
Conserved Region 3 of Human Papillomavirus 16 E7...
11
Conserved Region 3 of Human Papillomavirus 16 E7 Contributes to Deregulation of the Retinoblastoma Tumor Suppressor Biljana Todorovic, a,b Katherine Hung, a,b Paola Massimi, c Nikita Avvakumov, a,b * Frederick A. Dick, b,d Gary S. Shaw, d Lawrence Banks, c and Joe S. Mymryk a,b,e Departments of Microbiology and Immunology, a Biochemistry, d and Oncology, e Western University, London, Ontario, Canada; London Regional Cancer Program, London Health Sciences Centre, London, Ontario, Canada b ; and International Centre for Genetic Engineering and Biotechnology, Trieste, Italy c The human papillomavirus (HPV) E7 oncoprotein binds cellular factors, preventing or retargeting their function and thereby making the infected cell conducive for viral replication. A key target of E7 is the product of the retinoblastoma susceptibility lo- cus (pRb). This interaction results in the release of E2F transcription factors and drives the host cell into the S phase of the cell cycle. E7 binds pRb via a high-affinity binding site in conserved region 2 (CR2) and also targets a portion of cellular pRb for deg- radation via the proteasome. Evidence suggests that a secondary binding site exists in CR3, and that this interaction influences pRb deregulation. Additionally, evidence suggests that CR3 also participates in the degradation of pRb. We have systematically analyzed the molecular mechanisms by which CR3 contributes to deregulation of the pRb pathway by utilizing a comprehensive series of mutations in residues predicted to be exposed on the surface of HPV16 E7 CR3. Despite differences in the ability to in- teract with cullin 2, all CR3 mutants degrade pRb comparably to wild-type E7. We identified two specific patches of residues on the surface of CR3 that contribute to pRb binding independently of the high-affinity CR2 binding site. Mutants within CR3 that affect pRb binding are less effective than the wild-type E7 in overcoming pRb-induced cell cycle arrest. This demonstrates that the interaction between HPV16 E7 CR3 and pRb is functionally important for alteration of the cell cycle. P apillomaviruses are a group of DNA viruses that infect the skin and mucosal tissues of most vertebrates. More than 100 hu- man papillomavirus (HPV) types have been identified, but far more are presumed to exist (21). A subset of HPVs is associated with lesions that frequently progress to cancer, and these HPVs are classified as high-risk types. Persistent infection by high-risk HPVs is related to 99.7% of all human cervical cancer cases (20), other genitourinary cancers, and a growing number of oral can- cers (31). Although recently developed vaccines protect individu- als from the two most frequently carcinogenic HPV types, HPV16 and HPV18, they offer limited protection against other cancer- causing HPVs and provide no benefit to individuals with a preex- isting history of infection (13, 28, 48). The development of other therapies to treat HPV-induced malignancy may be facilitated by a greater understanding of the mechanisms of virally mediated cellular transformation. The viral E6 and E7 oncoproteins are consistently expressed in HPV-induced cancers and are necessary to maintain malignant cell growth (2, 4, 37, 49, 51). Repression of their transcription by reexpression of the viral E2 regulatory protein induces rapid growth arrest and senescence of cervical cancer cells, suggesting that these cancer cells are addicted to E6 and E7 (33, 34). The HPV E7 protein is a multifunctional oncoprotein that interacts with a multitude of cellular factors (3, 6, 8, 11, 53). One of the main activities of E7 is to induce terminally differentiated cells to enter the cell cycle. This is accomplished in part by E7’s association with the product of the retinoblastoma susceptibility locus (pRb) and the related pocket protein family members p107 and p130 (25). The interaction of E7 with pRb is essential for transformation and abrogation of the antiproliferative signals in cervical cancer (26, 47). One of the ways in which pRb has an essential role in prolif- eration control is through binding to and regulating its key effec- tors, the E2F family of transcription factors. E2Fs coordinate the transcription of genes that are necessary for cell cycle progression (10, 24, 43). During the cell cycle, as cells progress from the G 1 to S phase, the sequential phosphorylation of pRb by cyclin/cyclin- dependent kinase complexes causes the release of E2F from pRb and activation of genes required for the entry into S phase (19, 41, 50). The E7 protein contains three conserved regions (CR), termed CR1, CR2, and CR3. CR1 and CR2 are thought to be intrinsically disordered, while CR3 has a defined three-dimensional structure comprised of a unique 12132 fold (40, 45). All three re- gions are required for abrogation of epithelial cell quiescence and contribute to cellular transformation (5, 26, 47). The role of the LxCxE motif within CR2 is well established and is necessary for the abrogation of antiproliferative signals and oncogenic transforma- tion (26, 47). The LxCxE motif functions as a high-affinity binding site for pRb and related family members. However, other regions of E7 are also important for deregulating the pRb pathway, and E7 is known to exert its activity on pRb through multiple mecha- nisms. First, E7 binds the hypophosphorylated form of pRb in complex with E2F and blocks the association of E2F and pRb. Although the LxCxE motif in CR2 is necessary for interference with pRb-E2F binding (14, 44), a requirement for both the CR1 and CR3 regions has also been established (35). Precisely how these two regions contribute is not known. Second, E7 inactivates a fraction of the cellular pool of pRb by targeting it for protea- Received 27 June 2012 Accepted 18 September 2012 Published ahead of print 26 September 2012 Address correspondence to Joe S. Mymryk, [email protected]. * Present address: Nikita Avvakumov, Laval University Cancer Research Center, Hôtel-Dieu de Québec, Quebec City, Quebec, Canada. Copyright © 2012, American Society for Microbiology. All Rights Reserved. doi:10.1128/JVI.01637-12 December 2012 Volume 86 Number 24 Journal of Virology p. 13313–13323 jvi.asm.org 13313 on July 10, 2018 by guest http://jvi.asm.org/ Downloaded from
Transcript of Conserved Region 3 of Human Papillomavirus 16 E7...

Conserved Region 3 of Human Papillomavirus 16 E7 Contributes toDeregulation of the Retinoblastoma Tumor Suppressor
Biljana Todorovic,a,b Katherine Hung,a,b Paola Massimi,c Nikita Avvakumov,a,b* Frederick A. Dick,b,d Gary S. Shaw,d Lawrence Banks,c
and Joe S. Mymryka,b,e
Departments of Microbiology and Immunology,a Biochemistry,d and Oncology,e Western University, London, Ontario, Canada; London Regional Cancer Program, LondonHealth Sciences Centre, London, Ontario, Canadab; and International Centre for Genetic Engineering and Biotechnology, Trieste, Italyc
The human papillomavirus (HPV) E7 oncoprotein binds cellular factors, preventing or retargeting their function and therebymaking the infected cell conducive for viral replication. A key target of E7 is the product of the retinoblastoma susceptibility lo-cus (pRb). This interaction results in the release of E2F transcription factors and drives the host cell into the S phase of the cellcycle. E7 binds pRb via a high-affinity binding site in conserved region 2 (CR2) and also targets a portion of cellular pRb for deg-radation via the proteasome. Evidence suggests that a secondary binding site exists in CR3, and that this interaction influencespRb deregulation. Additionally, evidence suggests that CR3 also participates in the degradation of pRb. We have systematicallyanalyzed the molecular mechanisms by which CR3 contributes to deregulation of the pRb pathway by utilizing a comprehensiveseries of mutations in residues predicted to be exposed on the surface of HPV16 E7 CR3. Despite differences in the ability to in-teract with cullin 2, all CR3 mutants degrade pRb comparably to wild-type E7. We identified two specific patches of residues onthe surface of CR3 that contribute to pRb binding independently of the high-affinity CR2 binding site. Mutants within CR3 thataffect pRb binding are less effective than the wild-type E7 in overcoming pRb-induced cell cycle arrest. This demonstrates thatthe interaction between HPV16 E7 CR3 and pRb is functionally important for alteration of the cell cycle.
Papillomaviruses are a group of DNA viruses that infect the skinand mucosal tissues of most vertebrates. More than 100 hu-
man papillomavirus (HPV) types have been identified, but farmore are presumed to exist (21). A subset of HPVs is associatedwith lesions that frequently progress to cancer, and these HPVs areclassified as high-risk types. Persistent infection by high-riskHPVs is related to 99.7% of all human cervical cancer cases (20),other genitourinary cancers, and a growing number of oral can-cers (31). Although recently developed vaccines protect individu-als from the two most frequently carcinogenic HPV types, HPV16and HPV18, they offer limited protection against other cancer-causing HPVs and provide no benefit to individuals with a preex-isting history of infection (13, 28, 48). The development of othertherapies to treat HPV-induced malignancy may be facilitated bya greater understanding of the mechanisms of virally mediatedcellular transformation.
The viral E6 and E7 oncoproteins are consistently expressed inHPV-induced cancers and are necessary to maintain malignantcell growth (2, 4, 37, 49, 51). Repression of their transcription byreexpression of the viral E2 regulatory protein induces rapidgrowth arrest and senescence of cervical cancer cells, suggestingthat these cancer cells are addicted to E6 and E7 (33, 34). The HPVE7 protein is a multifunctional oncoprotein that interacts with amultitude of cellular factors (3, 6, 8, 11, 53). One of the mainactivities of E7 is to induce terminally differentiated cells to enterthe cell cycle. This is accomplished in part by E7’s association withthe product of the retinoblastoma susceptibility locus (pRb) andthe related pocket protein family members p107 and p130 (25).The interaction of E7 with pRb is essential for transformation andabrogation of the antiproliferative signals in cervical cancer (26,47). One of the ways in which pRb has an essential role in prolif-eration control is through binding to and regulating its key effec-tors, the E2F family of transcription factors. E2Fs coordinate thetranscription of genes that are necessary for cell cycle progression
(10, 24, 43). During the cell cycle, as cells progress from the G1 toS phase, the sequential phosphorylation of pRb by cyclin/cyclin-dependent kinase complexes causes the release of E2F from pRband activation of genes required for the entry into S phase (19,41, 50).
The E7 protein contains three conserved regions (CR), termedCR1, CR2, and CR3. CR1 and CR2 are thought to be intrinsicallydisordered, while CR3 has a defined three-dimensional structurecomprised of a unique �1�2�1�3�2 fold (40, 45). All three re-gions are required for abrogation of epithelial cell quiescence andcontribute to cellular transformation (5, 26, 47). The role of theLxCxE motif within CR2 is well established and is necessary for theabrogation of antiproliferative signals and oncogenic transforma-tion (26, 47). The LxCxE motif functions as a high-affinity bindingsite for pRb and related family members. However, other regionsof E7 are also important for deregulating the pRb pathway, and E7is known to exert its activity on pRb through multiple mecha-nisms. First, E7 binds the hypophosphorylated form of pRb incomplex with E2F and blocks the association of E2F and pRb.Although the LxCxE motif in CR2 is necessary for interferencewith pRb-E2F binding (14, 44), a requirement for both the CR1and CR3 regions has also been established (35). Precisely howthese two regions contribute is not known. Second, E7 inactivatesa fraction of the cellular pool of pRb by targeting it for protea-
Received 27 June 2012 Accepted 18 September 2012
Published ahead of print 26 September 2012
Address correspondence to Joe S. Mymryk, [email protected].
* Present address: Nikita Avvakumov, Laval University Cancer Research Center,Hôtel-Dieu de Québec, Quebec City, Quebec, Canada.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01637-12
December 2012 Volume 86 Number 24 Journal of Virology p. 13313–13323 jvi.asm.org 13313
on July 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

some-mediated degradation (9, 32, 36). Both the LxCxE motif andCR1 are necessary for efficient reduction of pRb steady-state levels(29, 38), while the role of CR3 has remained largely elusive. Onestudy has shown that small changes within CR3 do not disrupt thepRb degradation capacity of E7 (35), but it has also been hypoth-esized that CR3 contributes to pRb degradation due to the obser-vation that mutations in this region lead to reduced associationwith the cullin 2 E3 ubiquitin ligase complex (36, 38). Binding ofthe cullin 2 complex to E7 is mediated via its Zer1 subunit and iscurrently the only known mechanism by which HPV16 E7 targetspRb for destruction by the proteasome (36, 53). As the interactionof cullin 2 is exclusive to HPV16 E7, the mechanism by whichother HPV E7 types degrade pRb remains unclear (36, 53). Lastly,strong evidence has been presented that E7 CR3 contributes tobinding of pRb independently from the LxCxE motif, but thefunctional significance of this interaction is unknown (15, 40, 46).
Despite the large number of biological activities that are attrib-uted to CR3, the role of this domain of E7 in deregulating the pRbpathway is still unclear. The study presented here was aimed atdetermining the role of E7 CR3 sequences in deregulating the pRbpathway. We examined the involvement of surface-exposed resi-dues of HPV16 E7 CR3 in binding pRb, targeting it for degrada-tion, and influencing pRb’s ability to induce cell cycle arrest. Al-though CR3 is necessary for association with the cullin 2 complexand specific CR3 mutants affect cullin 2 interaction, none of thesemutants were impaired for pRb degradation. However, we iden-tified two specific patches on the surface of CR3 which contributeto pRb binding independently of the LxCxE motif in CR2. Addi-tionally, we show for the first time that the CR3-pRb interaction isfunctionally important in overcoming pRb-induced cell cycle ar-rest.
MATERIALS AND METHODSPlasmids. The surface-exposed mutants of E7 were previously generated bysite-directed mutagenesis, and their construction has been described (52). ForpRb degradation assays, E7 mutations were subcloned into the BamHI andXhoI restriction sites of a modified pCMV-Neo-Bam mammalian expressionvector. Constructs for del21-24 and C58G/C91G as well as C91G mutationswere obtained from D. Galloway (Fred Hutchinson Cancer Research Center,University of Washington) and K. H. Vousden (National Cancer Institute atFrederick, Frederick, MD), respectively, and subcloned as necessary. For deg-radation assays carried out in H1299 cells, full-length E7 mutants were sub-cloned into the pCMV-Neo-Bam expression vector with a tandem N-termi-nal flag-hemagglutinin tag using BamHI and XhoI restriction sites. Forexpression in yeast, full-length E7 was cloned into a modified pJG4-5� vector(Clontech) using EcoRI and SalI or XhoI sites (52) or, in the case of theHPV16 E7 CR3 region, using PvuII and XhoI. HPV6, HPV11, and HPV18CR3 regions were PCR amplified using the following primers: HPV6-F, CGAATTCCGAAGTGGACGGACAAGATTCA; HPV6-R, GATCCTCGAGTTAGGTCTTCGGTGCGCAG; HPV11-F, CGAATTCAAGGTGGACAAACAAGACGCA; HPV11-R, TATGTCGACTTATGGTTTTGGTGCGCAGATGGG; HPV18-F, CGAATTCGATGGAGTTAATCATCAACAT; andHPV18-R, CCGCTCGAGTTACTGCTGGGATGC. PCR products werecloned into EcoRI and XhoI or SalI sites of the modified pJG4-5�. Forexpression of green fluorescent protein (GFP)-fused constructs, eitherfull-length or PCR-amplified fragments of E7 were cloned into EcoRI andXbaI sites of pCAN-myc-EGFP. Either the wild-type or mutant CR3 re-gion of HPV16 E7 (residues 39 to 98) was subcloned from their respectivepJG4-5� plasmid into pCAN-myc-EGFP. Alternatively, fragments of res-idues 1 to 39, 1 to 57, and 39 to 58 were first PCR amplified using thefollowing primers: 1-39 F, GTCGAATTCATGCATGGAGATACACCTACATTGC; 1-39 R, ATTATCTAGAATCTCGAGTTAATCTATTTCATCC
TCCTC; 1-57 F, CGAATTCGTAATCATGCATGGAGATAC; 1-57 R, CCTCTCGAGCTAAAAGGTTACAATATTGTAATG; 39-58 F, ATTAGAATTCGATGGTCCAGCTGGACAA; and 39-58 R, ATTATCTACAATCTCGAGTTAACAAAAGGTTACAATATT. They were subsequently clonedinto the EcoRI and XbaI sites of pCAN-myc-EGFP. For recombinant pro-tein production and purification, the CR3 region of each E7 construct wascloned into the pGEX4T1 (Invitrogen) vector that was modified to con-tain the tobacco etch virus (TEV) protease recognition sequence betweenthe glutathione S-transferase (GST) tag and the protein of interest. ThepCMV-pRb, pGEX4T1-pRbC, and pScodon-pRbABC constructs werepreviously described (12, 22, 23). Residues 379 to 792 of pRb were PCRamplified and cloned into the BamHI site of pGEX4T1. pBB14-Us9-EGFPhas been described previously (39). pCMV-HA-cullin 2 was a gift from P.Branton (McGill University, Montreal, Canada); pcDNA3-HA-pRb was agift from J. DeCaprio (Dana Farber Cancer Institute, Harvard MedicalSchool, Boston, MA), and pcDNA3-His-�gal has been described previ-ously (42). PSH1834 reporter plasmid for yeast two-hybrid analyses wasrecovered from the EGY48 yeast strain and contains eight operator se-quences that respond to LexA. pCMV-HA-p21 plasmid has been de-scribed previously (16).
Cell culture, transfection, and pRb degradation assay. HumanSaos2, HT1080, and H1299 cells were maintained in Dulbecco’s modifiedEagle’s medium (DMEM) supplemented with 10% fetal bovine serumand penicillin-streptomycin (100 U/ml). H1299 control and cullin 2knockdown (KD) cells have been previously described (17, 18) and weremaintained supplemented with 1 �g/ml of puromycin. For pRb degrada-tion assays, Saos2 cells were seeded into 6-well plates at 3 � 105 cells perwell and transfected 24 h later with 2 �g of pRb with or without 0.2 �g ofE7 expression plasmid and with 0.1 �g of green fluorescent protein (GFP)or �-galactosidase (�-gal) expression plasmid as a control using Fu-geneHD (Roche) according to the instructions provided by the manufac-turer, unless otherwise noted. The amount of plasmid DNA was balancedwith empty pCMV vector where necessary. The amounts of pRb, GFP,and actin were assessed by Western blotting 48 h posttransfection. Assess-ment of pRb degradation in H1299 control or cullin 2 KD cells was carriedout similarly, where 0.2 �g of HA-pRb expression plasmid was cotrans-fected with an increasing amount of wild-type or mutant Flag-HA-taggedE7-expressing plasmid (20, 200, or 2,000 ng) using X-tremeGENE HP(Roche). The amounts of pRb, E7, and actin were determined 24 h post-transfection by Western blotting. For coimmunoprecipitation experi-ments, HT1080 cells were seeded into 10-cm plates at 2 � 106 cells perplate and transfected 24 h later with 8 �g of total DNA with X-tremeGENEHP (Roche) according to the manufacturer’s instructions. Twenty-four hposttransfection cells were collected for coimmunoprecipitation experi-ments. For cycloheximide treatment experiments, HT1080 cells wereseeded into 6-well plates 24 h before transfection. For each mutant, thecells were transfected with 2 �g of E7 pCMV-Neo-Bam expression plas-mid along with 0.1 �g of GFP expression plasmid using X-tremeGENEHP. At 24 h posttransfection, the cells were treated with 0.5 �g/�l cyclo-heximide for 0, 15, 30, and 60 min.
Antibodies. The following antibodies were used: rat anti-hemagglutinin(anti-HA) (Roche) at 1:2,000 and rabbit anti-glucose-6-phosphate dehy-drogenase (anti-G6PD), used as a loading control for yeast samples, at1:80,000 (Sigma). For detection of pRb in the degradation assay, mouseanti-pRb (G3-245; BD Pharmingen) was used at 1:500 or mouse anti-pRbhybridoma lysate (clone C36) at 1:4. GFP was detected using rabbit anti-GFP (Living Colors; Clontech) at 1:2,000. Cullin 2 was detected usingrabbit polyclonal anti-cul2 antibody at 1:10,000 (Bethyl Laboratories).Myc-tagged constructs were detected with mouse anti-myc hybridomalysate (clone 9E10) used at 1:200. Mouse anti-�-gal (Promega) and rabbitanti-actin (Sigma) were used at 1:2,000. Mouse anti-E7 (8C9 clone; Invit-rogen) was used at 1:200. Horseradish peroxidase (HRP)-conjugated goatanti-rabbit (Jackson Laboratories), goat anti-rat (Pierce), and rabbit anti-mouse (Jackson Laboratories) secondary antibodies were used.
Todorovic et al.
13314 jvi.asm.org Journal of Virology
on July 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Coimmunoprecipitation and Western blot analysis. Cells weretransfected in a 1:1 ratio of the myc-GFP fusion and hemagglutinin (HA)-tagged binding partner (cullin 2, pRb, or p21). Cells were harvested at 24h posttransfection by scraping and washed once with 1� phosphate-buff-ered saline (PBS). Cells were lysed in NP-40 (50 mM Tris, pH 7.8, 150 mMNaCl, 0.1% NP-40) lysis buffer supplemented with 1� mammalian pro-tease inhibitor cocktail (Sigma). Typically, 1 mg of cell lysate was mixedwith 100 �l of anti-myc hybridoma (clone 9E10) or 1 �l of anti-GFPantibody and 100 �l of 10% slurry of protein A-Sepharose resin (Sigma)and incubated at 4°C for 1 to 2 h with nutating. Immunoprecipitates werewashed three times with lysis buffer, resuspended in 2� lithium dodecylsulfate (LDS) sample buffer, and boiled for 5 min. Samples were thenseparated by SDS-PAGE, transferred to a polyvinylidene difluoride mem-brane (GE), and blocked in 5% nonfat milk in Tris-buffered saline-Tween 20.Western blot analyses were carried out with mouse anti-myc hybridomaclone (9E10) or rat monoclonal anti-HA (clone 3F10; Roche).
Yeast two-hybrid assay. Yeast two-hybrid analysis was performed instrain W303-1a (MATa leu2-3,112 his3-11,15 trp1-1 ura3-1 ade2-1, can1-100). Standard yeast culture medium was prepared using previously de-scribed methods (1). Yeast transformation was carried out using a modi-fied lithium acetate procedure (30). Yeast cells were transformed withthe reporter plasmid and pRb and E7 expression plasmids and thenplated on selective medium plates and grown at 30°C for 2 to 4 days.The method for assaying �-galactosidase activity in yeast has beendescribed previously (1).
GST pulldown assays. Recombinant GST-tagged E7 CR3 (residues 39to 98) and GST-tagged fragments of pRb (pRb-AB and pRb-C) were ex-pressed in and purified from the BL21-RIL strain of Escherichia coli andpRbABC from the BL21-Gold strain per protocols provided by the affinityresin manufacturer (Amersham). For GST pulldown experiments withGST-CR3, Saos2 cells were transfected with pRb expression plasmid andlysates prepared 48 h posttransfection in NP-40 lysis buffer. Approxi-mately 400 �g of Saos2 extract was used per reaction and mixed with anincreasing amount of GST-CR3, 600 �l of NP-40 lysis buffer, and 20 �l of50% glutathione Sepharose. Reaction mixtures were incubated at 4°C for1 h and then washed two times with the lysis buffer. Samples were resus-pended in 2� LDS sample buffer, boiled for 5 min, then separated by SDS-PAGE and examined by Western blotting for the amount of associated pRb.For GST pulldown assay with purified recombinant GST-pRbABC, -pRbAB,or -pRbC, HT1080 cells were transfected with myc-GFP-E7 39-98 expressionplasmid. Lysates were prepared 24 h posttransfection in NP-40 lysis buffer.Approximately 1 mg of lysate was mixed with an increasing amount of GST-pRb fragment and 20 �l of 50% glutathione Sepharose. Reaction mixtureswere incubated and samples processed as indicated above. Western blot anal-ysis was used to assess the amount of associated myc-GFP-E7 39-98 by blot-ting with anti-myc.
In vitro pRb binding assay. To obtain untagged E7 CR3 (residues 39to 98), purified recombinant GST-tagged wild-type E7 CR3 and mutantswere incubated with �0.2 �g/ml TEV protease overnight at 4°C. Thepurified protein concentration was determined using the Bio-Rad assayand was verified by running 0.5 �g of purified protein on a gel and silverstaining (SilverQuest staining kit; Invitrogen) prior to using the samplesin the assay. The ability of E7 CR3 to associate with GST-pRbABC wasassessed at protein concentrations well below saturation levels (see Fig.7A). Briefly, 0.6 �M E7 CR3 (residues 39 to 98) was incubated with 0.3�M GST-pRbABC at room temperature for 1 h in a total volume of 300 �lof phosphate buffer (20 mM phosphate, pH 7.0, 200 mM NaCl, 1% Tween20, 2 mM dithiothreitol) and 20 �l of 50% glutathione Sepharose. Sam-ples were washed three times with phosphate buffer, boiled in 2� LDSsample buffer for 5 min, and separated by SDS-PAGE. Gels were silverstained to determine the amount of associated E7 CR3.
Cell cycle analysis. A total of 6 � 105 or 1 � 106 Saos2 cells wereseeded into 6- or 10-cm dishes, respectively. Cells were transfected with1.5 �g of each Us9-EGFP expression plasmid (membrane-bound GFP)and pRb, as well as 3 �g of untagged, full-length E7 (pCMV-Neo-Bam
plasmids), with FugeneHD (Roche). Twenty-four h posttransfection, cellswere transferred to either 10- or 15-cm dishes (depending on the size ofthe original dish) using phosphate-buffered saline supplemented with 10mM EDTA (PBS-EDTA). Forty-eight h after the transfer to a new dish,cells were collected with PBS-EDAT solution and washed once with PBS.Cell pellets were resuspended in 300 �l of 50% fetal bovine serum (FBS)solution (in PBS) and fixed by dropwise addition of 900 �l of ice-cold 70%ethanol. Samples were incubated on ice for at least 2 h and then washedtwo times with PBS. Cells were resuspended in and stained with 300 �l ofpropidium iodide staining solution (1% FBS, 10 �g/ml propidium iodide,0.25 mg/ml RNase A in PBS). Cell cycle data were collected by flow cy-tometry on a FACSCalibur (BD Biosciences). DNA content was deter-mined based on propidium iodide staining. Data were analyzed usingFlowJo software, and the percentage of cells in G1 phase was determinedusing the Watson model.
RESULTSCR3 mutations. To examine the role that HPV16 E7 CR3 plays inpRb deregulation, we utilized the panel of putative surface-ex-posed mutants within CR3, here referred to as the surface-exposedmutants. These have been previously described by our group (52).Briefly, these mutations target residues whose side chains are pre-dicted to be at least 25% surface exposed but not those that wouldparticipate in stabilizing the structural features of E7 (40, 45, 52).Hence, these mutations were constructed with the dual aims ofpreserving the structure of the CR3 region while disrupting theinteraction surfaces available for cellular targets. In total we in-cluded in these studies 19 point mutants, one double mutant, andone mutant targeting the last three amino acids of CR3 (Fig. 1). Ascontrols, we also utilized the deletion mutant, del21-24, whichremoves the LxCxE motif in CR2, and the C58G/C91G mutant,which targets two critical zinc-coordinating cysteines in CR3 andis thought to grossly perturb the folded structure of this regionof E7.
Contribution of HPV16 E7 CR3 to pRb degradation. The Lx-CxE motif and certain residues within CR1 of E7 were previouslyshown to be important for pRb degradation (32, 35). Other stud-ies have also examined the role of CR3 and have found no contri-bution of any single residue to pRb degradation (7, 35). However,another study has shown that specific residues within CR3 con-tribute to cullin 2 complex binding, currently the exclusive mech-anism known to be used by HPV16 E7 to degrade pRb (36). Thesefindings led us to reexamine the role of CR3 in pRb degradation.Our first aim was to map the surface of CR3 which is required forpRb degradation utilizing the panel of surface-exposed mutants.Our approach was to test full-length wild-type E7 or E7 mutantsfor their ability to degrade pRb exogenously expressed in pRb-null
FIG 1 Schematic representation of HPV16 E7 and the position of mutationstargeting residues within CR3. The conserved regions (CR1, CR2, and CR3)are depicted as boxes. The amino acid sequences of CR3 and the mutationsused in this study are indicated. Also indicated are del21-24, which removesthe LxCxE motif in CR2 of E7, and the C58G/C91G mutant, which targetszinc-coordinating cysteine residues within CR3.
HPV16 E7 CR3 Deregulates pRb Function
December 2012 Volume 86 Number 24 jvi.asm.org 13315
on July 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
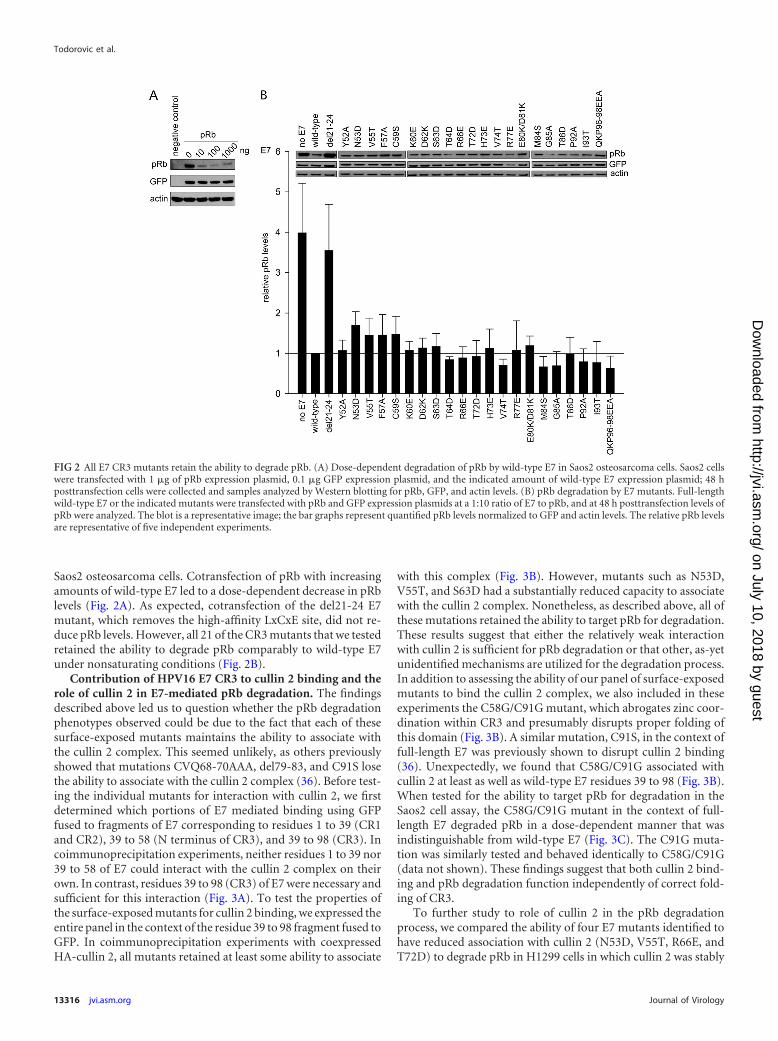
Saos2 osteosarcoma cells. Cotransfection of pRb with increasingamounts of wild-type E7 led to a dose-dependent decrease in pRblevels (Fig. 2A). As expected, cotransfection of the del21-24 E7mutant, which removes the high-affinity LxCxE site, did not re-duce pRb levels. However, all 21 of the CR3 mutants that we testedretained the ability to degrade pRb comparably to wild-type E7under nonsaturating conditions (Fig. 2B).
Contribution of HPV16 E7 CR3 to cullin 2 binding and therole of cullin 2 in E7-mediated pRb degradation. The findingsdescribed above led us to question whether the pRb degradationphenotypes observed could be due to the fact that each of thesesurface-exposed mutants maintains the ability to associate withthe cullin 2 complex. This seemed unlikely, as others previouslyshowed that mutations CVQ68-70AAA, del79-83, and C91S losethe ability to associate with the cullin 2 complex (36). Before test-ing the individual mutants for interaction with cullin 2, we firstdetermined which portions of E7 mediated binding using GFPfused to fragments of E7 corresponding to residues 1 to 39 (CR1and CR2), 39 to 58 (N terminus of CR3), and 39 to 98 (CR3). Incoimmunoprecipitation experiments, neither residues 1 to 39 nor39 to 58 of E7 could interact with the cullin 2 complex on theirown. In contrast, residues 39 to 98 (CR3) of E7 were necessary andsufficient for this interaction (Fig. 3A). To test the properties ofthe surface-exposed mutants for cullin 2 binding, we expressed theentire panel in the context of the residue 39 to 98 fragment fused toGFP. In coimmunoprecipitation experiments with coexpressedHA-cullin 2, all mutants retained at least some ability to associate
with this complex (Fig. 3B). However, mutants such as N53D,V55T, and S63D had a substantially reduced capacity to associatewith the cullin 2 complex. Nonetheless, as described above, all ofthese mutations retained the ability to target pRb for degradation.These results suggest that either the relatively weak interactionwith cullin 2 is sufficient for pRb degradation or that other, as-yetunidentified mechanisms are utilized for the degradation process.In addition to assessing the ability of our panel of surface-exposedmutants to bind the cullin 2 complex, we also included in theseexperiments the C58G/C91G mutant, which abrogates zinc coor-dination within CR3 and presumably disrupts proper folding ofthis domain (Fig. 3B). A similar mutation, C91S, in the context offull-length E7 was previously shown to disrupt cullin 2 binding(36). Unexpectedly, we found that C58G/C91G associated withcullin 2 at least as well as wild-type E7 residues 39 to 98 (Fig. 3B).When tested for the ability to target pRb for degradation in theSaos2 cell assay, the C58G/C91G mutant in the context of full-length E7 degraded pRb in a dose-dependent manner that wasindistinguishable from wild-type E7 (Fig. 3C). The C91G muta-tion was similarly tested and behaved identically to C58G/C91G(data not shown). These findings suggest that both cullin 2 bind-ing and pRb degradation function independently of correct fold-ing of CR3.
To further study to role of cullin 2 in the pRb degradationprocess, we compared the ability of four E7 mutants identified tohave reduced association with cullin 2 (N53D, V55T, R66E, andT72D) to degrade pRb in H1299 cells in which cullin 2 was stably
FIG 2 All E7 CR3 mutants retain the ability to degrade pRb. (A) Dose-dependent degradation of pRb by wild-type E7 in Saos2 osteosarcoma cells. Saos2 cellswere transfected with 1 �g of pRb expression plasmid, 0.1 �g GFP expression plasmid, and the indicated amount of wild-type E7 expression plasmid; 48 hposttransfection cells were collected and samples analyzed by Western blotting for pRb, GFP, and actin levels. (B) pRb degradation by E7 mutants. Full-lengthwild-type E7 or the indicated mutants were transfected with pRb and GFP expression plasmids at a 1:10 ratio of E7 to pRb, and at 48 h posttransfection levels ofpRb were analyzed. The blot is a representative image; the bar graphs represent quantified pRb levels normalized to GFP and actin levels. The relative pRb levelsare representative of five independent experiments.
Todorovic et al.
13316 jvi.asm.org Journal of Virology
on July 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

knocked down (Fig. 3E). Each mutant degraded pRb in cullin 2knockdown cells similarly to control cells, further suggesting thatE7 is utilizing a cullin 2-independent mechanism to degrade pRb.
CR3 binds pRb independently of the LxCxE motif in vitroand in vivo. Several reports have suggested that CR3 contributesto deregulation of pRb function by providing a secondary lower-affinity binding site. For example, full-length E7 binds pRb morestrongly than mutants retaining the LxCxE motif but lacking CR3(46). More recently, it was demonstrated in two independentstudies that CR3 binds pRb in vitro (15, 45). We first confirmedthat purified GST-CR3 of E7 was sufficient to independently pulldown pRb from cell lysates in a dose-dependent manner (Fig. 4A).Utilizing the reciprocal approach, we also demonstrated that arecombinant large pocket of pRb (GST-pRbABC) could associatewith GFP-CR3 from cell lysates obtained by overexpressing GFP-CR3 in HT1080 cells (Fig. 4B). Additionally, the small pocket ofpRb (pRbAB) was sufficient for this interaction, consistent with aprevious report (15). In contrast, the C terminus of pRb (pRbC)
was not sufficient to interact with CR3 of E7. As CR3 has neverbeen reported to interact with pRb in vivo, we examined the abilityof GFP-CR3 to coimmunoprecipitate HA-pRb. As expected, anN-terminal fragment of E7 (residues 1 to 57, containing the high-affinity LxCxE binding site for pRb) was found to associatestrongly with pRb by coimmunoprecipitation (Fig. 5A). Impor-tantly, the CR3 region of E7 (residues 39 to 98) was also sufficientto coimmunoprecipitate pRb, albeit at a substantially lower levelthan the full-length E7 protein or the LxCxE-containing N-termi-nal fragment. Yeast two-hybrid analysis utilizing E7 fragmentsspanning residues 1 to 57 or 39 to 98 as prey and full-length pRb asbait showed that CR3 also interacted with pRb independently ofthe N terminus. In these assays, residues 39 to 98 bound pRb withapproximately 30% of the activity observed with full-length E7(Fig. 5B).
CR3s from other HPV types also interact with pRb. Utilizingcoimmunoprecipitation experiments, we determined that full-length E7 from the high-risk HPV18 and low-risk HPV6 and
FIG 3 Contribution of cullin 2 complex to E7-induced pRb degradation. (A) CR3 of E7 is necessary and sufficient to bind cullin 2. HT1080 cells were transfectedwith an equal ratio of hemagglutinin (HA)-tagged cullin 2 expression plasmid to either myc-GFP or myc-GFP-fused E7 fragment (as indicated). Twenty-four hposttransfection, cell lysates were subjected to immunoprecipitation (IP) with anti-myc antibody, followed by Western blotting for cullin 2 with anti-HAantibody. (B) Cullin 2 binding capacity of CR3 mutants. HT1080 cells were cotransfected with HA-tagged cullin 2 and either myc-GFP or myc-GFP-fused E739-98 (wild type or indicated mutant). Coimmunoprecipitation was carried out as described above. (C) The C58G/C91G mutant retains the ability to degradepRb. Saos2 cells were transfected with 1 �g pRb, 0.1 �g �-galactosidase, and the indicated amounts of full-length wild-type or C58G/C91G mutant E7. Sampleswere collected 48 h posttransfection, and the steady-state levels of pRb were analyzed by Western blotting. %pRb indicates the relative amount of remaining pRbin each lane. (D) Cullin 2 levels in H1299 control and H1299 cullin 2 knockdown (KD) cells. The levels of endogenous cullin 2 present in H1299 control andH1299 cullin 2 KD cells were analyzed using anti-cul2 antibody. (B) Dependence of E7 on cullin 2 for pRb degradation. pRb degradation assays were conductedin cullin 2 KD and control cells for indicated E7 mutants, as described in Materials and Methods.
HPV16 E7 CR3 Deregulates pRb Function
December 2012 Volume 86 Number 24 jvi.asm.org 13317
on July 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

HPV11 also associated with pRb in mammalian cells cotrans-fected with E7 and pRb (Fig. 5C) or utilizing endogenous pRb(data not shown). This was expected, as each of the E7 proteinscontains a high-affinity LxCxE motif. We next tested whether theE7 CR3 regions from other HPV types could interact in vivo withpRb independently of the LxCxE binding site in CR2. In a yeasttwo-hybrid analysis, low-risk HPV6 and HPV11 E7 CR3 regionsalso interacted with pRb, whereas little or no activity was detected forthe high-risk HPV18 CR3 (Fig. 5D). Similar to the coimmunopre-cipitation experiments from mammalian cells, each of the full-lengthE7 proteins was also able to bind pRb in the yeast two-hybrid assay.These results indicate that while all of the E7 proteins tested couldbind pRb, the ability to interact with pRb via CR3 is not a universalproperty of all E7 proteins.
Mapping the binding surface for pRb in CR3. We initiallyused the yeast two-hybrid system as a rapid method to identifycandidate residues from HPV16 E7 that are involved in bindingpRb. We cloned our panel of mutants into a yeast two-hybrid preyvector as fragments expressing only residues 39 to 98 and assessedtheir capacity to bind pRb. With this approach, we identified anumber of mutants with statistically significant differences in pRbbinding. Specifically, V55T, F57A, R66E, M84S, and I93T wereincreased and Y52A, N53D, C59S, S63D, T64D, T72D, R77E, andG85A were decreased. The most significant increase in bindingwas observed for R66E, which displayed 10 times higher �-galac-tosidase activity than the wild-type CR3. In contrast, Y52A, N53D,C59S, and G85A all showed more than 50% reductions in activity(Fig. 6B). It should be noted that for the yeast two-hybrid analysis,CR3 mutants were previously assessed for any autoactivation po-
tential (52). Although R66E had greatly increased pRb bindingpotential in the yeast two-hybrid analysis, this was not due toenhanced autoactivation. To rule out the possibility that differen-tial expression played a role in the observed changes in bindingcapacities, we tested the majority of CR3 mutants which displayedincreased or decreased binding (mutations with P � 0.05) forexpression in yeast cells. All mutants with decreased binding ca-pacity were expressed comparably to the wild type except forS63D, which was expressed at a slightly lower level (Fig. 6C). Sim-ilarly, mutants with increased binding for pRb were expressedcomparably to the wild type, with the possibility that the slightincrease in binding seen with the D62K and I93T mutants could bethe result of increased expression. It should be noted that full-length E7 wild type or mutants were also assessed for the capacityto associate with pRb in a yeast two-hybrid analysis (Fig. 6A).With this approach, we found that none of the mutants exhibiteda substantially reduced ability to associate with pRb, suggestingthat defects in binding via CR3 are masked by the high-affinitypRb binding site within CR2.
Confirmation of the pRb binding properties of E7 CR3 mu-tants in vitro. Our initial analysis using the yeast two-hybrid sys-tem identified a number of residues that appeared to contribute tothe interaction of CR3 with pRb. Interestingly, most of these res-idues map to two independent patches on the surface of CR3.Specifically, residues Y52, N53, V55, F57, C59, S63, T64, R66, andT72 map to one region, referred to as patch 1; residues R77, M84,and G85 map to a second region, referred to as patch 2. However,results from yeast two-hybrid assays may be influenced by thepresence of additional factors in yeast. In order to conclusively
FIG 4 E7 CR3 interacts with pRb independently of the LxCxE motif in vitro. (A) GST-CR3 associates with pRb from cell lysates. Increasing amounts of GST-CR3(residues 39 to 98) were incubated with �400 �g of Saos2 cell lysate (previously transfected with pRb expression plasmid) and analyzed for the amount ofassociated pRb via Western blotting. (B) GFP-CR3 (residues 39 to 98) associates with the large and small pockets of pRb. HT1080 cells were transfected withmyc-GFP or myc-GFP-fused E7 39-98. Twenty-four h posttransfection, cell lysates were prepared and �1 mg was used in each GST pulldown reaction. Five �gof GST was incubated with 1 mg of myc-GFP-E7 39-98 lysate as a control. Five �g of GST-pRbABC (large pocket), GST-pRbAB (small pocket), and GST-pRbC(C terminus of pRb) was incubated with 1 mg of myc-GFP lysate as a control. Increasing amounts of GST-pRb fragments (0.5, 1, or 5 �g) were incubated with1 mg of myc-GFP-E7 39-98 lysate. Samples were washed and then analyzed by Western blotting for the amount of associated myc-GFP-E7 39-98. The bottompanel is the Ponceau stain of the membrane, illustrating the input levels of GST-pRb fragments. The left side of the Ponceau stain indicates the position and sizeof the ladder in kDa.
Todorovic et al.
13318 jvi.asm.org Journal of Virology
on July 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

determine if the identified residues contribute to pRb binding, weset up an in vitro GST pulldown assay utilizing the affinity-purifiedrecombinant GST-fused large pocket of pRb and increasing mo-lar amounts of purified wild-type CR3 (E7 residues 39 to 98). Inthis assay, CR3 associated with pRb in a dose-dependent man-ner (Fig. 7A). This analysis established appropriate proteinconcentrations for the assay, ensuring that the reactions werecarried out at levels of CR3 below saturation. For all subse-quent experiments, GST pulldown assays were set up with 0.3�M pRbABC and a 2-fold molar excess of wild-type or mutantCR3 (0.6 �M).
To confirm the contribution of patch 1 to pRb binding, wefocused on mutants of E7 with the most significantly altered
binding (P � 0.01) as determined by the yeast two-hybrid sys-tem (Fig. 6B). These included Y52A, N53D, V55T, F57A, C59S,S63D, T64D, R66E, and T72D. Of these mutants, all but R66E boundpRb at reduced levels in vitro (Fig. 7C and D). Taken together, thedata from these two independent types of analyses clearly indicatethat patch 1 contributes to pRb binding by CR3.
FIG 5 E7 CR3 interacts with pRb independently of the LxCxE motif in vivo.(A) E7 CR3 interacts with pRb in vivo. HT1080 cells were cotransfected withequal amounts of HA-tagged pRb and myc-GFP or myc-GFP-E7 fragments.Twenty-four h posttransfection, cell lysates were prepared and subjected toimmunoprecipitation with anti-myc antibody, followed by Western blottingfor HA-pRb. (B) HPV16 E7 CR3 interacts with pRb in a yeast two-hybridassay. Yeast cells were transformed with either empty vector, full-length E7, orthe two indicated E7 fragments as prey proteins, along with pRb as bait and aLexA-responsive �-galactosidase reporter plasmid. Following transformation,yeast cells were analyzed for �-galactosidase activity. Data are presented aspercent binding relative to that of full-length E7. (C) E7 proteins from otherHPV types also associate with pRb. HT1080 cells were cotransfected with equalamounts of HA-tagged pRb and myc-GFP or myc-GFP-E7 from HPV16,HPV18, HPV6, or HPV11. Twenty-four h posttransfection, immunoprecipi-tation and Western blotting were carried out as described above. (D) CR3regions from other HPV types also associate with pRb independently from theLxCxE motif. The C-terminal portions of HPV16, HPV18, HPV11, and HPV6were tested for the ability to associate with pRb similarly to what was describedfor panel B. Data are represented as experimentally determined �-galactosi-dase activity.
FIG 6 Identification of CR3 residues involved in binding pRb using the yeasttwo-hybrid system. (A) Yeast two-hybrid analysis of the binding capabilities offull-length E7 mutants and pRb. Yeast cells were transformed with full-lengthpRb expression plasmid as bait and full-length wild-type or mutant E7 as prey,in addition to the LexA-responsive �-galactosidase reporter plasmid. Data areshown relative to wild-type E7 and are representative of four independentexperiments. (B) Yeast two-hybrid analysis of the binding capabilities of E7CR3 mutants. Yeast cells were transformed with full-length pRb expressionplasmid as bait and wild-type or mutant fragment of E7 spanning residues 39to 98, in addition to the LexA-responsive �-galactosidase reporter plasmid.Data are represented relative to wild-type E7 39-98. **, P � 0.01; *, P � 0.05.(C) Expression level of E7 CR3 mutants in yeast cells. The same yeast culturesused for the analysis of the pRb-CR3 interaction were also analyzed by Westernblotting for E7 CR3 protein expression levels.
HPV16 E7 CR3 Deregulates pRb Function
December 2012 Volume 86 Number 24 jvi.asm.org 13319
on July 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Patch 2 overlaps residues previously reported to contribute topRb interaction, including R77, E80, and D81 (45). In agreement,E80K/D81K retained only �25% of the binding capacity for thelarge pocket of pRb in our in vitro tests. In contrast, R77E showeda substantial increase in pRb binding and G85A bound compara-bly to the wild type (Fig. 7C and D). These data suggest that neg-ative charges present within patch 2 contribute to pRb interaction,as previously proposed (45).
The CR3-pRb interaction is important for E7 to overcomepRb-induced cell cycle arrest. Although all surface-exposed mu-tants of E7 CR3 can target pRb for degradation, our data demon-strate that specific residues contribute to pRb binding. To deter-mine the functional significance of this interaction, we assessedpatch 1 and patch 2 mutants for their ability to overcome cell cyclearrest induced by reintroduction of pRb into pRb-null Saos2 cells.Reintroduction of pRb into Saos2 cells results in �80% of cellsarresting in the G1 phase of the cell cycle (Fig. 8A). Coexpressionof full-length wild-type E7 reverses this arrest, with �40% of thecells remaining in G1. The change in percentage of cells in G1 inthe presence or absence of wild-type E7 was normalized to 100%(Fig. 8A). Although the LxCxE deletion mutant del21-24 was im-paired for inducing G1 exit, it still retained 27% of wild-type ac-tivity. We tested all CR3 mutants that were assessed for pRb bind-ing in vitro for their ability to overcome this pRb-induced cell cyclearrest. Of those mutants located within patch 1, we found that allexcept C59S, R66E, and T72D had a significantly reduced abilityto overcome cell cycle arrest. Their activity ranged from 60 to 77%of that of wild-type E7. C59S and T72D showed a modest decreasein the ability to bind pRb in vitro but behaved similarly to wild-
type E7 in their ability to overcome cell cycle arrest. R66E retainedpRb binding in both the yeast two-hybrid and the in vitro studiesand overcame cell cycle arrest, similar to wild-type E7. This find-ing is consistent with a previous analysis of R66A in similar func-tional assays (35). Of the patch 2 mutants, R77E behaved likewild-type E7, but G85A and E80K/D81K retained only about 60%of wild-type capacity to overcome the G1 arrest. The reduced ability ofthe mutants to overcome the pRb-induced cell cycle arrest was notrelated to a reduction in their stability (Fig. 8B). We also tested se-lected mutants for their ability to interact with p21Cip1/WAF1 by coim-munoprecipitation, as this regulator of G1 exit has previously beenreported to interact with HPV16 E7 CR3 (27). However, no correla-tion was apparent between binding p21Cip1/WAF1 and ability to over-come a pRb-induced cell cycle arrest (Fig. 9). Taken together, CR3mutants with reduced ability to bind pRb typically were less able toovercome a pRb-induced cell cycle arrest.
DISCUSSION
Although the role of HPV E7 in cellular transformation and can-cer progression has been extensively studied, we still do not fullyunderstand how E7 deregulates one of its most vital interactingpartners, the retinoblastoma tumor suppressor (pRb). Precise as-sessment of E7-pRb protein interactions appears fundamental tounderstanding virally mediated subversion of cell cycle controland may allow novel shared features of viral and cellular pRbprotein interaction partners to be uncovered. The objective of thisstudy was to more precisely determine the role of E7 CR3 se-quences in deregulating the pRb pathway.
For these studies, we utilized an extensive panel of surface-
FIG 7 Mapping the pRb binding surface on CR3 in vitro. (A) CR3 associates with the large pocket of pRb (pRbABC) in a dose-dependent manner. GST-pRbABC(0.3 �M) was incubated with increasing amounts of purified CR3 (residues 39 to 98), and the amount of associated CR3 was determined by silver staining. (B)Representative gel image of CR3-pRb binding as quantified for panel C. (C) Patch 1 residues most significantly contribute to pRb binding. Purified wild-type CR3or the indicated mutant was assessed for the ability to bind pRb in a GST pulldown assay using 0.3 �M GST-pRbABC and 0.6 �M CR3. Data are presented relativeto wild-type protein and are representative of a minimum of three independent assays. **, P � 0.001; *, P � 0.032. (D) Based on the in vitro analysis, patch 1 andpatch 2 residues significantly contributed to pRb binding. Colored in blue are patch 1 residues Y52, N53, V55, F57, C59, S63, T64, and T72, as well as R77, E80,and D81 within patch 2.
Todorovic et al.
13320 jvi.asm.org Journal of Virology
on July 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

exposed mutants of HPV16 E7 CR3, which target residues that areaccessible for interaction with cellular partners (Fig. 1). Unlikeprevious work, we avoided using deletion mutants or mutantswhich target highly conserved, structurally important hydropho-bic residues of E7 CR3 (52). This approach allowed us to examinethe role of each individual residue on the surface of CR3 in dereg-ulating pRb function. Utilizing this panel of mutants, we firsttested whether any of the introduced changes on the surface of E7CR3 reduced its capacity to target pRb for degradation. Unexpect-edly, the entire panel of 21 surface-exposed mutants was as effec-tive as the wild-type HPV16 E7 protein in targeting pRb for deg-radation (Fig. 2B). The only mutant that was unable to induce pRbdegradation was del21-24, which cannot associate with pRb viathe LxCxE motif located in CR2. This is most consistent with aprevious report that even mutants with substantial deletions inCR3 were still able to destabilize pRb (35).
The ability of all of the CR3 mutants to degrade pRb was un-expected, as this region is necessary for interaction with the cullin2 E3 ubiquitin ligase which facilitates ubiquitination of pRb andsubsequent proteasome-mediated degradation (36, 53). In agree-ment with other studies, we show that CR3 confers associationwith cullin 2 (Fig. 3A). Our finding that mutations in the zinc-coordinating cysteine residues of CR3 still bind cullin 2 suggeststhat this interaction occurs independently of the correct folding ofCR3, perhaps via a linear interaction motif. Although some CR3mutations result in reduced binding capacity for cullin 2, thesemutants still degrade pRb as efficiently as wild-type E7 under non-saturating conditions. We found this to be the case in the Saos2degradation assay as well as in cells in which cullin 2 levels werediminished by stable expression of a short hairpin RNA targetingcullin 2. This suggests that a cullin 2-independent mechanism isutilized for pRb degradation. This is certainly possible, as the abil-ity of HPV16 E7 to bind cullin 2 is not shared by the E7 proteins ofany other HPV type so far tested (53).
Nearly 20 years ago, it was suggested that CR3 also functions asa secondary lower affinity binding site for pRb (46). However, thisinteraction was only recently confirmed by two independent re-
FIG 8 CR3-pRb interaction is functionally important for overcoming cellcycle arrest. (A) Mutants with reduced capacity to associate with pRb via CR3have defects in overcoming cell cycle arrest. Mutants analyzed for pRb bindingin vitro were also assessed for their ability to overcome cell cycle arrest inducedby reexpression of wild-type pRb in Saos2 cells. The percentage of cells in G1
phase of the cell cycle was determined by flow cytometry. Control cells repre-sent the normally cycling Saos2 population. Saos2 cells expressing pRb onlybut not E7 are labeled as pRb only. For all other samples, cells express pRb andeither the full-length wild-type or indicated mutant E7 protein. Data are rep-resentative of a minimum of three independent experiments. ***, P � 0.001;**, P � 0.01; and *, P � 0.05 relative to the wild-type E7 sample. The relativeactivity of E7 mutants in overcoming cell cycle arrest was calculated and isindicated below the bar graph. (B) Analysis of E7 mutant stability. HT1080
cells were transfected with the expression plasmid for the indicated E7 mutantstogether with GFP, and 24 h posttransfection they were treated with cyclohex-imide for 15, 30, or 60 min. The levels of E7 were analyzed by Western blotting.DMSO, dimethylsulfoxide.
FIG 9 Association of E7 mutants with p21Cip1/WAF1. HT1080 cells werecotransfected with expression plasmids for HA epitope-tagged p21 and wild-type full-length E7 or the indicated mutant fused to a myc-GFP tag. Twenty-four h posttransfection, the cells were lysed and immunoprecipitation wasperformed with anti-GFP antibody, followed by Western blotting.
HPV16 E7 CR3 Deregulates pRb Function
December 2012 Volume 86 Number 24 jvi.asm.org 13321
on July 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

ports (15, 45). Importantly, we have extended those in vitro stud-ies to demonstrate that the interaction of CR3 with pRb occurs inmammalian cells in vivo by utilizing coimmunoprecipitation ex-periments (Fig. 5A). We also found that CR3 from other HPVtypes, specifically the low-risk HPV11 and HPV6, associate withpRb (Fig. 5D). Interestingly, CR3 from HPV18 E7 did not bindpRb, suggesting that this interaction is present in some, but not all,E7 proteins. To establish the functional consequences of the pRb-CR3 interaction, we first sought to identify the surface of CR3 thatbinds pRb. We assessed our panel of surface-exposed CR3 mu-tants for their pRb binding properties by utilizing the yeast two-hybrid assay. This identified two specific patches on the surface ofCR3 that contribute to pRb binding. We confirmed these data invitro using purified recombinant proteins and GST pulldown ex-periments with the large pocket of pRb (pRbABC) fused to GSTand purified wild-type or mutant CR3. Although both yeast two-hybrid and in vitro interaction assays yield similar results for themajority of mutants, the two assays responded differently for asubset of the mutants. It is possible that additional factors presentin yeast but not in the fully recombinant system influence theresults. Nevertheless, these studies identified residues that com-prise patch 1, which forms one continuous surface on CR3 andsignificantly contributes to pRb binding (Fig. 7D). These includeresidues Y52, N53, V55, F57, C59, S63, T64, and T72. Consistentwith a previous report, we also identified residues within patch 2that also contribute to pRb binding (40). Although we tested othermutants of E7 that are found within the patch 2 region, we foundthat only R77E and E80K/D81K had an impact on binding. Thissuggests that negative charges in this region contribute to pRbbinding via electrostatic interactions, as previously proposed (45).
We have established that all CR3 mutants maintain the abilityto degrade pRb, but that specific residues in this region of E7 havereduced binding to pRb. We therefore wanted to determinewhether CR3 contributes functionally to deregulating the pRbpathway by targeting the remaining pools of undegraded pRb.Previous work has shown that CR3 of E7 is important for pRb-E2Fcomplex interference, but that small alterations in CR3 do not leadto significant changes in the ability of E7 to interfere with pRb-E2Fcomplex formation (35). Therefore, our approach was to assessthe role of the CR3-pRb interaction more globally by determiningthe ability of CR3 mutants with decreased pRb binding to over-come cell cycle arrest induced by reintroduction of pRb into pRb-null Saos2 cells. We found that there is a high degree of correlationbetween the ability of E7 to bind pRb via the CR3 region and theability of the corresponding full-length mutants to overcome cellcycle arrest. Most notably, mutations in residues within patch 1that exhibited the most significant decrease in pRb binding alsodisplayed a significant decrease in the ability to overcome pRb-induced cell cycle arrest. Additionally, mutants in patch 2 werealso examined. E80K/D81K had a reduced ability to overcome cellcycle arrest, which correlated with its reduced ability to bind pRbin vitro. Importantly, these data establish that the ability of HPV16E7 to induce pRb degradation is not sufficient to fully overcome apRb-induced cell cycle arrest. The data suggest that the remainingpool of undegraded pRb is still partially able to block cell cycleentry, and that this is overcome by the CR3-pRb interaction.
The cell cycle analysis also identified the G85A mutant as beinginteresting. Although this mutant binds pRb similarly to wild-typeE7 in vitro, it has a reduced ability to overcome cell cycle arrest.This mutant maintains the ability to interact with p21Cip1/WAF1
(Fig. 9), but it is possible that it has lost the capacity to interactwith another important cellular target which is necessary for E7 todrive the cells into the S phase of the cell cycle.
Taken together, we have identified a new surface of HPV16 E7that contributes to binding to pRb, which we have called patch 1.We also observed that negative charges located in patch 2 likelycontribute to electrostatic interactions with pRb. Our data illus-trate for the first time that the ability of E7 to bind pRb via CR3 isimportant in overcoming the cell cycle arrest functions of pRb. Inlight of these findings, we suggest that the CR3 domain of E7 is acandidate for targeted inactivation by small-molecule com-pounds. Specifically, development of small molecules that bindpatch 1 of CR3 may disrupt the ability of E7 to perturb pRb func-tion and have utility in the treatment of papillomas or HPV-in-duced cancers.
ACKNOWLEDGMENTS
This work was supported by a grant from the Canadian Institute of HealthResearch awarded to J.S.M. B.T. was supported by a Canadian Institute ofHealth Research doctoral award. G.S.S. was supported by the CanadianCancer Society (018414).
H1299 cullin 2 KD and control cells were generously provided by P.Branton and P. Blanchette.
REFERENCES1. Adams A, Gottschling DE, Kaiser CA, Stearns T. 1997. Methods in yeast
genetics. Cold Spring Harbor Press, Cold Spring Harbor, NY.2. Androphy EJ, Hubbert NL, Schiller JT, Lowy DR. 1987. Identification of
the HPV-16 E6 protein from transformed mouse cells and human cervicalcarcinoma cell lines. EMBO J. 6:989 –992.
3. Avvakumov N, Torchia J, Mymryk JS. 2003. Interaction of the HPV E7proteins with the pCAF acetyltransferase. Oncogene 22:3833–3841.
4. Banks L, et al. 1987. Identification of human papillomavirus type 18 E6polypeptide in cells derived from human cervical carcinomas. J. Gen. Vi-rol. 68(Pt 5):1351–1359.
5. Barbosa MS, et al. 1990. The region of the HPV E7 oncoprotein homol-ogous to adenovirus E1a and Sv40 large T antigen contains separate do-mains for Rb binding and casein kinase II phosphorylation. EMBO J.9:153–160.
6. Berezutskaya E, Bagchi S. 1997. The human papillomavirus E7 oncopro-tein functionally interacts with the S4 subunit of the 26 S proteasome. J.Biol. Chem. 272:30135–30140.
7. Bodily JM, Mehta KP, Cruz L, Meyers C, Laimins LA. 2011. The E7 openreading frame acts in cis and in trans to mediate differentiation-dependentactivities in the human papillomavirus type 16 life cycle. J. Virol. 85:8852–8862.
8. Bodily JM, Mehta KP, Laimins LA. 2011. Human papillomavirus E7enhances hypoxia-inducible factor 1-mediated transcription by inhibitingbinding of histone deacetylases. Cancer Res. 71:1187–1195.
9. Boyer SN, Wazer DE, Band V. 1996. E7 protein of human papillomavirus-16 induces degradation of retinoblastoma protein through the ubiq-uitin-proteasome pathway. Cancer Res. 56:4620 – 4624.
10. Brehm A, et al. 1998. Retinoblastoma protein recruits histone deacetylaseto repress transcription. Nature 391:597– 601.
11. Brehm A, et al. 1999. The E7 oncoprotein associates with Mi2 and histonedeacetylase activity to promote cell growth. EMBO J. 18:2449 –2458.
12. Cecchini MJ, Dick FA. 2011. The biochemical basis of CDK phosphory-lation-independent regulation of E2F1 by the retinoblastoma protein.Biochem. J. 434:297–308.
13. Centers for Disease Control and Prevention. 2010. FDA licensure ofbivalent human papillomavirus vaccine (HPV2, cervarix) for use in fe-males and updated HPV vaccination recommendations from the AdvisoryCommittee on Immunization Practices (ACIP). MMWR Morb. Mortal.Wkly. Rep. 59:626 – 629.
14. Chellappan S, et al. 1992. Adenovirus E1A, simian virus 40 tumor anti-gen, and human papillomavirus E7 protein share the capacity to disruptthe interaction between transcription factor E2F and the retinoblastomagene product. Proc. Natl. Acad. Sci. U. S. A. 89:4549 – 4553.
Todorovic et al.
13322 jvi.asm.org Journal of Virology
on July 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

15. Chemes LB, Sanchez IE, Smal C, de Prat-Gay G. 2010. Targetingmechanism of the retinoblastoma tumor suppressor by a prototypicalviral oncoprotein. Structural modularity, intrinsic disorder and phos-phorylation of human papillomavirus E7. FEBS J. 277:973–988.
16. Chen J, Saha P, Kornbluth S, Dynlacht BD, Dutta A. 1996. Cyclin-binding motifs are essential for the function of p21CIP1. Mol. Cell. Biol.16:4673– 4682.
17. Cheng CY, Blanchette P, Branton PE. 2007. The adenovirus E4orf6 E3ubiquitin ligase complex assembles in a novel fashion. Virology 364:36 – 44.
18. Cheng CY, et al. 2011. The E4orf6/E1B55K E3 ubiquitin ligase complexesof human adenoviruses exhibit heterogeneity in composition and sub-strate specificity. J. Virol. 85:765–775.
19. Classon M, Harlow E. 2002. The retinoblastoma tumour suppressor indevelopment and cancer. Nat. Rev. Cancer 2:910 –917.
20. Dawar M, Deeks S, Dobson S. 2007. Human papillomavirus vaccineslaunch a new era in cervical cancer prevention. CMAJ 177:456 – 461.
21. de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H.2004. Classification of papillomaviruses. Virology 324:17–27.
22. Dick FA, Dyson N. 2003. pRB contains an E2F1-specific binding domainthat allows E2F1-induced apoptosis to be regulated separately from otherE2F activities. Mol. Cell 12:639 – 649.
23. Dick FA, Sailhamer E, Dyson NJ. 2000. Mutagenesis of the pRB pocketreveals that cell cycle arrest functions are separable from binding to viraloncoproteins. Mol. Cell. Biol. 20:3715–3727.
24. Dyson N. 1998. The regulation of E2F by pRB-family proteins. Genes Dev.12:2245–2262.
25. Dyson N, Howley PM, Munger K, Harlow E. 1989. The human papil-loma virus-16 E7 oncoprotein is able to bind to the retinoblastoma geneproduct. Science 243:934 –937.
26. Edmonds C, Vousden KH. 1989. A point mutational analysis of humanpapillomavirus type 16 E7 protein. J. Virol. 63:2650 –2656.
27. Funk JO, et al. 1997. Inhibition of CDK activity and PCNA-dependentDNA replication by p21 is blocked by interaction with the HPV-16 E7oncoprotein. Genes Dev. 11:2090 –2100.
28. Future II Study Group. 2007. Quadrivalent vaccine against human pap-illomavirus to prevent high-grade cervical lesions. N. Engl. J. Med. 356:1915–1927.
29. Giarre M, et al. 2001. Induction of pRb degradation by the human pap-illomavirus type 16 E7 protein is essential to efficiently overcomep16INK4a-imposed G1 cell cycle arrest. J. Virol. 75:4705– 4712.
30. Gietz RD, Schiestl RH, Willems AR, Woods RA. 1995. Studies on thetransformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure.Yeast 11:355–360.
31. Gillison ML, et al. 2000. Evidence for a causal association between humanpapillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst.92:709 –720.
32. Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. 2001. Degrada-tion of the retinoblastoma tumor suppressor by the human papillomavi-rus type 16 E7 oncoprotein is important for functional inactivation and isseparable from proteasomal degradation of E7. J. Virol. 75:7583–7591.
33. Goodwin EC, DiMaio D. 2000. Repression of human papillomavirusoncogenes in HeLa cervical carcinoma cells causes the orderly reactivationof dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. U. S. A.97:12513–12518.
34. Goodwin EC, et al. 2000. Rapid induction of senescence in human cer-vical carcinoma cells. Proc. Natl. Acad. Sci. U. S. A. 97:10978 –10983.
35. Helt AM, Galloway DA. 2001. Destabilization of the retinoblastomatumor suppressor by human papillomavirus type 16 E7 is not sufficient toovercome cell cycle arrest in human keratinocytes. J. Virol. 75:6737– 6747.
36. Huh K, et al. 2007. Human papillomavirus type 16 E7 oncoprotein asso-ciates with the cullin 2 ubiquitin ligase complex, which contributes todegradation of the retinoblastoma tumor suppressor. J. Virol. 81:9737–9747.
37. Jeon S, Allen-Hoffmann BL, Lambert PF. 1995. Integration of humanpapillomavirus type 16 into the human genome correlates with a selectivegrowth advantage of cells. J. Virol. 69:2989 –2997.
38. Jones DL, Thompson DA, Munger K. 1997. Destabilization of the RBtumor suppressor protein and stabilization of p53 contribute to HPV type16 E7-induced apoptosis. Virology 239:97–107.
39. Kalejta RF, Brideau AD, Banfield BW, Beavis AJ. 1999. An integralmembrane green fluorescent protein marker, Us9-GFP, is quantitativelyretained in cells during propidium iodide-based cell cycle analysis by flowcytometry. Exp. Cell Res. 248:322–328.
40. Liu X, Clements A, Zhao K, Marmorstein R. 2006. Structure of thehuman papillomavirus E7 oncoprotein and its mechanism for inactiva-tion of the retinoblastoma tumor suppressor. J. Biol. Chem. 281:578 –586.
41. Longworth MS, Dyson NJ. 2010. pRb, a local chromatin organizer withglobal possibilities. Chromosoma 119:1–11.
42. Massimi P, Shai A, Lambert P, Banks L. 2008. HPV E6 degradation ofp53 and PDZ containing substrates in an E6AP null background. Onco-gene 27:1800 –1804.
43. Morris EJ, Dyson NJ. 2001. Retinoblastoma protein partners. Adv. Can-cer Res. 82:1–54.
44. Nevins JR. 1992. E2F: a link between the Rb tumor suppressor protein andviral oncoproteins. Science 258:424 – 429.
45. Ohlenschlager O, et al. 2006. Solution structure of the partially foldedhigh-risk human papilloma virus 45 oncoprotein E7. Oncogene 25:5953–5959.
46. Patrick DR, Oliff A, Heimbrook DC. 1994. Identification of a novelretinoblastoma gene product binding site on human papillomavirus type16 E7 protein. J. Biol. Chem. 269:6842– 6850.
47. Phelps WC, Munger K, Yee CL, Barnes JA, Howley PM. 1992. Struc-ture-function analysis of the human papillomavirus type 16 E7 oncopro-tein. J. Virol. 66:2418 –2427.
48. Romanowski B. 2011. Long term protection against cervical infectionwith the human papillomavirus: review of currently available vaccines.Hum. Vaccin. 7:161–169.
49. Schneider-Gadicke A, Schwarz E. 1987. Transcription of human papil-lomavirus type-18 DNA in human cervical carcinoma cell lines. Haema-tol. Blood Transfus. 31:380 –381.
50. Sherr CJ. 2000. The Pezcoller lecture: cancer cell cycles revisited. CancerRes. 60:3689 –3695.
51. Smotkin D, Wettstein FO. 1987. The major human papillomavirus pro-tein in cervical cancers is a cytoplasmic phosphoprotein. J. Virol. 61:1686 –1689.
52. Todorovic B, et al. 2011. Systematic analysis of the amino acid residues ofhuman papillomavirus type 16 E7 conserved region 3 involved indimerization and transformation. J. Virol. 85:10048 –10057.
53. White EA, et al. 2012. Systematic identification of interactions betweenhost cell proteins and E7 oncoproteins from diverse human papillomavi-ruses. Proc. Natl. Acad. Sci. U. S. A. 109:E260 –E267.
HPV16 E7 CR3 Deregulates pRb Function
December 2012 Volume 86 Number 24 jvi.asm.org 13323
on July 10, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from












![Human Papillomavirus Type 16 E7 Oncoprotein-induced ... · [CANCER RESEARCH 61, 2356–2360, March 15, 2001] Advances in Brief Human Papillomavirus Type 16 E7 Oncoprotein-induced](https://static.fdocuments.us/doc/165x107/605dd1c1b72c9c6f905bfd49/human-papillomavirus-type-16-e7-oncoprotein-induced-cancer-research-61-2356a2360.jpg)





