
Corrosion Mechanism of Q235A under 3.5% NaCl Salt Spray
6
Corrosion Mechanism of Q235A under 3.5% NaCl Salt Spray Jiantao Wang 1,2 , Bochen Jiang 2 and Jiangdong Cao 2 1 Merchant Marine College, Shanghai Maritime University, Shanghai, 201306, China 2 Jiangsu Shipping College, Nantong, 226010, China Low-carbon steel Q235A is the main material used in marine construction, but it experiences corrosion because of salt spray and seawater exposure. In this study, we investigated the corrosion performance of Q235A in the presence of 3.5% NaCl salt spray. We characterized the microstructure of the material using electron backscattered diffraction, scanning electron microscopy, and specimen weight loss. The phases and morphologies of the corrosion products were then analyzed after salt-fog corrosion testing. The degree of coverage and compactness of surface corrosion products gradually increased from the 24 to 96 hours in the salt spray environment. Under short-term corrosive conditions, surface corrosion products were primarily composed of a loose, porous layer of flocculent £ -FeOOH. With increasing time, the corrosion products gradually changed to a lump-like and block-like dense structure of ¢-FeOOH and ¡-FeOOH. The results of electrochemical polarization tests of the corroded steel samples showed that the £ -FeOOH might accelerate corrosion, and that ¢-FeOOH and ¡-FeOOH inhibit corrosion. [doi:10.2320/matertrans.MT-M2020053] (Received February 13, 2020; Accepted September 9, 2020; Published October 30, 2020) Keywords: corrosion, low-carbon steel, Q235A, NaCl, salt spray 1. Introduction Low-carbon steel Q235A, which has good plasticity, hot workability, and low cost, 1) is widely used in engineering. Following hot rolling, Q235A steel plates and pipes are suitable for the manufacture of various welded structural parts and machine parts. 2,3) However, the corrosion mechan- ism of the steel varies with the state of the service environment. 4,5) Consequently, extensive research has been conducted on the microstructure, corrosion, and protection of this material. For example, Wang et al. 6) examined solid- phase diffusion bonding between Q235A and austenitic stainless steel AISI304. They found that brittle compounds segregated to improve the performance of diffusion-welded joints when the welding temperature was controlled at 850°C. In another example, Hu et al. 7) studied the corrosion behavior of Q235A steel in reverse-osmosis water treatment equipment. They reported that corrosion under these service conditions is controlled by the diffusion of oxygen and corrosion products, in which a conductive Fe 3 O 4 layer forming accelerates the corrosion process. Chen et al. 8) studied a series of inhibitors that effectively retard the corrosion of Q235A at high concentration HCl. The compound H-NMR (D6-DMSO) shows 95.5% inhibition efficiency at 100 mg/L HCl. Li et al. 9) studied Q235A steel corrosion induced by chloride ions under stray current conditions and constructed a model based on an artificial neural network to accurately predict corrosion, as well as the corrosion potential and corrosion current density. Jianguo et al. 10) studied the polarization curves, corrosion processes, and mechanisms of Q235A steel under dry and wet cycling conditions. They reported the corrosion current of a dry-wet sample was 22.99 μA/cm 2 , and this was much higher than that of the bare steel (9.3 μA/cm 2 ). To improve the corrosion protection performance of Q235A steel, some researchers have treated steel surfaces with nickel-nanodiamond composite coatings or plasma-clad tungsten carbide particles or by laser cladding ultra-hard alloys. 11-13) The good welding features and mechanical properties (yield strength of 235 MPa, ultimate tensile strength of 410 MPa, and elongation of 23%) of Q235A steel make it a preferred material in the construction of shipping containers. However, in the marine environment, steel materials are constantly exposed to salt spray and seawater, making them prone to salt spray corrosion, reducing the strength and life service of the ship. Consequently, determining the corrosion of Q235A steel in a salt spray environment is important for the design and manufacture of the ship. In this study, we examined the corrosion products over time during a salt spray test and examined the effect of the corrosion products on the corrosion. 2. Experimental Procedures 2.1 Salt spray corrosion experiment The size of the metal samples used in the salt spray experiments was 10 mm © 10 mm © 5 mm, and the individ- ual metal samples were cleaned and weighed to the nearest 0.1 mg before testing. Test samples were cleaned with acetone to remove surface oil, polished with #1200 sandpaper, dried in the dry box, and soaked in pure ethanol. A corrosion test was conducted based on the ISO 9227- 2017 standard using a salt spray tester containing a 3.5% NaCl solution. The metal samples were subjected to salt-fog test for 24, 48, 72, and 96 hours. The pH of the salt-fog solution was 6.4, temperature of the sample chamber was at 35°C, salt spray settlement occurred at 1.0-2.0 mL/80 cm 2 ·h, and relative humidity of the chamber was 70%. After each test, the weight of the individual sample was recorded, and the average weight loss was determined from the four-sample experiment. 2.2 Electrochemical experiments Corroded steel samples were subjected to electrochemical experiments using the classical three-electrode electrochem- ical test system where the working electrode was the corroded Q235A steel sample. One side of the test sample was sealed with an epoxy coating, and the other side served as the working electrode with a 1 cm 2 working surface area. The reference electrode was a saturated calomel electrode, Materials Transactions, Vol. 61, No. 12 (2020) pp. 2342 to 2347 © 2020 The Japan Institute of Metals and Materials
Transcript of Corrosion Mechanism of Q235A under 3.5% NaCl Salt Spray

Corrosion Mechanism of Q235A under 3.5% NaCl Salt Spray
Jiantao Wang1,2, Bochen Jiang2 and Jiangdong Cao2
1Merchant Marine College, Shanghai Maritime University, Shanghai, 201306, China2Jiangsu Shipping College, Nantong, 226010, China
Low-carbon steel Q235A is the main material used in marine construction, but it experiences corrosion because of salt spray and seawaterexposure. In this study, we investigated the corrosion performance of Q235A in the presence of 3.5% NaCl salt spray. We characterized themicrostructure of the material using electron backscattered diffraction, scanning electron microscopy, and specimen weight loss. The phasesand morphologies of the corrosion products were then analyzed after salt-fog corrosion testing. The degree of coverage and compactness ofsurface corrosion products gradually increased from the 24 to 96 hours in the salt spray environment. Under short-term corrosive conditions,surface corrosion products were primarily composed of a loose, porous layer of flocculent £ -FeOOH. With increasing time, the corrosionproducts gradually changed to a lump-like and block-like dense structure of ¢-FeOOH and ¡-FeOOH. The results of electrochemical polarizationtests of the corroded steel samples showed that the £ -FeOOH might accelerate corrosion, and that ¢-FeOOH and ¡-FeOOH inhibit corrosion.[doi:10.2320/matertrans.MT-M2020053]
(Received February 13, 2020; Accepted September 9, 2020; Published October 30, 2020)
Keywords: corrosion, low-carbon steel, Q235A, NaCl, salt spray
1. Introduction
Low-carbon steel Q235A, which has good plasticity, hotworkability, and low cost,1) is widely used in engineering.Following hot rolling, Q235A steel plates and pipes aresuitable for the manufacture of various welded structuralparts and machine parts.2,3) However, the corrosion mechan-ism of the steel varies with the state of the serviceenvironment.4,5) Consequently, extensive research has beenconducted on the microstructure, corrosion, and protectionof this material. For example, Wang et al.6) examined solid-phase diffusion bonding between Q235A and austeniticstainless steel AISI304. They found that brittle compoundssegregated to improve the performance of diffusion-weldedjoints when the welding temperature was controlled at850°C. In another example, Hu et al.7) studied the corrosionbehavior of Q235A steel in reverse-osmosis water treatmentequipment. They reported that corrosion under these serviceconditions is controlled by the diffusion of oxygen andcorrosion products, in which a conductive Fe3O4 layerforming accelerates the corrosion process. Chen et al.8)
studied a series of inhibitors that effectively retard thecorrosion of Q235A at high concentration HCl. Thecompound H-NMR (D6-DMSO) shows 95.5% inhibitionefficiency at 100mg/L HCl. Li et al.9) studied Q235A steelcorrosion induced by chloride ions under stray currentconditions and constructed a model based on an artificialneural network to accurately predict corrosion, as well asthe corrosion potential and corrosion current density. Jianguoet al.10) studied the polarization curves, corrosion processes,and mechanisms of Q235A steel under dry and wet cyclingconditions. They reported the corrosion current of a dry-wetsample was 22.99 µA/cm2, and this was much higher thanthat of the bare steel (9.3 µA/cm2). To improve the corrosionprotection performance of Q235A steel, some researchershave treated steel surfaces with nickel-nanodiamondcomposite coatings or plasma-clad tungsten carbide particlesor by laser cladding ultra-hard alloys.1113)
The good welding features and mechanical properties(yield strength of 235MPa, ultimate tensile strength of
410MPa, and elongation of 23%) of Q235A steel make it apreferred material in the construction of shipping containers.However, in the marine environment, steel materials areconstantly exposed to salt spray and seawater, making themprone to salt spray corrosion, reducing the strength and lifeservice of the ship. Consequently, determining the corrosionof Q235A steel in a salt spray environment is important forthe design and manufacture of the ship. In this study, weexamined the corrosion products over time during a salt spraytest and examined the effect of the corrosion products onthe corrosion.
2. Experimental Procedures
2.1 Salt spray corrosion experimentThe size of the metal samples used in the salt spray
experiments was 10mm © 10mm © 5mm, and the individ-ual metal samples were cleaned and weighed to the nearest0.1mg before testing. Test samples were cleaned withacetone to remove surface oil, polished with #1200sandpaper, dried in the dry box, and soaked in pure ethanol.
A corrosion test was conducted based on the ISO 9227-2017 standard using a salt spray tester containing a 3.5%NaCl solution. The metal samples were subjected to salt-fogtest for 24, 48, 72, and 96 hours. The pH of the salt-fogsolution was 6.4, temperature of the sample chamber was at35°C, salt spray settlement occurred at 1.02.0mL/80 cm2·h,and relative humidity of the chamber was 70%. After eachtest, the weight of the individual sample was recorded, andthe average weight loss was determined from the four-sampleexperiment.
2.2 Electrochemical experimentsCorroded steel samples were subjected to electrochemical
experiments using the classical three-electrode electrochem-ical test system where the working electrode was thecorroded Q235A steel sample. One side of the test samplewas sealed with an epoxy coating, and the other side servedas the working electrode with a 1 cm2 working surface area.The reference electrode was a saturated calomel electrode,
Materials Transactions, Vol. 61, No. 12 (2020) pp. 2342 to 2347©2020 The Japan Institute of Metals and Materials

the auxiliary electrode was a platinum electrode, and theelectrolyte was 3.5% NaCl solution. The test was conductedusing a CS350 electrochemical workstation (Wuhan CorrTestInstruments Corp., Ltd., Wuhan, China). The test samplewas immersed in the test cell for 10 minutes, followed bypotentiodynamic polarization at a scan rate of 0.001V/s. Thescan was made in potential from corrosion potential tocatholic direction to ¹0.85V and then to anodic direction to¹0.40V.
2.3 Weight loss test and microstructure observationFor weight loss experiments, the corrosion products of the
retrieved samples were removed chemically by immersion ina specific solution (500mL HCl + 500mL distilled water +3.5 g hexamethylenetetramine) that was vigorously stirred for10 minutes at 25°C according to ISO 8407.
The microstructure of the Q235A was determined usingelectron backscattered diffraction (EBSD). The microstruc-ture of the material before and after corrosion was examinedusing scanning electron microscopy (SEM, JEOL JSM-6510,JEOL Ltd., Tokyo, Japan). The compositions of the corrosionproducts on the test sample surface were determined by X-raydiffraction (XRD, Rigaku Ultima IV, Rigaku Corporation,Tokyo, Japan). The analysis was performed using Cu-K¡ withan operating voltage of 40 kV, operating current of 50mA,scanning range of 10°90°, and scanning rate of 2°/minute.
3. Results and Discussion
3.1 Microstructural and morphology characterizationanalysis
The composition of the Q235A steel used in this study was
determined using X-ray fluorescence spectroscopy (Table 1).EBSD analysis was used to determine the test material’scrystal structure. The results confirmed that the steel wasmainly a body-centered cubic structure with a small amountof Fe3C. The grain size was asymmetrically distributed inthe range of ³335 µm, in which the larger size proportionmonotonously decreased (Fig. 1(a) and 1(b)). Ferrite wasthe main phase of Q235A and the elemental carbon meltedin ¡-Fe to form a second solid solution (Fig. 1(c)). Pearlitecrystals were mainly distributed along the material’s grainboundary and exhibited a lamellar and short columnarstructure.
Figure 2 shows the morphology of Q235A steel after thecorrosion in a salt-fog environment for different time periods.(a), (b), (c) and (d) in Fig. 2 were the morphologyappearances of corrosion for 24, 48, 72, and 96 hours,respectively. After 24 hours corrosion, the surface corrosionwas partially covered by corrosion products, and the rustlayer was lighter in color than that of the longer exposureperiod. After 48 hours corrosion, the coverage of the rustlayer expanded, becoming thick and darkened in color. After72 hours corrosion, the coverage area of the rust layerfurther expanded, becoming dark black with substantial edgecorrosion. After 96 hours corrosion, the rust layer completelycovered the surface. In this case, the corrosion products weremuch thicker, and became bright reddish-brown. In summary,
Table 1 Chemical composition of Q235A.
Fig. 1 Microstructure of Q235A: (a) EBSD grain orientation map; (b) Grain size; (c) Metal microstructure map; (d) Partially enlargedview of (c).
Corrosion Mechanism of Q235A under 3.5% NaCl Salt Spray 2343
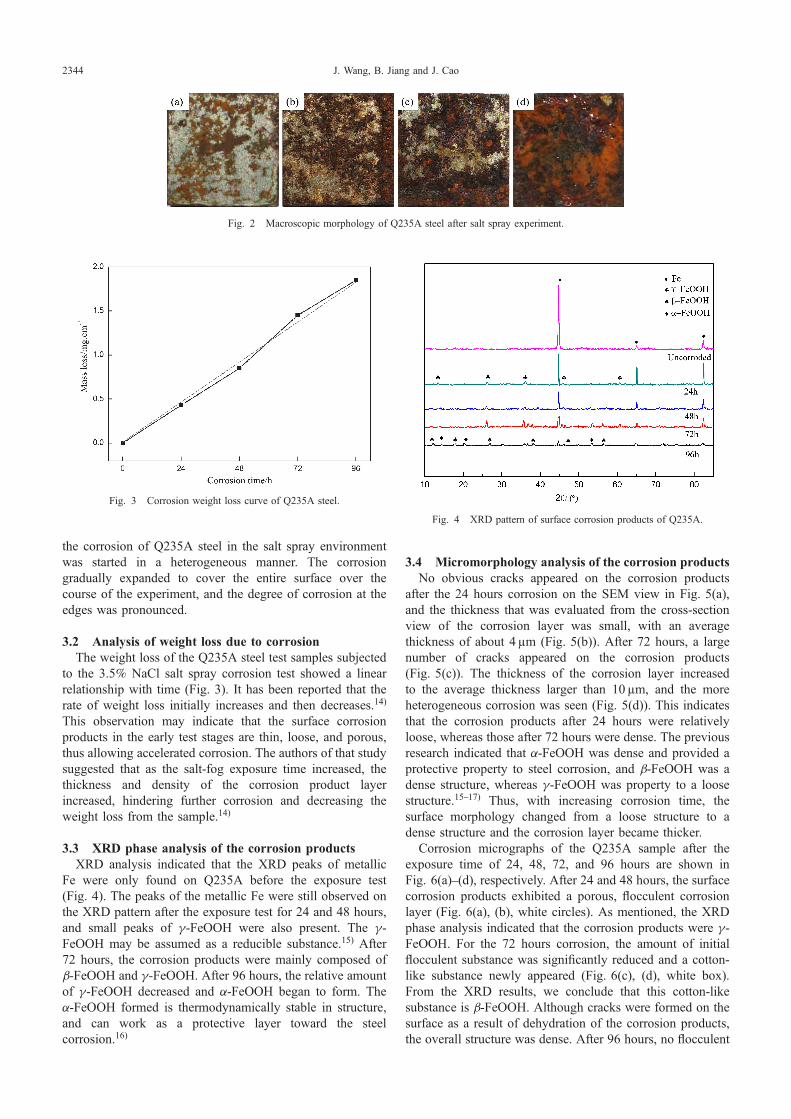
the corrosion of Q235A steel in the salt spray environmentwas started in a heterogeneous manner. The corrosiongradually expanded to cover the entire surface over thecourse of the experiment, and the degree of corrosion at theedges was pronounced.
3.2 Analysis of weight loss due to corrosionThe weight loss of the Q235A steel test samples subjected
to the 3.5% NaCl salt spray corrosion test showed a linearrelationship with time (Fig. 3). It has been reported that therate of weight loss initially increases and then decreases.14)
This observation may indicate that the surface corrosionproducts in the early test stages are thin, loose, and porous,thus allowing accelerated corrosion. The authors of that studysuggested that as the salt-fog exposure time increased, thethickness and density of the corrosion product layerincreased, hindering further corrosion and decreasing theweight loss from the sample.14)
3.3 XRD phase analysis of the corrosion productsXRD analysis indicated that the XRD peaks of metallic
Fe were only found on Q235A before the exposure test(Fig. 4). The peaks of the metallic Fe were still observed onthe XRD pattern after the exposure test for 24 and 48 hours,and small peaks of £-FeOOH were also present. The £-FeOOH may be assumed as a reducible substance.15) After72 hours, the corrosion products were mainly composed of¢-FeOOH and £-FeOOH. After 96 hours, the relative amountof £-FeOOH decreased and ¡-FeOOH began to form. The¡-FeOOH formed is thermodynamically stable in structure,and can work as a protective layer toward the steelcorrosion.16)
3.4 Micromorphology analysis of the corrosion productsNo obvious cracks appeared on the corrosion products
after the 24 hours corrosion on the SEM view in Fig. 5(a),and the thickness that was evaluated from the cross-sectionview of the corrosion layer was small, with an averagethickness of about 4 µm (Fig. 5(b)). After 72 hours, a largenumber of cracks appeared on the corrosion products(Fig. 5(c)). The thickness of the corrosion layer increasedto the average thickness larger than 10 µm, and the moreheterogeneous corrosion was seen (Fig. 5(d)). This indicatesthat the corrosion products after 24 hours were relativelyloose, whereas those after 72 hours were dense. The previousresearch indicated that ¡-FeOOH was dense and provided aprotective property to steel corrosion, and ¢-FeOOH was adense structure, whereas £-FeOOH was property to a loosestructure.1517) Thus, with increasing corrosion time, thesurface morphology changed from a loose structure to adense structure and the corrosion layer became thicker.
Corrosion micrographs of the Q235A sample after theexposure time of 24, 48, 72, and 96 hours are shown inFig. 6(a)(d), respectively. After 24 and 48 hours, the surfacecorrosion products exhibited a porous, flocculent corrosionlayer (Fig. 6(a), (b), white circles). As mentioned, the XRDphase analysis indicated that the corrosion products were £-FeOOH. For the 72 hours corrosion, the amount of initialflocculent substance was significantly reduced and a cotton-like substance newly appeared (Fig. 6(c), (d), white box).From the XRD results, we conclude that this cotton-likesubstance is ¢-FeOOH. Although cracks were formed on thesurface as a result of dehydration of the corrosion products,the overall structure was dense. After 96 hours, no flocculent
Fig. 2 Macroscopic morphology of Q235A steel after salt spray experiment.
Fig. 3 Corrosion weight loss curve of Q235A steel.
Fig. 4 XRD pattern of surface corrosion products of Q235A.
J. Wang, B. Jiang and J. Cao2344

corrosion substance was observed. The corrosion productswere primarily block-like and cotton-like substances (¢-FeOOH and ¡-FeOOH, respectively), and the corrosion layerwas very dense.
3.5 Electrochemical analysis of samples after corrosionThe corrosion potential became negative during the initial
stages of corrosion, and then became positive with increasedexposure time (Fig. 7). The corrosion potential of the 48hours corrosion sample was almost equal to that of 72 hours.The flat region of current density at potentials higher than¹0.5V was caused by the set-up of the electrochemicalworkstation. The corrosion current density increased with the
exposure time to 72 hours, and the increasing ratio slowed,and then slightly decreased between 72 and 96 hours (Table 2and Fig. 8). This indicated that the corrosion rate of the steelwas initially fast and then slowed with increased exposuretime. These results are consistent with those reportedpreviously in which the rate increased firstly and then slowedwith increased exposure time.18,19)
It has been assumed that the cathode reaction on bare steelwas controlled by the diffusion of oxygen in the neutral pHsolution, and that the anode reaction was controlled by chargetransfer on the steel surface. In initial corrosion, amorphousFe2+ and Fe3+ hydroxides, including anions, are formed;then, the amorphous compounds gradually change to mainly
Fig. 5 Surface morphology under salt spray corrosion: (a) 24 h, (c) 72 h; Cross-section morphology under salt spray corrosion: (b) 24 h,(d) 72 h.
Fig. 6 Corrosion micrograph of Q235A steel, the corrosion time of (a), (b), (c), (d) were 24, 48, 72 and 96 h, respectively.
Corrosion Mechanism of Q235A under 3.5% NaCl Salt Spray 2345

£-FeOOH.20) For the 24 h salt spray test sample, becausethe corrosion products were partially covered, the cathodereactions are assumed to be taken by the oxygen reductionand the reduction in corrosion products from Fe3+ toFe2+.21,22) For the 48, 72 and 96 h salt spray test samples,because of the increase in the amount of the corrosionproducts, the cathode reaction was dominated by thereduction reaction of the corrosion products and the corrosioncurrent density was gradually inhibited.
3.6 Corrosion mechanismFrom the results above presented, we divided the corrosion
process for this type of steel into the following steps (Fig. 9).The first step is the iron dissolution. Activation by chlorideions in the salt-fog accelerates iron dissolution to Fe2+,Fe(OH)+, and Fe(OH)2. In addition, because the solution onthe surface is changed from neutral to weakly acidic, thedissolution of bare iron is increased to produce moreFe2+.23,24) The main reactions in this process are presentedin eqs. (1) and (2):
Feþ 2Hþ ! Fe2þ þ H2 " ð1ÞFe2þ þ H2O ! FeðOHÞþ þ Hþ ð2Þ
The second step is the oxidation of dissolved divalentiron ions. In a neutral or weakly acidic environment, the maincorrosion product is £-FeOOH, with a smaller amount of¢-FeOOH. It has been suggested that Fe3O4 was formed inthe iron corrosion process;2527) however, we did not observeFe3O4 in the present study. The main reaction processes areshown in eqs. (3) and (4) as
2Fe2þ þ 1=2O2 þ 3H2O ! 2£-FeOOHþ 4Hþ ð3ÞFe3O4 þ O2 ! £-FeOOH ð4Þ
The third step is the conversion of iron oxyhydroxide.For the corroded Q235A, because of the presence of a rust
Fig. 7 Electrochemical polarization curve of Q235A after 3.5% NaCl saltspray test.
Table 2 Corrosion potential and corrosion current density of Q235A steel with the exposure time.
Fig. 8 The trend of corrosion current densities (icorr) of Q235A steel withthe exposure time.
Fig. 9 Schematic diagram of the corrosion process.
J. Wang, B. Jiang and J. Cao2346

layer, dissolved oxygen cannot be the main oxidizing agent.Rather, the corrosion product £-FeOOH acted as an oxidizingagent, and because of its porous structure, it accelerated thecorrosion.28,29) The main reaction processes are shown ineqs. (5)(7):
FeCl2 �����!H
þ
¢-Fe2ðOHÞ3Cl ð5Þ¢-Fe2ðOHÞ3Cl �! ¢-FeOOH ð6Þ
£-FeOOH �����!Hþor O2
¡-FeOOH ð7ÞBecause ¡-FeOOH is assumed to consist of smaller
particles than £-FeOOH and ¢-FeOOH, ¡-FeOOH works asa dense protective layer to the steel corrosion and thusimproves the steel’s corrosion resistance.
4. Conclusions
In a 3.5% NaCl salt spray corrosive environment, thedegree of corrosion of Q235A steel increased with exposuretime. During the first 24 hours, corrosion heterogeneouslyoccurred and the corrosion products gradually covered wholesurface. After 96 hours, thick and dense corrosion productsformed. With increasing salt spray exposure time, thecorrosion products on the steel gradually changed fromthe £-FeOOH to ¢-FeOOH and ¡-FeOOH. The £-FeOOHformed a flocculent layer, with a loose and porous structure,whereas the ¢-FeOOH and ¡-FeOOH were block-like andcotton-like in structure, respectively, and their structureswere dense. From the electrochemical polarization, £-FeOOHproduced after 24 hours of salt-fog exposure promotedcorrosion of the steel. After 96 hours, ¢-FeOOH and ¡-FeOOH corrosion products inhibited corrosion.
Acknowledgement
This work is supported by the Natural Science Foundationof Jiangsu Province for Universities and Colleges (No.19KJB430031) and the Nantong Science and TechnologyProject (No. GY12018032).
REFERENCES
1) L. Lin, F. Fan and X.D. Zhi: Appl. Mech. Mater. 274 (2013) 463466.
2) X. Yuan, Y. Xiang and X. Huang: Coal Mine Mach. 34(2) (2013) 125127.
3) K. Du and Z.H. Zhang: Phys. Chem. Test. 53 (2017) 2428.4) J. Li et al.: Therm. Process. Technol. 23(2) (2017) 115117.5) K. Otani, M. Sakairi and A. Kaneko: Mater. Trans. 57 (2016) 1539
1546.6) D. Wang and S. Xiu: Acta Metall. Sin. 53 (2017) 567574.7) J. Hu, S. Cao, L. Yin, Q. Liang and J. Xie: Anti-Corros. Methods
Mater. 59 (2012) 305310.8) G. Chen, H. Su, Y. Song, Y. Gao, J. Zhang, X. Hao and J. Zhao: Res.
Chem. Intermed. 39 (2013) 36693678.9) C. Wang, W. Li, Y. Wang, S. Xu and X. Yang: Constr. Build. Mater.
219 (2019) 164175.10) J.G. Liu, Y.T. Li and B.R. Hou: Adv. Mater. Res. 557559 (2012) 139
142.11) M. Liu et al.: Mater. Prot. 51(1) (2018) 6266.12) W. Li, Z. Ma and Z. Ding: Coal Technol. 37(11) (2018) 269271.13) Y. Fu, T. Qi, L. Zong and L. Zheng: China Mech. Eng. 28 (2017) 2378
2382.14) J. Zhang, L. Zuo, J. Jiang, H. Ma and D. Song: J. Chin. Soc. Corros.
Prot. 36 (2016) 363369.15) Y. Gao, Y. Huang, X. Meng, Y. Liao and R. Li: J. South China Univ.
Technol.: Nat. Sci. Ed. 45(9) (2017) 135141.16) L. Zhang, Z. Wang and C. Zhao: Equip. Environ. Eng. 11(1) (2014)
16.17) Y. Ma, Y. Li and F. Wang: Corros. Sci. 51 (2009) 9971006.18) H.J. Flitt and D.P. Schweinsberg: Corros. Sci. 47 (2005) 21252156.19) S. Xu, H. Wang and L. Su: Mater. Mech. Eng. 40(5) (2016) 8691.20) X. Peng, J. Wang, J. Wang, C. Shan, H. Jia, Z. Liu and H. Wang:
J. Chin. Soc. Corros. Prot. 33 (2013) 449454.21) D. Zhang, X. Gao, G. Su, L. Du, Z. Liu and J. Hu: J. Mater. Eng.
Perform. 26 (2017) 25992607.22) J. Dong, J.H. Dong, E. Han, C.M. Liu and W. Ke: Corros. Sci. Prot.
Technol. 18 (2006) 414417.23) X. Zhang, D. Liu, L.F. Wan and X. Li: Mech. Sci. Technol. 29 (2010)
10251030.24) H. Chen, X. Li and Y. Wei: J. Chin. Soc. Corros. Prot. 28(1) (2007) 17
19.25) B. Widyanto and S.W.S. Putri: Mater. Trans. 60 (2019) 732736.26) Y. Zheng, Y. Zou and J. Wang: Corros. Sci. Prot. Technol. 23(1) (2011)
9398.27) Z. Wang, Q. Yu, J. Chen, J. Wang, S. Xu and B. Hu: J. Chin. Soc.
Corros. Prot. 34 (2014) 5358.28) H. Liu, Y. Wei and Y. Sun: J. Mol. Catal. Chem. 226 (2005) 135140.29) K. Asami and M. Kikuchi: Corros. Sci. 45 (2003) 26712688.
Corrosion Mechanism of Q235A under 3.5% NaCl Salt Spray 2347


















