
Contribution of the Twin Arginine Translocation System to the … · consistently reduced....
9
INFECTION AND IMMUNITY, Sept. 2003, p. 4908–4916 Vol. 71, No. 9 0019-9567/03/$08.000 DOI: 10.1128/IAI.71.9.4908–4916.2003 Copyright © 2003, American Society for Microbiology. All Rights Reserved. Contribution of the Twin Arginine Translocation System to the Virulence of Enterohemorrhagic Escherichia coli O157:H7 Nathalie Pradel, 1 Changyun Ye, 1,2 Vale ´rie Livrelli, 3 Jianguo Xu, 2 Bernard Joly, 3 and Long-Fei Wu 1 * Laboratoire de Chimie Bacte ´rienne, UPR9043, IBSM, CNRS, F-13402 Marseille Cedex 20, 1 and Groupe de Recherche Pathoge ´nie Bacte ´rienne Intestinale, Universite ´ d’Auvergne Clermont-1, 63000 Clermont-Ferrand, 3 France, and Department of Diarrheal Diseases, National Institute of Communicable Diseases Prevention and Control, Chinese Center for Disease Control and Prevention, Beijing 102206, Peoples’ Republic of China 2 Received 5 March 2003/Returned for modification 30 May 2003/Accepted 17 June 2003 Shiga toxin-producing Escherichia coli O157:H7 is a major food-borne infectious pathogen. In order to analyze the contribution of the twin arginine translocation (TAT) system to the virulence of E. coli O157:H7, we deleted the tatABC genes of the O157:H7 EDL933 reference strain. The mutant displayed attenuated toxicity on Vero cells and completely lost motility on soft agar plates. Further analyses revealed that the tatABC mutation impaired the secretion of the Shiga toxin 1 (Stx1) and abolished the synthesis of H7 flagellin, which are two major known virulence factors of enterohemorrhagic E. coli O157:H7. Expression of the EDL933 stxAB 1 genes in E. coli K-12 conferred verotoxicity on this nonpathogenic strain. Remarkably, cytotoxicity assay and immunoblot analysis showed, for the first time, an accumulation of the holotoxin complex in the periplasm of the wild-type strain and that a much smaller amount of StxA 1 and reduced verotoxicity were detected in the tatC mutant cells. Together, these results establish that the TAT system of E. coli O157:H7 is an important virulence determinant of this enterohemorrhagic pathogen. Bacteria export numerous proteins across the cytoplasmic membrane via either the Sec machinery (26) or the twin argi- nine translocation (TAT) (also called membrane targeting and translocation [MTT]) system (41). The TAT system is different from the Sec pathway because of its unusual ability to transport folded proteins and even enzyme complexes into the periplasm. Most of the TAT substrates are cofactor-containing enzymes taking part in oxidation-reduction systems involved in energy conservation under anaerobic conditions. The essential tat genes are found in most bacterial genomes, including the pathogens Helicobacter pylori, Yersinia pestis, Vibrio cholerae, Salmonella enterica subsp. enterica serovar Typhi, Haemophilus influenzae, Mycobacterium tuberculosis, Staphylococcus aureus, and Pseudomonas aeruginosa (41). The functionality of the TAT pathway has been reported for Escherichia coli (32, 40), Zymomonas mobilis (12), Ralstonia eutropha (5), Bacillus sub- tilis (17), Pseudomonas stutzeri (14), and Streptomyces lividans (33). It has been demonstrated that the P. aeruginosa TAT system operates in parallel with the Sec machinery in the se- cretion of virulence factors via the type II secretion pathway (38). Further investigation has shown that the TAT system of P. aeruginosa contributes to virulence through the secretion of various factors associated with either pathogenesis or stress response (23). Recently, Ding and Christie reported that the TAT system is an important virulence determinant of the phy- topathogen Agrobacterium tumefaciens (10). Widespread E. coli is a major component of the normal intestinal flora of humans and other mammals and is usually harmless to the host. However, some specific E. coli strains represent primary pathogens with an enhanced potential to cause disease. Shiga toxin- or verotoxin-producing E. coli (STEC or VTEC, respectively) causes not only a broad range of symptoms in humans, including uncomplicated diarrhea, but also more severe diseases like hemorrhagic colitis (HC) and the often deadly hemolytic-uremic syndrome (HUS) (22). Among STEC, enterohemorrhagic E. coli (EHEC) serotype O157:H7 is the most important cause for HC and HUS. The number of O157:H7-related infections is increasing steadily worldwide, and this strain is considered to be both an emerging pathogen and a major threat to public health (19). Genomes of two EHEC O157:H7 isolates (EDL933 and Sakai) have been sequenced (13, 24). The genome of the EDL933 isolate con- tains about 177 so-called O-specific islands, which are DNA segments of more than 50 bp present only in EDL933 but are absent from the genome of the harmless MG1655 isolate of the K-12 strain (24). Some of the O-specific islands and the pO157 plasmid of O157:H7 encode virulence factors, including hemo- lysin, intimin, Shiga toxins 1 (Stx1) and 2 (Stx2), the specific H7 type of flagella, and other factors required for the virulence, such as the type II and type III secretion systems. Moreover, the tat genes of the K-12 strain, which encode the best-char- acterized prototype of bacterial TAT system, are conserved in the two EHEC genomes. However, it is unknown whether the TAT system contributes to the virulence unique to the EHEC. In this study, we knocked out the tatABC genes from the genome of the E. coli O157:H7 EDL933. Remarkably, the toxicity of the mutant was attenuated compared to the parental strain in the Vero cell assay. In addition, immunoblot analysis showed that the amount of Stx1 secreted by the mutant was * Corresponding author. Mailing address: Laboratoire de Chimie Bacte ´rienne, UPR9043, IBSM, CNRS, 31, chemin Joseph Aiguier, F-13402 Marseille cedex 20, France. Phone: 33-4-91164157. Fax: 33- 4-9171 8914. E-mail: [email protected]. 4908 on March 13, 2020 by guest http://iai.asm.org/ Downloaded from
Transcript of Contribution of the Twin Arginine Translocation System to the … · consistently reduced....

INFECTION AND IMMUNITY, Sept. 2003, p. 4908–4916 Vol. 71, No. 90019-9567/03/$08.00�0 DOI: 10.1128/IAI.71.9.4908–4916.2003Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Contribution of the Twin Arginine Translocation System to theVirulence of Enterohemorrhagic Escherichia coli O157:H7
Nathalie Pradel,1 Changyun Ye,1,2 Valerie Livrelli,3 Jianguo Xu,2 Bernard Joly,3and Long-Fei Wu1*
Laboratoire de Chimie Bacterienne, UPR9043, IBSM, CNRS, F-13402 Marseille Cedex 20,1 and Groupe deRecherche Pathogenie Bacterienne Intestinale, Universite d’Auvergne Clermont-1, 63000 Clermont-Ferrand,3
France, and Department of Diarrheal Diseases, National Institute of Communicable Diseases Preventionand Control, Chinese Center for Disease Control and Prevention,
Beijing 102206, Peoples’ Republic of China2
Received 5 March 2003/Returned for modification 30 May 2003/Accepted 17 June 2003
Shiga toxin-producing Escherichia coli O157:H7 is a major food-borne infectious pathogen. In order toanalyze the contribution of the twin arginine translocation (TAT) system to the virulence of E. coli O157:H7,we deleted the tatABC genes of the O157:H7 EDL933 reference strain. The mutant displayed attenuated toxicityon Vero cells and completely lost motility on soft agar plates. Further analyses revealed that the �tatABCmutation impaired the secretion of the Shiga toxin 1 (Stx1) and abolished the synthesis of H7 flagellin, whichare two major known virulence factors of enterohemorrhagic E. coli O157:H7. Expression of the EDL933 stxAB1genes in E. coli K-12 conferred verotoxicity on this nonpathogenic strain. Remarkably, cytotoxicity assay andimmunoblot analysis showed, for the first time, an accumulation of the holotoxin complex in the periplasm ofthe wild-type strain and that a much smaller amount of StxA1 and reduced verotoxicity were detected in the�tatC mutant cells. Together, these results establish that the TAT system of E. coli O157:H7 is an importantvirulence determinant of this enterohemorrhagic pathogen.
Bacteria export numerous proteins across the cytoplasmicmembrane via either the Sec machinery (26) or the twin argi-nine translocation (TAT) (also called membrane targeting andtranslocation [MTT]) system (41). The TAT system is differentfrom the Sec pathway because of its unusual ability to transportfolded proteins and even enzyme complexes into theperiplasm. Most of the TAT substrates are cofactor-containingenzymes taking part in oxidation-reduction systems involved inenergy conservation under anaerobic conditions. The essentialtat genes are found in most bacterial genomes, including thepathogens Helicobacter pylori, Yersinia pestis, Vibrio cholerae,Salmonella enterica subsp. enterica serovar Typhi, Haemophilusinfluenzae, Mycobacterium tuberculosis, Staphylococcus aureus,and Pseudomonas aeruginosa (41). The functionality of theTAT pathway has been reported for Escherichia coli (32, 40),Zymomonas mobilis (12), Ralstonia eutropha (5), Bacillus sub-tilis (17), Pseudomonas stutzeri (14), and Streptomyces lividans(33). It has been demonstrated that the P. aeruginosa TATsystem operates in parallel with the Sec machinery in the se-cretion of virulence factors via the type II secretion pathway(38). Further investigation has shown that the TAT system ofP. aeruginosa contributes to virulence through the secretion ofvarious factors associated with either pathogenesis or stressresponse (23). Recently, Ding and Christie reported that theTAT system is an important virulence determinant of the phy-topathogen Agrobacterium tumefaciens (10).
Widespread E. coli is a major component of the normal
intestinal flora of humans and other mammals and is usuallyharmless to the host. However, some specific E. coli strainsrepresent primary pathogens with an enhanced potential tocause disease. Shiga toxin- or verotoxin-producing E. coli(STEC or VTEC, respectively) causes not only a broad rangeof symptoms in humans, including uncomplicated diarrhea, butalso more severe diseases like hemorrhagic colitis (HC) andthe often deadly hemolytic-uremic syndrome (HUS) (22).Among STEC, enterohemorrhagic E. coli (EHEC) serotypeO157:H7 is the most important cause for HC and HUS. Thenumber of O157:H7-related infections is increasing steadilyworldwide, and this strain is considered to be both an emergingpathogen and a major threat to public health (19). Genomes oftwo EHEC O157:H7 isolates (EDL933 and Sakai) have beensequenced (13, 24). The genome of the EDL933 isolate con-tains about 177 so-called O-specific islands, which are DNAsegments of more than 50 bp present only in EDL933 but areabsent from the genome of the harmless MG1655 isolate of theK-12 strain (24). Some of the O-specific islands and the pO157plasmid of O157:H7 encode virulence factors, including hemo-lysin, intimin, Shiga toxins 1 (Stx1) and 2 (Stx2), the specific H7type of flagella, and other factors required for the virulence,such as the type II and type III secretion systems. Moreover,the tat genes of the K-12 strain, which encode the best-char-acterized prototype of bacterial TAT system, are conserved inthe two EHEC genomes. However, it is unknown whether theTAT system contributes to the virulence unique to the EHEC.
In this study, we knocked out the tatABC genes from thegenome of the E. coli O157:H7 EDL933. Remarkably, thetoxicity of the mutant was attenuated compared to the parentalstrain in the Vero cell assay. In addition, immunoblot analysisshowed that the amount of Stx1 secreted by the mutant was
* Corresponding author. Mailing address: Laboratoire de ChimieBacterienne, UPR9043, IBSM, CNRS, 31, chemin Joseph Aiguier,F-13402 Marseille cedex 20, France. Phone: 33-4-91164157. Fax: 33-4-9171 8914. E-mail: [email protected].
4908
on March 13, 2020 by guest
http://iai.asm.org/
Dow
nloaded from

consistently reduced. Expression of EDL933 stxAB1 genes inthe nonpathogenic E. coli K-12 strain confirmed the impair-ment of Stx1 translocation in the tat mutant. In addition, theTAT system was required for flagellin biosynthesis and themotility of EDL933. Together, these results showed that theTAT system is important for the synthesis of two major knownvirulence factors specific to EHEC O157:H7.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media. The EHEC reference strain used inthis study was the EDL933 serotype O157:H7 (ATCC 43895) provided by A.O’Brien. The E. coli O157 stx mutant is a laboratory stock strain at the Universityof Auvergne Clermont-1, which carries deletions in both the stx1A and stx2Agenes, as revealed by PCR. The nonpathogenic E. coli K-12 strains used in thisstudy are MC4100A (F� �(argF-lac)U169 araD139 rpsL150 (strR) thi flhD5301deoC1 ptsF25 relA1 fruA25 rbsR22 e14� LAM� ara�) and B1LK0A (same asMC4100A, �tatC) (30). Plasmid pKOBEG is a thermosensitive replicon thatcarries the � phage red��� operon expressed under the control of the arabinose-inducible PBAD promoter (9). The p8737 (tatABCD�) plasmid is described inreference 1.
The bacteria were routinely grown in Luria-Bertani (LB) broth, on LB plates,or in the minimal M9 media (20). Anaerobic growth was achieved normally instoppered bottles or tubes filled to the top. As required, ampicillin (100 �g/ml),chloramphenicol (50 �g/ml), kanamycin (50 �g/ml), L-arabinose (10 mM), trim-ethylamine N-oxide (TMAO; 1 mg/ml), glycerol (0.5%), ammonium molybdate(1 �M), or potassium selenite (1 �M) was added. Precultures were grown fromsingle colonies and used at 100-fold dilutions for inoculation of experimentalcultures.
Construction of the EDL933 �tatABC mutant. Replacement of the tatABCgenes on the E. coli O157:H7 EDL933 chromosome by the aphA-3 gene wasperformed according to the method of Ghigo et al. (http://www.pasteur.fr/recherche/unites/Ggb/Pcr3T.ppt) and Datsenko and Wanner (9). pKOBEG wasintroduced into the target strain EDL933 by selection for Cmr in a speciallow-salt, low-Mg2� broth (10 g of tryptone, 2.5 g of yeast extract, 0.25 g of NaCl,2.5 ml of 1 M KCl [pH 7.4]) at 30°C overnight. A fresh culture in the samemedium was inoculated from the overnight preculture by 100-fold dilution. Toinduce the recombinase expression from the � phage red��� operon onpKOBEG, 10 mM L-arabinose was added in this culture. The incubation wasperformed at 30°C with shaking until an optical density at 620 nm (OD620) of0.45 to 0.55 was reached, and then the cultures were placed in a water bath at42°C with shaking for 15 min to eliminate pKOBEG containing a thermosensi-tive replicon. Competent cells were prepared from these cultures. The aphA-3gene was amplified by PCR with the primers Kngbtataup (5-AACGTATAATGCGGCTTTGTTTAATCATCATCTACCACAGAGGAACATGTAAAGCCACGTTGTGTCTCAA-3) and Kngbtatcdn (5-ATATCAAACATCCTGTACTCCATATGACAACCGCCCTGACGGGCGGTTGATTAGAAAAACTCATCGAGCA-3) and the Taq DNA polymerase according to the manufacturer’sinstructions (Appligene-oncor, Illkrich, France). The amplified fragment carried,at the two ends, 50 bp (underlined) identical to the sequences from 1 to 50 bpupstream of the tatA gene and from 51 to 100 bp downstream of the tatC geneof E. coli EDL933. The PCR product was purified by using the QIAquick PCRpurification kit in accordance with the supplier’s instructions (QIAGEN S.A.,Courtaboeuf, France) and transformed, by electroporation, into EDL933 com-petent cells prepared as above and selected on LB-kanamycin plates. The Kmr
Cms phenotype of the recombinants was verified by the disk diffusion method onMueller-Hinton agar (BioMerieux, Marcy-L’Etoile, France), with disks pur-chased from Sanofi Diagnostic Pasteur (Marnes La Coquette, France). Thereplacement of the tatABC genes by aphA-3 was verified by PCR with theoligonucleotides tat1 (5-AACGTATAATGCGGCTTTGTTTAATCATCATCTACCACAGAGGAACATGT-3), tat2 (5-ATATCAAACATCCTGTACTCCATATGACAACCGCCCTGACGGGCGGTTGA-3), GBnp-3 (5-TTAGAAAAACTCATCGAGCA-3), and GB-5 (5-AGCCACGTTGTGTCTCAAAATC-3). Further verification was performed by digestion of the PCR products withthe SacII restriction enzyme. The general properties of the wild-type strainEDL933 and the mutant EDLtat were biochemically confirmed by using an API20E test (BioMerieux).
Cloning of the stxAB1 genes. The stxAB1 genes were amplified from the chro-mosome of E. coli O157:H7 isolate EDL933 by PCR with the primers stx1-1(5-ATGAATTCATATGAAAATAATTATTTTTAGAGT-3) and stx1-4 (5TCACTAAGCTTGTCGACGTCAACGAAAAATAACTTCGCT-3). The am-
plified fragment was digested with EcoRI and HindIII and cloned into thecorresponding sites of the pBAD24. The structure of the resulting plasmid,pSTX1AB, was verified by endonuclease digestion. Expression of stxAB1 geneswas under the tight control of the PBAD promoter.
Enzyme assays. To detect the TMAO and dimethyl sulfoxide (DMSO) reduc-tase activities, protein fractions were separated on nondenaturing polyacryl-amide (12.5%) gels. DMSO and TMAO reductase activities were visualized byan activity staining method which is based on a methyl viologen-linked TMAOreduction (31).
Cellular fractionation, electrophoresis, immunoblot, and mass spectrometry.To prepare cellular fractions, bacteria were grown anaerobically in 200 ml of LBmedium and the cells were harvested by centrifugation at 17,000 g for 10 min.Periplasm and spheroplasts were prepared by the lysozyme-EDTA-cold osmo-shock method (31). Spheroplasts were washed once and disrupted by sonicationin 2 ml of 40 mM Tris-HCl (pH 7.6), in a Branson Sonifier 450 in the continuousmode and with an output setting of 2 for 30 s. The supernatant filtrates preparedfrom the bacterial cells grown in brain heart infusion broth were precipitatedwith 10% trichloroacetic acid, and washed with acetone. Protein samples wereseparated by polyacrylamide gel electrophoresis in the presence (denaturing) orin the absence (nondenaturing) of sodium dodecyl sulfate (SDS) on 12.5% or15% acrylamide gels. Protein samples were electrotransferred, after the electro-phoresis, onto a polyvinylidene difluoride membrane and analyzed by immuno-blotting, by using the enhanced chemiluminescence method according to themanufacturer’s instructions (Amersham Biosciences). The images were digitizedand quantified with Kodak Image analysis software. Polyclonal antibodies againstStx were kindly provided by A. O’Brien. Anti-H7 flagellin sera were purchasedfrom Difco (Detroit, Mich.) and used at 1:10,000-fold dilution for immunoblotanalysis.
The two-dimensional gel electrophoresis (2DE) was performed according toAmersham Biosciences’ instructions. For mass spectrometry, protein sampleswere separated by 2DE and stained by Coomassie blue or silver staining and thespecific protein spots were excised. After crushing and washing of the excised gel,the proteinaceous material was reduced with dithiothreitol and alkylated withiodoacetamide in 100 mM NH4HCO3. Proteolytic digestion by trypsin was thenperformed overnight at 37°C. The supernatant was collected, the salts wereremoved by flow through an R2 Poros column, and the sample was analyzed bymass spectrometry. The protein was identified by using the ProFound software(http://129.85.19.192/profound_bin/). The identification significance was evalu-ated by the Est’dZ score, as described at http://129.85.19.192/profound/help.html.
Vero cell assay. The production of Shiga toxin was checked by a cytotoxicityassay on Vero cells (18). Vero cells (African green monkey kidney cells; ATCCCRL 1587) were grown at 37°C in Eagle basal medium (Seromed, Berlin, Ger-many) supplemented with 10% fetal calf serum (Seromed), 1% L-glutamine (LifeTechnologies, Paisley, Scotland), 2% penicillin-streptomycin-amphotericin B,and 1% minimal essential medium vitamin solution (Life Technologies) in anatmosphere of 5% CO2. Vero cells were cultivated as monolayers in 75-cm2
tissue culture flasks. The bacterial strains were inoculated into 10 ml of brainheart infusion broth (Biokar Diagnostics) and incubated at 37°C to an OD620 of1. After centrifugation of 1.5 ml at 12,000 g for 5 min, supernatant filtrateswere obtained with a 0.45-�m-pore-size filter (PolyLabo, Molsheim, France) andscreened for verotoxicity. Twofold serial dilutions of bacterial filtrates were donein 96-well flat-bottom microtiter plates (Nunc, Roskilde, Denmark) (50 �l perwell). A total of 50 �l of Eagle basal medium containing 105 Vero cells insuspension was added to each well. The culture plates were incubated for 24 h at37°C in a 5% CO2 atmosphere. After 24 h, the cell monolayers were washed withphosphate-buffered saline (PBS) (pH 7.4) (Seromed) and stained with a crystalviolet solution (1.3% crystal violet–5% ethanol in PBS). The verotoxin titer wasexpressed as the reciprocal of the highest sample dilution of culture filtrate whichcaused 50% cell detachment after 24 h of incubation, as judged by the dyeintensity and by microscopic observation. The stx mutant was used as a negativecontrol. Alternatively, 50 �l of each dilution of cellular fractions prepared fromK-12 strains expressing cloned stxAB1 genes were used in place of filtrates.
Motility assay. Motility assays were performed in 0.3% LB soft agar medium.Cell cultures were normalized to an OD600 of 0.9, and 10 �l of each strain wasinoculated onto the motility plates. Motility was examined after 24 h of incuba-tion at 37°C. At least three independent motility assays were carried out for eachstrain.
Extraction of flagella. Bacteria were recovered from 5 LB plates incubatedovernight at 37°C, centrifuged for 10 min at 5,000 g in PBS buffer (200 mMNaCl, 3 mM KCl, 1.5 mM KH2PO4, and 16 mM Na2HPO4 [pH 7.4]). The pelletwas resuspended in 10 ml of PBS and vortexed for 5 min. The suspension wascentrifuged for 15 min at 16,000 g. After recovery of the supernatant, SDS was
VOL. 71, 2003 TAT SYSTEM AND E. COLI O157:H7 VIRULENCE 4909
on March 13, 2020 by guest
http://iai.asm.org/
Dow
nloaded from

added (0.1% wt/vol). The solution was centrifuged at 11,000 g for 3 h at 4°C.The pellet was recovered in 400 �l of Tris buffer (40 mM, pH 8).
RESULTS AND DISCUSSION
TAT system of EHEC O157:H7 and construction of the�tatABC mutant of the EDL933 isolate. The genetic structureand composition of the tat genes can vary from genome togenome (41). The TAT apparatus of the nonpathogenic E. coliK-12 strain is encoded by the tatABCD operon and the tatEgene (32). TatA, TatB, and TatE share sequence homology attheir N termini, including one transmembrane segment and anadjacent amphipathic domain, whereas their C termini vary insequence and length (8). TatC is an integral membrane proteinwith apparently four transmembrane helices (11). The deletionof tatC leads to mislocation of all substrates analyzed anddisplays pleiotropic phenotypes (34). Therefore, TatC is essen-tial for TAT function. In contrast, TatD is a soluble proteinwith no discernible role in Tat-dependent protein export (4).We analyzed the genomes of the two E. coli O157:H7 isolates,EDL933 and Sakai, and found that they contained the sametatABCD operon and the tatE gene as the E. coli K-12 strain.In addition, the sequences of the TatA, TatB, and TatC pro-teins were 100% identical in all three genomes. Therefore, thebasic physiological function and the operation mechanism ofthe TAT system must be conserved in EHEC and nonpatho-genic E. coli. Remarkably, the tatE gene and the particulargenetic structure of the tat coding genes, i.e., the tatABCDoperon plus the tatE gene, is observed only for enteric bacteria,including E. coli, S. enterica serovar Typhi, V. cholerae, and Y.pestis (41). Therefore, the TAT system might provide an im-portant mechanism for adaptation of the environment of thegut.
In order to analyze the potential contribution of the TATsystem to the virulence of the EHEC, we constructed a tatmutant of the E. coli O157:H7 reference strain, EDL933, byreplacing the tatABC genes with the aphA-3 gene encodingkanamycin resistance. The deletion of the tatABC genes in themutant (EDLtat) was confirmed by PCR amplification and byendonuclease digestion. Using the tat1-tat2 oligonucleotidepair, the tatABC region of the wild-type parental strain wasamplified as an �1.7-kb fragment. In contrast, the replacementof the tatABC genes by aphA-3 resulted in a reduction of thesize of the corresponding PCR product to �1 kb in the recom-binant colonies analyzed. A SacII cleavage site is present onlyin the tatB gene. When subjected to the SacII digestion, thetatABC-carrying 1.7-kb fragment was cleaved into an �1.1-kbtatBC fragment and an �0.6-kb tatAB fragment, whereas the1-kb aphA-3 fragment remained intact, thus confirming thereplacement of the tatABC genes by aphA-3 in the chromo-some of the EDLtat mutant.
Three standard means for assaying TAT function in thenonpathogenic E. coli K-12 strain are to analyze the translo-cation of known TAT substrates, to test for anaerobic growthon TMAO-glycerol minimal media, and to inspect the mor-phology of the cells. SufI is one of the most used substrates inthe study of the TAT system (1, 35, 42). As shown in Fig. 1A,it was translocated into the periplasm of only the wild-typestrain but was absent from the periplasm of the �tatABC mu-tant. As a control, the translocation of the maltose binding
protein, MalE, was also analyzed. MalE is a typical substrate ofthe Sec pathway. Since MalE was normally translocated intothe periplasm of the tat mutant (Fig. 1A, lanes 3 and 4), the�tatABC mutation affected only the translocation of TAT sub-strates but had no effect on the Sec-dependent protein export.
Both membrane-bound DMSO reductase and periplasmicTMAO reductase support anaerobic growth on TMAO (40).The interruption of the tat genes of the harmless E. coli K-12strain abolishes the translocation of these two enzymes but hasonly a slight effect on the enzymatic activity. Because these twoenzymes are mislocated in the cytoplasm and cannot form theanaerobic respiratory chains, the K-12 tatB and tatC mutantsare incapable of anaerobic growth with TMAO as the soleelectron acceptor (6, 40). Like the E. coli K-12 tat mutants, theE. coli O157:H7 �tatABC mutant could not grow on the min-imal glycerol-TMAO medium, but the parental wild-type straingrew normally under the same conditions (data not shown).Surprisingly, neither TMAO reductase activity nor the TMAOreductase protein (TorA) was detected in the E. coli O157:H7strain EDL933 (Fig. 1B and C, lanes 1 and 4). The genomes ofthe E. coli O157:H7 strains EDL933 and Sakai contain the fullyconserved tor operon. In addition, we confirmed, by PCR, thepresence of the torA gene in the EDL933 stock strain used in
FIG. 1. Phenotype of the EDL933-derived �tatABC mutant. Peri-plasmic fractions (3 �g of protein each, all lanes in panel A, lanes 1 to3 in panels B and C) and spheroplasts (30 �g of protein each, lanes 4to 6 in panels B and C) of the E. coli O157:H 7 EDL933 (wild type[WT]), its derivative EDLtat mutant (�tatABC), and the E. coli K-12strain (K-12) were resolved on a native polyacrylamide (12.5%) gel.SufI and MalE were resolved on SDS denaturing gels and detected byimmunoblotting with polyclonal antisera against these two proteins,respectively (A). The DMSO reductase (DMSOR) and TMAO reduc-tase (TMAOR) activities were visualized by activity staining (B). Thegel was then analyzed by immunoblotting with polyclonal antiseraagainst the TMAO reductase (C) (TorA). Light microscope phase-contrast images of EDL933 (WT) and the EDLtat mutant (�tatABC)are presented in panel D.
4910 PRADEL ET AL. INFECT. IMMUN.
on March 13, 2020 by guest
http://iai.asm.org/
Dow
nloaded from
this study (data not shown). Therefore, the depletion of theTMAO reductase might result from a mutation in the regula-tory genes or in the torA structure gene and be specific to theEDL933 stock strain used in this study. This result is consistentwith the high genomic diversity of EHEC O157:H7, as revealedby analysis of 1,102 isolates in Japan (36). The growth of thewild-type EDL933 strain used in this study on the minimalmedium is thus supported only by the DMSO reductase. In-terestingly, DMSO reductase activities were absent from boththe periplasm and the spheroplasts of the �tatABC mutant(Fig. 1B, lanes 2 and 5). These results showed that the �tatABCmutation impairs both the translocation and the activity of theDMSO reductase of E. coli O157:H7 EDL933. Therefore, thedeletion of the tatABC genes seems to have much more severeconsequence in EDL933 than in the K-12 strain.
In addition to the failure of growth on minimal medium, tatmutations in the K-12 strain seem to affect the late stage ofcellular division. Thus, the mutant cells form chains with var-ious lengths depending on the tat mutants (34). Similarly, wefound that the �tatABC mutant exhibited an elongated shapeand formed chains, in contrast to the single-cell short bacilli ofwild-type strains (Fig. 1D). Together, these results confirmedthe phenotype expected for the �tatABC mutation.
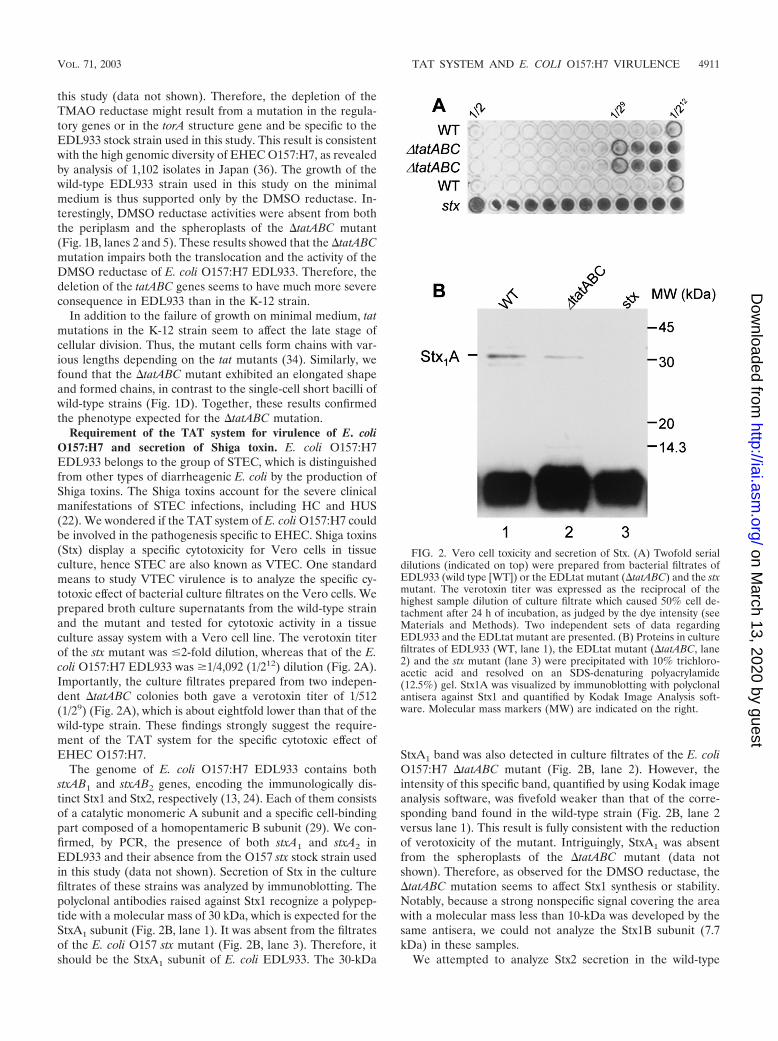
Requirement of the TAT system for virulence of E. coliO157:H7 and secretion of Shiga toxin. E. coli O157:H7EDL933 belongs to the group of STEC, which is distinguishedfrom other types of diarrheagenic E. coli by the production ofShiga toxins. The Shiga toxins account for the severe clinicalmanifestations of STEC infections, including HC and HUS(22). We wondered if the TAT system of E. coli O157:H7 couldbe involved in the pathogenesis specific to EHEC. Shiga toxins(Stx) display a specific cytotoxicity for Vero cells in tissueculture, hence STEC are also known as VTEC. One standardmeans to study VTEC virulence is to analyze the specific cy-totoxic effect of bacterial culture filtrates on the Vero cells. Weprepared broth culture supernatants from the wild-type strainand the mutant and tested for cytotoxic activity in a tissueculture assay system with a Vero cell line. The verotoxin titerof the stx mutant was �2-fold dilution, whereas that of the E.coli O157:H7 EDL933 was �1/4,092 (1/212) dilution (Fig. 2A).Importantly, the culture filtrates prepared from two indepen-dent �tatABC colonies both gave a verotoxin titer of 1/512(1/29) (Fig. 2A), which is about eightfold lower than that of thewild-type strain. These findings strongly suggest the require-ment of the TAT system for the specific cytotoxic effect ofEHEC O157:H7.
The genome of E. coli O157:H7 EDL933 contains bothstxAB1 and stxAB2 genes, encoding the immunologically dis-tinct Stx1 and Stx2, respectively (13, 24). Each of them consistsof a catalytic monomeric A subunit and a specific cell-bindingpart composed of a homopentameric B subunit (29). We con-firmed, by PCR, the presence of both stxA1 and stxA2 inEDL933 and their absence from the O157 stx stock strain usedin this study (data not shown). Secretion of Stx in the culturefiltrates of these strains was analyzed by immunoblotting. Thepolyclonal antibodies raised against Stx1 recognize a polypep-tide with a molecular mass of 30 kDa, which is expected for theStxA1 subunit (Fig. 2B, lane 1). It was absent from the filtratesof the E. coli O157 stx mutant (Fig. 2B, lane 3). Therefore, itshould be the StxA1 subunit of E. coli EDL933. The 30-kDa
StxA1 band was also detected in culture filtrates of the E. coliO157:H7 �tatABC mutant (Fig. 2B, lane 2). However, theintensity of this specific band, quantified by using Kodak imageanalysis software, was fivefold weaker than that of the corre-sponding band found in the wild-type strain (Fig. 2B, lane 2versus lane 1). This result is fully consistent with the reductionof verotoxicity of the mutant. Intriguingly, StxA1 was absentfrom the spheroplasts of the �tatABC mutant (data notshown). Therefore, as observed for the DMSO reductase, the�tatABC mutation seems to affect Stx1 synthesis or stability.Notably, because a strong nonspecific signal covering the areawith a molecular mass less than 10-kDa was developed by thesame antisera, we could not analyze the Stx1B subunit (7.7kDa) in these samples.
We attempted to analyze Stx2 secretion in the wild-type
FIG. 2. Vero cell toxicity and secretion of Stx. (A) Twofold serialdilutions (indicated on top) were prepared from bacterial filtrates ofEDL933 (wild type [WT]) or the EDLtat mutant (�tatABC) and the stxmutant. The verotoxin titer was expressed as the reciprocal of thehighest sample dilution of culture filtrate which caused 50% cell de-tachment after 24 h of incubation, as judged by the dye intensity (seeMaterials and Methods). Two independent sets of data regardingEDL933 and the EDLtat mutant are presented. (B) Proteins in culturefiltrates of EDL933 (WT, lane 1), the EDLtat mutant (�tatABC, lane2) and the stx mutant (lane 3) were precipitated with 10% trichloro-acetic acid and resolved on an SDS-denaturing polyacrylamide(12.5%) gel. Stx1A was visualized by immunoblotting with polyclonalantisera against Stx1 and quantified by Kodak Image Analysis soft-ware. Molecular mass markers (MW) are indicated on the right.
VOL. 71, 2003 TAT SYSTEM AND E. COLI O157:H7 VIRULENCE 4911
on March 13, 2020 by guest
http://iai.asm.org/
Dow
nloaded from

strain and the �tatABC mutant by immunoblotting. The anti-Stx2 polyclonal antisera did not reveal any specific band in theculture supernatants of these strains (data not shown). It mightbe due to poor production or release of Stx2 under the condi-tions used. The production and release of both Stx1 and Stx2are dependent on bacteriophage induction (21, 39). However,only the stxAB1 is expressed at the constitutive level indepen-dent of phage induction (39). Under the growth conditionsused in this study, the stxAB1 genes might be expressed atuninduced basic levels, whereas the Stx2 synthesized might betoo low to be detected.
Synthesis and export of Stx1 in the nonpathogenic E. coliK-12 strain. The stx genes are located in the late-phase regionof the Stx-converting phages, upstream and in the same tran-scriptional orientation as the S gene that encodes the holinnecessary for host lysis (37). It has been demonstrated recentlythat phage-mediated lysis regulates the quantity of Stx1 pro-duced and provides a mechanism for Stx1 release (39). How-ever, unlike Stx2, growth in low-iron media stimulates Stx1production from the iron-regulated promoter but does not leadto host lysis. Therefore, Wagner et al. proposed the contribu-tion of unknown phage-independent mechanisms, for example,the type II secretion system, to Stx1 release in vivo under thesecircumstances (39). Interestingly, in this study we detected theStxA1 subunit in the culture supernatant of E. coli O157:H7
grown under noninduced conditions, thus confirming thephage-independent release of Stx1. Most importantly, thequantity of StxA1 detected in the culture supernatant of the�tatABC mutant was reduced fivefold compared to the wild-type strain, which was paralleled by the attenuation of viru-lence of the mutant in a Shiga toxin-specific in vitro assay.Since StxA1 was absent from the spheroplasts of the �tatABCmutant, it is impossible to distinguish an effect of the �tatABCmutation on Shiga toxin secretion from that on Shiga toxinsynthesis or stability in the EDLtat mutant.
To better understand the effect of the �tatABC mutation onStx1 synthesis and export, we cloned the stxAB1 genes ofEDL933 and studied Stx1 export in the nonpathogenic E. coliK-12 strain. The wild-type K-12 MC4100A strain does notcontain lambda phage; release of Stx1 via phage-mediated lysiswas thus impossible. In addition, the expression of clonedstxAB1 genes was under the tight control of the PBAD pro-moter; we could dissociate Stx1 export from its synthesis. StxA1
was undetectable by immunoblot in the culture filtrates of boththe wild-type strain and the B1LK0A (�tatC) mutant (Fig. 3A,lanes 8 and 9). The anti-Stx1 antisera recognized two bands inthe periplasm of the wild-type strain harboring the plasmidpStx1AB. The bigger one was found also in the control straincarrying the vector pBAD24 (Fig. 3A, lanes 1 and 2). In con-trast, the smaller one was specific for MC4100A/pSTX1AB
FIG. 3. Synthesis and translocation of Stx1 in nonpathogenic E. coli K-12 strains. Culture filtrates and cellular fractions were prepared fromthe wild-type K-12 MC4100A strain (WT) and its B1LK0A �tatC derivative containing pStx1AB grown with arabinose. Samples were eithersubjected to electrophoresis on SDS denaturing gels and analyzed by immunoblotting with antibodies against Stx1 (A) or assayed for verotoxicity(B) as described in the legend to Fig. 2 and in Materials and Methods. Two sets of independent verotoxicity assays gave the same results. Onlyone set is presented here. NS, nonspecific band.
4912 PRADEL ET AL. INFECT. IMMUN.
on March 13, 2020 by guest
http://iai.asm.org/
Dow
nloaded from

and had a molecular mass (30 kDa) expected for StxA1. Im-portantly, this specific band was barely detectable in theperiplasm of the �tatC mutant expressing the cloned stxAB1
genes (Fig. 3A, lane 3). A nonspecific band was detected at themigration position expected for StxA1 in the spheroplasts ofthe three strains analyzed (Fig. 3A, lanes 4 to 6). Because ofthe poor resolution on the gels, it is unclear whether StxA1 waspresent in the spheroplasts.
The location of Stx1 expressed in nonpathogenic E. coli K-12strains was further analyzed by verotoxicity assay. As a control,neither the wild type nor the �tatC mutant transformed withvector pBAD24 exhibited cytotoxicity (Fig. 3B, lines 7 and 8).In contrast, cytotoxicity was detected in the 218-fold dilution ofthe periplasm of the wild-type strain expressing Stx1 (Fig. 3B,line 3). Therefore, expression of the stxAB1 genes is sufficientto allow the synthesis of active Shiga toxin in nonpathogenic E.coli. In addition, it shows, for the first time, the periplasmiclocation of Stx1. Similarly, Shiga toxin was also detected in theperiplasm of the �tatC mutant, but it was about 16-fold lessactive than that of the wild-type strain (Fig. 3B, line 4 versusline 3). The Vero cell cytotoxicity was found in 215- and 211-fold dilutions of the extracts prepared from the spheroplasts ofthe wild type and the �tatC mutant, respectively (Fig. 3B, lines5 and 6). A weak cytotoxicity was detected in the eightfolddilution of the filtrates of the wild type and the �tatC mutantexpressing the stxAB1 genes (Fig. 3B, lines 1 and 2). The de-tected cytotoxicity in the filtrates probably resulted fromphage-independent cell lysis.
The Stx1A protein and verotoxicity were significantly re-duced in both the O157:H7 and K-12 tat mutants. In the K-12strain, the Stx1A synthesis was induced by arabinose from thePBAD promoter of the pBAD24 plasmid. This promoter hasbeen widely used for expressing endogenous or heterologousgenes in K-12 tat mutants, and no effect of the tat mutations onthe transcription from this promoter has been observed (7, 30).Therefore, the reduction of Stx1A in the tat mutants must bedue to a posttranscriptional event, probably proteolysis. It hasbeen observed that various TAT substrates are degraded whenthe TAT system is nonfunctional or the translocation is medi-ated by defective signal peptides (7). It is necessary to removethese polypeptides, since accumulation of folded proteins inthe cytoplasm may have severe consequences.
The Shiga toxin consists of an enzymatically active A subunitand five identical B subunits (29). The B subunits are respon-sible for the binding of toxin to glycolipid receptor of specifictarget eucaryotic cells. After endocytosis, the A subunit inhib-its protein synthesis by releasing an adenine base from the 28SrRNA of the 60S ribosomal subunit. Since the spheroplasts ofMC4100A/pStx1AB and B1LK0A/pStx1AB displayed Verocell toxicity, the assembly of subunits A and B to form activeholotoxin may occur in the cytoplasm. Because it is capable ofexporting an enzyme complex of up to 100 kDa (28), the Tatsystem would be the most suitable known apparatus to exportthe �70-kDa holotoxin into the periplasm. At present, wecould not exclude an in vitro assembly of the holotoxin afterbreaking the cells. Nevertheless, much less Shiga toxin wasdetected in the periplasm of the tat mutant than in the wild-type strain. The holotoxin accumulated mainly in the peri-plasm, which is consistent with the fact that the K-12 strainsused here are not lysogens of lambda phage and lack the
plasmid-borne type II secretion system. The residual verotox-icity found in the filtrates might result from phage-independentlysis. Together, these results showed that the TAT system playsa very important role in secretion or stability of one majorknown virulence factor of EHEC, although a direct effect ofthe tat mutation on Stx1 secretion could not be established.
Effect of the �tatABC mutation on protein export into theperiplasm of the O157:H7 strain. The proteins exported by theTAT system are synthesized with twin arginine signal peptides.The twin arginine signal peptides resemble Sec-dependent sig-nal peptides considering their overall structures but possess atwin arginine motif in the N region, a weakly hydrophobic Hregion, and a Sec-avoidance signal composed of basic residuesaround the H region. Increasing evidence suggests that signalpeptides without the conserved RR motifs can also mediateTAT pathway translocation (15, 16, 25). Therefore, the crite-rion of the presence of the twin arginine motif in the signalpeptide seems not to be sufficient for identifying the proteinsthat are exported by the TAT system. Interestingly, Stx1Bpossesses a double lysine motif in the signal peptide, which iscompatible with a TAT-dependent translocation under certaincontexts (16, 25). It is possible that the TAT pathway might beinvolved, directly or indirectly, in the translocation of virulencefactors synthesized with the signal peptide without the twinarginine motif in E. coli O157:H7. To assess this hypothesis, wecompared the periplasmic protein profile of the wild-typeEDL933 strain with that of the �tatABC derivative by 2DE.The results showed that at least 8 proteins were absent from, orpresent in a significantly reduced amount, in the periplasm ofthe mutant (Fig. 4). Six of them were identified by mass spec-trometry. They included four periplasmic substrate-bindingproteins of ABC transporters, the hyperosmotically inducibleprotein (OsmY) and the H7 flagellin.
The periplasmic substrate-binding proteins are oligopeptidebinding protein (OppA), lysine-arginine-ornithine binding pro-tein (KRObp), galactose-glucose binding protein (GGbp), andthe putative third arginine binding protein (3Rbp). The pred-icated signal peptide of OppA contains a KR motif, and thethree others contain a KK motif. These motifs are compatiblewith TAT-dependent translocation (16, 25). The absence ofthese proteins from the periplasm of the �tatABC mutantwould suggest a TAT-dependent translocation, but it needs tobe confirmed. Although periplasmic substrate binding proteinsare required for chemotactic responses, contributions of theproteins identified in this study to the virulence of E. coliO157:H7 are unknown. osmY gene expression is induced byhyperosmotic stress, and interruption of osmY increases bac-terial sensitivity to hyperosmotic stress (43). Adaptation tohigh osmolarity plays an important role for the survival ofpathogens in hosts. The TAT system might contribute indi-rectly to the adaptation to high osmolarity.
The �tatABC mutation affects flagellar biogenesis. In con-trast to the substrate binding proteins and OsmY, the H7flagellin is an established virulence factor. In addition to therequirement for motility of the pathogen in the hosts, increas-ing evidence suggests that flagella also enhance pathogenicityeither by promoting adherence to host tissues or by directlyactivating host inflammatory signaling pathways (27). Flagellin,encoded by fliC, is the major structural protein of the flagellaof gram-negative bacteria, and it specifies the H antigen of E.
VOL. 71, 2003 TAT SYSTEM AND E. COLI O157:H7 VIRULENCE 4913
on March 13, 2020 by guest
http://iai.asm.org/
Dow
nloaded from

coli (27). Sequence analysis of FliC from pathogenic E. colistrains with distinct H antigens has revealed that their N- andC-terminal regions are largely conserved, whereas the centralregion is highly diverse and distinguishes EHEC O157:H7 orO55:H7 from EHEC O55:H6, O127:H6, and O142:H6 (27). Inthis study, the mass spectrometry analysis identified not onlypeptides located in the N- and C-terminal regions but alsothose specific to H7 flagellin in the central region. Recently, ithas been reported that the H7 flagellin of E. coli O157:H7activates epithelial cell mitogen-activated protein kinase andNF-�B pathways leading to interleukin-8 secretion (3). There-fore, the H7 flagellin is one of the major virulence factorsspecific to EHEC O157:H7.
Flagellum assembly is a sequential process including a suc-cessive secretion of proteins through the flagellar assemblyapparatus by a type III mechanism to the distal end, thus
adding protein subunits from the most proximal cell to themost distal cell. Intriguingly, FliC was detected in theperiplasm of the wild-type EDL933 strain but was absent fromthe �tatABC derivative (Fig. 4B). The periplasmic fractionswere prepared by the lysozyme-EDTA-cold osmoshockmethod. Lysozyme is a muramidase and is used to destroy thepeptidoglycan in this protocol. Since flagella are held by P ringand L ring located, respectively, in the peptidoglycan and outermembrane layers, this protocol might destabilize the flagellaand thus release the flagellin into the periplasmic fraction. Totest this hypothesis and to study the impact of the �tatABCmutation on flagellin synthesis, flagella were extracted fromthe wild-type strain and the �tatABC mutant. Cellular fraction-ation was performed after the extraction. These fractions wereresolved on SDS-denaturing gels, proteins were visualized byCoomassie blue staining, and flagellin was detected by immu-
FIG. 4. Comparison of periplasmic protein profiles obtained by 2DE. Periplasmic fractions (100 �g of protein each) of EDL933 (wild type[WT]) or its �tatABC derivative were separated first on 7-cm immobilized pH gradient strips (pH 3 to 10 in panel A, pH 3 to 6 in panel B) andthen on SDS denaturing polyacrylamide (12.5%) gels, and proteins were visualized by Coomassie blue staining. Molecular masses (MW, inkilodaltons) and pH ranges are indicated. The spots of which the counterparts are absent or present with significantly reduced quantity in themutant periplasmic fractions are indicated by white circles. The spots identified by mass spectroscopy are as follows: OppA (oligopeptide bindingprotein, Est’dZ score of 2.30, 47% covering), Asg2 (asparaginase type II, Est’dZ score of 2.26, 48% covering), RKObp (arginine-lysine-ornithine-binding protein, Est’dZ score of 2.27, 68% covering [pH 3 to 10], Est’dZ score of 2.37, 70% covering [pH 3 to 6]), 3Rbp (arginine third transportsystem periplasmic binding protein, Est’dZ score of 2.25, 26% covering), OsmY (hyperosmotically inducible protein, Est’dZ score of 2.31, 41%covering), FliC (flagellin, Est’dZ score of 2.14, 67% covering), and GGbp (galactose-glucose binding protein, Est’dZ score of 1.06, 25% covering).
4914 PRADEL ET AL. INFECT. IMMUN.
on March 13, 2020 by guest
http://iai.asm.org/
Dow
nloaded from

noblotting. As shown in Fig. 5A1, a strong protein band with amolecular mass expected for flagellin (�66 kDa) was detectedin the flagellar fraction of the wild-type strain (Fig. 5A1, lane5), indicating the high efficiency of flagellum extraction. Incontrast, no obvious flagellin band was revealed in the flagellarfraction of the mutant (Fig. 5A1, lane 6). Moreover, moreproteins were detected in the mutant fraction than in thecorresponding fraction of the wild-type strain (Fig. 5A1, lane 6versus lane 5), which must result from the leakage of periplas-mic proteins of the EDL933 �tatABC mutant, as observed forthe nonpathogenic E. coli K-12 tat mutants (34). Remarkably,immunoblot analysis showed that flagellin was detected only inthe flagellar fraction of the wild-type strain, but it was absentfrom the periplasm and the spheroplasts of the wild-type strainand all fractions of the mutant (Fig. 5A2). These resultsshowed that lysozyme treatment could release flagella into theperiplasmic fractions and, most importantly, suggested that the�tatABC mutation may affect flagellin synthesis or stability.
Consistent with the absence of flagellin, the �tatABC mutantcompletely lost motility on soft agar plates (Fig. 5B). In addi-tion, the introduction of the p8757 plasmid carrying the K-12tatABC operon restored the motility of the EDLtat mutant.Therefore, the absence of flagellin and the loss of the motilityof the mutant must be the consequence of the �tatABC mu-tation.
The expression of over 50 genes that encode structural sub-units of flagella, regulatory proteins, motor force generators,and the chemosensory machinery is hierarchically controlledbased on three groups of promoters corresponding to theirtemporal requirement during the flagellum assembly process(2). Secretion and polymerization of FliC to form the flagellarfilament are among the latest events, and the fliC gene isexpressed from a class III promoter depending on an alterna-tive sigma factor, 28 (2). Flagellin was not detected in the�tatABC mutant. From this result, it is unclear whether FliCwas synthesized at an extremely lower level in the mutant or ifit was degraded due to the absence of a functioning TATsystem. Analysis of the flagellar protein sequences revealedthat the processed FliP signal peptide contains a twin argininemotif. FliP is an integral membrane protein located in the basalbody of the flagellar assembly apparatus and is essential for theflagellar protein secretion (2). It is tempting to speculate thatthe �tatABC mutation might affect the FliP membrane inser-tion. FlgM is an anti- 28 factor, and its secretion through theflagellar assembly apparatus outside of the cells triggers tran-scription from the class III promoters, including that of fliC(2). The impairment of FliP membrane insertion could resultin the inhibition of the fliC gene expression via the accumula-tion of FliM in the cytoplasm. At present, we have assessed thishypothesis.
Concluding remarks. Recently, it has been reported that theTAT system of P. aeruginosa is essential for the export ofphospholipases, proteins involved in pyoverdine-dedicatediron uptake, anaerobic respiration, osmotic stress defense, mo-tility, and biofilm formation (23, 38). In addition, the corre-sponding tat mutant cells are attenuated for virulence. Simi-larly, the TAT system of the phytopathogen A. tumefacienscontributes to flagellar biogenesis, chemotactic responses, andvirulence (10). In this study, we revealed for the first time thatthe TAT system is required for the secretion of Shiga toxin andsynthesis of the H7 flagellin, which are two of the major viru-lence factors specific to EHEC O157:H7. Together, these find-ings indicate that the TAT system is an important virulencedeterminant of both human pathogens and phytopathogens.Since the tat coding genes are absent from animal genomes(41), it could represent a potential target for novel antimicro-bial compounds.
ACKNOWLEDGMENTS
This work was supported by the Programme de Recherches Avan-cees from AFCRST (PRA B99-03 to L.-F.W.), by the Basic ResearchProgram from the Ministry of Science and Technology, Beijing, Peo-ples’ Republic of China (no. G1999054101 to J.X.), and by the Min-istere de l’Education Nationale de la Recherche et de la Technologie(EA2348 to V.L.). C.Y. was supported by a postdoctoral fellowshipfrom the French Ministry of Research and Education.
We thank J. M. Ghigo for the plasmids and A. D. O’Brien for theantisera against Stx1 and Stx2 and for the EDL933 strain.
FIG. 5. Impact of the �tatABC mutation on FliC synthesis andcellular motility. Periplasmic fractions, spheroplasts, and flagellar frac-tions of the E. coli O157:H7 EDL933 (wild type [WT]) or its derivativeEDLtat mutant (�tatABC) were resolved on SDS denaturing poly-acrylamide (12.5%) gels and visualized either by Coomassie bluestraining (A1) or immunoblotting with polyclonal antisera against theH7 flagellin by an enhanced chemiluminescent procedure (A2). Flagel-lar fractions were prepared as described in Materials and Methods.The periplasmic fractions and the spheroplasts were prepared from thecells after flagellar extraction. The quantities of fractions loaded ongels correspond to those prepared from 15 mg (flagella) or 0.5 mg(periplasmic fractions and the spheroplasts) (wet weight) of cells. FliCrepresents flagellin. (B) Motility assay plates of EDL933 (WT), EDL-tat (�tatABC), and EDLtat complemented by the K-12 tatABC operon(�tatABC/p8737).
VOL. 71, 2003 TAT SYSTEM AND E. COLI O157:H7 VIRULENCE 4915
on March 13, 2020 by guest
http://iai.asm.org/
Dow
nloaded from

REFERENCES
1. Alami, M., D. Trescher, L.-F. Wu, and M. Muller. 2002. Separate analysis oftwin-arginine translocation (Tat)-specific membrane binding and transloca-tion in Escherichia coli. J. Biol. Chem. 277:20499–20503.
2. Aldridge, P., and K. T. Hughes. 2002. Regulation of flagellar assembly. Curr.Opin. Microbiol. 5:160–165.
3. Berin, M. C., A. Darfeuille-Michaud, L. J. Egan, Y. Miyamoto, and M. F.Kagnoff. 2002. Role of EHEC O157:H7 virulence factors in the activation ofintestinal epithelial cell NF-kappaB and MAP kinase pathways and theupregulated expression of interleukin 8. Cell. Microbiol. 4:635–648.
4. Berks, B. C., F. Sargent, and T. Palmer. 2000. The Tat protein exportpathway. Mol. Microbiol. 35:260–274.
5. Bernhard, M., B. Friedrich, and R. A. Siddiqui. 2000. Ralstonia eutrophaTF93 is blocked in tat-mediated protein export. J. Bacteriol. 182:581–588.
6. Bogsch, E., F. Sargent, N. R. Stanley, B. C. Berks, C. Robinson, and T.Palmer. 1998. An essential component of a novel bacterial protein exportsystem with homologues in plastids and mitochondria. J. Biol. Chem. 273:18003–18006.
7. Chanal, A., C. L. Santini, and L.-F. Wu. 2003. Specific inhibition of thetranslocation of a subset of Escherichia coli TAT substrates by the TorAsignal peptide. J. Mol. Biol. 327:563–570.
8. Chanal, A., C.-L. Santini, and L.-F. Wu. 1998. Potential receptor function ofthree homologous components, TatA, TatB and TatE, of the twin-argininesignal sequence-dependent metalloenzyme translocation pathway in Esche-richia coli. Mol. Microbiol. 30:674–676.
9. Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromo-somal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad.Sci. USA 97:6640–6645.
10. Ding, Z., and P. J. Christie. 2003. Agrobacterium tumefaciens twin-arginine-dependent translocation is important for virulence, flagellation, and chemo-taxis but not type IV secretion. J. Bacteriol. 185:760–771.
11. Gouffi, K., C. L. Santini, and L.-F. Wu. 2002. Topology determination andfunctional analysis of the Escherichia coli TatC protein. FEBS Lett. 525:65–70.
12. Halbig, D., T. Wiegert, N. Blaudeck, R. Freudl, and G. A. Sprenger. 1999.The efficient export of NADP-containing glucose-fructose oxidoreductase tothe periplasm of Zymomonas mobilis depends both on an intact twin-argininemotif in the signal peptide and on the generation of a structural export signalinduced by cofactor binding. Eur. J. Biochem. 263:543–551.
13. Hayashi, T., K. Makino, M. Ohnishi, K. Kurokawa, K. Ishii, K. Yokoyama,C. G. Han, E. Ohtsubo, K. Nakayama, T. Murata, M. Tanaka, T. Tobe, T.Iida, H. Takami, T. Honda, C. Sasakawa, N. Ogasawara, T. Yasunaga, S.Kuhara, T. Shiba, M. Hattori, and H. Shinagawa. 2001. Complete genomesequence of enterohemorrhagic Escherichia coli O157:H7 and genomic com-parison with a laboratory strain K-12. DNA Res. 8:11–22.
14. Heikkila, M. P., U. Honisch, P. Wunsch, and W. G. Zumft. 2001. Role of theTat transport system in nitrous oxide reductase translocation and cyto-chrome cd1 biosynthesis in Pseudomonas stutzeri. J. Bacteriol. 183:1663–1671.
15. Hinsley, A. P., N. R. Stanley, T. Palmer, and B. C. Berks. 2001. A naturallyoccurring bacterial Tat signal peptide lacking one of the ‘invariant’ arginineresidues of the consensus targeting motif. FEBS Lett. 497:45–49.
16. Ize, B., F. Gerard, M. Zhang, A. Chanal, R. Voulhoux, T. Palmer, A. Filloux,and L.-F. Wu. 2002. In vivo dissection of the Tat translocation pathway inEscherichia coli. J. Mol. Biol. 317:327–335.
17. Jongbloed, J. D., U. Martin, H. Antelmann, M. Hecker, H. Tjalsma, G.Venema, S. Bron, J. M. van Dijl, and J. Muller. 2000. TatC is a specificitydeterminant for protein secretion via the twin-arginine translocation path-way. J. Biol. Chem. 275:41350–41357.
18. Karmali, M. A., M. Petric, C. Lim, R. Cheung, and G. S. Arbus. 1985.Sensitive method for detecting low numbers of verotoxin-producing Esche-richia coli in mixed cultures by use of colony sweeps and polymyxin extractionof verotoxin. J. Clin. Microbiol. 22:614–619.
19. Mead, P. S., L. Slutsker, V. Dietz, L. F. McCaig, J. S. Bresee, C. Shapiro,P. M. Griffin, and R. V. Tauxe. 1999. Food-related illness and death in theUnited States. Emerg. Infect. Dis. 5:607–625.
20. Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring HarborLaboratory, Cold Spring Harbor, N.Y.
21. Muhldorfer, I., J. Hacker, G. Keusch, D. Acheson, H. Tschape, A. Kane, A.
Ritter, T. Olschlager, and A. Donohue-Rolfe. 1996. Regulation of the Shiga-like toxin II operon in Escherichia coli. Infect. Immun. 64:495–502.
22. Nataro, J. P., and J. B. Kaper. 1998. Diarrheagenic Escherichia coli. Clin.Microbiol. Rev. 11:142–201.
23. Ochsner, U. A., A. Snyder, A. I. Vasil, and M. L. Vasil. 2002. Effects of thetwin-arginine translocase on secretion of virulence factors, stress response,and pathogenesis. Proc. Natl. Acad. Sci. USA 99:8312–8317.
24. Perna, N. T., G. Plunkett, III, V. Burland, B. Mau, J. D. Glasner, D. J. Rose,G. F. Mayhew, P. S. Evans, J. Gregor, H. A. Kirkpatrick, G. Posfai, J.Hackett, S. Klink, A. Boutin, Y. Shao, L. Miller, E. J. Grotbeck, N. W. Davis,A. Lim, E. T. Dimalanta, K. D. Potamousis, J. Apodaca, T. S. Ananthara-man, J. Lin, G. Yen, D. C. Schwartz, R. A. Welch, and F. R. Blattner. 2001.Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature409:529–533.
25. Pradel, N., C.-L. Santini, C. Ye, L. Fevat, F. Gerard, M. Alami, and L.-F. Wu.2003. Influence of tat mutations on the ribose-binding protein translocationin E. coli. Biochem. Biophys. Res. Commun. 306:786–791.
26. Pugsley, A. P. 1993. The complete general secretory pathway in gram-neg-ative bacteria. Microbiol. Rev. 57:50–108.
27. Reid, S. D., R. K. Selander, and T. S. Whittam. 1999. Sequence diversity offlagellin (fliC) alleles in pathogenic Escherichia coli. J. Bacteriol. 181:153–160.
28. Rodrigue, A., A. Chanal, K. Beck, M. Muller, and L.-F. Wu. 1999. Co-translocation of a periplasmic enzyme complex by a hitchhiker mechanismthrough the bacterial Tat pathway. J. Biol. Chem. 274:13223–13228.
29. Sandvig, K. 2001. Shiga toxins. Toxicon 39:1629–1635.30. Santini, C.-L., A. Bernadac, M. Zhang, A. Chanal, B. Ize, C. Blanco, and
L.-F. Wu. 2001. Translocation of jellyfish green fluorescent protein via theTat system of Escherichia coli and change of its periplasmic localization inresponse to osmotic up-shock. J. Biol. Chem. 276:8159–8164.
31. Santini, C. L., B. Ize, A. Chanal, M. Muller, G. Giordano, and L.-F. Wu.1998. A novel Sec-independent periplasmic protein translocation pathway inEscherichia coli. EMBO J. 17:101–112.
32. Sargent, F., E. G. Bogsch, N. R. Stanley, M. Wexler, C. Robinson, B. C.Berks, and T. Palmer. 1998. Overlapping functions of components of abacterial Sec-independent protein export pathway. EMBO J. 17:3640–3650.
33. Schaerlaekens, K., M. Schierova, E. Lammertyn, N. Geukens, J. Anne, andL. Van Mellaert. 2001. Twin-arginine translocation pathway in Streptomyceslividans. J. Bacteriol. 183:6727–6732.
34. Stanley, N. R., K. Findlay, B. C. Berks, and T. Palmer. 2001. Escherichia colistrains blocked in Tat-dependent protein export exhibit pleiotropic defects inthe cell envelope. J. Bacteriol. 183:139–144.
35. Stanley, N. R., T. Palmer, and B. C. Berks. 2000. The twin arginine consensusmotif of Tat signal peptides is involved in Sec-independent protein targetingin Escherichia coli. J. Biol. Chem. 275:11591–11596.
36. Terajima, J., H. Izumiya, S. Iyoda, K. Tamura, and H. Watanabe. 2002. Highgenomic diversity of enterohemorrhagic Escherichia coli isolates in Japanand its applicability for the detection of diffuse outbreak. Jpn. J. Infect. Dis.55:19–22.
37. Unkmeir, A., and H. Schmidt. 2000. Structural analysis of phage-borne stxgenes and their flanking sequences in Shiga toxin-producing Escherichia coliand Shigella dysenteriae type 1 strains. Infect. Immun. 68:4856–4864.
38. Voulhoux, R., G. Ball, B. Ize, M. L. Vasil, A. Lazdunski, L. F. Wu, and A.Filloux. 2001. Involvement of the twin-arginine translocation system in pro-tein secretion via the type II pathway. EMBO J. 20:6735–6741.
39. Wagner, P. L., J. Livny, M. N. Neely, D. W. Acheson, D. I. Friedman, andM. K. Waldor. 2002. Bacteriophage control of Shiga toxin 1 production andrelease by Escherichia coli. Mol. Microbiol. 44:957–970.
40. Weiner, J. H., P. T. Bilous, G. M. Shaw, S. P. Lubitz, L. Frost, G. H. Thomas,J. Cole, and R. J. Turner. 1998. A novel and ubiquitous system for mem-brane targeting and secretion of cofactor-containing proteins. Cell 93:93–101.
41. Wu, L.-F., B. Ize, A. Chanal, Y. Quentin, and G. Fichant. 2000. Bacterialtwin-arginine signal peptide-dependent protein translocation pathway: evo-lution and mechanism. J. Mol. Microbiol. Biotechnol. 2:179–189.
42. Yahr, T. L., and W. T. Wickner. 2001. Functional reconstitution of bacterialTat translocation in vitro. EMBO J. 20:2472–2479.
43. Yim, H., and M. Villarejo. 1992. osmY, a new hyperosmotically induciblegene, encodes a periplasmic protein in Escherichia coli. J. Bacteriol. 174:3637–3644.
Editor: J. T. Barbieri
4916 PRADEL ET AL. INFECT. IMMUN.
on March 13, 2020 by guest
http://iai.asm.org/
Dow
nloaded from


















