
CROUZET TEMPERATURE CONTROLLERS. CT 48 CTD 43, CTD 46, & CTH 46 CTD 24 MIC 48.
Upload
nguyendieuCategory
view
264download
1
Common Technical Document (CTD)
Gerd Bode. M.D., Ph. D. Consultant in preclinical Sciences
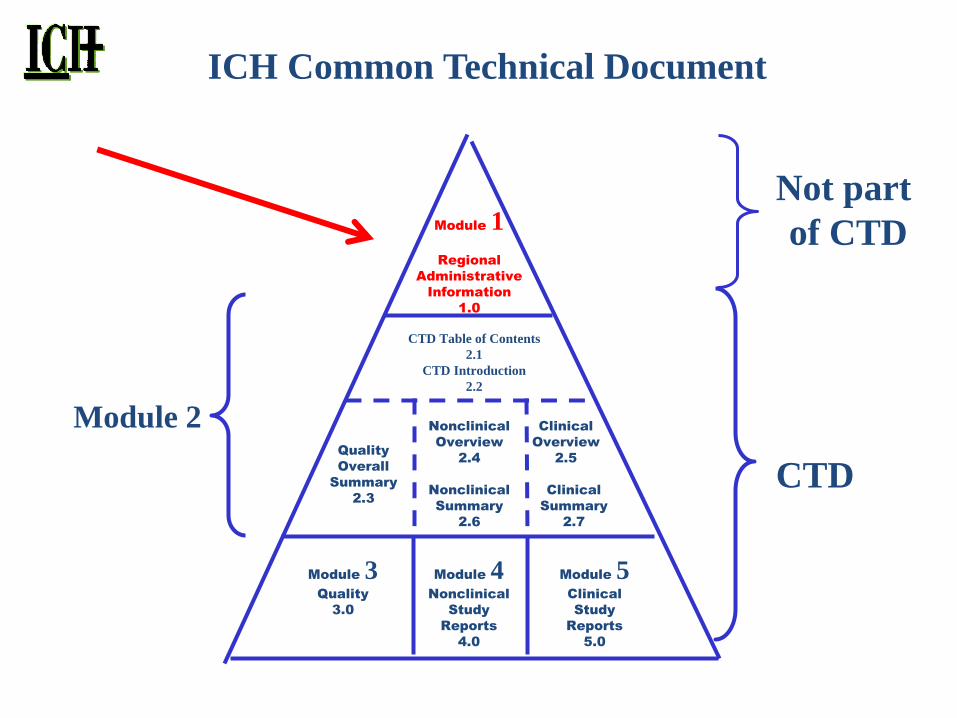
Module 1
Regional
Administrative
Information
1.0
Nonclinical
Overview
2.4
Nonclinical
Summary
2.6
Clinical
Overview
2.5
Clinical
Summary
2.7
Quality
Overall
Summary
2.3
Module 3
Quality
3.0
Module 4
Nonclinical
Study
Reports
4.0
Module 5
Clinical
Study
Reports
5.0
Not part
of CTD
CTD
Module 2
ICH Common Technical Document
CTD Table of Contents
2.1
CTD Introduction
2.2

Common Technical Document Safety (update in 2006)
• Modul 1, regional
• Modul 2, global
– 2.4 Non clinical Overview
– 2.6 Non clinical Summaries • written
• tabulated
• Modul 4 = Study Reports

EU CTD Module 1 Module 1 Table of Content
1.0 Cover Letter
1.1 Comprehensive Table of Contents
1.2 Application Form
1.3 Product Information
1.3.1 SPC, Labelling and Package Leaflet
1.3.2 Mock-up
1.3.3 Specimen
1.3.4 Consultation with Target Patient Groups
1.3.5 Product Information already approved in the Member States
1.3.6 Braille
1.4 Information about the Experts
1.4.1 Quality
1.4.2 Non-Clinical
1.4.3 Clinical

ICH CTD Modul 1 cont. 1.5 Specific Requirements for Different Types of Applications
1.5.1 Information for Bibliographical Applications
1.5.2 Information for Generic, ‘Hybrid’ or Bio-similar Applications
1.5.3 (Extended) Data/Market Exclusivity
1.5.4 Exceptional Circumstances
1.5.5 Conditional Marketing Authorisation
1.6 Environmental Risk Assessment 1.6.1 Non-GMO 1.6.2 GMO
1.7 Information relating to Orphan Market Exclusivity
1.7.1 Similarity
1.7.2 Market Exclusivity
1.8 Information relating to Pharmacovigilance
1.8.1 Pharmacovigilance System
1.8.2 Risk-management System
1.9 Information relating to Clinical Trials
1.10 Information relating to Paediatric Responses to Questions
Additional Data

Module 1
Regional
Administrative
Information
1.0
Nonclinical
Overview
2.4
Nonclinical
Summary
2.6
Clinical
Overview
2.5
Clinical
Summary
2.7
Quality
Overall
Summary
2.3
Module 3
Quality
3.0
Module 4
Nonclinical
Study
Reports
4.0
Module 5
Clinical
Study
Reports
5.0
Not part
of CTD
CTD
Module 2
Common Technical Document
CTD Table of Contents
2.1
CTD Introduction
2.2
Non-clinical Overview
2.4

History Comparison between previous application
formats and the CTD
European Nonclinical Expert Report
similar to
ICH Nonclinical Overview

about 30 pages
For References: use internationally accepted standards or Chemical Abstracts
Citations to the Tabulated Summaries: Table XX, Study/Report Number
Formal Aspects for Non-clinical Overview

Nonclinical Overview
Nonclinical Documentation
Literature
Efficacy Documentation
Quality Documentation
Labeling Guideline for Assessors
GLP Status Guidelines
General Aspects
Nonclinical Documentation
Literature
Efficacy Documentation
Labeling Guideline for Assessors
GLP Status Guidelines

ICH No. Title Step
S1A The Need for Carcinogenicity Studies of Pharmaceuticals
Step 5
S1B Carcinogenicity: Testing for Carcinogenicity of Pharmaceuticals
Step 5
S1C (R1) Carcinogenicity: Dose selection for carcinogenicity studies of pharmaceuticals
Step 5
Guidelines - Safety

ICH No. Title Step
S2A Genotoxicity: Guidance on Specific Aspects of Regulatory Genotoxicity Tests for Pharmaceuticals
Step 5
S2B Genotoxicity: A Standard Battery for Genotoxicity Testing of Pharmaceuticals
Step 5
Guidelines - Safety (cont.)
S2(R) : Guidance on Genotoxicity Testing and Data Interpretation for Pharmaceuticals intended for human Use. Step 2, 2008

ICH No. Title Step
S3A Toxicokinetics: A Guidance for Assessing Systemic Exposure in Toxicology Studies
Step 5
S3B PharmacoKinetics: Guidance for Repeated Dose Tissue Distribution Studies
Step 5
S4A Duration of Chronic Toxicity Testing in Animals (Rodent and non Rodent Toxicity Testing)
Step 5
S5A Reproductive Toxicology: Detection of Toxicity to Reproduction for Medicinal Products
Step 5
S5B (M) Reproductive Toxicology: Toxicity on Male Fertility (Modification)
Step 5
S6 Preclinical Safety Evaluation of Biotechnology-Derived Products
Step 5
Guidelines - Safety (cont.)
S5(R2): Detection of Toxicity to Reproduction for Medicinal Products & Toxicity to Male Fertility

Guidelines - Safety (cont.)
ICH No. Title Step
S7A Safety Pharmacology Studies for Human Pharmaceuticals
Step 5
S7B Non-Clinical Studies for Assessing Risk of Repolarisation – Associated Ventricular Tachyarrhythmia for Human Pharmaceuticals
Step 5
S8 Immunotoxicology Studies Step 5
S9 Nonclinical Development of Anticancer Pharmaceuticals
Step 1
S9 agreed on step 4 in October 2009: Implementation in summer 2010.

<Co->Rapporteur
Day 70 Critical Assessment Report
NON-CLINICAL
<Invented Name>
<(Active Substance)>
EMEA/H/C/{nnnn}/{nnn}/{nnn}
Applicant:
Rapporteur:
Co-Rapporteur:
Start of the procedure:
Date of this report:
Deadline for comments:

D70 QUESTIONNAIRE
To be completed by <Co->Rapporteur
THE PRE-SUBMISSION PHASE
Did the applicant make contact with you prior to submission of the
application? Yes No 1
If yes, were any major issues identified? Yes No 2
THE DOSSIER (Indicate in the continous scale 1: “strongly disagree” to 10: “strongly agree”)
Was the dossier presented in a satisfactory way? (e.g layout,
organisation of data etc)
1 2 3 4 5 6 7 8 9 10
- Quality --------------------------------> 3
- Non Clinical --------------------------------> 4- Clinical --------------------------------> 5
Were all important data/analysis included in the the dossier thus
making benefit risk assessment easy?1 2 3 4 5 6 7 8 9 10
- Quality ---------------------------------> 6- Non Clinical ---------------------------------> 7- Clinical ---------------------------------> 8
Was the “scientific overview” (expert report) sufficiently critical? 1 2 3 4 5 6 7 8 9 10
- Non Clinical ---------------------------------> 9
- Clinical --------------------------------> 10
Was the “summary” useful for the critical assessment? 1 2 3 4 5 6 7 8 9 10
- Quality --------------------------------> 11
- Non Clinical --------------------------------> 12
- Clinical --------------------------------> 13
Was the quality of the individual study reports / scientific data
satisfactory?1 2 3 4 5 6 7 8 9 10
- Quality --------------------------------> 14
- Non Clinical --------------------------------> 15- Clinical --------------------------------> 16

Overview of the nonclinical testing Strategy Pharmacology
Pharmacokinetics
Toxicology
Integrated overview and conclusions
List of literature citations
Content and Structural Format

Concept for the development of individual testing strategies
Potential areas of risk (in humans) which have to be experimentally
clarified
Main features of risk identification
in planned clinical studies
Basic principles to be considered
Appropriate nonclinical testing strategy for risk assessment (in humans) and for every
single step of medicinal products development
K. Olejniczak, Günzel P.and Bass R.;Preclinical Testing Strategies;
Drug Information Journal 35; 321-336, (2001)

Absorption
Distribution
Biotransformation
Excretion
Protein binding
Assays
PK models
Pharmacokinetic parameter
Part of Tox. studies
Main data: AUC, cmax, tmax
Dose-dependancy Dose linearity
Accumulation, Enzyme Induction
Inter-species comparison of systemic exposure + metabolites (including humans)
Pharmacokinetics Toxicokinetics

Review and discuss characteristics of toxic effects:
• onset in which targets
• type and severity of toxicity
• duration of toxic effects and time dependance,
• dose-dependency, dose linearity, bell-shaped
• degree of reversibility (or irreversibility)
• species-, subspecies or gender-related differences
• predictivity for humans
Toxicology

• toxic symptoms / exaggerated pharmacological effects
• causes of death, when?
• pathological findings
• genotoxic activity: strategy of testing,
chemical structure, its mode of action, and
its relationship to known genotoxic compounds
• female and male fertility, embryo-fetal development, peri/post-
natal toxicity
• consequences of use before and during pregnancy
and during lactation
Toxicology (contin.)

carcinogenic potential:
concerns from chemical structure of compound,
relationship to known carcinogens,
genotoxic potential,
exposure data for parent + (genotoxic) metabolites
extrapolation of carcinogenic risk to humans.
Speicies-specificity? Any epidemiologic data available?
local tolerance
other toxicity studies
studies to clarify special problems,
immunotoxicity
Toxicology

Overview = Critical Assessment
• advantages/disadvantages (PD vs. Tox)
• strict adherence to guidelines , deviation from guidelines: GLP, ICH etc.
• explaining effects, mechanisms, relevant predictive species, metabolites,
• indications, partient population, juvenile
• experiences with class
• recommendations for labeling

Module 1
Regional
Administrative
Information
1.0
Nonclinical
Overview
2.4
Nonclinical
Summary
2.6
Clinical
Overview
2.5
Clinical
Summary
2.7
Quality
Overall
Summary
2.3
Module 3
Quality
3.0
Module 4
Nonclinical
Study
Reports
4.0
Module 5
Clinical
Study
Reports
5.0
Not part
of CTD
CTD
Module 2
ICH Common Technical Document
CTD Table of Contents
2.1
CTD Introduction
2.2
Non-clinical Summary
2.6

Modul 2.6: Organisation of the Common Technical Document for the
Registration of Pharmaceuticals for Human Use
Nonclinical Summary 1. Pharmacology
– a. Written Summary
– b. Tabulated Summary
2. Pharmacokinetics – a. Written Summary
– b. Tabulated Summary
3.Toxicology – a. Written Summary
– b. Tabulated Summary

Objectives:
• to assist authors to prepare the nonclinical pharmacology, pharmacokinetics, and toxicology written summaries in an acceptable format
• not intended to indicate what studies are required
• but providing an appropriate format for the nonclinical data acquired.
• CTD = Placeholder
Nonclinical Written Summaries


Advice for Deviation from Guideline:
• no guideline can cover all eventualities
• common sense and a clear focus on needs of regulatory authority assessor are best to construct an acceptable document
• therefore, modify format , if needed
• aim: provide best possible presentation:
– to facilitate the understanding
– to evaluate the results

• No formal limit for length of the Nonclinical Written Summaries
• Recommendation: total length of 3 Nonclinical Written Summaries in general
100-150 pages
Length of Nonclinical Written Summaries

Module 2.6: Nonclinical Tabulated Summaries
Pharmacology, Pharmacokinetics, and Toxicology
34 Templates
31 Examples

Tabulated Summaries: Pharmacology
• Example for Pharmacology Overview
• Example for Safety Pharmacology

1 Pharmacology Overview Test Article:
Type of Study
Test
System
Method of
Administration
Study
Number
Location
Vol.
Page
1.1 Primary
Pharmacodynamics
1.2 Secondary
Pharmacodynamics
1.3 Safety Pharmacology
1.4 Pharmacodynamic Drug
Interactions

1.3 Safety Pharmacology Test Article:
OrganSystemsEvaluated
Species/Strain
Method ofAdmin.
Doses(mg/kg)
Genderand No.per Group Noteworthy Findings
GLPCompliance
StudyNumber

3A Toxicology Overview Test Article:
Type of Study Speciesand Strain
Method ofAdministration
Durationof Dosing Doses (mg/kg
a)
GLPCompliance
TestingFacility
StudyNumber
LocationVol. Page
3.1 Single-DoseToxicity
3.2 Repeat-DoseToxicity
3.3 Genotoxicity
3.4 Carcinogenicity
3.5 Reproductive &DevelopmentalToxicity
3.6 Local Tolerance
3.7 OtherToxicity Studies

Module 1
Regional
Administrative
Information
1.0
Nonclinical
Overview
2.4
Nonclinical
Summary
2.6
Clinical
Overview
2.5
Clinical
Summary
2.7
Quality
Overall
Summary
2.3
Module 3
Quality
3.0
Module 4
Nonclinical
Study
Reports
4.0
Module 5
Clinical
Study
Reports
5.0
Not part
of CTD
CTD
Module 2
Common Technical Document
CTD Table of Contents
2.1
CTD Introduction
2.2
Modul 4 Study Reports

The Organisation of Module 4: Nonclinical Study Reports
• The appropriate location for individual-animal data is in the study report or as an appendix to the study report. – 4.1 Table of Contents
– A Table of Contents should be provided that lists all of the nonclinical study reports and gives the location of each study report in the Common Technical Document. • Study Reports • The study reports should be presented in the following order:
– 4.2 Pharmacology • 4.2.1 Primary Pharmacodynamics • 4.2.2 Secondary Pharmacodynamics • 4.2.3 Safety Pharmacology • 4.2.4 Pharmacodynamic Drug Interactions

Module 4: Nonclinical Study Reports
4.3 Pharmacokinetics
– 4.3.1 Analytical Methods and Validation Reports (if separate reports are available)
– 4.3.2 Absorption
– 4.3.3 Distribution
– 4.3.4 Metabolism
– 4.3.5 Excretion
– 4.3.6 Pharmacokinetic Drug interactions (nonclinical)
– 4.3.7 Other Pharmacokinetic Studies

Module 4: Nonclinical Study Reports
4.4 Toxicology 4.4.1 Single-Dose Toxicity (in order by species, by route)
4.4.2 Repeat-Dose Toxicity (in order by species, by route, by duration; including supportive toxicokinetics evaluations)
4.4.3 Genotoxicity
4.4.3.1 In vitro
4.4.3.2 In vivo (including supportive toxicokinetics evaluations)
4.4.4 Carcinogenicity (including supportive toxicokinetics evaluations)
4.4.4.1 Long-term studies (in order by species; including range- finding studies that cannot appropriately be included under repeat-dose toxicity or pharmacokinetics)
4.4.4.2 Short- or medium-term studies (including range-finding studies that cannot appropriately be included under repeat- dose toxicity or pharmacokinetics)
4.4.4.3 Other studies

Module 4: Safety Study Reports cont.
4.4.5 Reproductive and Developmental Toxicity (including range- finding studies and supportive toxicokinetics evaluations) (If modified study designs are used, the following subheadings shouldbe modified accordingly.)
4.4.5.1 Fertility and early embryonic development
4.4.5.2 Embryo-fetal development
4.4.5.3 Prenatal and postnatal development, including maternal function
4.4.5.4 Studies in which the offspring (juvenile animals) are dosed and/or further evaluated.
4.4.6 Local Tolerance

Module 4: Nonclinical Study Reports cont.
4.4.7 Other Toxicity Studies (if available)
4.4.7.1 Antigenicity
4.4.7.2 Immunotoxicity
4.4.7.3 Mechanistic studies (if not included elsewhere)
4.4.7.4 Dependence
4.4.7.5 Metabolites
4.4.7.6 Impurities
4.4.7.7 Other
4.5 Key Literature References

•Summary of CTD :
CTD: - internationally agreed format for applications,
- accepted by regulatory authorities of EU, USA and Japan. Today globally.
Objectives: - to save time and resources and
- to facilitate regulatory review and communication.
CTD: - no information about the content of a dossier,
- no indication which studies and data are required for MAA.
Regional requirements: - may affect the content of the dossier submitted in each region, - therefore, CTD not always identical for all regions.
The Overall organisation of the Common Technical Document:
- Applicants should not modify the outlined format, - if deemed necessary: justify modifications.

Recommendations for Industry
1. multidisciplinary cooperation necessary
2. start collecting facts + critical comments early on for Summaries + Overview
3. integrate relevant data from Quality and Efficacy into Non- Clinical Safety Section
4. Prepare Tabulated Summaries at same time as Reports
5. Consider regulatory requirements
6. Clarify with EMEA handling of deviations

I Regional
Administrative
Information
Module 5 Module 3 Module 4
Nonclinical
Overview
Nonclinical
Overvies Clinical
Nonclinical
Summaries


















