
Cognitive and motor dysfunction in the early phase of...
85
Cognitive and motor dysfunction in the early phase of Parkinson’s disease Magdalena Eriksson Domellöf Department of Pharmacology and Clinical Neuroscience Department of Community Medicine and Rehabilitation Umeå University, Sweden
Transcript of Cognitive and motor dysfunction in the early phase of...

Cognitive and motor dysfunction in the early phase of Parkinson’s disease
Magdalena Eriksson Domellöf
Department of Pharmacology and Clinical Neuroscience
Department of Community Medicine and Rehabilitation
Umeå University, Sweden

Responsible publisher under swedish law: the Dean of the Medical Faculty
This work is protected by the Swedish Copyright Legislation (Act 1960:729)
ISBN: 978-91-7459-767-7
ISSN: 0346-6612
Elektronisk version tillgänglig på http://umu.diva-portal.org/
Tryck/Printed by: Print och Media, Umeå University, Umeå, Sweden 2013
Cover photo: Erik Domellöf

To Erik, Edith and Karin


I
Table of Contents Table of Contents I Abstract IV Abbreviations VI Svensk sammanfattning (Summary in Swedish) VII Original papers IX Introduction 1
Parkinson’s disease 1 Definition and diagnosis 1 Symptoms 2
Cardinal signs 2 Other motor symptoms 3 Non motor symptoms 3
Epidemiology 4 Histopathology and etiology 4 Pathology, biochemistry and physiology 5 Treatment of motor symptoms 6
Levodopa 6 Decarboxylase and COMT inhibitors 6 Dopamine agonist and MAO-B inhibitors 6 Treatment in later disease stages 7
Cognitive impairment 7 Cognitive impairment in PD 7
Executive functions 7 Memory 8 Attention 9 Visuospatial skills 9 Language function 9 PD-MCI 9 PDD 10
Epidemiology of cognitive impairment in PD 10 Risk factors for cognitive impairment in PD 12 Neuropathology of cognitive impairment in PDD 12 Genes and cognition in PD 13 Treatment of cognitive impairment and other non-motor features in PD 14 Cognitive motor relationship 15 Rationale of the thesis 15 Aims 17
Materials and methods 18 Study population 19 Non-participation 19 Assessment tools 23

II
Assessment of motor function 23 Assessment of cognitive function 23 Diagnosis 25 PD 26 Cognitive impairment 27 MCI 28 Dementia 28 Ethics 28 Statistics 29 Empirical studies 29 Paper I 29 Paper II 30 Paper III 31 Paper IV 33
Results 35 Paper I 35 Paper II 36 Paper III 36 Paper IV 38
Discussion 40 Main findings 40 Impaired cognitive functions 41 Proportions of MCI and Dementia 42 MCI 42 Dementia 42 Higher dementia and MCI rates 42 Predictive value of MDS-Task Force MCI criteria for PDD 43 Education, age, disease duration and gender aspects 44 Cognitive motor relationship 44 The association of bradykinesia with impaired working memory and executive
function 45 The association of posture and gait disturbances with impaired visuospatial
function and visuospatial memory 45 The effect of dopaminergic medication 47 Ethics 48 Methodological considerations 48 Study design 48 Loss to follow up and missing values 49 Statistics 49 Diagnosis of PD 50 UPDRS 50 SWEDDs 50 MCI and Dementia 50

III
Motor fluctuations 51 Neuropsychological testing 51 Control group 52 Future implications 52
Conclusions 54 Acknowledgements 55 References 57

IV
Abstract
Background: Parkinson’s disease (PD) is a chronic and progressive
neurodegenerative disease. The diagnosis is based on a combination of the
motor signs: tremor, bradykinesia, rigidity and postural abnormalities. Mild
Cognitive Impairment (MCI) is common early in the disease and a large
proportion of patients with PD develop dementia (PDD). Associations
between motor symptoms and cognitive decline have been suggested but the
results are inconclusive due to differences in the selection of participants and
variables tested. Large population based studies with comprehensive
neuropsychological investigation in newly diagnosed cases with PD followed
prospectively are rare. The aim of this thesis was to improve characterization
and understanding of cognition in PD, and to explore the relationship to
motor impairment in the early phase of PD. Methods: All new patients with
suspected idiopathic parkinsonism in the catchment area (142 ooo
inhabitants) were examined during a period of five years and four months.
Among other investigations, a comprehensive neuropsychological evaluation
was carried out in 119 of 148 patients with PD together with 30 age matched
healthy controls. Assessments were repeated after one three and five years.
Results: Patients performed worse than healthy controls in a majority of
neuropsychological tests. MCI at the time of diagnosis were found in 36%
according to recently published MCI criteria. Thirty% were cognitively
impaired using another definition. One fourth of the patients developed PDD
within five years after diagnosis and 25 % of those with MCI at baseline
reversed back to normal cognition. Age and MCI were significant predictors
of dementia. Education was an independent predictor for severe cognitive
dysfunction at diagnosis but did not predict PDD. Patients with MCI
converting to PDD had worse performance on visuospatial function,
semantic fluency, episodic memory, mental flexibility and conceptual
thinking. There were no differences in cognitive performance between
patients with predominant Postural and Gait Disturbances (PIGD) and the
tremor dominant subtype at the baseline investigation and belonging to the
PIGD subgroup at baseline did not predict PDD. Dementia converters
declined more rapidly than non-converters in posture/gait function.
Associations between bradykinesia and measures of executive functions and
working memory were found, and between posture and gait disturbances
and visuospatial function. Some of these associations were persistent after
one year. Patients receiving the dopamine agonist pramipexole performed
significantly worse on a measure of verbal fluency at the one year follow up.
Conclusions: The differences in proportions of cognitively impaired in the
different studies emphasize the value of joint criteria for PD-MCI. Even
when using such criteria, a substantial proportion of patients revert back to

V
normal function. The increase in motor disability in patients with PDD could
have several different causes that need to be further investigated. Associated
motor and cognitive dysfunctions could reflect common pathophysiological
processes in partly shared networks. Both dopaminergic and non-
dopaminergic motor and cognitive functions seems to be involved in PDD
which suggests that pharmacological treatment in PD needs to go beyond the
scope of dopaminergic deficiency in search for new therapies that would also
be effective for non-motor symptoms.

VI
Abbreviations
α-synuclein Alpha-synuclein
ß-amyloid Beta-amyloid
BBB Blood Brain Barrier
BVMT Brief Visuospatial Memory Test
BNT Boston Naming Test
COWAT Controlled Oral Word Assessment Tool
CBD Corticobasal Degeneration
DA Dopamine agonists
DBL Dementia with Levy Bodies
FCSRT Free and Cued Selective Reminding Test
fMRI Functional Magnetic Resonance Imaging
FP-CIT 123I-N (omega)-flouropropyl-2-ß-carbomethoxyl-3-ß-(4-
iodophenyl) nortropane
H&Y Hoehn & Yahr
ID Indeterminate
MADRS Montgomery-Åsberg Depression Rating Scale
MCI Mild Cognitive Impairment
MDS Movement Disorder Society
MMSE Mini-Mental State Examination
MSA Multiple System Atrophy
NYPUM Ny (New)-Parkinson Umeå
NPV Negative Predictive Value
PD Parkinson’s Disease
PDD Parkinson’s Disease Dementia
PIGD Postural Instability and Gait Disturbances
PPV Positive Predictive Value
PSP Progressive Supranuclear Palsy
UPDRS Unified Parkinson’s Disease Rating Scale
SD Standard Deviation
SE Standard Error

VII
Svensk sammanfattning (Summary in Swedish)
Parkinsons sjukdom (PS) är en kronisk och progressiv neurodegenerativ
sjukdom. Den kliniska diagnosen bygger på en kombination av motoriska
symptom: skakningar (tremor), rörelsehämning (bradykinesi), muskelstelhet
(rigiditet) samt balans och gångsvårigheter. Förekomsten av PS ökar med
ålder. Hos individer över 60 år är PS den vanligaste neurodegenerativa
sjukdomen efter Alzheimers sjukdom.
Kognitiva nedsättningar är vanliga redan vid tidig fas av PS och en stor del
utvecklar Parkinson demens. Kognitiva nedsättningar vid PS ger alvarliga
konsekvenser med ökad dödlighet och ökat beroende samt ökad stress för
vårdgivare Det är särskilt viktigt med en bättre beskrivning och förståelse för
kognitiva nedsättningar i tidig fas av sjukdomen eftersom det är då
interventioner med neuroprotektiva mediciner eller andra behandlingar
troligen ger bäst resultat. Att hitta specifika relationer mellan olika typer av
motoriska problem och kognitiva nedsättningar skulle kunna bidra till nya
idéer angående de underliggande patofysiologiska mekanismerna för
motoriska och kognitiva problem vilket kan ge en ökad förståelse för de
underliggande orsakerna.
De flesta tidigare studier som har undersökt kognitiv funktion vid PS har
varit tvärsnittsstudier och/eller inkluderat patienter i olika skeden av
sjukdomen. Associationer mellan motorproblem och kognitiva nedsättningar
har föreslagits men resultaten är även där ofullständiga på grund av
olikheter i de studerade patientgrupperna och vilka variabler som studerats.
Stora populationsbaserade studier av kognitiva problem vid PS med
omfattande neuropsykologisk utredning följda över tid är få. Därför var
målet med den aktuella avhandlingen att förbättra förståelsen av kognitiva
nedsättningar vid PS samt utforska relationen mellan kognition och motorik
i sjukdomens tidiga skede.
Detta gjordes genom en populationsbaserad cohort där målet var att
undersöka alla med misstänkt parkinsonism i upptagningsområdet för
Norrlands Universitets sjukhus. Alla inkluderade patienter genomgick
mängder av undersökningar mellan 1 januari 2004 och 31 april 2009, där
119 av 148 patienter med PS tillsammans med 30 friska kontroller
genomgick omfattande neuropsykologisk utredning vid studiens början samt
efter ett, tre och fem år.
I enlighet med tidigare studier presterade patienter med PS sämre än friska
kontroller på mängder av neuropsykologiska test. Andelen som

VIII
klassificerades som kognitivt nedsatta varierade mellan studierna mycket på
grund av att olika kriterier användes: 36 % när nyligen publicerade kriterier
anpassade till PS användes samt 30 % när en annan definition användes. En
fjärdedel av ursprungsgruppen utvecklade demens under uppföljningstiden
medan en fjärdedel av de med kognitiv nedsättning vid första mättillfället
återgick till normal kognition under uppföljningstiden. Ålder och MCI var
starka prediktorer för demens. Utbildningsnivå var starkt kopplat till
kognitiv nedsättning vid första mättillfället men predicerade inte senare
utveckling av demens. Patienter med kognitiv nedsättning som senare
utvecklade demens presterade sämre än de med kognitiv nedsättning som
inte utvecklade demens på flera olika kognitiva tester.
Det var inte någon skillnad i kognitiv funktion mellan patienter med
övervägande posturala problem och gång svårigheter (PIGD) jämfört med de
med övervägande Tremor. Att ha övervägande PIGD problem vid första
mättillfället predicerade inte utvecklingen av demens. Patienter som
utvecklade demens under uppföljningstiden fick å andra sidan mer posturala
problem och gångsvårigheter under uppföljningstiden än de som inte
utvecklade demens. Mönstret var liknande för bradykinesi och rigiditet, men
inte för tremor. Kopplingen mellan bradykinesi och sämre resultat på
arbetsminnestest, mental flexibilitet samt exekutiva funktioner samt mellan
posturala/gång problem och visuospatial funktion och visuospatialt minne
hittades. Vissa av dessa kopplingar visade sig också förändras parallellt efter
ett år. Det fanns inga signifikanta förändringar på de neuropsykologiska
testerna efter ett år men patienter som fick dopaminagonisten pramipexol
visade sig få sämre resultat på verbalt flöde vid ettårsuppföljningen.
Sammanfattningsvis så har denna studie visat olikheter i andelen kognitivt
nedsatta beroende på vilka kriterier som användes vilket betonar värdet av
en gemensamt kriterier för vad som utgör kognitiva nedsättningar vid PS.
Försämringen av motoriska problem i patienter med parkinson demens kan
bero på flera olika anledningar och behöver utredas ytterligare.
Kopplingarna mellan motoriska och kognitiva funktioner kan spegla
gemensamma patofysiologiska processer i delvist delade nätverk. Slutligen
så har vi visat att både traditionellt dopaminerga funktioner samt icke
dopaminerga funktioner på olika sätt är i kopplade till Parkinson demens
vilket ger en indikation till att interventioner vid PS behöver se bortom det
dopaminerga nätverken.

IX
Original papers
I. Elgh E, Domellöf M, Linder J, Edström M, Stenlund H, Forsgren L.
Cognitive functions in early Parkinson’s disease: a population based study.
European Journal of Neurology 2009:16:1278-1284
II. Domellöf ME, Elgh E, Forsgren L. The relation between cognition and
motor dysfunction in drug naïve newly diagnosed patients with Parkinson’s
disease. Movement Disorders. 2011:26:2183-2189
III. Domellöf ME, Forsgren L, Elgh E. Persistence of associations between
cognitive impairment and motor dysfunction in the early phase of
Parkinson’s disease. Journal of Neurology. 2013:9:2228-2236
IV. Domellöf ME, Forsgren L, Ekman U, Elgh E. Cognitive function in the
early phase of Parkinson’s disease, a longitudinal follow-up. Manuscript.
Papers are reprinted in this thesis with the kind permission of the
respective publisher.

X

1
Introduction
Parkinson’s disease (PD) is a chronic and progressive neurodegenerative
disease whose diagnosis is based on a combination of the motor signs:
tremor, bradykinesia, rigidity and postural abnormalities. The incidence of
PD increases with age and it is the most common neurodegenerative disease
next to Alzheimer’s disease (AD) in people over the age of 60 years. As life
expectancy increases and people over 60 years of age is the fastest growing
age group [1], the number of patients with PD will most likely increase.
PD was initially described by Dr James Parkinson in “An essay of the shaking
palsy” [2]. The description of the disorder was similar to how we describe it
today, as a progressive disease due to probable degenerative pathology in the
central nervous system. The main symptoms were characterized by resting
tremor, flexed posture and shuffling gait. The intellect and senses were
described as being intact. It was not until the beginning of the 20th century
that the first reports of mental deterioration in PD came (see Pollock and
Hornabrook for review) [3], and not until the 1970’s that dementia was
considered an important part of the clinical picture [4]. Today it is well
established that a substantial proportion of patients with PD develop
Parkinson’s Disease Dementia (PDD) [5] and that mild cognitive impairment
(MCI) is common at the time of diagnosis [6–8]. Despite this PD is still
primarily described as a movement disorder with treatment mostly focusing
on the reduction of motor symptoms.
Attempts have been made to find predictors for cognitive decline and
dementia in PD. Associations between motor symptoms and cognitive
decline have been suggested but the results are inconclusive due to
differences in the selection of participants and variables tested. Prospective
studies in large cohorts of well-defined newly diagnosed patients with PD
assessed with extensive neuropsychological protocols are rare [6–8]. This
thesis presents data from the NYPUM study where cognitive function in
early stages of PD and its association to motor function has been explored
both cross-sectionally and longitudinally.
Parkinson’s disease
Definition and diagnosis
PD is one of several disorders with parkinsonism. The most commonly used
diagnostic criteria for parkinsonism is the United Kingdom Parkinson’s
Disease Society Brain Bank (UK PDSBB) definition which is also used in this

2
thesis. It requires bradykinesia plus at least one of the following symptoms:
tremor, muscular rigidity or postural abnormalities for diagnosis [9].
Parkinsonism can be idiopathic, i.e. of unknown cause, and includes PD and
the atypical forms referred to as Parkinson plus syndromes or atypical
parkinsonism: Multiple System Atrophy (MSA), Progressive Supranuclear
Palsy (PSP), Cortico Basal Degeneration (CBD) and Dementia with Levy
Bodies (DLB). Parkinsonism that is secondary to known cause is classified as
secondary parkinsonism, e.g. treatment with neuroleptic drugs.
Diagnostic accuracy of PD is a big problem. Clinical-pathological studies
report incorrect diagnosis of PD in 10-24% of patients with advanced PD
[10]. This is due to the existence of several similar conditions with
parkinsonism but also due to the heterogeneity of PD both in presentation
and in response to medication. Clinical signs that may differentiate PD from
other parkinsonian disorders are: more often an asymmetrical onset of
motor symptoms, the presence of rest tremor and a positive clinical response
to dopaminergic medication [9].
No radiological methods can readily distinguish between the different
parkinsonian disorders. Nuclear imaging methods such as single photon
computed tomography (SPECT) or positron emission tomography (PET) can
reveal decreased dopaminergic nerve terminals in both PD and Parkinson
plus syndromes but do not distinguish between them [11]. A definite
diagnosis can only be confirmed with autopsy.
Symptoms
Cardinal signs
Bradykinesia is the main feature of parkinsonism and is characterized by
slowness or weakened and reduced amplitude of movements. Bradykinesia is
a collective term for hypokinesia and akineisa. Hypokinesia refers to reduced
spontaneous movement such as arm swing and reduced facial expression.
Akinesia refers to difficulty in initiating movement, which is more common
in the later stages of PD.
Tremor seen in PD is a slow (4-6 hertz) rest tremor, often initially unilateral.
It is common that the tremor increases with anxiety and affect. Other types
of tremor such as postural and action tremor can also be present.
Rigidity (stiffness) refers to an increased resistance to passive bending and
stretching of the patients arm. If the muscle tone varies it can give rise to

3
what is called the “cogwheel” phenomena with jerky movements during
bending of the limb.
Postural instability and gait disturbances are often present in PD. They are
characterized by slow pace while walking, small shuffling steps, balance
problems and gives an increased risk of falling. Postural instability is more
common in the later stages of PD and is believed to be due to impaired
postural reflexes.
Other motor symptoms
Apart from the motor symptoms that are used in the diagnostic procedure
there are several other common motor features such as impaired articulatory
ability (dysarthria), soft speech resulting from lack of coordination in the
vocal musculature (hypophonia), swallowing difficulties (dysphagia) and
drooling (sialorrhoea). All combined these are referred to as bulbar
functions.
In later stages of the disease motor complications and freezing of gate are
common phenomena. Motor complications are usually a result of long term
treatment and consist of motor fluctuations and dyskinesias. Motor
fluctuations refer to decline in motor performance usually in the wearing off
phase of the medication cycle. Sometimes with disease progression there can
also be off-periods that seem to appear more or less at random [12].
Dyskinesias are involuntary movements that are presented as stereotypic,
choreatic or dystonic movements. They usually appear at peak dose (when
the L-dopa has reach the plateau) and more rarely when the drug levels rise
or fall [12].
Non motor symptoms
Apart from the motor features in PD there are a range of non-motor features
and some of them are common. Non-motor symptoms in PD include
neuropsychiatric symptoms with depression, apathy, hallucinations,
cognitive impairment and dementia, sleep disorders with restless legs, REM-
sleep behavior disorder, excessive daytime somnolence, vivid dreaming and
insomnia, autonomic symptoms with bladder disturbances, orthostatic
hypotension and sweating, sensory symptoms with olfactory disturbance,
pain and visual dysfunction and gastrointestinal symptoms with
constipation. Most of the non-motor features develop after motor onset but
there are a few that can be present years before diagnosis: olfactory
dysfunction, mild dysautonomia as well as mood and sleep disturbances [13].

4
Epidemiology
The prevalence of PD in people over 65 years of age is around 1-2% and
increases from 0.6% in the ages 65-69 to 2.8% in the ages 85-89 [14]. The
cumulative incidence (which can be regarded as the lifetime risk) of PD up to
89 years of age is close to three% [15]. Early onset PD, i.e. clinical signs
developing before the age of 50 constitutes only three-four% of the PD
population [15, 16].
The incidence of PD in high quality studies ranges from 8.4 to 20.0 per 100
000 with a mean of 14.5 per 100 000 (95 % CI, 12.2-17.3) [17]. The mean age
at symptom onset is in the late 60’s and population based studies report age
at diagnosis to around 70 years of age [8, 15]. Most studies have reported
slightly higher prevalence for PD in men, but there are also studies reporting
no differences between men and women (see de Lau et al for review) [18].
Histopathology and etiology
Most cases with PD have an unknown etiology. Both genetic and
environmental factors have been implicated and PD is most likely caused by
an interaction of genetic, aging and environmental factors. Environmental
factors that have been linked to PD are lifestyle, dietary and occupational
exposures, such as increased risk with pesticide exposure or protective effect
of substances such as caffeine and smoking [18].
There are subsets of patients of about 10% that report a positive family
history [18]. Some families show an autosomal dominant inheritance
pattern, others a recessive inheritance pattern. Through the genetic forms of
PD, the hope is to unravel biological processes that are also of importance for
the vast majority with sporadic PD.
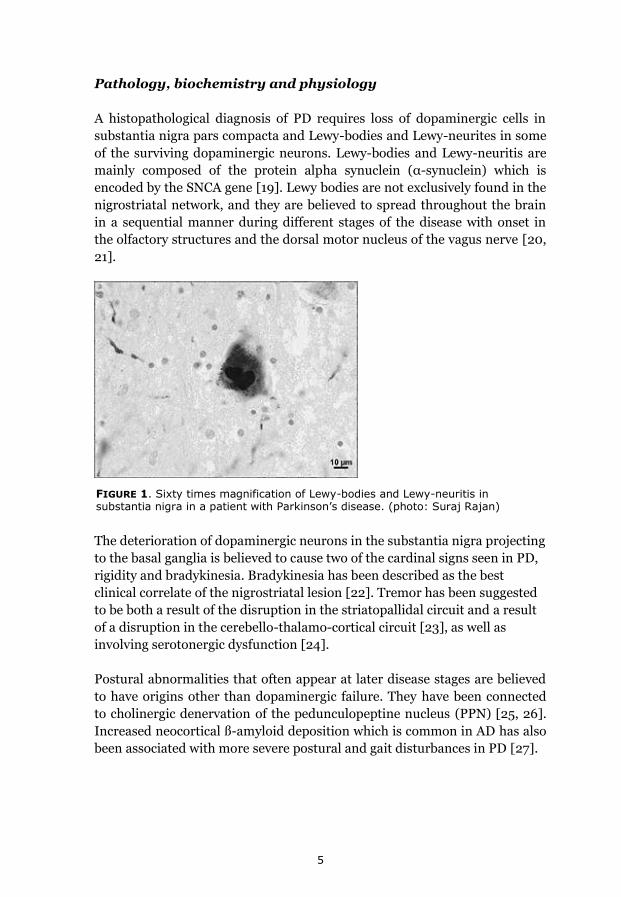
5
FIGURE 1. Sixty times magnification of Lewy-bodies and Lewy-neuritis in substantia nigra in a patient with Parkinson’s disease. (photo: Suraj Rajan)
Pathology, biochemistry and physiology
A histopathological diagnosis of PD requires loss of dopaminergic cells in
substantia nigra pars compacta and Lewy-bodies and Lewy-neurites in some
of the surviving dopaminergic neurons. Lewy-bodies and Lewy-neuritis are
mainly composed of the protein alpha synuclein (α-synuclein) which is
encoded by the SNCA gene [19]. Lewy bodies are not exclusively found in the
nigrostriatal network, and they are believed to spread throughout the brain
in a sequential manner during different stages of the disease with onset in
the olfactory structures and the dorsal motor nucleus of the vagus nerve [20,
21].
The deterioration of dopaminergic neurons in the substantia nigra projecting
to the basal ganglia is believed to cause two of the cardinal signs seen in PD,
rigidity and bradykinesia. Bradykinesia has been described as the best
clinical correlate of the nigrostriatal lesion [22]. Tremor has been suggested
to be both a result of the disruption in the striatopallidal circuit and a result
of a disruption in the cerebello-thalamo-cortical circuit [23], as well as
involving serotonergic dysfunction [24].
Postural abnormalities that often appear at later disease stages are believed
to have origins other than dopaminergic failure. They have been connected
to cholinergic denervation of the pedunculopeptine nucleus (PPN) [25, 26].
Increased neocortical ß-amyloid deposition which is common in AD has also
been associated with more severe postural and gait disturbances in PD [27].

6
Treatment of motor symptoms
Available treatments are mainly focused on reducing motor symptoms. They
have no effect on underlying pathophysiological processes and consequently
do not have curative or disease modifying effects. The most commonly used
treatments are pharmacological interventions with drugs that affect the
dopamine system, so called dopaminergic drugs.
Levodopa
The first drug available for treatment of PD was levodopa; it was developed
in the early 1960s based on the findings of the Swedish Nobel prize winner,
professor Arvid Carlson and early clinical trials by Ehringer and
Hornykiewicz [28, 29]. Dopamine cannot pass through the blood brain
barrier (BBB) but levodopa (L-dopa), a precursor of dopamine, can. L-dopa
is taken up by dopaminergic neurons and decarboxylates into dopamine
presynaptically. When released it binds to both D1 class receptors (including
D1 and D5) and D2 class receptors (including D2, D3 and D4)
postsynaptically. Dopamine that does not bind to postsynaptic receptors is
taken up into the presynaptic cell by the dopamine transporter.
Decarboxylase and COMT inhibitors
To prevent levodopa from being metabolized to dopamine in the periphery
before passing the BBB and entering the brain, levodopa is combined with
decarboxylase inhibitors (carbidopa or benserazide) and sometimes also
with catechol-O-methyl transferase (COMT) inhibitors (entacapone or
tolcapone). The result is that more of the drug enters the brain, and
peripheral side effects are reduced (e.g. nausea, hypotension).
Dopamine agonist and MAO-B inhibitors
Other dopaminergic drugs are dopamine agonists and monoamine oxidase
(MAO) B inhibitors. MAO-B metabolizes dopamine in the dopaminergic
neuron and inhibition of the enzyme increases the availability of dopamine.
Dopamine agonists (DA) act directly on the postsynaptic system. The various
DA used have different receptor profiles. The commonly used non ergot
dopamine agonists, such as pramipexole and ropinirole have a high affinity
for D2 class receptors [30]. DA is today often used as monotherapy in early
phase of PD, mostly in younger patients, in order to reduce/delay motor
complications. Although use of DA early in the disease gives the benefits of
delaying the start of motor fluctuations accompanied with the use of L-dopa
there are other side-effects of DA such as higher risk of hallucinations,
orthostatic hypotension, excess daytime sleepiness, sleep attacks and
impulse control disorder [31].

7
Treatment in later disease stages
In later stages of the disease a portable pump delivering apomorphine
subcutaneously or levodopa delivered intestinally can be used for continuous
dopaminergic stimulation to reduce variation in motor performance (motor
fluctuations and dyskinesias). An antagonist on the glutamate N-methyl-D-
aspartate (NMDA) receptor (amantadine) can also be tried for treatment of
dyskinesias and fluctuations. Neurosurgical interventions with targeted
lesions in the brain in advanced cases with PD have largely been replaced by
Deep Brain Stimulation (DBS). Most DBS operations for PD target the
subthalamic nucleus which has a central role in the motor circuits that are
disturbed in PD.
Cognitive impairment
Cognition can be defined as higher order brain functions that help the
individual to interact with the environment in a successful way. Sometimes
the cognitive functions do not work as intended, due to functional or
structural impairment. This can lead to dysfunction for the individual. These
disturbances can be reversible if caused by stress, medication, depression, or
sleep deprivation. They can also be due to degenerative processes which can
only be treated symptomatically. There are different ways to describe and
measure human cognition. No cognitive tests measures only one cognitive
domain and tests usually tap into different cognitive functions. A poor result
in a single test can be due to damage in various parts of the brain.
Cognitive impairment in PD
The profile of cognitive impairment in PD varies between individuals and is
as heterogenic as the rest of the clinical picture, probably because of the
diverse nature of the underlying pathology. Already in the early stages of the
disease a wide range of cognitive impairments have been demonstrated [6–
8], e.g. in memory, visuospatial function and executive functions. Some
suggest that executive problems are related to the early phase of the disease
due to an altered dopaminergic tone in the frontal cortex. On the other hand
impairment in visuospatial dysfunction and semantic verbal production
display an involvement of temporal and posterior structures. The
visuospatial dysfunction has been suggested to be related to later disease
stages and subsequent development of dementia [32].
Executive functions
Executive functions include a set of abilities that control and regulate other
cognitive functions. It can be described as our ability to plan, perform
abstract reasoning, solve problems, focus despite disturbances and shift

8
focus when appropriate. Executive functions have been linked to distributed
networks with an interaction between prefrontal and subcortical regions.
Cognitive decline in PD has sometimes been described as resembling the
pattern seen in frontal lobe patients with mainly frontally mediated attention
and executive problems [33]. Executive functions are affected both in PD
and PDD and have been suggested to be more affected in PDD than in AD
(the most common dementia disorder)[34].
Memory
Human memory consists of multiple systems. A basic distinction can be
made between short-term or working memory and long-term memory
(LTM). Working memory temporarily hold information while LTM refers to
the ability to store information [35]. Working memory has been shown to be
distinctly different from LTM. Connections between dopamine, aging and
cognition [36], especially working memory processes have been suggested
[37].
LTM can be subdivided into declarative and non-declarative memory. In
turn declarative LTM can be separated into semantic and episodic memory.
Semantic memory refers to a network of associations and concepts of basic
knowledge about the world. Episodic memory refers to information about
personally experience of past events. Different memory processes involved
are encoding, consolidation and retrieval of information (Figure 2.).
Disturbances of any of the three memory components, i.e. encoding,
consolidation or retrieval, can result in memory failure.
The medial temporal lobes are involved in acquisition and retrieval of new
episodic memories [38] . Consolidation of new memories has been linked to
hippocampus and surrounding structures [39].
FIGURE 2. Overview of memory processes

9
Frontally mediated cognitive processes that are engaged in working memory
and executive function are believed to be collaborating in long term memory
as well, especially in free recall [40]. In a test situation episodic memory is
often assessed by asking a person to recall or recognize information learned
at the time. Episodic memory impairment is present in PDD although some
studies claim that it is less severe than in AD [41]. Some suggest that
memory impairment in PD is more related to a frontally mediated retrieval
deficit than to an encoding problem. In PDD there is evidence of recognition
deficiencies as well (see Emre et al for review) [34].
Attention
Attention is a multidimensional function that involves processes that focus,
select, divide, sustain and inhibit behavior. Attention is important for all
cognitive skills and, in tests, especially difficult to separate from working
memory and executive functions. The term attention has been used
interchangeably with executive functions in some prior PD studies [34].
Visuospatial skills
Visuospatial function includes mental imagery and navigation, distance and
depth perception and visuospatial construction. It is the ability to
understand visual representations and their spatial relationships and is
governed by several different pathways originating from parietal cortex
(Occipito-parietal, parieto prefrontal, parieto pre-motor and parieto-medial
temporal) (see Kravitz et al for review) [42]. Both constructional abilities and
visuospatial function without the demands of fine motor control have been
shown more affected in PDD than in AD [34].
Language function
Language can be described as the capacity for acquiring and using complex
systems of communication. Language functions include abilities such as
reading, arithmetic, oral and written word production and comprehension.
Common test for language function are verbal fluency and word
comprehension test. Some studies use tests of verbal fluency as a measure of
language function whereas other includes it to measure executive functions.
Patients with PDD are considered to have less language impairment than
patients with AD [34].
PD-MCI
The concept of Mild Cognitive Impairment (MCI) was developed by Petersen
et al. [43] to detect individuals with an increased risk of developing AD. It is
defined as a transition stage between normal aging and dementia. The
original criterion was focused on memory impairment but it was later
suggested that also patients with impairment in other cognitive domains

10
could be at risk for developing dementia [44]. Studies have applied
Petersen’s MCI criteria on patients with PD with various results. To create
conformity between studies and clinicians within the field of PD a task force
commissioned by the Movement Disorder Society adapted the MCI criteria
to fit the specific cognitive profile seen in PD [45] and constructed guidelines
to assist in the MCI classification. The PD-MCI criteria are based on a
combination of literature review and expert consensus.
PDD
Dementia is defined as a progressive, irreversible deterioration of cognitive
function. The pathologies behind dementia disorders can be different types
of neurodegeneration and vascular lesions. The definition of dementia in the
Diagnostic and Statistical Manual of Mental Disorders IV is an overall
decline in intellectual function including difficulties with language, simple
calculations, planning and judgment, and motor skills. Furthermore a loss of
memory that is severe enough to interfere with activities of daily living that is
not due to physical decline. PDD require onset of motor symptoms at least
one year before the onset of dementia [34]. The one year rule is to
differentiate between PDD and DLB.
Epidemiology of cognitive impairment in PD
Five to seven percent of adults over the age of 60 are demented [46]. Of the
dementia population three-four% have PDD [47]. The proportion of patients
with PDD in PD populations varies between 28% and 90% [48] depending
on selection of participants and criteria used.
In community based studies of PDD the annual incidence rate has been
around 100 per 1000 person years [49–51] which corresponds to around
10% of the PD population developing dementia each year. The annual
incidence rate of dementia in population based studies with newly diagnosed
patients with PD has been 38.7 per 1000 person-years (95% CI, 23.9-59.3)]
[32]. See table 1 for longitudinal studies of newly diagnosed patients with
PD.
The proportion of patients with MCI in studies of newly diagnosed patients
with PD has ranged between 19 and 36% [6–8, 52, 53] with a higher
proportion in community-based samples. In adults older than 65 years in the
general population the proportion of MCI has ranged between three and 19%
[54].
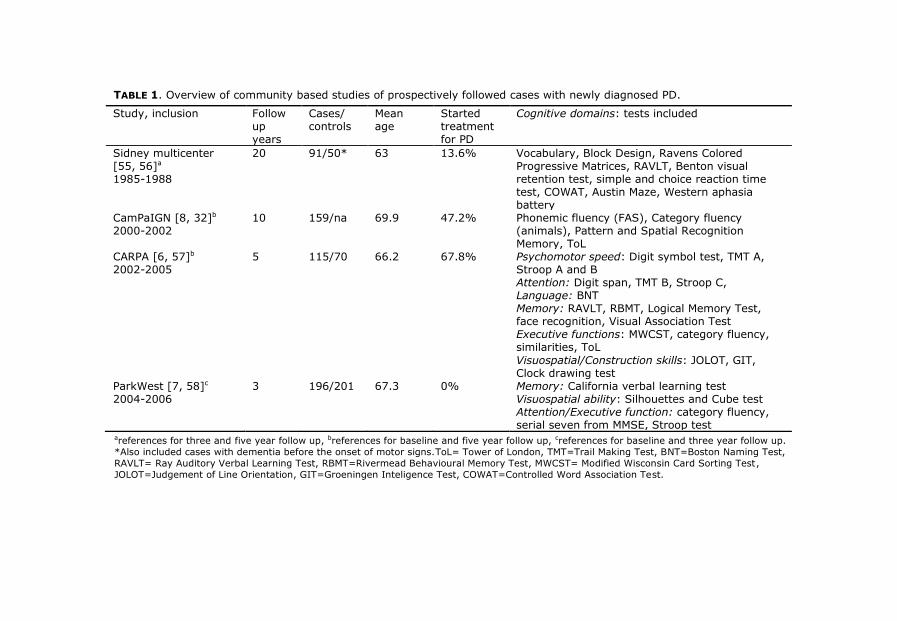
11
TABLE 1. Overview of community based studies of prospectively followed cases with newly diagnosed PD.
Study, inclusion Follow up years
Cases/ controls
Mean age
Started treatment for PD
Cognitive domains: tests included
Sidney multicenter [55, 56]a
1985-1988
20 91/50* 63 13.6% Vocabulary, Block Design, Ravens Colored Progressive Matrices, RAVLT, Benton visual retention test, simple and choice reaction time test, COWAT, Austin Maze, Western aphasia battery
CamPaIGN [8, 32]b
2000-2002 10 159/na 69.9 47.2% Phonemic fluency (FAS), Category fluency
(animals), Pattern and Spatial Recognition Memory, ToL
CARPA [6, 57]b 2002-2005
5 115/70
66.2 67.8% Psychomotor speed: Digit symbol test, TMT A, Stroop A and B Attention: Digit span, TMT B, Stroop C, Language: BNT Memory: RAVLT, RBMT, Logical Memory Test, face recognition, Visual Association Test Executive functions: MWCST, category fluency, similarities, ToL
Visuospatial/Construction skills: JOLOT, GIT, Clock drawing test
ParkWest [7, 58]c
2004-2006 3 196/201 67.3 0% Memory: California verbal learning test
Visuospatial ability: Silhouettes and Cube test Attention/Executive function: category fluency, serial seven from MMSE, Stroop test
areferences for three and five year follow up, breferences for baseline and five year follow up, creferences for baseline and three year follow up.
*Also included cases with dementia before the onset of motor signs.ToL= Tower of London, TMT=Trail Making Test, BNT=Boston Naming Test,
RAVLT= Ray Auditory Verbal Learning Test, RBMT=Rivermead Behavioural Memory Test, MWCST= Modified Wisconsin Card Sorting Test,
JOLOT=Judgement of Line Orientation, GIT=Groeningen Inteligence Test, COWAT=Controlled Word Association Test.

12
Risk factors for cognitive impairment in PD
Identification of clinical factors that predict development of dementia are
important for clinical practice and disease management [59]. Age [55],
depressive symptoms, specific neuropsychological impairments [60], specific
motor impairment, male sex, fewer years of education, visual hallucination,
REM sleep disorder and orthostatic hypotension have all been associated
with an increased risk of PDD. For MCI older age, motor disease severity,
non-tremor dominant motor phenotype and fewer years of education have
been reported as associated factors.
Older age is one of the primary clinical features predicting PDD [61, 62].
Early presence of frontal executive problems has been pointed out to be a
predictor for development of PDD [63]. Recent research has started to
evaluate this and connects the development of PDD also to posterior
cognitive problems such as visuospatial, verbal function and episodic
memory (see Kehagia, et al. for review) [64]. Preliminary results suggest that
PD-MCI with posterior cognitive deficits predicts a shorter time to PDD [45].
Patients with predominantly postural instability and gait difficulties (PIGD)
have been suggested to have a faster rate of cognitive decline [65]. However,
following patients with early PD for up to 10 years have not rendered in the
same results [66]. Some mean that it is change from tremor dominant
subtype to PIGD dominant subtype that is a predictor for developing PDD
[67].
Neuropathology of cognitive impairment in PDD
The neuropathology underlying cognitive decline and PDD is heterogeneous
and studies exploring pathological correlates of cognitive impairment in PD
have rendered conflicting results [68].
Cortical Lewy bodies and Lewy neuritis have been shown to be the most
significant correlate of dementia in PD [69]. α-synuclein pathology in the
parahippocampal gyrus and anterior cingulate gyrus is more pronounced in
PDD than in PD [70, 71]. Some individuals have pronounced α-synuclein
burden without being demented, which suggests that α-synuclein burden by
itself is not sufficient for dementia. Other factors such as cognitive reserve or
brain plasticity might determine the cognitive performance in relation to the
α-synuclein burden (See Irvin et al for review) [59]. Furthermore, some
patients with PDD express only small amounts of α-synuclein. Cholinergic
deficits have been seen in patients with PDD that have less pronounced
cortical and limbic Lewy bodies and Lewy neuritis.

13
Some studies claim that less than 10% of PDD cases have coexisting AD [72,
73], whereas other have reported proportions as high as 30-40% [69, 74, 75].
For example low CSF ß amyloid levels, have been linked to development of
PDD [76]. Differences between demented and non-demented patients have
also been found in the distribution of neurofibrillary tau pathology (also
common in Alzheimer disease). The spread for non-demented patients was
restricted to the entorhinal areas whereas neurofibrillary tangles had spread
to the rest of the limbic system, lateral temporal areas and beyond in
patients with PDD [21].
The contribution of vascular lesions to PDD is not well studied but some
indications of higher degrees of vascular lesions in PDD than PD without
dementia have been found [77]. A cross-sectional study combining the
cortical Lewy, ß-amyloid and tau stages have shown that this combination
perfectly discriminates between demented and none demented PD [76].
Genes and cognition in PD
Familial associations to the development of PDD have been reported [78].
However, not much is known about how genes contribute to cognitive
impairment and PDD and only a few of the findings have been linked to
biological processes likely to be involved in cognitive impairment and/or
PDD
The genetic polymorphism Catechol-O-methyl-transferase (COMT) gene
(Val158Met) has been suggested to have an impact on executive functions
through its link to the dopamine system but has not been connected to PDD
[32]. Monoamine oxidase (MAO) has also been suggested to be a candidate
gene linked to executive heterogeneity in PD.
Some of the familial forms of PD are linked to PDD. The gene SNCA coding
for α-synuclein on chromosome four can be multiplied and cause increases
in the concentration of α-synuclein. Triplicated cases are more often than
duplicated cases associated with cognitive decline and dementia with a more
rapid progression. A polymorphism of the DYRK1A gene has been connected
to the PDD and LBD [79] through effects on a kinase that phosphorylates α-
synuclein and amyloid precursor protein. Mutations in the ATP13A2 gene
results in a rare genetic variant that can cause young onset parkinsonism
(Kutor-Rakeb syndrome: PARK 9). These patients become demented and the
predominant pathology is atrophy with iron accumulation in basal ganglia
[80]. The MAPT H1/H1 genotype seems to have a strong influence on
cognitive functions, especially posterior cortical cognitive impairments and
give a higher risk of developing PDD [81].

14
Treatment of cognitive impairment and other non-motor
features in PD
Treatment for non-motor and non-dopaminergic symptoms in PD has
started to be evaluated because of the recent focus on the disabling effect of
the non-dopaminergic and non-motor features of the disease.
Cholinesterase inhibitors have been shown to have positive effect for
cognitive dysfunction and PDD, but also for behavioural disturbances and
activities of daily living in patients with PDD [82]. Rivastigmine is the
cholinesterase inhibitor with strongest evidence as an effective treatment of
PDD [83]. Special characteristics of responders are hard to find but those
with visual hallucinations [84] and those with elevated levels of
homocysteine [85] seem to respond especially well.
For psychotic symptoms in PD a gradual reduction of antiparkinson
medication is recommended [86]. If neuroleptic treatment is needed,
atypical forms (e.g. clozapine) shows the best effect/side effect profile for PD
patients [87].
Treating depression in patients with PD is complicated. A recent meta-
analysis concluded that there is insufficient evidence to suggest selective
serotonin reuptake inhibitors (SSRI), serotonin and norepinephrine
reuptake inhibitors (SNRIs), pramipexole or pergolide for treatment of
depression in patients with PD [88]. According to the same meta-analysis
the treatment with the best effect on reducing depressive symptoms in PD
was tricyclic antidepressants (TCAs).
Dopaminergic medication has been suggested to have both detrimental and
positive effects on cognitive functions. One explanation for this is that the
relationship between dopamine levels and cognitive performance is believed
to follow an inverted U-shaped curve, where both low and high levels of
dopamine cause impaired cognitive performance [89]. Individuals with
initially low levels of dopamine are believed to improve performance with
intake of dopaminergic drugs while patients with higher levels decline in
performance due to excessive levels of dopamine. Different functions are
mediated by different brain networks and affected differently by
dopaminergic depletion. In early PD for example, the loss of dopaminergic
neurons in striatum is most prominent in putamen and dorsal caudate, both
involved in the motor and dorsolateral circuit. The ventral striatum, involved
in limbic and orbitofrontal circuits is mostly intact.

15
High levels of dopamine agonists and levodopa are believed to have a
negative effect on reversal learning, decision-making and impulse control
[64]. The dopamine agonist pramipexole, has been suggested to have a more
harmful effect than pergolide [64]. Some studies have suggested that short
term use of the dopamine agonist pramipexole might cause decline in short
term verbal memory, attentional-executive functions and verbal fluency
[90]. A slight cognitive decline in semantic verbal fluency, executive
functions, verbal learning and memory has been shown shortly after
subthalamic deep brain stimulation [91].
Cognitive motor relationship
The association of motor control and cognitive function has been studied in
children [92], patients with brain injury [93] and patients with
neurodegenerative disorders [94]. One goal in linking motor and cognitive
function is to find which aspects of cognitive and motor functions are
processed by a given area or brain network [95]. Associations between
cognitive and motor function have been found in the elderly and in patients
with neurological disease. These associations can be unspecific reflecting co-
existence of common age-related syndromes or reflecting a more widespread
pathology affecting both motor and cognitive structures. They can also be
specific as in the association of specific cognitive and motor functions being
governed by the same neural networks.
Apart from the relation between PIGD subtype and PDD [67, 96], other
cognitive motor relationships have been suggested in PD. Early findings
reported bradykinesia and rigidity [4] to be associated with PDD and the
severity of bradykinesia has been connected with visuospatial reasoning and
psychomotor speed [97]. Others found no motor cognitive relationships [98,
99]. The results are inconclusive due to differences in the selection of
participants and variables tested.
Rationale of the thesis
Cognitive impairment in PD has severe consequences with increased
mortality, nursing home placement and caregiver stress [100]. A better
characterization and understanding of cognition in the early phase of the
disease is particularly important as this is the phase when early intervention
with potential neuroprotective drugs or other therapies is likely to be most
effective. Studying motor and cognitive relationships in early stages of the
disease is important to be able to estimate the risk of early development of
dementia or other type of cognitive problems with regard to motor function.
Also finding relationships between specific motor and cognitive functions

16
may provide new ideas about the underlying pathophysiological processes
for the motor and non-motor functions in PD.
Most studies on cognitive function in PD have been retrospective, cross
sectional, including a mixture of incident and prevalent cases or been biased
towards younger cases. Good descriptions of cognition in the early phase of
PD in unselected study populations followed prospectively were almost
completely lacking [8] when this study was initiated [15]. Prospective studies
in large cohorts of well-defined newly diagnosed patients with PD assessed
with extensive neuropsychological protocols can help connect cognitive
motor associations to specific functions rather than global cognitive and
motor decline and perhaps dissociate predictive clinical factors from
associated factors.

17
Aims
The aim of this thesis was to the improve characterization and
understanding of cognition in early phases of PD, to investigate clinical
determinants of cognitive decline and dementia and to explore what aspects
of cognitive function are connected to different motor functions. Further
aims were to investigate the variability of cognitive performance in PD in
patients followed prospectively from time of diagnosis and during the
following five years.
1. To describe the character and predictors of cognitive dysfunction in a
population based cohort with drug naive newly diagnosed PD. (Paper I)
2. To explore the relationship between cognition and motor dysfunction in a
population based cohort with drug naive newly diagnosed PD. (Paper II)
3. To explore if motor and cognitive variables associated at baseline change
in parallel after one year. (Paper III)
4. To report the magnitude of cognitive change one year after diagnosis and
investigate if different types of dopaminergic medication have an impact on
the results. (Paper III)
5. To explore the five-year course of cognitive functions in a prospectively
followed cohort of patients with PD and search for predictors for PDD (Paper
IV).
5. To explore the difference in the evolution of motor function in patients
that develop PDD compared to those who do not develop PDD (Paper IV).
18
Materials and methods
This thesis is based on data from the NYPUM-project (new parkinsonism in
Umeå), a population-based study of idiopathic parkinsonism addressing
etiological, diagnostic and prognostic factors. The patients were recruited
from southern part of Västerbotten County in northern Sweden with a
catchment area of 142 000 inhabitants.
Patients were included from January 1st 2004 to April 30th 2009 and
classified in to different forms of parkinsonism (PSP, MSA, CBG and LBD)
according to established clinical criteria. Cases were investigated extensively
at the time of presentation and repeatedly during follow up which was
between 4.5-9 years except for those who died. To avoid selection bias cases
were identified prospectively during the inclusion period and through many
sources to make case identification as complete as possible. A total of 185
patients with idiopathic parkinsonism were identified.
FIGURE 3. Map of the investigation area (Pantzare Information AB, Luleå,
Sweden)

19
Study population
The study populations participating in the studies of this thesis are presented
below. The differences in participants between the studies are due to sample
selection and change in diagnosis during follow up. Paper I is based on the
first four years of inclusion whereas paper II, III and IV include the whole
sample. Figure 4 (a, b, c, d) describes the flow chart of participants in each
paper.
Thirty age and sex matched controls based on the 50 first patients included
in the study were recruited. The controls were recruited by advertisements in
the local newspaper or among friends and family of the PD participants.
Requirements for controls were that they needed to be healthy with no
neurological disorders and normal neurological examination, and have a
normal FP-CIT scan.
Non-participation
About 20% (Paper I 21%, Paper II-IV 19%) of the patients declined to
participate in the neuropsychological assessment at baseline. They were
significantly older (79 vs. 69 years, p<0.001), had various medical
conditions, such as blindness, deafness, severe cardiac disease and had
higher scores on the UPDRS part III (35 vs. 26, p<0.001) and scored worse
on the MMSE (27.5 vs. 28.7; p=0.04).
There were also a substantial amount of patients that did not participate at
follow up. This loss was bigger for the neuropsychological evaluation than for
the study as a whole. Patients that did not participate in neuropsychological
testing at follow-up were older, had fewer years of education and higher
scores on the UPDRS. Furthermore, they performed worse on all
neuropsychological tests at baseline except a test of language.
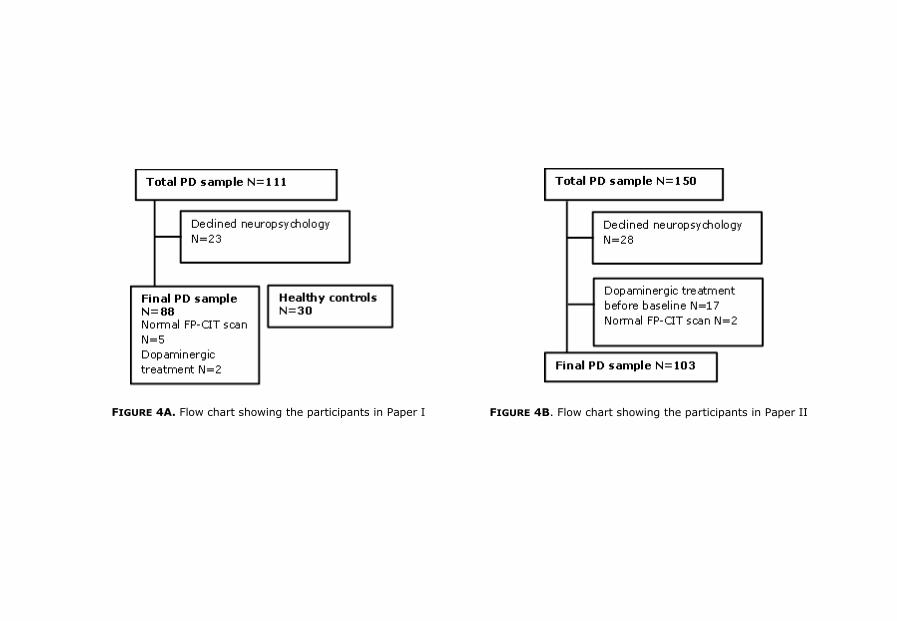
20
FIGURE 4A. Flow chart showing the participants in Paper I FIGURE 4B. Flow chart showing the participants in Paper II
21
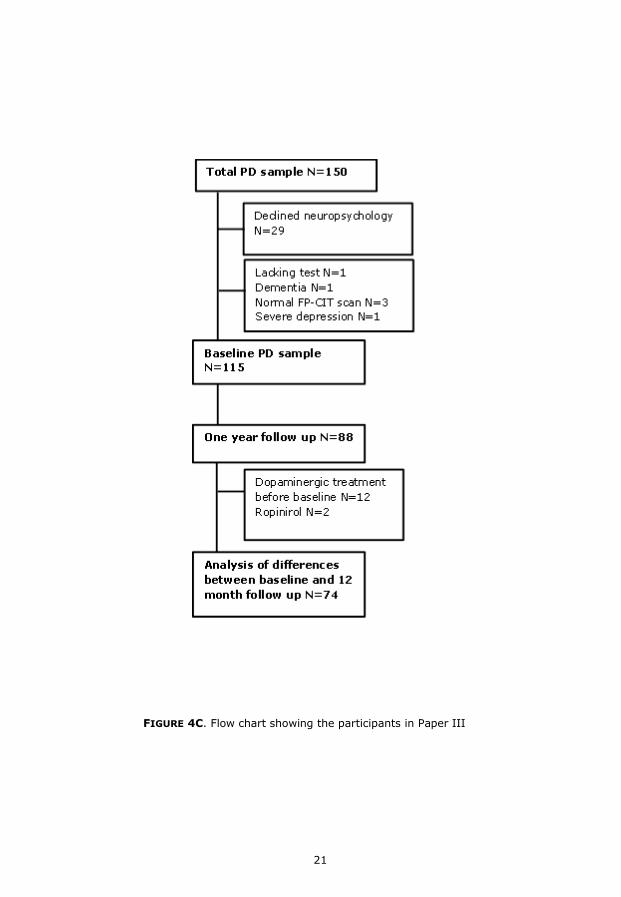
FIGURE 4C. Flow chart showing the participants in Paper III
22
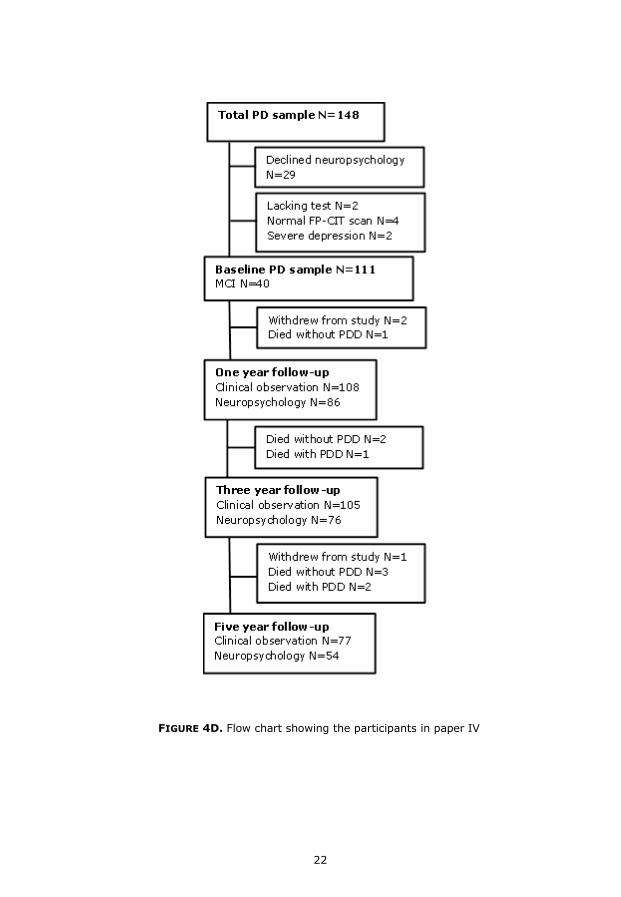
FIGURE 4D. Flow chart showing the participants in paper IV

23
Assessment tools
Global cognitive ability was measured with the Mini-mental State
Examination (MMSE) [101]. Depression was assessed with the Montgomery
and Åsberg Depression Rating Scale (MADRS); participants with scores over
eight were considered to have a mild depression and severe depression if
scores were 18 or over [102]. Dopamine transporter (DAT) Single-photon
emission computed tomography (SPECT)-imaging using 123I-Ioflupane ([123I]
FP-CIT) was performed within 1-2 months following inclusion. Levodopa
Equivalent Dose (LED) was calculated based on the conversion factors
suggested by Tomlinson et al [103].
Assessment of motor function
The severity of parkinsonism was measured by the Unified Parkinson’s
Disease Rating Scale (UPDRS) [9] and the Hoehn and Yahr staging.
Variables of the different motor-scores were calculated from the UPDRS III
[104]: the sum of UPDRS III items 20 and 21 for tremor, item 22 for rigidity,
the sum of items 24, 25, 26, and 31 for bradykinesia, the sum of items 27, 28,
29, and 30 for the posture and gait score (arising from chair, posture, gait
and postural stability), and the sum of items 18 and 19 for the bulbar score
(speech and facial expression) [105].
Previously published divisions of motor subtype were used to divide patients
into groups based on predominant motor feature: postural instability and
gait disturbances (PIGD), tremor or indeterminate phenotypes [106]. A
patient was classified as having tremor dominant PD if mean tremor score
divided by mean PIGD score was >=1.5, PIGD subtype if mean tremor score
divided by mean PIGD score was <=1.0 and indeterminate subtype if mean
tremor score divided by mean PIGD score was between >1.0 and <1.5.
Assessment of cognitive function
Cognitive functions were thoroughly investigated at the time of inclusion
(baseline) and after one, three, and five years. The patients were tested at
their optimal motor stage. The tools used were neuropsychological tests of
verbal and non-verbal episodic memory, working memory, attention,
psychomotor function, visuospatial function and executive function. Table 2
shows the total battery of neuropsychological tests with an explanation of
what cognitive domain each test was intended to measure and how the test
was executed. The tests are listed in the order of presentation.

24
The tests were chosen to assess important cognitive functions within a
reasonable amount of time. Since motor function is affected in PD, tests that
are not affected by motor function were chosen when possible. All tests
included in the battery are frequently used both in research and in clinical
practice and have good validity and reliability. Most of the tests have age and
education adjusted norms.
Instruments were chosen to minimise the effect of repeated testing, e.g.
parallel versions. Tests that did not have alternate forms or could be affected
by close repeated measurements were excluded from the 12-month follow-up
[107].
The assessments were performed by trained research assistants with
backgrounds in psychology and neuroscience supervised by a consultant in
clinical neuropsychology. To assure that the assessments would be coherent
within and between testers they were instructed to perform the assessment
in accordance with the standardized protocol of the different tests. They
practiced before they performed the first testing accompanied by a senior
tester during one or more testing and performed an assessment on a patient
during supervision with feedback. The test protocols were checked for errors.
Patients were encouraged to participate in all neuropsychological tests. For
some participants this was impossible due to tiredness or technical issues.
The tests with most missing cases were the WCST (n=15), logical memory
(n=9) and logical memory delayed (n=11). Missing data varied between one
and six for the other neuropsychological variables at baseline. Cases were
omitted if they had missing values on most tests. Otherwise we conducted
pairwise deletion of the data.

25
Neuropsychological test Cognitive domain Execution
1. Finger tapping test Psychomotor speed Tap left and right index
finger during 10 s
2. Free and Cued Selective
Reminding test
(FCSRT)[108]
Episodic memory Cued and free recall
after encoding 16 words
x3
3. Mental Control from
WMS[109]
Conceptual tracking Counting back and forth
and reciting alphabet
4. Trail Making Test A
(TMT A)[110]
Attention/psychomotor
speed
Drawing lines between
numbers
5. Trail Making Test B
(TMT B)[110]
Attention/psychomotor
speed
Drawing lines
alternating between
numbers & letters
6. Benton Judgement of Line
Orientation (JOLOT)[111]
Visuospatial function Judging the slope of two
lines
7. Digit span forward and
backward[109]
Working memory/
attention
Repeating numbers
forward and backward
8. FCSRT delayed recall Episodic memory Cued and free recall
after a delay
9. Boston Naming Test (BNT) Verbal naming Picture naming
10. Brief Visuospatial Memory
Test [112]
Visuospatial memory Drawing 6 figures after
10 min encoding x3
11. Associative Learning Test
(WMS)
Episodic memory Remembering word
pairs x3
12. Logical memory, immediate
recall (WMS)
Episodic memory Repeating short story
directly after encoding
13. Controlled Oral Word
Association Test
(COWAT)[113], phonemic
Verbal function/executive
function
Naming as many words
beginning with the letter
F,A or S, during one
minute
14. COWAT, categories Verbal function/executive
function
Naming as many words
from a given category:
animals, colours or
fruits
15. BVMT delayed recall Visuospatial memory Drawing encoded figures
after a 25 minute delay
16. Logical memory, delayed
recall
Episodic memory Reciting short story
after delay
17. Wisconsin Card Sorting
Test-computer version
Executive function Matching cards based on
unknown rules
TABLE 2. Total battery of neuropsychological tests in order of presentation.

26
Diagnosis
PD
PD was diagnosed according to the United Kingdom Parkinson’s Disease
Society Brain Bank (UK PDSBB) criteria for definite PD, requiring at least
three supportive criteria. Cases with 1-2 supportive criteria were classified as
probable PD. All cases were checked against diagnostic criteria for MSA
[114], PSP [115],CBD [116]and DLB [117]. In cases that fulfilled the
diagnostic criteria for multiple syndromes we classified the patient with the
atypical diagnosis. Definite PD and probable PD are included in all studies.
Since the diagnosis changed at follow up for a small number of patients, the
latest diagnosis available at the time of statistical analysis for each paper was
used.
BOX 1. UK PDSBB clinical diagnostic criteria for PD
Step 1. Diagnosis of parkinsonism Bradykinesia and at least one of the following:
a. Muscular rigidity
b. 4-6 hz tremor
c. Postural instability
Step 2. Exclusion criteria for PD
History of repeated strokes and head
injuries
History of definite encephalitis
Oculogyric crises
Neuroleptic treatment at onset of
symptoms
More than one affected relative
Sustained remission
Strictly unilateral features after three
years
Supranuclear gaze palsy
Cerebellar signs
Early severe autonomic
involvement**
Early severe dementia with
disturbances of memory and
language
Babinski sign
Presence of cerebral tumor or
communicating hydrocephalus
Negative response to large dose of
levodopa
MTPT exposure
Step 3. Supportive prospective
criteria for PDD. Three or more
required for PD definite
Unilateral onset
Rest tremor
Progressive disorder
Persistent asymmetry affecting the
side of onset most
Excellent response to levodopa
Severe levodopa induced chorea
Levodopa response for 5 years or
more
Clinical course of 10 years or more
*We excluded the criterion ”more than one affected relative” since it is now known that PD can have a genetic background **Defined as symptomatic orthostatic blood pressure fall or presence of urinary incontinence within 12 months of baseline visit.

27
Cognitive impairment
In Paper I a cognitive domain was considered to be impaired if more than
half of single test results within that domain were below cutoff level. Lezak et
al have presented a classification system for ability levels based on a
statistically defined range of scores [118]. Average performance is according
to this classification: mean +/-0.6 SD low averages, -0.6 to -1.3 SD
borderline, -1.3 to – 2.0 SD impaired. We chose -1.5 SD as a cut-off level
score for lowered test performance, which is commonly accepted in clinical
practice.
Paper I Paper IV
Cognitive domain Neuropsychological test Neuropsychological test
Episodic memory FCSRT free and cued recall
Logical Memory and Paired
Associative Learning from WMS
(healthy controls)
BVMT
FCSRT free recall
BVMT total
BVMT delayed
Working memory Digit span from WAIS III
Digit Span backwards
TMT B
Attention TMT A and B
Executive functioning WCST
Mental control from WMS
WCST total errors
WCST preservative errors
Animal fluency
Visuospatial
functioning
The Benton Judgment of Line
orientation
The Benton Judgment of Line
orientation
Verbal function Boston naming test
Controlled Oral Word Association
(FAS and categories)
Boston naming test
TABLE 3. Neuropsychological measures used for classification of cognitive
impairment (Paper I) and MCI according to MDS Task force criteria (Paper IV).
FCST=Free and cued selective reminding test, WMS=Weschler memory scale,
BVMT=Brief Visuospatial Memory Test,TMT Trail Making Test, WCST=Wisconsin Card
Sorting Test, BNT=Boston Naming Test

28
MCI
To classify patients as MCI in Paper IV the MDS Task Force criteria were
used [45]. The task force guidelines require a diagnosis of PD based on the
UK PD Brain Bank criteria [119], gradual decline in cognitive ability reported
by either patient, informant or observed by clinician, cognitive deficits on
either formal neuropsychological testing or a scale of global cognitive
abilities, and cognitive deficits that is not sufficient to interfere significantly
with functional independence, no dementia and a minimum of two
neuropsychological tests with 1-2 SD below the mean value of established
norms or a control group, and subjective cognitive complaints. As in Paper I,
1.5 SD below established norms were used to detect impaired domains in
Paper IV.
To capture subjective cognitive complaints from decline from premorbid
level, information on self-perceived cognitive decline or information from
family or friends were gathered through a semi structured short
questionnaire given to the patient when enrolling in the study. The
questionnaire addressed the patient and/or family members experience of
cognitive decline, a question of how the patient function cognitively was
asked before the neuropsychological assessment and information regarding
self-perceived memory and concentration from the Parkinson’s Disease
Questionnaire 39 (PDQ-39) [120].
Dementia
A diagnosis of PDD was made according to published criteria through a
consensus between three of the authors in paper IV (MED, LF and UE) [34].
The PDD diagnosis requires a diagnosis of PD a minimum of one year prior
to the onset of dementia and cognitive deficiency severe enough to affect
Activities of Daily Living (ADL). All available information were used
including medical files, neuropsychological testing, temporal cognitive
decline measured by repeated neuropsychological testing or MMSE, and
interview by the study nurse and information from family members.
Ethics
Written, informed consent was obtained from all participants and healthy
controls. The study was approved by the Regional Ethics Committee at Umeå
University.

29
Statistics
All statistical analyzes were two tailed and p-values under 0.05 were
considered significant for most analyzes. The Statistical Package for the
Social Sciences (SPSS) versions 15, 19 and 21 and the Predictive Analytics
Software (PASW) Statistics 17.0 were used for statistical analysis. Depending
on the distribution of scores parametric and non-parametric tests was used
where appropriate. The residuals of the regression models were explored to
see if they fulfilled the assumptions of normality, linearity and
homoscedasticity.
Empirical studies
Paper I
Patients with newly diagnosed PD were compared to healthy controls on the
total battery of cognitive tests. Patients and controls were also classified as
being impaired in different cognitive domains. Neuropsychological
investigations were performed in 88 of 111 classified as PD. Only two had
received pharmacological treatment for PD before the neuropsychological
testing (low doses for a few weeks). Two individuals did not complete enough
tests to make it possible to judge impairment in different domains. Table 2
shows the tests that were used for each domain. Those impaired in one or
more domains were compared to those with normal function. Furthermore,
those with two or more impaired domains were compared to the rest.
Characteristics of this sample are presented in table 4.
Patients
n=88
mean (SD, range)
Healthy controls
n=30
mean (SD, range)
P-value
Sex (female/male) 39/49 14/16 0.825
Age, years 68.1 (9.3) 68.2 (6.6) 0.979
Education, years 9.9 (4.1) 11.5 (3.5) 0.055
MMSE 28.7 (1.4,24-30) 29.1 (0.8,28-30) 0.074
Mild depression a n (%) 14 (16%) 0 <0.001
UPDRS III 23.8 (9.7,5-48) 0
Dopaminergic medication 2 0
FP-CIT scan normal 5 30
TABLE 4. Characteristic of the population in Paper I
MMSE= Mini Mental State Examination, MADRS=Montgomery and Åsbergs Rating
Scale;UPDRS= Unified Parkinson’s Disease Rating Scale, DAT= dopamine transporter. aMADSR 9-17.Reprinted from Paper I with kind permission from the copyright holders.

30
TABLE 5. Characteristic of the sample in Paper II.
PIGD=Postural impairment and gait disturbances, MMSE= Mini Mental State Examination,
MADRS=Montgomery and Åsberg Rating Scale; UPDRS= Unified Parkinson’s Disease Rating
Scale. Reprinted from Paper II with kind permission from the copyright holders.
Independent two-tailed t-tests were used to analyze differences in
demographics, clinical characteristics and cognitive tests raw scores between
patients and controls. For adjusted p-value linear regressions were
performed with age, gender, years of education and psychomotor function as
covariates for each cognitive variable. Patients being impaired in one or
more cognitive domain were compared to those with intact cognition on
demographic and clinical variables analyzed with Student’s t-test or Mann-
Whitney U-test. Binary logistic regression was performed between patients
with two or more cognitive domains impaired for the purpose of exploring
predictors for cognitive impairments. Demographic and clinical variables
that significantly differed between the groups in independent analyses were
included as covariates in the model: years of education, disease duration,
bradykinesia, facial expression, rigidity and total UPDRS III subscore.
Paper II
The associations between cognitive and motor functions were assessed.
Furthermore, a comparison between PIGD/Tremor/Indeterminate
phenotypes was made for baseline cognitive variables. Neuropsychological
assessments were performed in 122 of 150 patients that fulfilled the
diagnostic criteria for PD. Seventeen patients that started their dopaminergic
treatment before performing the neuropsychological investigation and two
with a normal FP-CIT scan were not included in the study. Thus 103 patients
were included (Table 5).
Total
n=103
mean (SD)
PIGD
n=55
mean (SD)
Tremor n=35
mean (SD)
Indeterminate
n=13
mean (SD)
P-
value
Sex (f/m) 41/62 20/35 15/20 6/7 0.731
Age (years) 68.4 (9.2) 69.5 (8.7) 63.0 (9.9) 69.3 (9.4) 0.492
Years of
education
9.9 (4.1) 10.4 (4.9) 9.4 (2.9) 9.3 (3.2) 0.894
Disease
duration
(months)
22 (23) 20 (16) 27 (33) 19 (11) 0.866
MMSE 28.8 (1.3) 28.6 (1.4) 29.0 (1.1) 25.5 (1.1) 0.496
UPDRS III 24.8 (10.6) 27.5 (10.5) 21.1 (9.9) 23.5 (10.3) 0.020

31
Differences in demographic, clinical and cognitive characteristics between
the PIGD, tremor and indeterminate group were analyzed with two-tailed
ANOVA, Kruskal-Wallis, chi-square or Fisher’s exact test when appropriate.
The nonparametric Spearman’s rho was used to explore correlations
between demographic, motor and neuropsychological variables. Multiple
linear regression analysis was performed to explore if the relationships
found in the correlation analysis between motor and cognitive variables were
unique or affected by other variables. To meet the assumptions of normality
and reduce skewing and outlier influence, some variables were transformed
with square root transformation or logarithm transformation. Most variables
were approximately normally distributed after transformation.
Paper III
The persistence of associations between motor score and cognitive functions
were investigated together with the magnitude of cognitive change between
baseline and follow-up for the total group and for patients receiving different
dopaminergic medication (pramipexole ± levodopa and without dopamine
agonists). Data from baseline and 12 month assessment were used.
Neuropsychological assessments were performed in 122 of 150 patients that
fulfilled the diagnostic criteria for PD. Six patients were excluded due to: not
enough tests performed (n=1), having dementia at baseline (n=1), normal
SPECT FP CIT scan (n=3) or severe depression according to MADRS (n=1).
Of the remaining 115 patients 88 (76%) participated in the 12 month
neuropsychological follow up. Twelve patients had started their medication
for parkinsonism before the baseline evaluation and were therefore excluded
from the analysis of the different effects of dopaminergic medication. Five
patients did not receive either levodopa or dopamine agonist at follow-up.
Two patients that received ropinirole instead of pramipexole were excluded
from the medication analysis (Table 6).
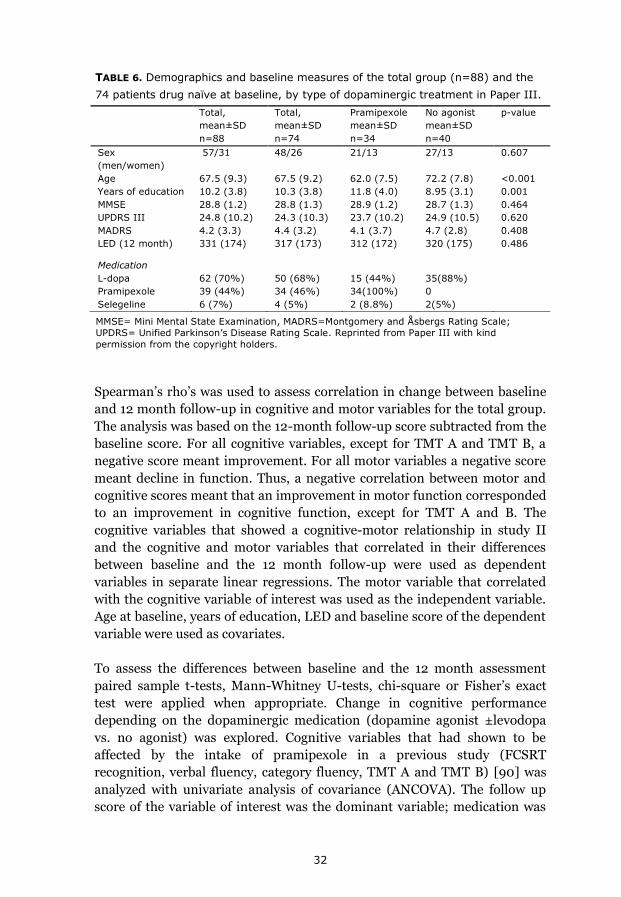
32
Spearman’s rho’s was used to assess correlation in change between baseline
and 12 month follow-up in cognitive and motor variables for the total group.
The analysis was based on the 12-month follow-up score subtracted from the
baseline score. For all cognitive variables, except for TMT A and TMT B, a
negative score meant improvement. For all motor variables a negative score
meant decline in function. Thus, a negative correlation between motor and
cognitive scores meant that an improvement in motor function corresponded
to an improvement in cognitive function, except for TMT A and B. The
cognitive variables that showed a cognitive-motor relationship in study II
and the cognitive and motor variables that correlated in their differences
between baseline and the 12 month follow-up were used as dependent
variables in separate linear regressions. The motor variable that correlated
with the cognitive variable of interest was used as the independent variable.
Age at baseline, years of education, LED and baseline score of the dependent
variable were used as covariates.
To assess the differences between baseline and the 12 month assessment
paired sample t-tests, Mann-Whitney U-tests, chi-square or Fisher’s exact
test were applied when appropriate. Change in cognitive performance
depending on the dopaminergic medication (dopamine agonist ±levodopa
vs. no agonist) was explored. Cognitive variables that had shown to be
affected by the intake of pramipexole in a previous study (FCSRT
recognition, verbal fluency, category fluency, TMT A and TMT B) [90] was
analyzed with univariate analysis of covariance (ANCOVA). The follow up
score of the variable of interest was the dominant variable; medication was
Total,
mean±SD
n=88
Total,
mean±SD
n=74
Pramipexole
mean±SD
n=34
No agonist
mean±SD
n=40
p-value
Sex
(men/women)
57/31 48/26 21/13 27/13 0.607
Age 67.5 (9.3) 67.5 (9.2) 62.0 (7.5) 72.2 (7.8) <0.001
Years of education 10.2 (3.8) 10.3 (3.8) 11.8 (4.0) 8.95 (3.1) 0.001
MMSE 28.8 (1.2) 28.8 (1.3) 28.9 (1.2) 28.7 (1.3) 0.464
UPDRS III 24.8 (10.2) 24.3 (10.3) 23.7 (10.2) 24.9 (10.5) 0.620
MADRS 4.2 (3.3) 4.4 (3.2) 4.1 (3.7) 4.7 (2.8) 0.408
LED (12 month) 331 (174) 317 (173) 312 (172) 320 (175) 0.486
Medication
L-dopa 62 (70%) 50 (68%) 15 (44%) 35(88%)
Pramipexole 39 (44%) 34 (46%) 34(100%) 0
Selegeline 6 (7%) 4 (5%) 2 (8.8%) 2(5%)
TABLE 6. Demographics and baseline measures of the total group (n=88) and the
74 patients drug naïve at baseline, by type of dopaminergic treatment in Paper III.
MMSE= Mini Mental State Examination, MADRS=Montgomery and Åsbergs Rating Scale;
UPDRS= Unified Parkinson’s Disease Rating Scale. Reprinted from Paper III with kind
permission from the copyright holders.
33
TABLE 7. Baseline characteristics for the total group and for MCI+ and MCI- in
Paper IV.
UPDRS=Unified Parkinson’s Disease Rating Scale, MADRS=Montgomery and Asberg
Depression Rating Scale (MADRS), MMSE= Mini Mental State Examination
entered as between subject factors and the baseline score of the variable of
investigation, age, years of education and LED were entered as covariates.
Interaction effects between medication and the covariates were checked
before the full factorial models were executed. Further, the proportion that
declined in variables with significant differences in the different medication
groups were calculated and a Chi square test was performed to evaluate if the
groups differed (two or more words less at follow-up vs. those with
improvement or smaller change). U-shaped associations between motor and
cognitive variables were checked separately for each medication group in a
regression model with cognitive variables as dependent variable, squared
change in motor score (motor²) as independent variable and change in
motor score as covariate.
Paper IV
Neuropsychological investigations were performed in 119 of 150 patients that
fulfilled the diagnostic criteria for PD. Eight were excluded, four had normal
presynaptic dopamine uptake, two could not perform tests and two had
severe depression. Thus 111 patients with PD were included. There were 17
(15 %) participants that had started their dopaminergic treatment before the
baseline assessment and these had been on dopaminergic medication for an
average of three months. Baseline, one year, three and five year follow up
data for the patients were included. (Table 7)
Total MCI-
(n=71)
MCI+
(n=40)
p-value
Age 68.7 (9.4) 67.4 (9.4) 71.0 (9.1) 0.048
Years of education 10.1 (4.1) 10.6 (4.4) 9.3 (3.5) 0.088
Sex (m/f) 68/43 38/33 30/10 0.026
Duration (months) 22.2 (22) 22 (22) 21 (15) 0.673
MMSE 28.7 (1.3) 29.1 (0.9) 28.0 (1.6) <0.001
MADRS 4.3 (3.4) 3.8 (3.5) 5.2 (3.1) 0.053
Started dopaminergic
treatment
15% 13% 20% 0.304
UPDRS III 25.4 (10.9) 23.4 (10.3) 28.9 (11.0) 0.012

34
Proportions and the annual incidence of dementia with a 95% confidence
interval (CI) were calculated. The time of dementia were estimated as the
midpoint between assessments that dementia was diagnosed. Person-years
at risk were calculated as the total follow up time until dementia or study end
for those not developing dementia. Incidence was calculated as the number
of cases with dementia divided by the total number of person years at risk
during follow up. To study the association of age, education, PIGD sub-type
and MCI to the development of dementia a Cox-proportional hazard model
was performed.
Patients with MCI (MCI+) that converted to dementia were compared to
patients with MCI who did not convert on baseline-demographics or
cognitive and -motor measurements. Student’s t-test, Mann-Whitney U-tests
and chi-square tests were used when appropriate.
Population average regression models for correlated data i.e. Generalized
Estimation Equations (GEE) were applied to investigate the association of
dementia to the evolvement of motor scores. GEE was chosen because it can
model non-normal distributed data and take non-independent
measurements such as repeated measurements or clustered data into
consideration. It is also flexible with unbalanced and missing data [121]. An
autoregressive model with Tweedie log link function was used. The
dependent variable was the motor score (posture/gait, bradykinesia, tremor,
rigidity or bulbar) in five separate models. The factors were set to dementia
status (dementia converters and non-converters) and follow-up (baseline,
one year, three years and five years). No dementia and baseline testing were
set to be the redundant parameters to which the other parameters were
compared. Covariates were age at baseline and LED. The significance level
was set to p<0.01 because of five dependent measures being tested.
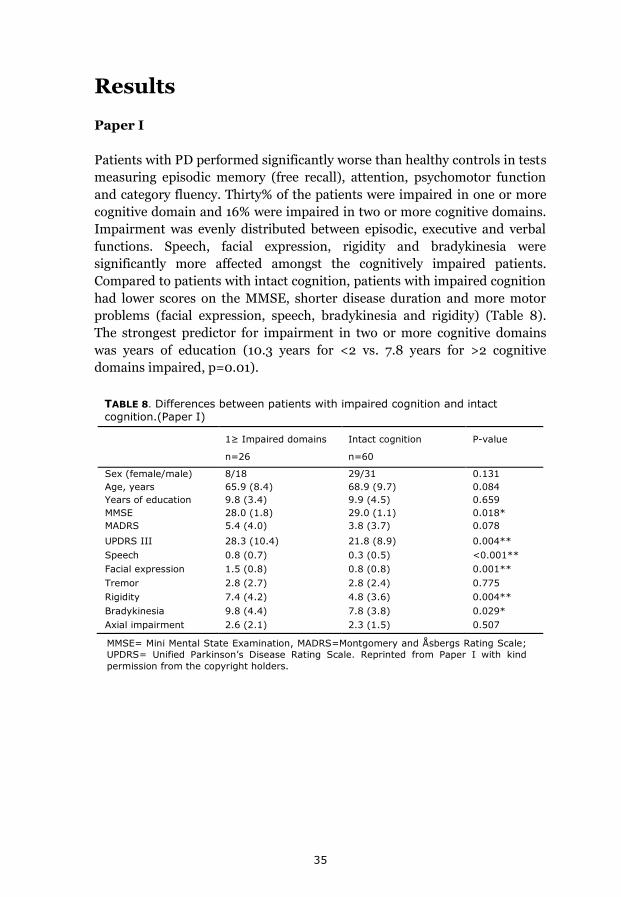
35
TABLE 8. Differences between patients with impaired cognition and intact cognition.(Paper I)
MMSE= Mini Mental State Examination, MADRS=Montgomery and Åsbergs Rating Scale;
UPDRS= Unified Parkinson’s Disease Rating Scale. Reprinted from Paper I with kind
permission from the copyright holders.
Results
Paper I
Patients with PD performed significantly worse than healthy controls in tests
measuring episodic memory (free recall), attention, psychomotor function
and category fluency. Thirty% of the patients were impaired in one or more
cognitive domain and 16% were impaired in two or more cognitive domains.
Impairment was evenly distributed between episodic, executive and verbal
functions. Speech, facial expression, rigidity and bradykinesia were
significantly more affected amongst the cognitively impaired patients.
Compared to patients with intact cognition, patients with impaired cognition
had lower scores on the MMSE, shorter disease duration and more motor
problems (facial expression, speech, bradykinesia and rigidity) (Table 8).
The strongest predictor for impairment in two or more cognitive domains
was years of education (10.3 years for <2 vs. 7.8 years for >2 cognitive
domains impaired, p=0.01).
1≥ Impaired domains
n=26
Intact cognition
n=60
P-value
Sex (female/male) 8/18 29/31 0.131
Age, years 65.9 (8.4) 68.9 (9.7) 0.084
Years of education 9.8 (3.4) 9.9 (4.5) 0.659
MMSE 28.0 (1.8) 29.0 (1.1) 0.018*
MADRS 5.4 (4.0) 3.8 (3.7) 0.078
UPDRS III 28.3 (10.4) 21.8 (8.9) 0.004**
Speech 0.8 (0.7) 0.3 (0.5) <0.001**
Facial expression 1.5 (0.8) 0.8 (0.8) 0.001**
Tremor 2.8 (2.7) 2.8 (2.4) 0.775
Rigidity 7.4 (4.2) 4.8 (3.6) 0.004**
Bradykinesia 9.8 (4.4) 7.8 (3.8) 0.029*
Axial impairment 2.6 (2.1) 2.3 (1.5) 0.507
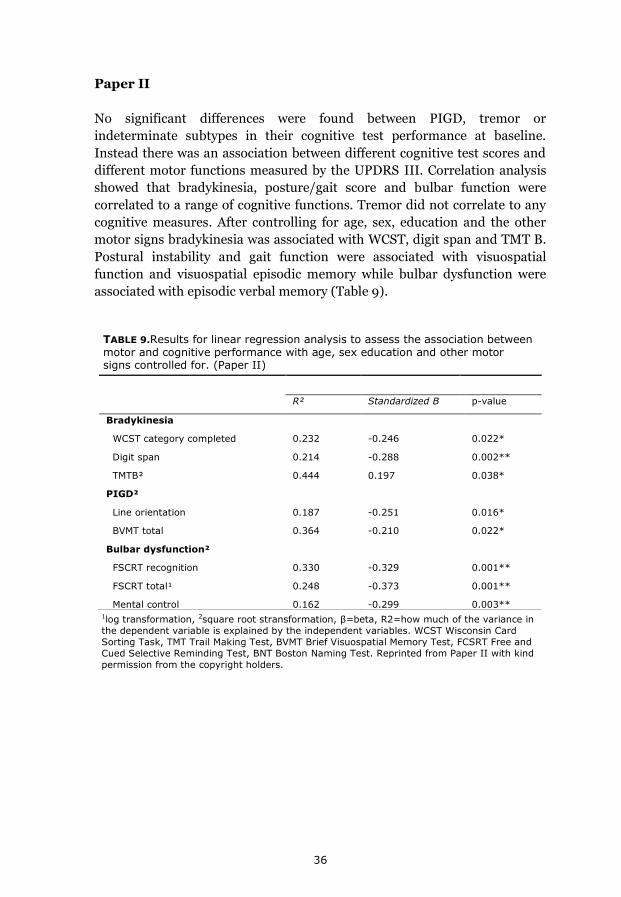
36
TABLE 9.Results for linear regression analysis to assess the association between motor and cognitive performance with age, sex education and other motor signs controlled for. (Paper II)
1log transformation, 2square root stransformation, β=beta, R2=how much of the variance in
the dependent variable is explained by the independent variables. WCST Wisconsin Card
Sorting Task, TMT Trail Making Test, BVMT Brief Visuospatial Memory Test, FCSRT Free and
Cued Selective Reminding Test, BNT Boston Naming Test. Reprinted from Paper II with kind
permission from the copyright holders.
Paper II
No significant differences were found between PIGD, tremor or
indeterminate subtypes in their cognitive test performance at baseline.
Instead there was an association between different cognitive test scores and
different motor functions measured by the UPDRS III. Correlation analysis
showed that bradykinesia, posture/gait score and bulbar function were
correlated to a range of cognitive functions. Tremor did not correlate to any
cognitive measures. After controlling for age, sex, education and the other
motor signs bradykinesia was associated with WCST, digit span and TMT B.
Postural instability and gait function were associated with visuospatial
function and visuospatial episodic memory while bulbar dysfunction were
associated with episodic verbal memory (Table 9).
R² Standardized Β p-value
Bradykinesia
WCST category completed 0.232 -0.246 0.022*
Digit span 0.214 -0.288 0.002**
TMTB² 0.444 0.197 0.038*
PIGD²
Line orientation 0.187 -0.251 0.016*
BVMT total 0.364 -0.210 0.022*
Bulbar dysfunction²
FSCRT recognition 0.330 -0.329 0.001**
FSCRT total¹ 0.248 -0.373 0.001**
Mental control 0.162 -0.299 0.003**
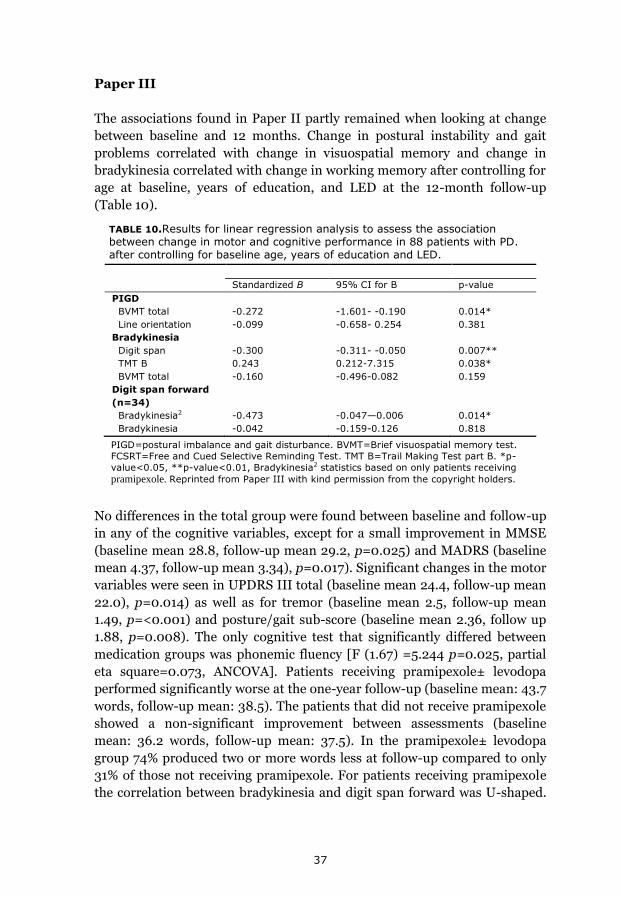
37
Paper III
The associations found in Paper II partly remained when looking at change
between baseline and 12 months. Change in postural instability and gait
problems correlated with change in visuospatial memory and change in
bradykinesia correlated with change in working memory after controlling for
age at baseline, years of education, and LED at the 12-month follow-up
(Table 10).
No differences in the total group were found between baseline and follow-up
in any of the cognitive variables, except for a small improvement in MMSE
(baseline mean 28.8, follow-up mean 29.2, p=0.025) and MADRS (baseline
mean 4.37, follow-up mean 3.34), p=0.017). Significant changes in the motor
variables were seen in UPDRS III total (baseline mean 24.4, follow-up mean
22.0), p=0.014) as well as for tremor (baseline mean 2.5, follow-up mean
1.49, p=<0.001) and posture/gait sub-score (baseline mean 2.36, follow up
1.88, p=0.008). The only cognitive test that significantly differed between
medication groups was phonemic fluency [F (1.67) =5.244 p=0.025, partial
eta square=0.073, ANCOVA]. Patients receiving pramipexole± levodopa
performed significantly worse at the one-year follow-up (baseline mean: 43.7
words, follow-up mean: 38.5). The patients that did not receive pramipexole
showed a non-significant improvement between assessments (baseline
mean: 36.2 words, follow-up mean: 37.5). In the pramipexole± levodopa
group 74% produced two or more words less at follow-up compared to only
31% of those not receiving pramipexole. For patients receiving pramipexole
the correlation between bradykinesia and digit span forward was U-shaped.
Standardized B 95% CI for B p-value
PIGD
BVMT total -0.272 -1.601- -0.190 0.014*
Line orientation -0.099 -0.658- 0.254 0.381
Bradykinesia
Digit span -0.300 -0.311- -0.050 0.007**
TMT B 0.243 0.212-7.315 0.038*
BVMT total -0.160 -0.496-0.082 0.159
Digit span forward
(n=34)
Bradykinesia2 -0.473 -0.047—0.006 0.014*
Bradykinesia -0.042 -0.159-0.126 0.818
TABLE 10.Results for linear regression analysis to assess the association between change in motor and cognitive performance in 88 patients with PD. after controlling for baseline age, years of education and LED.
PIGD=postural imbalance and gait disturbance. BVMT=Brief visuospatial memory test.
FCSRT=Free and Cued Selective Reminding Test. TMT B=Trail Making Test part B. *p-
value<0.05, **p-value<0.01, Bradykinesia2 statistics based on only patients receiving
pramipexole. Reprinted from Paper III with kind permission from the copyright holders.

38
That is, patients with less bradykinesia at follow-up either improved or
deteriorated in their working memory performance.
Paper IV
Twenty-seven patients with PD (24%) developed dementia within five years,
corresponding to an annual incidence of 56.0 per 1000 person-years (95%
CI, 37.2-80.3). Forty patients (36%) had MCI at baseline of which 22 (55%)
transitioned to dementia, corresponding to an annual incidence of 148 per
1000 person-years (95% CI, 94.9-215.0) in patients with MCI. Five (7%)
patients with normal cognition at diagnosis transitioned to dementia
corresponding to an annual incidence of 15 per 1000 person-years (95% CI,
4.9-34.6). Seven patients with MCI at baseline reversed back to normal
cognition and three patients shifted between MCI and normal cognition
between assessments. The sensitivity for MCI to predict dementia in early
stages of PD was 81%, the specificity 79%, the PPV 55% and the NPV 92%.
MCI converters performed significantly poorer than non-converters on
several cognitive measures at baseline: MMSE, FCSRT free recall, BVMT,
mental control, line orientation, animal fluency, and TMT B.
MCI (B=-2.486, Exp(B)=0.083, CI for Exp(B) 0.031-0.225, p<0.001) and
baseline age (B=1.077, Exp(B)=2.936, CI for Exp(B) 1.23-6.99, p=0.003)
were both significant predictors for dementia. Education and motor subtype
at baseline were not significant predictors of dementia. None of the patients
with the tremor subtype at the latest visit had developed dementia
There were significant negative changes for posture and gait impairment
(Wald Chi-Square 23.862 p<0.001), bulbar dysfunction (Wald Chi-Square
20.627, p<0.001), and bradykinesia (Wald Chi-Square 54.963, p<0.001)
during the follow-up period. Dementia converters had more bradykinesia
(Wald Chi-Square 25.035, p<0.001), rigidity (Wald Chi-Square 20.376,
p<0.001), bulbar dysfunction (Wald Chi-Square 12.557, p<0.001) as well as
posture and gait impairment (Wald Chi-Square 16.231, p<0.001). There was
an interaction effect between follow-up and dementia for posture and gait
(Wald Chi-Square 14.523, p=0.002); i.e. patients developing PDD differed
from PD patients that did not develop dementia in their evolvement of
posture and gait impairment (Figure 6a). The interaction effect for follow up
and dementia were close to significant for bradykinesia (figure 6b) between
baseline and three years (p=0.02), and for rigidity (figure 6c) between
baseline and five years (p=0.01) but not significant for the total follow up
period (rigidity: Wald Chi-Square 8.214, p=0.042, bradykinesia: Wald Chi-
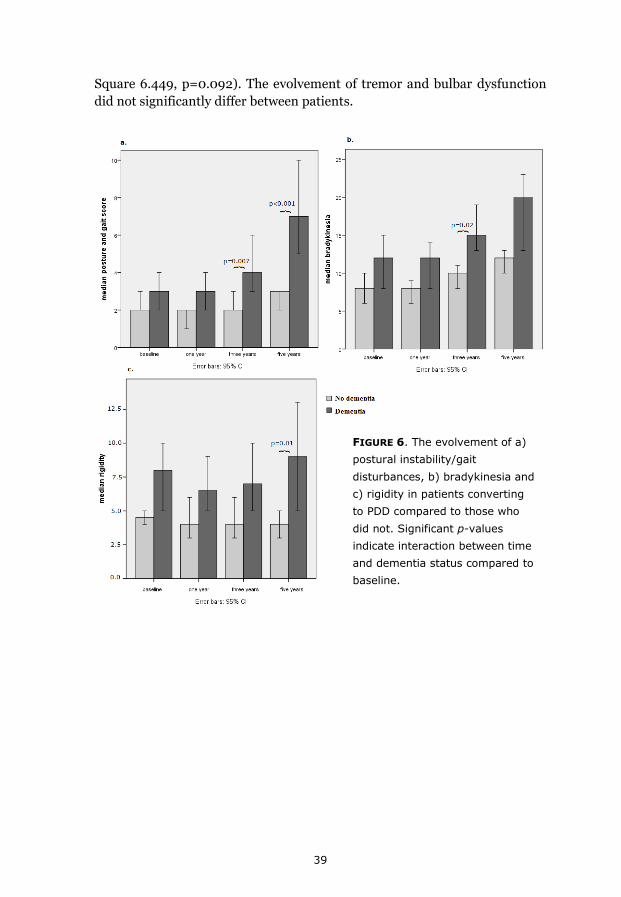
39
Square 6.449, p=0.092). The evolvement of tremor and bulbar dysfunction
did not significantly differ between patients.
FIGURE 6. The evolvement of a)
postural instability/gait
disturbances, b) bradykinesia and
c) rigidity in patients converting
to PDD compared to those who
did not. Significant p-values
indicate interaction between time
and dementia status compared to
baseline.

40
Discussion
Main findings
The main focus of this thesis is cognitive function in early PD and its
association to motor disturbances. In line with previous studies [6–8, 98] we
found, in comparison with healthy controls, that cognitive impairment is
common in newly diagnosed patients with PD and that a range of functions
are affected already at diagnosis. Patients with cognitive impairment at
baseline had significantly more motor problems according to the UPDRS III,
than those with intact cognition, and education was an independent
predictor for impairment in two or more cognitive domains at baseline but
did not predict dementia within five years.
Associations were found between bradykinesia and worse performance on
tasks measuring working memory and executive functions as well as between
posture/gait dysfunction and lower scores on tests measuring visuospatial
function and visuospatial memory. Some of these associations persisted also
after one year.
MCI at the time of diagnosis was found in 36% when diagnosed by recently
published MDS Task Force criteria (Paper IV). Thirty% were classified as
having cognitive impairment when using slightly different criteria (Paper I).
One fourth of the patients developed PDD within five years after diagnosis.
Age at diagnosis and MCI according to MDS Task Force criteria were
significant predictors of dementia. There were 25 % of those classified as
having MCI at baseline that converted to normal cognition or reversed back
and forth over the 1.5 SD limit between assessments.
Patients with MCI converting to PDD within five years had worse
performance on several cognitive tests at baseline: visuospatial function,
semantic fluency, episodic memory, conceptual tracking and mental
flexibility; but not in tests measuring executive function, working memory,
verbal phonemic fluency and language function.
There were no differences in baseline measures of cognitive function
between PIGD, tremor and indeterminate phenotype. PIGD phenotype at
baseline did not predict dementia within five years. None developed
dementia among those having a tremor dominant phenotype. Worsening of
posture/gait was more pronounced in patients developing dementia than in
patients that did not. A similar pattern was seen in bradykinesia and rigidity
but not for tremor and bulbar dysfunction.

41
Patients treated with the dopamine agonist pramipexole performed
significantly worse on a measure of verbal fluency at the one year follow up
compared with patients not treated with the drug.
Impaired cognitive functions
Compared with healthy controls, patients with PD performed significantly
worse already at the time of diagnosis on tests measuring attention, semantic
fluency and episodic memory (free recall). Attention/working memory and
episodic memory were the domains mostly affected followed by executive,
visuospatial and verbal function. This shows a diversity of cognitive decline
within patients with PD, in line with previous studies [6].This could be
expected considering the heterogeneity of the disease.
A diverse pattern of differences in baseline test performance between
patients with MCI converting to dementia compared with those not
converting to dementia was shown in Paper IV. WCST, a measure of
executive function, together with working memory, phonemic fluency and
verbal naming did not significantly differ between MCI converters compared
with non-converters at baseline. Interestingly many of the same tests did not
differ between patients and controls in Paper I.
Previous research has connected executive impairment [122, 123], and
memory [123] as predictors of PDD. Neuropsychological measures with a
more posterior cortical basis such as pentagon copying and semantic fluency
have been suggested to be predictors of dementia [124]. A recent study
comparing MCI converting to dementia within two years with those that did
not convert proposed that frontal lobe associated performance together with
atrophy in both frontostriatal areas and cholinergic structures, and not
visuospatial function, were predictors of dementia in PD-MCI [125].
Tests that significantly differed between MCI converters and non-converters
in Paper IV were tests measuring visuospatial function and semantic fluency
in line with Williams-Gray et al. [66], but also tests measuring episodic
memory free and delayed recall, conceptual tracking, and mental flexibility
where the latter tests are believed to be more frontally mediated.
Together this shows a diverse picture of cognitive function associated with
PDD with a range of different cognitive functions that are different between
MCI converters and non-converters. We need to look at both the striatal
network in combination with a more widespread cortical network in the
search for predictors for PDD.

42
Proportions of MCI and Dementia
MCI
The proportion of patients that were classified as being cognitively impaired
differed somewhat between Paper I (30%) and Paper IV (36%). Different
classification procedures with stricter criteria and removal of TMT A and
TMT B from the classification battery in Paper I and the use of MDS Task
Force criteria in paper IV were the reasons for this. The MDS Task Force
criteria had not yet been published when paper I was assembled. The
differences of results within the same study population depending on which
classification is used show that MCI classification relies on the tests included
and the cut-off values used and emphasizes the value of joint criteria for PD-
MCI, in research and in clinical practice.
Proportions of MCI in newly diagnosed cases with PD published prior to the
MDS Task Force criteria have ranged between 19 to 36% [6–8, 52, 53, 126].
The proportions of patients classified as MCI according to the MDS Task
Force criteria in newly diagnosed PD have been 25 to 35% [57, 58, 127]. This
makes the proportion of 36% in Paper IV at the higher range.
Dementia
In cross-sectional studies the prevalence of dementia has been around 30%
and increases over 20 years to 76% [128]. The majority of studies have been
based on prevalent PD cases with different disease duration. Following
patients from disease onset gives a more accurate estimate of dementia rates
and give a better base for determining the dementia risk. Studies on
prospectively studied newly diagnosed cases with PD are rare, and the
proportion of patients developing PDD in such studies within five years has
been 17% [32, 129] [incidence 38.7 per 1000 person-years (95% CI, 23.9-
59.3)] [32]. This makes the proportion of patients developing dementia
(24.3%) and the incidence rate (56 per 1000 person-years) slightly higher
than reported in previous studies on newly diagnosed cases, but longer
follow up times have rendered incidence rates similar to our study [54.7 per
1000 person-years (95% CI, 35.4-74.1)] [66].
Higher dementia and MCI rates
There could be several reasons for the high dementia and MCI rates in this
cohort. Cognition was mapped extensively and was more thoroughly
investigated than in most other longitudinal studies [58, 60]. Our patients
were older [57] and they had less education [57, 58]. Furthermore, the

43
annual incidence of PD reported from the NYPUM cohort is among the
highest reported in the literature [15]. This could be due to meticulous case
finding strategies that might also influence the proportion of patients with
cognitive impairment.
Predictive value of MDS-Task Force MCI criteria for PDD
The MDS Task Force MCI criteria were found to have a relatively good
sensitivity and specificity where 22 of 40 (55%) patients with MCI developed
dementia within 5 years. There are two longitudinal studies on newly
diagnosed patients with PD using MDS Task Force guidelines for MCI [57,
58]. A three year follow up where ten out of 37 (27%) patients with MCI
converted to dementia [58] and a five-year follow-up where 11 out of 43
(26%) converted [57]. The three-year study had an annual incidence rate of
dementia of 98.9 per 1000 person-years in patients with MCI at diagnosis.
After three years there were 11 out 40 patients with MCI at baseline that had
converted to dementia in our study and only one of 71 of those with normal
cognition.
Half of the patients that were classified as having MCI at baseline did not
develop dementia within five years. With prolonged follow-up the PPV might
increase as more patients with MCI may develop dementia, the NPV will
most likely decrease as more patients classified as having normal cognition
at baseline will probably also convert to dementia.
During follow-up 25% that were initially classified as MCI at baseline
reversed to normal cognitive function or shifted between normal cognition
and MCI between follow-up. In those cases the MCI at baseline might be due
to reversible causes, such as nervousness, depression or sleep deprivation,
rather than neurodegenerative. In some cases the cognitive improvement
could also be due to the intake of dopaminergic medication [130].
Previous studies in the general population have shown a reversion rate from
MCI to normal cognition between 13-41% [131–133]. A recently published
study using MDS Task Force criteria reported a reversion rate from MCI to
normal cognition to around 20% [58]. A study from Norway suggests that
patients with PD classified as MCI both at baseline and after 12 months have
a higher proportion of MCI converting to dementia as well as a lower
proportion of MCI reversing back to normal cognition than for those having
MCI at baseline or at the 12-month follow-up [58].

44
Education, age, disease duration and gender aspects
In Paper I shorter length of education was a predictor for having more than
two cognitive domains impaired. Less education did not predict the
development of dementia. Previous longitudinal studies on non PD
participants have found that short education does not increase the risk of
developing dementia [134, 135] but Kryscio et al. [134] found that education
significantly predicted transition from normal cognition to MCI.
Our finding of shorter disease duration among those with impaired cognitive
function in Paper I suggests an aggressive PD phenotype with cognitive
decline early in the disease. However in Paper IV we found no significant
differences in disease duration between those with MCI at baseline
compared to the rest. Disease duration did not differ between those
developing dementia compared with those who did not. This is in line with a
prospective population-based study that through separating general age
from age at motor onset showed that patients with PD have a similar
dementia age regardless of when they first are presented with motor
symptoms [136].
The only significant gender difference found were the difference between
how many were classified as MCI according to the MDS Task Force criteria
where 75% of those with MCI were male. The reason for this is hard to grasp
since this has not been shown in previous studies. There could be gender
differences in whether to agree to participate in the neuropsychological
testing if one experiences cognitive decline. One explanation could be that
female patients with PD have been suggested to more often be presented
with the tremor dominant phenotype [137]. However, there were no
significant differences in motor subtypes between male and female
participants in the present study.
Cognitive motor relationship
A pattern of motor cognitive associations already in early stages of PD could
be depicted from the connection found between motor and cognitive
measures, and the different motor and cognitive variables associated with
MCI and dementia. Bradykinesia was associated with worse working
memory performance and executive functions whereas postural instability
and gait disturbances were associated with visuospatial memory and
visuospatial function. Tremor showed no association with any cognitive
measures either at baseline or in the change over time. Bradykinesia, rigidity
and postural and gait disturbances were all more severe in those with MCI

45
and those developing PDD. Furthermore, there was a more pronounced
worsening of motor functioning over time in those developing dementia.
Some cross-sectional studies have investigated the cognitive motor
relationship in PD [4, 97, 98, 138]. Most of the earlier studies have been
rather small and included prevalent cases with various disease duration that
have received dopaminergic treatment of various duration. It can be
problematic to demonstrate motor cognitive relationships in medicated
patients since dopaminergic medication has shown to affect motor and
cognitive abilities differently. Correlation between the UPDRS motor score
and a range of neuropsychological measures, but not visuospatial function
has been reported [139], although a recent study using objective measures of
motor dysfunction connected visuospatial function to postural instability in
PD [94].
The association of bradykinesia with impaired working memory
and executive function
Both bradykinesia and decline in executive functions and attention/working
memory are common in PD. Bradykinesia [22], working memory [37] and
executive functions have been linked to the frontostriatal brain network and
further linked to nigrostriatal dysfunction. The association of bradykinesia
with executive dysfunction and attention/working memory dysfunction
found in Paper II supports the possibility of joint underlying networks.
Patients classified as having cognitive impairment (Paper I and Paper IV)
had more severe bradykinesia at baseline which suggests dopaminergic
involvement in MCI in early stages of PD. A recent cross-sectional functional
magnetic resonance imaging (fMRI) study of early PD linked cognitive
impairment in PD to frontostriatal dysfunction by showing under
recruitment in dorsal caudate nucleus and bilateral anterior cingulate while
performing an updating task in patients with PD-MCI [140].
Patients that developed PDD had more bradykinesia and rigidity than those
not developing dementia. This also indicates early frontostriatal
dopaminergic involvement in PDD.
The association of posture and gait disturbances with impaired
visuospatial function and visuospatial memory
Significant associations between postural and gait scores and visuospatial
memory and visuospatial function were found and further confirmed in the

46
association between change in posture and gait score and visuospatial
memory in Paper III.
PIGD/tremor and indeterminate subtype did not significantly differ in any
cognitive tests at diagnosis. The evolvement of postural instability and gait
scores differed between those developing dementia compared with those
who did not, with a significantly more pronounced decline in those
developing dementia. We showed in line with previous results that PIGD
subtype at the time of diagnosis did not predict the development of dementia
[66]. Together this shows that PIGD phenotype does not seem to be a
predictor of cognitive impairment in early stages of PD or a predictor of
developing dementia. It rather seems to be a parallel process with decline in
visuospatial function and memory and the development of PDD. In the
general population poor gait and cognition have been connected [141].
Dementia and falls often coexist and gait impairments have been related to
the severity of cognitive impairments [142].
Previous studies have shown that PIGD phenotype at diagnosis predicts
worse outcome in PD [66, 143]. PIGD has also been shown more common
among patients with worse cognitive performance [144] and has been
identified as a risk factor for dementia [96]. Both previous studies [96, 144]
included cases in various disease stages that had started their dopaminergic
medication before the first assessment. In line with our results is a study by
Alves et al (2006), which suggested that changing from tremor to PIGD
subtype is a risk factor of dementia and that having a persistent tremor
phenotype was protective of dementia [67].
A recent study found a connection between increased neocortical ß-amyloid
deposit and higher PIGD features in PDD which could possibly be one
underlying factor explaining the association of postural instability and gait
disturbances with impairment in visuospatial function and memory and later
development of dementia [27]. A specific association of gait dysfunction,
cognitive impairment and reduced cholinergic activity has been described
with several lines of evidence (see Ambioni et al. for review) [145].
Other explanations could be the association of degeneration of the
pedunculopontine nucleus (PPN) and the lateral pontine tegmentum area
(rich in acetylcholine) in gait disturbances [146]. Impaired cholinergic
integrity of the PPN has been associated with frequent falls in PD [147]

47
The effect of dopaminergic medication
Study III showed that there were some differences in change in cognitive
functions over one year (phonemic fluency) and the association of the change
between motor and cognitive functions (digit span forward and
bradykinesia) in patients receiving the dopamine agonist pramipexole
compared to patients who did not. Since we did not compare different
agonist we cannot tell whether our finding is a specific effect for pramipexole
or an effect that could also be found in other DA. Pramipexole is believed to
have a high affinity for D3 receptors [148] which could be connected to both
findings. A possible explanation for the decline in verbal fluency for patients
receiving DA could be that D3 auto receptors have been shown to contribute
to the presynaptic regulation of tonically released dopamine which leads to a
moderate inhibitory action on locomotion [148]. If this is also the case for
cognitive function, this could result in an inhibitory effect on word
production.
The U-shaped association for change in bradykinesia and working memory
could possibly be explained by the U-shaped association between dopamine
levels and cognitive functions. Bradykinesia has been described as the best
measure of the nigrostriatal lesion [22] and is related to the dorsal striatum
that is severely dopamine depleted in the early stages of the disease. There is
a high density of D3 auto receptors in the ventral striatum that is less
dopamine depleted early on in the disease. Therefore the U-shaped
association for patients receiving DA perhaps indicates that pramipexole is
more likely to impair working memory by overdosing than other types of
medication.
These findings have to be interpreted with caution. A lot of comparisons
were made and the findings could because of this be spurious. Still the
finding could be indicative of a possible influence of DA on certain aspects of
cognition that needs to be further investigated. In line with our result Brusa
et al. showed in 20 right handed patients with PD that pramipexole
produced significant impairment of short term verbal memory, attentional
executive functions and verbal fluency [90]. None of the previous studies
that have investigated the more prolonged effect of DA (6-24 months) has
included patients that received pramipexole as monotherapy. Patients
receiving pramipexole differed from the other patients in age and baseline
performance which could have influenced the results. This was because the
decision on the type of dopaminergic medication used was made by the
treating physician based on personal experience and the common practice of
using dopamine agonist in younger onset patients (before 65-70 years of
age).

48
Ethics
Studying elderly clinical populations in which a large proportion of the
patients eventually will develop dementia renders certain demands of ethical
considerations. Both cognitively impaired and borderline cognitively
impaired patients with PD have impairments in their decisional capacity
measured by the MacArthur Competence Assessment Tool for Clinical
Research [149]. This might be especially problematic when enrolling patients
with MCI whose reduced decisional capacity might be less obvious. It is also
important to note that many of the patients will change in their decisional
capacity during the course of the study. When enrolling patients in a
prospective study with repeated investigations it can be hard for the patient
to grasp how the extensive assessments will interfere with their daily life at
present, but even more so in the future, especially for a patient that is not
functioning at an optimal cognitive level.
Since the NYPUM study is implemented at a hospital there is close contact
between clinic and science. A clinical study where many health care persons
are involved can make the participants feel pressure to give consent and
continue in the research project. It is important to make clear that the
decision whether to remain in the study or not should under no
circumstances influence the future care of the patient.
Studies have shown that incidental findings of significant clinical importance
in fMRI research are around one-two % [150]. There is probably an even
higher proportion in elderly populations. This highlights the importance of
having a plan for taking care of possible findings and for bringing back the
results to the patient. One example of an ethical challenge could be whether
the participant should be told that they are a part of a risk group.
Methodological considerations
Study design
The main strength of the study is the population-based approach with a close
to complete identification of cases (making generalization of results possible)
and an extensive assessment of neuropsychological functions within a few
weeks of diagnosis. This allows testing of almost all patients prior to the
introduction of pharmacological treatment with dopaminergic agents. The
neuropsychological evaluation is repeated after one, three and five years and
can provide knowledge of the prognosis of cognitive function in PD. It also
makes it possible to identify factors at diagnosis that are predictive of
different outcomes and thereby can imply causality. This is in contrast to

49
cross-sectional investigations that only can confirm associations. One
limitation is the lack of histopathologically confirmed diagnosis.
Loss to follow up and missing values
A methodological problem seen in longitudinal studies of the elderly is a
cohort bias, i.e. selective loss to follow-up. There are many drop-outs, people
are old and die, they have a chronic progressive disease that affects them to
such a degree that they are unable to participate at follow-up. In the present
study already at the baseline testing, the dropouts differed from those
participating in the neuropsychological investigation. This is a problem when
describing the true progression for the entire population with PD. The
picture provided here is probably a little bit too optimistic. In the group that
developed dementia there were only five patients performing the five year
neuropsychological follow up. Therefore describing the longitudinal change
in the cognitive measures would be misguiding. Fortunately there were more
evaluations of these participants and most of them remained in the study
allowing evaluation of cognitive function/dementia even when they did not
perform the extensive neuropsychological test battery. This particular loss to
follow-up is hard to avoid but one way of minimizing the skewed loss to
follow-up is to perform home visits for the patients who are too tired to come
to the hospital.
There is also loss of individual measures due to tiredness and technical
issues. WCST was the test with most missing values which could be one
explanation why the result of WCST did not differ between the groups. We
did not omit patients who had not performed isolated tests. Since the
missing values were rather few for most variables, and distributed in
different cases, we used pairwise deletion of the data (except for a few cases).
The drawback with this method is that the parameters of the model will be
based on different set of data. The positive side is that we do not lose as
much power as we would have if all cases would have been omitted.
Statistics
A lot of variables and comparisons were used in the studies which increases
the risk for type I errors. We did not want to exclude possible true
relationships by adjusting for alpha error inflation by applying the strict
Bonferroni adjustment [151]. The risk for Dementia in PD seems to be
exponential, i.e. there is an exponential increase of new dementia cases with
age. This is problematic when using the Cox proportional Hazard model.

50
Diagnosis of PD
The number of patients with PD at baseline is different in all the four papers.
This is due to differences in the number of patients that had been included at
the time of the particular paper and to changes in diagnosis in some patients
as the disease progressed. Since the diagnosis of PD is more accurate as time
progresses and diagnostic criteria that are based on what happens
prospectively, e.g. the response to dopaminergic treatment, the diagnosis is
destined to change in some individuals.
UPDRS
The total score of UPDRS III is a validated and reliable tool for measuring
the progression of motor dysfunction in PD. The division into the different
motor components (e.g. bradykinesia, rigidity etc.), even though used in
several studies [106], is not validated as used in the thesis. UPDRS is a
clinical scale that relies solely on the investigators judgment. An alternative
to the clinical measures could be to use quantitative measures of the
different motor functions which could also enable description of other
aspects of motor functions. This could give a more precise picture into what
part of movement that is correlated with different cognitive functions.
SWEDDs
In patients clinically diagnosed with PD four-15% have normal imaging of
dopaminergic function (Scans Without Evidence of Dopaminergic Deficit
[SWEDDs]) [152]. SWEDDs patients are not likely to progress in severity or
develop motor and non-motor complications and it is highly likely that they
do not have PD at all [153]. We have treated the SWEDDs differently in the
different papers. In Paper I SWEDDs were included in the analysis. In Paper
II-IV the SWEDDs were excluded. The reason for this is that we did not have
as many prospective scans in Paper I and therefore we could not tell if the
normal datscan was an expression of early disease or if they were actually
true SWEDDs. Patients in Papers II –IV had been repeatedly scanned for
three to five years when the studies were assembled and therefore the
SWEDD classification was considered more stable.
MCI and Dementia
We tried to find variables that differed between MCI that developed
dementia and those who did not as a way of dissociating patients developing
early dementia from the rest of the group at diagnosis. The question that we
hoped to answer was how patients with MCI that converted to dementia in

51
the early phase differed from those who did not. It is likely that additional
patients from the MCI group will develop dementia therefore the specificity
and sensitivity for MCI is only for the early phase of PD, i.e. up to five years
after diagnosis.
Motor fluctuations
The occurrence of dyskinesias and motor fluctuations is common, and in the
long term management most patients will be affected by those treatment-
related side effects [154]. There is some evidence that cognitive performance
together with arousal state might be affected during motor fluctuations in
PD [155]. This is important when studying patients with PD for a longer
period of time. In the present study the ambition was to avoid testing if
substantial dyskinesias or other wearing-of symptoms were present. In the
cases where this was impossible or if the testing for some reason was
performed anyway, this was noted in the test protocol. Since the study
population was in the early phase of PD, dyskinesias and motor fluctuations
were minor problems.
Neuropsychological testing
Different neuropsychological tests aim to capture certain aspects of cognition
and manage to do so to a certain degree. However, it is complicated to apply
a reductionist view when looking at cognition since different cognitive
functions does not operate separately from each other. This is important to
have in mind when using standardized neuropsychological tests. It is obvious
in the PD literature that researchers are using different cognitive tests to
measure the same function. There are also examples were different studies
claim that a certain task measures different abilities. This is one contributing
factor to the various results between studies.
Some of the tests used in the studies require motor function such as both
TMT A and B. TMT B but not TMT A is correlated with bradykinesia which
indicates that it is likely that it is mental flexibility that contributes to the
association. BVMT is a test that requires drawing which can indeed be
influenced by especially tremor. According to the instructions for the test, for
example shaky handwriting should be overlooked in the judgment of
correctness of the figures.
The aim for the present study was to assemble a test battery that captured
different functional levels and could be performed even when functions start
to deteriorate. This is important to be able to measure change and at the
same time avoid ceiling as well as floor effects. It is also important for

52
minimizing the loss of study patients over time. If tests get too complicated
when functions deteriorate it can be hard to assess patients with severe
cognitive decline.
Control group
The small control group is a weakness of the study. The control group was
assessed with the UPDRS III, but associations between motor and cognitive
scores could not be calculated in the control group, as the controls had scores
of zero, at baseline and later. Thus it was not possible to see if the pattern of
association differed between the control group and the PD group. It has been
discussed whether to use norm values based on a control group or normative
values from other cohorts. Well-developed normative values taken from big
cohorts strictly following the standardization of the tests in similar age
groups that are being studied provide adequate data for comparison with the
study group of interest. It may be difficult to find such norm values,
especially with the tests in an assessment battery taken from different
sources. The optimal solution would be a large sample of controls tested
under the same conditions as the clinical subjects under investigation.
Future implications
The papers in the thesis discuss cognitive dysfunction in early stages of PD
and its relation to motor function for baseline and follow-up data up to five
years after diagnosis. We have so far completed the eight-year follow-up for
patients recruited during the first two years. This will allow investigations of
baseline measures that predict early onset and late onset dementia. Later we
will also receive autopsy results that will confirm the clinical diagnosis of PD
and enable identification of cellular signatures associated with the
development of dementia.
With longer follow-up we can study patients maintaining cognitive function.
This will make it possible to investigate what distinguishes patients with
preserved cognitive function from the large proportion of patients that
decline.
Combining different motor and cognitive measures as predictors for
dementia might display subtypes that go beyond the scope of studying
cognition and motor functions separately. Studying patients with postural
and gait impairment together with impairment in visuospatial memory and
function might give a better prediction for future outcome. Furthermore,
investigating objective measures of movement might provide a more specific
pattern of deficit connected to specific cognitive functions.

53
Blood samples and cerebrospinal fluid (CSF) from patients and controls will
enable genotyping and metabolomic profiling which could assist in the
interpretation of the motor and cognitive associations discussed in the
present thesis as well as finding predictors for cognitive decline and
dementia.

54
Conclusions
The findings in this thesis confirm results from previous studies, namely that
a large proportion of patients are cognitively impaired already at the time of
diagnosis with a range of cognitive functions being affected. The present
study is unique in that it combines being truly community-based with
employing an extensive neuropsychological test battery with repeated
investigation. All patients within the study are referred to the same
neurological department. This resulted in almost a complete recruitment of
affected cases all in early disease stages.
The findings show a diverse picture of cognitive and motor functions with
both dopaminergic and non-dopaminergic motor and cognitive functions
associated with cognitive impairment and PDD. This indicates that we need
to look at both the striatal network in combination with a more widespread
cortical network in the search for mechanisms behind cognitive impairment
in PD. The associations found between motor and cognitive dysfunction
could reflect shared underlying pathology in motor and cognitive
disturbances.
The differences in proportions of cognitively impaired in Paper I and Paper
IV were mostly due to the use of different criteria. This shows that MCI
classification is largely determined by the tests included in diagnosis and the
cut-off values used. This emphasizes the value of joint criteria for PD-MCI,
both in research and in clinic. Even when using joint criteria there is a
substantial amount of patients that reverts back to normal cognitive function
at follow-up.
There could be several explanations for increased motor severity in patients
with PDD. Patients with more posture and gait difficulties could be more
advanced in their disease progression, i.e. dementia and postural and gait
difficulties may represent a more advanced disease stage. Alternative
explanations could be that patients with dementia for unknown reasons
respond less well to medication compared with patients without dementia
and therefore have worse and declining motor performance. The parallel
decline in motor and cognitive functions might also affect each other (e.g.
less physical activity due to motor dysfunction resulting in a more inactive
life which leads to accelerating cognitive decline which in turn results in
worsening of motor performance). To answer these questions further studies
that are specifically designed to answer these questions are needed.

55
Acknowledgements
There are several people I would like to thank that in different ways have
contributed to this thesis.
First of all, I would like to express my gratitude to my supervisors Lars
Forsgren and Eva Elgh. Lars for giving me the opportunity of being a
doctoral student within the NYPUM-project and for always replying fast to
my queries, small or large! Eva for recruiting me to the NYPUM-project and
for great support along the way!
I would also like to thank all participants of the NYPUM project that with
great effort endured the neuropsychological examination on top of all the
other investigations, year after year.
A big thank you goes out to those helping us collecting the
neuropsychological data: Theres Åberg, Urban Ekman, Sofia Nording,
Carina Berg, John Wallert Holmqvist, Ida Andersson, Anton Andersson
and Neda Dimova Bränström. You all did a tremendous job!
Apart from great help with the data collection I would like to give special
thanks to Sofia and Urban. Sofia for great help with the project while I was
on maternity leave and for being a wonderful friend! Urban for rewarding
discussions on MCI, dementia, manuscripts, and everything in between.
Being a doctoral student within the NYPUM project became more fun when
you joined the crew!
Also, I would like to thank everyone else involved in the NYPUM project. A
special thanks to Jörgen Andersson for extracting data from the NYPUM
database whenever I needed it and filling in for me when writing the thesis
did not allow me to fulfill my project duties, and Mona Edström for doing a
great job with coordinating the project.
Thank you all great “brains” at the UFBI for inspiring lab meetings, with a
special thanks to Mikael Anderson for being tremendously patient when
helping me with SPM (even though I unfortunately did not include fMRI-
data in my thesis)
I’m further grateful to my colleagues at the Department of Geriatrics, past
and present! Even though I might have come across as an odd bird in your
“fika”-room, you have all showed tremendous hospitality. On top of learning

56
a lot about geriatric research when attending your seminars, I’ve had
wonderful discussions and laughs in the “fika”-room.
Loads of love to all of my friends who with walks, long conversations on the
telephone, trips to the beach, yoga sessions, coffee, laughs and food have
been tremendous support throughout this process, you know who you are!
A loving thank you to my family: Rose Eriksson that apart from being my
wonderful mother also did the proofreading of this thesis. Nils Klingberg
and Johan Eriksson for your brotherly support. My father Sigvard Eriksson,
I wish you were still with us. Karin and Edith for simply being the joy of my
life! Last but not least I want to thank Erik for taking a tremendous
responsibility for family duties during the last couple of months and on top
of that being patient with all my worries! I truly love you!

57
References
1. World population aging, Economic and social affairs, UN [http://www.un.org/en/development/desa/population/theme/ageing/index.shtml]
2. Parkinson J. An Essay on the Shaking Palsy. London: Sherwood, Neely, and Jones; 1817:66.
3. Pollock M, Hornabrook RW. The prevalence, natural history and dementia of Parkinson’s disease. Brain 1966, 89:429–448.
4. Marttila RJ, Rinne UK. Dementia in Parkinson’s disease. Acta Neurol Scand 1976, 54:431–441.
5. Aarsland D, Beyer MK, Kurz MW. Dementia in Parkinson’s disease. Curr Opin Neurol 2008, 21:676–82.
6. Muslimovic D, Post B, Speelman JD, Schmand B. Cognitive profile of patients with newly diagnosed Parkinson disease. Neurology 2005, 65:1239–45.
7. Aarsland D, Brønnick K, Larsen JP, Tysnes OB, Alves G. Cognitive impairment in incident, untreated Parkinson disease: the Norwegian ParkWest study. Neurology 2009, 72:1121–6.
8. Foltynie T, Brayne CEG, Robbins TW, Barker RA. The cognitive ability of an incident cohort of Parkinson’s patients in the UK. The CamPaIGN study. Brain 2004, 127:550–60.
9. Fahn S, Elton R. Recent Development in Parkinson’s Disease. New York: Mcmillan Health Care Information; 1987:153–164.
10. Hughes AJ, Ben-shlomo Y, Daniel SE, Lees AJ. What features improve the accuracy of clinical diagnosis in Parkinson’s disease: A clinicopathologic study. Neurology 1992, 57:34–38.
11. Mo SJ, Linder J, Forsgren L, Larsson A, Johansson L, Riklund K. Pre- and postsynaptic dopamine SPECT in the early phase of idiopathic parkinsonism: a population-based study. Eur J Nucl Med Mol Imaging 2010, 37:2154–64.
12. Fox SH, Lang AE. Levodopa-related motor complications--phenomenology. Mov Disord 2008, 23:509–14.

58
13. Tolosa E, Gaig C, Santamaría J, Compta Y. Diagnosis and the premotor phase of Parkinson disease. Neurology 2009, 72:12–20.
14. De Rijk MC, Launer LJ, Berger K, Breteler MM, Dartigues JF, Baldereschi M, et al. Prevalence of Parkinson’s disease in Europe: A collaborative study of population-based cohorts. Neurology 2000, 54:21–23.
15. Linder J, Stenlund H, Forsgren L. Incidence of Parkinson’s disease and parkinsonism in northern Sweden: a population-based study. Mov Disord 2010, 25:341–8.
16. Van Den Eeden SK. Incidence of Parkinson’s Disease: Variation by Age, Gender, and Race/Ethnicity. Am J Epidemiol 2003, 157:1015–1022.
17. Caslake R, Taylor K, Scott N, Gordon J, Harris C, Wilde K, et al. Age-, gender-, and socioeconomic status-specific incidence of Parkinson’s disease and parkinsonism in North East Scotland: The PINE study. Parkinsonism Relat Disord 2013, 19:515–521.
18. De Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol 2006, 5:525–535.
19. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302:841.
20. Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 2004, 318:121–34.
21. Braak H, Bohl JR, Müller CM, Rüb U, de Vos RAI, Del Tredici K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord 2006, 21:2042–51.
22. Vingerhoets FJ, Schulzer M, Calne DB, Snow BJ. Which clinical sign of Parkinson’s disease best reflects the nigrostriatal lesion? Ann Neurol 1997, 41:58–64.
23. Helmich RC, Hallett M, Deuschl G, Toni I, Bloem BR. Cerebral causes and consequences of parkinsonian resting tremor: a tale of two circuits? Brain 2012, 135:3206–3226.

59
24. Doder M, Rabiner EA, Turjanski N, Lees AJ, Brooks DJ. Tremor in Parkinson’s disease and serotonergic dysfunction: An 11C-WAY 100635 PET study. Neurology 2003, 60:601–605.
25. Bohnen NI, Müller MLTM, Koeppe R a, Studenski S a, Kilbourn M a, Frey K a, et al. History of falls in Parkinson disease is associated with reduced cholinergic activity. Neurology 2009, 73:1670–6.
26. Yarnall A, Rochester L, Burn DJ. The interplay of cholinergic function, attention, and falls in Parkinson’s disease. Mov Disord 2011, 26:2496–503.
27. Müller MLTM, Frey KA, Petrou M, Kotagal V, Koeppe RA, Albin RL, et al. β-Amyloid and postural instability and gait difficulty in Parkinson’s disease at risk for dementia. Mov Disord 2013, 28:296–301.
28. Carlsson A. The occurence, distribution and physiological role of catecholamines in the nervous system. Pharmacol Rev 1959:490–493.
29. Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behaviour in diseases of the extrapyramidal system. Klin Wochenschr 1960:1236–1239.
30. Moustafa AA, Herzallah MM, Gluck MA. Dissociating the Cognitive Effects of Levodopa versus Dopamine Agonists in a Neurocomputational Model of Learning in Parkinson’s Disease. Neurodegener Diseas 2013, 11:102–11.
31. Lang AE. When and how should treatment be started in Parkinson disease? Neurology 2009, 72:39–43.
32. Williams-Gray CH, Evans JR, Goris A, Foltynie T, Ban M, Robbins TW, et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain 2009, 132:2958–69.
33. Taylor AE, Saint-Cyr JA, Lang AE. Frontal lobe dysfunction in Parkinson’s disease. Brain 1986, 109:845–883.
34. Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 2007, 22:1689–707.

60
35. Baddeley A, Hitch G. Working Memory. In Psychol Learn Motiv. Edited by Bower G. New York: Academic Press; 1974:47–89.
36. Bäckman L, Nyberg L, Lindenberger U, Li S-C, Farde L. The correlative triad among aging, dopamine, and cognition: current status and future prospects. Neurosci Biobehav Rev 2006, 30:791–807.
37. Bäckman L, Nyberg L, Soveri A, Johansson J, Andersson M, Dahlin E, et al. Effects of working-memory training on striatal dopamine release. Science 2011, 333:718.
38. Squire LR, Stark CEL, Clark RE. The medial temporal lobe. Annu Rev Neurosci 2004, 27:279–306.
39. Squire LR. Memory and the hippocampus: A synthesis from findings with rats, monkeys, and humans. Psychol Rev 1992, 99:195–231.
40. Fletcher PC. Frontal lobes and human memory: Insights from functional neuroimaging. Brain 2001, 124:849–881.
41. Aarsland D. Performance on the dementia rating scale in Parkinson’s disease with dementia and dementia with Lewy bodies: comparison with progressive supranuclear palsy and Alzheimer's disease. J Neurol Neurosurg Psychiatry 2003, 74:1215–1220.
42. Kravitz DJ, Saleem KS, Baker CI, Mishkin M. A new neural framework for visuospatial processing. Nat Rev Neurosci 2011, 12:217–30.
43. Petersen RC, Smith GE, Waring SC, Ivnik RJ, Kokmen E, Tangelos EG. Aging, memory, and mild cognitive impairment. Int psychogeriatrics IPA 1997, 9:65–69.
44. Winblad B, Palmer K, Kivipelto M, Jelic V, Fratiglioni L, Wahlund L-O, et al. Mild cognitive impairment--beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med 2004, 256:240–6.
45. Litvan I, Goldman JG, Tröster AI, Schmand BA, Weintraub D, Petersen RC, et al. Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Mov Disord 2012, 27:349–56.

61
46. Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement 2013, 9:63–75.
47. Aarsland D, Zaccai J, Brayne C. A systematic review of prevalence studies of dementia in Parkinson’s disease. Mov Disord 2005, 20:1255–63.
48. Janvin CC, Aarsland D, Larsen JP. Cognitive predictors of dementia in Parkinson’s disease: a community-based, 4-year longitudinal study. J Geriatr Psych Neur 2005, 18:149–54.
49. Aarsland D, Andersen K, Larsen JP, Lolk A, Nielsen H, Kragh-Sørensen P. Risk of dementia in Parkinson’s disease: a community-based, prospective study. Neurology 2001, 56:730–6.
50. Marder K, Tang MX, Cote L, Stern Y, Mayeux R. The frequency and associated risk factors for dementia in patients with Parkinson’s disease. Arch Neurol 1995, 52:695–701.
51. Hobson P, Gallacher J, Meara J. Cross-sectional survey of Parkinson’s disease and parkinsonism in a rural area of the United Kingdom. Mov Disord 2005, 20:995–8.
52. Dalrymple-Alford JC, Livingston L, MacAskill MR, Graham C, Melzer TR, Porter RJ, et al. Characterizing mild cognitive impairment in Parkinson’s disease. Mov Disord 2011, 26:629–636.
53. Hoops S, Nazem S, Siderowf AD, Duda JE, Xie SX, Stern MB, et al. Validity of the MoCA and MMSE in the detection of MCI and dementia in Parkinson disease. Neurology 2009, 73:1738–1745.
54. Gauthier S, Reisberg B, Zaudig M, Petersen RC, Ritchie K, Broich K, et al. Mild cognitive impairment. Lancet 2006, 367:1262–1270.
55. Reid WG, Hely MA, Morris JG, Broe GA, Adena M, Sullivan DJ, et al. A longitudinal of Parkinson’s disease: clinical and neuropsychological correlates of dementia. J Clin Neurosci 1996, 3:327–33.
56. Hely MA, Morris JG, Rail D, Reid WG, O’Sullivan DJ, Williamson PM, et al. The Sydney Multicentre Study of Parkinson’s disease: a report on the first 3 years. J Neurol Neurosurg Psychiatry 1989, 52:324–328.

62
57. Broeders M, de Bie RMA, Velseboer DC, Speelman JD, Muslimovic D, Schmand B. Evolution of mild cognitive impairment in Parkinson disease. Neurology 2013, 81:346–52.
58. Pedersen KF, Larsen JP, Tysnes O-B, Alves G. Prognosis of mild cognitive impairment in early Parkinson disease: the Norwegian ParkWest study. JAMA Neurol 2013, 70:580–6.
59. Irwin DJ, Lee VM-Y, Trojanowski JQ. Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci 2013, 14:626–36.
60. Williams-Gray CH, Foltynie T, Brayne CEG, Robbins TW, Barker R a. Evolution of cognitive dysfunction in an incident Parkinson’s disease cohort. Brain 2007, 130:1787–98.
61. Kempster PA, O’Sullivan SS, Holton JL, Revesz T, Lees AJ. Relationships between age and late progression of Parkinson’s disease: a clinico-pathological study. Brain 2010, 133:1755–62.
62. Levy G, Schupf N, Tang M-X, Cote LJ, Louis ED, Mejia H, et al. Combined effect of age and severity on the risk of dementia in Parkinson’s disease. Ann Neurol 2002, 51:722–9.
63. Woods SP, Tröster AI. Prodromal frontal/executive dysfunction predicts incident dementia in Parkinson’s disease. JINS 2003, 9:17–24.
64. Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurol 2010, 9:1200–13.
65. Burn DJ, Rowan EN, Allan LM, Molloy S, O’Brien JT, McKeith IG. Motor subtype and cognitive decline in Parkinson’s disease, Parkinson's disease with dementia, and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2006, 77:585–9.
66. Williams-Gray CH, Mason SL, Evans JR, Foltynie T, Brayne C, Robbins TW, et al. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J Neurol Neurosurg Psychiatry 2013, 84:1258–64.
67. Alves G, Larsen JP, Emre M, Wentzel-Larsen T, Aarsland D. Changes in motor subtype and risk for incident dementia in Parkinson’s disease. Mov Disord 2006, 21:1123–30.

63
68. Svenningsson P, Westman E, Ballard C, Aarsland D. Cognitive impairment in patients with Parkinson’s disease: diagnosis, biomarkers, and treatment. Lancet Neurol 2012, 11:697–707.
69. Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, Van Deerlin V, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 2012, 72:587–98.
70. Kövari E, Gold G, Herrmann FR, Canuto A, Hof PR, Bouras C, et al. Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol 2003, 106:83–8.
71. Mattila PM, Rinne JO, Helenius H, Dickson DW, Röyttä M. Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol 2000, 100:285–290.
72. Braak H, Rüb U, Jansen Steur ENH, Del Tredici K, de Vos RAI. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 2005, 64:1404–10.
73. Apaydin H, Ahlskog JE, Parisi JE, Boeve BF, Dickson DW. Parkinson Disease NeuropathologyLater-Developing Dementia and Loss of the Levodopa Response. Arch Neurol 2002, 59:102–112.
74. Hurtig HI, Trojanowski JQ, Galvin J, Ewbank D, Schmidt ML, Lee VM-Y, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 2000, 54:1916–1921.
75. Galvin JE, Pollack J, Morris JC. Clinical phenotype of Parkinson disease dementia. Neurology 2006, 67:1605–11.
76. Compta Y, Pereira JB, Ríos J, Ibarretxe-Bilbao N, Junqué C, Bargalló N, et al. Combined dementia-risk biomarkers in Parkinson’s disease: A prospective longitudinal study. Parkinsonism Relat Disord 2013, 19:717–724.
77. Jellinger KA, Attems J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol 2008, 115:427–36.
78. Kurz MW, Schlitter AM, Larsen JP, Ballard C, Aarsland D. Familial occurrence of dementia and parkinsonism: a systematic review. Dement Geriatr Cogn Disord 2006, 22:288–95.

64
79. Jones EL, Aarsland D, Londos E, Ballard C. A pilot study examining associations between DYRK1A and α-synuclein dementias. Neurodegener Dis 2012, 10:229–31.
80. Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 2006, 38:1184–91.
81. Setó-Salvia N, Clarimón J, Pagonabarraga J, Pascual-Sedano B, Campolongo A, Combarros O, et al. Dementia risk in Parkinson disease: disentangling the role of MAPT haplotypes. Arch Neurol 2011, 68:359–64.
82. Rolinski M, Fox C, Maidment I, McShane R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson's disease. Cochrane database Syst Rev 2012, 3:CD006504.
83. Seppi K, Weintraub D, Coelho M, Perez-Lloret S, Fox SH, Katzenschlager R, et al. The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the non-motor symptoms of Parkinson’s disease. Mov Disord 2011, 26:S42–80.
84. Burn D, Emre M, McKeith I, De Deyn PP, Aarsland D, Hsu C, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord 2006, 21:1899–907.
85. Barone P, Burn DJ, van Laar T, Hsu C, Poewe W, Lane RM. Rivastigmine versus placebo in hyperhomocysteinemic Parkinson’s disease dementia patients. Mov Disord 2008, 23:1532–40.
86. Zahodne LB, Fernandez HH. Pathophysiology and Treatment of Psychosis in Parkinson’s Disease. Drugs Aging 2008, 25:665–682.
87. Pollak P. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry 2004, 75:689–695.
88. Liu J, Dong J, Wang L, Su Y, Yan P, Sun S. Comparative Efficacy and Acceptability of Antidepressants in Parkinson’s Disease: A Network Meta-Analysis. PLoS One 2013, 8:e76651.
89. Cools R, D’Esposito M. Inverted-U-shaped dopamine actions on human working memory and cognitive control. Biol Psychiatry 2011, 69:113–25.

65
90. Brusa L, Bassi A, Stefani A, Pierantozzi M, Peppe A, Caramia MD, et al. Pramipexole in comparison to l-dopa: a neuropsychological study. J Neural Transm 2003, 110:373–80.
91. Parsons TD, Rogers SA, Braaten AJ, Woods SP, Tröster AI. Cognitive sequelae of subthalamic nucleus deep brain stimulation in Parkinson’s disease: a meta-analysis. Lancet Neurol 2006, 5:578–588.
92. Diamond A. Close Interrelation of Motor Development and Cognitive Development and of the Cerebellum and Prefrontal Cortex. Child Dev 2000, 71:44–56.
93. Sosnoff JJ, Broglio SP, Ferrara MS. Cognitive and motor function are associated following mild traumatic brain injury. Exp Brain Res 2008, 187:563–71.
94. Amboni M, Barone P, Iuppariello L, Lista I, Tranfaglia R, Fasano A, et al. Gait patterns in parkinsonian patients with or without mild cognitive impairment. Mov Disord 2012, 27:1536–43.
95. Georgopoulos AP. Neural aspects of cognitive motor control. Curr Opin Neurobiol 2000, 10:238–241.
96. Burn DJ, Rowan EN, Allan LM, Molloy S, O’Brien JT, McKeith IG. Motor subtype and cognitive decline in Parkinson’s disease, Parkinson's disease with dementia, and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2006, 77:585–9.
97. Mortimer JA, Pirozzolo FJ, Hansch EC, Webster DD. Relationship of motor symptoms to intellectual deficits in Parkinson disease. Neurology 1982, 32:133–133.
98. Cooper JA, Sagar HJ, Jordan N, Harvey NS, Sullivan E V. Cognitive impairment in early, untreated parkinson’s disease and its relationship to motor disability. Brain 1991, 114:2095–2122.
99. Hietanen M, Teräväinen H. Cognitive performance in early Parkinson’s disease. Acta Neurol Scand 2009, 73:151–159.
100. Aarsland D, Larsen JP, Tandberg E, Laake K. Predictors of nursing home placement in Parkinson’s disease: A population-based, prospective study. .

66
101. Folstein M, Folstein S, Mchugh P. Mini-mental state- practical method for grading cognitive state of patients for clinician. J Psychiatr Res 1975, 12:189–198.
102. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Brit J Psychiatry 1979, 134:382–389.
103. Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov Disord Off J Mov Disord Soc 2010, 25:2649–2653.
104. Louis ED Cote L, Alfaro B, Mejia H, Marder K TMX. PRogression of parkinsonian signs in parkinson disease. Arch Neurol 1999, 56:334–337.
105. Uc EY, McDermott MP, Marder KS, Anderson SW, Litvan I, Como PG, et al. Incidence of and risk factors for cognitive impairment in an early Parkinson disease clinical trial cohort. Neurology 2009, 73:1469–1477.
106. Jankovic J, McDermott M, Carter J, Gauthier S, Goetz C, Golbe L, et al. Variable expression of Parkinson’s disease: a base-line analysis of the DATATOP cohort. The Parkinson Study Group. Neurology 1990, 40:1529–34.
107. Benedict RH, Zgaljardic DJ. Practice Effects During Repeated Administrations of Memory Tests with and Without Alternate Forms. Volume 20. Psychology Press; 1998:339–352.
108. Buschke H. Selective reminding for analysis of memory and learning. J Verb Learn Verb Behav 1973, 12:543–550.
109. Wechsler D. Wechsler Adult Intelligence Scale, 3rd edition(WAIS III): Test Manual, 3rd Edn. Psychology. New York; 1997.
110. Reitan RM. Validity of the Trail Making Test as an indicator of organic brain damage. Percept Mot Ski 1958, 8:271–276.
111. Benton AL. Contributions to Neuropsychological Assessment: A Clinical Manual. Oxford University Press; 1994:159.
112. Benedict RHB, Schretlen D, Groninger L, Dobraski M, Shpritz B. Revision of the Brief Visuospatial Memory Test: Studies of Normal Performance, Reliability and Validity. Psychol Assess 1996, 8:145–153.

67
113. Spreen O, Strauss E. A Compendium of Neuropsychological Tests: Administration , Norms , and Commentary , Third Edition. Volume 2nd. Oxford University Press; 1991:1240.
114. Gilman S, Low P., Quinn N, Albanese A, Ben-Shlomo Y, Fowler C., et al. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci 1999, 163:94–98.
115. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): Report of the NINDS-SPSP International Workshop. Neurology 1996, 47:1–9.
116. Litvan I, Bhatia KP, Burn DJ, Goetz CG, Lang AE, McKeith I, et al. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003, 18:467–86.
117. McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005, 65:1863–72.
118. Lezak M, Howiesson D, Loring D. Neuropsychological Assessment. New York: Oxford University Press; 2004:158–160.
119. Gibb W, Lees A. The relevence of the Lewy body to the pathogenesis of ideopathic Parkinson’s disease. J Neurosurg psychiatry 1988, 51:745–752.
120. Peto V, Jenkinson C, Fitzpatrick R, Greenhall R. The development and validation of a short measure of functioning and well being for individuals with Parkinson’s disease. Qual Life Res 1995, 4:241–248.
121. Hardin JW, Hilbe JM. Generalized Estimating Equations. CRC Press; 2013:261.
122. Janvin CC, Larsen JP, Aarsland D, Hugdahl K. Subtypes of mild cognitive impairment in Parkinson’s disease: progression to dementia. Mov Disord Off J Mov Disord Soc 2006, 21:1343–9.
123. Levy G, Jacobs DM, Tang M-X, Côté LJ, Louis ED, Alfaro B, et al. Memory and executive function impairment predict dementia in Parkinson’s disease. Mov Disord 2002, 17:1221–6.

68
124. Williams-Gray CH, Mason SL, Evans JR, Foltynie T, Brayne C, Robbins TW, et al. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J Neurol Neurosurg Psychiatry 2013, 84:1258–64.
125. Lee JE, Cho KH, Song SK, Kim HJ, Lee HS, Sohn YH, et al. Exploratory analysis of neuropsychological and neuroanatomical correlates of progressive mild cognitive impairment in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2013:jnnp–2013–305062–.
126. Caviness JN, Driver-Dunckley E, Connor DJ, Sabbagh MN, Hentz JG, Noble B, et al. Defining mild cognitive impairment in Parkinson’s disease. Mov Disord 2007, 22:1272–1277.
127. Marras C, Armstrong MJ, Meaney CA, Fox S, Rothberg B, Reginold W, et al. Measuring mild cognitive impairment in patients with Parkinson’s disease. Mov Disord 2013.
128. Aarsland D, Kurz MW. The epidemiology of dementia associated with Parkinson’s disease. Brain Pathol 2010, 20:633–9.
129. Broeders M, Velseboer DC, De Bie R, Speelman JD, Muslimovic D, Post B, et al. Cognitive Change in Newly-Diagnosed Patients with Parkinson’s Disease: A 5-Year Follow-up Study. J Int Neuropsychol Soc JINS 2013:1–14.
130. Cools R. Dopaminergic modulation of cognitive function-implications for L-DOPA treatment in Parkinson’s disease. Neurosci Biobehav R 2006, 30:1–23.
131. Boyle PA, Wilson RS, Aggarwal NT, Tang Y, Bennett DA. Mild cognitive impairment: risk of Alzheimer disease and rate of cognitive decline. Neurology 2006, 67:441–5.
132. Manly JJ, Tang M-X, Schupf N, Stern Y, Vonsattel J-PG, Mayeux R. Frequency and course of mild cognitive impairment in a multiethnic community. Ann Neurol 2008, 63:494–506.
133. Larrieu S, Letenneur L, Orgogozo JM, Fabrigoule C, Amieva H, Le Carret N, et al. Incidence and outcome of mild cognitive impairment in a population-based prospective cohort. Neurology 2002, 59:1594–1599.
134. Kryscio RJ, Schmitt FA, Salazar JC, Mendiondo MS, Markesbery WR. Risk factors for transitions from normal to mild cognitive impairment and dementia. Neurology 2006, 66:828–32.

69
135. Cobb JL, Wolf PA, Au R, White R, D’Agostino RB. The effect of education on the incidence of dementia and Alzheimer’s disease in the Framingham Study. Neurology 1995, 45:1707–1712.
136. Aarsland D, Kvaløy JT, Andersen K, Larsen JP, Tang MX, Lolk A, et al. The effect of age of onset of PD on risk of dementia. J Neurol 2007, 254:38–45.
137. Haaxma CA, Bloem BR, Borm GF, Oyen WJG, Leenders KL, Eshuis S, et al. Gender differences in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2007, 78:819–24.
138. Mayeux R, Stern Y. Intellectual dysfunction and dementia in Parkinson disease. Adv Neurol 1983, 38:211–27.
139. Riggeal BD, Crucian GP, Seignourel P, Jacobson CE, Okun MS, Rodriguez R l, et al. Cognitive decline tracks motor progression and not disease duration in Parkinson patients. Neuropsychiatr Dis Treat 2007, 3:955–8.
140. Ekman U, Eriksson J, Forsgren L, Mo SJ, Riklund K, Nyberg L. Functional brain activity and presynaptic dopamine uptake in patients with Parkinson’s disease and mild cognitive impairment: a cross-sectional study. Lancet Neurol 2012, 11:679–687.
141. Montero-Odasso M, Verghese J, Beauchet O, Hausdorff JM. Gait and cognition: a complementary approach to understanding brain function and the risk of falling. J Am Geriatr Soc 2012, 60:2127–36.
142. Van Iersel MB, Hoefsloot W, Munneke M, Bloem BR, Olde Rikkert MGM. Systematic review of quantitative clinical gait analysis in patients with dementia. Z Gerontol Geriatr 2004, 37:27–32.
143. Post B, Merkus MP, de Haan RJ, Speelman JD. Prognostic factors for the progression of Parkinson’s disease: a systematic review. Mov Disord 2007, 22:1839–51.
144. Verbaan D, Marinus J, Visser M, van Rooden SM, Stiggelbout a M, Middelkoop H a M, et al. Cognitive impairment in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2007, 78:1182–7.
145. Amboni M, Barone P, Hausdorff JM. Cognitive contributions to gait and falls: Evidence and implications. Mov Disord 2013, 28:1520–33.

70
146. Devos D, Defebvre L, Bordet R. Dopaminergic and non-dopaminergic pharmacological hypotheses for gait disorders in Parkinson’s disease. Fundam Clin Pharmacol 2010, 24:407–21.
147. Müller MLTM, Bohnen NI. Cholinergic dysfunction in Parkinson’s disease. Curr Neurol Neurosci Rep 2013, 13:377.
148. Beaulieu J-M, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 2011, 63:182–217.
149. Karlawish J, Cary M, Moelter ST, Siderowf A, Sullo E, Xie S, et al. Cognitive impairment and PD patients’ capacity to consent to research. Neurology 2013, 81:801–7.
150. Mamourian A. Incidental Findings on Research Functional MR Images: Should We Look? AJNR Am J Neuroradiol 2004, 25:520–522.
151. Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology 1990, 1:43–46.
152. Marek K, Jennings D, Seibyl J. Dopamine agonists and Parkinson’s disease progression: what can we learn from neuroimaging studies. Ann Neurol 2003, 53:160–166.
153. Schwingenschuh P, Ruge D, Edwards MJ, Terranova C, Katschnig P, Carrillo F, et al. Distinguishing SWEDDs patients with asymmetric resting tremor from Parkinson’s disease: a clinical and electrophysiological study. Mov Disord 2010, 25:560–9.
154. Marsden CD, Parkes JD. Success and problems of long-term levodopa therapy in Parkinson’s disease. Lancet 1977, 309:345–349.
155. Brown RG, Marsden CD, Quinn N, Wyke MA. Alterations in cognitive performance and affect-arousal state during fluctuations in motor function in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1984, 47:454–465.

1


















