fa05 CSE 182
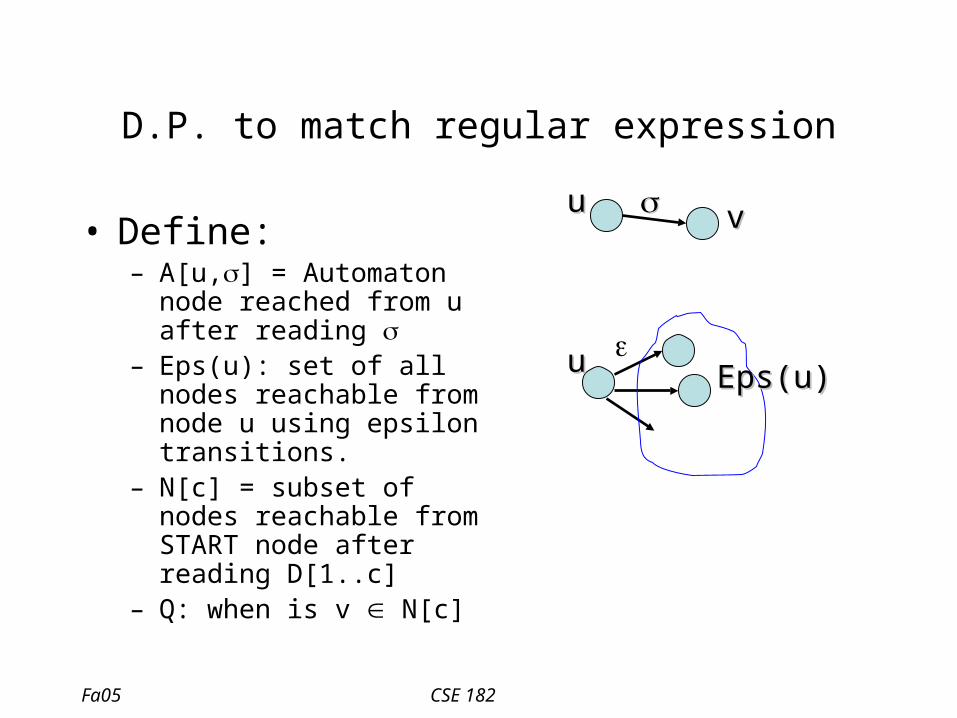
Alg. For matching R.E.
• If D[1..c] is accepted by the automaton RA
– There is a path labeled D[1]…D[c] that goes from START to END in RA
– There is a path labeled D[1]..D[c-1] from START to node u, and a path labeled D[c] from u to the END
D[1] .. D[c-1]
D[c]
u
Fa05 CSE 182
D.P. to match regular expression
• Define:– A[u,] = Automaton
node reached from u after reading
– Eps(u): set of all nodes reachable from node u using epsilon transitions.
– N[c] = subset of nodes reachable from START node after reading D[1..c]
– Q: when is v N[c]
uu vv
uu Eps(u)Eps(u)

Fa05 CSE 182
• Q: when is v N[c]?• A: If for some u N[c-1], w = A[u,D[c]],
• v {w}+ Eps(w)
D.P. to match regular expression

Fa05 CSE 182
Algorithm

Fa05 CSE 182
The final step
• We have answered the question:– Is D[1..c] accepted by R?– Yes, if END N[c]
• We need to answer – Is D[l..c] (for some l, and some c) accepted
by R
€
D[l..c]∈ R ⇔ D[1..c]∈ Σ∗R

Fa05 CSE 182
A structural view of proteins

Fa05 CSE 182
CS view of a protein
• >sp|P00974|BPT1_BOVIN Pancreatic trypsin inhibitor precursor (Basic protease inhibitor) (BPI) (BPTI) (Aprotinin) - Bos taurus (Bovine).
• MKMSRLCLSVALLVLLGTLAASTPGCDTSNQAKAQRPDFCLEPPYTGPCKARIIRYFYNAKAGLCQTFVYGGCRAKRNNFKSAEDCMRTCGGAIGPWENL

Fa05 CSE 182
Protein structure basics

Fa05 CSE 182
Side chains determine amino-acid type
QuickTime™ and aTIFF (Uncompressed) decompressorare needed to see this picture.QuickTime™ and aTIFF (Uncompressed) decompressorare needed to see this picture.
• The residues may have different properties.• Aspartic acid (D), and Glutamic Acid (E) are acidic
residues
Fa05 CSE 182
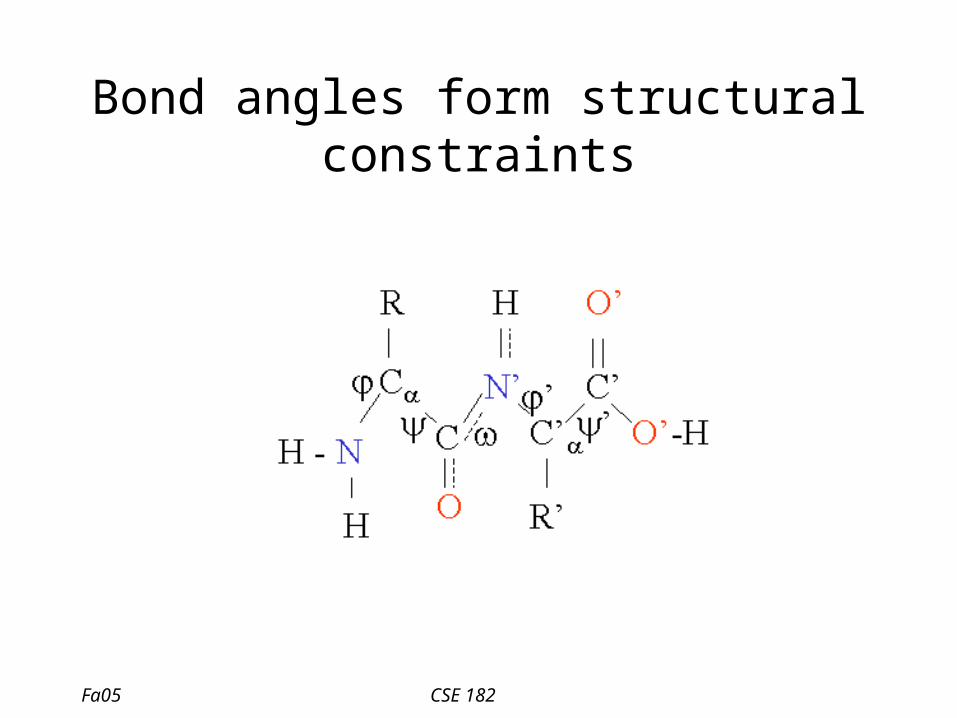
Bond angles form structural constraints

Fa05 CSE 182
Various constraints determine 3d structure
• Constraints– Structural constraints due to physiochemical
properties– Constraints due to bond angles– H-bond formation
• Surprisingly, a few conformations are seen over and over again.

Fa05 CSE 182
Alpha-helix
• 3.6 residues per turn• H-bonds between 1st
and 4th residue stabilize the structure.
• First discovered by Linus Pauling
Fa05 CSE 182
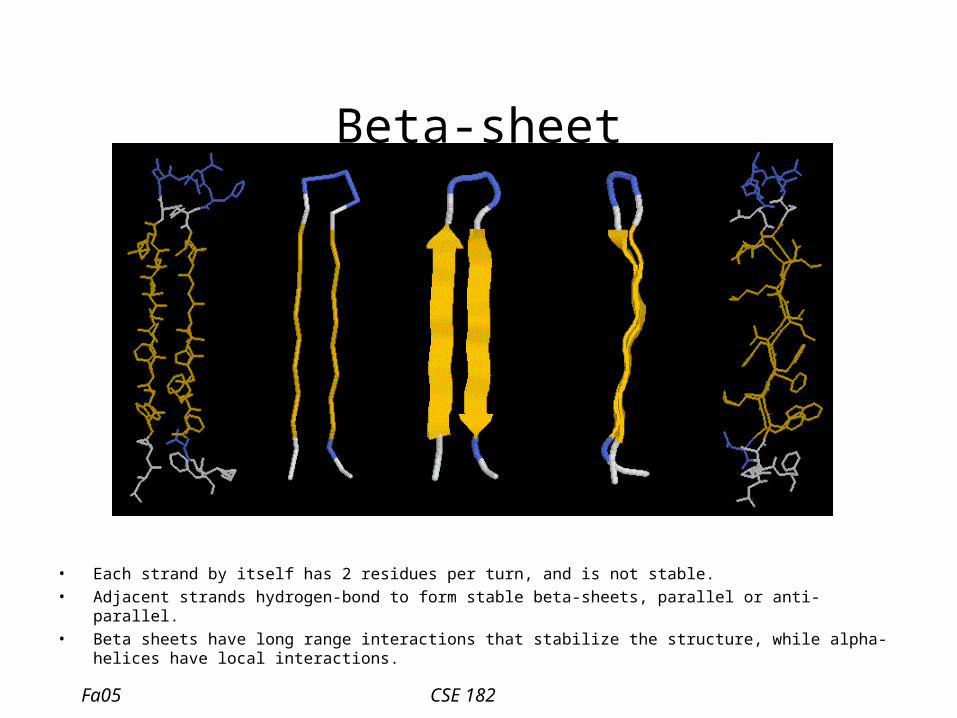
Beta-sheet
• Each strand by itself has 2 residues per turn, and is not stable.• Adjacent strands hydrogen-bond to form stable beta-sheets, parallel or anti-parallel.• Beta sheets have long range interactions that stabilize the structure, while alpha-helices have local
interactions.

Fa05 CSE 182
Domains
• The basic structures (helix, strand, loop) combine to form complex 3D structures.
• Certain combinations are popular. Many sequences, but only a few folds

Fa05 CSE 182
3D structure• Predicting tertiary structure is an important
problem in Bioinformatics.• Premise: Clues to structure can be found in
the sequence.• While de novo tertiary structure prediction is
hard, there are many intermediate, and tractable goals.

Fa05 CSE 182
Protein Domains• An important realization (in the last decade) is that proteins have a
modular architecture of domains/folds.• Example: The zinc finger domain is a DNA-binding domain.• What is a domain?
– Part of a sequence that can fold independently, and is present in other sequences as well
Fa05 CSE 182
Proteins containing zf domains
How can we find a motif corresponding to a zf domain

Fa05 CSE 182
Domain review• What is a domain?• How are domains expressed
– Motifs (Regular expression & others)– Multiple alignments– Profiles– Profile HMMs

Fa05 CSE 182
Databases of protein domains

Fa05 CSE 182
PROSITE
http://us.expasy.org/prosite/

Fa05 CSE 182

Fa05 CSE 182

Fa05 CSE 182
http://hmmer.wustl.edu

Fa05 CSE 182
HMMER programs
• Hmmalign– Align a sequence to an HMM
• Hmmbuild– Build a model from a multiple alignment
• Hmmemit– Emits a probabilistic sequence from an HMM
• Hmmpfam– Search PFAM with a sequence query
• Hmmsearch– Search a sequence database with an HMM query

Fa05 CSE 182

Fa05 CSE 182
Post-translational modification
• Residues undergo modification, usually by addition of a chemical group.
• Key mechanism for signal transduction, and many other cellular functions
• Some modifications might require single residues (Ex: phosphorylation). Others might require a pattern

Fa05 CSE 182
Fa05 CSE 182
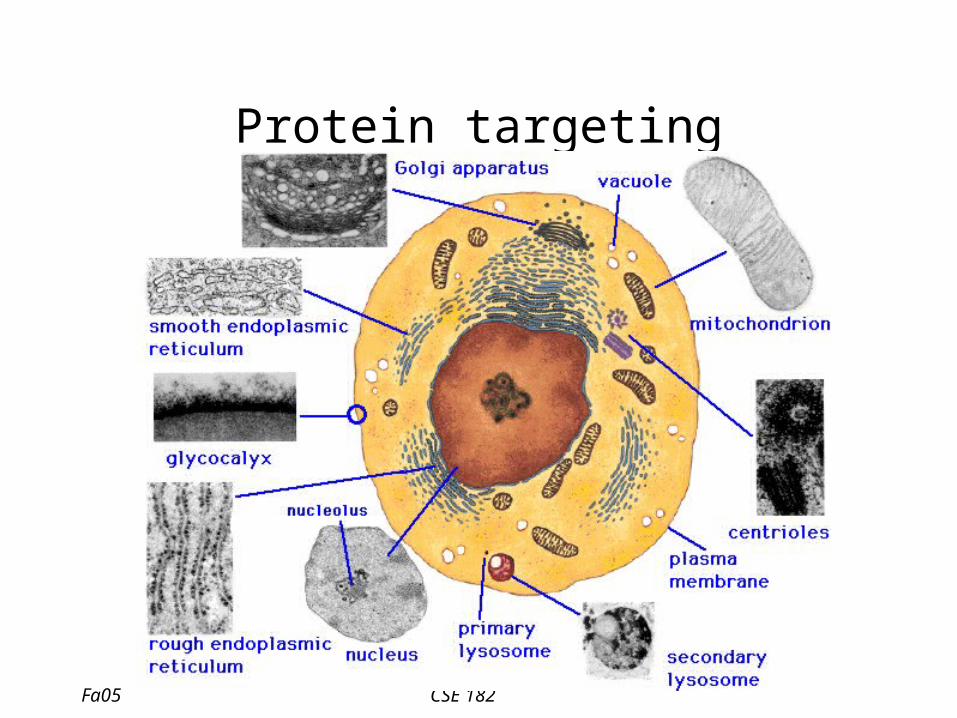
Protein targeting

Fa05 CSE 182
Protein targeting
• In 1970, Gunter Blobel showed that proteins have an N-terminal signal sequence which directs proteins to the membrane.
• Proteins have to be transported to other organelles: nucleus, mitochondria,…
• Can we computationally identify the ‘signal’ which distinguishes the cellular compartment?

Fa05 CSE 182
• For transmembrane proteins, can we predict the transmembrane, outer, and inner regions?

Fa05 CSE 182

Fa05 CSE 182
Multiple alignment tools

Fa05 CSE 182
Tools for secondary structure prediction
• Each residue must be given a state: Helix, Loop, Strand• HMMs/Neural networks are used to predict

Fa05 CSE 182
Next topic: Gene finding


















